

ABSTRACT
Immunoglobulin A nephropathy (IgAN) is a common glomerulonephritis partially correlated with mucosal immune system dysfunction. Progressive renal failure occurs in many patients, with about 30–50% of the patients with IgAN developing end-stage kidney disease (ESKD). Many treatments have been used for decades, despite uncertainty about their effectiveness and the ideal dose. Randomised controlled trials reported that systemic glucocorticoids can be an effective treatment for patients with persistent and significant proteinuria despite renin-angiotensin system inhibitors use possibly causing systemic side effects. The primary focus of IgAN management should be based on optimised supportive care, including renin-angiotensin system (RAS) blockade and now SGLT2 inhibitors. The novel targeted-release formulation (TRF) of budesonide has been tested to reduce the adverse events of systemic steroids by delivering the drug to the distal ileum. The local efficacy of TRF-budesonide may represent a novel and promising approach to treating IgAN. Two clinical trials showed that TRF-budesonide could significantly reduce proteinuria and haematuria and possibly preserve renal function while significantly reducing the side effects. However, the limited number of treated patients and the relatively short follow-up suggest caution before considering budesonide superior to the current six-months steroid pulses scheme. Long-term data on the efficacy and safety of TRF budesonide are awaited, together with the design of trials with a head-to-head comparison with systemic steroids before considering TRF-budesonide as the standard of care treatment for IgAN nephropathy.
Keywords: budesonide, IgA nephropathy, proteinuria, steroids, targeted-release formulation
INTRODUCTION
IgA nephropathy (IgAN) is the most common glomerulonephritis in the world and a major cause of end-stage kidney disease (ESKD) [1–4]. Proteinuria at follow-up and time-averaged proteinuria are the best assessed primary determinant of the rate of progression to ESKD demonstrated today [5, 6]. More recently, the International IgAN Prediction Tool has become available for predicting the risk of a 50% decline in eGFR or ESKD after biopsy [7].
According to the Kidney Disease Improving Outcome (KDIGO) recommendations, patients with proteinuria >1 g/day despite at least 90 days of optimised supportive care have a high risk of progressive loss of kidney function and may be considered for a six-month course of glucocorticoid therapy [8]. However, glucocorticoid use in IgAN is controversial despite being used for decades because not all patients benefit from the treatment. A few patients may develop severe side effects, usually when high-dose glucocorticoids are used.
This review will consider the mechanisms of action and the results of randomised studies with systemic glucocorticoids, and the potential therapeutic role of target-release formulation (TRF) budesonide, a new topical corticosteroid, and other topical formulations.
SYSTEMIC GLUCOCORTICOIDS
Mechanisms of action
Circulating levels of endogenous cortisol begin to increase between 2 and 4 a.m. peak a few minutes after awakening, and then decline through the day, reaching a nadir between 11 p.m. and 1 a.m. [9]. The half-life of cortisol is short (about 66 minutes). Synthetic glucocorticoids have longer half-lives and may be classified according to their half-life into short-acting (prednisone and derivates, deflazacort), with plasma half-lives of 60–200 minutes, intermediate (paramethasone and triamcinolone), with plasma half-lives of about 300 minutes, and long-acting (dexamethasone and betamethasone), with a half-life of up to 48 hours.
The genomic effects of glucocorticoids depend on the doses, availability, and affinity of the glucocorticoid receptors (GRs). GR undergoes a conformational change that triggers translocation into the nucleus upon binding glucocorticoids. Here, GRs bind to specific GC response elements of the target genes that control the transcription of anti-inflammatory genes. The gene transcription is called transactivation [10]. The process of impaired transcription is called transrepression [11]. GRs may exert pleiotropic effects in different organs; in the human kidney glomeruli, all glomerular cells can express GRs [12].
Non-genomic effects depend on interactions of glucocorticoids with a non-classical membrane-bound GR [13]; they are characterised by rapid onset (seconds to minutes) and short duration of action (60–90 minutes).
Glucocorticoids can elicit a variety of adverse events that are usually dose- and time-dependent. Many adverse events can be attributed to the ability of the GRs to transactivate genes producing metabolic effects. Instead, the beneficial anti-inflammatory effects are mainly due to transrepression.
Toxicity prevention
Measures to reduce glucocorticoid toxicity include using a single morning dose or alternate-day therapy to respect the circadian rhythm of cortisol and reduce pituitary adrenal suppression. A hypocaloric and low-salt diet, a supplement of calcium and Vitamin D3 400 U.I. daily, physical activity and smoke avoidance may prevent many of the side effects [14]. Bisphosphonate could be added for osteoporosis prevention, but rarely may cause focal and segmental glomerulosclerosis. Using formulations that exert non-genomic effects can reduce the risk of side effects. In this respect, intravenous megadoses of glucocorticoids may downregulate glucocorticoid receptors and exert non-genomic effects [14–17].
The rationale for use in IgA nephropathy
The mechanistic interpretation of IgAN is based on four-hit pathogenesis. The first hit is represented by circulating IgA1, which have an increased concentration of abnormally glycosylated molecules. IgA1 molecules are synthesised in the gut and tonsil, and respiratory mucous membranes, eliciting cytokine-driven aberrant mucosal immune responses [18]. The second hit, which is necessary for the development of the disease, is represented by an auto-antibody response (IgA or IgG class) to the abnormal IgA1 molecules, resulting in the formation of immune complexes (third hit). Circulating immune complexes can accumulate in the mesangium and activate complement through the lectin and alternative pathways, inducing the proliferation of mesangial cells and the secretion of extracellular matrix, cytokines, and chemokines, eventually resulting in kidney injury (fourth hit) (Fig. 1) [19].
Figure 1:
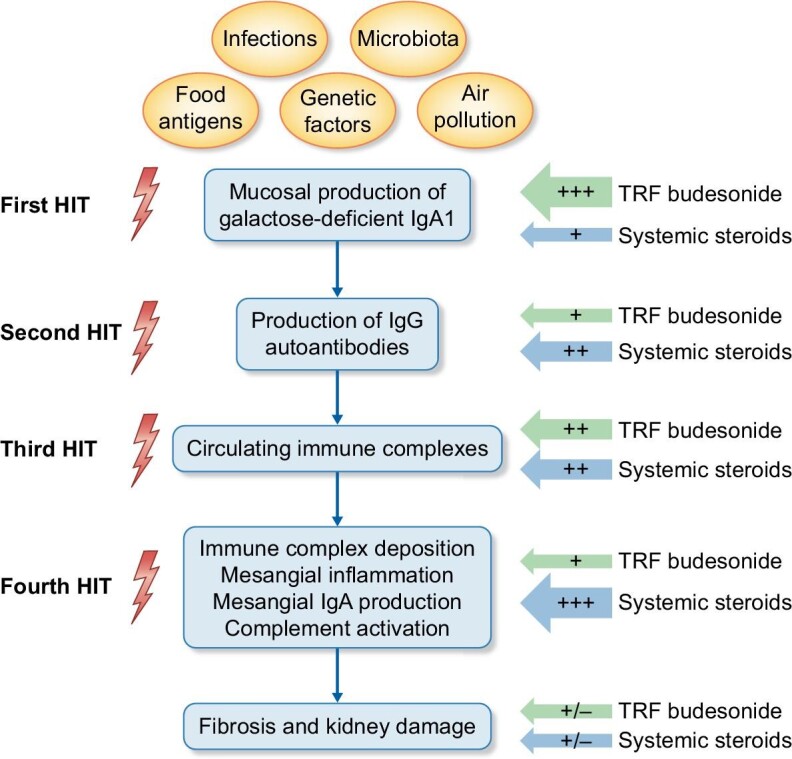
The four-hit hypothesis in the pathogenesis of IgA nephropathy and possible sites of actions of systemic steroids and target released formulation budesonide.
The pathogenesis of IgAN is made even more complex by recent observations that IgA-secretory plasmablasts can migrate from the mucosae to the kidney [18] and produce serum IgA against mesangial antigens, such as βII-spectrin [18, 20].
Systemic glucocorticoids may exert anti-inflammatory and immunosuppressive actions. Most effects are mediated by repressing the activity of key transcription regulators, such as kappa-light-chain-enhancer of activated B cells and activator protein 1, and other transcription factors [21]. Because GRs are almost ubiquitously expressed, glucocorticoids can exert systemic and local effects. In the kidney, glucocorticoids not only may directly interfere with the overproduction of inflammatory mediators, but may also exert protective effects on podocytes via the GR [22]. Systemic glucocorticoids may reduce the formation of autoantibodies towards abnormal IgA1 molecules. In this respect, patients treated with systemic steroids or immunosuppressants had much lower levels of IgA-IgG immunocomplexes than untreated ones [23]. On the contrary, total plasma IgA levels were not significantly changed during treatment [23], possibly suggesting that systemic corticosteroids have little effect on mucosal production of antibodies towards aberrant IgA.
RESULTS FROM CLINICAL TRIALS
Randomised clinical trials (RCTs) showed the efficacy and good tolerance of steroid regimen based on a short course of methylprednisolone pulses (MPP) followed by low-dose oral prednisone [24, 25].
In a long-term RCT, 86 patients with biopsy-proven IgAN, proteinuria between 1 to 3.5 g/day, and serum creatinine levels of ≤1.5 mg/dL were randomised to symptomatic treatment or to receive three intravenous MPP at the beginning of month 1, 3, and 5 followed by alternate-day oral prednisone, 0.5 mg/48 hour, for six months [26]. In this study, the supportive care with renin-angiotensin system (RAS) inhibitors was incomplete for historical reasons and thus not comparable with more recent glucocorticoid trials. The 10-year kidney survival was 97% in the treated group vs 53% in the controls (P = .0005) [27]. Proteinuria decreased from 1.9 g/day to 0.6 g/day (P < .001) in treated patients, increasing from 1.7 g/day to 2.0 g/day in the control arm. Excepting one patient who developed diabetes, the other patients assigned to glucocorticoids did not experience any significant side effects during follow-up. Of note, only 2 of 43 patients randomised to steroid therapy had a relapse of mild proteinuria over the ten-year follow-up [27]. They were successfully retreated with the same schedule.
In another Italian RCT, 97 IgAN patients with proteinuria >1 g/day and an estimated glomerular filtration rate (eGFR) >50 ml/min/1.73 m2 were randomised to receive ramipril alone or ramipril plus daily high-dose oral prednisone 1 mg/day for 60 days, then gradually tapered off [28]. After a follow-up till 96 months, 13/49 (26.5%) controls in the monotherapy group reached the primary outcome (doubling of serum creatinine or ESKD) compared with 2/48 (4.2%) in the prednisone group. The mean rate of eGFR decline was higher and proteinuria lower in the monotherapy group than in the prednisone one (P = .013). Adverse events were mild in both groups [28].
In a small pilot study, 63 patients were randomly assigned to either cilazapril (RAS inhibitor) alone or prednisone plus cilazapril. The combination therapy provided additional benefits compared with the RAS inhibitor alone [29].
In the Supportive Versus Immunosuppressive Therapy for the Treatment of Progressive IgA Nephropathy (STOP) trial, 162 participants with proteinuria >0.75 g/day were assigned to supportive care with RAS inhibitors or RAS inhibitors plus three MPP at the start of months 1, 3, 5 and prednisolone 0.5 mg/kg/48 hours up to 6 months [1]. Participants with an eGFR between 30 and 59 ml/min/1.73 m2 were given oral prednisolone, 40 mg/day tapered to 10 mg/day over the first three months plus cyclophosphamide, 1.5 mg/kg/day for three months, then azathioprine, 1.5 mg/kg/day up to month 36. After three years of follow-up, 14 participants in the immunosuppression group had complete clinical remission versus four in the supportive care group (P = .001). However, there was no difference between the two groups in the decrease of eGFR >15 ml/min/1.73 m2 (28% vs 26%). Serious adverse events were similar in the two groups (29 versus 33). Malignancies developed in two participants who received cyclophosphamide and azathioprine. In a second report, the follow-up of the STOP trial was extended up to 10 years (median 7.4 years) [30]: ESKD occurred in 17 patients randomised to supportive care and 20 with additional immunosuppression. Also, the rates of eGFR loss over 40% and annual eGFR loss did not differ between groups. The two study groups had similar numbers of serious adverse events. Two patients died in the supportive care group, and three in that with additional immunosuppression.
It is difficult to understand the lack of GFR protection despite the remission of proteinuria reported in the STOP trial. However, the finding should also be interpreted considering the slow progression rate of the control group. In a retrospective analysis of the European Validation Study of the Oxford Classification of IgAN (VALIGA) study, 184 participants who received glucocorticoids and RAS inhibitors were matched with 184 patients who received only RAS inhibitors [31]. Glucocorticoids significantly reduced proteinuria and the rate of kidney function decline and increased renal survival in patients with proteinuria >1 g/day. These benefits extended to participants with an eGFR ≤50 ml/min/1.73 m2, and the benefits increased proportionally with the level of proteinuria and the time elapsed after randomisation [31].
The Therapeutic Effects of Steroids in IgA Nephropathy Global (TESTING) trial assessed the efficacy and safety of oral methylprednisolone compared to placebo in 503 Chinese patients with IgAN treated with RAS inhibitors [32]. Participants were randomised to methylprednisolone (0.6–0.8 mg/kg/day; maximum, 48 mg/day) or placebo for two months, with subsequent weaning over 4–6 months. After randomising 262 patients, recruitment was discontinued because of excess severe adverse events: 14.7% in the methylprednisolone group vs 3.2% in the placebo group (P = .001). The dose of methylprednisolone was reduced (0.4 mg/kg/day, maximum 32 mg/day, weaning by 4 mg/day/month), and antibiotic prophylaxis for pneumocystis pneumonia was added for subsequent participants. Over a mean follow-up of 4.2 years, the primary renal outcome measure (ESKD or a 40% decrease in eGFR) occurred in 74 participants (28.8%) in the methylprednisolone group compared with 106 (43.1%) in the placebo group (P < .001). Serious adverse events were more frequent with high-dose methylprednisolone vs placebo (10.9% vs 2.8%) [32].
These RCTs showed that steroid therapy added to supportive therapy induced a significantly higher remission rate than supportive care alone. However, the TESTING trial reported more side effects in patients given steroids alone [32], and the STOP study found severe adverse events when adding cytotoxic drugs [29]. Ethnicity, age, gender, degree of kidney function, duration and entity of proteinuria, genetic factors, and inflammatory status were not the same in the different trials. Also, monitoring the participants and respecting measures to prevent side effects are not described. Finally, the type of glucocorticoid administration is of paramount importance. Previous studies concluded that a course of three MPP followed by alternate-day prednisone proved effective and safe with a favourable safety profile. Recently, another Chinese RCT compared a full-dose prednisone regimen (0.8–1.0 mg/kg for two months, then tapered off over four months) with three IV MPP at day 1 and 90 plus alternate-day prednisone (15 mg/48 hours) in 87 patients with biopsy-proven IgAN and proteinuria between 1 and 3.5 g/day [33]. The number of complete remissions at 18 months was similar (60% vs 56%), but 21% of the patients in the oral prednisone group presented infections vs 8% in MPP. Weight gain (19% vs 4%) and Cushing syndrome (18% vs 3%) were also more frequent in the prednisone group [33].
Whether the degree of activity or chronicity at kidney biopsy can predict response to steroid treatment is still an open question. In the VALIGA study, the whole MEST score significantly predicted the long-term renal outcome; the C score (crescents) was an outcome predictor only in untreated patients [34]. More recently, Itami et al. [35]. showed that only activity indexes were associated with response to steroid therapy, whereas chronicity ones were not [36]. Other authors did not confirm a change in the predictive capability of the MEST score in those receiving steroids [37].
Finally, according to meta-analyses [38, 39, 40], glucocorticoids can significantly reduce the risk of proteinuria and increase kidney survival in IgAN patients, although at risk of adverse reactions.
TARGETED THERAPY
Recent research has explored new ways of administering steroids in IgAN to optimise effectiveness while minimising side effects. Advances in the pathophysiology of IgAN have provided a theoretical basis for local treatment of the disease. As mentioned above, a subset of B cells generates IgA1 molecules lacking galactose residues (Gd-IgA1). These abnormal glycosylated IgA1 molecules, which circulate in greater amounts in IgAN patients, are mainly produced by lymphoid follicles at the level of Peyer's patches of the ileum [41]. Since Gd-IgA1 plays a significant role in IgAN pathogenesis, using a drug that selectively targets the ileum in correspondence with Peyer's patches is a reasonable approach.
Budesonide is a second-generation synthetic glucocorticoid with minimal systemic absorption. The molecule does not contain any fluorine atoms and possesses an unsymmetrical 16 alpha, 17 alpha-acetal structure; this structure increases its topical anti-inflammatory activity [42]. It has a much higher potency than prednisone (on a weight basis).
Budesonide has been used for decades for the topical treatment of inflammatory bowel diseases or asthma. Its delivery to the gut, release site, and relative timing varies with different formulations.
Targeted Release Formulation budesonide
The Targeted Release Formulation (TRF) (Nefecon®) has been developed recently. Thanks to the TARGIT starch capsule technology, the drug is delivered precisely to the distal ileum and proximal colon, where Peyer plaque density is highest. For this reason, theoretically, it acts in a targeted way at one of the two primary sites involved in initiating and fuelling the pathogenetic mechanisms of the disease. The efficacy and safety of TRF budesonide were first tested in a phase 2 double-blind placebo RCT, the NEFIGAN study (The Effect of Nefecon in Patients with Primary IgA Nephropathy at Risk of Developing End-stage Renal Disease) [43]. It enrolled 150 patients aged >18 with biopsy-confirmed primary IgAN and persistent proteinuria at maximum tolerated dose of RAS inhibitor. Participants were randomised to TRF-budesonide 16 mg/day, 8 mg/day, or placebo. The primary outcome was the mean change from baseline in urinary protein to creatinine ratio (UPCR) after nine months of treatment. Considering the two doses combined (16 mg/day plus 8 mg/day), patients randomised to TRF-budesonide had a 24.4% decrease in mean UPCR from baseline; this change was statistically significant in comparison to placebo. When the two dosages were analysed separately, the decrease in UPCR at nine months was statistically significant only for the 16 mg/day dose but of borderline significance for the lower dose (Fig. 2). On the contrary, patients who received a placebo had an increase in mean UPCR of 2.7%. Overall, the incidence of adverse events was similar in the three groups: Of note, two patients receiving TRF-budesonide at 16 mg/day experienced deep vein thrombosis and unexplained deterioration in kidney function. Two other patients developed diabetes.
Figure 2:

Percentage decrease in urinary protein to creatinine ratio in phase-2 (Targeted-release budesonide versus placebo in IgA nephropathy study, NEFIGAN [43]) and phase-3 (Efficacy and Safety of Nefecon in Patients with Primary IgA Nephropathy, NefIgArd [44]) trials with TRF-budesonide (16 mg/day) or a placebo after nine months of therapy and at the end of the follow-up period at 12 months.
More recently, the findings of the first part of a phase-3 study with TRF budesonide were published [44]. The Efficacy and Safety of Nefecon in Patients with Primary IgA Nephropathy (NefIgArd) trial (NCT03643965) was a multicentre, double-blind, randomised, placebo-controlled study [44]. In this first part, 199 IgAN patients with UPCR ≥0.8 g/g or proteinuria ≥1 g/day despite optimised supportive care, an eGFR of ≥35 to ≤90 ml/min/1.73 m2 were randomised to receive either budesonide 16 mg/day or a placebo for 9 months. Like phase-2 data, TRF-budesonide significantly decreased UPCR compared to placebo (27% lower in the budesonide group). Patients receiving budesonide also experienced a slower decline in eGFR with a 3.87 ml/min/1.73 m2 difference versus placebo, possibly due to hyperfiltration occurring in patients receiving budesonide as a consequence of some systemic absorption of the drug. Interestingly, three months after treatment discontinuation, UACR was still decreasing in the budesonide group (–48% compared to placebo). The most common side effects reported during the study were hypertension, peripheral oedema, muscle spasms, and acne. Two patients receiving budesonide with prediabetes at baseline developed type 2 diabetes during the study. Mild to moderate infections were reported similarly in the two groups. A recent press release from Callidatas (15 May 2023) reported some preliminary data on part B of the study regarding the complete trial population of 360 subjects over a longer follow-up period [45]. The trial met its primary endpoint on eGFR total slope compared to placebo with a long-lasting effect after treatment discontinuation. Similarly, a long-lasting reduction of UPCR was observed.
Other topical formulations
Other enteric formulations of budesonide have been used in IgAN, obtaining significant proteinuria reduction [46, 47]. However, none were tested in randomised clinical trials, and doses were not comparable to that tested with TRF budesonide. Moreover, considering they have a different kinetic release time in the gut, they do not necessarily act at Peyer's patches as TRF budesonide.
Considering that mucosal aberrant IgA antibodies are not produced only by the gut but also by the tonsils, the topical administration of steroids to the upper airways may also have some effects on IgAN. Recently, Sun et al. reported about 142 patients with biopsy-proven IgAN, who were randomised to either fluticasone propionate aerosol plus RAS blockade or RAS blockade alone; those randomised to fluticasone propionate aerosol had significantly lower proteinuria levels than those in the control group [48].
CONCLUSIONS
There is consensus that glucocorticoids should be reserved for IgAN patients at risk of rapid progression to ESKD. The type of administration and proper patient selection and monitoring influence their efficacy and safety. Based on the current knowledge, TRF budesonide at a dose of 16 mg/day could be indicated as first-line treatment for patients with mild-to-moderate forms of IgAN and those at risk of developing severe side effects associated with systemic glucocorticoids. Treatment duration should be of nine months (i.e. the one presented to the Food and Drug Administration (FDA) and European Medicine Agency (EMA)). Both agencies gave conditional approval but with slight differences: for the FDA, TRF budesonide is indicated for reducing proteinuria in IgAN patients with baseline UPCR >2.0 g/g, for EMA, the treatment indication is IgAN at risk of rapid disease progression with a UPCR ≥1.5 g/g.
Till now, TRF budesonide has been tested only in comparison with a placebo. But it is important to know whether it is really safer and possibly more effective than systemic glucocorticoids. TRF budesonide does not act in the kidney unless a small amount is absorbed systemically. However, TRF budesonide is much more potent than systemic steroids, so even a tiny amount of the absorbed drug could have clinical relevance. Conversely, systemic steroids have little effect on the gut compared to TRF budesonide.
Is it enough to halt the pathogenetic mechanism of the disease at the very beginning, or is it also necessary to decrease its activity at the kidney level? And again, how can one understand which mechanism is predominant in the single patient in the absence of reliable biomarkers?
Another important aspect is the safety of TRF budesonide, as cushingoid side effects were two times higher than those observed in the placebo group [45], suggesting that the systemic effects of TRF budesonide are not negligible.
Only an RCT comparing the efficacy and safety of TRF budesonide versus MPP and alternate-day prednisone or only oral moderate dose methylprednisolone could verify these hypotheses and indicate the best treatment strategy for proteinuric patients with IgAN resistant to RAS blockade (and SGLT2 inhibitors).
The availability of markers of disease activity in everyday clinical practice, such as the levels of aberrant IgA levels (Gd-IgA1) or IgG/IgA autoantibodies, could become an additional help over proteinuria for selecting the patients more suitable to a given treatment strategy, verifying disease activity during follow-up and the need of retreatment.
The pharmaceutical possibilities for IgAN are widening at an incredible speed (Fig. 3). Thanks to new rules for drug approval by the FDA and EMA for rare or ‘orphan’ diseases, several new drugs are now under clinical development for treating IgA nephropathy. These include agents inhibiting the complement system, the GAS6-AXL signalling pathway, anti-A proliferation-inducing ligand (APRIL), anti-B-lymphocyte stimulator (BLYS) monoclonal antibodies, tyrosine kinase and NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome inhibitors. The second line of treatment includes agents acting on the mechanism of CKD progression, such as sparsentan, and finerenone. They should be considered as an additive treatment in combination with systemic steroids or budesonide or as maintenance therapy in the long term. When these therapeutic possibilities become available for clinical use, clinicians will have to confront the difficult choice of selecting the right patient for the right drug. For all the agents, including TRF budesonide, the price could play an essential role in their penetration into the market.
Figure 3:

Present and future treatment options for IgA nephropathy. The figure summarises the main treatment options for IgAN for immunosuppression, modulation, or, more generally, nephroprotection. Some of these strategies are approved for clinical use in IgAN patients with a specific indication (TRF-budesonide and sparsentan). Others are approved for clinical use for CKD patients, including IgAN, or only for patients with type 2 diabetes (finerenone). Many new drugs are under clinical development for treating IgAN (blue boxes). aAPRIL, a proliferation-inducing ligand; bBLYS, B-lymphocyte stimulator protein; cSGLT2, sodium-glucose transporter 2; dsparsentan is approved for clinical use by the FDA and is under evaluation by EMA, while atresantan is under clinical development.
Contributor Information
Francesco Locatelli, Department of Nephrology and Dialysis, Alessandro Manzoni Hospital, Lecco, Italy.
Lucia Del Vecchio, Department of Nephrology and Dialysis, Sant’ Anna Hospital, ASST Lariana, Como, Italy.
FUNDING
This paper was published as part of a supplement funded by an educational grant from Otsuka America Pharmaceutical, Inc.
DATA AVAILABILITY STATEMENT
No new data were generated or analysed in support of this research.
CONFLICT OF INTEREST STATEMENT
F.L. has no conflict of interest to declare in the present article. L.DV. has been National Leader for the PROTECT study. She received speaker fees at meetings indirectly supported by Vifor Pharma and Astra Zeneca, Bayer. C.P. has no conflict of interest to declare.
REFERENCES
- 1. Coppo R, D'Amico G.. Factors predicting progression of IgA nephropathy. J Nephrol 2005;18:503–12. [PubMed] [Google Scholar]
- 2. Lee H, Kim DK, Oh KHet al. Mortality of IgA nephropathy patients: a single center experience over 30 years. PLoS One 2012;7:e51225. 10.1371/journal.pone.0051225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moriyama T, Tanaka K, Iwasaki Cet al. Prognosis in IgA nephropathy: 30-year analysis of 1,012 patients at a single center in Japan. PLoS One 2014;9:e91756. 10.1371/journal.pone.0091756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarrick S, Lundberg S, Welander Aet al. Mortality in IgA nephropathy: a nationwide population-based cohort study. JASN 2019;30:866–76. 10.1681/ASN.2018101017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barbour SJ, Cattran DC, Espino-Hernandez Get al. Identifying the ideal metric of proteinuria as a predictor of renal outcome in idiopathic glomerulonephritis. Kidney Int 2015;88:1392–401. 10.1038/ki.2015.241 [DOI] [PubMed] [Google Scholar]
- 6. Coppo R, Lofaro D, Camilla RRet al. Risk factors for progression in children and young adults with IgA nephropathy. An analysis of 261 cases from the VALIGA European cohort. Pediatr Nephrol 2017;32:139–50. 10.1007/s00467-016-3469-3 [DOI] [PubMed] [Google Scholar]
- 7. Barbour SJ, Coppo R, Zhang Het al. Evaluating a new international risk-prediction tool in IgA nephropathy. JAMA Intern Med 2019;179:942–52. 10.1001/jamainternmed.2019.0600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rovin BH, Adler SG, Barratt Jet al. Executive summary of the KDIGO 2021 Guideline for the management of glomerular diseases. Kidney Int 2021;100:753–79. 10.1016/j.kint.2021.05.015 [DOI] [PubMed] [Google Scholar]
- 9. Debono M, Ghobadi C, Rostami-Hodjegan Aet al. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009;94:1548–54. 10.1210/jc.2008-2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kadmiel M, Cidlowski JA.. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013;4:518–30. 10.1016/j.tips.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Granner DK, Wang JC, Yamamoto KR.. Regulatory actions of glucocorticoid hormones: from organisms to mechanisms. Adv Exp Med Biol 2015;872:3–31. 10.1007/978-1-4939-2895-8_1 [DOI] [PubMed] [Google Scholar]
- 12. Yan K, Kudo A, Hirano Het al. Subcellular localisation of glucocorticoid receptor protein in the human kidney glomerulus. Kidney Int 1999;56:65–73. 10.1046/j.1523-1755.1999.00503.x [DOI] [PubMed] [Google Scholar]
- 13. Ponticelli C, Glassock RJ.. Prevention of complications from use of conventional immunosuppressants: a critical review. J Nephrol 2019;32:851–70. 10.1007/s40620-019-00602-5 [DOI] [PubMed] [Google Scholar]
- 14. Panettieri RA, Schaafsma D, Amrani Yet al. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci 2019;40:38–49. 10.1016/j.tips.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrease J, Tripmacher R, Weltrich Ret al. Effect of glucocorticoid therapy on glucocorticoid receptors in children with autoimmune diseases. Pediatr Res 2001;49:130–5. 10.1203/00006450-200101000-00025 [DOI] [PubMed] [Google Scholar]
- 16. Liu L, Wang YX, Zhou Jet al. Rapid non-genomic inhibitory effects of glucocorticoids on human neutrophil degranulation. Inflamm Res 2005;54:37–41. 10.1007/s00011-004-1320-y [DOI] [PubMed] [Google Scholar]
- 17. Sinha A, Bagga A.. Pulse steroid therapy. Indian J Pediatr 2008;75:1057–66. 10.1007/s12098-008-0210-7 [DOI] [PubMed] [Google Scholar]
- 18. Currie EG, Coburn B, Porfilio EAet al. Immunoglobulin A nephropathy is characterised by anticommensal humoral immune responses. JCI Insight 2022;7:e141289. 10.1172/jci.insight.141289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coutinho AE, Chapman KE.. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 2011;335:2–13. 10.1016/j.mce.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nihei Y, Haniuda K, Higashiyama Met al. Identification of IgA autoantibodies targeting mesangial cells redefines the pathogenesis of IgA nephropathy. Sci Adv 2023;9:eadd6734. 10.1126/sciadv.add6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knoppova B, Reily C, King RGet al. Pathogenesis of IgA nephropathy: current understanding and implications for development of disease-specific treatment. JCM 2021;10:4501. 10.3390/jcm10194501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agrawal S, He JC, Tharaux PL.. Nuclear receptors in podocyte biology and glomerular disease. Nat Rev Nephrol 2021;17:185–204. 10.1038/s41581-020-00339-6 [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Lv J, Liu Pet al. Poly-IgA complexes and disease severity in IgA nephropathy. CJASN 2021;16:1652–64. 10.2215/CJN.01300121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Imbasciati E, Gusmano R, Edefonti Aet al. Controlled trial of methylprednisolone pulses and low-dose oral prednisone for the minimal change nephrotic syndrome. BMJ 1985;291:1305–8. 10.1136/bmj.291.6505.1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ponticelli C, Zucchelli P, Passerini Pet al. Methylprednisolone plus chlorambucil as compared with methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic membranous Nephropathy Treatment Study Group. N Engl J Med 1992;327:599–603. 10.1056/NEJM199208273270904 [DOI] [PubMed] [Google Scholar]
- 26. Pozzi C, Bolasco PG, Fogazzi GBet al. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet North Am Ed 1999;353:883–7. 10.1016/S0140-6736(98)03563-6 [DOI] [PubMed] [Google Scholar]
- 27. Pozzi C, Andrulli S, Del Vecchio Let al. Corticosteroid effectiveness in IgA nephropathy: long-term results of a randomised, controlled trial. J Am Soc Nephrol 2004;15:157–63. 10.1097/01.ASN.0000103869.08096.4F [DOI] [PubMed] [Google Scholar]
- 28. Manno C, Torres DD, Rossini Met al. Randomised controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol Dial Transplant 2009;24:3694–701. 10.1093/ndt/gfp356 [DOI] [PubMed] [Google Scholar]
- 29. Rauen T, Eitner F, Fitzner Cet al. Intensive supportive care plus immunosuppression in IgA nephropathy N. N Engl J Med 2015;373:2225–36. 10.1056/NEJMoa1415463 [DOI] [PubMed] [Google Scholar]
- 30. Rauen T, Wied S, Fitzner Cet al. After ten years of follow-up, no difference between supportive care plus immunosuppression and supportive care alone in IgA nephropathy. Kidney Int 2020;98:1044–52. 10.1016/j.kint.2020.04.046 [DOI] [PubMed] [Google Scholar]
- 31. Tesar V, Troyanov S, Bellur Set al. Corticosteroids in IgA nephropathy: a retrospective analysis from the VALIGA study. J Am Soc Nephrol 2015;26:2248–58. 10.1681/ASN.2014070697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lv J, Wong MG, Hladunewich MAet al. Effect of oral methylprednisolone on decline in kidney function or kidney failure in patients with IgA nephropathy: the TESTING randomised clinical trial. JAMA 2022;327:1888–98. 10.1001/jama.2022.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y, Fu R, Gao Jet al. Effect of pulsed intravenous methylprednisolone with alternative low-dose prednisone on high-risk IgA nephropathy: a 18-month prospective clinical trial. Sci Rep 2022;12:255. 10.1038/s41598-021-03691-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coppo R, D'Arrigo G, Tripepi Get al. ERA-EDTA Immunonephrology Working Group. Is there long-term value of pathology scoring in immunoglobulin A nephropathy? A validation study of the Oxford Classification for IgA Nephropathy (VALIGA) update. Nephrol Dial Transplant 2020;35:1002–9. 10.1093/ndt/gfy302 [DOI] [PubMed] [Google Scholar]
- 35. Itami S, Moriyama T, Miyabe Yet al. A novel scoring system based on Oxford Classification indicating steroid therapy use for IgA nephropathy. Kidney International Reports 2022;7:99–107. 10.1016/j.ekir.2021.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schimpf JI, Klein T, Fitzner Cet al. Renal outcomes of STOP-IgAN trial patients in relation to baseline histology (MEST-C scores). BMC Nephrol 2018;19:328. 10.1186/s12882-018-1128-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoon CY, Chang TI, Kang EWet al. Clinical usefulness of the Oxford classification in determining immunosuppressive treatment in IgA nephropathy. Ann Med 2017;49:217–29. 10.1080/07853890.2016.1252058 [DOI] [PubMed] [Google Scholar]
- 38. Natale P, Palmer SC, Ruospo Met al. Immunosuppressive agents for treating IgA nephropathy. Cochrane Database Syst Rev 2020;3:CD003965. 10.1002/14651858.CD003965.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Z, Yang Y, Jiang SMet al. Efficacy and safety of immunosuppressive treatment in IgA nephropathy: a meta-analysis of randomised controlled trials. BMC Nephrol 2019;20:333. 10.1186/s12882-019-1519-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang P, Wang Q, Xie Cet al. Efficacy and safety of agents in IgA nephropathy: an update network meta-analysis. Kidney Blood Press Res 2018;43:1890–7. 10.1159/000496000 [DOI] [PubMed] [Google Scholar]
- 41. Meng H, Ohtake H, Ishida Aet al. IgA production and tonsillar focal infection in IgA nephropathy. J Clin Exp Hematopathol 2012;52:161–70. 10.3960/jslrt.52.161 [DOI] [PubMed] [Google Scholar]
- 42. Brattsand R, Thalén A, Roempke Ket al. Influence of 16 alpha, 17 alpha-acetal substitution and steroid nucleus fluorination on the topical to systemic activity ratio of glucocorticoids. J Steroid Biochem 1982;16:779–86. 10.1016/0022-4731(82)90035-8 [DOI] [PubMed] [Google Scholar]
- 43. Fellström BC, Barratt J, Cook Het al. NEFIGAN Trial Investigators Targeted-release budesonide versus placebo in IgA nephropathy (NEFIGAN) patients: a double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017;389:2117–27. 10.1016/S0140-6736(17)30550-0 [DOI] [PubMed] [Google Scholar]
- 44. Barratt J, Lafayette R, Kristensen Jet al. NefIgArd Trial Investigators Results from part A of the multicenter, double-blind, randomised, placebo-controlled NefIgArd trial. Kidney Int 2023;103:391–402. 10.1016/j.kint.2022.09.017 [DOI] [PubMed] [Google Scholar]
- 45. https://www.prnewswire.com/news-releases/calliditas-announces-primary-endpoint-successfully-met-in-phase-3-nefigard-trial-evaluating-nefecon-in-iga-nephropathy-301769754.html (18 June 2023, date last accessed).
- 46. Lopez-Martinez M, Torres I, Bermejo Set al. Enteric budesonide in transplant and native IgA nephropathy: real-world clinical practice. Transpl Int 2022;35:10693. 10.3389/ti.2022.10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ismail G, Obrişcă B, Jurubiţă Ret al. Budesonide versus systemic corticosteroids in IgA Nephropathy: a retrospective, propensity-matched comparison. Medicine (Baltimore) 2020;99:e21000. 10.1097/MD.0000000000021000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun L, Zi X, Wang Zet al. The clinical efficacy of fluticasone propionate combined with ACEI/ARB in the treatment of immunoglobulin A nephropathy. BMC Nephrol 2023;24:63. 10.1186/s12882-023-03106-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this research.