

Abstract
The vaccinia virus H2R gene (VACWR 100) is conserved in all sequenced members of the poxvirus family and encodes a protein with a predicted transmembrane domain and four invariant cysteines. A recombinant vaccinia virus, in which expression of the H2 protein is stringently regulated, was unable to replicate without inducer. However, under nonpermissive conditions, all stages of virus morphogenesis appeared normal and extracellular virions were detected at the tips of actin tails. Nevertheless, virus did not spread to neighboring cells nor did syncytia form after low-pH treatment. Purified -H2 and +H2 virions from cells infected in the absence or presence of inducer, respectively, were indistinguishable in microscopic appearance and contained the same complement of major proteins, though only +H2 virions were infectious. The -H2 virions bound to cells, but their cores did not penetrate into the cytoplasm. In addition, exogenously added -H2 virions were unable to mediate the formation of syncytia after low-pH treatment. In contrast, virions lacking the A27 (p14) protein, which was previously considered to have an essential role in fusion, penetrated cells and induced extensive syncytia. The properties of H2, however, are very similar to those recently reported for the A28 protein. Moreover, coimmunoprecipitation experiments indicated an interaction between H2 and A28. Therefore, H2 and A28 are the only proteins presently known to be specifically required for vaccinia virus entry and are likely components of a fusion complex.
The mechanism by which poxviruses penetrate cells is not understood, and the little that we know comes from studies of vaccinia virus, the prototype of this large family. The slow progress in the field can be attributed to the complexity of poxviruses, making it difficult to determine which of the numerous known or predicted membrane proteins are involved. Poxviruses are linear double-stranded DNA viruses that replicate exclusively in the cytoplasm (22). The genome of vaccinia virus contains nearly 200 genes, about one-quarter of which are conserved in all members of the family. During its reproduction cycle, vaccinia virus produces several related infectious forms with different outer membranes. The most abundant infectious particle, known as the intracellular mature virion (IMV), is composed of a compact core surrounded by a lipoprotein membrane. The core contains the viral genome, enzymes involved in mRNA synthesis and modification, and proteins with presumed structural roles. There is uncertainty as to how the lipoprotein membrane destined to become the outer coat of IMV is formed and whether it consists of one or two closely apposed lipoprotein bilayers (12, 27). Most IMV remain within the cytoplasm of the intact cell and are released upon cell lysis. Some IMV undergo wrapping by a double membrane derived from trans-Golgi or endosomal cisternae to form intracellular enveloped virions (IEV) (11, 33, 40). The IEV are transported on microtubules to the periphery of the cell, where the outer IEV and plasma membranes fuse (23, 37, 44). In this process, the IEV lose one outer membrane but still contain an additional membrane relative to IMV. Electron microscopic images suggest that some IMV obtain the extra membrane by budding directly through the plasma membrane (21, 41). Most extracellular virions, called cell-associated enveloped virions (CEV), adhere to the cell surface, including the tips of actin-containing microvilli (3, 38). Enveloped virions that dissociate from the cell are called extracellular enveloped virions (EEV) (24). CEV and EEV contain the same membrane shells and mediate cell-to-cell and longer-range spread, respectively.
The outer membranes of IMV versus those of CEV and EEV have different viral protein components and apparently bind different, although unidentified, cell surface receptors (42). The viral proteins encoded by the A27L, D8L, and H3L genes enhance IMV attachment to cells by binding to proteoglycans (5, 14, 19). Entry of IMV occurs by fusion with the plasma membrane or vesicles formed by surface invaginations in a pH-independent manner (4, 8, 17), although nonfusion mechanisms have also been considered (20). In addition, entry events appear to activate certain cell signaling pathways (6, 18, 20). The partial inhibition of EEV infection by lysosomotropic agents suggests that endocytosis followed by acid disruption of the EEV outer membrane occurs, perhaps followed by fusion of the released IMV with the vesicle membrane (15, 42). The fusion of cells at a late stage of infection, triggered by exposure to low pH, may mimic the latter process by disrupting the outer membrane of enveloped particles on the cell surface (8, 10). Low pH also triggers cell-cell fusion after large numbers of purified IMV are adsorbed to cells, a process known as fusion from without (10).
Recently, we demonstrated that the entry of IMV and spread of CEV depend on the A28 protein, a component of the IMV membrane, and concluded that all forms of vaccinia virus, regardless of their outer coat, use a common mechanism of cell penetration (35). The latter study also provided the first evidence linking cell penetration and acid-triggered cell-cell fusion, as the A28 protein is required for both processes. A28 is not required for virus assembly (34), however, which distinguishes it from other essential IMV membrane proteins. Here we show that vaccinia virus encodes a second protein, H2, with functional characteristics of A28. Thus, A28 and H2 are each required for cell entry and cell-cell fusion. In contrast, IMV lacking the A27 protein, though referred to as a fusion protein (10, 43), was able to mediate acid-triggered fusion from without and enter cells.
MATERIALS AND METHODS
Cells and viruses.
Standard procedures were used for the preparation and maintenance of BS-C-1 cells (ATTC CCL-26) and for the propagation and titration of vaccinia virus (9). IMV was purified twice through a 36% sucrose cushion and banded on a 25-to-40% sucrose gradient as described elsewhere (35).
Protein sequence analysis.
The nonredundant protein sequence database (National Center for Biotechnology Information, National Institutes of Health, Bethesda, Md.) was searched using the BLASTP program, and iterative searches were performed using the PSI-BLAST program (2). Multiple sequence alignments were constructed using the CLUSTAL-X program (39). Protein secondary structure was predicted using the PHD program, with multiple sequence alignments submitted as queries (31).
Construction of vH2i.
vH2i was derived from vT7lacOI (1), which contains an isopropyl-β-d-thiogalactoside (IPTG)-inducible bacteriophage T7 RNA polymerase gene and the Escherichia coli lac repressor gene inserted into the nonessential thymidine kinase locus. The promoter of the H2R gene was replaced with an E. coli lac operator-regulated T7 promoter by homologous recombination using a PCR product that also contained the open reading frame (ORF) encoding enhanced green fluorescent protein (EGFP) regulated by the vaccinia virus synthetic early-late promoter. Plaques containing recombinant virus were identified by EGFP expression using an inverted fluorescence microscope and clonally purified in the presence of IPTG.
Electron microscopy.
BS-C-1 cells were infected with 10 PFU of virus per cell for 1 h at 37°C and incubated in the absence or presence of 100 μM IPTG. The infected cells were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, washed in 0.1 M sodium cacodylate buffer, postfixed with reduced osmium tetroxide, and washed in buffer. Cells were dehydrated in a series of ethyl alcohol dilutions, 50, 70, and 100%, followed by incubation in propylene oxide. The cells were then embedded in EMbed 812. Sections were obtained using the Leica Ultracut S ultramicrotome. Thin sections were stained with 7% uranyl acetate in 50% ethanol and then with 0.01% lead citrate and analyzed on the Philips CM100 transmission electron microscope.
Fluorescence microscopy.
HeLa cells were grown on coverslips, infected with vaccinia virus at a multiplicity of 5 PFU per cell, incubated for the times indicated in the figure legends, and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS). The cells were stained with antibody before or after permeabilization with 0.1% Triton X-100 in PBS followed by 5 μg of diamidino-2-phenylindole dihydrochloride (Molecular Probes)/ml for 5 min. Images were collected on a Leica TCS-NT/SP2 inverted confocal microscope system with an attached argon ion laser (Coherent Inc.).
Sources of antibodies.
Anti-B5 rat monoclonal antibody (MAb) 19C2 (33), anti-L1 MAb 7D11 (46), and anti-A4 rabbit polyclonal antibody (7) were used as primary antibodies for fluorescence microscopy. Cy-5-conjugated anti-rat donkey antibody (Jackson Immunoresearch), fluorescein isothiocyanate-conjugated goat anti-mouse (Jackson Immunoresearch), and Rhodamine Red-X-conjugated goat anti-rabbit antibody (Jackson Immunoresearch) were used as secondary antibodies. Peroxidase-conjugated antihemagglutinin (anti-HA) rat MAb (Roche Applied Science) and peroxidase-conjugated anti-V5 mouse MAb (Invitrogen) were used for Western blotting. Agarose-conjugated anti-HA MAb (Roche Applied Science) and agarose-conjugated anti-V5 mouse MAb (Sigma) were used for immunoprecipitation.
RESULTS
Conservation of H2 in all sequenced poxviruses.
The vaccinia virus H2R ORF (designated VACWR 100 in the WR strain) encodes a 21.5-kDa protein that is highly conserved in all sequenced poxviruses. Based on iterative database searches with the PSI-BLAST program, no significant sequence similarity between H2 and any nonpoxvirus protein was detected. All poxvirus H2 orthologs contain a predicted transmembrane domain, located 30 amino acids from the N terminus, and four conserved cysteines (Fig. 1). Secondary structure analysis suggested several β-strands alternating with α-helices (Fig. 1). The nucleotide sequence upstream of the H2R gene has characteristics of a typical late promoter. Taken together, these conserved features suggested to us that H2 is an IMV membrane protein, and its C-terminal domain with two potential intramolecular disulfide bonds localizes on the surface of the virion.
FIG. 1.

Alignment of H2 ORF orthologs from representative poxviruses. The alignment includes one representative sequence from each genus of Chordopoxvirinae and the two complete Entomopoxvirinae sequences. Virus abbreviation, name, and genus are as follows: VAC, vaccinia virus, Orthopoxvirus; MYX, myxoma virus, Leporipoxvirus; LSDV, lumpy skin disease virus, Capripoxvirus; SPV, swinepox virus, Suipoxvirus; YMTV, Yaba monkey tumor virus, Yatapoxvirus; BPSV, bovine papular stomatitis virus, Parapoxvirus; MCV, molluscum contagiosum virus, Molluscipoxvirus; FPV, fowlpox virus, Avipoxvirus; AMV, Amsacta moorei entomopoxvirus, Entomopoxvirus B; MSV, Melanoplus sanguinipes entomopoxvirus, Entomopoxvirus B. Invariant amino acid residues are shaded grey, and conserved cysteines are indicated additionally by asterisks. TM, predicted transmembrane domain. The predicted secondary structure (SS), consisting of several putative β-strands (E) alternating with α-helices (H), is shown.
H2 is essential for vaccinia virus reproduction and plaque formation.
The high conservation of the H2R gene in all poxviruses implied that it has an essential role. To investigate the function of the H2 protein, we constructed a recombinant vaccinia virus called vH2i, in which the synthesis of H2 transcripts is regulated by the E. coli lac operator system. vH2i encodes the lac repressor controlled by a vaccinia virus dual early-late promoter, the bacteriophage T7 RNA polymerase regulated by a vaccinia virus late promoter and lac operator, and the H2R ORF under the control of the T7 promoter and lac operator (Fig. 2A). In the absence of IPTG, the lac repressor inhibits two consecutive steps: the expression of T7 polymerase and transcription of the H2R ORF, contributing to stringent repression. An EGFP expression cassette was coinserted to allow detection of recombinant virus, which was plaque purified and propagated in the presence of IPTG.
FIG. 2.

Construction and characteristics of recombinant vaccinia virus vH2i. (A) Diagram of relevant portions of the vH2i genome. The locations of the H1L, H2R, H3L, and EGFP ORFs are shown. Additional abbreviations: P11, a vaccinia virus late promoter; P7.5, a vaccinia virus early-late promoter; PE/L, a vaccinia virus early-late synthetic promoter; PT7, bacteriophage T7 promoter; lacO, E. coli lac operator; lacI, E. coli lac repressor ORF; T7 pol, bacteriophage T7 RNA polymerase ORF. (B) Plaque formation and virus spread. BS-C-1 cell monolayers were infected with vH2i in the absence (upper panels) or presence (lower panels) of 100 μM IPTG. Cells were incubated for 48 h and then stained with crystal violet (left) or examined by fluorescence microscopy, with infected cells appearing green (right). (C) Effect of IPTG on the replication of vH2i under one-step growth conditions. Replicate samples of BS-C-1 cell monolayers were infected with 5 PFU of vH2i per cell, incubated for 1 h at 37°C, washed three times, and incubated further at the same temperature in the presence or absence of 100 μM IPTG. Cells were harvested at the indicated times postinfection (p.i.), and the virus titers were determined by plaque assay in the presence of 100 μM IPTG.
When cells were infected with vH2i in the absence of IPTG, single infected cells were detected by their green fluorescence but plaques failed to form (Fig. 2B). In contrast, normal plaques developed in the presence of IPTG (Fig. 2B). The inability of vH2i to spread from cell to cell in the absence of H2 induction could be due to a block in the formation or spread of infectious virus. To differentiate between these possibilities, we compared the yield of vH2i in the presence and absence of IPTG under one-step growth conditions (Fig. 2C). No increase in the amount of infectious vH2i was detected in the absence of IPTG during a 24-h period. In contrast, vH2i increased >100-fold during the same period of time in the presence of 100 μM IPTG. This result showed that the defect in the production of infectious virus caused by repression of H2 was sufficient to account for the absence of plaque formation.
Intracellular and extracellular virions are made in the absence of H2.
A block in morphogenesis could account for the failure of vH2i to produce infectious virus in the absence of IPTG. To investigate this possibility, thin sections of cells infected with vH2i in the presence or absence of IPTG were analyzed by transmission electron microscopy. No defect in morphogenesis was found. Normal-looking immature and mature viral structures, including IMV, IEV, and CEV, were observed in the absence (Fig. 3) as well as the presence (data not shown) of IPTG.
FIG. 3.

Electron microscopic images of cells infected with vH2i in the absence of inducer. BS-C-1 cell monolayers were infected with vH2i in the absence of IPTG for 24 h, fixed, and embedded in Epon, and ultrathin sections were prepared. (A) IMV in the cytoplasm; (B) IEV close to the cell surface; (C) CEV on the cell surface. The bar indicates the scale, in nanometers.
Confocal microscopy was used to determine whether the CEV were associated with actin tails, which are important for cell-to-cell spread of vaccinia virus. HeLa cells were infected with vH2i in the presence or absence of IPTG and stained with a MAb to the B5 protein, an outer membrane component of CEV. The cells were then permeabilized and stained with phalloidin to detect actin. CEV were present at the tips of actin-containing microvilli in the presence or absence of IPTG (Fig. 4), indicating that the defect in spread of vH2i could not be attributed to a failure at this step. Thus far, the phenotype of vH2i resembled the recently described phenotype of vA28i (35), as both conditional lethal viruses produced morphologically normal but noninfectious IMV and CEV.
FIG. 4.

Visualization of vaccinia virus on the surface of cells infected with vH2i in the absence and presence of IPTG. HeLa cells were infected with vH2i either in the presence (+IPTG) or absence (-IPTG) of inducer. After 15 h, the cells were fixed and stained with anti-B5 rat MAb followed by Cy-5-conjugated anti-rat antibody (green). The cells were then permeabilized prior to staining filamentous actin with rhodamine phalloidin (red) and DNA with diamidino-2-phenylindole dihydrochloride (blue). The arrows point to CEV at the tips of actin tails.
The H2R protein is required for low-pH-triggered fusion of infected cells.
Extensive fusion of infected cells can be induced by a brief low-pH exposure at a late time after infection. This process, confusingly named fusion from within, is actually mediated by progeny virions that have been released onto the cell surface. Any treatment or mutation that prevents the formation of virions or their delivery to the cell surface also prevents acid-mediated cell fusion (3, 32, 45). We previously found, however, that fusion from within did not occur when A28 expression was repressed, even though normal amounts of CEV were at the cell surface (35). To determine whether repression of H2 produced a similar defect, we infected cells with vH2i in the presence or absence of IPTG and briefly exposed the cells to pH 5.5 at 18 h after infection. While cells infected in the presence of IPTG formed large syncytia, there were no syncytia in the absence of IPTG (Fig. 5). These results indicated that the fusion of infected cells depends on the expression of H2.
FIG. 5.

Inability of cells infected with vH2i in the absence of IPTG to undergo low-pH-triggered fusion (fusion from within). BS-C-1 cell monolayers were infected with 5 PFU of vH2i per cell in the presence or absence of IPTG for 18 h at 37°C. At that time, the medium was replaced with pH 7.4 (data not shown) or 5.5 buffer for 2 min at 37°C. The incubation was then continued in regular medium for 3 h at 37°C. The cells were fixed, stained with Hoechst reagent to visualize DNA, and examined by phase-contrast (Phase) and fluorescence (Hoechst) microscopy.
Characterization of noninfectious IMV made in the absence of H2.
Until this point in the study, all infections were done with vH2i that had been propagated in the presence of IPTG and therefore contained H2 protein. We refer to such virions as +H2. However, when cells are infected with vH2i in the absence of IPTG, the progeny virions lack H2, and we refer to these as -H2. To further analyze the role of H2, we infected cells with vH2i in the presence or absence of IPTG and purified virus particles by centrifuging them successively through two sucrose cushions and banding them on a sucrose gradient. Both the +H2 and the -H2 IMV, produced in the presence and absence of IPTG, respectively, formed sharp bands in the middle of the gradient, with very similar opalescence and optical density at 260 nm (OD260). The specific infectivity determined by plaque assay, however, was about 100-fold lower for -H2 virions than for +H2 virions. Nevertheless, the negatively stained -H2 and +H2 virions were indistinguishable by electron microscopy (Fig. 6). In addition, no difference in the major proteins of -H2 and +H2 virions was detected by Coomassie blue staining of sodium dodecyl sulfate (SDS)-polyacrylamide gels (Fig. 7). The staining procedure is not sensitive enough to detect H2, which is a relatively minor protein component. Unexpectedly, a major 32-kDa band of vaccinia virus WR was absent in both -H2 and +H2 virions. This extra band in WR virus was identified as the nonessential protein D8 by mass spectroscopy of tryptic peptides. Since D8 was not expressed in the absence or presence of IPTG, the loss of this protein could not account for the phenotype of the mutant virus. We sequenced the D8R ORF of vH2i and found a deletion of one adenine in a run of seven adenines, which caused a frameshift after lysine 108 and premature termination. In contrast, this mutation was not present in the parental vlacOI virus or in five other independent vH2i clones. Importantly, the other clones with an intact D8R ORF were also unable to spread from cell to cell in the absence of IPTG. Therefore, the spontaneous mutation of the nonessential D8 gene did not have a significant effect on the phenotype.
FIG. 6.

Electron microscopic images of IMV purified from cells infected with vH2i in the absence (-H2) or presence (+H2) of IPTG. IMV, purified by two centrifugations through sucrose cushions and banding on a sucrose gradient, were adsorbed to grids, washed with water, and stained with 7% uranyl acetate in 50% ethanol for 30 s. The bar indicates the scale in nanometers.
FIG. 7.
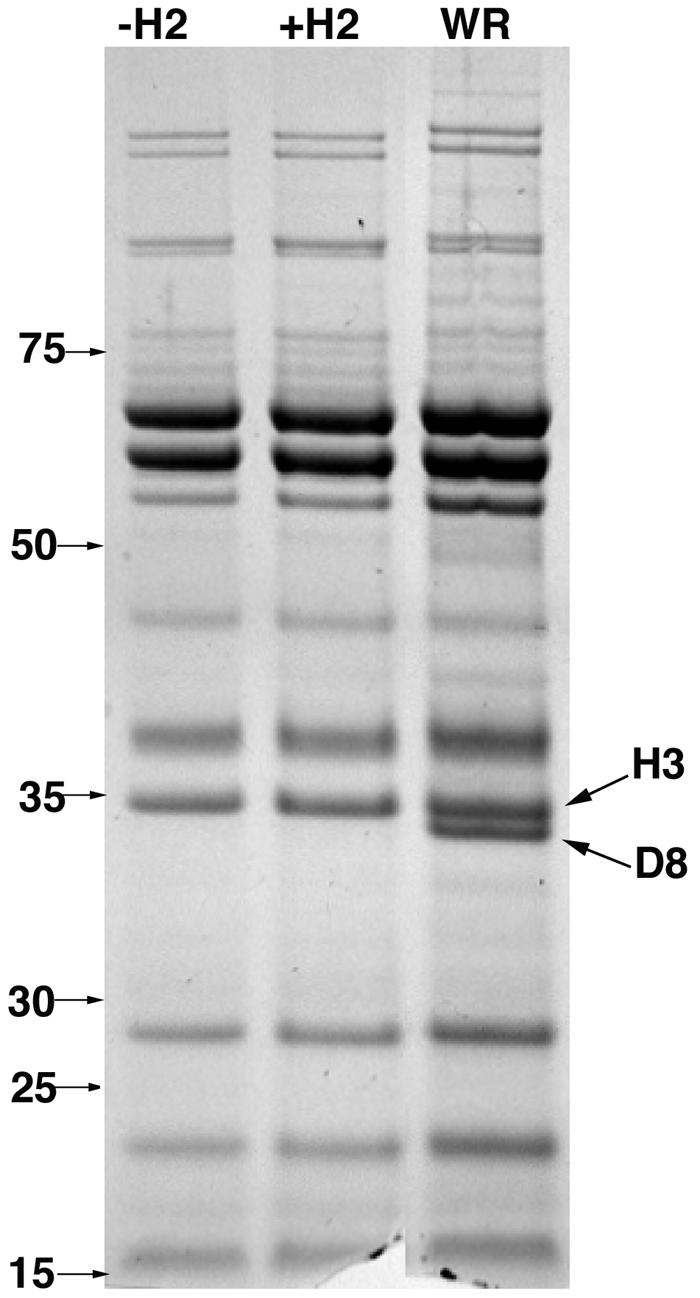
SDS-polyacrylamide gel electrophoresis analysis of IMV purified from cells infected with vH2i in the absence (-H2) or presence (+H2) of IPTG. Equal amounts (based on OD260) of sucrose gradient-purified IMV were resolved in 4-to-12% NuPAGE-SDS gels and stained with Coomassie blue. The masses and positions of marker proteins are shown on the left. The bands corresponding to the H3 and D8 proteins are shown on the right.
H2 is required for virus penetration.
We showed previously that virions lacking A28 can bind to cells but that the cores fail to enter (35). A similar experiment was carried out with purified virions produced in the presence or absence of H2. First, HeLa cells were incubated with -H2 or +H2 virions at 4°C to allow binding. Unattached virus was removed by washing, and the cells were either fixed and processed or incubated for an additional 2.5 h at 37°C to allow cores to penetrate into the cytoplasm. Cycloheximide was present to prevent breakdown of cores and cytopathic effects. The cells were fixed, permeabilized, and stained with a MAb to the L1 IMV membrane protein and an antibody to the A4 core protein to detect IMV on the cell surface and intracellular cores, respectively. This procedure depends on the inability of core antibody to penetrate IMV even after fixation and permeabilization (42). Many punctate A4 antibody-stained cores (red) were seen in cells incubated with infectious +H2 virions, but only rare cores were seen after incubation with noninfectious -H2 virions (Fig. 8) or after incubation of either virion preparation at 4°C (data not shown). In contrast, punctate L1 antibody-staining IMV (green) were seen on the surface of cells in all preparations (Fig. 8). As expected, there was almost no costaining of red cores and green virions (Fig. 8). The presence of noninfectious -H2 virions on the cell surface and the absence of intracellular cores pinpointed the defect to the step of membrane uncoating and penetration.
FIG. 8.

Inability of IMV purified from cells infected with vH2i in the absence of IPTG to penetrate cells. Replicate HeLa cell monolayers on coverslips were infected with 5 PFU of purified +H2 virions per cell or the equivalent OD260 of purified -H2 virions in the presence of 300 μg of cycloheximide per ml. After 1 h adsorption at 4°C, the cells were washed and either fixed or incubated further at 37°C for 2.5 h. Cells were fixed, permeabilized, and double stained with anti-A4 rabbit polyclonal antibody and anti-L1 mouse MAb followed by fluorescein isothiocyanate-conjugated goat anti-mouse (green) and Rhodamine Red-X-conjugated goat anti-rabbit (red) antibody, respectively. DNA was stained with 4′,6′-diamidino-2-phenylindole (blue). Cells were imaged by fluorescence microscopy.
The H2 protein is required for fusion from without.
The term fusion from without is used to describe the formation of syncytia following the adsorption of large numbers of IMV and brief low-pH treatment. BS-C-1 cells were incubated for 1 h at 4°C with 300 PFU per cell of purified infectious +H2 or the same OD260 of noninfectious -H2 IMV. After virus adsorption, the cells were briefly exposed to low or neutral pH at 37°C, followed by incubation with regular medium for 3 h at 37°C in the presence of cycloheximide to prevent any cytopathic effects. Although cultures that had been incubated with +H2 virions and exposed to low pH had extensive syncytia, no fusion was observed in cells that had been incubated with -H2 virions under the same conditions (Fig. 9). As expected, no fusion was observed without low-pH treatment (data not shown). Thus, H2 is required for vaccinia virus-induced fusion from without as well as from within.
FIG. 9.

Inability of IMV purified from cells infected with vH2i in the absence of IPTG to mediate fusion from without. BS-C-1 cell monolayers were incubated with 300 PFU of purified +H2 virions per cell or the equivalent OD260 of purified -H2 virions. After 1 h of adsorption at 4°C, the cells were washed, treated for 2 min with pH 7.4 or 5.5 buffer, and then incubated in regular medium with 300 μg of cycloheximide per ml for 3 h at 37°C. Cells were fixed, stained with Hoechst reagent, and examined by phase-contrast and fluorescence microscopy.
A27 is not required for fusion from without.
It had been reported earlier that virions deficient in the p14 IMV membrane protein (encoded by the A27L ORF of the Copenhagen strain of vaccinia virus or VACWR150 of the WR strain) exhibited reduced fusion from without (43). The low amount of fusion detected was attributed to the leakiness of the inducible mutant and residual A27 in the virus particle. The availability of an A27L deletion mutant (B. Ward, unpublished data), which also contained the A4 protein (VACWR123) fused to yellow fluorescent protein (YFP), allowed us to investigate whether A27 was truly required for fusion. The A27L deletion mutant (vA4-YFP/ΔA27) and the parent vA4-YFP virus were purified by sucrose cushion and gradient sedimentation. At the same OD260, the two viruses made a similar number of plaques, arguing against a defect in cell entry, although the plaques made by the deletion mutant were much smaller, consistent with a defect in wrapping of IMV and formation of CEV (30; Ward, unpublished). In Fig. 10, the tiny plaques made by the A27L deletion mutant on day 9 are compared with plaques produced by parental virus on day 3. Importantly, both the A27L deletion mutant and the A4-YFP virus produced characteristic syncytia in a fusion-from-without assay (Fig. 10). Using twofold dilutions, we found that the same amount of virus measured either by infectivity or by the OD260 was needed to induce complete syncytia of the cell monolayer. The ability of -A27 virions to induce syncytia was in marked contrast to the inability of -H2 virions to produce any syncytia even at higher virus concentrations.
FIG. 10.

Plaque formation and fusion from without mediated by virions lacking the A27 protein. (A) BS-C-1 cell monolayers were incubated with 200 PFU of purified vA4-YFP or vΔA27/A4-YFP virions per cell and treated as described in the legend to Fig. 9. (B) BS-C-1 cell monolayers were infected with vA4-YFP (upper panel) or vΔA27/A4-YFP (lower panel) virions, incubated in semisolid medium containing methyl cellulose for 3 or 9 days, respectively, and then stained with crystal violet.
H2 is an IMV membrane protein associated with A28.
The similar phenotypes of the inducible H2 and A28 mutants under nonpermissive conditions led us to investigate whether they are associated with each other. In a previous study (34), we constructed two recombinant vaccinia viruses containing HA epitope-tagged A28 ORFs. A28-HA is regulated by the normal A28 promoter in vA28-HA and by an IPTG-inducible promoter in vA28-HAi. For the present study, we appended a V5 tag to the C terminus of the H2 ORF in both of the constructs, forming vA28-HA/H2-V5 and vA28-HAi/H2-V5. IMV lacking A28-HA or containing A28-HA were purified from cells infected with vA28-HAi/H2-V5 in the absence or presence of IPTG, respectively. H2-V5 was detected by Western blotting of IMV with or without A28-HA, indicating that the latter is not required for the incorporation of H2 into virus particles. In the absence of A28-HA, most of the H2-V5 was solubilized with NP-40 and dithiothreitol, suggesting a membrane association (Fig. 11A). Relatively less H2-V5 was solubilized from virions that contained A28-HA, leading us to suspect that the two proteins are physically associated (Fig. 11A).
FIG. 11.
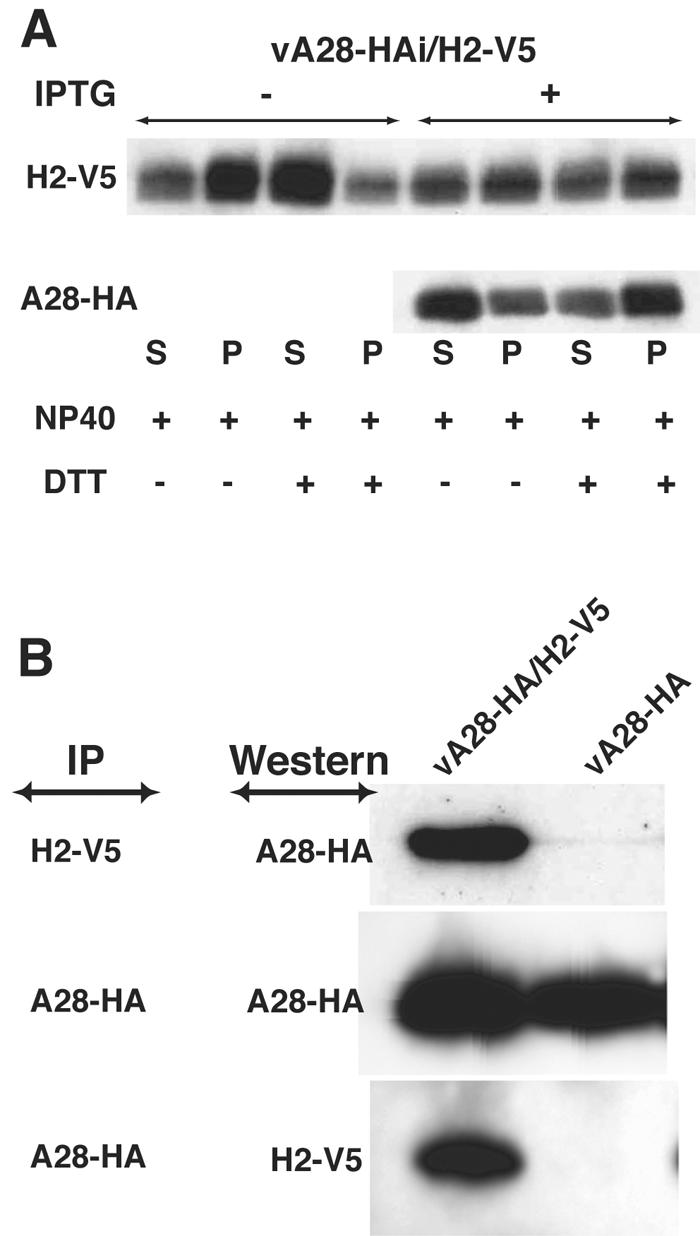
Association of H2 and A28. (A) BS-C-1 cells were infected with vA28-HAi/H2-V5 in the absence or presence of IPTG, and virions lacking A28-HA and containing A28-HA, respectively, were purified by sedimentation through two sucrose cushions and a sucrose gradient. The virions were incubated with NP-40 or NP-40 plus dithiothreitol and centrifuged to separate the soluble membrane and insoluble core fractions (34). The H2-V5 and A28-HA proteins were then detected by Western blotting using MAbs to the epitope tags conjugated to peroxidase. (B) BS-C-1 cells were infected with vA28-HA/H2-V5 or vA28-HA for 20 h, and the cells were lysed with 1% NP-40 in PBS. The detergent-soluble fraction was incubated with agarose beads to remove proteins that bind nonspecifically and then with anti-V5 MAb or anti-HA MAb immobilized on agarose beads. The bound proteins were analyzed by Western blotting with anti-HA or anti-V5 MAb conjugated to peroxidase.
To test for an interaction between H2 and A28, cells were infected with vA28-HA/H2-V5 or vA28-HA, and the proteins in the NP-40-soluble fraction were incubated with agarose beads to remove proteins that bind nonspecifically and then with anti-V5 MAb or anti-HA MAb immobilized on agarose beads. The bound proteins were analyzed by Western blotting with anti-HA MAb or anti-V5 MAb. The V5 MAb coprecipitated A28-HA in cells infected with vA28-HA/H2-V5 but not from the control cells infected with vA28-HA (Fig. 11B). Correspondingly, H2-V5 was detected by Western blotting of proteins that immunoprecipitated with an anti-HA MAb (Fig. 11B). These data indicated that H2 and A28 are associated with each other in infected cells.
DISCUSSION
We investigated the role of H2 by constructing a recombinant vaccinia virus in which the gene was stringently regulated by the E. coli lac operator system. The mutant had a conditional lethal phenotype but no defect in morphogenesis. Under nonpermissive conditions, immature and mature virions formed, and the latter were wrapped and transported to the plasma membrane, where they exited the cells as CEV at the tips of actin tails, which ordinarily facilitate cell-to-cell spread. Nevertheless, there was no cell-to-cell spread of CEV, and purified IMV were noninfectious. The defective virions attached to cells, but the cores could not penetrate into the cytoplasm. In addition, the IMV were incapable of inducing low-pH-triggered cell fusion. Each of these properties of the inducible H2 virus corresponded to those of the previously described inducible A28 virus. Thus, both proteins are needed for cell penetration and fusion. The structural organization of the H2 protein with a transmembrane domain and four invariant cysteines suggests that H2 has a membrane topology similar to that of A28 (34), with intramolecular disulfide bonds in the C-terminal domain on the surface of IMV. Alkylation experiments were consistent with a single reactive cysteine, likely to be the nonconserved one on the short N-terminal side of the transmembrane domain (T. Senkevich, unpublished data). Our finding that the two proteins coimmunoprecipitated from infected cell extracts suggested that they are part of a complex. Nevertheless, A28 expression was not required for the insertion of H2 in the IMV membrane.
The trimeric A27 protein, also called p14, is a component of the IMV membrane and had been implicated in cell fusion (10, 13, 28, 29, 43). The availability of an A27L deletion mutant (Ward, unpublished), however, allowed us to demonstrate unambiguously that purified IMV lacking A27 enters cells and mediates fusion from without. Furthermore, unlike A28 and H2, the A27L gene is not conserved in all poxviruses and has no predicted transmembrane domain. Antibody to A27 (28) and soluble A27 (14) might indirectly prevent virus entry and fusion by steric effects and binding to cell surface proteoglycans (5, 13, 43), respectively. The requirement of the A27 protein for wrapping IMV (30; Ward, unpublished) may explain the inability of A27 null mutants to mediate fusion from within, because that process requires CEV on the cell surface as well as the small plaque phenotype.
In common with A28 and H2, the L1 protein is an IMV transmembrane protein that is expressed late in infection, has intramolecular disulfide bonds, is essential for virus reproduction, and is conserved in all poxviruses (25, 26, 36). The finding that a MAb to L1 prevents virus penetration is also consistent with an involvement at this step of infection (16, 46). The latter finding, however, could be due to steric effects of antibody and does not provide direct evidence that L1 is required for entry. More direct evidence has been difficult to obtain, because mutation of the L1R gene results in a block in virus assembly (26). Thus, it has not been possible to obtain L1-deficient virions to test their binding and entry properties.
Although the initial step of interaction with cell receptors may be different for IMV and for CEV and EEV, our findings indicate that the final step of penetration involves the same A28- and H2-mediated mechanism. Since A28 and H2 are components of the IMV membrane, the outer CEV and EEV envelope would have to be removed so as to expose the underlying IMV membrane prior to fusion. Furthermore, the requirement of both A28 and H2 for penetration and low-pH-induced fusion argues strongly that the two processes are related. The ability of IMV to enter at neutral pH suggests that the fusion process can be activated in more than one way. We suspect that low pH induces massive cooperative fusion events, enabling efficient syncytium formation. Further studies are needed to determine the mechanism of fusion and the cellular receptor proteins involved.
Acknowledgments
We thank Andrea Weisberg for electron microscopy, Owen Schwartz for help with confocal microscopy, Norman Cooper for maintenance of cell lines, Brian Ward for the A27 deletion mutant, and Mariano Esteban and Alan Schmaljohn for antibodies.
REFERENCES
- 1.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blasco, R., and B. Moss. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 66:4170-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, A., and D. H. Metz. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J. Gen. Virol. 32:275-282. [DOI] [PubMed] [Google Scholar]
- 5.Chung, C.-S., J.-C. Hsiao, Y.-S. Chang, and W. Chang. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Magalhaes, J. C., A. A. Andrade, P. N. Silva, L. P. Sousa, C. Ropert, P. C. Ferreira, E. G. Kroon, R. T. Gazzinelli, and C. A. Bonjardim. 2001. A mitogenic signal triggered at an early stage of vaccinia virus infection: implication of MEK/ERK and PKA in virus multiplication. J. Biol. Chem. 276:38353-38360. [DOI] [PubMed] [Google Scholar]
- 7.Demkowicz, W. E., J. S. Maa, and M. Esteban. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J. Virol. 66:386-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doms, R. W., R. Blumenthal, and B. Moss. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, N.Y.
- 10.Gong, S. C., C. F. Lai, and M. Esteban. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 178:81-91. [DOI] [PubMed] [Google Scholar]
- 11.Hiller, G., and K. Weber. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollinshead, M., A. Vanderplasschen, G. L. Smith, and D. J. Vaux. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsiao, J. C., C. S. Chung, and W. Chang. 1998. Cell surface proteoglycans are necessary for A27L protein- mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 72:8374-8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsiao, J. C., C. S. Chung, and W. Chang. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichihashi, Y. 1996. Extracellular enveloped vaccinia virus escapes neutralization. Virology 217:478-485. [DOI] [PubMed] [Google Scholar]
- 16.Ichihashi, Y., and M. Oie. 1996. Neutralizing epitopes on penetration protein of vaccinia virus. Virology 220:491-494. [DOI] [PubMed] [Google Scholar]
- 17.Janeczko, R. A., J. F. Rodriguez, and M. Esteban. 1987. Studies on the mechanism of entry of vaccinia virus into animal cells. Arch. Virol. 92:135-150. [DOI] [PubMed] [Google Scholar]
- 18.Leao-Ferreira, L. R., R. Paes-De-Carvalho, F. G. De Mello, and N. Moussatche. 2002. Inhibition of vaccinia virus replication by adenosine in BSC-40 cells: involvement of A(2) receptor-mediated PKA activation. Arch. Virol. 147:1407-1423. [DOI] [PubMed] [Google Scholar]
- 19.Lin, C. L., C. S. Chung, H. G. Heine, and W. Chang. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locker, J. K., A. Kuehn, S. Schleich, G. Rutter, H. Hohenberg, R. Wepf, and G. Griffiths. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell 11:2497-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meiser, A., C. Sancho, and J. Krijnse Locker. 2003. Plasma membrane budding as an alternative release mechanism of the extracellular enveloped form of vaccinia virus from HeLa cells. J. Virol. 77:9931-9942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 23.Moss, B., and B. M. Ward. 2001. High-speed mass transit for poxviruses on microtubules. Nat. Cell Biol. 3:E245-E246. [DOI] [PubMed] [Google Scholar]
- 24.Payne, L. G. 1980. Significance of extracellular virus in the in vitro and in vivo dissemination of vaccinia virus. J. Gen. Virol. 50:89-100. [DOI] [PubMed] [Google Scholar]
- 25.Ravanello, M. P., and D. E. Hruby. 1994. Characterization of the vaccinia virus L1R myristylprotein as a component of the intracellular virion envelope. J. Gen. Virol. 75:1479-1483. [DOI] [PubMed] [Google Scholar]
- 26.Ravanello, M. P., and D. E. Hruby. 1994. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virus assembly. J. Virol. 68:6401-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Risco, C., J. R. Rodriguez, C. Lopez-Iglesias, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez, J. F., R. Janeczko, and M. Esteban. 1985. Isolation and characterization of neutralizing monoclonal antibodies to vaccinia virus. J. Virol. 56:482-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez, J. F., E. Paez, and M. Esteban. 1987. A 14,000-Mr envelope protein of vaccinia virus is involved in cell fusion and forms covalently linked trimers. J. Virol. 61:395-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez, J. F., and G. L. Smith. 1990. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 18:5347-5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rost, B., C. Sander, and R. Schneider. 1994. PHD—an automatic mail server for protein secondary structure prediction. Comput. Appl. Biosci. 10:53-60. [DOI] [PubMed] [Google Scholar]
- 32.Sanderson, C. M., F. Frischknecht, M. Way, M. Hollinshead, and G. L. Smith. 1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 79:1415-1425. [DOI] [PubMed] [Google Scholar]
- 33.Schmelz, M., B. Sodeik, M. Ericsson, E. J. Wolffe, H. Shida, G. Hiller, and G. Griffiths. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans-Golgi network. J. Virol. 68:130-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 78:2348-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 78:2357-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. USA 99:6667-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith, G. L., B. J. Murphy, and M. Law. 2003. Vaccinia virus motility. Annu. Rev. Microbiol. 57:323-342. [DOI] [PubMed] [Google Scholar]
- 38.Stokes, G. V. 1976. High-voltage electron microscope study of the release of vaccinia virus from whole cells. J. Virol. 18:636-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tooze, J., M. Hollinshead, B. Reis, K. Radsak, and H. Kern. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163-178. [PubMed] [Google Scholar]
- 41.Tsutsui, K. 1983. Release of vaccinia virus from FL cells infected with the IHD-W strain. J. Electron Microsc. 32:125-140. [PubMed] [Google Scholar]
- 42.Vanderplasschen, A., M. Hollinshead, and G. L. Smith. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79:877-887. [DOI] [PubMed] [Google Scholar]
- 43.Vazquez, M. I., and M. Esteban. 1999. Identification of functional domains in the 14-kilodalton envelope protein (A27L) of vaccinia virus. J. Virol. 73:9098-9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ward, B. M., and B. Moss. 2004. Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J. Virol. 78:2486-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolffe, E. J., S. N. Isaacs, and B. Moss. 1993. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 67:4732-4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53-63. [DOI] [PubMed] [Google Scholar]