

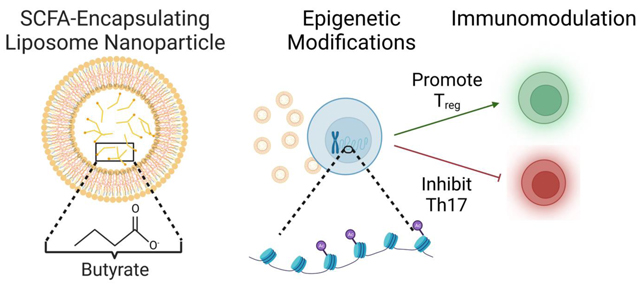
Abstract
Short chain fatty acids (SCFAs) are major metabolic products of indigestible polysaccharides in the gut and mediate the function of immune cells to facilitate homeostasis. The immunomodulatory effect of SCFAs has been attributed, at least in part, to the epigenetic modulation of immune cells through the inhibition the nucleus-resident enzyme histone deacetylase (HDAC). Among the downstream effects, SCFAs enhance regulatory T cells (Treg) over inflammatory T helper (Th) cells, including Th17 cells, which can be pathogenic. Here, we characterize the potential of two common SCFAs – butyrate and pentanoate – in modulating differentiation of T cells in vitro. We show that butyrate but not pentanoate exerts a concentration dependent effect on Treg and Th17 differentiation. Increasing the concentration of butyrate suppresses the Th17-associated RORγt and IL-17 and increases the expression of Treg-associated FoxP3. To effectively deliver butyrate, encapsulation of butyrate in a liposomal carrier, termed BLIPs, reduced cytotoxicity while maintaining the immunomodulatory effect on T cells. Consistent with these results, butyrate and BLIPs inhibit HDAC and promote a unique chromatin landscape in T cells under conditions that otherwise promote conversion into a pro-inflammatory phenotype. Motif enrichment analysis revealed that butyrate and BLIP mediated suppression of Th17 associated chromatin accessibility corresponded with a marked decrease in bZIP family transcription factor binding sites. These results support the utility and further evaluation of BLIPs as an immunomodulatory agent for autoimmune disorders that are characterized by chronic inflammation and pathogenic inflammatory T cells.
Keywords: short chain fatty acids, epigenetic modulation, T cells, immunomodulation
GRAPHICAL ABSTRACT
INTRODUCTION
T cells are an important immune cell subset that are normally protective, prevent disease and destroy pathogens. However, T cells can contribute to autoimmune disease by causing or exacerbating inflammation1,2. While the disease etiology can be complex, an imbalance between effector and regulatory T cell function is a major contributor to pathogenic inflammation3,4. Among T cell subsets that have been associated with inflammatory disease are T helper (Th)-17 T cells expressing retinoic acid-related orphan receptor gamma t (RORγt)5. On the other hand, regulatory T cells (Treg), characterized by their expression of forkhead box P3 (FoxP3), are widely recognized as important inhibitors of autoimmunity6. However, Treg can be insufficiently effective in autoimmune disorders7.
Suppressing inflammation by inhibiting immune cell activation or neutralizing select pro-inflammatory cytokines can be effective. However, immunosuppression also increases the risk of infections and immunosurveillance disorders, including cancer8,9. Therefore, therapies that restore immunoregulation could augment the existing standard-of-care without increasing the associated iatrogenic risk10. Among the endogenous regulators of immune function, short chain fatty acids (SCFAs) are metabolic products of gut microbiota and important immunomodulators that promote Treg11–13. Butyrate, a 4 carbon SCFA, and pentanoate, a 5 carbon SCFA, are anti-inflammatory immunomodulators which are among the major SCFA products of anaerobic fermentation of polysaccharide-based fibers in the gut14–16. Recent studies have demonstrated that SCFAs modulate gene expression in immune cells, including T cells, by inhibition of nucleus-resident enzyme histone deacetylase (HDAC) and binding to G-protein coupled receptors (GPCR) associated with cells in the gut14,17. By inhibiting HDAC, butyrate and pentanoate increase acetylation of lysine residues on histone proteins, thereby loosening the chromatin structure and facilitating access of certain transcription factors18. Both HDAC inhibition and GPCR binding promote the differentiation of CD4+ T cells towards Treg over Th1719. Under homeostatic conditions, SCFAs can promote Treg and have the potential to potently enhance Treg while suppressing Th17. These characteristics support the use of SCFAs as potential therapies in inflammatory disorders. However, the oral administration of free SCFAs is limited by the need for frequent dosing and potential challenges with patient compliance. To this end, a formulation for enhancing SCFA delivery could facilitate control over the timing, location, and specificity of immunomodulation.
Seeking to harness the therapeutic potential of SCFAs for immunomodulation, we first assessed the potential of butyrate and pentanoate to enhance Treg using SKG mice, a model which recapitulates key features of human inflammatory arthritis arising from pathogenic imbalance between Treg and Th1720–22. We show that butyrate, but not pentanoate, has a concentration-dependent effect on enhancing Treg and suppressing Th17 cell differentiation and secretion of associated cytokines. Encapsulation of butyrate in a liposomal carrier, termed butyrate liposomes (BLIPs), permitted effective cellular uptake with minimal toxicity, while retaining Treg upregulation and inflammatory Th17 suppression compared to butyrate. ATACseq revealed that both butyrate and BLIPs modulated the accessibility of Th17 associated loci and diminished basic leucine zipper (bZIP) binding motifs relative to cells exposed to Th17-inducing conditions alone, confirmed epigenetic modulation as a potential mechanism of action. The results support the utility of butyrate as an immunomodulatory agent in the treatment of inflammatory disease.
METHODS AND MATERIALS
In vitro murine Th17 differentiation assay
Murine pan CD4+ T cells were isolated from 8–14 week old male or female SKG mice via magnetic depletion using the mouse CD4+ T cell Isolation kit (Miltenyi Biotech ref: 130–104-454) according to the manufacturer’s instructions. Isolated CD4+ T cells were plated in 96 well plates coated with anti-mouse anti-CD3 and anti-CD28 antibodies at 100,000 cells/well in RPMI 1640 (Gibco ref: 31800–022, lot: 2338416) supplemented with non-essential amino acids (Gibco ref: 11140–050, lot: 2188976), sodium pyruvate (Corning ref: 25–000-CI, lot: 08620003), and beta-mercaptoethanol (Gibco ref: 21985–023, lot: 2060762). To induce Th17 differentiation CD4+ T cells were stimulated with αCD3 and αCD28 (adsorbed at 5 μg/mL each in PBS at 37º C for 3 hours) and IL-6 (50 ng/mL) (ref: 200–06, lot: 031916–1 B2321), TGF-β1 (5 ng/mL) (100–21, lot: 1218209 H1919), IL-23 (5 ng/mL) (200–23, lot: 0712S227 K1119) and IL-1β (20 ng/mL) (200–01B, lot: 0606B95 B2421) were added at the time of plating with butyrate, pentanoate, BLIPs, or blank liposomes. Assays with blank liposomes were conducted using splenocytes from male SKG mice. All cytokines were recombinant human cytokines purchased from Peprotech.
Flow cytometry analysis
The cells were stimulated with PMA and Ionomycin (Cell Stimulation Cocktail, Invitrogen, Cat. No. 00–4970-03, lot: 2430454) for 4 hours. Cytokine transport was blocked with Brefeldin A (Invitrogen, Cat. No. 00–4506-51). After staining using a Fixable Viability Kit (Biolegend, lot: B333785) and for the CD4 marker (cat:1540–26, lot:F2212-T406C), the cells were fixed and permeabilized (Invitrogen ref: 00–5523-00 , lot: 2333698) followed by staining with fluorochrome-conjugated anti-mouse antibodies against RORγt (clone:B2D, lot: 2304447), IL-17 (clone:eBio17B7, lot: 2142931), IFN-γ (clone: XMG1.2, lot: 2289505), and Foxp3 (clone:FJK-16s, lot: 2199652) at 1:800 dilution. IL-17, FoxP3, RORγt and IFN-γ expression was quantified using an Attune NxT flow cytometer. Data was analyzed using FlowJo software.
Multiplex cytokine analysis
Cells were centrifuged for 5 min at 350 rcf, and 150 uL media was removed from each well for multiplex cytokine analysis. IL-10, TNF, IFN-γ, IL-4, IL-2 and IL-17 expression were quantified using a BD Mouse Th1/Th2/Th17 Cytometric Bead Array (CBA) kit (BD ref: 560485, lot: 1229218) according to manufacturer’s instructions. After preparation of the CBA samples, protein concentrations were quantified using an Attune NxT flow cytometer.
Butyrate liposome (BLIP) preparation
BLIP were synthesized by a thin film hydration method. 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) (Avanti ref: 870273C-25mg, lot: 870273C-25MG-A-064) and DSPE-PEG-Mal (MW: 2000) (Laysan Bio, lot: 165–07) were dissolved in 500z μL chloroform at 12.5 mg/mL. The chloroform was evaporated off by rotary evaporation, leaving a thin lipid film. The film was then hydrated by 3 mL of 1M butyrate in PBS for BLIPs, and PBS for blank liposomes. The solution was sonicated using a probe sonicator (Fisher Scientific, FB120) for 5 minutes at 80% amplitude with 1 second on and 2 seconds off. BLIP diameter and PDI was characterized by dynamic light scattering with a Malvern Zetasizer (ZEN3600).
Fluorescent BLIPs were synthesized as described above with the addition of 5 uL of Cy5 acid dissolved in DMSO at 10 mg/mL (Broad Pharm, BP-22274,lot CY5–1G-1) to chloroform solution prior to rotary evaporation. Control free Cy5 was prepared by evaporating off DMSO from the same amount of Cy5 acid and solubilizing the thin film via sonication in 3 mL of PBS.
HDAC Activity Assays
HDAC activity was quantified using a Flurorometric HDAC Activity Assay Kit (Abcam, ab156064). For assays assessing HDAC activity of HDACs extracted from cultured T cells, T cells were cultured as previously described in Th17 conditions with the addition of either nothing, blank liposomes (500 μg/mL), BLIPs (500 μg/mL) or butyrate (0.5 mM). Nuclear proteins were extracted after 4 days of culture by pooling 2 million cells together per treatment condition and using the Nuclear Extraction Kit (Abcam, ab113474) according to manufacturer’s instructions with the omission of the peptidase inhibition cocktail (PIC) to prevent downstream inhibition of HDAC activity in the HDAC Activity Assay. Whole protein concentration of nuclear extracts were determined via absorbance on a Nanodrop at 280 nm, and samples were normalized for total protein concentration. Samples were then used in the HDAC Activity Assay according to manufacturer’s instructions.
For HDAC inhibition assay, manufacturer supplied HeLa cell HDAC extract was used, and the assay was run according to manufacturer’s protocol. Inhibitory function of BLIPs (500 μg/mL), blank liposomes (500 μg/mL) and butyrate (0.5 mM) were assayed. Manufacturer supplied Trichostatin A was used as a known HDAC inhibitor control.
ATAC-Seq Preparation and Sequencing
Cells used in ATAC-Seq experiments were subjected to the same procedure as described in the “In vitro mouse Th17 differentiation assay” above. On day 3, 105 cells were removed from media and prepared for ATAC-Seq using the Active Motif ATAC-Seq kit (Active Motif, Cat No. 53150) according to manufacturer instructions. Briefly, isolated cells were washed once with ice-cold PBS and subsequently resuspended in ATAC Lysis Buffer on ice to isolate the nuclei. The lysis buffer was removed, cells were washed, and nuclei were incubated with tagmentation mix at 37°C for 30 minutes, after which DNA was isolated and purified. DNA was then amplified via PCR using thei provided indexed primers according to manufacturer’s instructions. SPRI cleanup beads were then used to purify the amplified DNA prior to sequencing. Samples were sequenced at the La Jolla Institute for Allergy and Immunology using an Illumina NovaSeq sequencer (NovaSeq 6000 S10OD025052).
ATAC-Seq Analysis
Analysis of raw reads from sequencing of ATAC-seq prepared libraries was performed as follows: FASTQ files were trimmed using NGmerge23, and then aligned to mm10 reference genome using bowtie224 with the parameters (-k 4 -X 2000 –very sensitive). Reads corresponding to ENCODE blacklisted regions25, mitochondrial genome, and chrY were removed. Reads were then filtered for uniquely mappable reads with mapping quality >= 30 using SAMtools26, and duplicate reads were removed using MarkDuplicates from Picard tools. Peaks were then called using MACS227 with the parameters (-g mm -q 0.05 –nomodel –nolambda –keep-dup all –call-summits –shift −100 –extsize 200). Summits of all peaks were then extended to regions of 500 bp, and the 500 bp peaks from the groups being compared were merged to get consensus peaks via BEDtools28 slop and merge routines. BAM files following removal of PCR duplicates were then converted to bed files and the reads for each region of the consensus peaks were counted using BEDtools coverage. Normalized coverage tracks for each group were created by merging BAM files of replicates withing each group, then running deeptools29 bamCoverage with the parameters (-bs 10 –effectiveGenomeSize 2652783500 –normalizeUsing RPKM -e 200). Counts were normalized and differential ATAC-seq analysis was performed using DESeq230. Differentially accessible regions (DARs) were filtered using FDR-adjusted p-values <= 0.05 and fold-change >=2. For each pairwise comparison, motif enrichment analysis was performed by inputting all DARs into HOMER to identify known motifs for transcription factor binding sites enriched in peaks31. These results were filtered for known motifs with log p-value <= −35, with coverage >= 10% and fold increase of at least 1.5 over background coverage.
RESULTS
Butyrate suppresses induction of Th17 and enhances Treg in inflammatory conditions
To test the effect of butyrate on enhancing Treg, we used an ex vivo polarization assay in which SKG CD4+ T cells were subject to Th17 polarizing conditions using cytokine supplementation (Fig. 1a). CD4+ splenocytes were stimulated and plated with Th17 polarizing cytokines IL-6, TGF-β1, IL-1β, and IL-23, and immunophenotypes were analyzed after four days, as described in the materials and methods section. We found that IL-17A expression, referred to henceforth just as IL-17, was increased 15-fold in when compared to expanding CD4+ T cells using IL-2 alone (Fig. S1a,b). In a subset of conditions, butyrate was added to the T cell culture medium, ranging from 0.5 mM to 3 mM.
Figure 1. Butyrate suppresses induction of Th17 and enhances Treg in inflammatory conditions.

a) Schematic of in vitro cell culture assessing effects of butyrate concentration on Th17 and Treg differentiation from CD4+ mouse T (mT) cells. b) Representative flow plot of cell viability of CD4+ mT cells in Th17 inducing conditions. (c-d) quantification of c) fold expansion d) % live CD4+ mT cells in Th17 inducing conditions. e) representative flow plot and f) quantification of FoxP3 expression in CD4+ mT cells in inflammatory conditions. (g-i) g) representative flow plot and quantification of h) RORγt and i) IL-17 expression in CD4+ mT cells in the aforementioned culture conditions. Data in c,d,f,h,i are the mean ± S.D. of representative experiments. Statistical analyses in c,d,f,h,i were performed using one-way ANOVA with a post-hoc Dunnet’s multiple comparison test.
1 mM butyrate did not suppress expansion in Th17 polarizing conditions, however reduced mean viability from 84% to 71% [p = 0.004]. The highest concentration of butyrate, 3 mM, reduced mean viability to 46% [p < 0.0001]. Expansion was reduced but not statistically significant (Figs. 1b, c, d). 1 mM Butyrate increased FoxP3 expression to 23% compared to 10% in the control [p=0.003] (Figs. 1e, f). Additional butyrate supplementation did not have a significant additional effect on FoxP3 or CD4 expression. Distinct populations of RORγt+ and IL-17+ cells were observed by flow cytometry (Fig. 1g). RORγt was suppressed in a concentration dependent manner, with mean expression of 69% in the control (0 mM butyrate), 50% at 0.5 mM [p = 0.003], 43% at 1 mM [p = 0.002] and 21% at 3 mM [p = 0.0008] butyrate (Fig. 1h). 0.5 mM butyrate and 1 mM butyrate did not significantly reduce IL-17 expression compared to the control. However, addition of 3 mM butyrate reduced IL-17 expression from 18% to 10% [p = 0.03] (Fig. 1i).
Pentanoate modulates FoxP3 and IL-17 expression in CD4+ T cells
To test the effect of pentanoate on T cell expansion and differentiation we cultured CD4+ splenocytes in Th17-inducing conditions and concentrations of pentanoate from 0.5 mM to 3 mM for 4 days, and quantified immunophenotypes by flow cytometry on day 4 (Fig. 2a). Pentanoate reduced CD4+ T cell expansion across all conditions and reduced cell viability at concentrations of 1 mM and greater (Figs. 2b–d). Pentanoate reduced FoxP3 at all concentrations (Figs. 2e, f). Pentanoate at 1mM reduced RORγt expression to 57% [p < 0.0001], similar to pentanoate at 3 mM where expression was reduced to 55% [p < 0.0001], compared to 82% in Th17 polarizing conditions alone (Fig. 2g, h). IL-17 expression was reduced in all pentanoate conditions, with 15% of cells expressing IL-17 in the 0.5 mM pentanoate condition and 11% in the 1 mM pentanoate condition, compared to 18% in Th17 polarizing conditions alone (Fig. 2i).
Figure 2. Pentanoate modulates FoxP3 and IL-17 expression in CD4+ T cells.

a) Schematic of in vitro cell culture assessing effects of pentanoate concentration on Th17 and Treg differentiation from CD4+ mT cells. b) Representative flow plot of cell viability of CD4+ mT cells in Th17 inducing conditions. (c-d) quantification of c) fold expansion d) % live CD4+ mT cells in Th17 inducing conditions. e) representative flow plot and f) quantification of FoxP3 expression in CD4+ mT cells in inflammatory conditions. (g-i) g) representative flow plot and quantification of h) RORγt and i) IL-17 expression in CD4+ mT cells in the aforementioned culture conditions. Data in c,d,f,h,I are the mean ± S.D. of representative experiments. Statistical analyses in c,d,f,h,I were performed using one-way ANOVA with a post-hoc Dunnet’s multiple comparison test.
Butyrate modulates CD4+ T cell cytokine secretion in in vitro inflammatory conditions
We selected butyrate for further characterization of Treg enhancement and assessed the effect on cytokine production by CD4+ T cells in Th17 polarizing conditions (Fig. 3a). We collected cell culture supernatant from the experiment described above and analyzed cytokine concentration using a Multiplex Cytokine Bead Array. We found that there was an optimal concentration of butyrate that enhanced IL-10, a cytokine associated with anti-inflammatory effects. 0.5 mM and 1 mM butyrate increased IL-10 concentration to ~6000 [p = 0.07] and ~6600 pg/mL [p = 0.02] respectively, compared to ~3700 pg/mL in the untreated group (Fig. 3b). Consistent with the results from intracellular staining, butyrate suppressed IL-17 most effectively at 2 and 3 mM, reducing the concentration to 1400 [p = 0.004] and 700 pg/mL [p < 0.002] respectively, compared to 7800 pg/mL in the control group (Fig. 3c). To characterize whether butyrate affected production of other cytokines, we quantified IFN-γ, a Th1-associated cytokine and IL-4, a Th2-associated cytokine. Butyrate induced IFN-γ secretion of 327 pg/mL in the control, and upregulated IFN-γ secretion to 1404 pg/mL at 0.5 mM [p = 0.003] and 2228 pg/mL at 1 mM [p < 0.0001] (Fig. 3d). No effect was observed in IL-4, IL-2, and TNF secretion (Fig. 3e–g).
Figure 3. Butyrate modulates CD4+ T cell cytokine secretion in in vitro inflammatory conditions.

a) Schematic of in vitro cell culture assessing effects of butyrate on CD4+ mT cell cytokine secretion in inflammatory conditions. (b-g) Quantification of b) IL-10, c) IL-17, d) IFN-γ, e) IL-4, f) IL-2 and g) TNF cytokine secretion as analyzed by multiplex assay in vitro. Data in b-g represented as the mean ± S.D. of representative experiments. Statistical analyses in b-g were performed using one-way ANOVA with a post-hoc Dunnet’s multiple comparison test.
Butyrate-loaded liposomes maintain Treg immunomodulatory effects while improving viability
To facilitate delivery of butyrate, we synthesized butyrate-loaded PEGylated liposomes (BLIPs) using a thin film hydration method. We selected a water-soluble phospholipid – 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol) (DSPE-PEG) and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), commonly used materials in liposome formulations32–34(Fig. 4a, b). After dissolving the lipids in chloroform, the solvent was allowed evaporate and generate a lipid film which was hydrated with 1M sodium butyrate. The hydrated lipid film was then sonicated to generate liposomal particles, which were dialyzed to remove unencapsulated butyrate, and the final solution was stored at 4°C (Fig. 4c). The z-average of the BLIP diameter was determined by dynamic light scattering to be ~230 nm with a polydispersity index (PDI) of 0.236. To determine if the liposomal formulation improved uptake, Cyanine 5 (Cy5) was encapsulated into BLIPs to create fluorescent BLIPs (Fluo-BLIPs. Fluo-BLIPs or free Cy5 solubilized via sonication without lipids was cultured with splenocytes. After 5 hours, CD4+ T cells were analyzed using flow cytometry to examine Cy5 uptake (Figure S2a). CD4+ T cells from splenocytes cultured with Fluo-BLIPs exhibited a higher mean fluorescent intensity on the Cy5 channel and a greater fraction of Cy5+ CD4+ T cells (68 ± 5%) compared to free Cy5 (53 ± 1%) (Fig. S2b,c).
Figure 4. Butyrate-loaded liposomes maintain Treg immunomodulatory effects while improving viability.

a) Chemical structure of components of PEGylated Butyrate Liposome (BLIP) formulation. b) Schematic of BLIP synthesis. c) Intensity of BLIP particles as measured by dynamic light scattering. d) Schematic of in vitro cell culture assessing effects of BLIP concentration on Th17 and Treg differentiation from CD4+ mT cells. e) Representative flow plot of cell viability of CD4+ mT cells in Th17 inducing conditions. (f-g) quantification of f) fold expansion and g) % live CD4+ mT cells in Th17 inducing conditions. h) representative flow plot and i) quantification of FoxP3 expression in CD4+ mT cells in inflammatory conditions. (j-l) j) Representative flow plot and quantification of k) RORγt and l) IL-17 expression in CD4+ mT cells in the aforementioned culture conditions. (m-o) m) Representative flow plot and quantification of n) FoxP3 and o) IL-17 expression in mT cells from male mice differentiated in Th17-polarizing conditions treated as indicated. Data in f,g,i,k,l,n,o are the mean ± S.D. of representative experiments. Statistical analyses in f,g,I,k,l were performed using one-way ANOVA with a post-hoc Dunnet’s multiple comparison test. Statistical analysis in n, o were performed using one-way ANOVA with post-hoc Tukey test for multiple comparison.
To assess whether BLIPs enhance Treg and suppress Th17, we tested concentrations between 0.05 μg/mL to 500 μg/mL in Th17-polarizing CD4+ cell cultures (Fig. 4d). BLIPs reduced expansion of CD4+ T cells in a concentration dependent manner, similar to butyrate. At 500 μg/mL, expansion was reduced to 3.6 fold, compared to 7.1 fold in the control [p = 0.005]. 500 μg/mL marginally reduced viability, to 73%, compared to 78% in the BLIP-free control [p = 0.002] (Figs. 4e, f, g). A distinct population of FoxP3+ cells was observed by flow cytometry (Fig. 4h). FoxP3 expression was upregulated in a concentration dependent manner, with 500 μg/mL BLIP resulting in mean expression of 22%, compared to 14% in the control group [p = 0.003] (Fig. 4i). Both RORγt and IL-17 were suppressed in a concentration dependent manner by BLIPs, with RORγt reducing from 60% in the control to 50% at 50 μg/mL [p = 0.0004] and 29% at 500 μg/mL [p < 0.0001] (Figs. 4j, k). IL-17 was reduced from 23% in the control to 17% at 5 ug/mL [p < 0.0001] and 9.4% at 500 ug/mL [p < 0.0001] (Fig. 4l).
As phosphatidylcholines are known to have immunomodulatory properties, we sought to determine if the liposome formulation itself contributed to the immunomodulatory activity of the BLIPs. We conducted the Th17 polarization assay and treated cells with blank liposomes, 0.5 mM butyrate, or BLIPs. Blank liposomes alone at 500 mg/ml suppressed IL-17 expression (Fig. 4m–o). However, only BLIPs enhanced FoxP3 expression significantly over Th17 only conditions.
Butyrate reduces HDAC activity and alters chromatin accessibility of Th17-associated genes
To evaluate the effect of different treatments on HDAC inhibition, we performed HDAC activity assays comparing butyrate, BLIPs and blank liposomes. T cells were differentiated in Th17 inducing conditions alone, or in combination with either 0.5 mM butyrate, 500 μg/mL BLIPs, or 500 μg/mL blank liposomes. Whole nuclear proteins were isolated and assayed for HDAC activity. HDAC activity was comparable between T cells in the Th17 polarizing conditions and T cells treated with blank liposomes (Fig. 5a,b). 0.5 mM butyrate reduced HDAC activity compared to Th17 inducing conditions alone. HDAC activity in T cells treated with 500 μg/mL BLIP was the lowest among the tested groups. We also confirmed the effects in another cell type by testing supplier provided HDAC isolated from HeLa cells. HDAC inhibition by 500 μg/mL BLIP and 0.5 mM butyrate was comparable to Trichostatin A (TSA), a known pan-HDAC inhibitor, while blank liposomes did not inhibit HDAC activity (Fig. S3a).
Figure 5. Butyrate affects chromatin accessibility of Th17-associated genes.

(a,b) HDAC activity readouts from nuclear protein extracts from groups of mT cells treated as indicated represented as a) standardized absorbance values over time after addition of nuclear extracts and b) average rate of reaction between 39–76 minutes, after reaction rate stabilized. c) Normalized ATAC-seq coverage at the Foxp3, Rorc, and Il17a loci in average representations of the 0.5 mM butyrate (B0.5, red), 500 μg/mL BLIPs (BLIPs, purple), and Th17 polarization only conditions (Th17, blue). d) Scatter plots of ATAC-seq counts per peak comparing cells from the B0.5 condition to the Th17 condition. Red corresponds to differentially accessible regions (DARs) enriched in the B0.5 condition, blue corresponds to DARs enriched in the Th17 condition, and grey corresponds to regions that are accessible in both conditions, but not significantly different. e) Boxplots of ATAC-Seq counts per peak from B0.5 and Th17 conditions at common (grey) or differentially accessible regions enriched in either the B0.5 (red) or the Th17 group (blue) from the comparison in Fig. 5b. f) Heatmap of select Th17 and Treg associated genes in B0.5 and Th17 conditions with dendrograms showing relatedness of samples (columns) and individual genes (rows). (g-i) g) Scatter plots of ATAC-seq counts per peak, h) boxplots of ATAC-seq counts per peak, and i) heatmap of select Th17 and Treg associated genes comparing cells from the BLIPs (purple) and Th17 (blue) conditions. (j-l) j) Scatter plots of ATAC-seq counts per peak, k) boxplots of ATAC-seq counts per peak, and l) heatmap of select Th17 and Treg associated genes comparing cells from the BLIPs (purple) and B0.5 (red) conditions. m) Motif enrichment analysis comparing peaks pairwise between cells treated with BLIPs, B0.5, and Th17, with enrichment of motifs quantified as -log(P) value. Data in a are the mean ± S.D. of n = 2 absorbance values per group standardized by subtracting the average absorbance value of the no enzyme control from the sample absorbance value. Data in b represent mean ± S.D.. Scales in c are as follows: Foxp3 [0–200], Rorc [0–400], Il17a [0–130]; data in f, i, l represent the integrated ATAC signal across each known gene promoter region ranked as z-scores using data across each row; data in m represent motifs with an enrichment log p-value less than −35 found in 10% or more regions with coverage showing a fold increase of at least 1.5 over background coverage in at least one pairwise comparison. Statistical analysis in a was performed using one-way ANOVA with post-hoc Tukey test for multiple comparisons on area under the curve.
As histone modifications play important roles in determining chromatin accessibility, we next sought to assess whether butyrate and BLIP epigenetically modified the chromatin landscape of CD4+ T cells in Th17-polarizing conditions, we performed the assay for transposable-accessible chromatin with high-throughput sequencing (ATAC-Seq). Differences in chromatin accessibility between cells treated with 0.5 mM butyrate, BLIPs, and Th17 polarization only were observed at several regions, including Treg and Th17 associated loci such as Foxp3, Rorc, and Il17a (Fig. 5c). When comparing 0.5 mM butyrate to the untreated Th17-polarizing control, we found 26000+ differentially accessible regions (DARs), 13000+ associated with Th17 polarization alone and 12000+ with 0.5 mM butyrate (Fig. 5d, e). Chromatin accessibility was downregulated by 0.5 mM butyrate at the Il17, Il21 and Rorc gene loci, which are associated for Th17 differentiation (Fig. 5f). Downregulation by 0.5 mM butyrate was also observed at the Treg-associated Foxp3 and Il10 gene loci. When comparing chromatin accessibility between BLIPs and Th17-polarizing conditions alone, 9000+ DARs were found, with 4728 regions more accessible in the BLIP condition and 4635 regions more accessible in the Th17-polarizing condition alone (Fig. 5g, h). Chromatin accessibility to the Th17-associated gene loci for Il17, Il21 and Rorc as well as the Treg- associated gene loci for Foxp3 and Il10 were downregulated by BLIPs compared to Th17-inducing conditions alone (Fig. 5i). Direct comparison of the BLIPs to the 0.5 mM butyrate revealed that the chromatin accessibility was similar between the two conditions, with only 943 DARs. BLIPs increased accessibility at 737 of those regions while 0.5 mM butyrate increased accessibility at 206 regions (Fig. 5j, k). 0.5 mM butyrate marginally downregulated gene accessibility to Il17, Il21, Rorc, Foxp3 and Il10 (Fig. 5l). However, when compared across all treatment groups and including Th1 associated markers Tbet and Ifng, samples from the BLIPs and 0.5 mM butyrate groups were much more similar than Th17-polarizing conditions alone (Fig. S4a). To determine if the changes in chromatin accessibility were dose dependent, we compared 0.5 mM butyrate to 1 mM butyrate, but found no significant differences between the groups (Fig. S4b, c). Motif enrichment analysis comparing cells treated with Th17 polarization alone to 0.5 mM butyrate revealed a striking reduction in bZIP family binding motifs in the 0.5 mM butyrate condition (Fig. 5m). Comparison of BLIPs to Th17 polarization alone showed a similar trend.
DISCUSSION
The regulation of the immune response by endogenously produced immunoregulators is important in maintaining health and preventing inflammatory disorders. Here, we demonstrated the immunoregulatory effect of SCFAs in modulating the differentiation of T cells into Treg in conditions that otherwise favor Th17. We show that butyrate upregulates FoxP3+ Treg and suppresses IL-17+/RORγt + Th17 cells in inflammatory conditions that promote Th17 polarization in naïve SKG CD4+ T cells. Butyrate exerted a concentration-dependent effect on transcription factor and intracellular protein expression and cytokine secretion. Pentanoate downregulated RORγt and IL-17 expression in a concentration dependent manner in the same in vitro conditions, however the effect was less pronounced than that of butyrate. We demonstrate that encapsulating butyrate within POPC/DSPE-PEG-Mal liposomes maintains cell viability while retaining preferential modulation of Treg/Th17 ratio. Both butyrate and BLIPs modulated chromatin accessibility in Th17-polarizing conditions, supporting the idea that butyrate can epigenetically modulate T cell differentiation in inflammatory conditions.
Butyrate upregulated Treg and downregulated Th17 in a concentration dependent manner. At the upper limit of concentrations tested, 2 mM and 3 mM, butyrate was cytotoxic, reducing cell viability by half. While RORγt was more effectively suppressed at these higher concentrations, significant differences in IL-17+ or FoxP3+ proportions were not observed compared to 0.5 and 1 mM. The concentration of butyrate which suppressed IL-17 and RORγt while upregulating FoxP3 and retaining cell viability appears to be 1 mM. Prior work has shown the effect of butyrate on T cell differentiation at lower concentrations, up to 1 mM35. Concentrations above 1 mM were not investigated, and Treg and Th17 differentiation was observed in their respective polarizing conditions. Cell viability was not quantified in this study. SCFA-mediated Treg modulation in inflammatory Th17-inducing conditions has not been characterized. Our results point towards potent effects of SCFAs in inflammatory environments, and a significant impact on cell viability by butyrate. On the other hand, the suppression of Th17 and upregulation of Treg by pentanoate was less pronounced compared to butyrate. In contrast to butyrate, pentanoate did not reduce cell viability, and did not enhance FoxP3. These data are consistent with the literature, in which pentanoate has been shown to primarily modulate metabolic function in T cells16 . Moreover, pentanoate has been shown to increase CD4+ T cell proliferation in the absence of inflammatory cytokines. Therefore, based on the results of this work, we selected butyrate over pentanoate for further evaluation.
Butyrate at 1 mM and 0.5 mM enhanced the production of the immunoregulatory cytokine IL-10, which is associated with Treg function. 1 mM butyrate maximized IL-10 production, suggesting that there could be an optimal concentration for Treg enhancement and suppression of Th17 differentiation. The differential effect of butyrate is further supported by the observation that higher concentration reduced IL-10 and IL-17, whereas 0.5 mM and 1 mM butyrate did not significantly reduce IL-17. Therefore, harnessing the anti-inflammatory properties of butyrate will likely be context dependent, as higher concentrations of butyrate suppress Th17 activity while lower concentrations enhance Treg activity. Furthermore, IFN-γ, which was upregulated at all concentrations of butyrate, has been shown to have a context-dependent effect on autoimmune inflammation36. IFN-γ can have both immunostimulatory and immunoregulatory effects37. It is associated with a helper 1 (Th1)-driven immune response which mediates protection against pathogens. IFN-γ has also been demonstrated to play a protective role in autoimmune disease models while enabling induced Treg cells to control immune responses in others. For example, ablation of IFN-γ signaling exacerbates autoimmune arthritis in mouse models of autoimmune arthritis, including SKG mice 38–40. IFN-γ has been shown to be essential for the conversion of CD4+ T cells to CD4+ Treg in inflammatory conditions in experimental autoimmune encephalomyelitis 41. In graft-versus-host disease IFN-γ expressing Treg have been demonstrated to be essential for preventing disease 42. Our results support that the delivery of butyrate would benefit from a disease-specific evaluation.
To assess whether encapsulating butyrate for effective delivery would preserve the modulation Th17/Treg in vitro, we encapsulated butyrate in a liposomal formulation, termed BLIPs. Liposomes have been extensively studied as drug delivery vehicles43,44. Here, for the first time, we have assessed the liposomal delivery of butyrate to modulate T cells. To formulate BLIPs, we used DSPE-PEG-Mal, a common PEGylated phospholipid widely used in various preparations, including in clinically approved drug formulations. While we assessed their effect in vitro, DSPE-PEG has been demonstrated to increase bioavailability of encapsulated drugs molecules over non-PEGylated formulations33. The mean size of BLIPs (~230 nm) is consistent with prior reports of similar liposomal formulations45. Due to challenges regarding detection of butyrate in vitro, we examined liposomal delivery of the model dye Cy5 and found that liposomal encapsulation improved delivery of the Cy5 relative to free dye solubilized via sonication without lipids. Additionally, the results from the HDAC activity and inhibition assays support the notion of enhanced delivery of butyrate as T cell isolated HDACs of T cells treated with BLIPs displayed enhanced inhibition relative to HDAC from T cells treated with 0.5 mM butyrate, while both BLIPs and 0.5 mM butyrate displayed similar capacities for HDAC inhibition when tested on HeLa isolated HDACs. This suggests that differences in the activity of HDAC isolated from treated T cells represents the ability of butyrate to access HDAC in cells rather than added inhibition due to the activity of liposomal compounds.
Phenotypic modulation by BLIPs was observed to be concentration dependent. BLIPs attain similar upregulation of FoxP3 and downregulation of IL-17 to butyrate, while maintaining cell viability. While the blank liposomal formulation, which consists of phosphatidylcholines POPC and DSPE-PEG, inhibited IL-17 expression, it did not increase FoxP3 expression to the same extent as BLIPs or demonstrate HDAC inhibitory activity. The observation of reduced IL-17 expression is consistent with prior work demonstrating the phosphatidylcholines are associated with immunoregulatory effects in animals and humans46–48. These results support that BLIPs are suitable for butyrate delivery.
BLIPs maintain the immunomodulatory activity butyrate while maintaining high viability of T cells compared to free butyrate. A possible reason is that exogenous butyrate can also induce signaling via G-protein-coupled receptors (GPCRs), such as GPR43, which has been demonstrated to have a potent anti-proliferative effect on CD4+ T cells which affects viability in vitro49. In contrast, internalized butyrate, delivered via BLIPs, could reduce GPCR signaling while potentially enhancing HDAC inhibition compared to free butyrate and thereby avoid a reduction in cell viability.
ATAC-seq analysis revealed that butyrate and BLIPs comparably modulated the chromatin accessibility of loci associated with both Treg and Th17 function, with only a small number of DARs arising between the two groups, but a significant number of DARs when either 0.5 mM butyrate or BLIP-treated cells were compared to Th17-inducing conditions alone. These observations are consistent with prior work that has demonstrated that butyrate promotes epigenetic changes via HDAC inhibition to induce a Treg phenotype50–52. Most changes we observed were reductions in the accessibility of Th17 associated loci. While chromatin accessibility at the Foxp3 and Il10 loci was unaffected by addition of butyrate, the expression of FoxP3 and IL-10, as analyzed by flow cytometry and multiplex analysis respectively, increased. The apparent difference between the enhancement of gene products without a change in chromatin accessibility at these loci suggests that the mechanism of butyrate-mediated upregulation of FoxP3 and IL-10 protein expression is not only due to changes in chromatin accessibility at the specific gene loci. We further assessed the effect of butyrate by conducting a motif enrichment analysis. Both butyrate and BLIPs significantly reduced the prevalence of several bZIP class motifs. Of these, it is notable that the AP-1 family transcription factors Basic leucine zipper transcription factor, ATF-like (BATF) and JunB, whose binding motifs were significantly downregulated by butyrate, have been demonstrated to play important roles in Th17 differentiation53–56.
Our work suggests that BLIPs could be an effective agent for therapeutic delivery of butyrate. In addition to selection of the SCFA, an important consideration is that there is an optimal concentration range for butyrate for enhancing immunomodulation while avoiding cytotoxicity. Therefore, achieving target dose in affected tissue via systemic administration, for example via oral administration, could be challenging. Instead, local delivery of SCFAs via BLIPs is likely to result in therapeutically relevant immunomodulation. Therefore, future work should focus on characterizing the local and systemic effects of BLIP administration in vivo in an inflammatory disease context.
Supplementary Material
Supplementary Figure S1. Representative Th17 differentiation Representative flow cytometry plots depicting IL-17 expression in pan CD4+ mT cells exposed to either a) Th17 inducing conditions as described in Methods and Materials or b) IL-2 and platebound anti-CD3 and anti-CD28.
Supplementary Figure S2. Liposome uptake quantification via Cy5 a) Representative flow cytometry histograms for uptake of Cy5 at 5 in CD4+ mT cells from splenocytes cultured with free Cy5 or Cy5 BLIPs. (b,c) Quantification of Cy5+ uptake by CD4+ T cells at 5 hours as b) Cy5+ fraction of CD4+ mT cells and c) MFI of CD4+ mT cells on the Cy5 channel. Data represent mean ± S.D. Statistical analysis were performed using Student’s t-test.
Supplementary Figure S3. Butyrate and BLIPs have comparable inhibitory effect on HDAC extract a) HDAC inhibition assay of agents acting on HDAC extract from manufacturer provided HeLa cells demonstrating inhibitory effect of blank liposomes, BLIPs and 0.5 mM butyrate relative to buffer only control (no HDACs), active HDAC control, and known HDAC inhibitor Trichostatin A. Data in a represent mean ± S.D. Statistical analysis was performed using one-way ANOVA with post-hoc Tukey test for multiple comparisons on area under the curve.
Supplementary Figure S4. Additional ATAC-seq comparisons of treatment groups and 0.5 mM butyrate and 1 mM butyrate treated cells a) Heatmap of select Th17, Treg and Th1 associated genes across B0.5, Th17, and BLIPs treatment groups with dendrograms showing similarity of samples (columns) and individual genes (rows). (b,c) ATAC-seq data comparing CD4+ T cells exposed to Th17 inducing conditions in combination with either 0.5 mM butyrate (B0.5) or 1 mM butyrate (B1.0) visualized as b) Scatter plot depicting common (grey) peaks. No peaks were found to be differentially accessible between the groups, so all points are grey. c) Box and whisker plots of common peaks in the B0.5 and B1.0 samples.
Acknowledgments:
The authors acknowledge technical assistance by the Joint Cell Isolation Core and the Joint Bioinformatics and Computational Core of the Microenvironment in Arthritis Resource Center (MARC) at UC San Diego.
Funding:
National Institutes of Health grant F31AR079921 (DAM)
National Institutes of Health grant T32AR064194
National Institutes of Health grant P30AR073761 (NB)
Arthritis National Research Foundation (NJS)
Hellman Fellowship (NJS)
All authors reviewed the data and analysis, provided input on the manuscript, and approved the submission.
Footnotes
Ethics approval and consent to participate:
All institutional and national guidelines for the care and use of laboratory animals were followed. No live animal or human studies were carried out by the authors for this article. Consent to participate is not applicable for this article.
Consent for publication:
No human studies were carried out by the authors for this article. Consent to publish is not applicable for this article.
Competing interests:
The authors have no relevant financial or non-financial interests to disclose
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials:
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. ATAC-Seq data in this publication have been deposited in the NCBI’s Sequence Read Archive (SRA) database and are accessible through BioProject accession number PRJNA914429.
REFERENCES
- 1.Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR & Vignali DAA The development and function of regulatory T cells. Cellular and Molecular Life Sciences 66, 2603–2622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paust S. & Cantor H. Regulatory T cells and autoimmune disease. Immunol Rev 204, 195–207 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Ohl K. & Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol 45, 344–355 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Littman DR & Rudensky AY Th17 and Regulatory T Cells in Mediating and Restraining Inflammation. Cell 140, 845–858 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Park H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6, 1133–1141 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakaguchi S. et al. Foxp3 + CD25 + CD4 + natural regulatory T cells in dominant self-tolerance and autoimmune disease. [DOI] [PubMed] [Google Scholar]
- 7.Pan W. et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat Commun 6, 7096 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Mortaz E. et al. Cancers Related to Immunodeficiencies: Update and Perspectives. Front Immunol 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vial T. Immunosuppressive drugs and cancer. Toxicology 185, 229–240 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Tabas I. & Glass CK Anti-Inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science (1979) 339, 166–172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vinolo MAR, Rodrigues HG, Nachbar RT & Curi R. Regulation of Inflammation by Short Chain Fatty Acids. Nutrients 3, 858–876 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cummings JH, Pomare EW, Branch WJ, Naylor CP & Macfarlane GT Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson WT, Dorn NC, Ogbonna DA, Bottini N. & Shah NJ Lipid-based regulators of immunity. Bioeng Transl Med 7, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dass NB et al. The relationship between the effects of short-chain fatty acids on intestinal motility in vitro and GPR43 receptor activation. Neurogastroenterology & Motility 19, 66–74 (2007). [DOI] [PubMed] [Google Scholar]
- 15.HAMER HM et al. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27, 104–119 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Luu M. et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol 8, 80–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu WS, Parmigiani RB & Marks PA Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene vol. 26 5541–5552 Preprint at 10.1038/sj.onc.1210620 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Wawman RE, Bartlett H. & Oo YH Regulatory T Cell Metabolism in the Hepatic Microenvironment. Front Immunol 8, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakaguchi S, Takahashi T, Hata H, Nomura T. & Sakaguchi N. SKG mice, a new genetic model of rheumatoid arthritis. Arthritis Res Ther 5, 10 (2003). [Google Scholar]
- 21.Hsieh W-C et al. PTPN2 links colonic and joint inflammation in experimental autoimmune arthritis. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svensson MND et al. Reduced expression of phosphatase PTPN2 promotes pathogenic conversion of Tregs in autoimmunity. Journal of Clinical Investigation 129, 1193–1210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaspar JM NGmerge: merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinformatics 19, 536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langmead B. & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amemiya HM, Kundaje A. & Boyle AP The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramírez F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz S. et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 38, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Che J, Okeke C, Hu Z-B & Xu J. DSPE-PEG: A Distinctive Component in Drug Delivery System. Curr Pharm Des 21, 1598–1605 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Gabizon A. & Martin F. Polyethylene Glycol-Coated (Pegylated) Liposomal Doxorubicin Rationale for Use in Solid Tumours. Drugs vol. 54 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Oberholzer T, Albrizio M. & Luisi PL Polymerase chain reaction in liposomes. Chem Biol 2, 677–682 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Kespohl M. et al. The microbial metabolite butyrate induces expression of Th1- associated factors in cD4+ T cells. Front Immunol 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivashkiv LB IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nature Reviews Immunology vol. 18 545–558 Preprint at 10.1038/s41577-018-0029-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood KJ & Sawitzki B. Interferon gamma: a crucial role in the function of induced regulatory T cells in vivo. Trends Immunol 27, (2006). [DOI] [PubMed] [Google Scholar]
- 38.Ortmann RA & Shevach EM Susceptibility to Collagen-Induced Arthritis: Cytokine-Mediated Regulation. Clinical Immunology 98, (2001). [DOI] [PubMed] [Google Scholar]
- 39.Hirota K. et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. Journal of Experimental Medicine 204, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SH, Kwon JY, Kim S-Y, Jung K. & Cho M-L Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci Rep 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z. Role of IFN-g in induction of Foxp3 and conversion of CD4+CD25- T cells to CD4+ Tregs. Journal of Clinical Investigation (2006) doi: 10.1172/JCI25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koenecke C. et al. IFN-γ Production by Allogeneic Foxp3 + Regulatory T Cells Is Essential for Preventing Experimental Graft-versus-Host Disease. The Journal of Immunology 189, (2012). [DOI] [PubMed] [Google Scholar]
- 43.Shah S, Dhawan V, Holm R, Nagarsenker MS & Perrie Y. Liposomes: Advancements and innovation in the manufacturing process. Adv Drug Deliv Rev 154–155, 102–122 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Antimisiaris SG et al. Overcoming barriers by local drug delivery with liposomes. Adv Drug Deliv Rev 174, 53–86 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Perez R. v. et al. SELECTIVE TARGETING OF KUPFFER CELLS WITH LIPOSOMAL BUTYRATE AUGMENTS PORTAL VENOUS TRANSFUSION-INDUCED IMMUNOSUPPRESSION1,2. Transplantation 65, 1294–1298 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Johnson WT, Dorn NC, Ogbonna DA, Bottini N. & Shah NJ Lipid-based regulators of immunity. Bioeng Transl Med 7, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broadhurst MJ et al. IL-22 + CD4 + T Cells Are Associated with Therapeutic Trichuris trichiura Infection in an Ulcerative Colitis Patient. Sci Transl Med 2, (2010). [DOI] [PubMed] [Google Scholar]
- 48.He B. et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kibbie JJ et al. Butyrate directly decreases human gut lamina propria CD4 T cell function through histone deacetylase (HDAC) inhibition and GPR43 signaling. Immunobiology 226, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arpaia N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furusawa Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, (2013). [DOI] [PubMed] [Google Scholar]
- 52.Arpaia N. & Rudensky AY Microbial metabolites control gut inflammatory responses. Proceedings of the National Academy of Sciences 111, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schraml BU et al. The AP-1 transcription factor Batf controls TH17 differentiation. Nature 460, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamazaki S. et al. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci Rep 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pham D, Silberger DJ, Hatton RD & Weaver CT Batf promotes and stabilizes Th17 cell development by antagonizing the actions of STAT5. The Journal of Immunology 202, 124.10 (2019). [Google Scholar]
- 56.Carr TM, Wheaton JD, Houtz GM & Ciofani M. JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation. Nat Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Representative Th17 differentiation Representative flow cytometry plots depicting IL-17 expression in pan CD4+ mT cells exposed to either a) Th17 inducing conditions as described in Methods and Materials or b) IL-2 and platebound anti-CD3 and anti-CD28.
Supplementary Figure S2. Liposome uptake quantification via Cy5 a) Representative flow cytometry histograms for uptake of Cy5 at 5 in CD4+ mT cells from splenocytes cultured with free Cy5 or Cy5 BLIPs. (b,c) Quantification of Cy5+ uptake by CD4+ T cells at 5 hours as b) Cy5+ fraction of CD4+ mT cells and c) MFI of CD4+ mT cells on the Cy5 channel. Data represent mean ± S.D. Statistical analysis were performed using Student’s t-test.
Supplementary Figure S3. Butyrate and BLIPs have comparable inhibitory effect on HDAC extract a) HDAC inhibition assay of agents acting on HDAC extract from manufacturer provided HeLa cells demonstrating inhibitory effect of blank liposomes, BLIPs and 0.5 mM butyrate relative to buffer only control (no HDACs), active HDAC control, and known HDAC inhibitor Trichostatin A. Data in a represent mean ± S.D. Statistical analysis was performed using one-way ANOVA with post-hoc Tukey test for multiple comparisons on area under the curve.
Supplementary Figure S4. Additional ATAC-seq comparisons of treatment groups and 0.5 mM butyrate and 1 mM butyrate treated cells a) Heatmap of select Th17, Treg and Th1 associated genes across B0.5, Th17, and BLIPs treatment groups with dendrograms showing similarity of samples (columns) and individual genes (rows). (b,c) ATAC-seq data comparing CD4+ T cells exposed to Th17 inducing conditions in combination with either 0.5 mM butyrate (B0.5) or 1 mM butyrate (B1.0) visualized as b) Scatter plot depicting common (grey) peaks. No peaks were found to be differentially accessible between the groups, so all points are grey. c) Box and whisker plots of common peaks in the B0.5 and B1.0 samples.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. ATAC-Seq data in this publication have been deposited in the NCBI’s Sequence Read Archive (SRA) database and are accessible through BioProject accession number PRJNA914429.