

Abstract
A functional interplay of bottom-up and top-down processing allows an individual to appropriately respond to the dynamic environment around them. These processing modalities can be represented as attractor states using a dynamical systems model of the brain. The transition probability to move from one attractor state to another is dependent on the stability, depth, neuromodulatory tone, and tonic changes in plasticity. However, how does the relationship between these states change in disease states, such as anxiety or depression? We describe bottom-up and top-down processing from Marr’s computational-algorithmic-implementation perspective to understand depressive and anxious disease states. We illustrate examples of bottom-up processing as basolateral amygdala signaling and projections and top-down processing as medial prefrontal cortex internal signaling and projections. Understanding these internal processing dynamics can help us better model the multifaceted elements of anxiety and depression.
Keywords: amygdala, anxiety, attractor states, bottom-up, top-down, depression, PFC
INTRODUCTION
According to the most recent National Institute of Mental Health reports on US adults, anxiety and depression have a lifetime prevalence of 30% and 20%, respectively.1,2 Treatment options remain unsatisfactory for a significant number of patients struggling with the disorders, due in part to a lack of understanding of the underlying mechanisms.3 In the last decades, neuroscience research has made important advances in gaining a better understanding of the mechanisms that could give rise to such disorders.
In this review, we provide an overview of the literature to answer three central questions about disease states in the brain:
What accounts for the brain’s ability to shift from a healthy to a diseased state?
How does this shift affect how stimuli are processed?
Which physical neural-dynamic changes drive this shift?
To address these questions, we draw from David Marr’s analytical model to propose a three-level computational-algorithmic-implementational framework centered around the hypothesis that shifts in top-down (TD) and bottom-up (BU) processing occur to produce anxiety and depression as distinct disease states.4 First, we describe how changes in the computational landscape (attractor state dynamics) account for these shifts in processing (Figure 1). Then, at the algorithmic level, we describe how stimulus and valence processing maintain these states and become disrupted (Figure 2). Lastly, we identify and summarize findings on key brain structures, including the amygdala and the prefrontal cortex (PFC) as representative centers of BU and TD processing, respectively, which at the circuitry level contribute to the biological implementation of these changes (Figure 3). Integrating research within each of these fields can allow us to come to a deeper understanding of mental health disorders and provide a path toward more efficacious treatments. Anxiety and depression are complex diagnoses; manifestations of anxious and depressive pathologies are diverse, as reflected in the diagnostic criteria specified in the most recent edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V). To define anxiety and depression as we discuss them in this review, we outline common symptomatic criteria delineated in the DSM-V, categorized by psychosocial and physiological deficits (Box 1).5 Other reviews on anxiety and depression discuss the dysfunction of dopaminergic valence circuits,6,7 the biological variable of sex,8 and the dysfunction of threat-related circuitry.9,10 It is worth noting that multifaceted disorders require multifaceted frameworks and thus we endeavor in our model to incorporate the collective computational, psychodynamic, and biological investigations into these illnesses (see Box 1).
FIGURE 1.
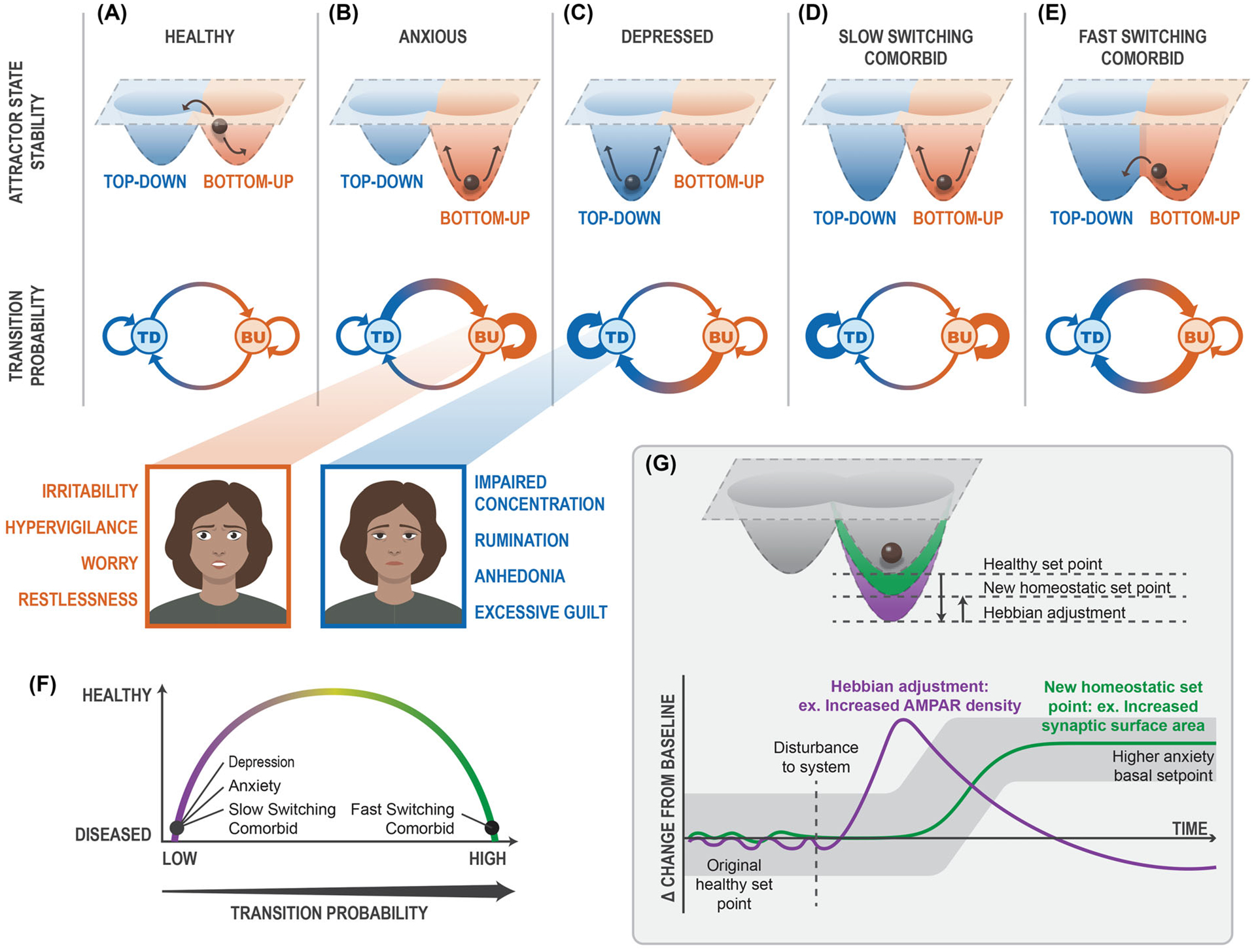
Attractor state dynamics of bottom-up, top-down processing. Bottom-up (orange) and top-down (blue) processing in (A) healthy, (B) anxious, (C) depressed, (D) slow-switching comorbid, and (E) fast-switching comorbid states are represented through attractor state dynamics (upper), where the ball indicates population activity at a given state with arrows showing average range of motion, and through the transition probability between states (lower), where the thicker arrows indicate higher transition probabilities. (F) Disordered transition probabilities result in different phenotype presentations. When the transition probability between TD/BU states is critically low, patients present anxiety, depression, or slow-switching comorbidity. When the transition probability between TD/BU states is critically high, patients present fast-switching comorbidity. (G, upper) Attractor state depth can change from Hebbian (purple) and homeostatic (green) plasticity changes. (G, lower) Plasticity changes are altered on different timescales; whereas Hebbian plasticity deviates from the basal setpoint, homeostatic plasticity resolves plastic deviations and, if needed, re-establishes the basal setpoint. Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic receptor; BU, bottom-up; TD, top-down.
FIGURE 2.
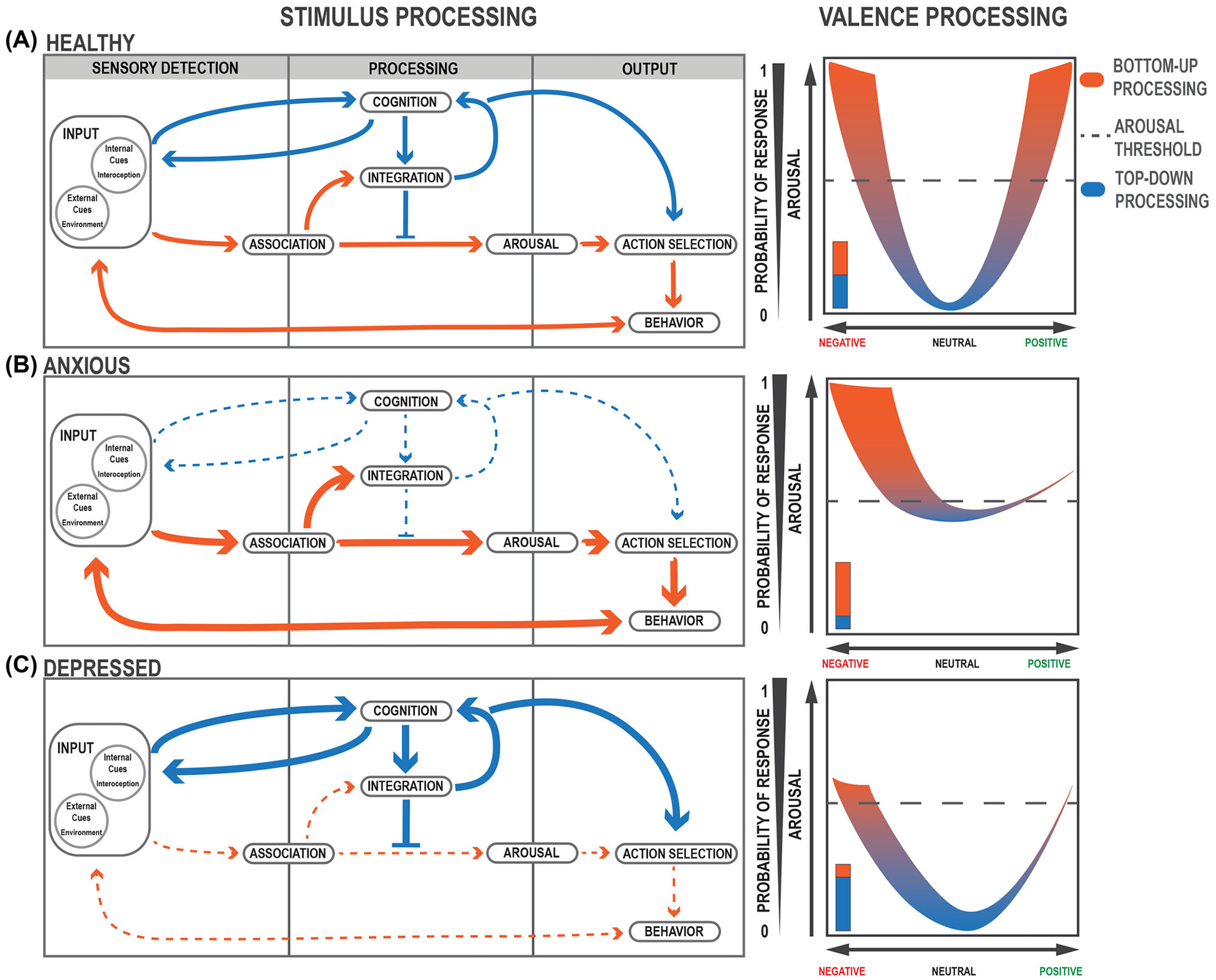
Interaction of bottom-up and top-down feedback loops during stimulus processing. (A–C, left) Flow of stimulus and valence processing in (A, left) healthy, (B, left) anxious, and (C, left) depressed states. Larger, thicker arrows indicate increased bias or activation. Dashed, thinner arrows indicate decreased activation. (A–C, right) Valence processing curves. (A, right) In a healthy state, valence processing corresponds appropriately to increases in arousal and positive–negative valuations of valence. The dashed line indicates the threshold of responding to a given stimulus. (B, right) In an anxious state, arousal is increased with a negative valence bias. (C, right) In a depressed state, arousal is decreased with a negative valence bias.
FIGURE 3.
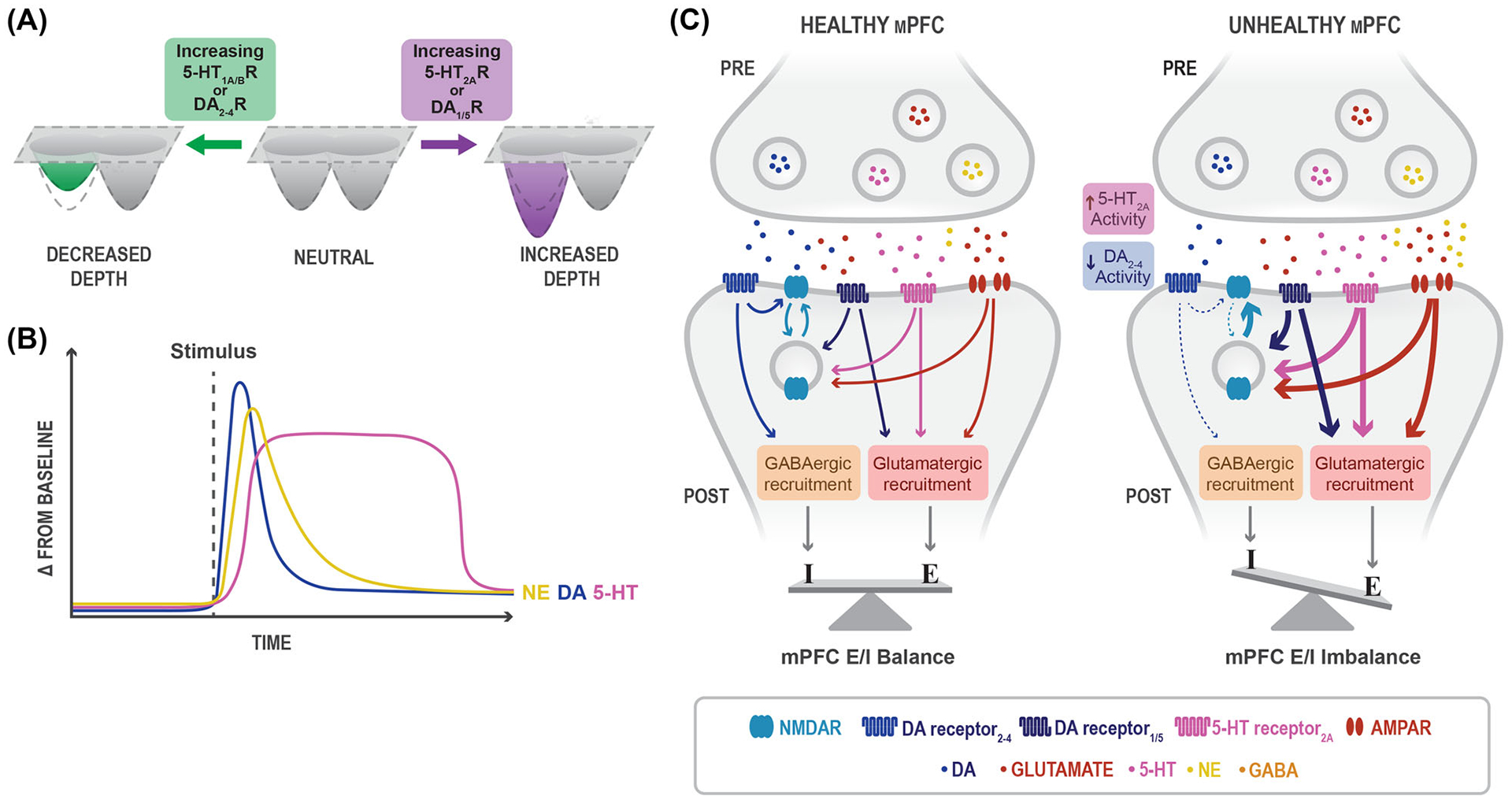
The effects of neuromodulation on the depth of attractor states and synaptic plasticity. (A) As neural populations receive higher bouts of 5-HT1A/B receptor activity and/or increased DA2–4 receptor activity, the depth of an attractor state decreases. As neural populations experience higher levels of DA1/5 receptor activity and/or increased levels of 5-HT2A receptor activity, the depth of an attractor state increases. (B) Variable release of neuromodulation across time after receiving a given stimulus. DA (blue) and NE (yellow) exhibit short encoding of stimulus. 5-HT (pink) imposes a slower, sustained release onto synapses. (C) In healthy mPFC synapses, 5-HT2A receptors (pink), D1-like receptors (dark blue), and AMPARs (red) interact to encourage NMDAR externalization. These receptors additionally recruit glutamatergic neurons (red). D2-like receptors (blue) internalize surface NMDARs and recruit GABAergic neurons (orange). Under unhealthy conditions in the mPFC, increased pressure from 5-HT2A and D2-like receptors (in addition to decreased pressure from D1-like receptors) causes increased externalization of NMDARs and glutamatergic neuron recruitment. The consequence of this synaptic change is an excitatory/inhibitory imbalance in the mPFC. Abbreviations: 5-HT, serotonin; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic receptor; DA, dopamine, E, excitation; GABA, gamma-aminobutyric acid; I, inhibition; mPFC, medial prefrontal cortex; NE, norepinephrine; NMDAR, N-methyl-D-aspartate receptor; POST, postsynaptic neuron; PRE, presynaptic neuron.
BOX 1.
| Generalized Anxiety Disorder (GAD) | Major Depressive Disorder (MDD) |
|---|---|
|
|
ATTRACTOR STATES ARE REPRESENTATIVE OF NEURAL NETWORK ACTIVITY
The brain is a dynamic system that depends on multiple hidden variables, of which we can only observe a minority. The ongoing activity of a single cell is governed by the combination of inputs: their amplitude, location, timing, and regulation of ion channels combined with slower signals from neuromodulation. On a larger scale, a cell receives inputs from tens of thousands of other neurons, whose connections and activity are unknown. Such a vast number of hidden variables in neural activity demand computational models that can represent biological activity accurately—understanding the brain as a dynamic system accounts for these hidden variables that cannot be observed.11–13 Such models contain “attractor states,” which represent states that a system gravitates toward regardless of initial conditions.11 As Antonio Damasio argues in his work, The feeling of what happens: Body and emotion in the making of consciousness, motivational senses of self emanate from a proto-self—a collection of patterns that summate and map the momentary homeostatic needs of an individual in many dimensions.14 Attractor states manifest as the computational foundations of the brain, upon which we construct our psychosocial identity. Attractor states are often likened to a bowl: if network activity was a ball, the ball would gravitate toward the deepest, or most stable, part of the bowl. Moreover, the deeper the bowl, the more energy it would require for the ball to roll out. This concept from mathematical systems can also be applied to biological systems, and this framework has been used in the context of mental health and disease.12,15–18 To advance our understanding of neural dynamics in the context of mental health, viewing the brain as an attractor state network allows us to make predictions based on past activity.
Mounting evidence suggests that neural ensembles demonstrate activity that is similar to attractor state models.11,18–22 An attractor network (distinct from the attractor state) is a type of dynamic network that evolves toward stability over time. To return to the bowl, an attractor state creates a malleable bowl, which can be deepened or otherwise augmented with significant, persistent changes in activity.23 For example, foam can be easily depressed with force, while removing your hand and pushing down on another area will change the location and depth of the depression. Many networks with continuous dynamics will develop attractor states that minimize energy in the system and are thus hyperstable.11,13,24 Population activity can and will deviate from an attractor state while processing different stimuli (noise in the network system); however, network activity will ultimately converge back to a stable state. The minimum level of energy in a system at which network activity converges is known as the basin of attraction.23,25 In neural populations with strong excitatory connections, multiple stable states can exist, all with basins of attraction.25 To classify the relative stability of mental states, we can use an attractor network to model the brain’s ability to transition between different forms of processing. Attractor states represent the first, computational level in our framework, in which the transition probabilities of network activity influence biological and behavioral phenotypes. We illustrate a multipoint attractor manifold to focus on specific components of anxious/depressive disease states in the brain. This framework does not invalidate the possibility for other attractor networks and their potential influence on network activity.
Patients with diagnosed anxiety and depression reveal different brain states as represented by functional MRI (fMRI) readouts, suggesting that anxiety and depression exist in separate attractor states.26 When comparing the response to sad facial expressions in patients with major depressive disorder (MDD) and generalized anxiety disorder, PFC activity is positively correlated with depressive symptoms, whereas it is negatively correlated with anxious symptoms. Human imaging studies indicate a significant relationship between anxious arousal and increased amygdalar–subcortical activity. In humans experiencing anhedonia, researchers observe increased limbic–paralimbic activity.26–28 Interestingly, areas with increased activity in anxiety had decreased activity in patients with depression and areas with decreased activity in anxiety had increased activity in patients with depression. The existence of these separate brain states supports the theory that there are bistable attractor states that can represent anxiety and depression (Figure 1A–E). As individuals exhibit an increased tendency toward TD or BU processing, attractor states reflect a similar geometry—the depth of one attractor state represents the increased likelihood that network activity will gravitate toward that type of processing.12 As we illustrate in Figure 1, healthy individuals can smoothly alternate between TD and BU processing states to modulate behavior appropriately. In pathologic conditions, the depth of the attractor states increases such that network activity is heavily biased toward either TD (in depression) or BU (in anxiety) (Figure 1B,C).
The concurrence of both anxiety and depression (comorbid anxious-depression) is very common, occurring in roughly half of cases and presenting an even greater challenge for treatment.2,29–31 It is possible that when the energy dynamics of one attractor state is disrupted, other states can be either compensatory or decompensatory. In our model, both attractor states are hyperstable in comorbid anxious-depression, leading to concentrated switching between pathological levels of TD and BU processing (Figure 1D–F). However, maladaptive stability may occur with different transition probabilities (Figure 1F). These differences in transition probability are termed in this review as a “slow-switching” comorbid state, where a person may be diagnosed with anxiety and depression over the course of their lifetime (Figure 1D), and “fast-switching” comorbid state, wherein a rapid state leads an individual to perceive symptoms of anxiety and depression simultaneously (Figure 1E).
PLASTICITY MODIFIES THE ARCHITECTURE OF ATTRACTOR STATES
Changes in the geometry of an attractor state may represent neural dynamic changes in firing rate or synaptic strength. Increased depth of attractor states has been biologically described by excitatory glutamatergic ion receptor function and spine concentration (Figure 1G).12 Experimental evidence demonstrates that reduced N-methyl-D-aspartate receptor (NMDAR)/α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic receptor (AMPAR) function in a given brain region influences processing capabilities, such as in associative learning and memory.32 To achieve an attractor state, neurons must retain a level of stability while being flexible enough to receive and transmit new information. This dynamic version of stability is achieved by neural plasticity, mechanisms by which neurons change in sensitivity and strength. Neural plasticity is generally described as two opposing mechanisms, Hebbian and homeostatic plasticity.33 Hebbian plasticity deviates neural activity from homeostasis and homeostatic plasticity encourages neural activity back to a predetermined setpoint34 (Figure 1G). These two types of plasticity happen on different timescales, Hebbian plasticity changes neurons acutely, whereas homeostatic plasticity occurs over a longer timescale (Figure 1G). Hebbian plasticity occurs when NMDARs in the synapse are activated, resulting in a signaling cascade wherein AMPAR density increases and the synapse becomes more sensitive to stimulation.35 Homeostatic plasticity is a form of metaplasticity, which affects neurons’ ability to undergo further synaptic changes based on previous plasticity. Homeostatic plasticity can consist of scaling receptors to different synapses, which in turn alters the sensitivity of the neuron.33 In the context of attractor networks, plasticity is the way that attractor states can change and stabilize their geometry. While the depth of attractor states is dependent on plasticity, changes in plasticity probability can be induced through long-term enhancement or depression of excitability via homeostatic plasticity.36
The mechanisms of Hebbian and homeostatic plasticity biologically influence the strength and identity of further plasticity.37 Hebbian plasticity modifies synaptic strength in the same direction of a given stimulus. For example, strong stimulation leads to an increase in synaptic strength resulting in long-term potentiation (LTP); conversely, low-frequency stimulation weakens synaptic strength and causes long-term depression.38,39 Homeostatic plasticity operates in contrast to Hebbian plasticity, such that the threshold for further Hebbian plasticity is modified. Because homeostatic plasticity intends to maintain a homeostatic setpoint for a neural network, it utilizes biological mechanisms to (A) acutely return activity to a baseline and (B) chronically modify a baseline to adapt to periods of high activity. As we illustrate in Figure 1G, a period of activity deviating from baseline increases the threshold for induction of LTP by methods of synaptic scaling or genetic augmentation of neuronal activity.34,37 The conclusion of homeostatic plasticity is thus a modification of thresholds for Hebbian plasticity and maintenance of a new setpoint following a period of high activity.
Homeostatic plasticity is capable of developing therapeutically beneficial setpoints as well as pathologic setpoints.3 Because plasticity is largely experience-dependent, changes in the external environment can dramatically modify the activity of a neural network and its consequent attractor network. One example of this potential is a change in social network and support. Individuals experiencing poor quantity and quality of social interaction also report large deteriorations in mental health.40 On a circuit level, social isolation acts with circuits historically correlated with depression in a parallel manner.40,41 These data suggest that social isolation encourages a new homeostatic setpoint, altering neural network activity, and hyperstabilizing a pathological attractor state. With this new setpoint, acute improvements of one’s social network would do little to permanently modify the firing rate at a population level in these circuits. Thus, acute intervention with no chronic effect will not improve mental health long-term. With improvements in the quantity and quality of social stimuli on a chronic scale, a new homeostatic level of firing rate is established, thus shifting a pathological attractor state into a salubrious one.
STIMULUS PROCESSING INVOLVES A BALANCE OF TOP-DOWN AND BOTTOM-UP
Changes in attractor state dynamics can result in major shifts in how stimuli are processed. At the algorithmic level, we distinguish between two feedback loops that, when balanced, allow for appropriate assessment and response to stimuli. The rapid and unidirectional BU feedback loop allows for immediate responses to sensory stimuli, whereas the slow and convoluted TD feedback loop allows for more complex assessments of the environment and fine-tuned behavior (Box 2). These loops proceed in three main steps: sensory detection, processing, and output (Figure 2A).
BOX 2.
| Bottom-up is the initial, rapid mode of processing that relies heavily on an immediate, less conscious, and more emotional reaction to salient stimuli from the environment. This type of processing is adaptive, allowing these to be rapidly and easily identifiable (e.g., being startled by a dog barking) | Top-down is a slow mode of processing that relies on a cognitive, context, prior-knowledge, and goal-driven approach to assessing stimuli. This type of processing acts as a filter to identify which of the initially perceived salient stimuli are relevant and deserving of attention and behavioral responses (e.g., recognizing the dog is behind a fence) |
Sensory detection
The first task of the system is to appropriately identify relevant information from an overwhelmingly stimulus-filled environment. Input signals, composed of both external (e.g., sounds from the environment) and internal cues (e.g., hunger), are detected by sensory processes in the body in a preattentive, automatic phase.42,43 Here, the brain must determine what is salient enough to bring to the forefront of attention and potentially act upon. For instance, studies have indicated that individuals have a latency bias toward identifying fearful stimuli among a background of nonfearful stimuli when compared to the reverse.44 Salient stimuli will meet an attention threshold at the first BU node: association (Figure 2A). Associative processes, both learned and innate, allow for valence assignment of the stimulus as positive (appetitive or rewarding), negative (threatening or aversive), or neutral.45 At the same time, input cues are sent to the central node of the TD loop: cognition, involved in conscious, goal-directed thinking.42,46
Processing
Integration receives inputs from both BU and TD centers to contextualize and assess relevant information about the stimulus. The BU loop allows for information concerning the valence and intensity of the stimulus to be transmitted directly to the arousal node to bring the body into a state of psychological and physiological alertness. The TD loop may counteract these efforts by filtering out stimuli that are irrelevant or unnecessary for goal-oriented behavior.47–49 This allows for disengagement from the stimulus to prevent an inappropriate response. The TD loop continuously incorporates new information from changing environments and involves multiple iterative, inner feedback loops to continue amending cognitive assessments of stimuli. A loud barking sound, for example, might initially prompt a startle response, triggered by BU processing, but as more relevant information surrounding this sound is integrated by TD processes, one would notice the dog is safely held by an owner on the leash.
Output and feedback
Action selection is again informed by a balanced interplay of both loops: arousal and cognition. Once the action has been selected, a behavioral response is produced, to move toward or away from an external stimulus (e.g., moving away from a spider) or to change an internal state (e.g., eating food). Action selection feeds back into the cognitive node allowing for future strategization and goal orientation. These actions reconfigure the set of stimuli presented to the system, completing the loop.
In a healthy state, these parallel processes allow for appropriate and evolutionarily adaptive behavioral responses that can change depending on the valence, intensity, and relevance of stimuli. The BU loop, driven by reflexive, innate, emotional responses to stimuli, allows for rapid, immediate processing to respond to very threatening or very rewarding stimuli (Figure 2). As stimuli valence intensifies (more positive or negative), arousal generally increases,45 resulting in a higher likelihood of a behavioral response. The TD loop prevents unnecessary increases in arousal levels by filtering out stimuli that are neutral and irrelevant (Figure 2A).9
MALADAPTIVE STIMULUS PROCESSING AND VALENCE DESTABILIZATION RESULT IN DISEASE STATES
Although there is heightened negative affect and bias toward evaluating stimuli as negative in both anxiety and depression,42,47,50 the manifestation of these diseases is often quite different. For instance, the prospect of interacting with a peer may provoke negative nervous or threatening feelings for someone experiencing anxiety, whereas for someone with depression, the same prospect, though also negatively evaluated, might induce feelings of fatigue. In Clark and Watson’s tripartite model of anxiety and depression, negative affect is the common link between anxiety and depression, and contributes to anxious-depressed comorbid states.50 In such comorbid states, increased attractor state depth results in maladaptive behavior and increased negative affect. In this review, we focus on the phenotypes identified in the tripartite model that distinguish these two disease states: increased physiological arousal in anxiety, and anhedonia and disengagement from the environment in depression.50 We propose that an imbalance in the two processing modes leads to these distinct disease phenotypes.
As reviewed in detail by Mogg and Bradley, in an anxious state, the system shifts overwhelmingly toward BU processing, without enough regulation from TD, disrupting goal-oriented behavior.51 This leads to excessive physiological and psychological arousal and increased sensitivity to environmental stimuli, prompting stimuli to be biased as threatening9,42,51,52 (Figure 2B). Stimuli that would commonly be perceived as neutral might trigger psychological symptoms associated with anxiety, including hypervigilance and worry, as well as physiological symptoms, such as increased heart rate, sweating, and muscle tension (Box 1). In depression, TD cognitive processes take control over emotional, reflexive ones, and overinhibit the BU loop. This can lead to the inability to engage appropriately with the environment or reach the arousal threshold to act upon relevant stimuli (Figure 2C),47,50,51 manifesting as a lack of motivation and anhedonia (Box 1). Within this model, bidirectional overcompensation in individuals with comorbid anxious-depression leads to a maladaptive alternation between BU- and TD-biased processing (Figure 1D–F).
Human studies show that emotion and attention processing activate distinct patterns of activity in the brain depending upon whether a TD- or BU-eliciting stimulus is presented.53,54 BU-inducing stimuli, such as images that can quickly provoke an automatic emotional response, have been shown to activate brain regions, such as the amygdala, whereas TD-inducing stimuli (words and imaginative activities) have implicated the PFC.53,54 We focus on the amygdala and PFC as representatives of BU and TD processing, respectively, and elucidate how the biological mechanisms implement chronic changes on the computational and algorithmic level.
ANXIETY DISORDERS CAN ORIGINATE FROM HYPERACTIVITY IN BOTTOM-UP REGIONS
BU processing at homeostatic levels is necessary for an animal to remain vigilant and cautious; however, pathological increases of activity in areas associated with adaptive anxiety processing can lead to maladaptive anxiety symptoms and behavior.9 Within the context of BU processing, the amygdala is a critical region in the brain for associative learning, BU processing, and arousal.44,54–56 The amygdala is also involved in processing changes in the internal state as an important interoception axis.57,58 What must be noted is the theorized bidirectional relationship between interoception and emotional arousal. As internal states change, emotions may arouse through conscious or subconscious evaluation of these homeostatic deviations.59 This interplay between internal state and emotional response reflects classic theories of emotion59,60 and illustrates how amygdala activity can simultaneously reflect interoceptive and emotional status.54,58 As evidenced by experiments in humans, patients with amygdalar insults struggle to generate physiological responses in anticipation of risky behavior or in response to losing in a gambling task.61 Further human behavior experiments show that associative processing related to emotion is also impaired—patients with amygdala lesions fail to recognize fearful emotions in faces, although they can decipher personal identity.62 Amygdala volume correlates positively with fearfulness in human subjects while conversely, bilateral damage to the amygdala is associated with decreased anxiety and fearfulness.63 The basolateral amygdala (BLA) specifically is a driving factor in anxiety,64 composing one-third of a tripartite group of regions with reciprocal connections that are heavily involved in emotional processing, including the ventral hippocampus, and the medial PFC (mPFC).65–67 We will focus specifically on the relationship between the mPFC and the BLA, due to their theorized roles in our algorithmic processing model and their respective involvement in emotional processing.
In human fMRI studies, the functional connectivity between the BLA and the mPFC has been correlated with higher levels of anxiety.68,69 Human fMRI investigations into different biotypes of mood disorders have elucidated that dysfunctional connectivity between the PFC and the amygdala increases anxiety.70 Rodent studies have specifically illustrated that the directionality between the BLA and the mPFC is relevant in mediating fear and anxiety-like behavior. From a processing perspective, amygdala projections to the mPFC encode motivational and arousal information, which can be modulated and filtered by the mPFC. These data are bolstered from rodent studies using imaging,71 electrophysiological,72,73 and optogenetic66,74 techniques, which have elucidated that BLA input to the mPFC is necessary for anxiety behaviors. Theta oscillations between the BLA and the mPFC synchronize in anxiogenic environments; oscillatory stimulation is sufficient to induce anxiety-like behaviors in rodents.66,75,76 Excitation of BLA projections to the mPFC produces anxiogenic effects, freezing responses, reduction in social interactions, and attenuation in cue-associated fear in mice; inhibition of BLA–mPFC projections facilitates social interaction and reduces freezing.74,77 The role of the BLA in fear learning fits the role of the association node, projecting rapid associative information to the integrative mPFC. Amygdala inputs drive feedforward inhibition of mPFC neurons by targeting parvalbumin (PV)-positive interneurons.72 These results suggest the associative information from the BLA impacts on neural ensembles in the mPFC, possibly by reducing background activity and allowing for appropriate neurons to integrate and direct behavior. When considering these results together, hyperexcitability in the BLA biases the BU pathway and represents an entrenched BU attractor state in anxiety disorders.
DEPRESSION MAY EMERGE FROM HYPERACTIVITY IN TOP-DOWN REGIONS
The mPFC has been historically linked to mood disorders and connectivity within this region has been implicated in depression. Encoding in the mPFC is attributed to reward learning,78 emotional memory,56 decision making,79 and moral assessment.80 Patients with vmPFC lesions exhibit impaired strategic decision making61 and intuitive moral judgment,81 suggesting that the mPFC plays a critical role in TD processing.82 Activity in the frontal cortices, specifically the anterior cingulate cortex, is associated with increased optimism and likelihood to imagine positive future outcomes, suggesting that the mPFC is required for long-term mediation that is characteristic of TD processing.83 Experimental and functional imaging studies have demonstrated dramatic changes focused on the PFC in depression.79,84,85 MDD has been associated with hyperactivation of the mPFC and reduced activation in reward-processing dopaminergic neurons in the nucleus accumbens (NAc).85–87 Deep brain stimulation targeting the PFC, namely, the subgenual cingulate region, resulted in profound antidepressant effects; however, others have struggled to replicate these results.88,89 What is also of importance are the connections between the mPFC and other regions, namely, the ventral tegmental area (VTA) and lateral habenula (LHb).90–93 The VTA is highly associated with reward and motivated behavior, whereas chronic inhibition of VTA–dopamine (DA) neurons induces depressive-like symptoms.94 Activity in the VTA is also susceptible to stress on different timescales—acute stress increases VTA neurons projecting to the mPFC, while chronic stress depresses VTA–mPFC activity.90,95
The LHb also regulates reward-prediction responses and has recently been implicated in regulating negative associations.92 Because of this unique signaling pattern, the LHb has been referred to as the brain’s “anti-reward center” and receives input from a diverse set of neurotransmitters.6,69 Increasing excitability of LHb neurons via optogenetic96 and genetic97 methods is sufficient to induce avoidance behavior, anhedonia, and other core depressive symptoms6,98 in rodent models. In contrast, clinical studies applying deep brain stimulation to inhibit the LHb observed antidepressive effects.99 The LHb receives primarily glutamatergic inputs from mPFC.92 Following the upstream encoding of reward and cognition, the mPFC–LHb circuitry is likely critical for behavioral control over negative affectation. Research investigating the mPFC, LHb, and VTA suggests a relationship in which decreasing transmission from the VTA leads to an excitation/inhibition (E/I) imbalance in the mPFC, which is reflected as hyperexcitability in projecting regions like the LHb (Figure 3C). Interestingly, the LHb projects back to the VTA, potentially constructing a TD processing loop that is hyperactive in negatively biased valence disorders like depression.99
OSCILLATORY ACTIVITY AS A DRIVER FOR TOP-DOWN/BOTTOM-UP BALANCE
Proper attention to stimuli relies on a healthy balance of TD and BU processing. In contrast, salient stimuli are processed in a BU fashion through sensory inputs, deliberate TD control of attention allows one to focus not only on vestibular senses but also on the long-term evaluation of these stimuli.100,101
Oscillatory activity in the brain refers to the rhythmic and synchronously phase-locked activity between brain regions.102 This synchrony allows for distant brain regions to communicate and hold long-term information for conscious problem-solving by linking the information in these regions via a temporal framework. In both humans and monkeys, local field potential (LFP) gamma frequency (25–140 Hz) coherence between the PFC and visual cortex drastically increases during attention to a visual stimulus, implicating a concerted role of the mPFC for driving long-term attention.103 This type of oscillatory activity also provides the PFC with the ability to represent a variety of distinct categories of information in concert, allowing for more complex working memory.104,105
Oscillatory synchrony between the amygdala and the mPFC may be critical for healthy emotional evaluation of stimuli and may help bias the brain into certain states. Theta (3–8 Hz) oscillations between these brain regions are enhanced during fear learning in both rodents and humans, and disruption of these theta rhythms affects fear extinction or recall depending on the theta phase.106–108 Similar frequencies between the PFC and the hippocampus regulate avoidance behaviors in rodents, and phase-locked stimulation at these frequencies also enhances avoidance.109,110 These data imply that when signals are in phase, synchronous activity is amplified to bind certain inputs together and encourage attractor state transitions. Additionally, if these oscillatory dynamics are disrupted, it may lead to maladaptive emotional processing and brain states. For example, asymmetric alpha (8–12 Hz) activity between the left and right prefrontal regions has been commonly found in patients with MDD.111–113 Reduced gamma and alpha activity is also found in patients with euthymic bipolar disorder, characterized by a lack of mood disturbances, suggesting a shift in oscillatory brain states during certain emotional phases.114,115 Enhancing certain rhythms may also aid in the transitions to healthier attractor states. Disrupting the mPFC through stress can lead to the reduced amygdala and VTA synchrony and increase emotional pathologies found in anxiety and depression.116 Recently, work has been done to map how oscillatory activity propagates throughout the brain rather than through a simple pair-wise comparison between two brain regions. The collection of factors (spectral power, synchrony, and phase directionality) as observed through LFPs comprises the conceptual “electome” framework. These electome factors describe how oscillatory activity may shift brain dynamics into system-wide states and how certain states have been shown to predict vulnerability to depression.117 These data suggest that oscillatory synchrony may be integral for healthy brain states, and disruption of this synchrony may lead to unstable transitions between these states.
Dysfunctions in oscillatory activity may lead to maladaptive transitional patterns between these states, leading to conditions that may either be stuck in one attractor versus the other, or with very shallow attractors enabling high transition probability manifesting as comorbidity with anxiety-related and depressive symptoms. As oscillatory activity in attention suggests an upper limit to how much information the brain can attend to at one time;104,105 such may be the case when patients experience symptoms related to both anxiety and depression. It may be difficult to imagine feeling both anxious and apathetic to a stimulus at once, but imbalanced oscillatory activity may lead to rapid transitions between these states, which is perceived as these symptoms being experienced at the same time in comorbidity.
NEUROTRANSMITTER BALANCES DRIVE ATTRACTOR AND MENTAL STATE STABILITY
Glutamate and gamma-aminobutyric acid (GABA) are the primary neurotransmitters involved in the excitation and inhibition of activity, respectively. Metabolically, glutamate acts as a precursor for GABA, which contributes greatly to controlling neural network dynamics through inhibitory action.118,119 GABAergic interneurons can be identified by their expression of somatostatin (SST) and PV, among other markers. Postmortem studies of patients with depression have shown a reduction of SST/PV interneurons in the PFC.120,121 Treatments with various antidepressants, electroconvulsive therapy, and cognitive behavioral therapy restore GABA levels in depressed subjects.122
While glutamate and GABA drive excitation and inhibition post-synaptically, they also contribute to and are subjected to changes in plasticity. GABAergic transmission in the mPFC constrains LTP, depending on the presence of either GABA-A or GABA-B receptors.123,124 GABA-A receptor (GABA-AR) mutant mice have significantly less surface expression of NMDAR and AMPARs and exhibit anhedonia and behavioral inhibition.125,126 Chemogenetic inhibition of GABAergic neurons in the mPFC is not only sufficient but necessary for antidepressant responses.127 Because GABAergic interneurons are responsible for controlling E/I within the cortex and excitatory projections, hyperactivation of GABAergic interneurons supports a model in which E/I imbalances cause anhedonia and other depressive symptoms119,128,129 (Figure 3C).
The E/I balance in the mPFC can be disrupted by a dearth of inhibitory GABAergic neurons. Our model hypothesizes that unregulated excitatory activity in the mPFC can lead to excitotoxicity in the region (leading to observed structural changes) and hyperexcitability in downstream projections like the LHb causing aversive behaviors.92 This hypothesis is further supported by human imaging studies that demonstrate a biotype of depression in which frontostriatal networks have pronounced hyperconnectivity in patients with more severe anhedonia and motor retardation.70 The consequences of this regional specificity are reflected in the differential effects of anxiety and depression drugs, specifically ketamine; recent studies have demonstrated that ketamine preferentially increases LTP in GABAergic interneurons in the mPFC, potentially rectifying the E/I imbalance.130 Glutamate causes the excitation of neurons, and excessive glutamate may drive excitotoxicity in the mPFC in the case of depression.124,131,132 Patients with MDD and postpartum depression have marked elevations of glutamate in the mPFC.124,132 Reciprocally, increased glutamatergic signaling from the BLA to the mPFC causes anxiety-like behavior. Glutamate intake at excitatory synapses can attune the polarity and magnitude of long-term plasticity changes, demonstrating that chronic increases in glutamate release can manipulate the basal setpoint of circuits, leading to hyperreactive neuronal populations.133 In both anxiety and depression, human and rodent studies demonstrate that increased glutamate is present in either the BLA or the mPFC, respectively. These tonic changes in glutamate can represent deepened attractor states and, thus, one can use neurotransmitter levels to model TD- and BU-biased processing modalities.
NEUROMODULATION IN TOP-DOWN AND BOTTOM-UP PROCESSING CIRCUITS INFLUENCES PLASTICITY AND MENTAL STATE
Fast ionic signals represent the movement of the brain state, but to shape the manifold upon which our activity travels, the brain must operate on other timescales, including using long-term affective signaling like neuromodulation. While an animal acutely responds to stimuli, the strategies by which it responds, and the lasting effect of cumulative stimuli must have a longer-term effect. The mood reflects the summative impact of expectations and reward outcomes.20 The metaphorical narrative surrounding mood is akin to the geometry of attractor state landscapes.134 Where the processing is how the ball moves across energy landscapes, mood describes the energetic limitations of the said landscape, or the dimensions of an attractor state. Experiences affect mood, which in turn is integrated to affect subsequent experience through neuromodulation and plasticity alterations.
There is a strong relationship between the level of expression of given neuromodulators and mental state. Neuromodulator release in the brain has been linked to anxiety and depression and provides insight into how mental illness is encoded.127 As computational theorists of attractor states argue, the stability of an attractor state is analogous to the level of plasticity in the region. Thus, the presence and identity of neuromodulators inform our model through an explanation of how selective changes in plasticity alter attractor state stability. We will be specifically focusing on the role of serotonin (5-HT), norepinephrine (NE), and DA, and their role in plasticity in TD and BU regions. We suggest a model in which regional dysfunctions of modulation drive anxiety and depression symptoms, entrenching hyperstable attractor states. In our model, we argue that anxiety is caused by a hyperstable attractor state entrenching BU-biased processing.135 This state is due to the hyperexcitability of the BLA and its glutamatergic projections, namely to the mPFC.72 Conversely, depression is caused by a hyperstable TD-biased attractor state landscape, in which an E/I imbalance is maintained by disordered neuromodulation in the mPFC.
DISORDERED NEUROMODULATION: SEROTONIN
5-HT has been historically linked to emotional behaviors, with a likely role in TD/BU processing motifs. In the amygdala, infusions of serotonin influence fear learning in mice. Microdialysis studies demonstrate that inescapable shock enhances 5-HT levels in the BLA from both conditioned stimulus and unconditioned stimulus presentations.136 Further investigations on fear learning demonstrate that 5-HT signaling is positively correlated with fear memories. Mice that overexpress 5-HT transporter (5-HTT), a protein that clears 5–HT from the extracellular space, demonstrate impaired fear learning, whereas the 5-HTT–underexpressed counterparts have impaired fear extinction. In primates, manipulations of 5-HT receptors affect trait anxiety-like phenotypes.137
The role of 5-HT in fear learning in the amygdala supports the hypothesis that 5-HT causes changes in plasticity. 5-HT release in the BLA depresses excitatory postsynaptic currents from outside inputs, likely via inhibitory GABAergic neurons.138,139 Simultaneously, the application of 5-HT increases the excitability of BLA pyramidal neurons—suggesting that 5-HT release increases excitability within the BLA.140 Activation of 5-HT2A receptors (5-HT2ARs) enhances NMDAR-mediated potentials, demonstrating that 5-HT2ARs facilitate NMDAR-dependent synaptic plasticity in the BLA.141 This model has been fortified by results demonstrating that chronic administration of selective serotonin reuptake inhibitors (SSRIs; e.g., fluoxetine) increases plasticity by decreasing the number of PV+ interneurons in the BLA.138,141 Establishing a 5-HT–induced hyperplastic state in the amygdala led to the elimination of fear memories—suggesting that 5-HT likely influences excitability in the amygdala, and increases in 5-HT or 5-HT-receptor sensitivity modulate anxiety states and likely emotional processing in general.136,140–142 In our TD/BU processing model, increases in amygdala sensitivity cause a BU processing bias. Our model would thus hypothesize that increases in 5-HT levels would cause a BU processing bias and increase spontaneous emotional processing. Indeed, SSRIs have historically been used to treat disorders of negative affect, including depression and anxiety.143 In both healthy and depressed individuals, chronic SSRI administration leads to a positive shift in valence processing, possibly via amygdalar circuits.139,144 Further studies investigating SSRI administration demonstrate that SSRI treatments (increasing levels of 5-HT in the synaptic cleft) enhance LTP from BU regions communicating to the mPFC.141 These results suggest that excess 5-HT signaling acutely mediates plasticity in mPFC.
While 5-HT facilitates excitability in the BLA, the mPFC both receives 5-HT inputs and exerts TD control of 5-HT expression in downstream regions. 5-HTergic inputs from the mPFC are complicated by the heterogeneous encoding of different 5-HT receptors.140 Human blood-oxygen-level-dependent (BOLD) fMRI studies reveal a positive relationship between 5-HT2A receptor activity and amygdala reactivity to anxious stimuli, which is inversely moderated by 5-HT1A receptor activity.145,146 5-HT1A and 5-HT2A receptors are localized in a relevant orientation–the inhibitory 5-HT1A receptor is located on the axon hillock, whereas the excitatory 5-HT2A receptors are located on the dendrites of glutamatergic neurons.145 Given this orientation, 5-HT1A receptors can effectively gate 5-HT2A receptor activity and PFC output, suggesting that 5-HT signaling in part modulates TD control from the mPFC to the amygdala.147 Thus, disordered localizations of 5-HTergic signaling could deleteriously influence the inhibitory brakes on fast, associative BU processing.
Studies investigating the dynamic changes in 5-HT signaling in the mPFC provide more insight into how 5-HT can mediate TD processing and plasticity. 5-HT2A receptor activation enhances NMDAR-dependent plasticity, strengthening associative memory.141 Rodent studies comparing responses to stressful experiences demonstrate that 5-HT sensitivity significantly increases as a response to stress in the mPFC, and high increases in DA efflux follow.136 Interestingly, 5-HT1A agonism decreases both 5-HT and DA release, further suggesting that low 5-HT1A receptor binding is required for TD mPFC processing.145 Whole-life knockout of 5-HT1A receptors in mice leads to a depression phenotype, suggesting that without the needed gating of 5-HT2A signaling, the mPFC exerts excessive TD control, leading to a phenotype that is hyporesponsive to stress.148,149
The relationship between 5-HT and DA signaling is supported by developmental studies, which show that lesioning of 5-HTergic projections to the mPFC increases DA release.150 These data and the aforementioned receptor data together suggest a model for 5-HT control of stress responses in the amygdala which projects excitatory input to the mPFC. Agonism of 5-HT1A receptors enhances DA signaling in the mPFC, suggesting a broader involvement between 5-HT and DA release.151 5-HT signaling likely encompasses a modulatory role in the mPFC, flattening attractor state transition probabilities and selectively altering plasticity, depending on the activated receptor (Figure 3A). Disordered localization of the 5-HT signal could disrupt this system and lead to pathological entrenchment of mental states like anxiety or depression.
DISORDERED NEUROMODULATION: DOPAMINE
Whereas 5-HT could flatten attractor state landscapes, DA signaling plays a role in the entrenchment of salient behaviors. DA computationally controls behavior in the mPFC by gating sensory input, manipulating memories, and relaying motor commands.152,153 This gating of activity has led to models that also suggest that DA modulates response to the saliency of reward.154,155 DA release has been theorized to reflect reward prediction errors (RPEs) in the mPFC.153 Inactivation of the mPFC in classical conditioning tasks with a differing probability of reward affects DAergic signaling when the reward is not guaranteed.78 DA signaling in the NAc core does conflict with RPE, as experimental results cannot replicate RPE in practice.155 Dysfunctional DA activity is correlated with problems in processing motivation, pleasure, and reward.95,152,156 Imaging studies in patients with social anxiety reflect a positive correlation between symptom severity and DA transporter proteins (DAT) availability in the amygdala and hippocampus.157,158 Moreover, 5-HT and DAT coexpression was significantly increased in the amygdala of patients with anxiety, suggesting that DA may also play a role in the progression of anxiety.
Patients with depression report difficulties with motivation (anhedonia) and reward processing, both of which DA is intimately involved in encoding in the mPFC. What is of relevance is the relationship between the mPFC and the VTA, a circuit that is historically linked to depression.95,159 Phasic stimulation of VTA-DA inputs to the mPFC also increases conditioned place preference.160 Alongside evidence that VTA-DA neurons lack activity in patients with depression, these data support a model in which a dearth of DA input to the mPFC enhances depressive symptoms. In mice with chronic stress-induced anhedonia, decreased levels of DA release have been measured from the VTA projections to the mPFC. In social defeat stress models of depression, mice initially exhibit a hyperexcitability of VTA-DA neurons, as compared to resilient mice that exhibit stable firing.95 What provides more insight into how DA signaling influences mPFC activity is the role of DA inputs in plasticity. Experimental enhancement of VTA-DA neuron excitability achieved an antidepressant effect through homeostatic plasticity.95 These results support a model in which homeostatic plasticity interacts with DA neurons in the brain to modulate depression states. Anxiety circuits can be similarly altered via chronic stimulation, altering excitability levels to a healthier homeostatic setpoint.161
DA exerts influence through D1-like (type 1/5) and D2-like (type 2–4) receptors, which causes increases and decreases in levels of the messenger molecule cyclic adenosine monophosphate (cAMP), respectively.162,163 Changes in cAMP retroactively recruit or inhibit NMDAR signaling; thus, DA signaling can bidirectionally alter plasticity. DA release, therefore, plays an important role in the modulation of GABA/glutamate signaling in the mPFC.163 Inactivation of DA release is sufficient to eliminate reinforcement learning behaviors normally attributed to GABA/glutamate signaling.163 These data ultimately support a theory of regional-specific alterations in DA signaling in the states of depression and anxiety.
5-HT and DA both mediate NMDAR recruitment and LTP in the amygdala, encouraging greater sensitivity in the BLA. Behaviorally, D1 agonism in the amygdala elicits anxiogenic effects, whereas antagonism elicits anxiolytic effects.164,165 D2 agonism/antagonism illustrates more mixed results, suggesting that D1 receptors in the BLA encode rapid associative processing in the BLA, whereas D2 signaling develops adaptive responses. D1 receptor activation in the BLA further dampens mPFC-induced inhibition, suggesting that the mPFC regulation of BLA activity is inversely dependent on DA levels in the BLA.165,166 These data suggest that excess D1 signaling could drive BLA excitability and, given the role of D1 receptors in increasing NMDAR conductance, deepens a BU-biased attractor state landscape.
The contributions of DA signaling in the mPFC are necessary for motivation and emotional processing; thus, our model attributes a lack of DA signaling to an imbalance between excitatory and inhibitory neurons in the mPFC, leading to anhedonia.167 DAergic control of the E/I balance in the mPFC is influenced upstream by 5-HTergic signaling, possibly causing an increase in inhibitory signaling and plasticity in the mPFC.167,168 Whereas 5-HTergic signaling seems to flatten attractor state landscapes, DAergic signaling defines and entrenches them.12 Recent rodent studies have shown that DA-dependent plasticity is occluded in the mPFC of rats susceptible to chronic mild stress, although this effect is reversed following the administration of ketamine.167 Following our tri-level model, these DAergic data suggest that disordered DA inputs to the mPFC would deleteriously increase the depth of a point attractor state, leading to excess TD control over other regions and depressive symptoms.
DISORDERED NEUROMODULATION: NOREPINEPHRINE
Functional DAergic signaling in the mPFC is not possible without intact NE release.169 DA and NE interact to regulate excitatory and inhibitory firing in the mPFC and maintain a delicate balance; too little DA/NE influence leads to memory impairment, whereas excess agonism of DA/NE receptors leads to excitotoxicity and dysfunction.169,170 Much like DA, NE is released in the amygdala and the mPFC in response to acute stimuli like anxiety or reward and is involved in sleep/wake cycling, stress, and fear responses171 (Figure 3B). Due to its involvement in processes that are so often disordered in anxiety and depression, human studies have found correlative links between disordered NE release and mental illness. NE transporter and 5-HTT polymorphisms are related to increased susceptibility for anxiety in human subjects.137,172,173 NE neuronal cell groups can activate the hypothalamic–pituitary–adrenal axis in stress responses, consolidate negative emotional memory through amygdalar and hippocampal projections, and control reward evaluation in the PFC.174,175 In the cortex, NE inhibits cAMP, prolonging potassium currents and stabilizing attractor state networks.176 In the amygdala, the release of NE has a similar role to 5-HT in regulating brain-derived-neurotrophic-factor-positive neurons, suggesting that it strengthens synaptic plasticity in BU processing regions as well.177 While NE release is necessary for processing reward in the mPFC, continuous NE release from chronic stress causes deleterious effects in cortical processing.
DA and NE affect the behavior of E/I neurons by reducing feedforward GABA inhibition and enhancing LTP.119 Increased levels of DAT and NE in patients with anxiety disorders bolster this interpretation that excess monoamine release causes downstream dysregulation of GABA.158 Injection of corticosterone into the amygdala induces anxiety-like behavior, imitating depressed GABAergic tone in the lateral amygdala.121 In addition to inducing a hypervigilant state, corticosterone increases the release of DA178 and NE179,180 in BU regions. Bidirectionally, the inhibition of GABA pyramidal neurons in the BLA promotes fear learning.181 Because GABAergic interneurons innervate and modulate glutamatergic outputs from the amygdala, reduction of GABA release triggered by dysfunctional neuromodulation may cause hyperexcitability of glutamatergic neurons.
Chronic release of NE via repeated traumatic stress can cause the consolidation of a lasting negative affective bias.182 Human subjects describe acute systemic infusions of NE as hyperarousal and later describe a “numbing” experience (when circulating NE is much lower, potentially representing NE “burnout”)175 (Figure 3B). As NE and 5-HT interact in anxiety, they also have a relationship in depression. When drug-naïve patients with depression were treated with an NE reuptake inhibitor (NRI) versus an SSRI, patients given SSRIs experienced a relapse of symptoms, whereas NRI patients did not.183 These data together with data demonstrating the role of NE in plasticity suggest a powerful effect of dysfunctional NE release, which acutely deepens attractor states. Following behavioral data in humans and rodents, it is possible that excessive NE release due to chronic stress could promote dysfunction in the mPFC and the amygdala, leading to an anxious-depressive model with two hyperstable attractor states.25 This model predicts behaviors that are typically seen in cases of unstable NE, DA, and 5-HT firing, including impairments in cognitive flexibility, working memory, hypervigilance, and negative valence bias. Recent research into the role of neurotensin demonstrates that it encodes valence assignment in the BLA by exerting a modular influence over synaptic plasticity in a valence-dependent manner.184 Given that patients with anxiety and depression suffer from negative valence bias, neurotensin signaling may be impaired in these mood disorders.184,185 Reciprocally, neurotensin inhibits DA-mediated suppression of VTA neurons,186 suggesting that disordered neurotensin modulation could impact VTA–DA signaling to the mPFC, causing disordered DAergic firing.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
While attractor state modeling argues how anxious and depressive states persist, the question still stands: how do these disease states develop, and what causes a transition into these states? Modeling of other natural phenomena, such as earthquakes, nuclear chain reactions, forest fires, and avalanches, all exhibit activity that can be described by power laws; neural activity can be modeled the same way.187,188 Neuronal processing requires the integration and redistribution of thousands of inputs with dynamics described as neuronal avalanches.187 This theory suggests that as a population nears a critical threshold, the observable behavior does not change until one unit exceeds threshold and causes many other units to do so in turn.189 This dynamism is described in the critical brain hypothesis, which suggests that neuron populations operate in the vicinity of the critical point of a phase transition, allowing for variable dynamics (through neuronal avalanches) from rest.190 While attractor states explain the stability and perpetuation of a given state, neuronal avalanches and the broader critical brain hypothesis explain the passage from one attractor state to another. In a healthy system, neuronal avalanches can elicit state transitions; however, in an unhealthy system, neuron populations would struggle to reach the criticality necessary for state transition. Neuronal avalanches have been observed in cortical populations using ex vivo microelectrode array recordings187 and in vivo electrode recordings;189 suggesting a role of brain criticality in TD processing, but it has yet to be investigated if neuronal avalanches are observed in subcortical regions.
As a class, mental health disorders present the largest economic burden to our society as well as being the least well-understood in terms of biological mechanisms. Many disparate subfields interface with mental health disorders, from computational psychology to neuropharmacology to modern circuit neuroscience, yet they are poorly integrated. Here, we have linked the computational, algorithmic, and implementational levels of investigation to connect the conceptual frameworks posited by each field. On a computational level, the function of the mind in health and disease can be compared to attractor states, analogous to brain states. On an algorithmic level, we explore how TD or BU brain states represent psychological processing pathways that are mediated by neural circuits. On an implementational level, we can probe the way synaptic changes can alter neural dynamics and shift brain states along multiple distinct parameters. Our review presents a framework through which to understand and decipher the differences observed in anxious and depressed subjects based on behavioral and neural readouts. Future studies should test the idea of a shift in processing modalities in more depth by directly measuring and comparing neural processes implicated in these two modes of anxious and depressed individuals while responding to various stimuli. Additionally, computational analyses of brain states in animal models may reveal the attractor networks mediating anxiety and depression.
To forge a path forward amidst the last frontier of our understanding—ourselves—we must integrate and synthesize the diverse perspectives for investigating psychiatric disease. This conceptual model is largely speculative and subject to evolution. Directed and focused investigation along multiple levels will enable the diagnosis of brain-based diseases using both behavior and brain activity as readouts.
OUTSTANDING QUESTIONS.
What happens on a chronic scale when you have a hyperstable state? Why are changes in homeostatic or metaplasticity not rebalancing attractor state geometry?
How do TD and BU attractor states interact with each other? Do these attractor states represent a part of a greater energy landscape in the brain, and what shape does that broader landscape take in health and disease?
How can we leverage our understanding of attractor states to develop individual treatments and/or predict disease biotypes?
How can we better model comorbid types of anxious-depression to further investigate neuromodulatory influence and plasticity change?
Are there shared circuits or a divergent point in processing that account for negative affective states in both anxiety and depression?
Major life changes in specific developmental periods make individuals highly susceptible for the development of mental illness. How does stress in specific developmental periods (e.g., childhood, adolescence) uniquely alter plasticity in cortical and limbic regions?
Are the changes in plasticity that we see in patients with anxiety and depression indicative of a genetic susceptibility to these illnesses?
ACKNOWLEDGMENTS
We thank Jeremy Delahanty and Anousheh Bakhti-Suroosh for helpful discussion. We thank Amy Cao for redrawing our illustrations. K.M.T. is an HHMI Investigator and the Wylie Vale chair at the Salk Institute for Biological Studies and this work was supported by funding from Salk, HHMI, Clayton Foundation, Kavli Institute for Brain and Mind, Dolby Family Fund, Blavatnik Family Foundation, R01-MH115920 (NIMH), R37-MH102441 (NIMH), and Pioneer Award DP1-AT009925 (NCCIH).
Funding information
National Institute of Mental Health, Grant/Award Numbers: R01-MH115920 (NIMH), R37-MH102441 (NIMH)
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Hasin DS, Sarvet AL, Meyers JL, Saha TD, Ruan WJ, Stohl M, & Grant BF (2018). Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry, 75, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessler RC, Angermeyer M, Anthony JC, De Graaf R, Demyttenaere K, Gasquet I, De Girolamo G, Gluzman S, Gureje O, Haro JM, Kawakami N, Karam A, Levinson D, Medina Mora ME, Oakley Browne MA, Posada-Villa J, Stein DJ, Adley Tsang CH, Aguilar-Gaxiola S, … Ustün TB (2007). Lifetime prevalence and age-of-onset distributions of mental disorders in the World Health Organization’s World Mental Health Survey Initiative. World Psychiatry, 6, 168–176. [PMC free article] [PubMed] [Google Scholar]
- 3.Tye KM (2014). Neural circuit reprogramming: A new paradigm for treating neuropsychiatric disease? Neuron, 83, 1259–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marr D, & Poggio T (1976). From understanding computation to understanding neural circuitry.
- 5.American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders.
- 6.Russo SJ, & Nestler EJ (2013). The brain reward circuitry in mood disorders. Nature Reviews Neuroscience, 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salamone JD, & Correa M (2012). The mysterious motivational functions of mesolimbic dopamine. Neuron, 76, 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bangasser DA, & Cuarenta A (2021). Sex differences in anxiety and depression: Circuits and mechanisms. Nature Reviews Neuroscience, 22, 674–684. [DOI] [PubMed] [Google Scholar]
- 9.Calhoon GG, & Tye KM (2015). Resolving the neural circuits of anxiety. Nature Neuroscience, 18, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dillon DG, Rosso IM, Pechtel P, Killgore WDS, Rauch SL, & Pizzagalli DA (2014). Peril and pleasure: An RDoC-inspired examination of threat responses and reward processing in anxiety and depression. Depression and Anxiety, 31, 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller P (2016). Dynamical systems, attractors, and neural circuits. F1000Research, 5, 992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rolls ET (2021). Attractor cortical neurodynamics, schizophrenia, and depression. Translational Psychiatry, 11, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang XJ (2009). Attractor network models. In Encyclopedia of neuroscience (pp. 667–679). Elsevier Ltd. [Google Scholar]
- 14.Damasio A (1999). The feeling of what happens: Body and emotion in the making of consciousness. Harcourt College Publishers. [Google Scholar]
- 15.Anticevic A, & Murray JD (2017). Computational psychiatry: Mathematical modeling of mental illness. Academic Press. [Google Scholar]
- 16.Chumbley JR, Dolan RJ, & Friston KJ (2008). Attractor models of working memory and their modulation by reward. Biological Cybernetics, 98, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dayan P, Dolan RJ, Friston KJ, & Montague PR (2015). Taming the shrewdness of neural function: Methodological challenges in computational psychiatry. Current Opinion in Behavioral Sciences, 5, 128–132. [Google Scholar]
- 18.Friston KJ (1997). Transients, metastability, and neuronal dynamics. Neuroimage, 5, 164–171. [DOI] [PubMed] [Google Scholar]
- 19.Anderson DJ (2016). Circuit modules linking internal states and social behaviour in flies and mice. Nature Reviews Neuroscience, 17, 692–704. [DOI] [PubMed] [Google Scholar]
- 20.Eldar E, Rutledge RB, Dolan RJ, & Niv Y (2016). Mood as representation of momentum. Trends in Cognitive Sciences, 20, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grove JCR, Gray LA, La Santa Medina N, Sivakumar N, Ahn JS, Corpuz TV, Berke JD, Kreitzer AC, & Knight ZA (2022). Dopamine subsystems that track internal states. Nature, 608, 374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leutgeb S, Leutgeb JK, Barnes CA, Moser EI, Mcnaughton BL, & Moser M-B (2005). Independent codes for spatial and episodic memory in hippocampal neuronal ensembles. Science, 309, 619–623. [DOI] [PubMed] [Google Scholar]
- 23.Rolls ET (2016). A non-reward attractor theory of depression. Neuroscience & Biobehavioral Reviews, 68, 47–58. [DOI] [PubMed] [Google Scholar]
- 24.Hopfield JJ (1982). Neural networks and physical systems with emergent collective computational abilities. Proceedings of the National Academy of Sciences of the United States of America, 79, 2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B, & Miller P (2020). Attractor-state itinerancy in neural circuits with synaptic depression. Journal of Mathematical Neuroscience, 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oathes DJ, Patenaude B, Schatzberg AF, & Etkin A (2015). Neurobiological signatures of anxiety and depression in resting-state fMRI. Biological Psychiatry, 77, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’angelo C, Mirijello A, Leggio L, Ferrulli A, Carotenuto V, Icolaro N, Miceli A, D’angelo V, Gasbarrini G, & Addolorato G (2008). State and trait anxiety and depression in patients with primary brain tumors before and after surgery: 1-year longitudinal study. Journal of Neurosurgery, 108, 281–286. [DOI] [PubMed] [Google Scholar]
- 28.Demenescu LR, Renken R, Kortekaas R, Van Tol M-J, Marsman JBC, Van Buchem MA, Van Der Wee NJA, Veltman DJ, Den Boer JA, & Aleman A (2011). Neural correlates of perception of emotional facial expressions in out-patients with mild-to-moderate depression and anxiety. A multicenter fMRI study. Psychological Medicine, 41, 2253–2264. [DOI] [PubMed] [Google Scholar]
- 29.Coplan JD (2015). Treating comorbid anxiety and depression: Psychosocial and pharmacological approaches. World Journal of Psychiatry, 5, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkins PE, Ducker I, Gooding R, James M, & Rutter-Eley E (2021). Anxiety and depression in a sample of UK college students: A study of prevalence, comorbidity, and quality of life. Journal of American College Health, 69, 813–819. [DOI] [PubMed] [Google Scholar]
- 31.Kaiser T, Herzog P, Voderholzer U, & Brakemeier E-L (2021). Unraveling the comorbidity of depression and anxiety in a large inpatient sample: Network analysis to examine bridge symptoms. Depression and Anxiety, 38, 307–317. [DOI] [PubMed] [Google Scholar]
- 32.Li F, & Tsien JZ (2009). Memory and the NMDA receptors. New England Journal of Medicine, 361, 302–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turrigiano GG, & Nelson SB (2004). Homeostatic plasticity in the developing nervous system. Nature Reviews Neuroscience, 5, 97–107. [DOI] [PubMed] [Google Scholar]
- 34.Galanis C, & Vlachos A (2020). Hebbian and homeostatic synaptic plasticity—Do alterations of one reflect enhancement of the other? Frontiers in Cellular Neuroscience, 14, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bear MF, & Malenka RC (1994). Synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology, 4, 389–399. [DOI] [PubMed] [Google Scholar]
- 36.Abraham WC (2008). Metaplasticity: Tuning synapses and networks for plasticity. Nature Reviews Neuroscience, 9, 387–387. [DOI] [PubMed] [Google Scholar]
- 37.Stanton PK (1996). LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus, 6, 35–42. [DOI] [PubMed] [Google Scholar]
- 38.Abbott LF, & Nelson SB (2000). Synaptic plasticity: Taming the beast. Nature Neuroscience, 3, 1178–1183. [DOI] [PubMed] [Google Scholar]
- 39.Andersen N, Krauth N, & Nabavi S (2017). Hebbian plasticity in vivo: Relevance and induction. Current Opinion in Neurobiology, 45, 188–192. [DOI] [PubMed] [Google Scholar]
- 40.Lee CR, Chen A, & Tye KM (2021). The neural circuitry of social homeostasis: Consequences of acute versus chronic social isolation. Cell, 184, 1500–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomes-Leal W (2021). Adult hippocampal neurogenesis and affective disorders: New neurons for psychic well-being. Frontiers in Neuroscience, 15, 594448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mogg K, & Bradley BP (1998). A cognitive-motivational analysis of anxiety. Behaviour Research and Therapy, 36, 809–848. [DOI] [PubMed] [Google Scholar]
- 43.Theeuwes J (2010). Top–down and bottom–up control of visual selection. Acta Psychologica, 135, 77–99. [DOI] [PubMed] [Google Scholar]
- 44.Öhman A, Flykt A, & Esteves F (2001). Emotion drives attention: Detecting the snake in the grass. Journal of Experimental Psychology: General, 130, 466–478. [DOI] [PubMed] [Google Scholar]
- 45.Tye KM (2018). Neural circuit motifs in valence processing. Neuron, 100, 436–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corbetta M, & Shulman GL (2002). Control of goal-directed and stimulus-driven attention in the brain. Nature Reviews Neuroscience, 3, 201–215. [DOI] [PubMed] [Google Scholar]
- 47.Lonigan CJ, Vasey MW, Phillips BM, & Hazen RA (2004). Temperament, anxiety, and the processing of threat-relevant stimuli. Journal of Clinical Child & Adolescent Psychology, 33, 8–20. [DOI] [PubMed] [Google Scholar]
- 48.Vanunu Y, Hotaling JM, Le Pelley ME, & Newell BR (2021). How top-down and bottom-up attention modulate risky choice. Proceedings of the National Academy of Sciences, 118, e2025646118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veerapa E, Grandgenevre P, El Fayoumi M, Vinnac B, Haelewyn O, Szaffarczyk S, Vaiva G, & D’hondt F (2020). Attentional bias towards negative stimuli in healthy individuals and the effects of trait anxiety. Scientific Reports, 10, 11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark LA, & Watson D (1991). Tripartite model of anxiety and depression: Psychometric evidence and taxonomic implications. Journal of Abnormal Psychology, 100, 316–336. [DOI] [PubMed] [Google Scholar]
- 51.Mogg K, & Bradley BP (2016). Anxiety and attention to threat: Cognitive mechanisms and treatment with attention bias modification. Behaviour Research and Therapy, 87, 76–108. [DOI] [PubMed] [Google Scholar]
- 52.Eysenck MW, Derakshan N, Santos R, & Calvo MG (2007). Anxiety and cognitive performance: Attentional control theory. Emotion, 7, 336–353. [DOI] [PubMed] [Google Scholar]
- 53.Mcrae K, Misra S, Prasad AK, Pereira SC, & Gross JJ (2012). Bottom-up and top-down emotion generation: Implications for emotion regulation. Social Cognitive and Affective Neuroscience, 7, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ochsner KN, Ray RR, Hughes B, Mcrae K, Cooper JC, Weber J, Gabrieli JDE, & Gross JJ (2009). Bottom-up and top-down processes in emotion generation: Common and distinct neural mechanisms. Psychological Science, 20, 1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janak PH, & Tye KM (2015). From circuits to behaviour in the amygdala. Nature, 517, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright P, Albarracin D, Brown RD, Li H, He G, & Liu Y (2008). Dissociated responses in the amygdala and orbitofrontal cortex to bottom-up and top-down components of emotional evaluation. Neuroimage, 39, 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gothard KM, & Fuglevand AJ (2022). The role of the amygdala in processing social and affective touch. Current Opinion in Behavioral Sciences, 43, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gründemann J, Bitterman Y, Lu T, Krabbe S, Grewe BF, Schnitzer MJ, & Lüthi A (2019). Amygdala ensembles encode behavioral states. Science, 364, eaav8736. [DOI] [PubMed] [Google Scholar]
- 59.Lang PJ (1994). The varieties of emotional experience: A meditation on James–Lange theory. Psychological Review, 101, 211–221. [DOI] [PubMed] [Google Scholar]
- 60.Schachter S, & Singer J (1962). Cognitive, social, and physiological determinants of emotional state. Psychological Review, 69, 379–399. [DOI] [PubMed] [Google Scholar]
- 61.Bechara A, Damasio H, Damasio AR, & Lee GP (1999). Different contributions of the human amygdala and ventromedial prefrontal cortex to decision-making. Journal of Neuroscience, 19, 5473–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adolphs R, Tranel D, Damasio H, & Damasio A (1994). Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature, 372, 669–672. [DOI] [PubMed] [Google Scholar]
- 63.Lilienfeld SO, Sauvigné KC, Reber J, Watts AL, Hamann S, Smith SF, Patrick CJ, Bowes SM, & Tranel D (2018). Potential effects of severe bilateral amygdala damage on psychopathic personality features: A case report. Personality Disorder, 9, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bach DR, Hoffmann M, Finke C, Hurlemann R, & Ploner CJ (2019). Disentangling hippocampal and amygdala contribution to human anxiety-like behavior. Journal of Neuroscience, 39, 8517–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Joyce MKP, Marshall LG, Banik SL, Wang J, Xiao D, Bunce JG, & Barbas H (2022). Pathways for memory, cognition and emotional context: Hippocampal, subgenual area 25, and amygdalar axons show unique interactions in the primate thalamic reuniens nucleus. Journal of Neuroscience, 42, 1068–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Padilla-Coreano N, Bolkan SS, Pierce GM, Blackman DR, Hardin WD, Garcia-Garcia AL, Spellman TJ, & Gordon JA (2016). Direct ventral hippocampal-prefrontal input is required for anxiety-related neural activity and behavior. Neuron, 89, 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Šimić G, Tkalčić M, Vukić V, Mulc D, Španić E, Šagud M, Olucha-Bordonau FE, Vukšić M, & R Hof P (2021). Understanding emotions: Origins and roles of the amygdala. Biomolecules, 11, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Etkin A, Prater KE, Schatzberg AF, Menon V, & Greicius MD (2009). Disrupted amygdalar subregion functional connectivity and evidence of a compensatory network in generalized anxiety disorder. Archives of General Psychiatry, 66, 1361–1372. [DOI] [PubMed] [Google Scholar]
- 69.Gold PW, & Kadriu B (2019). A major role for the lateral habenula in depressive illness: Physiologic and molecular mechanisms. Frontiers in Psychiatry, 10, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, Fetcho RN, Zebley B, Oathes DJ, Etkin A, Schatzberg AF, Sudheimer K, Keller J, Mayberg HS, Gunning FM, Alexopoulos GS, Fox MD, Pascual-Leone A, Voss HU, … Liston C (2017). Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nature Medicine, 23, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burgos-Robles A, Kimchi EY, Izadmehr EM, Porzenheim MJ, Ramos-Guasp WA, Nieh EH, Felix-Ortiz AC, Namburi P, Leppla CA, Presbrey KN, Anandalingam KK, Pagan-Rivera PA, Anahtar M, Beyeler A, & Tye KM (2017). Amygdala inputs to prefrontal cortex guide behavior amid conflicting cues of reward and punishment. Nature Neuroscience, 20, 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dilgen J, Tejeda HA, & O’donnell P (2013). Amygdala inputs drive feedforward inhibition in the medial prefrontal cortex. Journal of Neurophysiology, 110, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vouimba R-M, & Maroun M (2011). Learning-induced changes in mPFC–BLA connections after fear conditioning, extinction, and reinstatement of fear. Neuropsychopharmacology, 36, 2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Felix-Ortiz AC, & Tye KM (2014). Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. Journal of Neuroscience, 34, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pape H-C, & Pare D (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiological Reviews, 90, 419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stujenske JM, Likhtik E, Topiwala MA, & Gordon JA (2014). Fear and safety engage competing patterns of theta-gamma coupling in the basolateral amygdala. Neuron, 83, 919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klavir O, Prigge M, Sarel A, Paz R, & Yizhar O (2017). Manipulating fear associations via optogenetic modulation of amygdala inputs to prefrontal cortex. Nature Neuroscience, 20, 836–844. [DOI] [PubMed] [Google Scholar]
- 78.Starkweather CK, Gershman SJ, & Uchida N (2018). The medial prefrontal cortex shapes dopamine reward prediction errors under state uncertainty. Neuron, 98, 616–629.e6.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fuster JM (2017). Chapter 8 - Prefrontal cortex in decision-making: The perception–action cycle. In Dreher J-C, & Tremblay L (Eds.), Decision neuroscience (pp. 95–105). Academic Press. [Google Scholar]
- 80.Young L, Cushman F, Hauser M, & Saxe R (2007). The neural basis of the interaction between theory of mind and moral judgment. Proceedings of the National Academy of Sciences, 104, 8235–8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koenigs M, Young L, Adolphs R, Tranel D, Cushman F, Hauser M, & Damasio A (2007). Damage to the prefrontal cortex increases utilitarian moral judgements. Nature, 446, 908–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bechara A (2000). Emotion, decision making and the orbitofrontal cortex. Cerebral Cortex, 10, 295–307. [DOI] [PubMed] [Google Scholar]
- 83.Sharot T, Riccardi AM, Raio CM, & Phelps EA (2007). Neural mechanisms mediating optimism bias. Nature, 450, 102–105. [DOI] [PubMed] [Google Scholar]
- 84.Anderson KM, Collins MA, Kong R. u., Fang K, Li J, He T, Chekroud AM, Yeo BTT, & Holmes AJ (2020). Convergent molecular, cellular, and cortical neuroimaging signatures of major depressive disorder. Proceedings of the National Academy of Sciences, 117, 25138–25149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pizzagalli DA, & Roberts AC (2022). ‘Prefrontal cortex and depression’: Correction. Neuropsychopharmacology, 47, 609–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bittar TP, & Labonté B (2021). Functional contribution of the medial prefrontal circuitry in major depressive disorder and stress-induced depressive-like behaviors. Frontiers in Behavioral Neuroscience, 15, 699592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hare BD, & Duman RS (2020). Prefrontal cortex circuits in depression and anxiety: Contribution of discrete neuronal populations and target regions. Molecular Psychiatry, 25, 2742–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dunlop BW, & Mayberg HS (2017). Neuroimaging advances for depression. Cerebrum, 2017, cer-16–17. [PMC free article] [PubMed] [Google Scholar]
- 89.Mayberg HS, Lozano AM, Voon V, Mcneely HE, Seminowicz D, Hamani C, Schwalb JM, & Kennedy SH (2005). Deep brain stimulation for treatment-resistant depression. Neuron, 45, 651–660. [DOI] [PubMed] [Google Scholar]
- 90.Douma EH, & de Kloet ER (2020). Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neuroscience & Biobehavioral Reviews, 108, 48–77. [DOI] [PubMed] [Google Scholar]
- 91.Hu H (2016). Reward and aversion. Annual Review of Neuroscience, 39, 297–324. [DOI] [PubMed] [Google Scholar]
- 92.Hu H, Cui Y, & Yang Y (2020). Circuits and functions of the lateral habenula in health and in disease. Nature Reviews Neuroscience, 21, 277–295. [DOI] [PubMed] [Google Scholar]
- 93.Qi G, Zhang P, Li T, Li M, Zhang Q, He F, Zhang L, Cai H, Lv X, Qiao H, Chen X, Ming J, & Tian B (2022). NAc-VTA circuit underlies emotional stress-induced anxiety-like behavior in the three-chamber vicarious social defeat stress mouse model. Nature Communications, 13, 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai H-C, Finkelstein J, Kim S-Y, Adhikari A, Thompson KR, Andalman AS, Gunaydin LA, Witten IB, & Deisseroth K (2013). Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature, 493, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, Li X, Dietz DM, Pan N, Vialou VF, Neve RL, Yue Z, & Han M-H (2014). Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science, 344, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stamatakis AM, & Stuber GD (2012). Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nature Neuroscience, 15, 1105–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, Malinow R, Yates JR, & Hu H (2013). βCaMKII in lateral habenula mediates core symptoms of depression. Science, 341, 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jakobs M, Pitzer C, Sartorius A, Unterberg A, & Kiening K (2019). Acute 5 Hz deep brain stimulation of the lateral habenula is associated with depressive-like behavior in male wild-type Wistar rats. Brain Research, 1721, 146283. [DOI] [PubMed] [Google Scholar]
- 99.Browne CA, Hammack R, & Lucki I (2018). Dysregulation of the lateral habenula in major depressive disorder. Frontiers in Synaptic Neuroscience, 10, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buschman TJ, & Miller EK (2007). Top-down versus bottom-up control of attention in the prefrontal and posterior parietal cortices. Science, 315, 1860–1862. [DOI] [PubMed] [Google Scholar]
- 101.Miller EK, & Buschman TJ (2013). Cortical circuits for the control of attention. Current Opinion in Neurobiology, 23, 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Varela F, Lachaux J-P, Rodriguez E, & Martinerie J (2001). The brainweb: Phase synchronization and large-scale integration. Nature Reviews Neuroscience, 2, 229–239. [DOI] [PubMed] [Google Scholar]
- 103.Gregoriou GG, Gotts SJ, Zhou H, & Desimone R (2009). High-frequency, long-range coupling between prefrontal and visual cortex during attention. Science, 324, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siegel M, Warden MR, & Miller EK (2009). Phase-dependent neuronal coding of objects in short-term memory. Proceedings of the National Academy of Sciences of the United States of America, 106, 21341–21346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Warden MR, & Miller EK (2007). The representation of multiple objects in prefrontal neuronal delay activity. Cerebral Cortex, 17, i41–i50. [DOI] [PubMed] [Google Scholar]
- 106.Chen S, Tan Z, Xia W, Gomes CA, Zhang X, Zhou W, Liang S, Axmacher N, & Wang L (2021). Theta oscillations synchronize human medial prefrontal cortex and amygdala during fear learning. Science Advances, 7, eabf4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lesting J, Narayanan RT, Kluge C, Sangha S, Seidenbecher T, & Pape H-C (2011). Patterns of coupled theta activity in amygdala-hippocampal-prefrontal cortical circuits during fear extinction. PLoS ONE, 6, e21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Likhtik E, Stujenske JM, A Topiwala M, Harris AZ, & Gordon JA (2014). Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nature Neuroscience, 17, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Padilla-Coreano N, Canetta S, Mikofsky RM, Alway E, Passecker J, Myroshnychenko MV, Garcia-Garcia AL, Warren R, Teboul E, Blackman DR, Morton MP, Hupalo S, Tye KM, Kellendonk C, Kupferschmidt DA, & Gordon JA (2019). Hippocampal-prefrontal theta transmission regulates avoidance behavior. Neuron, 104, 601–610.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Siegle JH, & Wilson MA (2014). Enhancement of encoding and retrieval functions through theta phase-specific manipulation of hippocampus. eLife, 3, e03061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deslandes AC, De Moraes H, Pompeu FAMS, Ribeiro P, Cagy M, Capitão C, Alves H, Piedade RAM, & Laks J (2008). Electroencephalographic frontal asymmetry and depressive symptoms in the elderly. Biological Psychology, 79, 317–322. [DOI] [PubMed] [Google Scholar]
- 112.Henriques JB, & Davidson RJ (1991). Left frontal hypoactivation in depression. Journal of Abnormal Psychology, 100, 535–545. [DOI] [PubMed] [Google Scholar]
- 113.Kemp AH, Griffiths K, Felmingham KL, Shankman SA, Drinkenburg W, Arns M, Clark CR, & Bryant RA (2010). Disorder specificity despite comorbidity: Resting EEG alpha asymmetry in major depressive disorder and post-traumatic stress disorder. Biological Psychology, 85, 350–354. [DOI] [PubMed] [Google Scholar]
- 114.Başar E, Güntekin B, Atagün I, Turp Gölbaşı B, Tülay E, & Özerdem A (2012). Brain’s alpha activity is highly reduced in euthymic bipolar disorder patients. Cognitive Neurodynamics, 6, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Özerdem A, Güntekin B, Atagün I, Turp B, & Başar E (2011). Reduced long distance gamma (28–48 Hz) coherence in euthymic patients with bipolar disorder. Journal of Affective Disorders, 132, 325–332. [DOI] [PubMed] [Google Scholar]
- 116.Hultman R, Mague SD, Li Q, Katz BM, Michel N, Lin L, Wang J, David LK, Blount C, Chandy R, Carlson D, Ulrich K, Carin L, Dunson D, Kumar S, Deisseroth K, Moore SD, & Dzirasa K (2016). Dysregulation of prefrontal cortex-mediated slow-evolving limbic dynamics drives stress-induced emotional pathology. Neuron, 91, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hultman R, Ulrich K, Sachs BD, Blount C, Carlson DE, Ndubuizu N, Bagot RC, Parise EM, Vu M-AT, Gallagher NM, Wang J, Silva AJ, Deisseroth K, Mague SD, Caron MG, Nestler EJ, Carin L, & Dzirasa K (2018). Brain-wide electrical spatiotemporal dynamics encode depression vulnerability. Cell, 173, 166–180.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nasir M, Trujillo D, Levine J, Dwyer JB, Rupp ZW, & Bloch MH (2020). Glutamate systems in DSM-5 anxiety disorders: Their role and a review of glutamate and GABA psychopharmacology. Frontiers in Psychiatry, 11, 548505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sarawagi A, Soni ND, & Patel AB (2021). Glutamate and GABA homeostasis and neurometabolism in major depressive disorder. Frontiers in Psychiatry, 12, 637863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Godfrey KEM, Gardner AC, Kwon S, Chea W, & Muthukumaraswamy SD (2018). Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: A systematic review and meta-analysis. Journal of Psychiatric Research, 105, 33–44. [DOI] [PubMed] [Google Scholar]
- 121.Jie F, Yin G, Yang W, Yang M, Gao S, Lv J, & Li B (2018). Stress in regulation of GABA amygdala system and relevance to neuropsychiatric diseases. Frontiers in Neuroscience, 12, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Möhler H (2012). The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology, 62, 42–53. [DOI] [PubMed] [Google Scholar]
- 123.Fritschy J-M, & Panzanelli P (2014). GABAA receptors and plasticity of inhibitory neurotransmission in the central nervous system. European Journal of Neuroscience, 39, 1845–1865. [DOI] [PubMed] [Google Scholar]
- 124.Ghosal S, Hare BD, & Duman RS (2017). Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Current Opinion in Behavioral Sciences, 14, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, & Luscher B (2016). Bidirectional homeostatic regulation of a depression-related brain state by GABAergic deficits and ketamine treatment. Biological Psychiatry, 80, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, & Luscher B (2010). Gamma-aminobutyric acid-type A receptor deficits cause hypothalamic–pituitary–adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biological Psychiatry, 68, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Duman RS, Sanacora G, & Krystal JH (2019). Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron, 102, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hasler G, Buchmann A, Haynes M, Müller ST, Ghisleni C, Brechbühl S, & Tuura R (2019). Association between prefrontal glutamine levels and neuroticism determined using proton magnetic resonance spectroscopy. Translational Psychiatry, 9, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, & Deisseroth K (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature, 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gerhard DM, Pothula S, Liu R-J, Wu M, Li X-Y, Girgenti MJ, Taylor SR, Duman CH, Delpire E, Picciotto M, Wohleb ES, & Duman RS (2020). GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. Journal of Clinical Investigation, 130, 1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mcewen AM, Burgess DTA, Hanstock CC, Seres P, Khalili P, Newman SC, Baker GB, Mitchell ND, Khudabux-Der J, Allen PS, & Lemelledo J-M (2012). Increased glutamate levels in the medial prefrontal cortex in patients with postpartum depression. Neuropsychopharmacology, 37, 2428–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, Plitman E, Sano Y, Tarumi R, Elsalhy M, Katayama N, Ogyu K, Miyazaki T, Kishimoto T, Graff-Guerrero A, Meyer JH, Blumberger DM, Daskalakis ZJ, Mimura M, & Nakajima S (2019). Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Molecular Psychiatry, 24, 952–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Valtcheva S, & Venance L (2019). Control of long-term plasticity by glutamate transporters. Frontiers in Synaptic Neuroscience, 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rolls ET (2020). Attractor network dynamics, transmitters, and memory and cognitive changes in aging. In Heilman KM & Nadeau SE (Eds.), Cognitive changes and the aging brain (pp. 203–225). Cambridge University Press. [Google Scholar]
- 135.Fragale JEC, Khariv V, Gregor DM, Smith IM, Jiao X, Elkabes S, Servatius RJ, Pang KCH, & Beck KD (2016). Dysfunction in amygdala-prefrontal plasticity and extinction-resistant avoidance: A model for anxiety disorder vulnerability. Experimental Neurology, 275, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Amat J, Matus-Amat P, Watkins LR, & Maier SF (1998). Escapable and inescapable stress differentially alter extracellular levels of 5-HT in the basolateral amygdala of the rat. Brain Research, 812, 113–120. [DOI] [PubMed] [Google Scholar]
- 137.Quah SKL, Mciver L, Roberts AC, & Santangelo AM (2020). Trait anxiety mediated by amygdala serotonin transporter in the common marmoset. Journal of Neuroscience, 40, 4739–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bauer EP (2015). Serotonin in fear conditioning processes. Behavioural Brain Research, 277, 68–77. [DOI] [PubMed] [Google Scholar]
- 139.Dayer A (2014). Serotonin-related pathways and developmental plasticity: Relevance for psychiatric disorders. Dialogues in Clinical Neuroscience, 16, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bocchio M, Mchugh SB, Bannerman DM, Sharp T, & Capogna M (2016). Serotonin, amygdala and fear: Assembling the puzzle. Frontiers in Neural Circuits, 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen A, Hough CJ, & Li H (2003). Serotonin type II receptor activation facilitates synaptic plasticity via n-methyl-d-aspartate-mediated mechanism in the rat basolateral amygdala. Neuroscience, 119, 53–63. [DOI] [PubMed] [Google Scholar]
- 142.Man M-S, Mikheenko Y, Braesicke K, Cockcroft G, & Roberts AC (2012). Serotonin at the level of the amygdala and orbitofrontal cortex modulates distinct aspects of positive emotion in primates. International Journal of Neuropsychopharmacology, 15, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Locher C, Koechlin H, Zion SR, Werner C, Pine DS, Kirsch I, Kessler RC, & Kossowsky J (2017). Efficacy and safety of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and placebo for common psychiatric disorders among children and adolescents: A systematic review and meta-analysis. JAMA Psychiatry, 74, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cowen PJ, & Browning M (2015). What has serotonin to do with depression? World Psychiatry, 14, 158–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fisher PM, Price JC, Meltzer CC, Moses-Kolko EL, Becker C, Berga SL, & Hariri AR (2011). Medial prefrontal cortex serotonin 1A and 2A receptor binding interacts to predict threat-related amygdala reactivity. Biology of Mood & Anxiety Disorders, 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hornboll B, Macoveanu J, Rowe J, Elliott R, Paulson OB, Siebner HR, & Knudsen GM (2013). Acute 5-HT2A receptor blocking alters the processing of fearful faces in orbitofrontal cortex and amygdala. Journal of Psychopharmacology, 27, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Challis C, & Berton O (2015). Top-down control of serotonin systems by the prefrontal cortex: A path towards restored socioemotional function in depression. ACS Chemical Neuroscience, 6, 1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garcia-Garcia AL, Meng Q, Canetta S, Gardier AM, Guiard BP, Kellendonk C, Dranovsky A, & Leonardo ED (2017). Serotonin signaling through prefrontal cortex 5-HT1A receptors during adolescence can determine baseline mood-related behaviors. Cell Reports, 18, 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Savitz J, Lucki I, & Drevets WC (2009). 5-HT(1A) receptor function in major depressive disorder. Progress in Neurobiology, 88, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Niederkofler V, Asher TE, & Dymecki SM (2015). Functional interplay between dopaminergic and serotonergic neuronal systems during development and adulthood. ACS Chemical Neuroscience, 6, 1055–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bortolozzi A, Diaz-Mataix L, Scorza MC, Celada P, & Artigas F (2005). The activation of 5-HT receptors in prefrontal cortex enhances dopaminergic activity. Journal of Neurochemistry, 95, 1597–1607. [DOI] [PubMed] [Google Scholar]
- 152.Glimcher PW (2011). Understanding dopamine and reinforcement learning: The dopamine reward prediction error hypothesis. Proceedings of the National Academy of Sciences, 108, 15647–15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ott T, & Nieder A (2019). Dopamine and cognitive control in prefrontal cortex. Trends in Cognitive Sciences, 23, 213–234. [DOI] [PubMed] [Google Scholar]
- 154.Chen J, & Bruchas M (2021). Neuromodulation: A model for dopamine in salience encoding. Current Biology, 31, R1426–R1429. [DOI] [PubMed] [Google Scholar]
- 155.Kutlu MG, Zachry JE, Melugin PR, Cajigas SA, Chevee MF, Kelly SJ, Kutlu B, Tian L, Siciliano CA, & Calipari ES (2021). Dopamine release in the nucleus accumbens core signals perceived saliency. Current Biology, 31, 4748–4761.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang P, Gao T, Dong Z, Zhou C, Lai Y, Pan T, Liu Y, Zhao X, Sun X, Hua H, Wen G, Gao L, & Lv Z (2018). Neural circuitry among connecting the hippocampus, prefrontal cortex and basolateral amygdala in a mouse depression model: Associations correlations between BDNF levels and BOLD–fMRI signals. Brain Research Bulletin, 142, 107–115. [DOI] [PubMed] [Google Scholar]
- 157.Bahi A, & Dreyer J-L (2019). Dopamine transporter (DAT) knockdown in the nucleus accumbens improves anxiety- and depression-related behaviors in adult mice. Behavioural Brain Research, 359, 104–115. [DOI] [PubMed] [Google Scholar]
- 158.Hjorth OR, Frick A, Gingnell M, Hoppe JM, Faria V, Hultberg S, Alaie I, Månsson KNT, Wahlstedt K, Jonasson M, Lubberink M, Antoni G, Fredrikson M, & Furmark T (2021). Expression and co-expression of serotonin and dopamine transporters in social anxiety disorder: A multitracer positron emission tomography study. Molecular Psychiatry, 26, 3970–3979. [DOI] [PubMed] [Google Scholar]
- 159.Buchta WC, Mahler SV, Harlan B, Aston-Jones GS, & Riegel AC (2017). Dopamine terminals from the ventral tegmental area gate intrinsic inhibition in the prefrontal cortex. Physiological Reports, 5, e13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang S, Zhang Z, Gambeta E, Xu SC, Thomas C, Godfrey N, Chen L, M’dahoma S, Borgland SL, & Zamponi GW (2020). Dopamine inputs from the ventral tegmental area into the medial prefrontal cortex modulate neuropathic pain-associated behaviors in mice. Cell Reports, 31, 107812. [DOI] [PubMed] [Google Scholar]
- 161.Liu W-Z, Zhang W-H, Zheng Z-H, Zou J-X, Liu X-X, Huang S-H, You W-J, He Y, Zhang J-Y, Wang X-D, & Pan B-X (2020). Identification of a prefrontal cortex-to-amygdala pathway for chronic stress-induced anxiety. Nature Communications, 11, 2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Madadi Asl M, Vahabie A-H, & Valizadeh A (2019). Dopaminergic modulation of synaptic plasticity, its role in neuropsychiatric disorders, and its computational modeling. Basic and Clinical Neuroscience, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Speranza L, Di Porzio U, Viggiano D, De Donato A, & Volpicelli F (2021). Dopamine: The neuromodulator of long-term synaptic plasticity, reward and movement control. Cells, 10, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kröner S, Rosenkranz JA, Grace AA, & Barrionuevo G (2005). Dopamine modulates excitability of basolateral amygdala neurons in vitro. Journal of Neurophysiology, 93, 1598–1610. [DOI] [PubMed] [Google Scholar]
- 165.De La Mora MP, Gallegos-Cari A, Arizmendi-García Y, Marcellino D, & Fuxe K (2010). Role of dopamine receptor mechanisms in the amygdaloid modulation of fear and anxiety: Structural and functional analysis. Progress in Neurobiology, 90, 198–216. [DOI] [PubMed] [Google Scholar]
- 166.Takahashi H, Takano H, Kodaka F, Arakawa R, Yamada M, Otsuka T, Hirano Y, Kikyo H, Okubo Y, Kato M, Obata T, Ito H, & Suhara T (2010). Contribution of dopamine D1 and D2 receptors to amygdala activity in human. Journal of Neuroscience, 30, 3043–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lamanna J, Isotti F, Ferro M, Spadini S, Racchetti G, Musazzi L, & Malgaroli A (2022). Occlusion of dopamine-dependent synaptic plasticity in the prefrontal cortex mediates the expression of depressive-like behavior and is modulated by ketamine. Scientific Reports, 12, 11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bland ST, Hargrave D, Pepin JL, Amat J, Watkins LR, & Maier SF (2003). Stressor controllability modulates stress-induced dopamine and serotonin efflux and morphine-induced serotonin efflux in the medial prefrontal cortex. Neuropsychopharmacology, 28, 1589–1596. [DOI] [PubMed] [Google Scholar]
- 169.Xing B, Li Y-C, & Gao W-J (2016). Norepinephrine versus dopamine and their interaction in modulating synaptic function in the prefrontal cortex. Brain Research, 1641, 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Devoto P, Sagheddu C, Santoni M, Flore G, Saba P, Pistis M, & Gessa GL (2020). Noradrenergic source of dopamine assessed by microdialysis in the medial prefrontal cortex. Frontiers in Pharmacology, 11, 588160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Goddard AW, Ball SG, Martinez J, Robinson MJ, Yang CR, Russell JM, & Shekhar A (2010). Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depression and Anxiety, 27, 339–350. [DOI] [PubMed] [Google Scholar]
- 172.Baffa A, Hohoff C, Baune BT, Müller-Tidow C, Tidow N, Freitag C, Zwanzger P, Deckert J, Arolt V, & Domschke K (2010). Norepinephrine and serotonin transporter genes: Impact on treatment response in depression. NPS, 62, 121–131. [DOI] [PubMed] [Google Scholar]
- 173.Stockmeier CA (2003). Involvement of serotonin in depression: Evidence from postmortem and imaging studies of serotonin receptors and the serotonin transporter. Journal of Psychiatric Research, 37, 357–373. [DOI] [PubMed] [Google Scholar]
- 174.Berridge CW, & Spencer RC (2016). Differential cognitive actions of norepinephrine α2 and α1 receptor signaling in the prefrontal cortex. Brain Research, 1641, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Montoya A, Bruins R, Katzman M, & Blier P (2016). The noradrenergic paradox: Implications in the management of depression and anxiety. Neuropsychiatric Disease and Treatment, 12, 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tully K, & Bolshakov VY (2010). Emotional enhancement of memory: How norepinephrine enables synaptic plasticity. Molecular Brain, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chou D, Huang C-C, & Hsu K-S (2014). Brain-derived neurotrophic factor in the amygdala mediates susceptibility to fear conditioning. Experimental Neurology, 255, 19–29. [DOI] [PubMed] [Google Scholar]
- 178.Mcreynolds JR, Donowho K, Abdi A, Mcgaugh JL, Roozendaal B, & Mcintyre CK (2010). Memory-enhancing corticosterone treatment increases amygdala norepinephrine and Arc protein expression in hippocampal synaptic fractions. Neurobiology of Learning and Memory, 93, 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kao C-Y, Stalla G, Stalla J, Wotjak CT, & Anderzhanova E (2015). Norepinephrine and corticosterone in the medial prefrontal cortex and hippocampus predict PTSD-like symptoms in mice. European Journal of Neuroscience, 41, 1139–1148. [DOI] [PubMed] [Google Scholar]
- 180.Peng B, Xu Q, Liu J, Guo S, Borgland SL, & Liu S (2021). Corticosterone attenuates reward-seeking behavior and increases anxiety via D2 receptor signaling in ventral tegmental area dopamine neurons. Journal of Neuroscience, 41, 1566–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tipps M, Marron Fernandez De Velasco E, Schaeffer A, & Wickman K (2018). Inhibition of pyramidal neurons in the basal amygdala promotes fear learning. eNeuro, 5, ENEURO.0272–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jett JD, & Morilak DA (2013). Too much of a good thing: Blocking noradrenergic facilitation in medial prefrontal cortex prevents the detrimental effects of chronic stress on cognition. Neuropsychopharmacology, 38, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Moret C, & Briley M (2011). The importance of norepinephrine in depression. Neuropsychiatric Disease and Treatment, 7, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Li H, Namburi P, Olson JM, Borio M, Lemieux ME, Beyeler A, Calhoon GG, Hitora-Imamura N, Coley AA, Libster A, Bal A, Jin X, Wang H, Jia C, Choudhury SR, Shi X. i., Felix-Ortiz AC, De La Fuente V, Barth VP, … Tye KM (2022). Neurotensin orchestrates valence assignment in the amygdala. Nature, 608, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Boules M, Li Z, Smith K, Fredrickson P, & Richelson E (2013). Diverse roles of neurotensin agonists in the central nervous system. Frontiers in Endocrinology, 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stuhrman K, & Roseberry AG (2015). Neurotensin inhibits both dopamine- and GABA-mediated inhibition of ventral tegmental area dopamine neurons. Journal of Neurophysiology, 114, 1734–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Beggs JM, & Plenz D (2003). Neuronal avalanches in neocortical circuits. Journal of Neuroscience, 23, 11167–11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Herz AVM, & Hopfield JJ (1995). Earthquake cycles and neural reverberations: Collective oscillations in systems with pulse-coupled threshold elements. Physical Review Letter, 75, 1222–1225. [DOI] [PubMed] [Google Scholar]
- 189.Mariani B, Nicoletti G, Bisio M, Maschietto M, Oboe R, Leparulo A, Suweis S, & Vassanelli S (2021). Neuronal avalanches across the rat somatosensory barrel cortex and the effect of single whisker stimulation. Frontiers in Systems Neuroscience, 15, 709677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ezaki T, Fonseca Dos Reis E, Watanabe T, Sakaki M, & Masuda N (2020). Closer to critical resting-state neural dynamics in individuals with higher fluid intelligence. Communications Biology, 3, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]