

Abstract
First-generation tyrosine kinase inhibitors (TKIs) have been associated with good responses in non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR)-sensitizing mutations. However, this therapeutic strategy inevitably promotes resistance to TKIs. This study aimed to investigate the functional role and mechanism of proscillaridin A in NSCLC with or without EGFR mutations. Cellular function assays showed that proscillaridin A could inhibit cell proliferation, migration and invasion in vitro independent of EGFR mutation status. Real-time PCR of the human chromosome 17 α-satellite region revealed that proscillaridin A significantly suppressed tumour micrometastasis in vivo. In immunofluorescence experiments, we found that proscillaridin A decreased filopodia length in NSCLC cells. Furthermore, proscillaridin A also downregulated EGFR-Src-mediated cytoskeleton-related pathways, including FAK-paxillin signalling, which has been shown to promote cell filopodia formation by regulating small G-proteins. Therefore, we used the GST-PBD pull-down assay to demonstrate that proscillaridin A could decrease Cdc42 activity. Moreover, survival analyses of 591 lung adenocarcinoma patients from the GEO database indicated that the expression levels of Src and paxillin and the risk score of the gene signature based on these two factors were negatively correlated with overall survival and could be used as independent prognostic factors. In conclusion, we speculate that proscillaridin A inhibits lung cancer cell growth and motility by regulating EGFR-Src-associated pathways.
Keywords: Proscillaridin A, EGFR, Src, small GTPase, lung adenocarcinoma
Introduction
Lung cancer is currently the most commonly diagnosed cancer and the leading cause of cancer-related death worldwide, presenting a major health burden [1]. Approximately 85% of lung cancer cases are non-small cell lung cancer (NSCLC), which includes adenocarcinoma, squamous cell carcinoma and large cell carcinoma [2]. Carcinogenic mutations often lead to lung cancer progression, and most mutations, such as human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), KRAS, AKT1 and MEK mutations, cause constitutive kinase activation. These protein mutations are in the signal transduction pathway and often induce cancer cell growth and metastasis, which lead to poor prognosis [3]. Therefore, there are currently many targeted therapies for the abovementioned mutations that can inhibit tumour progression and enhance the patient’s response to the drug [4].
EGFR is a transmembrane tyrosine kinase receptor that activates signal transduction pathways that regulate cell proliferation, differentiation and survival [5]. In Asia, approximately 40-55% of NSCLC patients have EGFR mutations; in these patients, approximately 40-49% have exon 19 deletions, and the other 39-47% have L858R mutations [6]. These mutations cause constitutive kinase activation and induce normal cell oncogenesis. Recently, small molecular tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, have been the first-line medicine to treat NSCLC patients with exon 19 deletion and L858 mutation (first-generation TKIs). These patients have a good response to gefitinib and erlotinib treatment [7]. However, approximately 50% of patients after using first-generation TKIs have a secondary EGFR mutation in T790M [8]. Therefore, the development of a new targeted drug, including the second-generation TKIs afatinib [9] and third-generation osimertinib [10], has become a major goal in the treatment of lung cancer.
The Src gene is classified as a proto-oncogene due to its potential to promote cell growth and division and is involved in several signalling pathways within cells. These pathways include the focal adhesion kinase (FAK), phosphatidylinositol 3-kinase (PI3K), and signal transducer and activator of transcription 3 (STAT3) pathways [11,12]. Furthermore, a prior clinical study revealed that NSCLC patients with high Src expression levels had a poor prognosis [13]. In another previous study, the Src and EGFR family could form a complex and benefit cell transformation and cancer development [14]. Therefore, inhibition of Src and EGFR activity may provide a new therapeutic target for TKI-resistant NSCLC patients.
FAK is an intracellular tyrosine kinase signalling transducer, and its signalling is mediated by integrin and other cell surface receptors, such as EGFR [15]. In recent years, FAK was believed to enhance cell migration and invasion through signal transduction and to promote actin cytoskeleton reconstruction [16]. FAK has been shown to be highly expressed in many human cancers and is often associated with advanced disease as well as a poor prognosis in NSCLC patients [17,18]. Due to the mutual activation between EGFR and Src and the phosphorylation of FAK by Src [13,19], inhibiting EGFR activity may reduce FAK-mediated cell motility.
To develop new candidate EGFR inhibitors, we previously used a pharmacophore model and ELISA to screen the NCI-60 compound library [20] and identified proscillaridin A (NSC number: 7521) as a promising candidate. A previous study indicated that proscillaridin A could inhibit NSCLC growth by upregulating DR4 [21], but its effects on NSCLC migration, invasion and metastasis, as well as the mechanism of action, are still unclear. In this study, we investigated the ability of proscillaridin A to inhibit NSCLC progression and metastasis by in vitro and in vivo approaches. Our findings may provide a new lead compound for the development of anticancer drugs and provide a therapeutic strategy for the treatment of NSCLC patients in the future.
Materials and methods
Cell culture and drug treatment
To investigate the effect of proscillaridin A on NSCLC, four cell lines with different EGFR status were used in this study. The A549 (ATCC CCL-185, EGFRwild-type, gefitinib resistant) and H1975 (ATCC CRL-5908, EGFRL858R+T790M, gefitinib resistant) human lung adenocarcinoma cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), while the PC9 (EGFRexon 19 del, gefitinib sensitive) and PC9IR (EGFRexon 19 del, gefitinib resistant) cell lines were generously provided by Dr. Chih-Hsin Yang at NTU Hospital. All cell lines were cultured in RPMI-1640 media (Gibco, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified atmosphere of 5% CO2. Proscillaridin A, a compound used in the study, was obtained from the National Cancer Institute (NCI, Germantown, MD, USA) and was prepared as a 0.1 M stock solution in dimethyl sulfoxide (DMSO).
Cell viability assay
Cells were seeded in 96-well microplates at 4000 cells/well and incubated overnight to allow cell adherence. Cells were treated with proscillaridin A at different concentrations or for different times. The designated volume of PrestoBlue cell viability reagent (Invitrogen, Carlsbad, CA, USA) was added to each culture well to react with cells. Afterwards, the absorbance was measured at 570 nm (with 600 nm as the reference) using a Victor3 spectrophotometer (Perkin-Elmer, Santa Clara, CA, USA).
Colony formation assay
To conduct the anchorage-dependent growth assay, the H1975, A549, PC9, and PC9IR cells were suspended in RPMI and seeded at a density of 500 cells per well into six-well plates containing culture media and drug solution. Following a 7-10 day incubation period, the cells were washed with 1X PBS and fixed with methanol. A 0.05% crystal violet stain was applied to the fixed cells. For the anchorage-independent growth assay, six-well plates were precoated with 0.7% low melting point (LMP) agarose in RPMI supplemented with 10% FBS. PC9 and PC9IR cells were then seeded in the wells in 0.35% LMP agarose/RPMI with 10% FBS at a density of 1 × 103 cells per well. Once the LMP agarose solidified, the cells were treated with proscillaridin A and incubated for three weeks. The resulting colonies were stained with 0.5 mg/ml p-iodonitrotetrazolium violet, and colonies with a diameter greater than 0.5 mm were counted using an inverted microscope. Detailed methods for this procedure are available elsewhere [22].
Migration and invasion assay
A transwell apparatus with a polycarbonate membrane (8-μm pore size, 6.5-mm diameter; Corning Costar Corporation, MA, USA) coated with or without Matrigel (2.5 mg/ml; R&D systems, Minneapolis, USA) was used for transwell invasion and migration assays, respectively, as described previously [23]. The cells (H1975 and A549) were pretreated with proscillaridin A for 24 hrs and then seeded in upper wells (2 × 104 or 4 × 104 per well) containing serum-free medium, and the lower wells were filled with the same medium supplemented with 10% FBS. After 12 hrs (migration) or 18 hrs (invasion) of incubation, the cells in the upper wells and on the membrane were swabbed with a Q-tip, fixed with methanol, and stained with 10% Giemsa solution (Sigma Chemical, St. Louis, MO, USA). The cells that were attached to the lower surface of the polycarbonate filter were counted using a light microscope (magnification, × 200).
In vivo tumour micrometastasis analysis
Five-week-old advanced severe immunodeficiency (ASID) mice were purchased from the National Laboratory Animal Center (NLAC, Taipei, Taiwan). In order to investigate the effect of proscillaridin A on the in vivo metastasis of gefitinib-resistant NSCLC cells, H1975 cells (EGFRL858R+T790M, 1 × 106 live cells) were injected into mice through the tail vein. To confirm the effect of proscillaridin A on tumour migration, we separated mice injected with tumour cells for one week into two groups, one treated with 0.1% DMSO and the other treated with 4 mg/kg proscillaridin A. The former group (n=4) was intraperitoneally injected with 100 µl PBS with 0.1% DMSO every two days, and the latter group (n=5) was injected with proscillaridin A. After 8 weeks, the mice were sacrificed using CO2, and the lungs were harvested. The mouse experiments were approved by the Institutional Animal Care and Use Committee of National Chung Hsing University (IACUC Number: 110-117). The harvested lung tissues were then subjected to genomic DNA extraction using conventional protease K and phenol-chloroform extraction followed by isopropanol precipitation [24]. To detect micrometastases of human lung adenocarcinoma cells in mice, specific primers for the human chromosome 17 α-satellite region (Cr17) were used, as described previously [25]. The highly conserved actin sequence in humans and mice was used as a control: forward primer, 5’-TCAGATCATTGCTCCTCCTG-3’ and reverse primer, 5’-ACGATGGAGGGGCCGGACTC-3’. To detect the level of human Cr17, an ABI StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA, USA) with the SYBR Green approach (Applied Biosystems) was used. The method for calculating Cr17 DNA amount has been previously described [26].
Actin staining
Cells were plated onto 24-well chamber slides, and after cells adhered to slides, different concentrations of proscillaridin A were added and incubated for 48 hrs. Alexa Fluor 594 phalloidin (6.6 μM; Invitrogen) was used to visualize filamentous actin (F-actin). Nuclei were demarcated by 4’,6-diamidino-2-phenylindole (DAPI) staining. Cells were mounted onto slides and visualized by upright fluorescence microscopy (BX51; Olympus) at 1000 × magnification.
Western blotting
Western blotting was used to examine the protein phosphorylation and expression level in lung cancer cells before and after proscillaridin A treatment as described previously [27]. EGFR (A-10) and paxillin (D-9) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Phospho-Src (Tyr418) and FAK were purchased from Invitrogen. Phospho-EGFR (Tyr1068), phospho-FAK (Tyr576/Tyr577) and phospho-paxillin (Tyr118) were purchased from Cell Signaling Technology (Beverly, MA, USA), and the primary antibody to Src was produced in our laboratory (ATCC CRL-2651). GAPDH (Santa Cruz Biotechnology) was used as a loading control.
Rac1 and Cdc42 activity assay (GST-PBD pull down)
The Rac1 and Cdc42 activity assay was performed using a previously described method [28]. Briefly, cells were treated with proscillaridin A for 48 hrs, and equal volumes of cell lysates were incubated with 2 μg of homemade GST-tagged p21-binding domain (GST-PBD) fusion protein preloaded on glutathione sepharose 4B resin (GE healthcare, Pittsburgh, PA, USA) to pull down active Rac1 and Cdc42 protein at 4°C for 1 hr. After washing with lysis buffer three times, the captured proteins were eluted with 6X sample buffer and then subjected to western blotting to quantitate the activated Rac1 and Cdc42 with Rac1 (Upstate Biotechnology, Lake Placid, NY, USA) and Cdc42 (Santa Cruz) antibodies.
Survival analyses using gene expression and clinical data
To evaluate the effect of Src and paxillin expression levels on clinical prognosis, six published microarray datasets with information on 1002 lung cancer patients were obtained from the Gene Expression Omnibus (GEO) database under accession numbers GSE30219 [29], GSE37745 [30,31], GSE31546, GSE29013 [32], GSE31210 [33,34], and GSE50081 [35]. The clinical data were curated from the GEO Series Matrix files. After filtering out the profiles derived from normal lung tissue and nonlung adenocarcinoma samples, the remaining expression profiles from lung adenocarcinoma samples were used in the study (n=591). To reduce variation among the samples, normalization between microarrays was performed by using a quantile normalization method [36], and then the values were log-transformed to a base-2 scale. Subsequently, the mean of the expression levels detected by all probes for Src and paxillin were calculated and then subjected to survival analysis. On the other hand, according to a previous study [37], a risk score function was constructed as a gene signature through the linear combination of gene expression levels of Src and paxillin. The expression levels detected by probes for Src and paxillin were assessed for their association with overall survival using univariate Cox regression analysis. The probes significantly associated with survival were used to calculate a risk score by weighting the Cox regression coefficient. The median expression value and risk score served as cut-offs for patient classification. The log-rank test and Kaplan-Meier curve were applied to compare the overall survival probability between groups. The independent prognostic factors were evaluated using multivariate Cox regression analysis and considering the covariates of age, sex and stage. All statistical tests were two-tailed, and a P value of less than 0.05 was to indicate statistical significance.
Results
Cytotoxic effect of proscillaridin A on NSCLC
To determine the cytotoxicity of proscillaridin A on NSCLC, several NSCLC cell lines with different types of EGFR mutations were used, including PC9, PC9IR, H1975, and A549, as described in Materials and Methods. After exposure to different doses of proscillaridin A for 24, 48 and 72 hrs, the results showed that proscillaridin A could decreased cell viability in these four cell lines in a dose- and time-dependent manner (Figure 1A). The IC50 concentrations for each cell line at the indicated treatment times were also obtained and are illustrated below each bar graph. In addition, proscillaridin A inhibited anchorage-dependent colony formation in gefitinib-sensitive (PC9) and gefitinib-resistant (PC9IR, H1975 and A549) cells (Figure 1B). In anchorage-independent colony formation, since A549 and H1975 couldn’t form colonies in soft agar gel, only PC9 and PC9IR cells were used in this experiment. However, we still found that proscillaridin A could inhibit the formation of cell colonies both in gefitinib-sensitive and gefitinib-resistant NSCLC (Figure 1C). Furthermore, the effect of proscillaridin A and osimertinib on the cell viability of these four cell lines was compared. The results demonstrated that the inhibitory effect of proscillaridin A was more significant than that of osimertinib in each cell line (Figure 2A-D).
Figure 1.
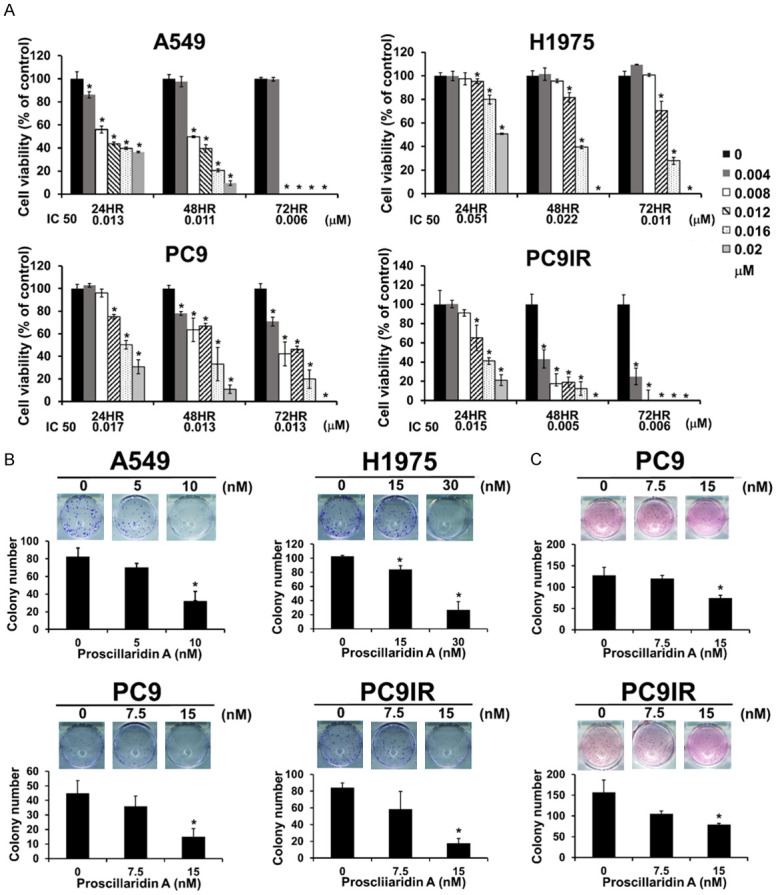
Effect of proscillaridin A on cell viability and clonogenicity in NSCLC cell lines with different EGFR statuses. A. Proscillaridin A cytotoxicity determined by cell viability assay of NSCLC cell lines with different EGFR statuses. These results are presented as percentages compared with the vehicle control (0 µM, 0.1% DMSO). The I.C. 50 at each time point is shown at the bottom of each bar chart. Each experiment was independent and repeated three times. B and C. Clonogenicity was determined by colony formation assay without or with LMP agarose. B. Anchorage-dependent colony formation (without agarose). Colonies with diameters ≥ 0.5 mm were counted. C. Anchorage-independent colony formation (with agarose). Colonies with diameters ≥ 0.5 mm were counted. Each experiment was independently performed in triplicate; 0 nM: 0.1% DMSO. *P < 0.05 compared with the vehicle control.
Figure 2.
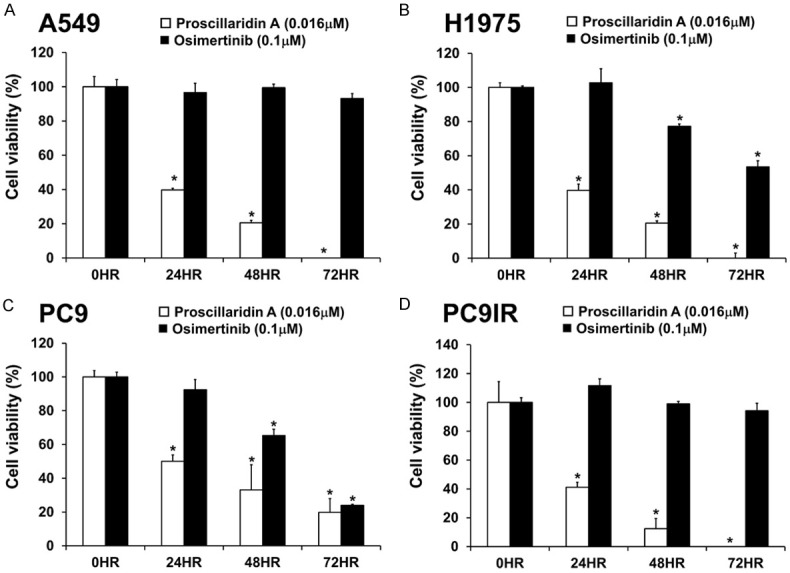
Comparison of the efficacies of proscillaridin A and osimertinib in NSCLC cell lines with wild-type EGFR and EGFR mutations. Lung cancer A549 (A), H1975 (B), PC9 (C), and PC9IR (D) cell lines were treated with 100 nM osimertinib and 16 nM proscillaridin A at the indicated time points. Cell viability was measured by the PrestoBlue assay, and the results are presented as percentages of the vehicle control (0 hr, 0.1% DMSO). Each experiment was independently performed in triplicate. The statistical analysis showed *P < 0.05 compared with the vehicle control.
Proscillaridin A inhibits cell motility and micrometastasis in NSCLC
To investigate the effect of proscillaridin A on the migration and invasion ability of NSCLC cells, we used transwell migration assays and Matrigel-based transwell invasion assays in A549 and H1975 cell lines. After treatment with proscillaridin A for 24 hrs, we performed a 12-hr Transwell migration assay and an 18-hr Matrigel-based Transwell invasion assay. We found that proscillaridin A inhibited NSCLC cell migration and invasion in a dose-dependent manner (Figure 3A and 3B). It is well known that the formation of lamellipodia and filopodia is closely related to the migration and invasion of cancer cells [38]; therefore, we used phalloidin staining to observe the change in cell morphology and detect filopodia and lamellipodia formation in NSCLC cells after treatment with proscillaridin A. After 48 hrs of exposure to proscillaridin A, our results indicated that proscillaridin A could inhibit filopodia formation but had little effect on lamellipodia formation in NSCLC cells (Figure 3C). Finally, to investigate the effect of proscillaridin A on tumour micrometastasis in vivo, H1975 cells were injected into ASID mice through the tail vein. The mice were randomly separated into the proscillaridin A-treated (I.P., 4 mg/kg every two days) and DMSO-treated groups. After eight weeks of proscillaridin A treatment, the mice were sacrificed to collect lung tissues. Then, genomic DNA extracted from mouse lung tissues was employed to perform real-time PCR using human Cr17-specific primers to detect micrometastasis. Each genomic DNA sample was independently used to perform real-time PCR experiments twice, and each experiment was performed in triplicate. The PCR data revealed that there was only a small amount of human-specific DNA in the treatment group, indicating that proscillaridin A inhibited the micrometastasis of human lung adenocarcinoma H1975 cells to the lungs of mice (Figure 3D).
Figure 3.
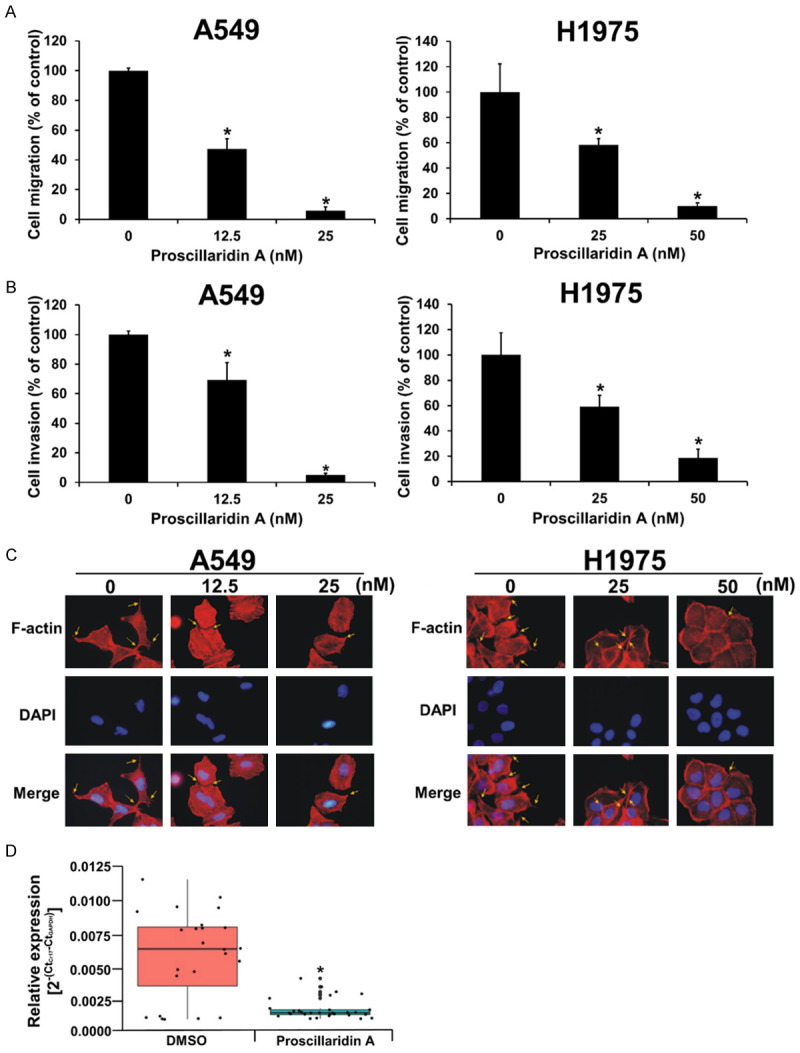
Inhibition of NSCLC cell migration, invasion and micrometastasis by proscillaridin A. A. Effect of proscillaridin A on NSCLC cell migration, as determined by a noncoated transwell assay. These results are presented as percentages compared with the vehicle control (0 nM, 0.1% DMSO). B. Effect of proscillaridin A on NSCLC cell invasion, as determined by Matrigel-coated transwell assay. These results are presented as percentages compared with the vehicle control (0 nM, 0.1% DMSO). C. Proscillaridin A inhibited NSCLC filopodia formation, as determined by phalloidin staining. The yellow arrows indicate the filopodia structure. Each experiment was independently performed in triplicate. D. Micrometastatic analysis, as measured by real-time PCR. The genomic DNA extracted from the lung tissues of the DMSO-treated group and drug-treated group mice was subjected to real-time PCR with human Cr17-specific primers. *P < 0.05 compared with the vehicle control.
Proscillaridin A inhibits the EGFR-related signalling pathway in NSCLC
To investigate whether proscillaridin A could inhibit the activity and the expression of EGFR and its related pathway proteins, two EGFR-TKI-resistant cell lines, A549 and H1975, were used in this experiment. Western blotting results showed that after treatment with proscillaridin A for 24 and 48 hrs, proscillaridin A inhibited EGFR activity and protein expression in A549 and H1975 cells in a dose-dependent manner. Similar results were observed for Src protein activity and expression levels at 24 or 48 hrs, implying crosstalk between EGFR and Src (Figure 4A). As EGFR may be involved in the interaction and activity of Src and FAK [39] and paxillin is downstream of FAK [40], we next investigated whether proscillaridin A could affect the EGFR-Src-FAK-paxillin axis. As expected, proscillaridin A significantly inhibited FAK and paxillin protein phosphorylation or expression at 24 or 48 hours, even in a dose-dependent manner (Figure 4B). These results suggest that proscillaridin A can decrease the activity of Src by inhibiting EGFR activity, thereby affecting the downstream signalling pathway of Src in NSCLC.
Figure 4.
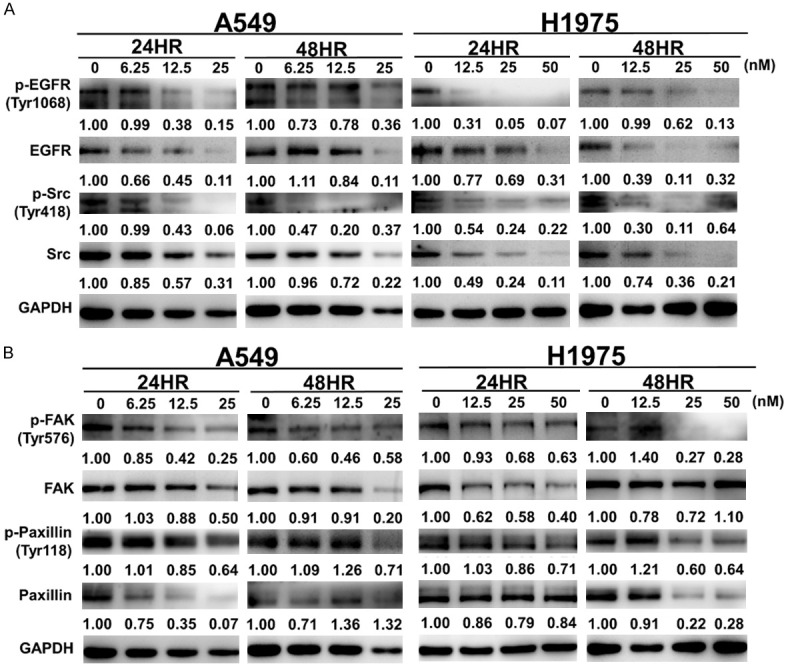
Western blot analysis of the EGFR-related pathway after proscillaridin A treatment. A549 and H1975 cells were treated with the designated concentrations of proscillaridin A for 24 and 48 hrs, and then the cell lysates were used to perform western blot assays with the indicated antibodies. 0 nM: 0.1% DMSO. A. Protein and phosphorylation levels of EGFR and Src. B. Protein and phosphorylation levels of FAK and paxillin. GAPDH was used as an internal control. Protein expression was quantified by ImageJ (NIH), and the result is shown just below the gel graph. Each experiment was performed independently in triplicate.
Proscillaridin A decreases the activity of the small-GTPase proteins Cdc42 and Rac1
Our above data indicated that proscillaridin A could significantly inhibit the formation of filopodia but slightly inhibit the formation of lamellipodia and decrease FAK and paxillin activity. In addition, a previous report showed that the FAK signalling pathway increased the activity of small-GTPase proteins such as Cdc42, Rac1 and RhoA and subsequently increased actin filament formation to enhance cell motility [16]. Therefore, it is reasonable to speculate that proscillaridin A could inhibit NSCLC motility through this pathway. To detect activated Rac and Cdc42, the purified GST-PBD fusion protein was employed to perform pull-down experiments. Our results revealed that after 48 hours of proscillaridin A treatment, the activity or protein level of Cdc42 and Rac was significantly reduced at the highest concentrations (Figure 5A).
Figure 5.
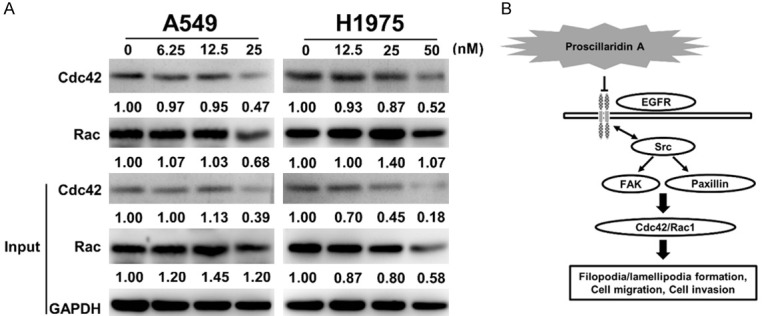
GST pull-down and western blot analyses of small GTPase protein activity after proscillaridin A treatment. A. A549 and H1975 cells were treated with the designated concentrations of proscillaridin A for 48 hrs, 0 nM; 0.1% DMSO only. The total protein and active form levels of Cdc42 and Rac were determined by western blot assay with the indicated antibodies. GAPDH was used as a loading control. Protein expression was quantified by ImageJ (NIH), and the result is shown directly below the gel plot. Each experiment was independently performed in triplicate. B. Diagram of the possible pathway regulated by proscillaridin A. This theoretical model proposes a potential mechanism by which proscillaridin A inhibits Cdc42/Rac activity through the EGFR-Src-paxillin-FAK axis, thereby reducing the formation of filopodia and lamellipodia and further resulting in decreased cell migration, invasion, and metastasis.
Src and paxillin expression was associated with the clinical outcome of lung adenocarcinoma patients
We extended our analysis by examining the Src and paxillin expression levels in tumour specimens from 591 patients with lung adenocarcinoma in GEO datasets. Initially, the correlation test through the calculation of the Pearson correlation coefficient showed that the log2-transformed gene expression levels of Src and paxillin were significantly positively correlated (r=0.60, P < 0.0001; Figure 6A). Further survival analyses indicated that the patients with high Src and paxillin expression levels exhibited shorter median overall survival than patients with low gene expression (P < 0.0001 and P < 0.0001, respectively; log-rank test; Figure 6B, 6C). Furthermore, a total of six Src probes and two paxillin probes with significant Cox regression coefficients were identified and used to calculate the risk score as a prognostic gene signature (Table 1). The risk score formula was (0.546 × expression level of 201087_at) + (0.823 × expression level of 211823_s_at) + (0.681 × expression level of 213324_at) + (0.625 × expression level of 221281_at) + (0.920 × expression level of 221284_s_at) + (0.402 × expression level of 237103_at) + (0.768 × expression level of 1558211_s_at) + (0.625 × expression level of 1565082_x_at). Subsequently, the analysis of the gene signature derived from data on 591 patients revealed that a high risk score was associated with a reduced median overall survival (P < 0.0001; log-rank test; Figure 6D). A multivariate Cox proportional hazards regression model with the covariates of sex, age and stage was used to assess the independent prognostic factors in the published cohort (n=591). All hazard ratios considering the effects of covariates indicated that high expression of Src and paxillin and a high gene signature risk score were significant independent risk factors (Table 2). The hazard ratio of the gene signature (HR=2.156) was higher than that of Src (HR=1.739) and paxillin (HR=1.872), implying that it had a greater impact on prognosis. Our data suggest that the gene signature consisting of Src and paxillin can predict the clinical outcome of patients with lung adenocarcinoma and is an independent prognostic factor.
Figure 6.
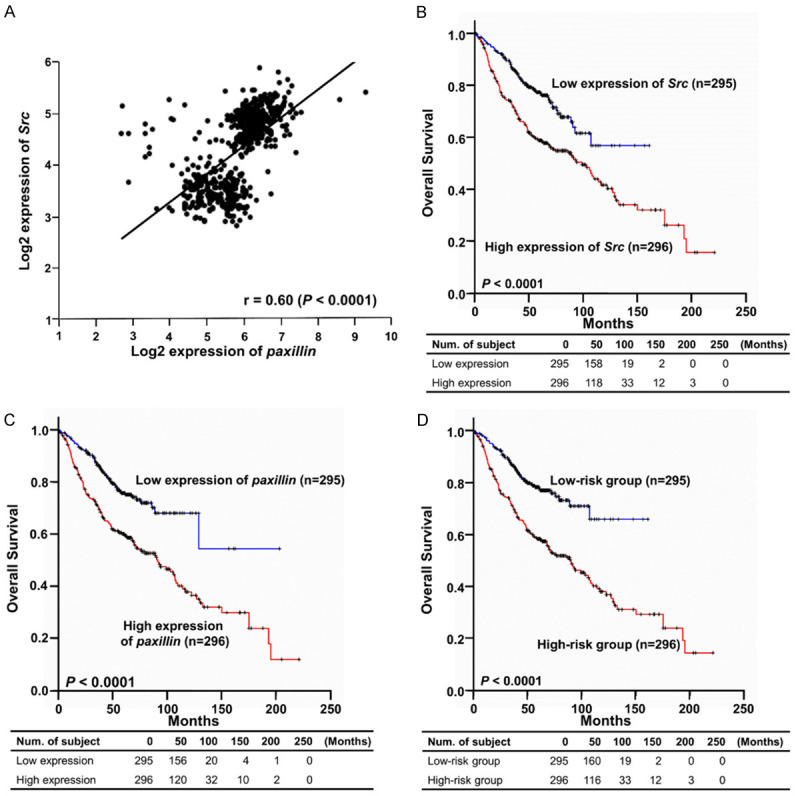
Kaplan-Meier estimates of the overall survival of patients with lung adenocarcinoma according to Src and paxillin expression levels. A total of 591 clinical samples derived from the six published NSCLC microarray datasets were used in this prognostic analysis. A. Correlation analysis of log2-transformed gene expression levels of Src and paxillin. B. An overall survival curve was generated based on the Src expression level. C. An overall survival curve was generated based on the paxillin expression level. D. An overall survival curve was generated based on the gene signature derived from the expression levels of Src and paxillin. The survival curves were estimated by the Kaplan-Meier method. The log-rank test was used to assess the difference between survival curves. A P value less than 0.05 was considered to indicate statistical significance.
Table 1.
Univariate Cox proportional hazards regression analysis of the overall survival of patients with lung adenocarcinoma according to the expression levels of individual probes for Src and paxillin
| Probe ID | Gene name | Cox coefficient | Hazard ratio | P-value |
|---|---|---|---|---|
| 213324_at | Src | 0.681 | 1.976 | < 0.0001 |
| 221281_at | Src | 0.625 | 1.868 | < 0.0001 |
| 221284_s_at | Src | 0.920 | 2.509 | < 0.0001 |
| 237103_at | Src | 0.402 | 1.495 | 0.0036 |
| 1558211_s_at | Src | 0.768 | 2.155 | < 0.0001 |
| 1565082_x_at | Src | 0.625 | 1.867 | < 0.0001 |
| 201087_at | paxillin | 0.546 | 1.726 | < 0.0001 |
| 211823_s_at | paxillin | 0.823 | 2.278 | < 0.0001 |
Table 2.
Multivariate Cox proportional hazards regression analysis of the overall survival of patients with lung adenocarcinoma
| Variable | Hazard ratio | 95% HR C.I. | P-value | |
|---|---|---|---|---|
| Overall survival based on the expression of Src | ||||
| Median of expression level | 1.739 | 1.291 | 2.344 | 0.0003 |
| Gender | 1.107 | 0.840 | 1.459 | 0.4717 |
| Age | 1.028 | 1.012 | 1.043 | 0.0003 |
| Stage (1.2 vs 3.4) | 2.063 | 1.254 | 3.396 | 0.0044 |
| Overall survival based on the expression of paxillin | ||||
| Median of expression level | 1.872 | 1.391 | 2.520 | 3.52e-05 |
| Gender | 1.183 | 0.901 | 1.552 | 0.2269 |
| Age | 1.028 | 1.013 | 1.043 | 0.0002 |
| Stage (1.2 vs 3.4) | 2.092 | 1.271 | 3.442 | 0.0037 |
| Overall survival based on the risk score | ||||
| Median of risk score | 2.156 | 1.586 | 2.932 | 9.38e-07 |
| Gender | 1.096 | 0.833 | 1.442 | 0.5140 |
| Age | 1.024 | 1.009 | 1.040 | 0.0015 |
| Stage (1.2 vs 3.4) | 2.202 | 1.339 | 3.622 | 0.0019 |
Discussion
EGFR mutations are very common in lung cancer patients in Asia and can promote the progression of lung cancer, so it is very urgent to develop therapeutics targeting EGFR mutations. Although tyrosine kinase inhibitors such as gefitinib and osimertinib are currently available, some patients have or acquire mutations that confer drug resistance. Therefore, there is still a need to develop novel therapeutic approaches. In this study, we found that proscillaridin A significantly inhibits the growth and invasion of NSCLC cell lines with wild-type EGFR or mutant EGFR.
Previous reports have shown that proscillaridin A has an antitumour effect in many cancers. For example, proscillaridin A inhibited breast cancer MCF-7 cell growth by repressing topoisomerase I and II [41]. It also inhibited cell proliferation and migration by activating GSK3β and disturbing EB1 protein accumulation at the microtubule plus-end and microtubule dynamics instability in glioblastoma [42]. Furthermore, proscillaridin A inhibited the growth of MYC-overexpressing leukaemia cells by globally inhibiting acetylation, resulting in downregulation of MYC protein expression [43]. Previous studies have also shown that the EGFR pathway can induce or participate in the expression or activity of MYC, causing cell proliferation and motility [44-46]. Therefore, based on the results of our study and previous studies, it is reasonable to speculate that proscillaridin A may regulate cell proliferation and migration through the EGFR-MYC pathway. In the future, whether the EGFR-MYC axis is involved in the inhibition of cell growth and mobility by proscillaridin A deserved further study. In hepatocellular carcinoma, proscillaridin A could induce cell autophagy and mitochondrial damage, leading to the inhibition of tumour progression [47]. In NSCLC, proscillaridin A induced cell apoptosis and inhibited cell growth through calcium-induced death receptor 4 upregulation [21]. Proscillaridin A also promoted ER and oxidative stress, inhibited Stat3 activity, and induced apoptosis in A549 NSCLC cells [48]. Although proscillaridin A can inhibit tumour cell growth in many cancers, few studies have shown that proscillaridin A can inhibit cell motility in NSCLC, including invasiveness and metastasis. Here, our study indicates that proscillaridin A inhibits cell growth and motility and alters cell morphology in NSCLC. On the other hand, we can also observe in cell viability experiments that proscillaridin A has similar inhibitory effects on PC9IR and A549 cells, while H1975 cells are more resistant. We speculate that although A549, PC9IR and H1975 are all gefitinib-resistant cells, H1975 cells show more resistant to proscillaridin A due to the presence of the T790M mutation. However, proscillaridin A still showed better inhibitory activity against H1975 compared to osimertinib. To our knowledge, this is the first report revealing that proscillaridin A can inhibit cell growth, migration, invasion, and metastasis, either in first-generation TKI-sensitive or TKI-resistant NSCLC cell lines.
The mutation frequency of EGFR is approximately 40-55% in Asian NSCLC patients; therefore, the activity of EGFR is an important factor in the treatment of NSCLC patients [49]. Activated EGFR can trigger downstream cell survival pathways, such as activation of AKT, ERK, and STAT3 signalling, leading to cell proliferation, migration, and anti-apoptosis [50]. For patients with EGFR-activating mutations, including L858R and exon 19 deletion, current treatment strategies are very effective using first- and second-generation TKIs [7]. Although first- and second-generation TKIs have a good response in EGFR mutation NSCLC patients, approximately 60% of patients develop drug-resistant mutations (T790M) after first- or second-generation TKI treatment [51]. To address this situation, a third-generation TKI (osimertinib) was developed, which has a good response to patients with secondary mutations, such as T790M [52]. However, there are still many problems in third-generation TKI treatment, such as other mutation points in EGFR, EGFR amplification, MET/HER2 amplification, MAPK or PI3K pathway activation, and fusion protein formation [53]. Therefore, the development of new candidate EGFR inhibitors is still urgent in the treatment of NSCLC. In our previous study, we found that proscillaridin A has potential EGFR inhibitory capacity in EGFR mutant cells (H1975) using pharmacophore modelling, molecular docking and ELISAs [20]. Furthermore, another previous report also revealed that proscillaridin A can inhibit EGFR activity in H1975 cells [21]. In this study, we found that proscillaridin inhibited EGFR activity in both EGFR-mutant NSCLC cells (H1975) and EGFR wild-type NSCLC cells (A549), and the IC50 value was lower than that of osimertinib. These results indicated that proscillaridin A may be a potential therapeutic compound in TKI-resistant NSCLC.
Src protein has been recognized as an important oncogenic protein that drives multiple signalling pathways, leading to the promotion of cancer cell motility, tumorigenesis, angiogenesis, and metastasis [54]. In a previous study, the collaboration between Src and activated EGFR was found to be essential for inducing cell transformation, which plays a crucial role in EGFR-driven oncogenesis [55]. Src has also been demonstrated to regulate cancer progression through several important pathways, including the PI3K/AKT, STAT3, MEK/ERK, JNK, FAK, paxillin, and p130cas pathways [56]. In addition, the Src-FAK signalling pathway, through the paxillin-ERK-p130cas axis, modulates the reorganization of the actin cytoskeleton, facilitating cell migration [57]. Several known Src inhibitors, such as dasatinib (BMS-354825), saracatinib (AZ-D0530), and ponatinib (AP24534), have been used as therapeutic agents, and their efficacy against solid tumours has been assessed in clinical trials [58]. Src inhibitors trigger apoptosis in different types of NSCLC cell lines and inhibit cell survival and EGFR-mediated malignant transformation [59]. Therefore, inhibition of Src and EGFR activity could be a new therapeutic strategy in NSCLC patients with EGFR mutations, especially those with EGFR TKI resistance. Our data showed that proscillaridin A not only inhibits EGFR activity but also suppresses Src protein phosphorylation.
FAK is a nonreceptor kinase, and its activation can drive many cell signalling pathways, such as cell growth, cell survival and cell migration [60]; FAK is usually overexpressed in many cancers and plays an essential role in tumour progression [60]. A previous study showed that autophosphorylation of FAK on tyrosine residue Y397 results in high affinity for Src, which further activates Src kinase and subsequently phosphorylates FAK at Y576 and Y577 within the FAK catalytic domain [61-63]. FAK-Src signalling can phosphorylate the downstream protein paxillin on Y118, thereby increasing cell motility and adhesion turnover [57]. When paxillin is phosphorylated, it can interact with Rho family GTPases to cause cellular actin remodelling, induce filopodia, invadopodia, and lamellipodia formation, and regulate cell migration [64]. In previous studies, it has been shown that Rho family GTPases are considered one of the key pathways regulating cell migration and invasion [65]. Cdc42 and Rac1 are members of the Rho family of GTPases, both of which cause actin polymerization and enhance cell motility. Activation of Cdc42 promotes filopodia formation, whereas lamellipodia formation requires activation of Rac1. Both of these structures enhance cell invasion and migration [66]. In our western blotting data, proscillaridin A inhibited FAK and paxillin activity or expression. Furthermore, proscillaridin A decreased Cdc42 and Rac protein levels or activity, thereby reducing filopodia formation and cell motility in NSCLC.
FAK and its related pathways play an important role in cancer progression, so it is feasible to develop therapeutic strategies targeting FAK. Few FAK inhibitors have been developed; for example, GSK2256098 is a FAK inhibitor that inhibits the growth and survival of pancreatic cancer cells [67]. Another FAK inhibitor, BI 853520, could also inhibit the growth, migration, and survival of breast cancer cells [68]. Although these FAK inhibitors are effective against cancer cells, they are currently only in phase 1 clinical trials, so the development of new FAK inhibitors is still an urgent need. Our results showed that proscillaridin A may indirectly or directly inhibit FAK and paxillin; therefore, proscillaridin A is a potential FAK inhibitor.
A previous study indicated that NSCLC patients with high Src activity or expression levels had a poor prognosis [13]. Another previous clinical investigation revealed that high expression of paxillin leads to a poor prognosis in patients with NSCLC [69]. However, no studies have shown that the gene signature of Src-paxillin can predict the prognosis of NSCLC patients. In our data, we analysed 591 lung adenocarcinoma patients from the GEO database and found that Src and paxillin expression levels were positively correlated. We also found that the prognosis of lung adenocarcinoma patients with high expression of Src or paxillin was poor. More importantly, the gene signature of Src-paxillin performed better in predicting NSCLC patient prognosis in terms of both p value and hazard ratio (Table 2). Taken together, our data suggest that proscillaridin A can inhibit cell growth by inhibiting EGFR activity to reduce the activity of the downstream proteins Src, FAK, and paxillin. Additionally, it decreases Cdc42 and Rac1 protein expression or activity, thereby reducing filopodia formation and ultimately inhibiting the motility of NSCLC cells (Figure 5B). Furthermore, we also found that the Src-paxillin gene signature may serve as an independent prognostic factor for predicting the prognosis of NSCLC patients.
Acknowledgements
This study was supported by grants from the National Science and Technology Council, Taiwan (MOST 103-2314-B-005-001-MY3 and MOST 110-2314-B-005-005-MY3).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637–658. doi: 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 4.Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(Suppl):21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 6.Li K, Yang M, Liang N, Li S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: perplexity and solution (Review) Oncol Rep. 2017;37:1347–1358. doi: 10.3892/or.2017.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 8.Ma C, Wei S, Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis. 2011;3:10–18. doi: 10.3978/j.issn.2072-1439.2010.12.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park K, Tan EH, O’Byrne K, Zhang L, Boyer M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, Shi Y, Kim SW, Laskin J, Kim DW, Arvis CD, Kolbeck K, Laurie SA, Tsai CM, Shahidi M, Kim M, Massey D, Zazulina V, Paz-Ares L. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17:577–589. doi: 10.1016/S1470-2045(16)30033-X. [DOI] [PubMed] [Google Scholar]
- 10.Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34. doi: 10.1186/s13045-016-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Rev Clin Exp Hematol. 2003;7:205–228. [PubMed] [Google Scholar]
- 12.Chang YM, Bai L, Yang J, Kung HJ, Evans C. Survey of Src activity and Src-related growth and migration in prostate cancer lines. Cancer Res. 2006;66:592–592. [Google Scholar]
- 13.Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, Ma L, Wistuba II, Johnson FM, Kurie JM. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170:366–376. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Oh D, Dubey AK, Yao M, Yang B, Groves JT, Sheetz M. EGFR family and Src family kinase interactions: mechanics matters? Curr Opin Cell Biol. 2018;51:97–102. doi: 10.1016/j.ceb.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 17.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 18.Dy GK, Ylagan L, Pokharel S, Miller A, Brese E, Bshara W, Morrison C, Cance WG, Golubovskaya VM. The prognostic significance of focal adhesion kinase expression in stage I non-small-cell lung cancer. J Thorac Oncol. 2014;9:1278–1284. doi: 10.1097/JTO.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Weng CW, Wei CH, Tsai JY, Lai YH, Chang GC, Chen JJW. Hybrid pharmacophore- and structure-based virtual screening pipeline to identify novel EGFR inhibitors that suppress non-small cell lung cancer cell growth. Int J Mol Sci. 2022;23:3487. doi: 10.3390/ijms23073487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li RZ, Fan XX, Duan FG, Jiang ZB, Pan HD, Luo LX, Zhou YL, Li Y, Yao YJ, Yao XJ, Leung EL, Liu L. Proscillaridin A induces apoptosis and suppresses non-small-cell lung cancer tumor growth via calcium-induced DR4 upregulation. Cell Death Dis. 2018;9:696. doi: 10.1038/s41419-018-0733-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai MF, Wang CC, Chang GC, Chen CY, Chen HY, Cheng CL, Yang YP, Wu CY, Shih FY, Liu CC, Lin HP, Jou YS, Lin SC, Lin CW, Chen WJ, Chan WK, Chen JJ, Yang PC. A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J Natl Cancer Inst. 2006;98:825–838. doi: 10.1093/jnci/djj229. [DOI] [PubMed] [Google Scholar]
- 23.Chen HW, Lee JY, Huang JY, Wang CC, Chen WJ, Su SF, Huang CW, Ho CC, Chen JJ, Tsai MF, Yu SL, Yang PC. Curcumin inhibits lung cancer cell invasion and metastasis through the tumor suppressor HLJ1. Cancer Res. 2008;68:7428–7438. doi: 10.1158/0008-5472.CAN-07-6734. [DOI] [PubMed] [Google Scholar]
- 24.Shen CY, Yu JC, Lo YL, Kuo CH, Yue CT, Jou YS, Huang CS, Lung JC, Wu CW. Genome-wide search for loss of heterozygosity using laser capture microdissected tissue of breast carcinoma: an implication for mutator phenotype and breast cancer pathogenesis. Cancer Res. 2000;60:3884–3892. [PubMed] [Google Scholar]
- 25.Becker M, Nitsche A, Neumann C, Aumann J, Junghahn I, Fichtner I. Sensitive PCR method for the detection and real-time quantification of human cells in xenotransplantation systems. Br J Cancer. 2002;87:1328–1335. doi: 10.1038/sj.bjc.6600573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen JJ, Yao PL, Yuan A, Hong TM, Shun CT, Kuo ML, Lee YC, Yang PC. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res. 2003;9:729–737. [PubMed] [Google Scholar]
- 27.Lai YH, Lin SY, Wu YS, Chen HW, Chen JJW. AC-93253 iodide, a novel Src inhibitor, suppresses NSCLC progression by modulating multiple Src-related signaling pathways. J Hematol Oncol. 2017;10:172. doi: 10.1186/s13045-017-0539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 29.Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, Laffaire J, de Reynies A, Beer DG, Timsit JF, Brambilla C, Brambilla E, Khochbin S. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra166. doi: 10.1126/scitranslmed.3005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Botling J, Edlund K, Lohr M, Hellwig B, Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Ponten F, Konig A, Fernandes O, Karlsson M, Helenius G, Karlsson C, Rahnenfuhrer J, Hengstler JG, Micke P. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin Cancer Res. 2013;19:194–204. doi: 10.1158/1078-0432.CCR-12-1139. [DOI] [PubMed] [Google Scholar]
- 31.Jabs V, Edlund K, Konig H, Grinberg M, Madjar K, Rahnenfuhrer J, Ekman S, Bergkvist M, Holmberg L, Ickstadt K, Botling J, Hengstler JG, Micke P. Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer. PLoS One. 2017;12:e0187246. doi: 10.1371/journal.pone.0187246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie Y, Xiao G, Coombes KR, Behrens C, Solis LM, Raso G, Girard L, Erickson HS, Roth J, Heymach JV, Moran C, Danenberg K, Minna JD, Wistuba II. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin Cancer Res. 2011;17:5705–5714. doi: 10.1158/1078-0432.CCR-11-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, Sarai A, Kawano S, Yamaguchi R, Miyano S, Yokota J. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi M, Yamaguchi R, Nakata A, Kohno T, Nagasaki M, Shimamura T, Imoto S, Saito A, Ueno K, Hatanaka Y, Yoshida R, Higuchi T, Nomura M, Beer DG, Yokota J, Miyano S, Gotoh N. Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS One. 2012;7:e43923. doi: 10.1371/journal.pone.0043923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Der SD, Sykes J, Pintilie M, Zhu CQ, Strumpf D, Liu N, Jurisica I, Shepherd FA, Tsao MS. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J Thorac Oncol. 2014;9:59–64. doi: 10.1097/JTO.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 36.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 37.Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ, Yang PC. Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci Rep. 2015;5:14273. doi: 10.1038/srep14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacquemet G, Hamidi H, Ivaska J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol. 2015;36:23–31. doi: 10.1016/j.ceb.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Parkin A, Man J, Timpson P, Pajic M. Targeting the complexity of Src signalling in the tumour microenvironment of pancreatic cancer: from mechanism to therapy. FEBS J. 2019;286:3510–3539. doi: 10.1111/febs.15011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 41.Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull. 2006;29:1493–1497. doi: 10.1248/bpb.29.1493. [DOI] [PubMed] [Google Scholar]
- 42.Berges R, Denicolai E, Tchoghandjian A, Baeza-Kallee N, Honore S, Figarella-Branger D, Braguer D. Proscillaridin A exerts anti-tumor effects through GSK3beta activation and alteration of microtubule dynamics in glioblastoma. Cell Death Dis. 2018;9:984. doi: 10.1038/s41419-018-1018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Da Costa EM, Armaos G, McInnes G, Beaudry A, Moquin-Beaudry G, Bertrand-Lehouillier V, Caron M, Richer C, St-Onge P, Johnson JR, Krogan N, Sai Y, Downey M, Rafei M, Boileau M, Eppert K, Flores-Diaz E, Haman A, Hoang T, Sinnett D, Beausejour C, McGraw S, Raynal NJ. Heart failure drug proscillaridin A targets MYC overexpressing leukemia through global loss of lysine acetylation. J Exp Clin Cancer Res. 2019;38:251. doi: 10.1186/s13046-019-1242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu YL, Chou RH, Wu CH, Wang YN, Chang WJ, Tseng YJ, Chang WC, Lai CC, Lee HJ, Huo L, Chen CH, Hung MC. Nuclear EGFR suppresses ribonuclease activity of polynucleotide phosphorylase through DNAPK-mediated phosphorylation at serine 776. J Biol Chem. 2012;287:31015–31026. doi: 10.1074/jbc.M112.358077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonamy C, Sechet E, Amiot A, Alam A, Mourez M, Fraisse L, Sansonetti PJ, Sperandio B. Expression of the human antimicrobial peptide β-defensin-1 is repressed by the EGFR-ERK-MYC axis in colonic epithelial cells. Sci Rep. 2018;8:18043. doi: 10.1038/s41598-018-36387-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao K, Wang Q, Wang Y, Huang K, Yang C, Li Y, Yi K, Kang C. EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017;406:12–21. doi: 10.1016/j.canlet.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 47.Luo M, Liu Y, Liu N, Shao W, Ming L, Liu J, Xie Y. Proscillaridin A inhibits hepatocellular carcinoma progression through inducing mitochondrial damage and autophagy. Acta Biochim Biophys Sin (Shanghai) 2021;53:19–28. doi: 10.1093/abbs/gmaa139. [DOI] [PubMed] [Google Scholar]
- 48.Maryam A, Mehmood T, Yan Q, Li Y, Khan M, Ma T. Proscillaridin A promotes oxidative stress and ER stress, inhibits STAT3 activation, and induces apoptosis in A549 lung adenocarcinoma cells. Oxid Med Cell Longev. 2018;2018:3853409. doi: 10.1155/2018/3853409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 50.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 51.Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–i19. doi: 10.1093/annonc/mdx703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang T, Zhou C. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients with EGFR inhibitor-resistant non-small cell lung cancer. Transl Lung Cancer Res. 2014;3:370–372. doi: 10.3978/j.issn.2218-6751.2014.08.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–737. doi: 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sirvent A, Benistant C, Roche S. Oncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancer. Am J Cancer Res. 2012;2:357–371. [PMC free article] [PubMed] [Google Scholar]
- 55.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci U S A. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–678. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 58.Belli S, Esposito D, Servetto A, Pesapane A, Formisano L, Bianco R. c-Src and EGFR inhibition in molecular cancer therapy: what else can we improve? Cancers (Basel) 2020;12:1489. doi: 10.3390/cancers12061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leung EL, Tam IY, Tin VP, Chua DT, Sihoe AD, Cheng LC, Ho JC, Chung LP, Wong MP. SRC promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular-targeted therapy. Mol Cancer Res. 2009;7:923–932. doi: 10.1158/1541-7786.MCR-09-0003. [DOI] [PubMed] [Google Scholar]
- 60.Yoon H, Dehart JP, Murphy JM, Lim ST. Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights. J Histochem Cytochem. 2015;63:114–128. doi: 10.1369/0022155414561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 62.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, Lopez E. Paxillin: a crossroad in pathological cell migration. J Hematol Oncol. 2017;10:50. doi: 10.1186/s13045-017-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–112. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warner H, Wilson BJ, Caswell PT. Control of adhesion and protrusion in cell migration by Rho GTPases. Curr Opin Cell Biol. 2019;56:64–70. doi: 10.1016/j.ceb.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, He DH, Zajac-Kaye M, Hochwald SN. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle. 2014;13:3143–3149. doi: 10.4161/15384101.2014.949550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tiede S, Meyer-Schaller N, Kalathur RKR, Ivanek R, Fagiani E, Schmassmann P, Stillhard P, Hafliger S, Kraut N, Schweifer N, Waizenegger IC, Bill R, Christofori G. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis. 2018;7:73. doi: 10.1038/s41389-018-0083-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu DW, Cheng YW, Wang J, Chen CY, Lee H. Paxillin predicts survival and relapse in non-small cell lung cancer by microRNA-218 targeting. Cancer Res. 2010;70:10392–10401. doi: 10.1158/0008-5472.CAN-10-2341. [DOI] [PubMed] [Google Scholar]