

Abstract
Infections with DNA viruses commonly result in the association of viral genomes and replication compartments with cellular nuclear substructures known as promyelocytic leukemia protein (PML) nuclear bodies or ND10. While there is evidence that viral genomes can associate with preexisting ND10, we demonstrate in this study by live-cell microscopy that structures resembling ND10 form de novo and in association with viral genome complexes during the initial stages of herpes simplex virus type 1 (HSV-1) infection. Consistent with previous studies, we found that the major ND10 proteins PML, Sp100, and hDaxx are exchanged very rapidly between ND10 foci and the surrounding nucleoplasm in live cells. The dynamic nature of the individual protein molecule components of ND10 provides a mechanism by which ND10 proteins can be recruited to novel sites during virus infection. These observations explain why the genomes and replication compartments of DNA viruses that replicate in the cell nucleus are so commonly found in association with ND10. These findings are discussed with reference to the nature, location, and potential number of HSV-1 prereplication compartments and to the dynamic aspects of HSV-1 genomes and viral products during the early stages of lytic infection.
Promyelocytic leukemia protein (PML) nuclear bodies (also known as ND10) are discrete nuclear substructures that have been implicated in many cellular processes, including transcription, DNA repair, chromatin metabolism, oncogenesis, and apoptosis (for reviews, see references 2, 6, 12, and 47). ND10 sites have also been closely linked with as yet incompletely understood aspects of infection by several DNA viruses. Parental viral genomes have a tendency to associate with ND10, and viral immediate-early gene transcription takes place at these sites (22, 23). Viral replication complexes (replication compartments) are also commonly associated with ND10 (reviewed in references 15 and 27). However, many such viruses also encode regulatory proteins that cause the disruption of ND10 (reviewed in reference 15), so the association between viral replication compartments and ND10 is clearest for mutant viruses that lack ND10 disruption activity.
The mechanisms that underlie the association of viral genomes with ND10 have not been clearly defined. Plasmids that contain a simian virus 40 (SV40) origin of DNA replication can become associated with ND10, but only if the SV40 large T antigen (which binds to the origin sequences and promotes DNA replication) is present (40). The association of herpes simplex virus type 1 (HSV-1) amplicon genomes with ND10 is increased if active viral transcription units are present in the amplicons (38, 41). These data suggest that the biological activity of exogenous DNA promotes its association with ND10, a conclusion supported by studies of sequences integrated into cellular chromatin that become associated with ND10 when bound to exogenous protein factors (42). However, none of these studies has been able to differentiate between the following three possible scenarios: (i) that viral genomes migrate through the nucleus until they encounter an ND10 structure, (ii) that ND10 structures themselves move to sites of foreign DNA-protein complexes, or (iii) that novel ND10 complexes are formed in association with such complexes.
While studying early events during HSV-1 infection, we noted that in cells at the edges of developing viral plaques in cell monolayers, dot-like accumulations of the HSV-1 transcriptional regulatory protein ICP4 frequently formed in an arc just inside the edges of the nuclei of newly infected cells (18, 19). Because ICP4 binds strongly to viral DNA and accumulates in viral replication compartments, this and other evidence supported the hypothesis that the foci represented ICP4 binding to parental HSV-1 genomes and very early viral replication compartments (16, 19). An examination of the location of the major ND10 component proteins PML, Sp100, and hDaxx in such cells infected by an ICPO-negative HSV-1 mutant revealed that they also had highly asymmetric distributions, accumulating in marked association with the ICP4 complexes. These asymmetric patterns of the ND10 proteins were in distinct contrast to the normal apparently random distribution of ND10 throughout the nucleus (18). We suggested that while viral genome complexes may migrate through the nucleus until an ND10 structure is encountered, in addition either ND10 sites themselves may migrate to the sites of the incoming viral complexes or ND10-like structures may be formed de novo in association with and in response to the viral genome complexes (18).
In this study, we provide direct evidence that the ICP4 complexes do indeed contain viral genomes, and we show by live-cell microscopy that the associated ND10-like complexes are formed by ND10 proteins being deposited to create new aggregates in association with the viral complexes rather than by the migration of preexisting intact ND10 structures. Fluorescence recovery after photobleaching (FRAP) and fluorescence lifetime in photobleaching (FLIP) experiments confirmed previous studies that demonstrated that ND10 component proteins are highly mobile, allowing a rapid exchange of protein molecules between existing ND10 foci and the surrounding nucleoplasm. This behavior of ND10 component molecules provides a mechanism that allows the formation of novel ND10 structures at diverse sites in the nucleus. We propose that the cell responds to incoming biologically active viral genomes in a manner that leads to the deposition of novel ND10 structures that are closely associated with the viral nucleoprotein complexes. This model explains why the replication compartments of nuclear replicating DNA viruses in general are so commonly observed to be in association with ND10.
MATERIALS AND METHODS
Viruses and cells.
The parental virus HSV-1 strain 17+ (wt HSV-1), the ICP0-null virus dl1403 (39), and virus dl0C4 expressing enhanced cyan fluorescent protein (ECFP)-linked ICP4 in the dl1403 ICP0-null background (16) were grown in BHK cells and titrated in U2OS cells. The viruses were used at the stated multiplicities of infection (MOIs) based on their PFU titers in U2OS cells. BHK cells were grown in Glasgow modified Eagle's medium containing 100 U of penicillin/ml and 100 μg of streptomycin/ml and supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. U2OS cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS) and antibiotics as described above. Human fetal foreskin fibroblast cells (HFFF-2; European Collection of Cell Cultures) were grown in Dulbecco's modified Eagle's medium supplemented with 10% FCS, 1% glutamine, and antibiotics as described above. The baculoviruses Ac.CMV.ECFP-PML (38) and Ac.CMV.EYFP-PML (19) express ECFP- and enhanced yellow fluorescent protein (EYFP)-linked PML (isoform IV) from the human cytomegalovirus promoter in mammalian cells. The baculoviruses Ac.CMV.EYFP-Sp100 and Ac.CMV.EYFP-Daxx express EYFP-linked versions of Sp100-A and hDaxx in mammalian cells (20).
Fluorescence in situ hybridization.
HFFF-2 cells were grown on 13-mm-diameter glass coverslips and then infected with wt HSV-1 at a multiplicity of infection of 10 PFU per cell. At various times thereafter, the cells were washed twice with phosphate-buffered saline (PBS) containing 1% FCS and then fixed by incubation for 5 min at −20°C with precooled 95% ethanol-5% glacial acetic acid. The cells were then washed three times with PBS-1% FCS and stored at 4°C until further use. The probes for in situ hybridization were cosmids containing a 39.7-kb segment of the HSV-1 genome between coordinates 24,699 and 64,405 or a 35.6-kb segment between coordinates 79,442 and 115,152 (gifts from Andrew Davison and Charles Cunningham, MRC Virology Unit). After a DNase I treatment, the probes were labeled by nick translation with Cy3-dCTP (Amersham) according to the manufacturer's protocol. The cells were prehybridized by incubation for 30 min at 37°C with 20 μl of hybridization buffer (50% formamide, 10% dextran sulfate, 4× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]) in a humidified microarray hybridization chamber (Camlab). The coverslips were then removed from the chamber and blotted dry. A probe was added to the hybridization buffer to a concentration of 1 ng/μl, after which each coverslip was incubated at 95°C for 2 min to denature the probe and the sample. The coverslips and hybridization mixture were then placed into the humidified chamber, and hybridization was continued overnight at 37°C. The cells were then washed for 5 min at 60°C with 2× SSC and once with 2× SSC at room temperature. After a further wash with PBS-1% FCS, the coverslips were incubated with the anti-ICP4 monoclonal antibody 58S for 1 h. After several washes, the cells were incubated with fluorescein isothiocyanate-conjugated sheep anti-mouse immunoglobulin G (Sigma) for another hour. The cells were washed several times, air dried, and then mounted on glass slides by the use of a glycerol-PBS mounting solution (CitiFluor). The samples were examined under a Zeiss LSM 510 confocal microscope with 488- and 543-nm laser lines, with each channel scanned separately under image capture conditions that eliminated channel overlap. The images were exported as TIFF files and then processed with Photoshop.
Immunofluorescence.
Fluorescence in situ hybridization was combined with immunofluorescence by performing the hybridization first as described above and then incubating the coverslips with relevant primary and secondary antibodies as described previously (18). ICP4 was detected with the monoclonal antibody 58S (36) and PML was detected with the rabbit serum r8 (4), with fluorescein isothiocyanate-conjugated sheep anti-mouse and anti-rabbit secondary antibodies, respectively. In simple immunofluorescence experiments, the cells were fixed with formaldehyde and prepared for the detection of ICP4 with the monoclonal antibody 58S or rabbit serum r74, and ICP0 was detected with the monoclonal antibody 11060 or rabbit serum r190, as described previously (18).
Live-cell microscopy.
Cells were grown in coverslipped glass-bottomed chambers and examined under a Zeiss Axiovert S100 microscope equipped with a motorized excitation filter wheel and a filter set designed to discriminate between EYFP and ECFP (Chroma). Images were captured with a Hamamatsu Orca charge-coupled device camera, with automated functions and image capture conditions controlled by AQM software (Kinetic Imaging). The microscope was maintained in a humidified chamber with temperature and CO2 control (EMBL Laboratories). Further details are given in previous publications (18, 19).
FRAP and FLIP experiments used the 510-nm laser line of a Zeiss LSM 510 META confocal microscope for EYFP bleaching and image detection, with areas of interest, bleaching, and time series image acquisition controlled by the LSM 510 software. The cells were maintained at 37°C by use of a heated stage and lens heater under the control of a thermostat. The medium was buffered with HEPES to maintain the pH. For FRAP, five small circular regions of interest that included individual ND10 foci were selected for bleaching (20 reiterations at full laser power). Thereafter, a sequence of 10 images at 20-s intervals was captured. Because ND10 foci moved during this period, larger regions of interest that included these foci in all images in the sequence were selected. Average pixel intensities were determined, from which the average pixel intensity of an unbleached region not including an ND10 structure (background value) was subtracted. The resultant average pixel intensity at each time point for each bleached structure was determined as a percentage of the initial value before bleaching. The average of these values for each bleached structure at each time point in a cell was calculated. Background values and values for a control unbleached ND10 structure remained constant throughout the time series (data not shown).
For FLIP experiments, two large rectangular regions of interest were selected to divide the nucleus into four bands. A series of seven bleach operations were performed on the two rectangular regions (20 reiterations at 100% laser power), with images captured immediately after and 20 s after each bleach operation. Analysis was conducted in a similar manner to that for FRAP experiments. Regions of interest defining the background, five individual ND10 structures in unbleached areas, and one ND10 structure in a bleached area were selected, and average pixel intensities minus the background were calculated for each structure at each time point. The data presented represent averages of two or three cells, with five individual unbleached foci analyzed for each. Background levels in the unbleached areas remained constant throughout the time series, while the intensity of ND10 foci within the bleached areas decreased by about 90% during the experiment (data not shown).
RESULTS
The ICP4 foci that form during the early stages of HSV-1 infection are commonly associated with viral genomes.
We have reported that ICP4 forms small discrete foci during the early stages of HSV-1 infection (18, 19). On the bases that ICP4 binds to viral DNA, that the number of ICP4 foci increases with increasing doses of input virus, and that many of the initial ICP4 foci later develop into replication compartments, we argued that at least a proportion of the ICP4 foci must represent an association of ICP4 with parental viral genomes (18, 19). We have now confirmed this conclusion directly by using in situ hybridization to detect HSV-1 DNA in combination with antibody detection of ICP4 in fixed HSV-1-infected cells. The majority of the ICP4 foci that formed early during wt HSV-1 strain 17 infection were strongly associated with viral foci that were detected by in situ hybridization (Fig. 1, row A). An examination of a large number of similar cells indicated that some of the nuclear DNA foci did not exhibit an ICP4 signal, while a minority of the ICP4 foci were apparently not associated with a DNA signal. These observations imply that there is a differential recruitment of ICP4 to individual viral genomes (perhaps dependent on their biological activities) and that occasionally ICP4 foci may form at sites that do not contain readily detectable viral DNA. Another general feature of these samples was that the colocalization of the ICP4 and DNA signals within structures containing both components was neither complete nor precise, suggesting that the compaction of DNA and accessibility to ICP4 were not uniform within the viral nucleoprotein foci. Although a treatment of the samples with RNase had no significant effect on these observations (data not shown), we cannot exclude that a proportion of the in situ hybridization signal was derived from viral transcription from the viral genome template.
FIG. 1.

ICP4 foci that form at early times of HSV-1 infection are commonly associated with viral DNA. (Row A) Combined in situ hybridization of HSV-1 DNA and immunostaining of ICP4 in an HFFF-2 cell 3 h after infection with wt HSV-1 (MOI, 10 PFU/cell), showing DNA, ICP4, and merged (DNA is red, and ICP4 is green) images. (Row B) A HFFF-2 cell at the edge of a developing dl1403 plaque was stained for PML and ICP4 (red and green, respectively, in the merged image), demonstrating the association of PML foci with the ICP4 foci that characteristically form just inside the nuclear periphery. (Row C) The ICP4 foci that form just inside the nuclear periphery in cells at the edge of a developing plaque are extensively associated with DNA (ICP4 is green and DNA is red in the merged image). (Row D) In such cells, foci of viral DNA inside and close to the edge of the nucleus are commonly associated with structures containing PML (DNA is red and PML is green in the merged image).
The number of genomic foci in individual cells frequently exceeded the nominal MOI based on the number of PFU applied to the cells. This is most likely explained by the fact that the particle-to-PFU ratio of the virus stocks averaged around 30 (16) and perhaps also because the eye is drawn to cells receiving more than the average number of genomes. Although similar viral genomic foci could be observed in cells treated with acycloguanosine to inhibit DNA replication (data not shown), an increase in size and a loss of the tight punctate character of the viral genomic foci in some cells that were not so treated suggested that DNA replication can be initiated in some cells even at early times of infection.
By 4 h postinfection, replication compartments that gave substantial hybridization signals had developed in many cells (data not shown). Again, incomplete colocalization of the viral DNA and ICP4 signals and differential intensities of the hybridization signal within a replication compartment were common, suggesting a lack of uniformity within these structures which was reminiscent of previous observations of the detailed distribution of HSV-1 DNA replication and capsid assembly proteins (10). Despite the incomplete colocalization of the ICP4 and in situ hybridization signals in HSV-1 replication compartments, however, the two signals were nonetheless tightly associated.
In a developing HSV-1 plaque on a cultured cell monolayer, progeny HSV-1 virus particles from productively infected cells infect neighboring cells at very high multiplicities. Under these conditions, virus particles spread between cells by direct cell contacts, and therefore the infection is spatially asymmetrical. Once virus particles have entered the newly infected cell, they are carried on microtubules towards the nucleus, and then the viral capsids congregate on the outside edge of the nuclear envelope, preferentially near the microtubule organizing center or on the side of the nucleus that faces the cell from which the virus originated (11, 33, 37). The viral DNA is then released and enters the nucleus, whereupon viral immediate-early (IE) gene transcription takes place. ICP4 also forms discrete foci in cells at the edges of developing plaques, and frequently these foci are arranged in arcs just inside the nuclear envelope (18, 19). Such arcs of ICP4 foci were also strongly associated with ND10-like structures when the degradation of PML and Sp100 was avoided by use of an HSV-1 virus which was defective in ICP0 (Fig. 1, row B) (18).
HFFF-2 cells were infected with the ICP0-null mutant virus dl1403 at a low MOI and then were analyzed by combined in situ hybridization and immunostaining. Cells at the edges of developing plaques commonly contained arcs of ICP4 foci that were intimately associated with viral DNA (Fig. 1, row C), and in turn the viral DNA foci were juxtaposed to ND10-like structures at a very high frequency (Fig. 1, row D). These data confirm that foci of ICP4 provide a reliable method for detecting the locations of viral genomes during HSV-1 infection, and they emphasize the observation that in the absence of ICP0, ND10-like complexes are found in association with asymmetrically distributed viral nucleoprotein complexes at the periphery of cells in a developing plaque.
Redistribution of PML to viral genomic foci also occurs in wild type HSV-1 infections.
It was important to demonstrate that the redistribution of PML into an asymmetric pattern associated with dl1403 genomes in cells at the edges of developing plaques also occurs during wt HSV-1 infection. This was a difficult experiment because of the disruption of ND10 and the degradation of PML that is induced by ICP0 at early times during wt HSV-1 infection. Nonetheless, an examination of wt HSV-1 plaques stained for PML and treated by in situ hybridization revealed cells in the very early stages of infection in which, in addition to PML in remaining ND10 structures, PML signals also appeared in the characteristic asymmetric pattern near the nuclear envelope and associated with viral DNA foci (Fig. 2, rows A and B). The cells at the edges of developing wt virus plaques varied in phenotype from those in which PML was in association with the viral genomic foci while much remained elsewhere in ND10 structures (Fig. 2, row A), through those in which the majority of PML was associated with peripheral wt viral genomes (Fig. 2, row B), to cells in which little or no PML was detectable. In cells retaining only small quantities of PML, the remaining PML foci could be observed in association with viral DNA complexes near the edge of the nucleus (Fig. 2, row C).
FIG. 2.

Redistribution of PML to foci associated with HSV-1 genomes during wt virus infection. HFFF-2 cells were infected at a low MOI with wt HSV-1, and then the following day, the samples were treated to detect viral DNA by in situ hybridization (red) and to detect PML by labeling with the rabbit serum r8 (green) (rows A, B, and C). Row D shows a cell from a similar sample that was costained for ICP0 (red) and ICP4 (green). The insets in the merged images show expanded views of selected regions of the nuclei to illustrate details typical of the association of the various structures. Further details are given in the text.
The above observations suggest that in the early stages of infection, ICP0 is likely to migrate not only to preexisting ND10 sites but also to the PML foci that develop in association with viral genomes. The activity of ICP0 would then lead to the disruption of these genome-associated PML foci in addition to the disruption of preexisting ND10. This hypothesis was supported by the observation of cells at the edges of wt virus plaques that were stained for ICP4 and ICP0 (Fig. 2, row D). Examinations of such samples revealed cells in which ICP0 was present in punctate foci throughout the nucleoplasm and also in association with peripherally located accumulations of ICP4. Note that careful observation was required to identify cells with this phenotype, probably because infection develops very rapidly in HFFF-2 cells at the edges of developing plaques. Once ICP0 has degraded PML and disrupted ND10, it becomes diffusely distributed in the nucleus and begins to accumulate in the cytoplasm. Therefore, the phenotypes illustrated for wt HSV-1 infections in Fig. 2 are transient because of the highly dynamic nature of both PML and ICP0. We must also emphasize the point that while phenotypes similar to those illustrated throughout this paper can be detected in other cell types, they are most easily observed in human fibroblast cells.
De novo induction of ND10-like complexes in association with HSV-1 genome complexes.
The phenomena of asymmetrical infection in a developing plaque and the maintenance of ND10 proteins in the absence of functional ICP0 were harnessed to develop a methodology to examine the dynamics of viral and ND10 proteins in live infected cells and therefore to distinguish among the three possible explanations for the common association between HSV-1 genome complexes and ND10 that were posed in the introduction.
To examine the dynamics of ND10 during the initial stages of HSV-1 infection, we coinfected HFFF-2 cells with an HSV-1 virus expressing ECFP-linked ICP4 in an ICP0-negative background (virus dl0C4) and a baculovirus expressing either EYFP-linked Sp100 or PML (20). Since the asymmetric effect was most clearly seen in cells at the edges of developing plaques (18), the cells were infected with dl0C4 at a low multiplicity, and then the following day an uninfected cell expressing EYFP-Sp100 and situated close to a developing plaque was selected for live-cell time-lapse microscopy. Since it could not be predicted when, or even if, the cell of interest would become infected, image capture sequences were allowed to run for several hours, with maintenance of the temperature and atmospheric control.
Figure 3 shows a selection of frames from an image sequence (see Video S1 in the supplemental material) that illustrate the changes in the Sp100 location in response to the initial stages of HSV-1 infection. Starting from a uniform distribution of ND10, Sp100 began to accumulate in multiple foci near the edge of the nucleus. The two panels at the bottom right in Fig. 3 show the increase in numbers of Sp100 foci at the right-hand end of the nucleus over a 90-min time period. Particularly towards the upper right-hand side of the illustrated nucleus, these foci developed into complex aggregates as time progressed. Importantly, an examination of sequential frames of the time series (see Video S1 in the supplemental material) indicated that the initial ND10 foci elsewhere in the uninfected cell did not move en masse to the edge of the nucleus, but rather they gradually lost fluorescence intensity, while the foci at the nuclear periphery correspondingly increased in both number and fluorescence intensity. This process began at an early stage of infection, before ECFP-linked ICP4 had accumulated to a sufficient quantity to be visible under the image capture conditions employed. Note that ICP4 was readily detectable by antibody staining in similar fixed cell samples (Fig. 1) (18, 19); the relatively inefficient detection of ECFP-ICP4 by live-cell microscopy was due to limitations in the detection of low-intensity ECFP signals above a relatively high background autofluorescence. However, once ECFP-ICP4 foci became visible (Fig. 4, top row), they appeared in close association with EYFP-Sp100 foci at the edge of the nucleus (Fig. 4, middle row), in the characteristic pattern that had previously been documented by analysis of the normal viral and endogenous cellular proteins in fixed cell samples (Fig. 1) (18, 19).
FIG. 3.

Time-lapse live-cell microscopy detecting the redistribution of Sp100 during the initial stages of HSV-1 infection. An uninfected cell expressing EYFP-Sp100 at the edge of a developing HSV-1 dl0C4 plaque was examined by image capture every 15 min. Selected frames from the full sequence are shown, with the times after initiation of the sequence indicated in minutes. The last two images in the third row illustrate expanded views of the right-hand part of the nucleus at the 0- and 90-min time points to demonstrate the increase in the total numbers of ND10-like foci. Selected frames showing the signal from ECFP-ICP4 expressed by the incoming virus are presented in Fig. 4.
FIG. 4.

(Top row) Selected images from the time series shown in Fig. 3 showing the ECFP signal from ECYP-ICP4. (Middle row) Merged color images showing EYFP-Sp100 (red) and ECFP-ICP4 (green) from the images in the top row and the corresponding images from the time series shown in Fig. 5. (Bottom row) EYFP-PML, ECFP-ICP4, and merged (PML is red, and ICP4 is green) images from the final time point of the time sequence depicted in Fig. 5.
Similar results were obtained for cells expressing EYFP-PML. In the example shown (Fig. 5; see Video S2 in the supplemental material), novel accumulations of PML began to form in the bottom right-hand portion of the nucleus at the 70-min time point, and then these foci increased in intensity at the apparent expense of the original foci in other parts of the nucleus. This image sequence shows the de novo induction of PML foci particularly clearly, since they appear in a region of the nucleus that at earlier time points contained no PML. The bottom row of Fig. 4 shows a double-labeled image from the last time point of the series, illustrating that the PML complexes that began to form at earlier time points (Fig. 5) were associated at later times with ICP4-defined viral replication compartments.
FIG. 5.

Time-lapse live-cell microscopy detecting the redistribution of PML during the initial stages of HSV-1 infection. An uninfected cell expressing EYFP-PML at the edge of a developing HSV-1 dl0C4 plaque was examined by image capture every 10 min. Selected frames from the full sequence are shown, with the times after initiation of the sequence indicated in minutes. The channels detecting PML and ICP4 from the last frame of the sequence are shown in Fig. 4.
Taken together, the results shown in Fig. 1 to 5 demonstrate that novel ND10-like complexes form at the sites of incoming viral genomes that have assembled into viral nucleoprotein complexes and that are destined to develop into replication compartments. Figure 2 demonstrates that this occurs in a normal wt virus infection in the presence of ICP0 and with normal endogenous levels of PML and other ND10 proteins. Therefore, the results of the live-cell experiments shown in Fig. 3 to 5 are not artifacts caused by the high-level expression of exogenous ND10 proteins, nor do they represent a phenomenon that is restricted to ICP0-negative HSV-1 infections.
PML, Sp100, and especially hDaxx molecules in ND10 are rapidly exchanged with protein molecules from elsewhere.
What is the mechanism by which ND10 proteins can relocate from existing ND10 structures to novel positions in association with viral nucleoprotein complexes? To investigate the spatial dynamics of selected ND10 proteins, we infected HFFF-2 cells with a baculovirus expressing EYFP-linked PML, Sp100, or hDaxx and then examined the dynamics of the protein molecules in ND10 structures by FRAP. Figures 6 and 7 show that hDaxx behaved very dynamically, with almost complete recovery of the fluorescence intensity in bleached ND10 within 20 s, indicating a rapid exchange of hDaxx molecules between ND10 structures and the surrounding environment. PML and Sp100 were less mobile, in both cases recovering about 30% of the fluorescence intensity that had been lost from bleached ND10 over a period of 3 min (Fig. 6 and 7). Complementary experiments using the FLIP technique gave consistent results, with a loss of the ND10 proteins from unbleached foci following repeated bleaching of other parts of the nucleus (Fig. 7). As in the FRAP experiments, PML and Sp100 were considerably less dynamic than hDaxx during FLIP, but there was an egress of at least 20% of the material from individual ND10 foci during a period of 3 to 4 min of repeated bleaching of neighboring regions (Fig. 7).
FIG. 6.

Analysis of dynamics of hDaxx, PML, and Sp100 by FRAP. (Top row) HFFF-2 cells were infected with Ac.CMV.EYFP-Daxx, and then the following day, the cells were examined by live-cell confocal microscopy. Two foci of hDaxx (arrows) were bleached by 20 reiterations of the 510-nm laser at full power, and then images were captured at 20-s intervals thereafter. The images before bleaching, immediately after bleaching, and at the first 20-s time point are shown. (Middle row) HFFF-2 cells were infected with Ac.CMV.EYFP-Sp100, and then the following day, the cells were examined by live-cell confocal microscopy. Five foci of Sp100 (arrows) were bleached by 20 reiterations of the 510-nm laser at full power, and then images were captured at 20-s intervals thereafter. The images before bleaching, immediately after bleaching, and at the 160-s time point are shown. (Bottom row) HFFF-2 cells were infected with Ac.CMV.EYFP-PML, and then the following day, the cells were examined by live-cell confocal microscopy. Five foci of PML (arrows) were bleached by 20 reiterations of the 510-nm laser at full power, and then images were captured at 20-s intervals thereafter. The images before bleaching, immediately after bleaching, and at the 160-s time point are shown.
FIG. 7.
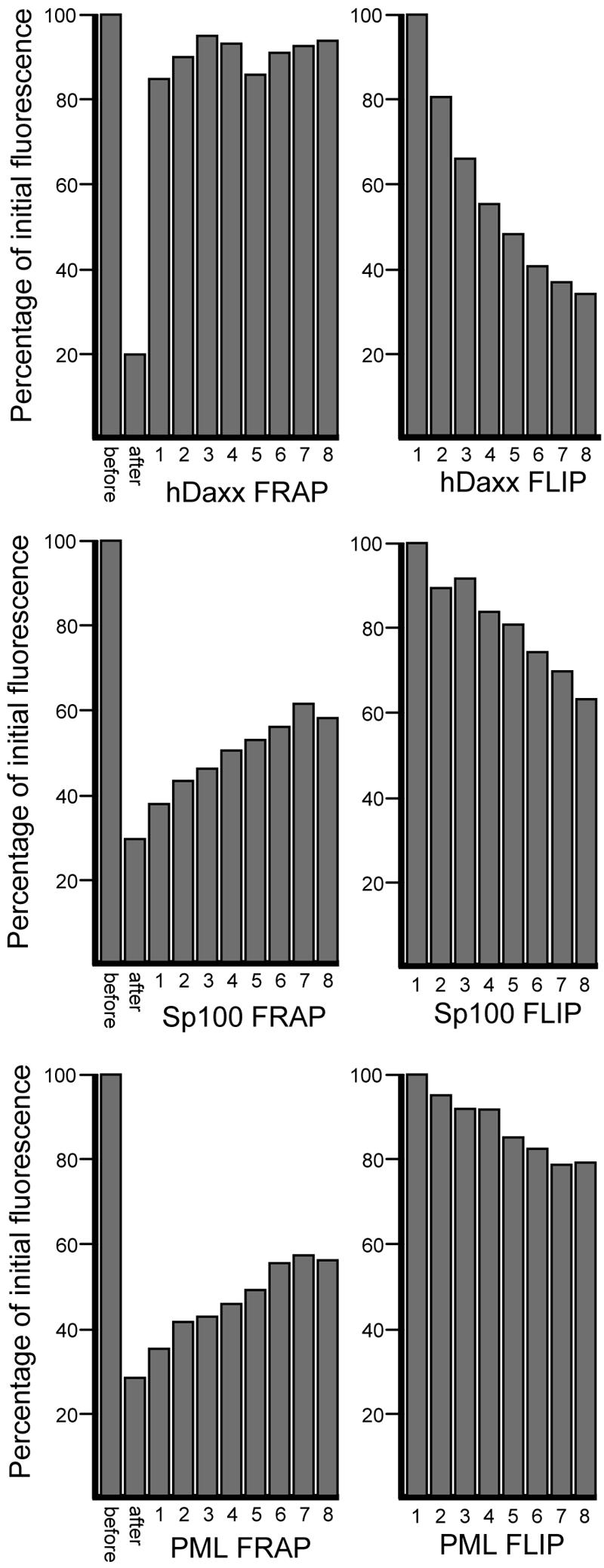
Quantification of the dynamics of hDaxx, PML, and Sp100 by FRAP and FLIP. The left graph of each pair of histograms shows a quantification of the complete data sets, for which selected images are shown in Fig. 6. Making allowance for the movement of the dots, the average fluorescence intensities in regions of interest containing the bleached foci were determined with LSM 510 software, and then the average intensities of a background region were subtracted. The percentages of initial fluorescence at each time point were calculated. The histograms show the average results at all time points of the series for all of the arrowed foci in Fig. 6. Control data from dots that had not been bleached remained constant throughout the period of the experiment (not shown). The right graph of each pair shows the FLIP data average for each protein, calculated as described in Materials and Methods.
These data are consistent with the results of previous studies (45) and demonstrate that individual PML, Sp100, and especially hDaxx molecules have a dynamic association with individual ND10 foci, with proteins both entering and exiting ND10 over periods of seconds or minutes. This process provides a simple mechanism by which ND10 proteins can relocate to sites of viral nucleoprotein complexes. In this model, individual ND10 protein molecules can be considered to have particular rates of association and dissociation, not only at ND10 structures but also at locations throughout the nucleoplasm, as represented by their diffuse background distribution. If the locations juxtaposed to viral nucleoprotein complexes at which ND10 proteins accumulate have a higher affinity for the ND10 proteins than sites elsewhere in the nucleus, then the natural dynamics of the proteins would inevitably lead to the accumulation of proteins at these novel foci at the expense of preexisting ND10, without the need to invoke any specific mechanism of transport or targeting.
DISCUSSION
Some of the hallmarks of ND10 structures are the rapid changes in morphology, composition, and number that they undergo in response to stresses such as virus infection, DNA damage, heat shock, and treatment with heavy metals, alkylating agents, and drugs that inhibit phosphatases, proteasomes, and transcription (1, 7, 8, 13, 15, 17, 26, 29, 30, 32). Live-cell studies have revealed that the large majority of ND10 foci have only limited movement in the nucleus (14, 31, 45), although a small subset of the bodies appear to be more mobile (31). It has been demonstrated that the disruption of ND10 that occurs during heat shock is due to the budding of microstructures from the parent body (14) and that chromatin damage also disrupts ND10 through a fission mechanism (13). Thus, there is evidence that ND10 foci can move either as complete structures or as microstructures that bud from them, but generally this is only over limited distances. On the other hand, it is clear that certain component protein molecules of ND10 are very highly mobile, with high rates of entry into and exit from ND10 structures. These include PML and Sp100 (Fig. 6 and 7) (45), CBP (5), and hDaxx (Fig. 6 and 7).
Since it was first observed that the parental genomes and replication compartments of several nuclear replicating DNA viruses are closely associated with ND10 structures (22, 23, 28; reviewed in references 15 and 27), the issues of how this occurs and the significance of the phenomenon have remained matters of debate. In this paper, we demonstrated that ND10-like structures can form de novo in response to and in association with incoming HSV-1 genomes and the resulting viral nucleoprotein complexes. This process is likely enabled by the spatially dynamic properties of several key components of ND10, including the PML protein itself. Our data do not exclude the possibility that parental viral genomes are able to migrate to sites adjacent to preexisting ND10, but they strongly suggest a simple explanation for the association with ND10 of the replication complexes of all relevant viruses examined thus far. Our data are also consistent with previous conclusions that transcriptional activity on HSV-1 genomes increases their association with ND10 and that viral genomes that are so associated are more likely to develop into a replication compartment (18, 38).
The preferential association of ND10 with specific cellular genes or chromosome sites has been observed in several situations, including regions of DNA replication in middle to late S phase (21), the major histocompatibility complex gene cluster on chromosome 6 (35), chromosomal regions with high transcriptional activities (21, 35, 44), and sites of integrated sequences that are at high copy numbers and to which foreign proteins are bound (42). A similar mechanism to that described here may be operating in at least some of these examples. In addition, there are several reports that demonstrate an association of PML foci with sites of DNA repair after DNA damage (1, 3, 7, 8, 24, 46). The biological activities occurring on the chromatin at these sites may be viewed as analogous in many respects to those on transcriptionally active viral genomes that are undergoing the initial stages of DNA replication. A dynamic recruitment of PML and other ND10 proteins to sites of DNA repair enabled by the efflux of these proteins from existing ND10 structures provides a ready explanation for their association with ND10-like foci.
Taken with the results of earlier related studies (18, 19), the results presented here have several important implications for our understanding of some basic features of HSV-1 infection. The directional nature of infection in cells at the edge of a developing plaque, the asymmetric distribution of viral genomes near the nuclear envelope, and the fact that replication compartments develop from these sites demonstrate the following points: (i) there is limited movement of the viral genomes within the nucleus once they have been released into the nucleoplasm; (ii) prereplication compartments, hitherto defined as a subset of the foci formed by ICP8 and other proteins (9, 25, 34, 43), may perhaps be most easily defined in the very early stages of infection by the association of ICP4 with parental viral genomes; and (iii) the observation of a very large number of small replication compartments forming near the nuclear envelope in cells at the edges of developing plaques demonstrates that HSV-1 DNA replication can initiate at abundant locations that may be defined only by the presence of the viral genome and its associated proteins, not by the preexisting nuclear architecture. This suggestion is in distinct contrast to the previously established view that replication compartments develop at a limited number of preexisting sites within the nucleus.
While it was widely assumed previously that HSV-1 genomes migrate through the nucleus until they encounter an ND10 structure and/or a specific location at which a replication compartment could begin to develop, upon reflection this model presents a significant problem. Even when packed exceptionally tightly within the capsid, the HSV-1 genome fills a space that has a diameter of approximately 90 nm. Once released, it is reasonable to assume that the viral DNA becomes much less densely packed, and after large numbers of viral and cellular protein molecules have become bound to the viral DNA, the whole complex must increase greatly in both size and mass. It is pertinent that these structures appear to be similar in size to preexisting ND10 foci, that is, with dimensions on the order of 1 μm (Fig. 1 and data not shown). As with ND10 and other nuclear substructures of a similar size, there are likely to be considerable restraints on the movement of such complexes through the nucleoplasm, particularly if they are to be targeted to specific preexisting sites where replication could commence. The model put forward in this paper presents a simple mechanism that circumvents these problems. Furthermore, the presence of viral genomes and nucleoprotein complexes near the nuclear envelope has obvious advantages since viral transcripts need be transported only a short distance from their sites of synthesis into the cytoplasm, and similarly, if the viral transcripts engage in translation soon after their arrival in the cytoplasm, then viral proteins that are targeted into the nucleus can be transported back rapidly (Fig. 8). This idea is consistent with the observation that in the very earliest stages of infection, ICP4 can first be detected in foci at the nuclear periphery (18).
FIG. 8.

Summary of a model for the spatial distribution of HSV-1 genomes and viral nucleoprotein complexes in relation to cellular ND10 proteins from the earliest stages of infection through the development of replication compartments. Further details are given in the text.
When considering the above ideas in comparison with previous data, it is pertinent that the observation of cells in developing plaques in human fibroblasts is perhaps the most natural system available for the study of virus infections of cultured cells, as the cells are not transformed and have been passaged only a limited number of times after being derived from human tissue; the virus particles have not been subjected to any manipulation, being freshly derived from the cells that initiated plaque formation; and the route of transmission by cell-to-cell spread is likely to be much more akin to that in a developing natural infection than the method of applying viruses from laboratory stocks onto the surface of a cell monolayer. These ideas are not in conflict with previous data, but the additional information gained from asymmetric infections allows more detailed analysis and subsequent subtle reinterpretation.
Taking all the data together, the events following the entry of the HSV-1 genome into the nucleus and proceeding to the point of replication compartment development are summarized in Fig. 8. The initial transcription of IE genes leads to the assembly of a transcriptionally active viral nucleoprotein complex close to the nuclear envelope. Subsequently, the preferential deposition of ND10 protein components leads to the assembly of ND10-like structures that are in association with the viral nucleoprotein complexes. Meanwhile, ICP0 is targeted to both preexisting ND10 foci and the novel ND10-like structures and causes their disruption and the degradation of PML. Viral replication compartments develop from the viral nucleoprotein complexes, having of course recruited all of the viral DNA replication proteins, as deduced in other studies (9, 10, 25, 43). A prediction that arises from this model is that initial IE gene transcription takes place at viral genomes that are not associated with ND10 proteins. This is consistent with previous data on the locations of SV40 transcriptional complexes (40).
This paper provides important insights into how PML-containing ND10-like structures may form and become associated with several different types of events involving DNA viruses and chromatin-based processes. The functional implications and an explanation for why this happens remain more elusive and may vary considerably for events involving transcription, apoptosis, DNA damage, DNA repair, viral transcription, and DNA replication. In particular, whether the underlying basis of these effects has positive or negative implications for the outcome of HSV-1 infection remains to be determined. Whatever the functions underlying PML and its associated proteins in these diverse activities, it is clear that the cell maintains a mechanism by which the ND10 constituent proteins can be deposited actively, rapidly, and efficiently at specific sites in response to cellular stresses and events occurring in the nucleus.
Supplementary Material
Acknowledgments
This work was supported by the Medical Research Council.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Barr, S. M., C. G. Leung, E. E. Chang, and K. A. Cimprich. 2003. ATR kinase activity regulates the intranuclear translocation of ATR and RPA following ionizing radiation. Curr. Biol. 13:1047-1051. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi, R., and P. P. Pandolfi. 2003. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22:9048-9057. [DOI] [PubMed] [Google Scholar]
- 3.Bischof, O., S. H. Kim, J. Irving, S. Beresten, N. A. Ellis, and J. Campisi. 2001. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell Biol. 153:367-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boddy, M. N., K. Howe, L. D. Etkin, E. Solomon, and P. S. Freemont. 1996. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13:971-982. [PubMed] [Google Scholar]
- 5.Boisvert, F. M., M. J. Kruhlak, A. K. Box, M. J. Hendzel, and D. P. Bazett-Jones. 2001. The transcription coactivator CBP is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol. 152:1099-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borden, K. L. 2002. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol. Cell. Biol. 22:5259-5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carbone, R., M. Pearson, S. Minucci, and P. G. Pelicci. 2002. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 21:1633-1640. [DOI] [PubMed] [Google Scholar]
- 8.Conlan, L. A., C. J. McNees, and J. Heierhorst. 2004. Proteasome-dependent dispersal of PML nuclear bodies in response to alkylating DNA damage. Oncogene 23:307-310. [DOI] [PubMed] [Google Scholar]
- 9.de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857-868. [DOI] [PubMed] [Google Scholar]
- 10.de Bruyn Kops, A., S. L. Uprichard, M. Chen, and D. M. Knipe. 1998. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology 252:162-178. [DOI] [PubMed] [Google Scholar]
- 11.Dohner, K., A. Wolfstein, U. Prank, C. Echeverri, D. Dujardin, R. Vallee, and B. Sodeik. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13:2795-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eskiw, C. H., and D. P. Bazett-Jones. 2002. The promyelocytic leukemia nuclear body: sites of activity? Biochem. Cell Biol. 80:301-310. [DOI] [PubMed] [Google Scholar]
- 13.Eskiw, C. H., G. Dellaire, and D. P. Bazett-Jones. 2004. Chromatin contributes to structural integrity of promyelocytic leukemia bodies through a SUMO-1-independent mechanism. J. Biol. Chem. 279:9577-9585. [DOI] [PubMed] [Google Scholar]
- 14.Eskiw, C. H., G. Dellaire, J. S. Mymryk, and D. P. Bazett-Jones. 2003. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 116:4455-4466. [DOI] [PubMed] [Google Scholar]
- 15.Everett, R. D. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20:7266-7273. [DOI] [PubMed] [Google Scholar]
- 16.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett, R. D., P. Lomonte, T. Sternsdorf, R. van Driel, and A. Orr. 1999. Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci. 112:4581-4588. [DOI] [PubMed] [Google Scholar]
- 18.Everett, R. D., G. Sourvinos, C. Leiper, J. B. Clements, and A. Orr. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 78:1903-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everett, R. D., G. Sourvinos, and A. Orr. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 77:3680-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett, R. D., and A. Zafiropoulos. 2004. Visualization by live-cell microscopy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol. 78:11411-11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grande, M. A., I. van der Kraan, B. van Steensel, W. Schul, H. de The, H. T. van der Voort, L. de Jong, and R. van Driel. 1996. PML-containing nuclear bodies: their spatial distribution in relation to other nuclear components. J. Cell Biochem. 63:280-291. [DOI] [PubMed] [Google Scholar]
- 22.Ishov, A. M., and G. G. Maul. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134:815-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishov, A. M., R. M. Stenberg, and G. G. Maul. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 138:5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurki, S., L. Latonen, and M. Laiho. 2003. Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization. J. Cell Sci. 116:3917-3925. [DOI] [PubMed] [Google Scholar]
- 25.Lukonis, C. J., J. Burkham, and S. K. Weller. 1997. Herpes simplex virus type 1 prereplicative sites are a heterogeneous population: only a subset are likely to be precursors to replication compartments. J. Virol. 71:4771-4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattsson, K., K. Pokrovskaja, C. Kiss, G. Klein, and L. Szekely. 2001. Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc. Natl. Acad. Sci. USA 98:1012-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maul, G. G. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20:660-667. [DOI] [PubMed] [Google Scholar]
- 28.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67-75. [DOI] [PubMed] [Google Scholar]
- 29.Maul, G. G., D. Negorev, P. Bell, and A. M. Ishov. 2000. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129:278-287. [DOI] [PubMed] [Google Scholar]
- 30.Maul, G. G., E. Yu, A. M. Ishov, and A. L. Epstein. 1995. Nuclear domain 10 (ND10) associated proteins are also present in nuclear bodies and redistribute to hundreds of nuclear sites after stress. J. Cell Biochem. 59:498-513. [DOI] [PubMed] [Google Scholar]
- 31.Muratani, M., D. Gerlich, S. M. Janicki, M. Gebhard, R. Eils, and D. L. Spector. 2002. Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat. Cell Biol. 4:106-110. [DOI] [PubMed] [Google Scholar]
- 32.Nefkens, I., D. G. Negorev, A. M. Ishov, J. S. Michaelson, E. T. Yeh, R. M. Tanguay, W. E. Muller, and G. G. Maul. 2003. Heat shock and Cd2+ exposure regulate PML and Daxx release from ND10 by independent mechanisms that modify the induction of heat-shock proteins 70 and 25 differently. J. Cell Sci. 116:513-524. [DOI] [PubMed] [Google Scholar]
- 33.Ojala, P. M., B. Sodeik, M. W. Ebersold, U. Kutay, and A. Helenius. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868. [DOI] [PubMed] [Google Scholar]
- 35.Shiels, C., S. A. Islam, R. Vatcheva, P. Sasieni, M. J. Sternberg, P. S. Freemont, and D. Sheer. 2001. PML bodies associate specifically with the MHC gene cluster in interphase nuclei. J. Cell Sci. 114:3705-3716. [DOI] [PubMed] [Google Scholar]
- 36.Showalter, S. D., M. Zweig, and B. Hampar. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP4. Infect. Immun. 34:684-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sourvinos, G., and R. D. Everett. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 21:4989-4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 40.Tang, Q., P. Bell, P. Tegtmeyer, and G. G. Maul. 2000. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J. Virol. 74:9694-9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang, Q., L. Li, A. M. Ishov, V. Revol, A. L. Epstein, and G. G. Maul. 2003. Determination of minimum herpes simplex virus type 1 components necessary to localize transcriptionally active DNA to ND10. J. Virol. 77:5821-5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsukamoto, T., N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont, and D. L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871-878. [DOI] [PubMed] [Google Scholar]
- 43.Uprichard, S. L., and D. M. Knipe. 1997. Assembly of herpes simplex virus replication proteins at two distinct intranuclear sites. Virology 229:113-125. [DOI] [PubMed] [Google Scholar]
- 44.Wang, J., C. Shiels, P. Sasieni, P. J. Wu, S. A. Islam, P. S. Freemont, and D. Sheer. 2004. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Biol. 164:515-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiesmeijer, K., C. Molenaar, I. M. Bekeer, H. J. Tanke, and R. W. Dirks. 2002. Mobile foci of Sp100 do not contain PML: PML bodies are immobile but PML and Sp100 proteins are not. J. Struct. Biol. 140:180-188. [DOI] [PubMed] [Google Scholar]
- 46.Xu, Z. X., A. Timanova-Atanasova, R. X. Zhao, and K. S. Chang. 2003. PML colocalizes with and stabilizes the DNA damage response protein TopBP1. Mol. Cell. Biol. 23:4247-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong, S., P. Salomoni, and P. P. Pandolfi. 2000. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2:E85-E90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.