

Abstract
The human immunodeficiency virus gp41 envelope protein mediates the entry of the virus into the target cell by promoting membrane fusion. In order to gain new insights into the viral fusion mechanism, we studied a 35-residue peptide pertaining to the loop domain of gp41, both in solution and membrane bound, by using infrared and fluorescence spectroscopy. We show here that the peptide, which has a membrane-interacting surface, binds and interacts with phospholipid model membranes and tends to aggregate in the presence of a membranous medium and induce the leakage of vesicle contents. The results reported in this work, i.e., the destabilization and fusion of negatively charged model membranes, suggest an essential role of the loop domain in the membrane fusion process induced by gp41.
Human immunodeficiency virus (HIV) is the causative agent of AIDS, which acts by killing the CD4 T cells of the host organism (15, 23, 32, 47). Membrane fusion, i.e., the attachment and consequent fusion of viral and cellular membranes, is mediated by envelope glycoproteins located on the outer surface of the viral membrane. The HIV surface glycoprotein is initially synthesized as a highly glycosylated precursor, gp160, which is later cleaved endoproteolytically by one cellular convertase into the surface protein gp120, which determines viral tropism through the cellular surface receptors, and the transmembrane protein gp41, which is responsible for the membrane fusion process. Both gp120 and gp41 remain noncovalently associated and supposedly trimeric in their native prefusogenic state (15, 23).
The native state of the HIV gp41 envelope protein is thought to be metastable by interacting with gp120 (32). For fusion and mixing of the viral and cellular contents to occur, gp41 must undergo a complex series of conformational changes, apparently triggered by the attachment of gp120 to CD4 and chemokine receptors of the target cell (13). It is thought that gp41 catalyzes membrane fusion through the induction of transient nonlamellar structures at the point where both bilayers merge (15, 23). The gp41 sequence is highly conserved and contains different functional regions within its ectodomain that are critical for membrane fusion (Fig. 1). A stretch of about 15 hydrophobic residues, named the fusion peptide (FP) and located at the N terminus of gp41, is believed to insert into and destabilize the membrane, thus facilitating viral and cell membrane fusion (4, 21). Consecutive to the FP, two heptad repeat regions, NHR and CHR, have been identified (22). Another essential segment for gp41-mediated fusion is the Trp-rich pretransmembrane domain (PTM) linking the CHR to the membrane-embedded anchor, TM (44). In the absence of lipid membranes, three NHR molecules fold into a central parallel triple-stranded α-helical coiled coil, with an outer layer of three antiparallel CHR α-helices wrapped antiparallel on the outside of this core and with each pair of NHR and CHR molecules connected by an immunogenic, protease-sensitive loop that reverses the polypeptide chain (6, 7, 48). This trimeric helical hairpin structure is thought to form at a late stage during the membrane fusion process (7). It has been proposed that there is a prehairpin intermediate in gp41-induced membrane fusion in which the N-terminal coiled coil is formed but the C-terminal helices are not packed (50). The transition from the prehairpin to the hairpin structure brings the two membranes into close proximity, driving fusion. Although much information has been gathered in recent years, we do not yet know the exact mechanisms of membrane fusion and the processes behind them.
FIG. 1.

(A) Schematic view of HIVHXB2 gp41 ectodomain showing the structural and functional regions as well as their relative lengths. The fusion peptide (FP), the loop, the N- and C-terminal heptad repeats (NHR and CHR), the pretransmembrane stretch (PTM), and the transmembrane (TM) domain were drawn approximately to scale. The residue numbers correspond to their positions in gp160 of the HXB2 HIV strain. The amino acid composition of the gp41 fragment studied in this work is also indicated. (B) gp41 hairpin conformation scheme showing the membranotropic regions of gp41 (see reference 17).
While the FP and the PTM are essential for perturbation of the membrane, other regions of gp41 are also involved in the interaction and destabilization of the viral and host membranes. It was shown recently that additional segments of the gp41 ectodomain bind to and partition the surfaces of phospholipid model membranes, change their conformation, and induce the formation of nonlamellar structures, indicating that these segments may play an essential role in the viral fusion process in addition to the other membrane-perturbing domains (12, 14, 26, 33, 34, 35, 38, 44, 45). These regions might interact with biological membranes, contributing to the merging of the viral envelope and the cell membrane. Since it is desirable to identify critical gp41 segments that might be responsible for the fusion process, we have studied a peptide of 35 amino acids, analogous to residues 579 to 613 of the HXB2R strain, comprising part of the conserved immunodominant region of the gp41 ectodomain and the C-terminal region of the NHR region (Fig. 1). The purposes of the present work were to study this gp41 fragment both in aqueous solution and in the presence of different membrane model systems composed of different phospholipid mixtures, to evaluate its incorporation and location in membrane model systems, and to study its effect on the integrity and phase behavior of the membrane. The results reported here suggest that this gp41 fragment binds and interacts with negatively charged membranes and therefore that this region of the gp41 protein, together with the FP and PTM domains, might have a direct role in the conformational change of the gp41 subunit and subsequent fusion activation.
MATERIALS AND METHODS
Materials.
The HIV envelope protein gp41 fragment (Arg-Ile-Leu-Ala-Val-Glu-Arg-Tyr-Leu-Lys-Asp-Gln-Gln-Leu-Leu-Gly-Ile-Trp-Gly-Cys-Ser-Gly-Lys-Leu-Ile-Cys-Thr-Thr-Ala-Val-Pro-Trp-Asn-Ala-Ser, with N-terminal acetylation and C-terminal amidation) corresponding to amino acids 579 to 613 of HIVHXB2R was obtained from Genemed Synthesis, San Francisco, Calif. The peptide was purified by reverse-phase high-performance liquid chromatography to >95% purity, and its composition and molecular mass were confirmed by amino acid analysis and mass spectroscopy. Since trifluoroacetate has a strong infrared absorbance at approximately 1,673 cm−1, which interferes with the characterization of the peptide amide I band (46), residual trifluoroacetic acid, used both in peptide synthesis and in the high-performance liquid chromatography mobile phase, was removed by several lyophilization-solubilization cycles in 10 mM HCl (53). Cholesterol (Chol), 1,2-dimyristoyl-sn-glycero-phosphatidylcholine (DMPC), 1,2-dimyristoyl-d54-sn-glycero-3-phosphocholine (DMPCd), 1,2-dimyristoyl-sn-glycero-phosphatidylglycerol (DMPG), 1,2-dimyristoyl-sn-glycero-phosphatidic acid (DMPA), egg sphingomyelin (SM), egg phosphatidylcholine (EPC), egg phosphatidylglycerol (EPG), egg phosphatidic acid (EPA), and bovine brain phosphatidylserine (BPS) were obtained from Avanti Polar Lipids (Birmingham, Ala.). Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (N-Rh-PE), N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (NBD-PE), 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS), and p-xylene-bis-pyridiniumbromide (DPX) were obtained from Molecular Probes (Eugene, Oreg.). Deuterium oxide (99.9% by atom), Triton X-100, EDTA, and HEPES were purchased from Sigma (St. Louis, Mo.). All other chemicals were commercial samples of the highest purity available. Water was double distilled and deionized in a Millipore system (Madrid, Spain).
Hydrophobic moments, hydrophobicity, and interfaciality.
Hydrophobic moment calculations were performed according to the method of Eisenberg et al. (17, 18), and the scale for calculating hydrophobic moments was taken from the work of Engelman et al. (19). Hydrophobicity and interfacial values, i.e., whole-residue scales for the transfer of an amino acid of an unfolded chain into the membrane hydrocarbon palisade and the membrane interface, respectively, were obtained from http://blanco.biomol.uci.edu/hydrophobicity_scales.html (49, 51).
Sample preparation.
Aliquots containing appropriate amounts of lipid in chloroform-methanol (2:1 [vol/vol]) were placed in a test tube, the solvents were removed by evaporation under a stream of O2-free nitrogen, and finally, traces of solvents were eliminated under a vacuum in the dark for more than 3 h. A preweighed amount of freeze-dried protein was suspended by the addition of an appropriate volume of buffer containing 20 mM HEPES, 50 mM NaCl, and 0.1 mM EDTA, pH 7.4 (in either D2O or H2O [see below]). The protein solution was then added to the tube containing the dried lipid to obtain a final lipid/peptide molar ratio of 15:1, unless otherwise stated, and the suspension was vortexed at about 5°C above the transition temperature of the phospholipid to obtain multilamellar vesicles. The mixture was freeze-thawed twice in order to ensure complete homogenization of the sample and a maximization of contacts between the peptide and the phospholipid and then was incubated for 10 min at 55°C with occasional vortexing. The freeze-thaw cycle was repeated again, and the suspension was then centrifuged at 12,000 × g for 15 min in order to remove any peptide that was not bound to the phospholipids in the membrane. The freeze-thaw, incubation at 55°C, and centrifugation steps were repeated once more in order to remove the unbound protein. The pellet was resuspended in either D2O or H2O buffer and used for measurements. Except for temperature studies, all data were obtained at 25°C. The phospholipid and peptide concentrations were measured by previously described methods (5, 16).
Infrared measurements.
Infrared measurements were conducted essentially as described previously (24). Fourier transform infrared spectra were obtained in a Nicolet 520 Fourier transform infrared spectrometer equipped with a deuterated triglycine sulfate detector and a sample shuttle accessory in order to average the background spectra among the sample spectra over the same time period. Subtraction of the buffer spectra, determinations of frequencies at the center of gravity, and band-narrowing strategies were applied as previously reported (24). Protein secondary structure elements were quantified from a curve-fitting analysis by band decomposition of the original amide I′ band after spectral smoothing (for details, see reference 24). Briefly, for each component, the following three parameters were considered: band position, band height, and bandwidth. The number and position of component bands were obtained through deconvolution, and for decomposing the amide I′ band, gaussian components were used. The curve-fitting procedure was accomplished in two steps: in the first one, the band position was fixed, allowing the width and height to approach final values, and in the second one, band positions were allowed to change. When necessary, these two steps were repeated. Band decomposition was performed with SpectraCalc software (Galactic Industries, Salem, Mass.). The fitting result was evaluated visually by overlapping the reconstituted overall curve onto the original spectrum and by examining the residual obtained by subtracting the fit from the original curve. The procedure gave differences of <2% in band areas after the artificial spectra were subjected to the curve-fitting procedure. The frequency positions of the band centers were independently evaluated by second-derivative procedures, as they were always very close to the positions found by deconvolution.
Fluorescence measurements.
Fluorescence measurements were performed essentially as described previously (12). For binding and fluorescence anisotropy experiments using the intrinsic fluorescence of Trp, small unilamellar vesicles were used (12). Large unilamellar vesicle (LUV) liposomes were used to study vesicle aggregation, lipid mixing, and leakage, and they were prepared by the extrusion method (25) by the use of polycarbonate filters with a pore size of 0.1 μm (Nuclepore, Pleasanton, Calif.). The buffer used to prepare LUV liposomes for assays of vesicle leakage also contained 25 mM ANTS and 90 mM DPX. Nonencapsulated fluorescent probes were separated from the vesicle suspension through a Sephadex G-75 filtration column (Pharmacia, Uppsala, Sweden) and were eluted at room temperature with a buffer containing 10 mM HEPES, 130 mM NaCl, and 0.1 mM EDTA, pH 7.4. Steady-state fluorescence measurements were taken as described previously (12, 24). The excitation and emission wavelengths were 295 and 359 nm, respectively, for observing peptide fluorescence. All data were corrected for background intensities and progressive dilution, but emission spectra were not corrected for photomultiplier wavelength dependence. Fluorescence anisotropies were determined according to the following equation (12):
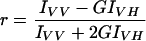 |
where IVV and IVH are the fluorescence intensities, with the subscripts indicating the vertical (V) or horizontal (H) orientation of the excitation and emission Glan-Thompson polarizers. The instrumental factor G (G = IHV/IHH) was determined by measuring the polarized components of fluorescence of the protein or probes with horizontally polarized excitation. Partition coefficients were obtained according to the following equation:
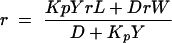 |
where rL and rW are anisotropies in the lipid and aqueous phases, respectively, Y = ΦL/ΦW, D = 1/[(γ[L]) − 1], and ΦL and ΦW are the quantum yields in both phases. Leakage was assayed by treating the probe-loaded liposomes (final lipid concentration, 0.1 mM) with appropriate amounts of peptide in a fluorometer cuvette. Changes in fluorescence intensity were recorded at excitation and emission wavelengths of 350 and 510 nm, respectively. One hundred percent release was achieved by adding Triton X-100 to the cuvette at a final concentration of 0.1% (wt/wt). Leakage was quantified on a percent basis according to the following equation:
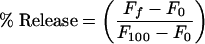 |
with Ff being the equilibrium value of fluorescence after the peptide addition, F0 being the initial fluorescence of the vesicle suspension, and F100 being the fluorescence value after the addition of Triton X-100. Peptide-induced vesicle lipid mixing was measured as described previously (43) by the use of LUV liposomes. The concentration of both fluorescent probes, NBD-PE and N-Rh-PE, was 0.6%. Labeled and unlabeled LUV vesicles at a ratio of 1:9 were placed in a cuvette at a final lipid concentration of 0.1 mM and were treated with appropriate amounts of peptide. Fluorescence was measured at excitation and emission wavelengths of 470 and 585 nm, respectively. We were not able to distinguish between complete fusion or hemifusion due to the ability of the peptide to induce leakage of the vesicle contents.
RESULTS
To study the ability of the gp41 peptide fragment to interact with membranes, we obtained fluorescence anisotropy values in the presence of phospholipid model membranes at different lipid/peptide ratios (Fig. 2A). In the presence of model membranes, the anisotropy values of the gp41 fragment increased upon increasing the lipid/peptide ratio, with the limiting value being about 0.20 to 0.23, indicating a significant motional restriction of the Trp moieties of the peptide at a relatively high lipid/protein ratio (28). However, differences in the anisotropy values of the gp41 fragment were observed when it was in the presence of the zwitterionic phospholipid EPC and in the presence of the negatively charged phospholipids BPS, EPG, and EPA (Fig. 2A). This was further corroborated by a change in the emission frequency maximum of Trp, as shown in Fig. 2B. In solution, the peptide had an absorbance maximum at 295 nm and an emission maximum at 354 nm when excited at the absorbance maximum, indicating that the Trp residues of the peptide were in a hydrophilic environment. In the presence of increasing concentrations of EPC, the emission maximum of the Trp changed from 354 to 351 nm (Fig. 2B). However, in the presence of increasing concentrations of negatively charged phospholipids, the emission maximum changed from 354 to about 345 nm for EPG and from 354 to about 343 nm for EPA and BPS, i.e., there was a difference of about 9 to 11 nm for negatively charged phospholipids (Fig. 2B). Since upon an increase in the environmental hydrophobicity the Trp fluorescence emission increases and the emission maximum frequency decreases (28), the shift of the emission maximum of Trp by about 11 and 9 nm in the presence of BPS and EPA liposomes and EPG liposomes, respectively, suggested that Trp sensed a low-polarity environment (entered a hydrophobic environment) in the presence of liposomes containing negatively charged phospholipids, but to a much lesser extent in the presence of zwitterionic liposomes. The peptide, having a theoretical isoelectric point of 9.1, has a charge of about +1.91 at pH 7.4, as used in this work. EPG, EPA, and BPS, in contrast to the zwitterionic phospholipid EPC, are negatively charged phospholipids, so the binding force of the peptide to liposomes containing those phospholipids had an apparently electrostatic origin. In these experiments, the phospholipid/peptide molar ratio reached a very high value (2,300:1), and the spectral contribution of free peptide was negligible. The change in anisotropy values of the Trp fluorescence of the peptide allowed us to obtain its apparent partition coefficient, Kp. Using a γ value of 0.519 M−1 (41), we obtained Kp values of 1.9 × 102 ± 0.39 × 102 for EPC, 1.56 × 103 ± 0.4 × 103 for EPA, 1.55 × 103 ± 0.6 × 103 for EPG, and 3.2 × 103 ± 1.4 × 103 for BPS, indicating that the peptide was bound to the surfaces of negatively charged phospholipid-containing model membranes with a high affinity. Similar Kp values have been found for other peptides pertaining to the gp41 protein in the presence of negatively charged phospholipid-containing membranes (12, 41).
FIG. 2.

Fluorescence anisotropy (A) and fluorescence emission maxima (B) of the gp41 fragment in the presence of increasing concentrations of small unilamellar vesicles composed of EPG (⋄), EPA (•), BPS (▴), and EPC (□).
In order to further explore the possible interaction of the gp41 fragment with phospholipid model membranes, we studied the effect of the peptide on the release of encapsulated fluorophores. For these experiments, we used unsaturated phospholipids instead of saturated ones because of the large amount of variability in the basal fluorescence of the latter; nevertheless, the results were qualitatively similar for both unsaturated and saturated phospholipids. Figure 3 shows the results obtained with different liposome compositions, namely EPC, EPA, EPG, BPS, and a mixture of DMPA, DMPC, SM, and Chol at a molar ratio of 57:29:7:7. The peptide hardly exerted any effect on liposomes made of EPC, but significant leakage values were observed for LUVs composed of negatively charged phospholipids, with higher values for EPG-containing liposomes than for other liposomes (Fig. 3A). We also studied phospholipid mixing, as shown in Fig. 3B, for which we again found significant values for EPG-containing liposomes. The curvature degree, i.e., the capability of obtaining nonlamellar phases, was higher for EPG than for the other phospholipids used in this work, which might explain why we observed lipid mixing for EPG-containing liposomes but not for the other liposome types studied. The peptide also induced liposome aggregation (results not shown), as observed by the increase in light scattering for liposomes composed of either EPG, EPA, or BPS, but not for those composed of EPC, and only at a very low ratio of lipid to peptide, i.e., 5:1.
FIG. 3.

Effect of gp41 fragment on leakage data at 25°C for LUV contents (A) and phospholipid mixing (B) for different lipid compositions and different lipid-to-peptide molar ratios. ⋄, EPG; •, EPA; ▴, BPS; □, EPC; ×, DMPA-DMPC-SM-Chol.
The existence of structural changes induced by membrane binding was investigated by looking at the infrared amide I′ band located between 1,700 and 1,600 cm−1 (1). The infrared spectrum of the amide I′ region of the fully hydrated peptide in D2O buffer at 25°C and pH 7.4 is shown in Fig. 4A. The spectrum was formed by different underlying components that gave rise to a broad and asymmetric band with a maximum at about 1,642 cm−1. The maximum of the band did not change significantly upon increasing the temperature, indicating a high degree of conformational stability of the peptide in solution. It was also apparent that no aggregation of the peptide took place, since no band was observed at 1,618 to 1,622 cm−1 (1). In order to study the peptide when it was effectively bound to the phospholipid membrane, we prepared all samples containing both phospholipids and the peptide by mixing and washing the unbound peptide as described in Materials and Methods. The infrared spectra in the amide I′ and C=O regions of samples prepared in this way and containing the peptide and unsaturated phospholipid, either EPC, EPA, or EPG, are shown in Fig. 4B, C, and D, respectively. Whereas a small quantity of peptide was bound to EPC liposomes (Fig. 4B), a significant amount was bound to either EPA or EPG (Fig. 4C and D, respectively). When EPC was used, the peptide was recovered almost completely in the supernatant (not shown), indicating that the peptide was only bound in significant amounts to negatively charged membranes and not to zwitterionic phospholipids. As shown in the deconvolved spectra (Fig. 4C and D, dotted lines), two bands, at about 1,622 and 1,685 cm−1, with the former band being more intense than the latter, can be discerned. The infrared spectra in the amide I′ and C=O regions of samples containing the gp41 fragment and model membranes composed of saturated phospholipids, either DMPA, DMPCd and DMPG at a molar ratio of 1:1, or DMPC, DMPA, SM, and Chol at a molar ratio of 57:29:7:7, are shown in Fig. 4E, F, and G, respectively. In all cases, a significant amount of peptide was bound to the model membrane systems, as found previously for membranes containing negatively charged phospholipids. The band envelope of the amide I′ band of the peptide bound to these model membranes was different from that found for the pure peptide in solution since, as found above, two bands, at about 1,622 and 1,685 cm−1, were well resolved (Fig. 4E, F, and G). The frequencies of the 1,622- and 1,685-cm−1 bands and their relative intensities indicate the presence of extended β-strands with strong intermolecular interactions (1). This was in contrast to the peptide in solution, for which no bands at 1,622 and 1,685 cm−1 were observed (Fig. 4A). It is interesting that the bandwidth at the half-height of the amide I′ band envelope when the peptide was bound to the membranes increased 4 to 5 cm−1 compared to the peptide in solution. The increase in bandwidth indicates the presence of additional structures contributing to the broader envelope, more flexibility of the peptide, or both.
FIG. 4.

Infrared (solid lines) and deconvolved (dotted lines) spectra of the amide I′ and C=O regions of samples containing the gp41 fragment in solution (A) and in the presence of EPC (B), EPA (C), EPG (D), DMPA (E), DMPCd and DMPG at a molar ratio of 1:1 (F), and DMPC, DMPA, SM, and Chol at a molar ratio of 57:29:7:7 (G). For the unsaturated samples, the spectra shown are those obtained at 25°C, whereas for the saturated ones, the spectra correspond to those recorded at 10°C above the Tm of the phospholipid mixture. The phospholipid/peptide molar ratio was 15:1 in all samples.
To observe the underlying components of the broad amide I′ band, we applied self-deconvolution and derivative methods to the original envelope (1, 24). For the peptide in solution, we identified different component bands at frequencies of about 1,680, 1,667, 1,652, 1,638, 1,623, and 1,605 cm−1, with the 1,638-cm−1 band being the main one (Fig. 5A). The results of the decomposition of the amide I′ band of the gp41 fragment in the presence of three different model membrane systems, i.e., DMPA, DMPCd and DMPG at a molar ratio of 1:1, and DMPC, DMPA, SM, and Chol at a molar ratio of 57:29:7:7, are shown in Fig. 5B, C, and D, respectively, in which it is also possible to distinguish component bands with similar frequencies to those found for the peptide in solution, i.e., bands at about 1,680, 1,667, 1,652, 1,638, 1,623, and 1,605 cm−1. To assign the component bands to specific structural features and to estimate the percentage of each component, we decomposed the amide I′ infrared band as described in Materials and Methods. The most significant difference between the amide I′ band in solution and in the presence of model membranes was the change in the intensity of the 1,623-cm−1 band: the change was about 20% for the peptide in solution and about 27 to 30% for the peptide in the presence of model membranes (Table 1).
FIG. 5.

Amide I′ band decomposition of gp41 fragment spectra in solution (A) and in the presence of DMPA (B), DMPC, DMPA, SM, and Chol at a molar ratio of 57:29:7:7 (C), and DMPCd and DMPG at a molar ratio of 1:1, all of which were at a phospholipid/peptide molar ratio of 15:1 in D2O medium at 10°C above the Tm of the phospholipid mixture. The original spectra (solid lines), the component bands (dotted lines), and the differences between the original and fitted spectra (dotted lines with increased spacing) are shown. In the upper part of each panel, the deconvolved original (solid line) and fitted (dotted line) spectra are overlapped (see the text for details).
TABLE 1.
Secondary structure content of gp41 peptide fragment, as determined by infrared spectroscopya
| Peptide environment | % α-Helix (∼1,650-1,655 cm−1) | % β-Sheet (1,625-1,640 cm−1) | % 310-Helix (∼1,655-1,670 cm−1) | % β-Turn (1,670-1,685 cm−1) | % β-Aggregate (∼1,620-1,625 cm−1) |
|---|---|---|---|---|---|
| In solution | 24 | 31 | 15 | 10 | 20 |
| DMPA | 23 | 25 | 16 | 10 | 26 |
| DMPC-DMPA-SM-Chol | 19 | 27 | 14 | 11 | 29 |
| DMPA-DMPCd | 20 | 25 | 15 | 9 | 30 |
Values are rounded off to the nearest integer.
We also studied the effects of the gp41 fragment on the phase transitions of DMPA, DMPCd-DMPG, and a mixture of DMPC, DMPA, SM, and Chol by infrared spectroscopy. The temperature dependence of the CH2 symmetric frequency of pure DMPA is shown in Fig. 6A, in which a highly cooperative change at approximately 48°C is shown, corresponding to the gel-to-liquid crystalline phase transition, Tm, of the phospholipids (30). In the presence of the peptide, a small decrease in the gel-to-liquid crystalline phase transition temperature was observed, but the frequencies above and below the Tm were similar (Fig. 6). No significant effect was observed for the DMPC-DMPA-SM-Chol mixture in the presence of the peptide. However, for the sample composed of DMPCd and DMPG, although no change was observed for the Tm of the mixture, the frequency above but not below the Tm decreased, indicating that the incorporation of the peptide increased the proportion of trans isomers, i.e., the hydrocarbon chains of the phospholipids were more ordered in the presence of the peptide below the main phase transition.
FIG. 6.

Temperature dependence of the CH2 (A, B, and D) and CD2 (C) symmetric stretching frequency in the presence (▪) and absence (○) of the gp41 fragment in phospholipid samples containing DMPA (A), DMPC, DMPA, SM, and Chol at a molar ratio of 57:29:7:7 (B), and DMPCd and DMPG at a molar ratio of 1:1 (C and D). The lipid-to-peptide molar ratio was 15:1.
After deconvolution, the broad C=O carbonyl band presented two components, at 1,742 and 1,727 cm−1, which represented the sn-1 and sn-2 C=O groups in their dehydrated and hydrated forms, respectively (30). The frequencies of these two components are not affected by temperature, but their relative intensities change depending on the physical state of the phospholipid bilayer (30). For pure DMPA, the frequency at the maximum of the C=O vibration band presented one transition at approximately 48°C, since the 1,742-cm−1 component had a higher intensity than the 1,727-cm−1 component below Tm and the opposite above it (Fig. 7A). In the presence of the gp41 fragment, the two components of the C=O carbonyl band did not change in frequency compared to the pure phospholipid, but they changed in relative intensity. This change in intensity gave rise to a slightly broad transition, and significantly, the maximum of the frequency above the Tm was higher in the presence of the peptide than in its absence (Fig. 7A). The same effect was observed for the other phospholipid model membranes, DMPCd-DMPG and DMPC-DMPA-SM-Chol, in the presence of the gp41 fragment, i.e., there was a slightly broad transition and a higher frequency of the maximum of the C=O carbonyl band (Fig. 7A). The presence of the gp41 fragment did not induce any significant differences in the frequency and width of the acyl chain CH2 scissoring and the PO2− double bond stretching bands appearing at 1,468 and 1,220 cm−1, respectively, compared with the pure phospholipids, indicating that there were no differences in packing and hydration between the pure phospholipid and the mixtures containing the peptide (not shown).
FIG. 7.

Temperature dependence of the C=O stretching band frequency (A) and the maximum of the amide I′ band (B) for samples containing the gp41 fragment and different phospholipid mixtures at a phospholipid-to-peptide molar ratio of 15:1. Samples contained DMPA (• and ○), DMPCd-DMPG (▪ and □), and DMPC-DMPA-SM-Chol (▴ and ▵). Filled symbols represent the frequencies of samples containing both phospholipid and peptides, whereas open symbols represent those of the pure lipid mixtures.
The temperature dependence of the maximum of the amide I′ band of the gp41 fragment in solution can be observed in Fig. 7B. At low temperatures, the frequency maximum was about 1,639 cm−1 and increased to about 1,645 cm−1 at the highest temperature studied. In the presence of DMPC-DMPA-SM-Chol, the frequency was about 1,633 cm−1 at low temperatures and decreased to about 1,625 cm−1 at higher ones, whereas in the presence of either DMPA or DMPCd-DMPA, it was about 1,625 cm−1 at all temperatures (Fig. 7B). It was possible to distinguish a small increase in the intensity of the broad component appearing at about 1,646 cm−1 for the peptide in the presence of DMPA (Fig. 4E), but the maximum of the band remained the same throughout all of the temperature range studied. Moreover, no defined transitions were observed in the amide I′ band which could be concomitantly ascribed to that observed for the phospholipid in the same sample (Fig. 7B). A similar behavior was observed for the gp41 fragment in the presence of DMPCd-DMPG (Fig. 4F and 7B). However, it was possible to observe a different behavior of the peptide in the presence of the mixture composed of DMPC, DMPA, SM, and Chol, since the maximum of the band decreased from about 1,633 at 20°C to about 1,625 cm−1 at 70°C, i.e., a decrease of about 9 cm−1 (Fig. 7B). This decrease in frequency was accompanied by an increase in the intensity of the band at about 1,622 cm−1 (Fig. 4G), indicating an increase in the extended β-strands upon an increase in the temperature.
DISCUSSION
Membrane fusion reactions are involved in many important biological processes, but strong electrostatic, hydration, and steric repulsion forces represent large energetic barriers (11). In biological systems, these barriers are overcome by fusion proteins, with the simplest membrane fusion reaction being the one produced by the entry of enveloped viruses into host cells (3, 15, 23). Different structural conformational changes induced by a complex series of protein-protein and protein-phospholipid interactions occur in fusion proteins. However, little is known about how these conformational changes drive membrane apposition and how they overcome the energy barrier for membrane fusion (29). Several lines of evidence indicate that, in addition to classical fusion peptides, different regions of fusion proteins, including the HIV gp41 ectodomain, are essential for membrane fusion (8, 12, 27, 37, 40, 45). Therefore, destabilization of the lipid bilayer appears to be the result of the interaction of different segments of fusion proteins with the membrane and therefore might play a critical role in membrane fusion. Understanding the factors that determine the specificity and stability of the metastable (native) and stable (fusogenic) conformations is required for an understanding of the mechanisms of viral membrane fusion and consequent viral entry into cells (3).
Many studies have been performed to examine the interactions of synthetic peptides mimicking the N-terminal fusion peptide and the Trp-rich proximal membrane regions of HIV gp41. However, fewer studies have been done on the interaction of other segments of the gp41 protein with membranes. We have recently shown the presence of different membranotropic regions within the gp41 ectodomain, i.e., the FP, the proximal region connecting the FP to the NHR, the PTM, and the loop (33). Moreover, an analysis of the hydrophobic and interfacial properties of the gp41 ectodomain surface (not shown) identified the already known membranotropic sequences in the gp41 ectodomain, i.e., the FP, PTM, and TM domains, and strongly suggests the presence of a stretch with interfacial hydrophobicity characteristics in the loop region. In this study, we have focused on the possible roles of the loop region in the membrane fusion process by studying a peptide which originated from this region. This 35-amino-acid peptide binds with a high affinity to negatively charged membranes, with a binding force of an apparently electrostatic origin. We previously found similar binding affinities for shorter peptides pertaining to the loop region of the gp41 protein in the presence of negatively charged phospholipid-containing model membranes (12). However, significantly, the 35-amino-acid peptide studied here is also capable of binding, although with a lower affinity, to membranes containing zwitterionic phospholipids (see above). After binding, the Trp residue resides in an environment with a low dielectric constant showing significant motional restriction, confirming that this portion of the peptide enters a hydrophobic environment but remains located at the surface of the membrane, as indicated by the absence of significant effects on the frequency of the CH2 and CD2 stretching bands of the hydrocarbon chains of the phospholipids. The strong binding of the peptide to negatively charged membranes was also confirmed by infrared spectroscopy. Only the amide I′ band corresponding to the bound peptide was present in model membranes composed of negatively charged phospholipid-containing membranes, being nearly absent in model membranes formed by DMPC.
The infrared spectra of the amide I′ region of the fully hydrated peptide in D2O buffer at 25 and 70°C were very similar, indicating the stability of its conformation in solution, even at high temperatures. However, when the peptide was bound to negatively charged phospholipids, its conformation changed, as the intensity of the 1,622-cm−1 band, assigned to oligomeric structures (10), increased significantly. Although we have worked with phospholipid model membranes, the binding to the surface and the modulation of the phospholipid biophysical properties which take place when the peptide is bound to the membrane, i.e., partitioning into the membrane surface, aggregation, and perturbation of the bilayer architecture, may be related to the conformational changes which occur upon binding of the HIV-1 envelope glycoprotein to its receptors. The change in conformation and the possible formation of oligomeric forms in the presence of membranes may indicate the propensity of the peptide to self-assemble and suggest that these changes might be part of the structural transition that transforms gp41 from the inactive to the active state, which is most probably the dominant form during membrane fusion (9). It should be taken into account that the location of the peptide at the surface of the membrane may effectively reduce the head-group area of the phospholipid and promote the formation of nonbilayer phases (3, 12).
It has previously been suggested that hairpin folding drives the fusion reaction by initiating pore formation, but it was recently shown that fusion pore formation occurs prior to hairpin formation and that hairpins might be required for pore stabilization (31). Folding into a hairpin would bring the apposing membranes together, align the target membrane fusion domains with the transmembrane region of the viral envelope, and imply a direct interaction of the two membrane-proximal regions with their respective membrane targets (42). Since membrane fusion can be described by a succession of steps, i.e., the apposition of membranes, hemifusion of the outer leaflets, pore formation, and pore enlargement, exposed regions of the prehairpin intermediates should be responsible for the very first steps of fusion until pore initiation. Membrane fusion requires membrane destabilization, and many studies have shown that different regions of the gp41 ectodomain can significantly perturb membranes (8, 12, 27, 33, 37, 40, 45). The gp41 peptide fragment that we studied binds to, interacts with, and perturbs phospholipid model membranes, and at the same time, tends to aggregate in a membranous environment. Therefore, it would be tempting to consider that the region where this sequence resides in the native protein plays a significant role in the fusion mechanism induced by gp41. Moreover, it is supposed that the β-sandwich structure of the gp120 inner domain is associated with the gp41 loop by specific hydrophobic interactions before gp120 interacts with its receptors (52). The loop domain of gp41, exposed after gp120 interacts with its receptor, may bind to and destabilize the host cell membrane as well as stabilize the trimeric helical hairpin after it is formed (36). It should also be noted that the energy required for the conformational changes which take place during this process may be provided by membrane binding (36). Interestingly, the study of different mutations at residues located in the loop region suggested that the loop region has a direct role in the events that take place in the membrane fusion process, possibly affecting both the formation of fusion pores and their expansion (2).
Taking into account all of these data, we have summarized in a simplified model the roles of the different membranotropic regions of gp41 (Fig. 8). Following the triggering of the envelope glycoproteins by receptors and coreceptors, the gp41 protein, which is free to oscillate, inserts the FP, most likely in the β-sheet conformation (39), into the target membrane, joining them together. gp41 monomers interact with both membranes through the FP, the PTM, and the loop; at the same time that this interaction is taking place, it might be capable of inducing the self-assembly of gp41 oligomers at the membrane surface where the fusion site will be located (20). Even so, the two interacting faces of gp41 might interact with their adjacent membranes before or concomitantly with hairpin formation, i.e., the NHR and CHR domains have the multiple functions of binding to membranes, destabilizing them, and folding into the hairpin conformation in coordination with the loop, PTM, and FP regions (31, 42). The change in free energy associated with the structural changes taking place should be sufficient to cause lipid mixing and membrane fusion. The existence of specific lipid domains (rafts) in the membranes may be important at this step (42). In this way, the locations of complexes of gp41 would define a specific interfacial hydrophobicity distribution on the membrane surface and thus drive fusion pore enlargement (29). This interfacial hydrophobicity would also be responsible for the tendency of segments derived from rafts to oligomerize at the membrane surface (see above).
FIG. 8.

Schematics of the hypothetical intermediates of gp41-induced membrane fusion. Dark segments represent membranotropic regions of gp41. The different segments which intervene in the fusion process are shown as cylinders but are not necessarily helical. The formation of gp41 complexes at the membrane surface would create fusion-competent monomers and oligomers, the folding of gp41 into hairpins would align membrane-associated TM and FP domains, and the loop would be placed near the membrane surface and would participate along with the PTM and FP domains in the fusion process; the oligomerization of hairpins should stabilize the pore at the fusion site, leading to complete fusion.
The HIV entry process is considered an attractive target for chemotherapeutic intervention, as blocking HIV entry into target cells leads to a suppression of viral infectivity, viral replication, and the cytotoxicity induced by virus-cell contacts (15, 54). The importance of identifying drugs that prevent membrane fusion is emphasized by the success of drug combination regimens for the treatment of AIDS, which are mainly directed towards inverse transcriptase and viral proteases. Several compounds are being developed to specifically target each of the steps leading to virus entry, and some compounds have reached early clinical development. However, other problems, such as long-term toxicity, cost, and the emergence of multiple drug-resistant strains, remain far from being solved (54). Resistance would be minimized, though not eliminated, by the use of entry inhibitors in combination with existing reverse transcriptase and protease inhibitors. Therefore, the inhibition of membrane fusion by a direct action on gp120 and/or gp41 is increasing in importance as an additional approach either to directly combat HIV infection or to prevent its spread. An understanding of the structural features of the prefusogenic and fusogenic intermediates is thus very important because gp41 appears to be an attractive drug target. The results reported in this work sustain the notion that peptides originating from the loop region of gp41 are capable of modifying the biophysical properties of phospholipid membranes, providing an additional driving force for the merging of viral and target cell membranes. In conclusion, the data shown in this work suggest that this region might play a significant role in the membrane fusion mechanism induced by gp41.
Acknowledgments
This work was supported by grants BMC2002-00158 (Ministerio de Ciencia y Tecnología, Spain) and LSHB-CT-2003-503480 (European Union project “Targeting Replication and Integration of HIV” [TRIoH]) to J.V. R.P. and M.R.M. are recipients of predoctoral fellowships from Ministerio de Educación y Ciencia, Spain.
REFERENCES
- 1.Arrondo, J. L., A. Muga, J. Castresana, and F. M. Goñi. 1993. Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 59:23-56. [DOI] [PubMed] [Google Scholar]
- 2.Bär, S., and M. Alizon. 2004. Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J. Virol. 78:811-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blumenthal, R., M. J. Clague, S. R. Durell, and R. M. Epand. 2003. Membrane fusion. Chem. Rev. 103:53-69. [DOI] [PubMed] [Google Scholar]
- 4.Bosch, M. L., P. L. Earl, K. Fargnoli, S. Picciafuoco, F. Giombini, F. Wong-Staal, and G. Franchini. 1989. Identification of the fusion peptide of primate immunodeficiency viruses. Science 244:694-697. [DOI] [PubMed] [Google Scholar]
- 5.Böttcher, C. S. F., C. M. Van Gent, and C. Fries. 1961. A rapid and sensitive sub-micro phosphorus determination. Anal. Chim. Acta 24:203-204. [Google Scholar]
- 6.Caffrey, M., M. Cai, J. Kaufman, S. J. Stahl, P. T. Wingfield, D. G. Covell, A. M. Gronenborn, and G. M. Clore. 1998. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 17:4572-4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. [DOI] [PubMed] [Google Scholar]
- 8.Chan, D. C., C. T. Chutkowski, and P. S. Kim. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 95:15613-15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang, D. K., S. F. Cheng, and V. D. Trivedi. 1999. Biophysical characterization of the structure of the amino-terminal region of gp41 of HIV-1. Implications on viral fusion mechanism. J. Biol. Chem. 274:5299-5309. [DOI] [PubMed] [Google Scholar]
- 10.Chehín, R., M. Thorolfsson, P. M. Knappskog, A. Matínez, T. Flatmark, J. L. R. Arrondo, and A. Muga. 1998. Domain structure and stability of human phenylalanine hydroxylase inferred from infrared spectroscopy. FEBS Lett. 422:225-230. [DOI] [PubMed] [Google Scholar]
- 11.Chernomordik, L. V., and M. M. Kozlov. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175-207. [DOI] [PubMed] [Google Scholar]
- 12.Contreras, L. M., F. J. Aranda, F. Gavilanes, J. M. González-Ros, and J. Villalaín. 2001. Structure and interaction with membrane model systems of a peptide derived from the major epitope region of HIV protein gp41: implications on viral fusion mechanism. Biochemistry 40:3196-3207. [DOI] [PubMed] [Google Scholar]
- 13.Dimitrov, A. S., X. Xiao, D. S. Dimitrov, and R. Blumenthal. 2000. Early intermediates in HIV-1 envelope glycoprotein-mediated fusion triggered by CD4 and co-receptor complexes. J. Biol. Chem. 276:30335-30341. [DOI] [PubMed] [Google Scholar]
- 14.Dimitrov, A. S., S. S. Rawat, S. Jiang, and R. Blumenthal. 2003. Role of the fusion peptide and membrane-proximal domain in HIV-1 envelope glycoprotein-mediated membrane fusion. Biochemistry 42:14150-14158. [DOI] [PubMed] [Google Scholar]
- 15.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777-810. [DOI] [PubMed] [Google Scholar]
- 16.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6:1948-1954. [DOI] [PubMed] [Google Scholar]
- 17.Eisenberg, D., R. M. Weiss, and T. C. Terwilliger. 1982. The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299:371-374. [DOI] [PubMed] [Google Scholar]
- 18.Eisenberg, D., E. Schwarz, M. Komaromy, and R. Wall. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179:125-142. [DOI] [PubMed] [Google Scholar]
- 19.Engelman, D. M., T. A. Steitz, and A. Goldman. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Chem. 15:321-353. [DOI] [PubMed] [Google Scholar]
- 20.Follis, K. E., S. J. Larson, M. Lu, and J. H. Nunberg. 2002. Genetic evidence that interhelical packing interactions in the gp41 core are critical for transition of the human immunodeficiency virus type 1 envelope glycoprotein to the fusion-active state. J. Virol. 76:7356-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallaher, W. R. 1987. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell 50:327-328. [DOI] [PubMed] [Google Scholar]
- 22.Gallaher, W. R., J. M. Ball, R. F. Garry, M. C. Griffin, and R. C. Montelaro. 1989. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res. Hum. Retrovir. 5:431-440. [DOI] [PubMed] [Google Scholar]
- 23.Gallo, S. A., C. M. Finnegan, M. Viard, Y. Raviv, A. Dimitrov, S. S. Rawat, A. Puri, S. Durell, and R. Blumenthal. 2003. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 1614:36-50. [DOI] [PubMed] [Google Scholar]
- 24.Giudici, M., R. Pascual, L. de la Canal, K. Pfüller, U. Pfüller, and J. Villalaín. 2003. Interaction of viscotoxins A3 and B with membrane model systems: implications to their mechanism of action. Biophys. J. 85:971-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hope, M. J., M. B. Bally, G. Webb, and P. R. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 812:55-65. [DOI] [PubMed] [Google Scholar]
- 26.Kliger, Y., S. G. Peisajovich, R. Blumenthal, and Y. Shai. 2000. Membrane-induced conformational change during the activation of HIV-1 gp41. J. Mol. Biol. 301:905-914. [DOI] [PubMed] [Google Scholar]
- 27.Kliger, Y., S. A. Gallo, S. G. Peisajovich, I. Muñoz-Barroso, S. Avkin, R. Blumenthal, and Y. Shai. 2001. Mode of action of an antiviral peptide from HIV-1. Inhibition at a post-lipid mixing stage. J. Biol. Chem. 276:1391-1397. [DOI] [PubMed] [Google Scholar]
- 28.Lakowicz, J. R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Plenum Press, New York, N.Y.
- 29.Lentz, B. R., V. Malinin, M. E. Haque, and K. Evans. 2000. Protein machines and lipid assemblies: current views of cell membrane fusion. Curr. Opin. Struct. Biol. 10:607-615. [DOI] [PubMed] [Google Scholar]
- 30.Mantsch, H. H., and E. N. McElhaney. 1991. Phospholipid phase transitions in model and biological membranes as studied by infrared spectroscopy. Chem. Phys. Lipids 57:213-226. [DOI] [PubMed] [Google Scholar]
- 31.Markosyan, R. M., F. S. Cohen, and G. B. Melikyan. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell 14:926-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markovic, I., and K. A. Clouse. 2004. Recent advances in understanding the molecular mechanisms of HIV-1 entry and fusion: revisiting current targets and considering new options for therapeutic intervention. Curr. HIV Res. 2:223-234. [DOI] [PubMed] [Google Scholar]
- 33.Moreno, M. R., R. Pascual, and J. Villalaín. 2004. Identification of membrane-active regions of the HIV-1 envelope glycoprotein gp41 using a 15-mer gp41-peptide scan. Biochim. Biophys. Acta 1661:97-105. [DOI] [PubMed] [Google Scholar]
- 34.Peisajovich, S. G., R. F. Epand, M. Pritsker, Y. Shai, and R. M. Epand. 2000. The polar region consecutive to the fusion peptide participates in membrane fusion. Biochemistry 39:1826-1833. [DOI] [PubMed] [Google Scholar]
- 35.Peisajovich, S. G., and Y. Shai. 2001. SIV gp41 binds to membranes both in the monomeric and trimeric states: consequences for the neuropathology and inhibition of HIV infection. J. Mol. Biol. 311:249-254. [DOI] [PubMed] [Google Scholar]
- 36.Peisajovich, S. G., L. Blank, R. F. Epand, R. M. Epand, and Y. Shai. 2003. On the interaction between gp41 and membranes: the immunodominant loop stabilizes gp41 helical hairpin conformation. J. Mol. Biol. 326:1489-1501. [DOI] [PubMed] [Google Scholar]
- 37.Poumbourios, P., W. el Ahmar, D. A. McPhee, and B. E. Kemp. 1995. Determinants of human immunodeficiency virus type 1 envelope glycoprotein oligomeric structure. J. Virol. 69:1209-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sackett, K., and Y. Shai. 2002. The HIV-1 gp41 N-terminal heptad repeat plays an essential role in membrane fusion. Biochemistry 41:4678-4685. [DOI] [PubMed] [Google Scholar]
- 39.Sackett, K., and Y. Shai. 2003. How structure correlates to function for membrane associated HIV-1 gp41 constructs corresponding to the N-terminal half of the ectodomain. J. Mol. Biol. 333:47-58. [DOI] [PubMed] [Google Scholar]
- 40.Salzwedel, K., J. T. West, and E. Hunter. 1999. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 73:2469-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos, N. C., M. Prieto, and M. A. Castanho. 1998. Interaction of the major epitope region of HIV protein gp41 with membrane model systems. A fluorescence study. Biochemistry 37:8674-8682. [DOI] [PubMed] [Google Scholar]
- 42.Shnaper, S., K. Sackett, S. A. Gallo, R. Blumenthal, and Y. Shai. 2004. The C- and the N-terminal regions of glycoprotein 41 ectodomain fuse membranes enriched and not enriched with cholesterol, respectively. J. Biol. Chem. 279:18526-18534. [DOI] [PubMed] [Google Scholar]
- 43.Struck, D. K., D. Hoekstra, and R. E. Pagano. 1981. Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20:4093-4099. [DOI] [PubMed] [Google Scholar]
- 44.Suárez, T., S. Nir, F. M. Goñi, A. Sáez-Cirión, and J. L. Nieva. 2000. The pre-transmembrane region of the human immunodeficiency virus type-1 glycoprotein: a novel fusogenic sequence. FEBS Lett. 477:145-149. [DOI] [PubMed] [Google Scholar]
- 45.Suárez, T., W. R. Gallaher, A. Agirre, F. M. Goñi, and J. L. Nieva. 2000. Membrane interface-interacting sequences within the ectodomain of the human immunodeficiency virus type 1 envelope glycoprotein: putative role during viral fusion. J. Virol. 74:8038-8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surewicz, W. K., H. H. Mantsch, and D. Chapman. 1993. Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment. Biochemistry 32:389-394. [DOI] [PubMed] [Google Scholar]
- 47.Turner, B. G., and M. F. Summers. 1999. Structural biology of HIV. J. Mol. Biol. 285:1-32. [DOI] [PubMed] [Google Scholar]
- 48.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wimley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426-430. [DOI] [PubMed] [Google Scholar]
- 49.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319-365. [DOI] [PubMed] [Google Scholar]
- 50.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770-9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842-848. [DOI] [PubMed] [Google Scholar]
- 52.York, J., and J. H. Nunberg. 2004. Role of hydrophobic residues in the central ectodomain of gp41 in maintaining the association between human immunodeficiency virus type 1 envelope glycoprotein subunits gp120 and gp41. J. Virol. 78:4921-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, Y. P., R. N. A. H. Lewis, R. S. Hodges, and R. N. McElhaney. 1992. FTIR spectroscopic studies of the conformation and amide hydrogen exchange of a peptide model of the hydrophobic transmembrane alpha-helices of membrane proteins. Biochemistry 31:11572-11578. [DOI] [PubMed] [Google Scholar]
- 54.Zwick, M. B., E. O. Saphire, and D. R. Burton. 2004. gp41: HIV's shy protein. Nat. Med. 10:133-134. [DOI] [PMC free article] [PubMed] [Google Scholar]