

Abstract
Hepatocellular carcinoma (HCC) represents a lethal cancer, and most HCC cases occur in the fibrotic or cirrhotic livers. Hepatic stellate cells (HSCs), the main effector cells of liver fibrosis, could secret biological contents to maintain liver inflammation. Herein, we aimed to identify the key transcription factor secreted by extracellular vesicles (EVs) derived from HSCs and explored its oncogenic mechanism. The activated HSC cell line LX-2 was co-cultured with HCC cells with or without the EVs release inhibitor GW4869. The effects of co-culture with HSC on HCC cell proliferation, migration, invasion, and epithelial-to-mesenchymal transition were analyzed. Co-culture with activated LX-2 enhanced HCC cell growth and motility, while GW4869 inhibited the pro-carcinogenic effect of HSC, suggesting that HSC promoted HCC progression through the secretion of EVs. HSC-derived EVs carried the key oncogenic transcription factor PRDM16, and uptake of EVs-derived PRDM16 by HCC cells activated the NOTCH1-mediated Notch signaling pathway. Knocking down PRDM16 in EVs or blocking Notch signaling in HCC cells significantly inhibited the tumor-promoting effects of HSC-derived EVs. Our study demonstrates that HSC-derived EVs activate the NOTCH1-mediated Notch signaling pathway in HCC cells by carrying PRDM16, leading to HCC progression.
Keywords: Hepatocellular carcinoma, hepatic stellate cells, extracellular vesicles, PRDM16, notch signaling pathway
Introduction
Hepatocellular carcinoma (HCC), the most frequent primary liver cancer, has been recognized as a leading cause of death among patients with cirrhosis, and its incidence is expected to increase in the future [1]. HCC is highly therapy-resistant and thus difficult to treat, and the improvements in the outcomes of patients receiving systemic therapies have been modest [2]. Thus, novel therapies for HCC remain an unmet medical need. Hepatic stellate cells (HSC) are a subpopulation of the HCC tumor microenvironment and activated HSCs transform into myofibroblast-like cells to expedite fibrosis in response to liver injury or chronic inflammation, leading to cirrhosis and HCC development [3,4]. It has been noted that molecules secreted by HSC promote epithelial-mesenchymal transition (EMT) in HCC cells, enabling migration and invasion [5]. Therefore, understanding the molecular mechanism of EMT in HCC might generate more desirable targets for anti-tumor therapy.
Extracellular vesicles (EVs) are membrane-derived vesicles released by a vast network of cell types, including hepatocytes, HSC, and immune cells in normal and pathological conditions [6,7]. EVs are heterogeneous and can contain cell-derived biomolecules, such as proteins, lipids, RNAs, and microRNAs [8]. EVs have shown great potential for treating HCC and there are still many unsolved questions regarding their role in HCC progression [9]. For instance, it has been reported that increased HSCs-derived exosomal microRNA-148a-3p suppressed HCC tumorigenic function in vitro and in vivo [10]. Therefore, we anticipated that there also exists an oncogene that is responsible for the role of HSCs-derived EVs in HCC.
Human HSC lines provide valuable new tools in the study of liver disease, and LX-2 cells possess advantages for their viability in serum-free media and high transfectability [11]. Integrated bioinformatics prediction revealed that positive regulatory domain I-containing protein 16 (PRDM16) was a transcription factor differentially expressed in HCC cells co-cultured with LX-2 cells. PRDM16 is involved in a spectrum of biological processes, including cell fate determination and development, and regulates transcription through intrinsic chromatin-modifying activity or by complexing with histone-modifying or other nuclear proteins [12]. PRDM16 expression has been used to predict the outcome of patients with cytogenetically normal acute myeloid leukemia [13]. In this study, we examined the expression and prognostic value of PRDM16 in HCC and its actions in HSC-derived EVs and explored its downstream effectors using both bioinformatics prediction and experimental validation in vitro and in vivo. Our study would provide a basis for potential new therapeutic strategies targeting HCC.
Materials and methods
Cell lines and cell culture
HSC cell line LX-2 (CL-0560) and HCC cell lines HuH-7 (CL-0120) and Li-7 (CL-0139) were all purchased from Procell (Wuhan, Hubei, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and 1% streptomycin/penicillin at 37°C with 5% CO2. A 36-h TGF-β (2 ng/mL) treatment was used to induce LX-2 activation, and untreated cells were used as a control. The short hairpin RNA of PRDM16 (sh-PRDM16 1, 2, 3#) was transfected into LX-2 cells using Lipofectamine 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to the manufacturer’s protocol. Subsequent experiments were performed 48 hours later. HCC cells were treated with NOTCH1 inhibitor Tangeretin (HY-N0133, MedChemExpress, Monmouth Junction, NJ, USA) at a concentration of 15 μg/mL or control dimethylsulfoxide (DMSO) for 24 h.
Transfection of shRNA for PRDM16 (sh-PRDM16 1, 2, 3#) was performed using Lipofectamin2000 (Thermo Fisher) according to the manufacturer’s protocol. LX-2 cells were seeded into 24-well plates at 2 × 105 cells/well and cultured to a 90% confluence. Lipofectamin2000 reagent (5 μL) was diluted by 50 μL of Opti-MEM® Medium (Thermo Fisher), and 5 μg of shRNA was diluted by 250 μL of Opti-MEM® Medium, respectively. The diluted DNA (50 μL) was incubated with diluted Lipofectamine 2000 Reagent (50 μL) at room temperature for 5 min to form a DNA-lipid complex. The DNA-lipid complex was added to the cells at 50 μL/well, and the cells were collected for subsequent experiments after incubation at 37°C for 48 h. The loop sequence was CTCGAG. The sequences of the shRNAs used (sh-PRDM16 1, 2, 3#) were as follows: sh-PRDM16 1#: ATTGACTGCAGAGTCTATTTACTCGAGTAAATAGACTCTGCAGTCAAT; sh-PRDM16 2#: CGGTGACGTTGTAAATAATATCTCGAGATATTATTTACAACGTCACCG; sh-PRDM16 3#: ACGAGAAAGAAGACTCTTATTCTCGAGAATAAGAGTCTTCTTTCTCGT.
Immunofluorescence
LX-2 cells (1 × 104) treated with or without TGF-β were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% TritonX-100 for 10 min, and sealed with 5% bovine serum albumin (BSA) for 30 min at room temperature. The cells were subsequently incubated with Alexa Fluor® 488 fluorescently coupled α-SMA primary antibody (1:100, ab202295, Abcam, Cambridge, UK) overnight at 4°C. The nuclear labeling was conducted using 4’,6-diamidino-2-phenylindole (DAPI; 50 μg/mL) for 10 min at room temperature in the dark. The staining results were observed by fluorescence microscopy (Carl Zeiss, Oberkochen, Germany).
Co-culture experiments
Serum-derived EVs were removed by centrifuging the DMEM complete medium at 100,000 g for 18 h. HCC cell lines (4 × 105) and LX2 (1 × 105) were resuspended in EVs-depleted medium and seeded in the apical and basolateral chambers of Transwell cell culture inserts (0.4 µm pore size; BD Biosciences, San Jose, CA, USA), respectively, with or without 20 µM GW4869 (HY-19363, MedChemExpress) for a 24-h routine incubation.
Cell counting kit-8 (CCK-8)
The treated HCC cells were seeded at a 3000 cells/well density into 96-well plates and cultured to the indicated time points (0, 24, 48, 72 h). Subsequently, 10 μL of CCK8 solution (CK04, Dojindo Laboratories, Kumamoto, Japan) was added for 2-h incubation. The cell proliferation was measured by measuring the optical density (OD) value at 450 nm using a microplate reader.
5-Ethynyl-2’-deoxyuridine (EdU) assay
Cell-Light Apollo 643 Stain Kit (C10371-2, Guangzhou RiboBio Co., Ltd., Guangzhou, Guangdong, China) was used to assess the in vitro DNA synthesis activity of HCC cells. The treated HCC cells were treated with 50 μM EdU for 2 h at room temperature, fixed by 4% paraformaldehyde for 20 min, and permeabilized using 0.5% TritonX-100 for 10 min. The cells were treated with 1 × Apollo 643 fluorescent dye for 30 min at room temperature in the dark. The nuclei were further incubated in the dark for 30 min at room temperature using 1 × Hoechst33342 reaction solution, and the staining results were observed by microscopy.
Transwell assay
For migration studies, treated HCC cells (1 × 105) were resuspended in serum-free DMEM and seeded into the apical chamber of Transwell inserts (8 μm pore size, BD Biosciences), while the basolateral chamber was supplemented with a complete medium containing 10% FBS. After 24 h of incubation, the non-migrated cells in the apical chamber were removed with a cotton swab, and the migrated cells on the lower surface were fixed with 4% paraformaldehyde for 10 min and stained with 0.1% crystal violet for 15 min (both at room temperature). For invasion studies, the apical chamber of the Transwell insert was pre-coated with Matrigel, and the remaining steps were the same as for migration studies.
Protein extraction and western blot
Total proteins were extracted from the cells by lysing the cells on ice with RIPA Lysis Buffer (E-BC-R327, Elabscience Biotechnology Co., Ltd., Wuhan, Hubei, China), and the main components were 50 mM Tris (pH = 7.4), 150 mM NaCl, 1% TritonX-100, 1% sodium deoxycholate, 1 mM EDTA, 0.1% sodium dodecyl sulfate (SDS), 10 mM sodium fluoride, 1 mM sodium orthovanadate, and 1 mM PMSF. The supernatant was collected after centrifugation at 4000 g for 10 min at 4°C, and the protein concentration in the supernatant was quantified by BCA Protein Colorimetric Assay Kit (E-BC-K318-M, Elabscience). Equal amounts of proteins (20 μg) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. After being blocked by 5% BSA at room temperature for 2 h, the membranes were probed overnight at 4°C with primary antibodies, including β-Tubulin (1:500, ab6046, Abcam), E-cadherin (1:1000, #3195, Cell Signaling Technologies, Beverly, MA, USA), Vimentin (1:1000, #5741, Cell Signaling Technologies), Snail (1:1000, ab216347, Abcam), PRDM16 (1:1000, ab85874, Abcam), CD81 (1:1000, ab109201, Abcam), CD9 (1:1000, ab236630, Abcam), TSG101 (1:1000, ab30871, Abcam), Calnexin (1:1000, ab133615, Abcam), NOTCH1 (1:2000, 10062-2-AP, ProteinTech Group, Chicago, IL, USA), HES5 (1:1000, 22666-1-AP, ProteinTech Group), HES1 (1:1000, #11988, Cell Signaling Technologies). Appropriate secondary horseradish peroxidase-conjugated secondary antibody was applied for a 1-h incubation at room temperature. The results were visualized using an ECL luminescence reagent (abs920, Absin, Shanghai, China).
Isolation and identification of HSC-derived EVs
HSC-derived EVs were obtained as previously described [14]. Serum-derived EVs were removed by centrifuging the complete medium at 100,000 g for 18 h. Activated LX-2 cells were subsequently cultured with an EVs-depleted medium for 24 h. The culture supernatant was then centrifuged at 300 g for 5 min, at 1200 g for 20 min, and at 10,000 g for 30 min to remove cells and debris. The supernatant was ultracentrifuged at 100,000 g for 60 min at 4°C to precipitate EVs. EVs were washed twice in phosphate-buffered saline (PBS) and recovered by centrifugation at 100,000 g for 1 h. EVs proteins were measured using the BCA Protein Colorimetric Assay Kit (Elabscience) and stored at -80°C. The cells were treated with 20 μg/mL of EVs in subsequent experiments. The expression of CD9, CD81, TSG101, and Calnexin in EVs was examined by western blot assay to verify the purity of EVs. The morphology of EVs was observed using an H-7650 transmission electron microscope (TEM, Hitachi, Tokyo, Japan), and the particle size of EVs was determined by Nanosight NS300 (Malvern Panalytical, Malvern, UK).
The uptake of EVs
PKH67 working solution (50 μL, DL22066, Duolaimi, Wuhan, Hubei, China) was added to 20 μg of EVs resuspended in PBS and incubated for 10 min at room temperature in the dark. The excess stain was removed by centrifugation at 100000 g for 1 h, and the bottom sediment was resuspended with PBS. HCC cells were treated with 20 μg/mL of PKH67-labeled EVs for 24 h. Subsequently, the EVs not taken up by the cells were removed by washing with PBS, and the cells were fixed with 4% paraformaldehyde. Nuclei were labeled by incubating with 0.1 μg/mL DAPI for 10 min at room temperature in the dark. The uptake of EVs by the cells was subsequently observed under fluorescence microscopy, and the average fluorescence intensity of PKH67 was quantified by Image J.
RNA extraction and reverse transcription-quantitative PCR (RT-qPCR)
TRIGene Reagent (P118-05, GenStar BioSolutions, Beijing, China) was used to extract total RNA from cells or EVs. RNA was reverse transcribed into cDNA by PrimeScript RT Reagent Kit with gDNA Eraser (RR047B, Takara Holdings Inc., Kyoto, Japan), and qPCR reaction was performed by TB Green Premix Ex Taq II (RR82WR, Takara) on a Bio-rad CFX96 fluorescent quantitative PCR instrument. The PCR amplification program consisted of an initial denaturation step at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 30 s. The results were normalized to GAPDH expression. Relative changes in expression were calculated according to the 2-ΔΔCt method. Gene-specific primer sequences are as follows: PRDM16 primers: 5’-CAGCCAATCTCACCAGACACCT-3’; 5’-GTGGCACTTGAAAGGCTTCTCC-3’; NOTCH1 primers: 5’-GGTGAACTGCTCTGAGGAGATC-3’; 5’-GGATTGCAGTCGTCCACGTTGA-3’; HES1 primers: 5’-GGAAATGACAGTGAAGCACCTCC-3’; 5’-GAAGCGGGTCACCTCGTTCATG-3’; HES5 primers: 5’-TCCTGGAGATGGCTGTCAGCTA-3’; 5’-CGTGGAGCGTCAGGAACTGCA-3’; GAPDH primers: 5’-GTCTCCTCTGACTTCAACAGCG-3’; 5’-ACCACCCTGTTGCTGTAGCCAA-3’.
Luciferase reporter assay
The reporter gene plasmid pNOTCH1-TA-Luc (D4229, Beyotime Biotechnology Co., Ltd., Shanghai, China) containing multiple NOTCH1 binding sites was used to detect the transcriptional activity of NOTCH1 and the Notch signaling pathway activity. The promoter fragment of NOTCH1 was synthesized (chr9: 136546049-136547048) and inserted into pGL3-Basic Vector (E1751, Promega Corporation, Madison, WI, USA) to construct NOTCH1 promoter luciferase reporter plasmids. The above plasmids were transfected into the treated cells by Lipofectamin2000 and detected using a Firefly Luciferase Reporter Gene Assay Kit (RG006, Beyotime) after 48 h.
Chromatin immunoprecipitation (ChIP)
The ability of PRDM16 to recruit to the NOTCH1 promoter was assayed by the SimpleChIP® Enzymatic ChIP Kit (#9003, Cell Signaling Technologies) according to the manufacturer’s protocol. ChIP was performed using digested chromatin from HCC cells and the primary antibody to PRDM16 (PA5-142015, Invitrogen Inc., Carlsbad, CA, USA) or goat IgG isotype control (Invitrogen), and 10% of the chromatin supernatant was used as Input. Purified DNA was analyzed by qPCR, and the enrichment of immunoprecipitated DNA was expressed as a percentage relative to Input. The primer sequence of the NOTCH1 promoter used was: forward primer: 5’-ATGGCAGGCATTTTGGACTC-3’ and reverse primer: 5’-CCAGAAAGCACAAACGGGTC-3’.
In vivo tumor model
Forty 6-week-old female BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animal studies were designed following the National Institutes of Health Animal Care and Use Guidelines and approved by the Animal Care Committee of the First Affiliated Hospital of Anhui Medical University.
Li-7 cells resuspended in 200 μL of PBS (1 × 106 cells/mice) were injected subcutaneously into the right abdomen of each mouse. One week after tumor injection, all mice were randomly divided into 5 groups (n = 8/group): the PBS, EVs-NC, EVs-KD, EVs-NC + DMSO, and EVs-NC + Tangeretin groups. PBS, EVs-NC, or EVs-KD were administrated via the tail vein once a week (100 μg/mice/week), respectively. The remaining two groups of mice were treated by intraperitoneal injection of 30 mg/kg of Tangeretin or DMSO, respectively [15], once a week while receiving tail vein injection of EVs-NC. The mice were monitored weekly for changes in tumor volume and tumor volume = long diameter × short diameter2/2. The mice were euthanized at week 4 by intraperitoneal injection of an overdose of sodium pentobarbital (150 mg/kg), and xenograft tumor tissues were collected.
Immunohistochemistry
The xenograft tumor tissues were paraffin-embedded and sectioned (5 μm). The sections were dewaxed and rehydrated, and endogenous peroxidase activity was blocked using 5% H2O2. Antigen retrieval was performed by microwave treatment in sodium citrate buffer (pH = 6.0). The sections were sealed at room temperature for 1 h in 10% normal goat serum, followed by incubation with primary antibodies to PRDM16 (CSB-PA872534LA01HU, Cusabio Biotech, Newark, DE, USA), NOTCH1 (1:500, 10062-2-AP, ProteinTech Group), HES5 (1:200, 22666-1-AP, ProteinTech Group), and HES1 (1:1000, #11988, Cell Signaling Technologies) overnight at 4°C. Then, the nuclei were incubated for 1 h at room temperature using peroxidase-labeled secondary antibodies, developed using DAB, and labeled with hematoxylin. Finally, the brownish-brown positively stained cells were observed by light microscopy (Zeiss).
Statistical analyses
Each procedure of all experiments was undertaken in triplicate. The data were exhibited as mean ± standard deviation and analyzed using GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA). One-way/two-way analysis of variance (ANOVA) was applied for multiple group comparisons (followed by either Tukey’s or Sidak’s multiple comparison tests). P values of less than 0.05 were considered significant.
Results
HSC promote the malignant biological behavior of HCC cells through EVs secretion
We activated the HSC cell line LX-2 in vitro by TGF-β treatment and observed that TGF-β induction led to a significant upregulation of the HSC activation marker protein α-SMA (Figure 1A). Subsequently, activated LX-2 cells were co-cultured with HCC cell lines by Transwell in the presence or absence of the EVs release inhibitor GW4869 for 24 h (Figure 1B). Co-culture with LX-2 resulted in a significant enhancement of the proliferation of HCC cells, as detected by CCK8 assay and EdU staining. The proliferative capacity of HCC cells induced by co-culture of LX-2 cells was significantly reduced after the addition of GW4869 (Figure 1C, 1D). The results of Transwell and western blot experiments showed that the migratory and invasive abilities of HCC cells co-cultured with LX-2 were significantly enhanced (Figure 1E, 1F), accompanied by increased expression of Snail and Vimentin proteins and diminished expression of E-cadherin (Figure 1G). By contrast, GW4869 treatment significantly inhibited LX-2-induced metastatic activity of HCC cells (Figure 1E-G).
Figure 1.
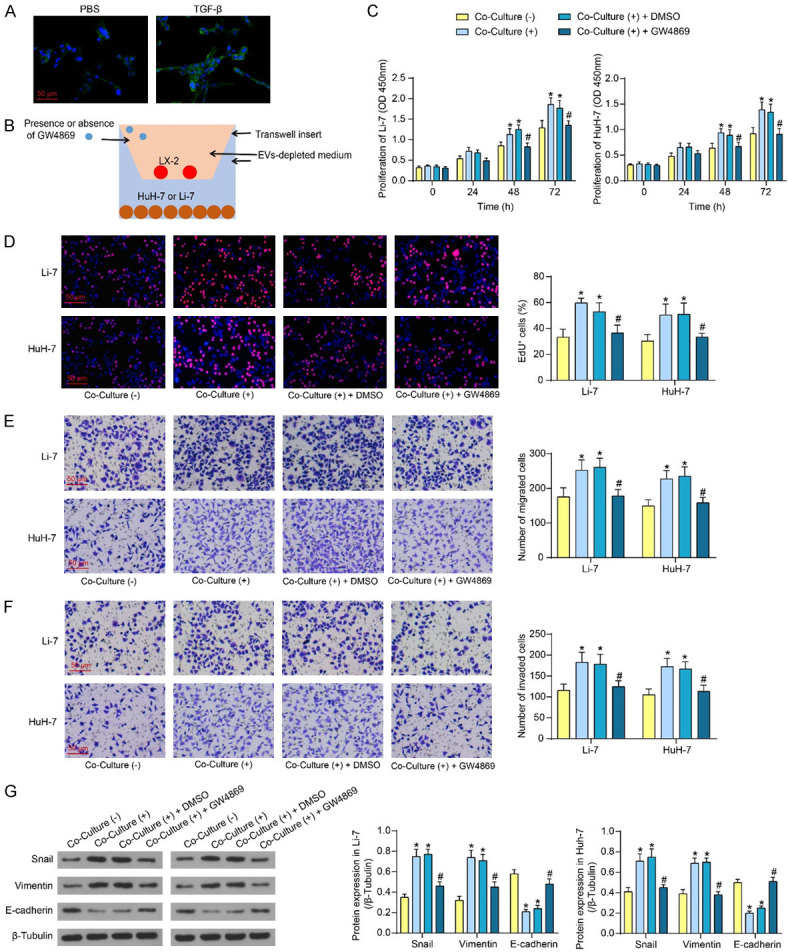
Blocking the secretion of EVs by LX-2 inhibits its effects on HCC cell biological behavior. A. Expression of activation marker protein α-SMA after TGF-β (2 ng/mL) treatment of LX-2 cells for 36 h detected by immunofluorescence. B. Schematic diagram of co-culture of activated HSC with HCC cells. C. The proliferative activity of HCC cells after co-culture with LX-2 in the presence or absence of GW4869 was measured using CCK8. D. EdU+ cells in HCC cells after co-culture with LX-2 in the presence or absence of GW4869 were measured using EdU staining. E. The migratory activity of HCC cells after co-culture with LX-2 in the presence or absence of GW4869 was measured using Transwell assay. F. The invasive activity of HCC cells after co-culture with LX-2 in the presence or absence of GW4869 was measured using Transwell assay. G. EMT-related protein expression in HCC cells after co-culture with LX-2 in the presence or absence of GW4869 was measured using western blot assays. The results represent means ± SD, *P < 0.05 vs Co-Culture (-); #P < 0.05 vs Co-Culture (+) + DMSO. All experiments were repeated at least three times, two-way ANOVA.
HSC increase the expression of PRDM16 in HCC cells through EVs
To further explore the molecular mechanisms of HSC-derived EVs, the GSE193660 dataset in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) was analyzed for differential transcriptome expression in HCC cells caused by co-cultured with LX-2. Benjamini & Hochberg (False discovery rate) was used to correct the p-value, and adjusted P < 0.01, |Log2 fold change| > 1 was set as the significance threshold to screen out the significantly differentially expressed genes (Figure 2A). The differentially expressed genes in GSE193660 were cross-screened with the human TF list downloaded from the HumanTFDB (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/). There were five transcription factors (ELF3, FOS, FOSB, TCF23, and PRDM16) in the intersection (Figure 2B). Among them, the effects of ELF3 [16], FOS [17], and FOSB [18] on HCC progression have been revealed, so TCF23 and PRDM16 were selected for further analysis.
Figure 2.
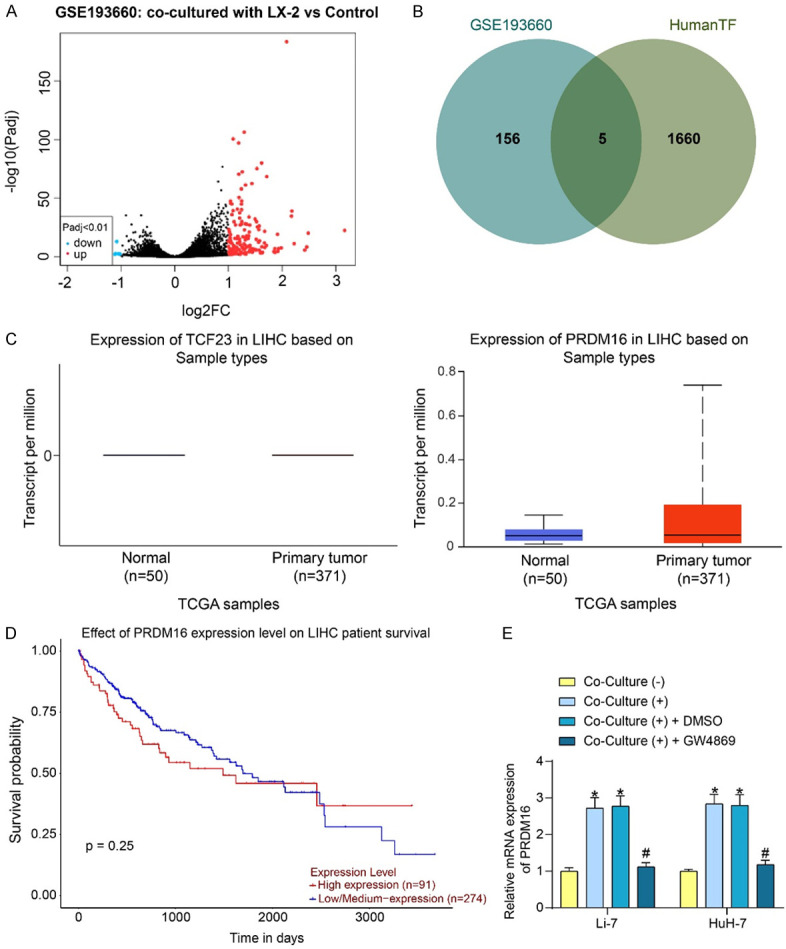
Co-culture with LX-2 results in elevated expression of PRDM16 in HCC cells. A. Analysis of transcriptome changes in HCC cells caused by co-cultured with LX-2 in the GSE193660 dataset. B. Differentially expressed transcription factors in HCC cells resulting from co-cultured with LX-2. C. TCF23 and PRDM16 expression in HCC patients predicted in the UALCAN database. D. The prognostic significance of PRDM16 expression on the survival of HCC patients was analyzed by the UALCAN database. E. PRDM16 mRNA expression in HCC cells after co-culture with LX-2 in the presence or absence of GW4869 was measured using RT-qPCR. The results represent means ± SD, *P < 0.05 vs Co-Culture (-); #P < 0.05 vs Co-Culture (+) + DMSO. All experiments were repeated at least three times, two-way ANOVA.
The expression of both in HCC was analyzed in UALCAN (https://ualcan.path.uab.edu/index.html) (Figure 2C). Only PRDM16 was found to be significantly altered in HCC, with abnormally high expression in tumor tissues, while TCF23 expression was very limited in both tumor and normal tissues. The prognostic significance of PRDM16 expression on patients’ survival was further analyzed using the UALCAN website. Patients with high PRDM16 expression had lower survival rates than those with low PRDM16 expression over a survival period of up to 2000 days (Figure 2D), which suggested the prognostic value of PRDM16.
A significant increase in PRDM16 expression was found in HCC cells co-cultured with LX-2 cells, whereas co-culture with LX-2 cells did not promote PRDM16 expression in HCC cells when GW4869 was present, as revealed by RT-qPCR (Figure 2E).
HSC-derived EVs are taken up by HCC cells by directly carrying PRDM16
It is not clear whether LX-2-derived EVs affect PRDM16 expression in HCC cells directly by carrying PRDM16 or indirectly by carrying other factors. For this purpose, we transfected shRNAs of PRDM16 into activated LX-2, and RT-qPCR and western blot were performed to detect the efficiency of shRNA-mediated inhibition (Figure 3A). Among them, sh-PRDM16 1# and 2# showed more pronounced inhibitory efficiency and were selected for subsequent experiments. Co-culture with LX-2 cells transfected with sh-PRDM16 1# and 2# significantly attenuated the expression of PRDM16 in LX-2-induced HCC cells (Figure 3B).
Figure 3.

LX-2 secretes EVs to carry PRDM16 into HCC cells. A. The knockdown efficiency of shRNAs for PRDM16 in LX-2 cells was measured using RT-qPCR and western blot analysis. B. Effect of co-culture with LX-2 cells with or without knockdown of PRDM16 on PRDM16 expression in HCC cells. C. Observation of the morphology of extracted EVs using TEM. D. The particle size of the extracted EVs using NTA. E. The expression of EVs marker proteins using western blot assays. F. PRDM16 mRNA expression in LX-2 and extracted EVs by RT-qPCR. G. Uptake of PKH67-labeled EVs by HCC cells observed under fluorescence microscopy and quantification of the average fluorescence intensity of PKH67. H. The effect of EVs treatment on PRDM16 expression in HCC cells was measured using RT-qPCR. The results represent means ± SD, *P < 0.05 vs sh-NC; #P < 0.05 vs LX-2 cells; &P < 0.05 vs EVs-NC; @P < 0.05 vs PBS. All experiments were repeated at least three times, one-way or two-way ANOVA.
A conditioned medium of LX-2 cells transfected with sh-NC, sh-PRDM16 1# and 2# was collected, and the derived EVs were isolated (named EVs-NC and EVs-KD 1# and 2#, respectively). The typical elliptical shape of EVs derived from LX-2 cells in these three groups was observed by TEM (Figure 3C). The particle size analysis by NTA showed that there was no significant difference between the three groups of EVs, which were mainly distributed in the range of 50-200 nm with a peak at about 100 nm (Figure 3D). Western blot assays showed that EVs expressed positive marker proteins CD9, CD81, and TSG101, but not negative marker protein Calnexin (Figure 3E), demonstrating the high concentration of extracted EVs. As expected, PRDM16 was highly enriched in LX-2-derived EVs-NC, while the level of PRDM16 was significantly attenuated in EVs-KD 1# and 2# (Figure 3F).
Three sets of EVs were labeled by PKH67, and the HCC cells were subsequently treated with them for 24 h. It was observed that HCC cells were able to take up EVs into the cytoplasm, and there was no significant difference in the amount of the two EVs taken up by the cells (Figure 3G). It was detected by RT-qPCR that EVs-NC treatment resulted in significantly higher PRDM16 expression in HCC cells compared with the PBS group, while EVs-KD 1# and 2#, in which PRDM16 expression was knocked down, failed to significantly increase PRDM16 expression in HCC cells (Figure 3H).
Knockdown of PRDM16 inhibits the malignant biological behavior of HCC cells induced by HSC-derived EVs
Co-culture with EVs-NC promoted the proliferative capacity of HCC cells, while EVs-KD 1# and 2# with knockdown of PRDM16 did not have this effect (Figure 4A). Similarly, EdU staining results showed that EVs-NC significantly promoted DNA synthesis activity in HCC cells, whereas EVs-KD 1# and 2# did not significantly affect the DNA synthesis activity (Figure 4B). EVs-NC treatment resulted in attenuated E-cadherin expression and promoted protein expression of Vimentin and Snail in HCC cells. However, EVs-KD 1# and 2# did not significantly promote EMT in HCC cells (Figure 4C). Finally, it was demonstrated by Transwell assay that EVs-NC carrying PRDM16 significantly promoted the migration and invasion of HCC cells, while the EVs-induced cellular mobility was significantly reversed by knocking down the PRDM16 expression (Figure 4D, 4E). EVs-KD 2# was used as EVs-KD for the following experiments.
Figure 4.
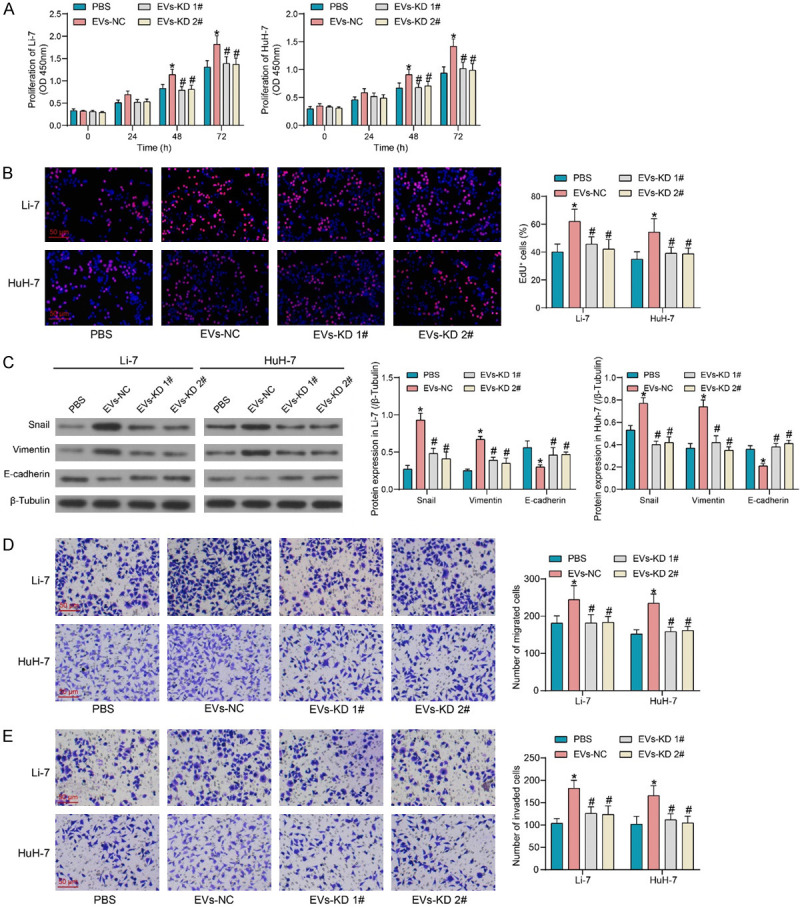
Encapsulated PRDM16 is responsible for the pro-carcinogenic effect of LX-2-derived EVs. A. The proliferative activity of HCC cells after EVs treatment was measured using CCK8. B. EdU+ cells in HCC cells after EVs treatment were measured using EdU staining. C. EMT-related protein expression in HCC cells after EVs treatment was measured using western blot assays. D. The migratory activity of HCC cells after EVs treatment was measured using Transwell assay. E. The invasive activity of HCC cells after EVs treatment was measured using Transwell assay. The results represent means ± SD, *P < 0.05 vs PBS; #P < 0.05 vs EVs-NC. All experiments were repeated at least three times, two-way ANOVA.
PRDM16 transmitted by EVs can induce NOTCH1 transcription
To investigate the downstream mechanisms of PRDM16, we downloaded the list of genes positively correlated with PRDM16 in HCC in UALCAN and performed KEGG pathway enrichment analysis (Figure 5A). We observed that genes co-expressed with PRDM16 were significantly enriched for the Hedgehog signaling, the Notch signaling, and the TGF-beta signaling pathways that are closely associated with cancer. Among the genes enriched to these pathways, NOTCH1 was the most outstanding factor in terms of relevance (Figure 5B). Therefore, we hypothesized that PRDM16 mediated the transcriptional activation of NOTCH1 to promote HCC progression through the Notch signaling pathway (Figure 5C).
Figure 5.

EVs-derived PRDM16 regulates the Notch signaling pathway in HCC cells. A. The KEGG enrichment analysis of genes positively correlated with PRDM16 in HCC. B. Correlation between NOTCH1 expression and PRDM16 expression in HCC. C. The schematic for the Notch signaling pathway. D. The effect of EVs treatment on Notch signaling pathway activity in HCC cells was measured using luciferase reporter assays. E. Effect of EVs treatment on the expression of NOTCH1 and the downstream factors HES1 and HES5 in HCC cells by RT-qPCR. F. The enrichment ability of PRDM16 on the NOTCH1 promoter was measured using ChIP-qPCR. G. The effect of PRDM16 carried by EVs on the transcriptional activity of the NOTCH1 promoter in HCC cells was measured using luciferase reporter assays. The results represent means ± SD, *P < 0.05 vs PBS; #P < 0.05 vs EVs-NC; &P < 0.05 vs IgG. All experiments were repeated at least three times, two-way ANOVA.
Notch signaling pathway activity was detected in cells by the luciferase reporter plasmid pNOTCH1-TA-Luc. It was observed that EVs-NC treatment significantly increased the luciferase activity of pNOTCH1-TA-Luc in HCC cells, and the Notch signaling pathway was overactivated. However, EVs-KD with knockdown of PRDM16 did not have this effect (Figure 5D). In addition, EVs-NC significantly promoted the expression of NOTCH1 in HCC cells and led to enhanced expression of HES1 and HES5, downstream genes of the Notch signaling pathway (Figure 5E).
PRDM16 was significantly enriched for the promoter fragment of NOTCH1, as detected by ChIP-qPCR experiments (Figure 5F). It was observed by luciferase reporter assay that EVs-NC significantly promoted the luciferase activity of NOTCH1 promoter reporter plasmid in HCC cells, and the transcription of the NOTCH1 promoter was increased. By contrast, EVs-KD with knockdown of PRDM16 cannot increase the luciferase activity of the NOTCH1 promoter compared to PBS (Figure 5G).
Blocking NOTCH1 signaling reverses HCC cell activity induced by HSC-derived EVs
HCC cells co-cultured with EVs-NC were further treated with Tangeretin, a NOTCH1 inhibitor, or control DMSO. Tangeretin significantly inhibited the protein expression of NOTCH1, HES1, HES5, Snail, and Vimentin, and promoted the protein expression of E-cadherin, as revealed by western blot assays (Figure 6A). It was found by Transwell assays that NOTCH1 signaling blocked by Tangeretin resulted in significant inhibition of EVs-NC-induced HCC cell migration and invasion (Figure 6B, 6C). In addition, CCK8 and EdU staining results showed that NOTCH1 inhibition also reduced HCC cell proliferation (Figure 6D, 6E).
Figure 6.

Tangeretin reverses the promoting effect of LX-2-derived EVs on HCC cell malignant phenotype. A. The effect of Tangeretin on Notch signaling pathway- and EMT-related protein expression in HCC cells. B. The migratory activity of HCC cells after Tangeretin treatment was measured using Transwell assay. C. The invasive activity of HCC cells after Tangeretin treatment was measured using a Transwell assay. D. The proliferative activity of HCC cells after Tangeretin treatment was measured using CCK8. E. EdU+ cells in HCC cells after Tangeretin treatment were measured using EdU staining. The results represent means ± SD, *P < 0.05 vs EVs-NC + DMSO. All experiments were repeated at least three times, two-way ANOVA.
HSC-derived EVs promote HCC progression by delivering PRDM16 and mediating the Notch signaling
A xenograft tumor model was constructed by subcutaneous injection of Li-7 cells into nude mice, which were treated with PBS, EVs-NC, EVs-KD, EVs-NC + DMSO, and EVs-NC + Tangeretin. We observed that EVs-NC accelerated the growth of subcutaneous xenografts, resulting in a significant increase in the weight of harvested xenografts, whereas the knockdown of PRDM16 (EVs-KD) did not significantly promote tumor growth. Furthermore, combined treatment with Tangeretin inhibited tumor growth (Figure 7A, 7B). Immunohistochemical assays were conducted to detect the expression of relevant proteins in xenograft tumors (Figure 7C-F). EVs-NC treatment significantly promoted PRDM16 expression in xenograft tissues and enhanced the expression of NOTCH1, HES1, and HES5. Combined Tangeretin treatment significantly inhibited the expression of NOTCH1, HES1, and HES5, despite no significant effect on PRDM16 expression. EVs-KD did not significantly affect the expression of the above factors compared to the PBS-treated group.
Figure 7.

LX-2-derived EVs affect HCC growth in vivo via PRDM16/Notch signaling. (A) The volume of xenografts in mice. (B) Weight of harvested xenografts in mice. Immunohistochemical detection of PRDM16 (C), NOTCH1 (D), HES1 (E), and HES5 (F) expression in xenograft tumors. The results represent means ± SD (n = 8), *P < 0.05 vs PBS; #P < 0.05 vs EVs-NC; &P < 0.05 vs EVs-NC + DMSO. One-way or two-way ANOVA.
Discussion
When the fibrosis is not resolved, the excessive deposition of scar tissue eventually contributes to cirrhosis and the loss of organ function, and the pivotal role of HSCs in liver fibrosis makes them critical therapeutic and diagnostic targets [19]. This study first uncovered that blocking EV secretion using GW4869 inhibited HCC cell proliferation, migration, invasion, and EMT after co-culture. Then, we identified that PRDM16 expression was upregulated in HCC patients and predicted an unsatisfactory prognosis. Further, it was revealed that the HCC cells exhibited reduced proliferation, migration, invasion, and EMT in response to co-cultured with HSC-EVs with PRDM16 knockdown. Lastly, we identified that PRDM16 in HSC-EVs activated the Notch signaling pathway via binding to the NOTCH1 promoter.
Myojin et al. revealed that LX-2 cells cultured with hepatoma cells enhanced hepatoma cell growth [20], indicating the tumor-promoting effects of HSC. It has been reported that HCC cells efficiently absorbed HSC-derived small EVs, providing an advantage in the treatment of HCC using HSC-EVs [21]. In the present study, we observed that the application of GW4869 mitigated the promoting effects of HSC-EVs on the malignant phenotype of HCC cells, suggesting the effects of HSC on HCC were mediated through the delivery of EVs. EMT is a multistep biological process in which epithelial cells change in plasticity by transient de-differentiation into a mesenchymal phenotype [22]. GW4869 inhibited the motivation of drug-resistant human microvascular endothelial cells on nasopharyngeal carcinoma progression by modulating EMT in vivo [23]. Moreover, hexokinase 1 from HSC-EVs promoted the progression of HCC [24]. In addition, exosomes from HSC were a conduit for the uptake of microRNA-214 by primary mouse hepatocytes [25]. These findings suggested that the cargoes of HSC-EVs mediated its oncogenic role.
To probe the possible molecular mechanism, we conducted integrated bioinformatics prediction using the GEO dataset and the HumanTFDB. Among the five transcription factors screened out, the effects of TCF23 and PRDM16 on HCC remained less explored. Considering TCF23 showed little relevance to HCC regarding expression profile, we chose PRDM16 for further study. Yamato et al. found that 5-year overall survival was significantly worse in adult patients with acute myeloid leukemia who had high PRDM16 expression (18% vs. 34%; P = 0.002) [26]. As for its functional role, Gudmundsson et al. observed that PRDM16 is a critical controller of long-term quiescence of hematopoietic stem cells [27]. However, as we described above, its action in HCC remains unanswered. Interestingly, PRDM16 expression was promoted in HCC cells co-cultured with HSC-EVs, while the knockdown of PRDM16 in HSC resulted in unaltered expression in HCC cells. As a consequence, HCC cells reverted to a more epithelial phenotype in response to PRDM16 depletion in HSC-EVs.
Subsequent pathway enrichment analysis showed that the genes positively correlated with PRDM16 were significantly enriched to oncogenic pathways, among them NOTCH1 was the most outstanding factor for correlation. Upon activation by different ligands, Notch signaling plays pleomorphic roles in HCC, affecting neoplastic growth, invasion capability, and stem-like properties [28]. The transcription of NOTCH1 has been reported to be regulated by multiple transcription factors in differentiation [29]. In the present study, we identified the binding relation between PRDM16 and the NOTCH1 promoter. Celastrus orbiculatus extract, isolated from the Celastrus orbiculatus Vine, has been suggested to downregulate the expression of NOTCH1 and HES1, thus inhibiting vasculogenic mimicry in HCC [30]. Furthermore, high expression of HES5 was closely associated with histological grade and metastasis, and positively correlated with proliferation marker Ki-67 in patients with HCC [31]. The HSC-EVs in the present study not only induced the expression of NOTCH1 in HCC cells but also stimulated the expression of HES1 and HES5, indicating its role in activating the Notch pathway. Tangeretin, a member of polymethoxyflavones, is mainly found in the peel of citrus fruits and exhibits wide bioactivities including antioxidant, anti-inflammatory, and neuroprotective effects [32]. Here, we observed that inhibition of NOTCH1 using Tangeretin mitigated tumor-promoting effects of HSC-EVs as PRDM16 knockdown in vitro and in vivo. However, there are other signaling pathways enriched by the co-expressed genes of PRDM16, and an examination of their activation and involvement in the oncogenic role of PRDM16 in HCC should be performed.
Conclusion
In conclusion, our current study demonstrated that PRDM16 was transmitted from HSC to HCC cells via EVs. HSCs-EVs enhanced HCC cell proliferation, migration, invasion, and EMT by activating NOTCH1-mediated Notch signaling via PRDM16 (Figure 8). The findings above might shed new insight into HCC treatment.
Figure 8.
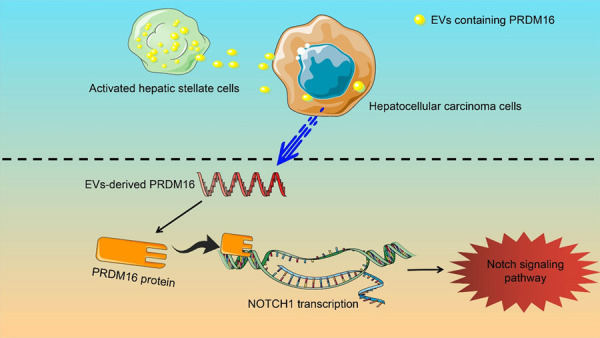
Schematic illustration. Activated HSC promote PRDM16-mediated transcriptional activation of NOTCH1 by secreting PRDM16-carrying EVs into HCC cells, which leads to Notch signaling pathway activation and HCC progression.
Acknowledgements
This work was supported by the Natural Science Foundation of Anhui Province (2108085MH289).
Disclosure of conflict of interest
None.
References
- 1.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616. doi: 10.1038/s41571-018-0073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry AE, Baldeosingh R, Lamm R, Patel K, Zhang K, Dominguez DA, Kirton KJ, Shah AP, Dang H. Hepatic stellate cells and hepatocarcinogenesis. Front Cell Dev Biol. 2020;8:709. doi: 10.3389/fcell.2020.00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang MC, Wang CJ, Liao PC, Yen CJ, Shan YS. Hepatic stellate cells secretes type I collagen to trigger epithelial mesenchymal transition of hepatoma cells. Am J Cancer Res. 2014;4:751–763. [PMC free article] [PubMed] [Google Scholar]
- 5.Ma H, Xie L, Zhang L, Yin X, Jiang H, Xie X, Chen R, Lu H, Ren Z. Activated hepatic stellate cells promote epithelial-to-mesenchymal transition in hepatocellular carcinoma through transglutaminase 2-induced pseudohypoxia. Commun Biol. 2018;1:168. doi: 10.1038/s42003-018-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz-Hernandez R, Rojas A, Gato S, Gallego J, Gil-Gomez A, Castro MJ, Ampuero J, Romero-Gomez M. Extracellular vesicles as biomarkers in liver disease. Int J Mol Sci. 2022;23:16217. doi: 10.3390/ijms232416217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Xu Y, Zhong H, Yuan H, Liang F, Liu J, Tang W. Extracellular vesicles in Inter-Kingdom communication in gastrointestinal cancer. Am J Cancer Res. 2021;11:1087–1103. [PMC free article] [PubMed] [Google Scholar]
- 8.Thietart S, Rautou PE. Extracellular vesicles as biomarkers in liver diseases: a clinician’s point of view. J Hepatol. 2020;73:1507–1525. doi: 10.1016/j.jhep.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Wang X, Zhang X, Shao T, Luo Y, Wang W, Han Y. Extracellular vesicles and hepatocellular carcinoma: opportunities and challenges. Front Oncol. 2022;12:884369. doi: 10.3389/fonc.2022.884369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Chen F, Huang P, Wang X, Zhou K, Zhou C, Yu L, Peng Y, Fan J, Zhou J, Lu Z, Hu J, Wang Z. Exosome-depleted MiR-148a-3p derived from hepatic stellate cells promotes tumor progression via ITGA5/PI3K/Akt axis in hepatocellular carcinoma. Int J Biol Sci. 2022;18:2249–2260. doi: 10.7150/ijbs.66184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chi J, Cohen P. The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol Metab. 2016;27:11–23. doi: 10.1016/j.tem.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Xiang X, Lu Q, Xu X, Cai P, Chen S, Pan J, Zeng Z. Prognostic impact of PRDM16 expression in acute myeloid leukemia with normal cytogenetics. Hematology. 2022;27:499–505. doi: 10.1080/16078454.2022.2066306. [DOI] [PubMed] [Google Scholar]
- 14.Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach JM, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, Bedina Zavec A, Benmoussa A, Berardi AC, Bergese P, Bielska E, Blenkiron C, Bobis-Wozowicz S, Boilard E, Boireau W, Bongiovanni A, Borras FE, Bosch S, Boulanger CM, Breakefield X, Breglio AM, Brennan MA, Brigstock DR, Brisson A, Broekman ML, Bromberg JF, Bryl-Gorecka P, Buch S, Buck AH, Burger D, Busatto S, Buschmann D, Bussolati B, Buzas EI, Byrd JB, Camussi G, Carter DR, Caruso S, Chamley LW, Chang YT, Chen C, Chen S, Cheng L, Chin AR, Clayton A, Clerici SP, Cocks A, Cocucci E, Coffey RJ, Cordeiro-da-Silva A, Couch Y, Coumans FA, Coyle B, Crescitelli R, Criado MF, D’Souza-Schorey C, Das S, Datta Chaudhuri A, de Candia P, De Santana EF, De Wever O, Del Portillo HA, Demaret T, Deville S, Devitt A, Dhondt B, Di Vizio D, Dieterich LC, Dolo V, Dominguez Rubio AP, Dominici M, Dourado MR, Driedonks TA, Duarte FV, Duncan HM, Eichenberger RM, Ekstrom K, El Andaloussi S, Elie-Caille C, Erdbrugger U, Falcon-Perez JM, Fatima F, Fish JE, Flores-Bellver M, Forsonits A, Frelet-Barrand A, Fricke F, Fuhrmann G, Gabrielsson S, Gamez-Valero A, Gardiner C, Gartner K, Gaudin R, Gho YS, Giebel B, Gilbert C, Gimona M, Giusti I, Goberdhan DC, Gorgens A, Gorski SM, Greening DW, Gross JC, Gualerzi A, Gupta GN, Gustafson D, Handberg A, Haraszti RA, Harrison P, Hegyesi H, Hendrix A, Hill AF, Hochberg FH, Hoffmann KF, Holder B, Holthofer H, Hosseinkhani B, Hu G, Huang Y, Huber V, Hunt S, Ibrahim AG, Ikezu T, Inal JM, Isin M, Ivanova A, Jackson HK, Jacobsen S, Jay SM, Jayachandran M, Jenster G, Jiang L, Johnson SM, Jones JC, Jong A, Jovanovic-Talisman T, Jung S, Kalluri R, Kano SI, Kaur S, Kawamura Y, Keller ET, Khamari D, Khomyakova E, Khvorova A, Kierulf P, Kim KP, Kislinger T, Klingeborn M, Klinke DJ 2nd, Kornek M, Kosanovic MM, Kovacs AF, Kramer-Albers EM, Krasemann S, Krause M, Kurochkin IV, Kusuma GD, Kuypers S, Laitinen S, Langevin SM, Languino LR, Lannigan J, Lasser C, Laurent LC, Lavieu G, Lazaro-Ibanez E, Le Lay S, Lee MS, Lee YXF, Lemos DS, Lenassi M, Leszczynska A, Li IT, Liao K, Libregts SF, Ligeti E, Lim R, Lim SK, Line A, Linnemannstons K, Llorente A, Lombard CA, Lorenowicz MJ, Lorincz AM, Lotvall J, Lovett J, Lowry MC, Loyer X, Lu Q, Lukomska B, Lunavat TR, Maas SL, Malhi H, Marcilla A, Mariani J, Mariscal J, Martens-Uzunova ES, Martin-Jaular L, Martinez MC, Martins VR, Mathieu M, Mathivanan S, Maugeri M, McGinnis LK, McVey MJ, Meckes DG Jr, Meehan KL, Mertens I, Minciacchi VR, Moller A, Moller Jorgensen M, Morales-Kastresana A, Morhayim J, Mullier F, Muraca M, Musante L, Mussack V, Muth DC, Myburgh KH, Najrana T, Nawaz M, Nazarenko I, Nejsum P, Neri C, Neri T, Nieuwland R, Nimrichter L, Nolan JP, Nolte-’t Hoen EN, Noren Hooten N, O’Driscoll L, O’Grady T, O’Loghlen A, Ochiya T, Olivier M, Ortiz A, Ortiz LA, Osteikoetxea X, Ostergaard O, Ostrowski M, Park J, Pegtel DM, Peinado H, Perut F, Pfaffl MW, Phinney DG, Pieters BC, Pink RC, Pisetsky DS, Pogge von Strandmann E, Polakovicova I, Poon IK, Powell BH, Prada I, Pulliam L, Quesenberry P, Radeghieri A, Raffai RL, Raimondo S, Rak J, Ramirez MI, Raposo G, Rayyan MS, Regev-Rudzki N, Ricklefs FL, Robbins PD, Roberts DD, Rodrigues SC, Rohde E, Rome S, Rouschop KM, Rughetti A, Russell AE, Saa P, Sahoo S, Salas-Huenuleo E, Sanchez C, Saugstad JA, Saul MJ, Schiffelers RM, Schneider R, Schoyen TH, Scott A, Shahaj E, Sharma S, Shatnyeva O, Shekari F, Shelke GV, Shetty AK, Shiba K, Siljander PR, Silva AM, Skowronek A, Snyder OL 2nd, Soares RP, Sodar BW, Soekmadji C, Sotillo J, Stahl PD, Stoorvogel W, Stott SL, Strasser EF, Swift S, Tahara H, Tewari M, Timms K, Tiwari S, Tixeira R, Tkach M, Toh WS, Tomasini R, Torrecilhas AC, Tosar JP, Toxavidis V, Urbanelli L, Vader P, van Balkom BW, van der Grein SG, Van Deun J, van Herwijnen MJ, Van Keuren-Jensen K, van Niel G, van Royen ME, van Wijnen AJ, Vasconcelos MH, Vechetti IJ Jr, Veit TD, Vella LJ, Velot E, Verweij FJ, Vestad B, Vinas JL, Visnovitz T, Vukman KV, Wahlgren J, Watson DC, Wauben MH, Weaver A, Webber JP, Weber V, Wehman AM, Weiss DJ, Welsh JA, Wendt S, Wheelock AM, Wiener Z, Witte L, Wolfram J, Xagorari A, Xander P, Xu J, Yan X, Yanez-Mo M, Yin H, Yuana Y, Zappulli V, Zarubova J, Zekas V, Zhang JY, Zhao Z, Zheng L, Zheutlin AR, Zickler AM, Zimmermann P, Zivkovic AM, Zocco D, Zuba-Surma EK. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Zheng L, Sun Y, Wang T, Wang B. Tangeretin enhances radiosensitivity and inhibits the radiation-induced epithelial-mesenchymal transition of gastric cancer cells. Oncol Rep. 2015;34:302–310. doi: 10.3892/or.2015.3982. [DOI] [PubMed] [Google Scholar]
- 16.Zheng L, Xu M, Xu J, Wu K, Fang Q, Liang Y, Zhou S, Cen D, Ji L, Han W, Cai X. ELF3 promotes epithelial-mesenchymal transition by protecting ZEB1 from miR-141-3p-mediated silencing in hepatocellular carcinoma. Cell Death Dis. 2018;9:387. doi: 10.1038/s41419-018-0399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakiri L, Hamacher R, Grana O, Guio-Carrion A, Campos-Olivas R, Martinez L, Dienes HP, Thomsen MK, Hasenfuss SC, Wagner EF. Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation. J Exp Med. 2017;214:1387–1409. doi: 10.1084/jem.20160935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu B, Yu M, Ma X, Sun J, Liu C, Wang C, Wu S, Fu P, Yang Z, He Y, Zhu Y, Huang C, Yang X, Shi Y, Qiu S, Sun H, Zhu AX, Zhou J, Xu Y, Zhu D, Fan J. IFNalpha potentiates anti-PD-1 efficacy by remodeling glucose metabolism in the hepatocellular carcinoma microenvironment. Cancer Discov. 2022;12:1718–1741. doi: 10.1158/2159-8290.CD-21-1022. [DOI] [PubMed] [Google Scholar]
- 19.Zivko C, Fuhrmann G, Luciani P. Liver-derived extracellular vesicles: a cell by cell overview to isolation and characterization practices. Biochim Biophys Acta Gen Subj. 2021;1865:129559. doi: 10.1016/j.bbagen.2020.129559. [DOI] [PubMed] [Google Scholar]
- 20.Myojin Y, Hikita H, Sugiyama M, Sasaki Y, Fukumoto K, Sakane S, Makino Y, Takemura N, Yamada R, Shigekawa M, Kodama T, Sakamori R, Kobayashi S, Tatsumi T, Suemizu H, Eguchi H, Kokudo N, Mizokami M, Takehara T. Hepatic stellate cells in hepatocellular carcinoma promote tumor growth via growth differentiation factor 15 production. Gastroenterology. 2021;160:1741–1754. e16. doi: 10.1053/j.gastro.2020.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Liu C, Zhou X, Long Q, Zeng H, Sun Q, Chen Y, Wu D, Liu L. Small extracellular vesicles containing miR-30a-3p attenuate the migration and invasion of hepatocellular carcinoma by targeting SNAP23 gene. Oncogene. 2021;40:233–245. doi: 10.1038/s41388-020-01521-7. [DOI] [PubMed] [Google Scholar]
- 22.Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798–808. doi: 10.1016/j.jhep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Huang L, Hu C, Chao H, Zhang Y, Li Y, Hou J, Xu Z, Lu H, Li H, Chen H. Drug-resistant endothelial cells facilitate progression, EMT and chemoresistance in nasopharyngeal carcinoma via exosomes. Cell Signal. 2019;63:109385. doi: 10.1016/j.cellsig.2019.109385. [DOI] [PubMed] [Google Scholar]
- 24.Chen QT, Zhang ZY, Huang QL, Chen HZ, Hong WB, Lin T, Zhao WX, Wang XM, Ju CY, Wu LZ, Huang YY, Hou PP, Wang WJ, Zhou D, Deng X, Wu Q. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022;4:1306–1321. doi: 10.1038/s42255-022-00642-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Charrier A, Zhou Y, Chen R, Yu B, Agarwal K, Tsukamoto H, Lee LJ, Paulaitis ME, Brigstock DR. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology. 2014;59:1118–1129. doi: 10.1002/hep.26768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamato G, Yamaguchi H, Handa H, Shiba N, Kawamura M, Wakita S, Inokuchi K, Hara Y, Ohki K, Okubo J, Park MJ, Sotomatsu M, Arakawa H, Hayashi Y. Clinical features and prognostic impact of PRDM16 expression in adult acute myeloid leukemia. Genes Chromosomes Cancer. 2017;56:800–809. doi: 10.1002/gcc.22483. [DOI] [PubMed] [Google Scholar]
- 27.Gudmundsson KO, Nguyen N, Oakley K, Han Y, Gudmundsdottir B, Liu P, Tessarollo L, Jenkins NA, Copeland NG, Du Y. Prdm16 is a critical regulator of adult long-term hematopoietic stem cell quiescence. Proc Natl Acad Sci U S A. 2020;117:31945–31953. doi: 10.1073/pnas.2017626117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giovannini C, Fornari F, Piscaglia F, Gramantieri L. Notch signaling regulation in HCC: from hepatitis virus to non-coding RNAs. Cells. 2021;10:521. doi: 10.3390/cells10030521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brooks YS, Ostano P, Jo SH, Dai J, Getsios S, Dziunycz P, Hofbauer GF, Cerveny K, Chiorino G, Lefort K, Dotto GP. Multifactorial ERbeta and NOTCH1 control of squamous differentiation and cancer. J Clin Invest. 2014;124:2260–2276. doi: 10.1172/JCI72718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jue C, Min Z, Zhisheng Z, Lin C, Yayun Q, Xuanyi W, Feng J, Haibo W, Youyang S, Tadashi H, Shintaro I, Shiyu G, Yanqing L. COE inhibits vasculogenic mimicry in hepatocellular carcinoma via suppressing Notch1 signaling. J Ethnopharmacol. 2017;208:165–173. doi: 10.1016/j.jep.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Zhu G, Shi W, Fan H, Zhang X, Xu J, Chen Y, Xu Z, Tao T, Cheng C. HES5 promotes cell proliferation and invasion through activation of STAT3 and predicts poor survival in hepatocellular carcinoma. Exp Mol Pathol. 2015;99:474–484. doi: 10.1016/j.yexmp.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Kozak J, Forma A, Czeczelewski M, Kozyra P, Sitarz E, Radzikowska-Buchner E, Sitarz M, Baj J. Inhibition or reversal of the epithelial-mesenchymal transition in gastric cancer: pharmacological approaches. Int J Mol Sci. 2020;22:277. doi: 10.3390/ijms22010277. [DOI] [PMC free article] [PubMed] [Google Scholar]