

Abstract
Serotonin N-acetyltransferase (arylalkylamine N-acetyltransferase [AANAT]) is the key enzyme in melatonin synthesis regulated by circadian rhythm. To date, our understanding of the oscillatory mechanism of melatonin has been limited to autoregulatory transcriptional and posttranslational regulations of AANAT mRNA. In this study, we identify three proteins from pineal glands that associate with cis-acting elements within species-specific AANAT 3′ untranslated regions to mediate mRNA degradation. These proteins include heterogeneous nuclear ribonucleoprotein R (hnRNP R), hnRNP Q, and hnRNP L. Their RNA-destabilizing function was determined by RNA interference and overexpression approaches. Expression patterns of these factors in pineal glands display robust circadian rhythm. The enhanced levels detected after midnight correlate with an abrupt decline in AANAT mRNA level. A mathematical model for the AANAT mRNA profile and its experimental evidence with rat pinealocytes indicates that rhythmic AANAT mRNA degradation mediated by hnRNP R, hnRNP Q, and hnRNP L is a key process in the regulation of its circadian oscillation.
Circadian rhythm is a fundamental biological phenomenon in living organisms (10, 41, 53). To date, efforts to understand the molecular mechanisms of circadian rhythm have focused mainly on transcriptional regulation. A number of studies show that autoregulatory transcriptional-posttranslational feedback loops are crucial for the rhythmic expression of clock-controlled genes (14, 30, 40, 41, 46). However, limited data on the posttranscriptional level are available (45). Since mRNA turnover has notable effects on the synthesis of specific proteins and provides the cell with flexibility in achieving rapid changes at the transcript level (9, 35, 50, 52), it is possible that posttranscriptional regulation functions in the rhythmic expression of circadian genes.
Recent evidence supports the existence of posttranscriptional mechanisms. In Drosophila, the degradation of Period (per) mRNA modulates its proper circadian fluctuation (49). The accelerated decay of mouse Per1 (mPer1) mRNA in a tau mutant is additionally suggestive of the presence of a posttranscriptional regulatory pathway (32). In transgenic experiments, the differences between the mRNA fluctuations of clock-controlled genes and reporters were tentatively accounted for by variations in their mRNA stability mediated by 3′ untranslated regions (3′UTRs) (22, 51). In computational modeling approaches, mRNA degradation is assumed in the construction of circadian clock models, although its role in rhythm formation is not currently clear (12, 31). Here, we postulate that dynamic mRNA degradation is essential for the formation of circadian rhythms in clock-controlled gene expression, and we support our theory with mathematical modeling and experimental evidence of rat serotonin N-acetyltransferase (arylalkylamine N-acetyltransferase [AANAT]) mRNA rhythms.
AANAT is a rate-limiting enzyme in the melatonin synthetic pathway that drives the daily rhythm in the level of circulating melatonin (4, 6, 28). The rat AANAT mRNA profile displays a low basal level during the daytime that increases up to 100-fold during the night and then decreases to the basal level with the onset of brightness (4, 29, 42). This nocturnal induction of AANAT mediated by adrenergic cyclic AMP (cAMP) signaling is explained by two major transcription factors, specifically, cAMP responsive element-binding protein (CREB) as an activator and inducible cAMP early repressor (ICER) as an inhibitor. The phosphorylated form of CREB (pCREB) rapidly activates AANAT, and mRNA accumulates up to peak levels until midnight, when the simultaneously increased level of ICER leads to the repression of AANAT and, ultimately, to the termination of mRNA synthesis (14, 40). AANAT mRNA degradation is possibly involved in its circadian oscillation patterns (6, 42). Here, we elucidate the molecular mechanisms of dynamic AANAT mRNA degradation, and propose a biological role in the circadian rhythmicity of clock-controlled genes.
MATERIALS AND METHODS
Plasmids.
In the absence of convenient restriction sites for cloning, standard PCR techniques were employed to amplify the desired sequences. The coding region of AANAT mRNA as a reporter gene was obtained by reverse transcription (RT)-PCR. The AANAT level decreases rapidly, with a half-life of ∼3.5 min, after sudden exposure to light during the night (16). Since AANAT is unstable in the absence of protein kinase A activation (29), it is an accurate reporter of the remaining mRNA level. In brief, total RNA prepared from nocturnal pineal glands was subjected to reverse transcription with oligo(dT) (Roche) as a primer. Amplification of cDNA was performed with Pfu polymerase (SolGent) and a primer pair specific for the AANAT coding region (5′ primer 5′-CGGGATCCATGTTGAGCATCCACCCCCTG-3′ and 3′ primer 5′-AAGAATTCCTC-AGCAGCCACTGTTCCTCC-3; accession number U38306 [flanking sequences for cloning purposes are underlined]). The amplified DNA fragment was digested with BamHI and EcoRI, cloned into pcDNA3.1/His C (Invitrogen), and designated pcNAT. Rat AANAT 3′UTR (3′N578) was obtained for in vitro binding assays between 3′UTRs and cell extracts, as described above. In brief, cDNA was amplified with two primers specific for 3′N578 (5′ primer 5′-GGGAATTCCCAAGCTGCGCACTTGGC-3′ and 3′ primer 5′-AATCTAGAGGGAACATAGCTGCTT-3′). The amplified DNA fragment was digested with EcoRI and XbaI, cloned into pBluescript SK(+) (pSK) (Stratagene), and designated pSK-3′N578. Where necessary, DNA with 5′- or 3′-protruding ends was treated with T4 DNA polymerase (Roche) or Klenow (Roche) to obtain blunt ends. pSK-3′N578 was treated with KpnI-EcoRI-T4 DNA polymerase to remove a 48-nucleotide (nt) region between the KpnI site flanking the T7 promoter and EcoRI site flanking the 3′UTR region, and it was self-ligated to create pSK′-3′N578. To construct the various deletion mutants, pSK′-3′N336, pSK′-3′n223 as shown in Fig. 2A, and various regions of AANAT 3′UTR were amplified by using standard PCR techniques with pSK′-3′N578 as a template. The sequences of the amplified fragments are shown in Fig. 1A. All of the PCR fragments are flanked by an EcoRI site at one end and an XbaI site at the other end. Following EcoRI-XbaI digestion, fragments were introduced into the corresponding restriction sites of plasmid pSK′. Sense strands were transcribed with T7 RNA polymerase (Roche). To generate chimeric reporter plasmids, as shown in Fig. 2A, various deletion mutants of AANAT 3′UTR were amplified by PCR and introduced into the EcoRI-XbaI site of pcNAT. For the construction of pcNAT-b3′UTR, AANAT 3′UTR was amplified from bovine AANAT 10-1B cDNA (accession number NM_177509) kindly provided by C. M. Craft (8) and introduced into the EcoRI-XbaI site of plasmid pcNAT, as described above. To create pFlag-hnRNP R, a full-length coding sequence of rat hnRNP R was obtained by PCR of a rat fetal brain cDNA library (Clontech), with primers based on the published sequences of human and mouse hnRNP Rs (23, 44). The coding region of hnRNP R was amplified with two primers specific for hnRNP R cDNA (5′ primer 5′-AAAGCTTATGGCTAATCAGGTGAA-3′ and 3′ primer 5′-AATCTAGACTACTTCCACTGTTGC-3′; accession number AF441128). The amplified DNA fragment was digested with HindIII and XbaI, and cloned into pFlag-CMV2 (Sigma). Rat hnRNP R cDNA has been newly cloned and deposited in GenBank with accession number AY184814. For the construction of pchnRNP L, plasmid pTM/hnRNP L (21) was treated with EcoRI and XhoI. The DNA fragment was inserted into the EcoRI-XhoI site of pcDNA3.1/His A (Invitrogen) to generate pchnRNP L. To construct pEGFP-hnRNP Q, pSK-hnRNP Q/synaptotagmin-binding cytoplasmic RNA-interacting protein (SYNCRIP) was treated with NheI-Klenow followed by Asp718. The DNA fragments were inserted into pEGFP-C1 (Clontech) treated with EcoRI-Klenow followed by Asp718. pSK-hnRNP Q/SYNCRIP was generated by ligation between pSK (HindIII-XbaI) and pRSETC9-15 treated with the corresponding restriction enzymes. For constructing pEGFP-hnRNP C1, pGAD424/hnRNP C1 (1-290) (25) was treated with NcoI-Klenow followed by BamHI. DNA fragments were inserted into pEGFP-C1 treated with BglII-Klenow followed by BamHI. All the constructs generated by amplification of cDNA were confirmed by sequencing.
FIG. 2.

Determination of the cellular protein-binding regions in rat AANAT 3′UTR. (A) A schematic diagram of reporter plasmid (pcNAT) and chimeric reporters containing AANAT 3′UTR (pcNAT-3′N578) or its deletion mutants are presented. The solid triangles indicate the endpoints of each deletion as shown in Fig. 1A. The names of the truncated 3′UTRs are indicated on the right. (B) CHO-K1 cells were transiently transfected with the reporter plasmids, incubated for 6 h, and incubated with actinomycin D for a further 5 h before harvesting. AANAT activities were determined as described in the legend to Fig. 1C. (C) UV cross-linking analyses between 32P-labeled riboprobes and cytoplasmic (Cyto) or nuclear (Nuc) lysates extracted from CHO-K1 cells. Molecular mass markers are indicated on the left in kilodaltons. The arrows on the right indicate the positions of RNA-protein complexes.
FIG. 1.

AANAT 3′UTR-mediated mRNA degradation. (A) Sequence of rat AANAT 3′UTR RNA (3′N578). The asterisks represent the translation stop codon. Putative poly(A) signals are underlined. The boundaries of deletion mutants shown in Fig. 2A are indicated by solid triangles. (B) Schematic representation of sequence homology comparisons for AANAT 3′UTRs among vertebrate species. Each 3′UTR sequence alignment was performed by using MultAlin (http://prodes.toulouse.inra.fr/multalin/multalin.html). Poly(A) signals are represented by closed squares, and their locations in rat AANAT 3′UTR are indicated as numbers. Accession numbers for the AANAT 3′UTRs shown are as follows: rat, U38306; mouse, AF004108; hamster, AF092100; chicken, U46502; bovine, NM_177509; sheep, U29663; monkey, U46661; and human, XM_008139. (C) Species-specific AANAT 3′UTRs determine their mRNA stability. CHO-K1 cells were transiently transfected with control plasmid pCMV · SPORT-β-gal and reporter plasmids pcNAT (open bar), pcNAT-b3′UTR (gray bar), or pcNAT-3′N578 (closed bar) as shown in Fig. 2A, incubated for 6 h, and then harvested (Act.D 0), or incubated with 5 μg of actinomycin D/ml for a further 5 h before harvesting (Act.D 5). AANAT activities were determined and normalized against β-galactosidase measurements as cpm/optical density. Values, shown as the means ± standard errors of the means (SEM) from duplicate experiments, are the AANAT activities in pcNAT-3′N578 or pcNAT-b3′UTR-transfected populations relative to those measured in pcNAT-transfected cells, to which a value of 100% was assigned. mRNA levels of reporters were determined by Northern blot analyses (Northern) with the AANAT coding region as a probe. The arrows on the right represent reporter mRNAs, NAT, and 3′UTR-containing NAT (NAT-3′N578 or NAT-b3′UTR). 18S rRNA (18S rRNA), as a control for variation in loading, was stained with ethidium bromide. (D) Kinetic analysis of rat AANAT 3′UTR-containing mRNAs. CHO-K1 cells were transiently transfected with reporter plasmids pcNAT (i) or pcNAT-3′N578 (ii) and incubated for 6 h. Total RNA was isolated from cells at the indicated times after the addition of actinomycin D (Act.D) and were subjected to Northern blot analysis using 32P-labeled DNA probes against the AANAT coding region and 18S rRNA. (iii) Signals were quantitated with a PhosphorImager, normalized to 18S rRNA signals; shown are means ± SEM. For each transfection condition, the AANAT/18S rRNA ratio at time zero was adjusted to 100%.
Experimental animals, primary pinealocyte cultures, and transfection.
The animals used in this study were maintained as described in a previous report (13). Rats were maintained in a controlled environment (12 h of light and 12 h of darkness, with lights on from zeitgeber time zero [ZT0] to ZT12). Pineal glands from rats were prepared according to a previous study (4). Dissociated pinealocytes were prepared by papain digestion and maintained as described previously (39) with minor modifications. Dissociated pinealocytes (3 × 105 cells) were maintained in Dulbecco's modified Eagle medium containing 10% fetal bovine serum, 2 mM glutamine, 10 μg of ascorbic acid/ml, and antibiotics on a poly-d-lysine-coated 48-well plate. To analyze the effects of AANAT 3′UTR-binding proteins on AANAT mRNA oscillation, adenovirus-mediated small interfering RNA (siRNA) transfections were performed as previously described (2). Thirty-six hours later, cells were stimulated with isoproterenol (ISO; 5 μM). For time course experiments with AANAT mRNA, cells were treated with ISO for up to 18 h.
Transient transfection, AANAT assay, and Northern blot analysis.
CHO-K1 cells were transfected by the electroporation method (1) at room temperature. In brief, CHO-K1 cells were transiently cotransfected with 0.3 μg of control plasmid pCMV · SPORT-β-gal (Invitrogen) and 2 μg of reporter plasmids, incubated for 6 h, and harvested or incubated with 5 μg of actinomycin D/ml for a further 5 h before harvesting. For coimmunoprecipitation, CHO-K1 cells were cotransfected with 10 μg of pFlag-hnRNP R or pFlag-CMV2 as a mock transfection and 1.5 μg of reporter plasmids (pcNAT or pcNAT-3′N578) and incubated for 18 h, followed by fractionation. For overexpression of rat AANAT 3′UTR-associated proteins, CHO-K1 cells were transiently transfected with 10 μg of pFlag-CMV2, pcDNA3.1/His C, or pEGFP-C1 as a mock transfection, 5 or 10 μg of pFlag-hnRNP R, pchnRNP L, pEGFP-hnRNP Q or pEGFP-hnRNP C1, and a combination of hnRNP R, hnRNP Q, and hnRNP L (3.3 μg each) and incubated for 18 h. Following secondary cotransfection with 0.3 μg of control plasmid and 2 μg of reporter plasmids, cells were incubated for a further 6 h before harvesting. The AANAT assay was performed as described previously (5). We prepared total RNA and extracted protein from rat pineal glands by using TRI Reagent (Molecular Research Center) according to the manufacturer's instructions. Northern blot analysis (26) was performed by using the AANAT coding region, hnRNP L, and hnRNP R as the probe and was normalized with ethidium bromide-stained 18S rRNA or 32P-labeled 18S cDNA (accession number, X0117) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) cDNA (accession number, NM_017008) probes as controls for variation in loading. The amount of reporter mRNA in the absence of actinomycin D treatment was defined as 100% (time zero), and actinomycin D-treated reporter mRNA levels were calculated as a percentage of the remaining mRNA.
Protein preparation, UV cross-linking, RNA affinity purification, and peptide sequencing.
The fractionation of pineal glands and CHO-K1 cells into cytoplasmic and nuclear extracts and purification of recombinant GST-hnRNP L were performed as described earlier (21). [32P]UTP-labeled RNA, unlabeled competitor RNA, and biotinylated RNA were transcribed in XbaI-linearized plasmids (see Fig. 2A) with T7 RNA polymerase (Roche). RNA-binding and UV cross-linking experiments were performed as described previously (26). Streptavidin-biotin RNA-affinity purification of AANAT 3′UTR-binding proteins was performed according to an earlier report (20, 27). In brief, cytoplasmic extracts prepared from rat pineal glands were incubated with or without biotinylated 3′N578 and subjected to streptavidin resin adsorption. For the competition assay, the cytoplasmic extract was preincubated with fivefold molar excess of competitor, 3′N578 RNA, prior to the addition of streptavidin resin. Resin-bound proteins were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver stained. Candidate bands were identified using matrix-assisted laser desorption ionization - time of flight (MALDI-TOF) mass spectrometry, as described earlier (38).
Antibody generation and coimmunoprecipitation.
To generate anti-hnRNP R serum, two male Sprague-Dawley rats were immunized using an N-terminal peptide specific for rat hnRNP R (amino acids 1 to 13) (44). Immunoblot analyses were performed by using a SUPEX kit (Neuronex, Kyungbuk, Republic of Korea), according to the manufacturer's instructions. To confirm the identity of the UV cross-linked complex, p82 and p68, as hnRNP R and hnRNP Q (37), and 3 μg of a polyclonal antibody against hnRNP Q (anti-SYNCRIP-N antibody; kindly provided by A. Mizutani, University of Tokyo, Tokyo, Japan) or anti-Flag monoclonal antibody (Sigma) as a control were added to the UV cross-linked samples after RNase cocktail treatment and were immunoprecipitated (26, 27). To determine if hnRNPs R, Q, and L interact with AANAT mRNA in vivo, coimmunoprecipitation reactions were performed with an anti-Flag antibody (20, 24). Cytoplasmic lysates of Flag-hnRNP R-overexpressing CHO-K1 cells with or without RNase A treatment were precleared with protein G-agarose beads and subjected to immunoprecipitation at 4°C with the antibody. Immunoprecipitates were directly resuspended in SDS loading buffer, subjected to SDS-PAGE, and analyzed by immunoblotting. AANAT mRNA was coimmunoprecipitated as described above. RNA associated with the antibody-antigen complexes was isolated by using TRI Reagent. Next, RNA was analyzed by RT-PCR with rat AANAT 3′UTR-specific primers (5′ oligonucleotide 5′-CCCAAGCTGCGCACT-TGG-3′ and 3′ oligonucleotide 5′-GGGAACATAGCTGCTTTA-3′).
SiRNA experiments.
The sequences of synthesized siRNAs (Dharmacon Research) were as follows: hnR283 siRNA targeted against hnRNP R, 5′-AGTGCATTTTTGTGGAG-3′ (corresponding to nt 283 to 301 relative to the start codon; accession number AY184814); hnL1066 siRNA against hnRNP L, 5′-TATGGCTTGGATCAATCTA-3′ (nt 1066 to 1084; accession number NM_001533); hnQ1089 siRNA against hnRNP Q, 5′-ACTGGAACGAGTGAAGAAG-3′ (nt 1089 to 1107, accession number, AY034483); and siRNA against luciferase as a mock transfection. Reporter plasmids and siRNAs were cotransfected into HEK-293T cells by using Metafectene (Biontex) according to the manufacturer's recommendations. Annealing and RNA interference analyses were performed as described in an earlier study (11).
Mathematical modeling.
The rhythmic profile of AANAT mRNA has been described with Michaelis-Menten kinetics. This approach was pioneered by Goodwin (19) and has since been used extensively for the quantitative study of gene expression (18). The actual concentrations of proteins that shape the AANAT mRNA time course are fit to periodic functions that have explicit time dependence. These functions were used to solve the kinetic equation of AANAT mRNA. The following function fits the concentration changes of pCREB and an average of hnRNP R, hnRNP Q, and hnRNP L, designated RQL.
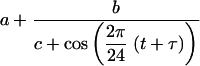 |
The least-squares parameters for pCREB are a = −0.83, b = 3.8, c = 1.6, and τ = 5.0, where τ may be converted to the period T by the relationship T = 12 − τ, and t represents zeitgeber time. For RQL, individual curves were fit for each trans-acting factor and averaged; for hnRNP R, a = 0.75, b = 0.17, c = 1.2, and τ = 4.8; for hnRNP L, a = 1.1, b = 2.2, c = 1.4, and τ = 2.2; and for hnRNP Q, a = 0.76, b = 0.14, c = 1.1, and τ = 5.5. The ICER time course was constructed with the same function, based on a previous observation (39). The parameters are a = 0.2, b = 2.1, c = 1.6, and τ = 2.0. The dynamic time course of AANAT mRNA is described with the following kinetic equation (34, 48):
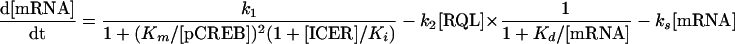 |
The first term on the right side (synthesis rate) is the Michaelis-Menten equation for competitive inhibition, whereby Km and Ki represent the half-maximal concentrations of substrate pCREB and the competitive inhibitor ICER, respectively. The square in this term accounts for the fact that pCREB forms a dimer before the initiation of transcription (33). The second term represents RQL-mediated mRNA degradation. The rate is proportional to the RQL concentration. AANAT mRNA undergoes a ∼100-fold increase, while the RQL increase remains at most 4-fold. Kd is the concentration at which half-maximal velocity is attained. The last term describes spontaneous decay due to RNA instability, whereby the decay time constant (1/ks) is calculated from the observed half-life of AANAT mRNA without 3′UTR (data not shown). The kinetic equation was iteratively solved with Mathematica software (Wolfram Research, Champaign, Ill.) by using the Levenberg-Marquardt algorithm until least-squares parameters were calculated: k1 = 1,809, k2 = 554, kS = 0.049, Km = 5.65, Ki = 3, and Kd = 110. Since the algorithm seeks only local minima, several initial values were tried until the smallest squares of errors were found.
RESULTS
AANAT 3′UTR regulates mRNA stability.
We analyzed the RNA sequences of the 578-nt 3′UTR (3′N578) of rat AANAT mRNA. Interestingly, two consensus polyadenylation [poly(A)] signals, located at 152 and 559 nt from the start site of 3′UTR, were analyzed. To determine the poly(A) signal functional in AANAT expression, Northern blot analysis was performed by using duplicate blots hybridized to two different probes, specifically, the AANAT coding region and the region encompassing 408 nt between the first and second poly(A) signal. Similar autoradiography results were obtained with both blots during the circadian cycle (data not shown). Our results confirmed that rat AANAT 3′UTR is 578 bases long.
Species-to-species variations in the AANAT mRNA profile (15) are mirrored in their 3′UTR characteristics (Fig. 1B). The UTRs of species with significant day-night mRNA fluctuations (such as mouse, hamster, and chicken) are similar in length and highly homologous to rat 3′UTR (3, 17, 43). In species with high basal levels of mRNA that fluctuate only marginally (as in ungulates and primates), UTRs are shorter than in rats but are highly homologous to the 5′ upstream sequences around the first putative poly(A) signal (6, 7, 8). This finding indicates that differences among the 3′UTRs may account for the remarkable variations in the AANAT mRNA profiles among species. To confirm this possibility, reporter (pcNAT) comprising the rat AANAT coding region was linked to either rat 3′UTR (pcNAT-3′N578) or bovine 3′UTR (pcNAT-b3′UTR) and transfected into CHO-K1 cells, which were subsequently examined for responsiveness to actinomycin D. Following transcription inhibition, cells transfected with pcNAT-3′N578 displayed a significant decrease in the AANAT mRNA level compared to those transfected with a construct comprising only the coding region without 3′UTR (pcNAT). In cells transfected with pcNAT-b3′UTR, no significant changes in the reporter level were observed, either with or without actinomycin D treatment. The reporter activities correlated well with the mRNA levels (Fig. 1C). These results suggest that the degradation of AANAT mRNA is directed by species-specific 3′UTR. To monitor the clearance rate of rat AANAT 3′UTR-containing reporter NAT-3′N578 mRNA, the decay curves were plotted over a 4-h actinomycin D treatment period (Fig. 1D, panel iii). The reporter mRNA without rat AANAT 3′UTR was stable with no significant degradation 4 h after actinomycin D (t1/2, over 4 h). In contrast, the NAT-3′N578 mRNA was rapidly degraded (t1/2 of ∼224 min). These results strongly suggest that rat AANAT 3′UTR is a determinant of mRNA stability. Accordingly, we hypothesized that the circadian fluctuation of rat AANAT mRNA is regulated by its mRNA degradation.
Determination of cis-acting elements in rat AANAT 3′UTR.
To determine which domains of rat AANAT 3′UTR function in mediating mRNA degradation, various chimeric reporters containing truncated derivatives of 3′UTR were generated (Fig. 2A). Cells transfected with both pcNAT-3′N578 and pcNAT-3′N336 displayed a significant decrease in AANAT activity compared to those transfected with pcNAT. In contrast, cells transfected with pcNAT-3′n223 highly homologous to bovine 3′UTR exhibited reporter activity similar to those transfected with pcNAT (Fig. 2B). Our results suggest that 336 nt from the 3′ end of the rat AANAT 3′UTR (3′N336) are required for rat AANAT mRNA destabilization.
To characterize the specific regions of rat AANAT 3′UTR that interact with RNA-binding proteins, deletion mutants of 3′N578 were generated to map the minimal boundaries (Fig. 2A). Truncated derivatives of 3′N578 were tested for their ability to bind cytoplasmic and nuclear CHO-K1 lysates by using UV cross-linking assays. The rat AANAT 3′UTR (3′N578) formed four major RNA-protein complexes with apparent molecular masses of 50, 68, 82, and 120 kDa in cytoplasmic lysates and four major complexes with apparent molecular masses of 61, 68, 82, and 120 kDa in nuclear extracts (Fig. 2C). To determine the specific interactions between 3′N578 and cellular proteins, competition analyses using UV cross-linking were performed with 5- or 20-fold molar excess of unlabeled 3′N578. Binding of all cytoplasmic and nuclear lysates to labeled 3′N578 was competed out by the unlabeled form, suggesting that interactions between cell lysates and 3′N578 is specific (data not shown). Since a similar binding pattern was observed in lysates of pineal glands (data not shown), CHO-K1 cells were used in further characterization studies, including in vitro binding assays and transfection analyses. Among all the proteins bound to 3′N578, 61-kDa protein (p61), p68, and p82 strongly formed complexes with 3′N336. In contrast, 3′n223 homologous to bovine AANAT 3′UTR bound only weakly to p61, p68, and p82 (Fig. 2C). Despite the possibility that other factors are involved in AANAT mRNA degradation, our data suggest that 3′N336-specific proteins p82, p68, and p61 play a key role in the process.
Identification of rat AANAT 3′UTR-binding proteins.
To identify the specific proteins that bind rat AANAT 3′UTR, cytoplasmic extracts prepared from rat pineal glands were subjected to biotin-streptavidin RNA-affinity purification. Proteins specific for rat AANAT 3′UTR p82, p68, and p61 were determined by competition analysis (Fig. 3A). MALDI-TOF mass spectrometry was employed to identify the three proteins. Specifically, p82 was identified as hnRNP R (accession number NP_005817), p68 was identified as hnRNP Q (accession number AKK59705), and p61 was identified as an isoform of 61-kDa hnRNP L (accession number NM_001533).
FIG. 3.


Identification of rat AANAT 3′UTR-binding proteins. (A) AANAT 3′UTR-binding proteins were purified from cytoplasmic extracts (Cyto) of pineal glands, fractionated by SDS-PAGE, and silver stained. Proteins identified by MALDI-TOF mass spectrometry are indicated on the right. (B) Confirmation of identified proteins. (i) Identified proteins were confirmed by immunoblot analysis using antibodies against hnRNP R, hnRNP Q, and hnRNP L. Cytoplasmic extract was loaded onto the Input lane. (ii) Nuclear extracts labeled by UV cross-linking with 32P-labeled 3′N336 were subjected to immunoprecipitation with antibodies against hnRNP Q or Flag as a control, separated by SDS-PAGE, and autoradiographed. (iii) Purified GST-hnRNP L was incubated with 32P-labeled riboprobes and subjected to UV cross-linking analysis. (iv) RNA pull-down experiments were performed with nuclear extracts of rat pineal glands and biotinylated RNAs corresponding to various fragments as described in the legend to Fig. 2A to show whether the identified trans-acting factors can interact with the AANAT 3′UTR. Immunoblot analysis was performed with specific antibodies as depicted on the right. (C) hnRNP R, hnRNP Q, and hnRNP L are associated with AANAT 3′UTR in vivo. CHO-K1 cells were cotransfected with pFlag-hnRNP R or pFlag-CMV2 as a mock transfection and reporter plasmids pcNAT or pcNAT-3′N578 and incubated for 18 h followed by fractionation. Cytoplasmic extracts with or without RNase A were incubated with the anti-Flag antibody and anti-HA antibody as a control. Antibody-antigen complexes were absorbed by protein G-agarose beads. Precipitated proteins were resolved by SDS-PAGE followed by immunoblot (IB) analyses with specific antibodies as depicted on the right. mRNAs associated with trans-acting factors in vivo were extracted and used as templates for RT-PCR analysis. The negative and positive controls (Control) for RT-PCR are shown on the left. The molecular sizes of cDNA bands are indicated on the left as kilobases.
The identities of the above three proteins were individually confirmed by immunoblot analyses. Among the three isoforms of hnRNP L with apparent molecular masses of 66, 61, and 56 kDa, the 61-kDa protein specifically interacted with AANAT 3′UTR (Fig. 3B, panel i). Interactions between the 3′UTR and hnRNP R and/or hnRNP Q were confirmed by immunoprecipitation of UV cross-linked proteins with 32P-labeled 3′N336, using anti-hnRNP Q antibody that recognizes both hnRNP R and hnRNP Q, as described previously (36, 37). Both hnRNP R and hnRNP Q were detected by the antibody, suggesting that they interact directly with the 3′UTR. No bands were detected when an anti-Flag monoclonal antibody was used as a negative control (Fig. 3B, panel ii). Direct interactions between hnRNP L and the 3′UTR were further confirmed by UV cross-linking with purified recombinant hnRNP L protein. Purified hnRNP L exhibited much stronger RNA-binding activity to labeled 3′N336 containing the hnRNP L-binding region than to probe 3′n223 containing no hnRNP L-binding domain (Fig. 3B, panel iii), consistent with deletion analysis (Fig. 2C). Moreover, direct binding of hnRNP R and hnRNP Q to the rat-specific AANAT 3′UTR (3′N336) was assessed by using purified GST-hnRNP R and GST-hnRNP Q and 32P-labeled riboprobes as described in the legend to Fig. 3C, panel iii. Both purified hnRNP R and hnRNP Q interacted strongly with 3′N336 but weakly with 3′n223. This result was correlated with deletion analysis (data not shown). The effect of the rat-specific AANAT 3′UTR (3′N336) on binding of trans-acting factors was confirmed by RNA pull-down assays. RNA pull-down experiments were performed by using nuclear extracts of rat pineal glands and biotinylated AANAT 3′UTRs. hnRNPs R, Q, and L were strongly associated with 3′N578 and 3′N336 containing rat-specific AANAT 3′UTR, whereas 3′n223 was weakly associated with the three proteins (Fig. 3B, panel iv).
To determine if the three proteins interact with rat AANAT 3′UTR in vivo, coimmunoprecipitation experiments were performed using cytoplasmic extracts from CHO-K1 cells cotransfected with pFlag-hnRNP R and pcNAT-3′N578. The immunoprecipitate was then analyzed for the existence of other proteins by immunoblotting. The results showed that all three proteins are coprecipitated by anti-Flag antibody and not by anti-HA antibody. Notably, these interactions were sensitive to RNase treatment, suggesting that coprecipitation of these proteins by anti-Flag antibody is due to their association with target mRNA and not due to direct protein-protein interactions. RT-PCR with AANAT 3′UTR-specific primers and cDNA sequencing revealed that rat AANAT 3′UTR was directly associated with the proteins in vivo (Fig. 3C). Taken together, these results confirm the identities of p61, p68, and p82 as hnRNP L, hnRNP Q, and hnRNP R, respectively, and indicate that the three proteins are directly associated with rat-specific AANAT 3′UTR in vivo.
Functional analyses of AANAT 3′UTR-binding proteins.
To test the function of hnRNP R, hnRNP Q, and hnRNP L as 3′UTR-regulatory molecules in AANAT mRNA degradation, we employed overexpression and knockdown approaches. Ectopic expression of the corresponding proteins in the cytoplasm was confirmed by immunoblotting with antibodies against Flag (Fig. 4A, panel i), Xpress (Fig. 4A, panel ii), and enhanced green fluorescent protein (EGFP) (Fig. 4A, panels iii and iv). Endogenous GAPDH, 18S rRNA, and mRNA from pcNAT were unaffected by the three regulatory proteins. Degradation of AANAT 3′UTR-containing mRNA (NAT-3′N578) was accelerated in the presence of hnRNP R, hnRNP Q, and hnRNP L in a concentration-dependent manner, but not in the presence of hnRNP C1. The level of reporter mRNA in each group was correlated with its enzymatic activity (Fig. 4A).
FIG. 4.



Overexpression of hnRNP R, hnRNP Q, and hnRNP L promotes destabilization of AANAT 3′UTR-containing mRNA. (A) CHO-K1 cells were transiently transfected with 10 μg of pFlag-CMV2 (i), pcDNA3.1/His C (ii), or pEGFP-C1 (iii and iv) as a mock transfection; 5 or 10 μg of pFlag-hnRNP R (i), pchnRNP L (ii), pEGFP-hnRNP Q (iii), or pEGFP-hnRNP C1 (iv); and 3.3 μg of hnRNP R plus 3.3 μg of hnRNP L plus 3.3 μg of hnRNP Q (R/Q/L) (i to iv). The cells were incubated for 18 h, secondarily cotransfected with 0.3 μg of control plasmid and 2 μg of either pcNAT (open bars) or pcNAT-3′N578 (closed bars), and incubated for a further 6 h before harvesting. Values, shown as the means ± SEM from duplicate experiments, are the AANAT activities in pcNAT-3′N578- or pcNAT-transfected populations relative to those measured in mock-cotransfected pcNAT-3′N578 or pcNAT, to which a value of 100% was assigned. The ectopic expression of the corresponding proteins in the cytoplasm was confirmed by immunoblotting with antibodies against Flag (i), Xpress (ii), EGFP (iii and iv), and GAPDH as a loading control. mRNA levels of reporters were determined as described in the legend to Fig. 1C. (B) hnRNP C1 as a mock transfection (i), hnRNP R (ii), hnRNP Q (iii), and hnRNP L (iv) were overexpressed as in Fig. 4A, incubated for 18 h, and then secondarily cotransfected with 0.3 μg of control plasmid and 2 μg of pcNAT-3′N578. Six hours after transfection, total RNA was isolated from cells at the indicated times after the addition of actinomycin D (Act. D) and was subjected to Northern blot analysis as described in the legend to Fig. 1D. (v) Signals were quantitated with a PhosphorImager and normalized to 18S rRNA signals; shown are means ± SEM. For each transfection condition, the AANAT/18S rRNA ratio at time zero was adjusted to 100%.
To monitor the clearance rate of NAT-3′N578 mRNA following overexpression of hnRNP R, hnRNP Q, and hnRNP L, the decay curves of reporter mRNAs were plotted over a 4-h actinomycin D treatment period, and half-lives were measured. Ectopic expression of the three proteins individually enhanced the degradation rate of NAT-3′N578 mRNA about twofold, while overexpression of hnRNP C1 as a negative control had no effect (t1/2 of ∼210 min) (Fig. 4B).
Transfection with siRNAs targeted against hnRNP R and hnRNP L mRNA efficiently reduced their mRNA and protein levels but not those of hnRNP Q, GAPDH, actin, and 18S rRNA (Fig. 5A and B). Knockdown of hnRNP R and hnRNP L stabilized NAT-3′N578 mRNA more than twofold but did not stabilize NAT mRNA, compared to cells transfected with mock siRNA (Fig. 5B). These results strongly suggest that hnRNP R, hnRNP Q, and hnRNP L play a key role in AANAT 3′UTR-mediated mRNA degradation.
FIG. 5.

Knockdown of trans-acting factors stabilizes AANAT 3′UTR-containing mRNA. (A and B) HEK-293T cells were cotransfected with siRNAs (mock, hnR283, hnL1066, and hnR283/hnL1066 siRNA) and with reporter plasmids pcNAT and pcNAT-3′N578. (A) The level and specificity of hnRNP R and hnRNP L depletion at 36 h after transfection was assessed by Northern and immunoblot analyses. (B) AANAT levels were determined by immunoblot (IB) analysis using antibodies against AANAT and GAPDH. Activities and mRNA levels of AANAT in pcNAT-3′N578- or pcNAT-transfected cells were determined as described in the legend to Fig. 4A.
Rhythmic expression of trans-acting factors and mathematical modeling for AANAT mRNA oscillation.
As reported previously (4, 42), the rat AANAT mRNA level reaches a maximum at approximately 5 h (ZT17) after the onset of darkness, to about 100 times that observed at late afternoon, and it declines to undetectable levels between ZT01 and ZT05 (Fig. 6A, panel i).
FIG. 6.


Rhythmic expression of trans-acting factors and mathematical modeling for AANAT mRNA oscillation. (A) (i) Characterization of the rhythmic expression of hnRNP R, hnRNP Q, and hnRNP L in rat pineal glands. Cell lysates extracted from rat pineal glands isolated at different time points as indicated were immunoblotted (IB) with antibodies against pCREB, hnRNP Q, hnRNP R, hnRNP L, AANAT, and 14-3-3 or GAPDH as loading controls. The rat AANAT mRNA levels were characterized over a 24-h period by Northern blot analysis (Northern). The open and closed bars indicate when lights were on and off, respectively. (ii and iii) Kinetics of pCREB, hnRNP R, hnRNP L, and hnRNP Q during the circadian cycle. The level of each factor was fitted with a minimal modification of the cosine function with a 24-h period. The plot fits concentration changes of [pCREB] (ii) and of an average of hnRNP R, hnRNP L, and hnRNP Q, called [RQL] (iii). Curves based on experimentally obtained data for 24 h are plotted twice. (B) Numerical fitting of circadian oscillation in rat AANAT mRNA using the functions pCREB, ICER, and RQL. (C) In silico analysis of the effect of RQL level on the AANAT mRNA rhythmicity. (i) The mRNA levels were simulated by assuming relative basal RQL levels of 1/4 (dotted line), 1 (normal level; thick line), and 5 (thin line), while the other parameters remained normal. The peak point of control data ([RQL] = 1) is marked by the intersection with the vertical dashed line. The effect of the RQL level on peak amplitude (open circles) (ii) and phase shift of peak time (closed circles) (iii) of rat AANAT mRNA is shown. The mRNA levels were simulated by using a differential equation of AANAT mRNA kinetics by removing rhythmicity of RQL level (iv) and by abolishing ICER (v) while other parameters remained normal. Thin and dotted lines represent normal and simulated AANAT mRNA levels, respectively.
We analyzed the expression modes of trans-acting factors to AANAT 3′UTR hnRNP R, hnRNP Q, and hnRNP L and the transcription factor pCREB, as well as their target AANAT, using the pineal glands employed for the Northern blot analyses. All of the factors displayed relatively high basal levels at all times, which peaked during the night. hnRNP R and hnRNP Q levels reached a maximum at ZT19 (approximately two times higher than that at ZT05). Maximal expression of hnRNP L was observed at ZT21 (more than sevenfold higher than that at ZT05). Levels of pCREB reached a maximum at ZT19, at over five times that at ZT05. The observed fluctuations in the AANAT protein level according to the circadian cycle correlated well with a previous report (16). In contrast, 14-3-3 and GAPDH levels remained unchanged (Fig. 6A, panel i). The results suggest that circadian oscillation of hnRNP R, hnRNP Q, and hnRNP L plays a role in dynamic AANAT mRNA degradation.
A mathematical model showing the collective effect of regulatory proteins in shaping rhythmic mRNA profile was constructed to clarify the role played by 3′UTR-mediated mRNA degradation. Previous qualitative models have explained AANAT mRNA oscillations in terms of an autoregulatory transcriptional feedback loop (14, 29). Here, we postulate that dynamic mRNA degradation mediated by 3′UTR and the corresponding trans-acting factors is essential for rhythm formation, and we construct a mathematical model based on this assumption. Thus, our kinetic equation for the dynamics of AANAT mRNA has one additional term accounting for active mRNA degradation. The mRNA oscillation was described with Michaelis-Menten kinetics assuming substrate saturation, since the increment in AANAT mRNA is more than 100-fold, but it is only about 4- to 5-fold in trans-acting factors. The kinetic equation was solved by using the periodic functions of the following regulatory proteins: the transcriptional activator pCREB, the repressor ICER (39), and three mRNA decay-mediating proteins, hnRNP R, hnRNP Q, and hnRNP L, which averaged together into a single function, RQL, for simplification (see Materials and Methods and Fig. 6A, panels ii and iii). Using this kinetic equation, we drew a model of the AANAT mRNA profile. The curve of rat AANAT mRNA is asymmetric, with a half-life of about 2.9 h. The skewed shape of the curve is due to the time lag between RQL and pCREB binding (Fig. 6B).
To confirm the accuracy of the model in describing the oscillations of AANAT mRNA, we created a profile of AANAT mRNA in an in silico ICER null mutation, and compared it with a previously described profile for ICER knockout mice (13). The in silico result was consistent with the experimental data showing slightly increased peak amplitude and no phase shift of AANAT mRNA profile (Fig. 6C, panel v), confirming that our kinetic equation is sufficient to explain the dynamics of AANAT mRNA during circadian rhythms.
Our mathematical model predicts the effects of RQL on the AANAT mRNA profile (Fig. 6C, panel i). A gradual increase in the RQL amount (from twofold to fivefold) results in a dramatic decrease in peak amplitude and advancement of peak time of AANAT mRNA. Decreasing RQL from one-half to one-eighth results in the opposite effect (Fig. 6C, panels ii and iii). Upon abolishing rhythmicity of the RQL level, the profile of AANAT mRNA displays increased peak amplitude and delayed peak time (Fig. 6C, panel iv). Upon elimination of the RQL function from the model, the mRNA profile displays saturation, which manifests as a small identical peak-to-peak oscillation riding on a dramatic accumulation of basal level (data not shown). These analyses suggest that the AANAT mRNA decay mediated by rhythmic trans-acting factors is essential for its circadian oscillation.
AANAT mRNA degradation regulates its circadian rhythmicity.
In formulating the theoretical model, we assumed that hnRNP R, hnRNP Q, and hnRNP L are key molecules in AANAT mRNA degradation. The model predicts that the rise and fall of three trans-acting factors modulates the AANAT mRNA profile. To test the above hypotheses experimentally, we employed knockdown by RNA interference in rat pinealocytes. Transfection with siRNAs against hnRNP R, hnRNP Q, and hnRNP L mRNA efficiently attenuated their protein levels, but not that of GAPDH (Fig. 7A, panel i). Treatment of rat pinealocytes with norepinephrine or β1-adrenergic agonist ISO elicits activation of cAMP signaling and melatonin biosynthesis in a mode similar to that seen with nocturnal pinealocytes (39). Time course experiments of AANAT mRNA in mock-transfected cells showed that AANAT mRNA level is undetectable in the absence of ISO treatment but reaches a maximum after 9 h of ISO stimulation and declines gradually thereafter. The AANAT mRNA level of siRNA-transfected pinealocytes reached a maximum at approximately 12 h after ISO treatment, with an approximately twofold increase compared to the mock-transfected cells (Fig. 7A, panel ii). Knockdown of hnRNP R, hnRNP Q, and hnRNP L elicited an increase of peak amplitude and a delay of peak time in the AANAT mRNA profile (Fig. 7B). These results are consistent with the in silico analyses shown in Fig. 6C, panel i.
FIG. 7.

AANAT mRNA degradation modulates its circadian oscillation. (A) (i) Pinealocytes were transfected with mock and hnR283/hnL1066/hnQ1089 siRNAs. The levels and specificities of hnRNP R, hnRNP Q, and hnRNP L depletion at 36 h after transfection were assessed by immunoblot analyses. (ii) Total RNA was isolated from pinealocytes at the indicated times (in hours) after the addition of ISO and was subjected to Northern blot analysis using 32P-labeled DNA probes against the AANAT coding region and ribosomal protein large subunit 32 (RPL32). (B) Kinetic analysis of AANAT mRNA in mock-transfected (open squares) or siRNA-transfected (closed squares) pinealocytes. The AANAT mRNA level was quantified with a densitometer and normalized against RPL32 mRNA from three independent experiments; shown are means ± SEM. The peak amount of AANAT mRNA (ISO, 9 h) in mock-transfected cells was defined as 100%. The peak time of AANAT mRNA in mock-transfected cells is marked by the vertical dashed line.
DISCUSSION
Circadian oscillation of AANAT mRNA is promoted by transcriptional and posttranscriptional regulations. The regulation of mRNA stability is an important mechanism that enables fine control of gene expression. Here, we show that the AANAT mRNA profile is regulated by a combination of cis-acting elements at species-specific 3′UTRs and their oscillatory partners hnRNP R, hnRNP Q, and hnRNP L, suggesting that dynamic AANAT mRNA degradation functions as a key process in the maintenance of its circadian rhythmicity.
Potent effect of AANAT 3′UTR on mRNA degradation.
Comparisons of AANAT 3′UTR sequences among species and transfection analysis suggest that the remarkable species-to-species differences in AANAT mRNA profiles during circadian rhythm are determined by their 3′UTRs. In ungulates and primates, AANAT mRNA levels remain constant during the 24-h period, but proteins show dramatic circadian rhythmicity consistent with those in rodents and birds. Transcriptional and posttranscriptional controls may be involved in the regulation of the latter (8, 15), whereas the constant level observed for ungulates and primates may be due to their stable mRNAs without hnRNP R-, hnRNP Q-, and hnRNP L-binding regions in the 3′UTR that mediate mRNA degradation. Therefore, these analyses suggest that the AANAT 3′UTRs control mRNA degradation and determine the species-specific profile of AANAT mRNA. In the course of this investigation, we examined other 3′UTRs of clock-controlled transcripts for mRNA degradation. Notably, the 3′UTRs of oscillatory clock mPer and mouse Cryptochrome mRNAs dramatically induced degradation as well (unpublished data). These results suggest that 3′UTR-mediated mRNA decay in clock-controlled oscillations acts as a general executor in the formation of circadian rhythmicity.
Cytoplasmic function for hnRNP R, hnRNP Q, and hnRNP L.
Our data indicate that the hnRNP R, hnRNP Q, and hnRNP L function as specific cytoplasmic regulatory proteins for AANAT mRNA degradation. This finding is interesting, considering the subcellular localization and the broad range of RNA-binding specificity that are general characteristics of hnRNPs (9). Our studies have revealed that hnRNPs R and L shuttle between the nucleus and the cytoplasm (data not shown), suggesting their multifunctional regulatory roles in both compartments (50). hnRNP R has been cloned (23, 44), but its biological role has not yet been elucidated. The hnRNP L protein regulates gene expression by mediating mRNA export, viral internal translation, and mRNA stability (9, 21, 46, 47, 50). hnRNP Q is a cytoplasmic counterpart of hnRNP R that binds preferentially to poly(A) RNA, both in vitro (36) and as a component of a protein complex that leads to translationally coupled c-fos mRNA degradation (20), mediates pre-mRNA splicing (37), and augments internal ribosomal entry site-mediated translation of hepatitis C virus mRNA (27).
Rhythmic expressions of hnRNP R, hnRNP Q, and hnRNP L in pineal glands are robust. This fact enables us to speculate that these proteins play an important role in framing the AANAT mRNA time course. Previous studies have shown that ICER is the most important down-regulator of the AANAT mRNA level (4, 14, 29). However, ICER knockout fails to disrupt the circadian rhythm of AANAT mRNA (13). Simulation studies reveal that abolishing hnRNP R, hnRNP Q, and hnRNP L is more potent than ICER knockout at the disruption of circadian rhythm. This finding is consistent with our observation that deleting the AANAT 3′UTR prolongs the half-life of its transcript to roughly 14 h (data not shown). Moreover, knockdown of hnRNP R, hnRNP Q, and hnRNP L in rat pinealocytes using RNA interference resulted in changes of phase and peak amplitude of AANAT mRNA level. This experimental evidence is consistent with in silico analyses. Despite the possibility that the expression and/or activity of other molecules for mRNA degradation can also follow circadian rhythm, our evidence here suggests that hnRNP R, hnRNP Q, and hnRNP L play a key role in the degradation of rat AANAT mRNA.
AANAT mRNA degradation during circadian rhythm.
The expression of hnRNP R, hnRNP Q, and hnRNP L is maintained at a significant basal level at all times except at nighttime, when they are increased. This increase is correlated to the expression mode of their substrate AANAT mRNA. Several lines of evidence support the possibility that basal or enhanced degradation of AANAT mRNA occurs via its 3′UTR during circadian rhythm. First, the level of reporter mRNA containing rat AANAT 3′UTR was much lower than that of the mRNA without the 3′UTR in the absence of transcriptional inhibitor, implying that 3′UTR-containing mRNA basally undergoes degradation even in the middle of active transcription, most likely due to endogenous levels of hnRNP R, hnRNP Q, and hnRNP L. Second, the level of the three trans-acting factors exists basally at all times and increases at ZT21 to a level more than threefold higher than that in late afternoon. It can be seen through our mathematical model of AANAT mRNA level that the accumulation of AANAT mRNA between ZT12 and ZT17 may still be possible during highly efficient transcription, because the amount of de novo synthesized AANAT mRNA overwhelms that of the degraded mRNA despite ongoing basal mRNA degradation. After repression of AANAT between ZT17 and ZT21, simultaneously enhanced levels of three trans-acting factors at ZT21 mediate a dramatic decline from the peak level of AANAT mRNA at ZT 21 to the undetectable level between ZT01 and ZT05. Since diurnal levels of pCREB and three trans-acting factors exist basally, the undetectable level of AANAT mRNA can be explained not only by transcriptional down-regulation, but also by mRNA degradation during daytime (ZT00 to approximately ZT12). Moreover, removing rhythmicity at the level of trans-acting factors induces increased peak amplitude and phase shift of AANAT mRNA. In conclusion, these analyses suggest that rhythmic AANAT mRNA decay mediated by expression of hnRNPs R, Q, and L at particular times give a distinct shape to the overall AANAT mRNA profile during circadian rhythm.
Acknowledgments
We thank G. Dreyfuss, D. C. Klein, K. Mikoshiba, C. M. Craft, and C. R. Astell for kindly providing the anti-hnRNP L antibody (4D11), anti-AANAT antibody, anti-SYNCRIP-N antibody, bovine AANAT cDNA 10-1B, and plasmid pRSETC9-15, respectively. We additionally thank J. B. Park for useful discussions on MALDI mass spectrometry, researcher J. E. Lee for performing the MALDI analysis, and Y. Kang, E. M. Hur, and K. S. Yi for critical reading of the manuscript.
This work was supported by the Brain Neurobiology Research Program, the National Research Laboratory Program, the Systems Bio-Dynamics Research Center of the Ministry of Science and Technology (MOST), and the Brain Korea 21 program of the Ministry of Education.
REFERENCES
- 1.Andreason, G. L., and G. A. Evans. 1988. Introduction and expression of DNA molecules in eukaryotic cells by electroporation. BioTechniques 6:650-660. [PubMed] [Google Scholar]
- 2.Baler, R., S. Covington, and D. C. Klein. 1997. The rat arylalkylamine N-acetyltransferase gene promoter: cAMP activation via a cAMP-responsive element-CCAAT complex. J. Biol. Chem. 272:6979-6985. [DOI] [PubMed] [Google Scholar]
- 3.Bernard, M., P. M. Iuvone, V. M. Cassone, P. H. Roseboom, S. L. Coon, and D. C. Klein. 1997. Avian melatonin synthesis: photic and circadian regulation of serotonin N-acetyltransferase mRNA in the chicken pineal gland and retina. J. Neurochem. 68:213-224. [DOI] [PubMed] [Google Scholar]
- 4.Borjigin, J., M. M. Wang, and S. H. Snyder. 1995. Diurnal variation in mRNA encoding serotonin N-acetyltransferase in pineal gland. Nature 378:783-785. [DOI] [PubMed] [Google Scholar]
- 5.Chae, H. D., T. J. Park, Y. K. Lee, T. G. Lee, and K. T. Kim. 1999. Rapid and simple measurement of serotonin N-acetyltransferase activity by liquid biphasic diffusion assay. Neurochem. Int. 35:447-451. [DOI] [PubMed] [Google Scholar]
- 6.Coon, S. L., P. H. Roseboom, R. Baler, J. L. Weller, M. A. Namboodiri, E. V. Koonin, and D. C. Klein. 1995. Pineal serotonin N-acetyltransferase: expression cloning and molecular analysis. Science 270:1681-1683. [DOI] [PubMed] [Google Scholar]
- 7.Coon, S. L., E. del Olmo, W. S. Young III, and D. C. Klein. 2002. Melatonin synthesis enzymes in Macaca mulatta: focus on arylalkylamine N-acetyltransferase (EC 2.3.1.87). J. Clin. Endocrinol. Metab. 87:4699-4706. [DOI] [PubMed] [Google Scholar]
- 8.Craft, C. M., J. Murage, B. Brown, and X. Zhan-Poe. 1999. Bovine arylalkylamine N-acetyltransferase activity correlated with mRNA expression in pineal and retina. Brain Res. Mol. Brain Res. 65:44-51. [DOI] [PubMed] [Google Scholar]
- 9.Dreyfuss, G., V. N. Kim, and N. Kataoka. 2002. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3:195-205. [DOI] [PubMed] [Google Scholar]
- 10.Dunlap, J. C. 1999. Molecular bases for circadian clocks. Cell 96:271-290. [DOI] [PubMed] [Google Scholar]
- 11.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2000. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 12.Forger, D. B., and C. S. Peskin. 2003. A detailed predictive model of the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 100:14806-14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foulkes, N. S., J. Borjigin, S. H. Snyder, and P. Sassone-Corsi. 1996. Transcriptional control of circadian hormone synthesis via the CREM feedback loop. Proc. Natl. Acad. Sci. USA 93:14140-14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foulkes, N. S., J. Borjigin, S. H. Snyder, and P. Sassone-Corsi. 1997. Rhythmic transcription: the molecular basis of circadian melatonin synthesis. Trends Neurosci. 20:487-492. [DOI] [PubMed] [Google Scholar]
- 15.Ganguly, S., S. L. Coon, and D. C. Klein. 2002. Control of melatonin synthesis in the mammalian pineal gland: the critical role of serotonin acetylation. Cell Tissue Res. 309:127-137. [DOI] [PubMed] [Google Scholar]
- 16.Gastel, J. A., P. H. Roseboom, P. A. Rinaldi, J. L. Weller, and D. C. Klein. 1998. Melatonin production: proteasomal proteolysis in serotonin N-acetyltransferase regulation. Science 279:1358-1360. [DOI] [PubMed] [Google Scholar]
- 17.Gauer, F., V. J. Poierl, M. L. Garidou, V. Simonneaux, and P. Pevet. 1999. Molecular cloning of the arylalkylamine N-acetyltransferase and daily variations of its mRNA expression in the Syrian hamster pineal gland. Brain Res. Mol. Brain Res. 71:87-95. [DOI] [PubMed] [Google Scholar]
- 18.Goldbeter, A. 2002. Computational approaches to cellular rhythms. Nature 420:238-245. [DOI] [PubMed] [Google Scholar]
- 19.Goodwin, B. C. 1965. Oscillatory behavior in enzymatic control processes. Adv. Enzyme Regul. 3:425-438. [DOI] [PubMed] [Google Scholar]
- 20.Grosset, C., C. Y. Chen, N. Xu, N. Sonenberg, H. Jacquemin-Sablon, and A. B. Shyu. 2000. A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell 103:29-40. [DOI] [PubMed] [Google Scholar]
- 21.Hahm, B., O. H. Cho, J. E. Kim, Y. K. Kim, J. H. Kim, Y. L. Oh, and S. K. Jang. 1998. Polypyrimidine tract-binding protein interacts with hnRNP L. FEBS Lett. 425:401-406. [DOI] [PubMed] [Google Scholar]
- 22.Hardin, P. E., J. C. Hall, and M. Rosbash. 1990. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343:536-540. [DOI] [PubMed] [Google Scholar]
- 23.Hassfeld, W., E. K. Chan, D. A. Mathison, D. Portman, G. Dreyfuss, G. Steiner, and E. M. Tan. 1998. Molecular definition of heterogeneous nuclear ribonucleoprotein R (hnRNP R) using autoimmune antibody: immunological relationship with hnRNP P. Nucleic Acids Res. 26:439-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holcik, M., and R. G. Korneluk. 2000. Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Mol. Cell. Biol. 20:4648-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim, J. H., B. Hahm, Y. K. Kim, M. Choi, and S. K. Jang. 2000. Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J. Mol. Biol. 298:395-405. [DOI] [PubMed] [Google Scholar]
- 26.Kim, J. H., K. Y. Paek, K. Choi, T.-D. Kim, B. Hahm, K.-T. Kim, and S. K. Jang. 2003. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol. Cell. Biol. 23:708-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, J. H., K. Y. Paek, S. H. Ha, S. Cho, K. Choi, C. S. Kim, S. H. Ryu, and S. K. Jang. 2004. A cellular RNA-binding protein enhances internal ribosomal entry site-dependent translation through an interaction downstream of the hepatitis C virus polyprotein initiation codon. Mol. Cell. Biol. 24:7878-7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein, D. C., and J. L. Weller. 1970. Indole metabolism in the pineal gland: a circadian rhythm in N-acetyltransferase. Science 169:1093-1095. [DOI] [PubMed] [Google Scholar]
- 29.Klein, D. C., S. Ganguly, S. L. Coon, Q. Shi, P. Gaildrat, F. Morin, J. L. Weller, T. Obsil, A. Hickman, and F. Dyda. 2003. 14-3-3 proteins in pineal photoneuroendocrine transduction: how many roles? J. Neuroendocrinol. 15:370-377. [DOI] [PubMed] [Google Scholar]
- 30.Lee, C., J. P. Etchegaray, F. R. A. Cagampang, A. S. I. Loudon, and S. M. Reppert. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107:855-867. [DOI] [PubMed] [Google Scholar]
- 31.Leloup, J. C., and A. Goldbeter. 2003. Toward a detailed computational model for the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 100:7051-7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowrey, P. L., K. Shimomura, M. P. Antoch, S. Yamazaki, P. D. Zemenides, M. R. Ralph, M. Menaker, and J. S. Takahashi. 2000. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288:483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayr, B., and M. Montminy. 2001. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2:599-609. [DOI] [PubMed] [Google Scholar]
- 34.McAdams, H. H., and A. Arkin. 1998. Simulation of prokaryotic genetic circuits. Annu. Rev. Biophys. Biomol. Struct. 27:199-224. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell, P., and D. Tollervey. 2000. mRNA stability in eukaryotes. Curr. Opin. Genet. Dev. 10:193-198. [DOI] [PubMed] [Google Scholar]
- 36.Mizutani, A., M. Fukuda, K. Ibata, Y. Shiraishi, and K. Mikoshiba. 2000. SYNCRIP, a cytoplasmic counterpart of heterogeneous nuclear ribonucleoprotein R, interacts with ubiquitous synaptotagmin isoforms. J. Biol. Chem. 275:9823-9831. [DOI] [PubMed] [Google Scholar]
- 37.Mourelatos, Z., L. Abel, J. Yong, N. Kataoka, and G. Dreyfuss. 2001. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J. 20:5443-5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park, J. B., J. H. Kim, Y. Kim, S. H. Ha, J. S. Yoo, G. Du, M. A. Frohman, P. G. Suh, and S. H. Ryu. 2000. Cardiac phospholipase D2 localizes to sarcolemmal membranes and is inhibited by α-actinin in an ADP-ribosylation factor-reversible manner. J. Biol. Chem. 275:21295-21301. [DOI] [PubMed] [Google Scholar]
- 39.Pfeffer, M., E. Maronde, C. A. Molina, H. W. Korf, and J. H. Stehle. 1999. Inducible cyclic AMP early repressor protein in rat pinealocytes: a highly sensitive natural reporter for regulated gene transcription. Mol. Pharmacol. 56:279-289. [DOI] [PubMed] [Google Scholar]
- 40.Reppert, S. M., and D. R. Weaver. 2001. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol. 63:647-676. [DOI] [PubMed] [Google Scholar]
- 41.Reppert, S. M., and D. R. Weaver. 2002. Coordination of circadian timing in mammals. Nature 418:935-941. [DOI] [PubMed] [Google Scholar]
- 42.Roseboom, P. H., S. L. Coon, R. Baler, S. K. McCune, J. L. Weller, and D. C. Klein. 1996. Melatonin synthesis: analysis of the more than 150-fold nocturnal increase in serotonin N-acetyltransferase messenger ribonucleic acid in the rat pineal gland. Endocrinology 137:3033-3044. [DOI] [PubMed] [Google Scholar]
- 43.Roseboom, P. H., M. A. Namboodiri, D. B. Zimonjic, N. C. Popescu, I. R. Rodriguez, J. A. Gastel, and D. C. Klein. 1998. Natural melatonin “knockdown” in C57BL/6J mice: rare mechanism truncates serotonin N-acetyltransferase. Brain Res. Mol. Brain Res. 63:189-197. [DOI] [PubMed] [Google Scholar]
- 44.Rossoll, W., A. K. Kroening, U. M. Ohndorf, C. Steegborn, S. Jablonka, and M. Sendtner. 2002. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum. Mol. Genet. 11:93-105. [DOI] [PubMed] [Google Scholar]
- 45.Sathyanarayanan, S., X. Zheng, R. Xiao, and A. Sehgal. 2004. Posttranslational regulation of Drosophila PERIOD protein by protein phosphatase 2A. Cell 116:603-615. [DOI] [PubMed] [Google Scholar]
- 46.Shearman, L. P., S. Sriram, D. R. Weaver, E. S. Maywood, I. Chaves, B. Zheng, K. Kume, C. C. Lee, G. T. van der Horst, M. H. Hastings, and S. M. Reppert. 2000. Interacting molecular loops in the mammalian circadian clock. Science 288:1013-1019. [DOI] [PubMed] [Google Scholar]
- 47.Shih, S. C., and K. P. Claffey. 1999. Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J. Biol. Chem. 274:1359-1365. [DOI] [PubMed] [Google Scholar]
- 48.Smolen, P., D. A. Baxter, and J. H. Byrne. 2000. Mathematical modeling of gene networks. Neuron 26:567-580. [DOI] [PubMed] [Google Scholar]
- 49.So, W. V., and M. Rosbash. 1997. Post-transcriptional regulation contributes to Drosophila clock gene mRNA cycling. EMBO J. 16:7146-7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilkinson, M. F., and A. B. Shyu. 2001. Multifunctional regulatory proteins that control gene expression in both the nucleus and the cytoplasm. Bioessays 23:775-787. [DOI] [PubMed] [Google Scholar]
- 51.Wilsbacher, L. D., S. Yamazaki, E. D. Herzog, E. J. Song, L. A. Radcliffe, M. Abe, G. Block, E. Spitznagel, M. Menaker, and J. S. Takahashi. 2002. Photic and circadian expression of luciferase in mPeriod1-luc transgenic mice in vivo. Proc. Natl. Acad. Sci. USA 99:489-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilusz, C. J., M. Wormington, and S. W. Peltz. 2001. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2:237-246. [DOI] [PubMed] [Google Scholar]
- 53.Young, M. W., and S. A. Kay. 2001. Time zones: a comparative genetics of circadian clocks. Nat. Rev. Genet. 2:702-715. [DOI] [PubMed] [Google Scholar]