

Abstract
PINCH1, an adaptor protein composed of five LIM domains, mediates protein-protein interactions and functions as a component of the integrin-integrin-linked kinase (ILK) complex. The integrin-ILK signaling complex plays a pivotal role in cell motility, proliferation, and survival during embryonic development of many animal species. To elucidate the physiological function of PINCH1 in mouse embryonic development, we have deleted the mouse PINCH1 gene by homologous recombination. Mice heterozygous for PINCH1 are viable and indistinguishable from wild-type littermates. However, no viable homozygous offspring were observed from PINCH1+/− intercrosses. Histological analysis of homozygous mutant embryos revealed that they had a disorganized egg cylinder by E5.5, which degenerated by E6.5. Furthermore, E5.5 PINCH1−/− embryos exhibited decreased cell proliferation and excessive cell death. We have also generated and analyzed mice in which PINCH1 has been specifically deleted in ventricular cardiomyocytes. These mice exhibit no basal phenotype, with respect to mouse survival, cardiac histology, or cardiac function as measured by echocardiography. Altogether, these data indicate that PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes.
Integrins mediate cell-cell and cell-matrix interactions and are required for embryonic development (13, 26, 41). Integrins, which contain a cytoplasmic and extracellular domain, link the cytoplasm to the extracellular matrix and play an important role in cytoskeletal organization and regulation of gene expression, which affects cell adhesion, migration, proliferation, differentiation, and survival (3, 16, 17, 23, 26, 27). Integrins transduce their signals by associating with adaptor proteins that interconnect the integrins to the cytoskeleton, cytoplasmic kinases, and transmembrane growth factor receptors. Proteins associating either directly or indirectly with the cytoplasmic tails of integrins modulates the ligand binding capacity of integrins, altering integrin adhesive function by an inside-out signaling mechanism (25, 30).
An essential binding partner of the integrin cytoplasmic domain is the integrin-linked kinase (ILK). ILK is an ankyrin (ANK) repeat protein containing a serine/threonine kinase domain, which interacts with the cytoplasmic tail of β1, β2, and β3 integrins (21). ILK couples integrins and growth factors to downstream signaling pathways, leading to the regulation of diverse processes, such as cell cycle progression, survival, division, and changes in morphology and spreading (9, 10, 49). Genetic studies of Drosophila melanogaster, Caenorhabditis elegans, and mice have provided direct evidence that ILK can function as an important mediator of the integrin-dependent signaling pathway (31, 38, 50). ILK is composed of four ANK repeats (21), a pleckstrin homology (PH)-like domain (11), and a C-terminal Ser/Thr kinase domain (21). The first ANK domain has been shown to bind to the two highly related LIM-domain-only proteins PINCH1 and -2 (4, 44, 51).
PINCH, which is composed of five tandemly arrayed LIM domains, has been suggested to play a role in integrin-ILK function (36, 48). LIM domains are double zinc finger structures that serve as protein binding interfaces (33, 39). It has been suggested that PINCH functions as a molecular scaffold that supports the assembly of a multiprotein complex at sites of integrin enrichment (48). Biochemical studies of human PINCH1 have identified ILK as a binding partner for the first LIM domain of PINCH (44) and the SH2-SH3 adaptor protein NCK2 as a partner for the fourth LIM domain (45). Both the colocalization of PINCH1 with integrins and its capacity to bind ILK and NCK2 provided the first hints that PINCH1 might play a role in recruitment of regulatory factors to integrin-rich sites and, therefore, contribute to the integrin signaling cascade (47, 49).
Genetic studies of C. elegans and Drosophila point to an essential role of PINCH as an adaptor protein in mediating integrin-ILK-dependent signaling (7, 24). The deletion of UNC-97, an orthologue of PINCH1, in C. elegans results in an embryonic-lethal phenotype called PAT (paralyzed and arrested elongation at the twofold stage) (24) resembling that of β-integrin/PAT-3 (15) or ILK/PAT-4 (31). In Drosophila muscle, PINCH displays a completely overlapping expression pattern with ILK and βPS integrin, prominently enriched at the muscle attachment sites (7). Flies deficient in PINCH1 (named stck in Drosophila) exhibit muscle detachment, similar to the phenotypes of ILK and PS-integrin (7, 28, 32, 50).
In this study, we report the phenotype of PINCH1 germ line-deficient mice and mice in which PINCH1 has been specifically deleted in ventricular cardiomyocytes. We show that PINCH1 is already detectable in blastocysts at approximately E3.5. PINCH1−/− embryos at E5.5 exhibit a disorganized egg cylinder, with decreased cell proliferation and excessive cell death, thus pointing to an important role of PINCH1 in the formation of egg cylinders. We also show that mice in which PINCH1 is specifically deleted in cardiomyocytes exhibit no basal phenotype with regard to mouse survival, cardiac histology, or cardiac function.
MATERIALS AND METHODS
Gene targeting.
A genomic PINCH1 fragment was isolated from a 129SVJ library (Stratagene) and used to construct the PINCH1 targeting vector by standard techniques as shown in Fig. 1A. Briefly, one loxP site was inserted into the second intron and a second loxP site along with the neo cassette flanked by frt sites was inserted into the third intron of the PINCH1 gene. The targeting vector was linearized with SalI and electroporated into R1 embryonic stem (ES) cells. Three hundred ninety-four G418-resistant ES clones were screened for homologous recombination by Southern blot analysis as described below.
FIG. 1.

Targeted generation of PINCH1-null mice. (A) Targeting strategy. A restriction map of the relevant genomic region of PINCH1 is shown on the top, the targeting construct is shown in the center, and the mutated locus after recombination is shown at the bottom. The targeting construct was generated by introducing one loxP site into the second intron and another loxP site and the neo cassette flanked by frt sites into the third intron of the PINCH1 gene. B, BamHI; S, SalI; X, XbaI. neo represents the neomycin resistance gene, arrowheads represent loxP sites, and the long boxes represent frt sites. (B and C) Detection of wild-type and targeted alleles by Southern blot analysis. DNAs from electroporated ES cells (B) and from the progeny of germ line-transmitted chimera mice (C) were digested with BamHI and analyzed by Southern blot analysis with the probe as shown in panel A. The 11- and 7.5-kb bands represent wild-type and mutant alleles, respectively.
Southern blot analysis.
DNA was extracted from G418-resistant ES cell clones as previously described (34). ES cell DNA was digested with BamHI, electrophoresed on a 0.8% (wt/vol) agarose gel, and subsequently blotted onto nitrocellulose. A 480-bp fragment, corresponding to the 5′ end of the right arm of the target vector, was generated by PCR with mouse genomic DNA and specific PINCH1 primers (forward primer, 5′-ATATGATAGGAGACACTATTCACG; reverse primer, 5′-AAGCTTCAGAAAGGACCTGT). The PCR product was subsequently radiolabeled using [32P]dATP by random priming (Invitrogen, San Diego, Calif.). DNA blots were hybridized with the radiolabeled probe and visualized by autoradiography. The wild-type allele is represented by a band of 11 kb, whereas a band of 7.5 kb represents the correctly targeted mutant allele.
Generation and genotyping of mice.
Two independent homologous recombinant clones were microinjected into blastocysts from C57BL/6J mice at the Transgenic Core Facility of the University of California, San Diego. Male chimeras were inbred with female Black Swiss mice to generate germ line-transmitted heterozygous mice with a neo cassette (PINCH1+/flox+neo). PINCH1+/flox+neo micewere crossed with protamine-Cre (Pro-Cre) mice (35), generating mice which were doubly heterozygous (Pro-Cre/PINCH1+/flox+neo). Cre expression in Pro-Cre mice is restricted to male germ cells undergoing spermatogenesis. Therefore, Pro-Cre/PINCH1+/flox+neo males were crossed to female breeders to generate germ line-heterozygous-null mutant offspring. Heterozygous mice were interbred to generate homozygous knockouts. Progeny from these crosses were genotyped by PCR with the wild-type-specific primers (forward primer, P1, CCCAGAAGGACTCTTTTATGAG; reverse primer, P2, CTTGGAGAAGAAGTACTCAGGT) and primers for the mutant allele (neo-specific primer, Pneo, AATGGGCTGACCGCTTCCTCGT; reverse primer, P3, CTTGGAGAAGAAGTACTCAGGT). To identify the genotype of the embryos at the peri-implantation stage, uterine deciduas around E5.5 were sectioned and stained with antibody specific for PINCH1 as described as below.
PINCH1+/flox+neo mice were crossed with FLPase deleter mice (37), which delete DNA sequences flanked by two frt sites in all cell types. This resulted in the generation of mice with a floxed PINCH1 allele no longer containing the neo cassette (PINCH1+/flox). PINCH1+/flox mice were subsequently intercrossed and crossed with MLC2v-Cre mice (5, 6) to generate mice which were homozygous floxed (PINCH1flox/flox) and doubly heterozygous for PINCH1 floxed allele and MLC2v-Cre allele (PINCH1+/flox+neo Cre). Interbreeding between both PINCH1flox/flox mice and the PINCH1+/flox+neo Cre mice was used to generate mice in which PINCH1 is specifically deleted in ventricular cardiomyocytes. To genotype PINCH1 floxed and MLC2v-Cre-positive alleles, PINCH1 primers (forward primer, P4, CCCAGAAGGACTCTTTTATGAG; reverse primer, P5, CTTGGAGAAGAAGTACTCAGGT) and Cre primers (forward primer, Pcre1, GTTCGCAAGAACCTGATGGACA; reverse primer, Pcre2, CTAGAGCCTGTTTTGCACGTTC) were used.
Reverse transcription-PCR (RT-PCR) analysis.
Total RNA was isolated from ES cells and embryos at E6.5 and E7.5 by using Trizol reagent (GIBCO BRL). First-strand cDNA synthesis was performed with the random primer and Superscript kit (Invitrogen). The cDNA was utilized as a PCR template to perform PCR by standard protocols. Specific primers for PINCH1 (forward, TCAAGAATGCTGGCAGACAC; reverse, ACACCAGGCCTTGTTGAGAG) were utilized.
In situ hybridization.
Whole-mount in situ hybridization was carried out with digoxigenin-labeled RNA probes as previously described (46). A murine RNA probe spanning the 1,000-bp fragment of PINCH1 cDNA was utilized.
Dissection and histological analysis of embryos.
Timed matings were conducted with interbreeding between PINCH1+/− mice. Females with copulation plugs were considered to be at embryonic development day 0.5 (E0.5) of gestation. Pregnant females were sacrificed at different time points of gestation, and the embryos were dissected from maternal tissue, examined, photographed, and genotyped by PCR. For histological preparations, embryos in decidua were fixed in 4% paraformaldehyde overnight at 4°C and destined for paraffin embedding. Serial sagittal sections were cut at 5 μm from paraffin blocks and stained with hematoxylin and eosin.
Immunostaining assays.
Five-micrometer sections were treated for 1 h with 5% bovine serum albumin in phosphate-buffered saline and subsequently incubated overnight at 4°C in a humidified chamber with a polyclonal antibody to PINCH1 (provided by A. Rearden, University of California, San Diego). After being washed with 0.25% Triton X-100 in phosphate-buffered saline, the sections were incubated with fluorescently labeled secondary antibodies. The specimens embedded in Vectashield mounting medium (Vector Laboratories) were analyzed under the fluorescence microscope.
BrdU labeling of embryos.
Pregnant females at E5.5 were injected intraperitoneally with bromodeoxyuridine (BrdU; Amersham-Pharmacia, Little Chalfont, Buckinghamshire, United Kingdom) and sacrificed 2 h later. Deciduae were removed and fixed in 4% paraformaldehyde overnight at 4°C. Five-micrometer sections were denatured with 2 N HCl, trypsinized, and incubated with a mouse monoclonal antibody to BrdU (Sigma; B 2531). Detection was performed utilizing a peroxidase ABC kit (Vector Laboratories) and 3,3-diaminobenzidine. BrdU-labeled cells were counted from sections. For quantitative analyses, representative sections from ventral, mid-, and dorsal levels of each embryo were utilized to count the total number of cells and the number that were labeled with BrdU, to give a proliferation index.
Apoptosis assays.
Implantation sites (from E5.5 to E6.5) were collected for apoptosis studies. To detect apoptotic cells, the terminal deoxynucleotidyltransferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed according to the manufacturer's instructions (fluorescein in situ cell death detection kit; Boehringer Mannheim, Mannheim, Germany). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain (Vector Laboratories). In addition to TUNEL staining, DNA fragmentation was verified under UV illumination by using the DAPI counterstain.
Echocardiographic analysis.
Mice were anesthetized with isofluorane and subjected to echocardiography as previously described (43).
RESULTS
Targeted disruption of PINCH1 gene in the mouse.
To explore the physiological role of PINCH1 in the mouse, we have generated a conditional allele of the PINCH1 gene by homologous recombination in ES cells. We and others have successfully generated multiple floxed alleles utilizing three loxP sites (tri-loxP) (5, 18). However, a drawback to the tri-loxP approach is that it can require two in vitro manipulations of ES cells, one for the original selection and one to excise the selectable neo gene. Removal of the neo gene is desirable, as its presence can disrupt expression of the targeted gene, preventing the floxed allele from being phenotypically neutral. However, additional in vitro manipulation of ES cells jeopardizes the likelihood of germ line transmission. To avoid this problem, we and others have capitalized on the recent development of another site-specific recombination system which has been used in transgenic mice, the frt-site/FLPase recombinase system, which does not overlap with the loxP/cre-mediated recombination. A combination of these two systems can be utilized to generate a floxed allele containing the neo gene flanked by frt sites. In this manner, the neo gene can be removed from the floxed allele in vivo by breeding a mouse containing the floxed allele to a mouse which expresses FLPase in all cells (37).
The targeting vector for PINCH1 was designed as shown in Fig. 1A, in which one loxP site was introduced into the second intron and a second loxP site as well as the neo cassette flanked by frt sites was introduced into the third intron of the PINCH1 gene. This targeting vector was linearized with SalI and introduced into R1 ES cells via electroporation. G418-resistant ES clones were screened for homologous recombination by Southern blot analysis. Of 394 ES cell clones analyzed (Fig. 1B), two had undergone homologous recombination. These two ES cell clones were independently injected into blastocysts and gave rise to chimera mice that were then used to breed mice which would be germ line-transmitting heterozygous mice with the neo cassette (PINCH1+/flox+neo). The PINCH1+/flox+neo mice were then bred to homozygosity for the conditional allele (PINCH1flox+neo/flox+neo). Although the PINCH1flox+neo/flox+neo mice can survive and seem normal, to avoid potential effects of the neo cassette on PINCH1 gene expression, we crossed PINCH1+/flox+neo mice with FLPase deleter mice (37), which deletes DNA sequences flanked by two frt sites in all cell types. This resulted in the generation of mice with a floxed PINCH1 allele no longer containing the neo cassette (PINCH1+/flox). PINCH1+/flox mice were subsequently intercrossed to generate mice which were homozygous floxed (PINCH1flox/flox). The PINCH1flox/flox mice were then crossed with MLC2v-Cre mice to generate mice in which PINCH1 is specifically deleted in ventricular cardiomyocytes.
PINCH1+/flox+neo mice were crossed with Pro-Cre mice (35), generating mice which were doubly heterozygous (Pro-Cre PINCH1+/flox+neo). Cre expression in Pro-Cre mice is restricted to male germ cells undergoing spermatogenesis. Therefore Pro-Cre PINCH1+/flox+neo males were crossed to female breeders to generate germ line-heterozygous-null mutant offspring.
The PINCH1 null mutation results in early embryonic lethality.
Heterozygous PINCH1 mutants survive and have no apparent phenotype. Heterozygous PINCH1 mice were interbred in an attempt to generate homozygous knockouts. However, the genotypic analysis of mice generated from interbred heterozygous PINCH1 mice failed to detect any homozygous null offspring in over 200 mice that were analyzed. Of the progeny from heterozygote intercrosses, 30% (n = 60) were wild type and 70% (n = 140) were heterozygous for the PINCH1 gene (Table 1). Approximately 50 homozygous knockout offspring would have been expected, if homozygous mice were to be viable. These results indicated that loss of PINCH1 function is associated with embryonic lethality.
TABLE 1.
Phenotypes and genotypes of offspring from PINCH+/− intercrosses
| Stage | No. of offspring
|
||||
|---|---|---|---|---|---|
| Phenotype
|
Genotype
|
||||
| Normal | Abnormal-resorbed | +/+ | +/− | −/− | |
| 1 mo old | 200 | 60 | 140 | 0 | |
| E9.5 | 36 | 9a | 12 | 24 | 0 |
| E8.5 | 32 | 8a | 10 | 22 | 0 |
| E7.5 | 20 | 6a | 10 | 20 | 0 |
| E6.5 | 20 | 4 | 7 | 13 | 4 |
| E5.5 | 20b | 5b | |||
No DNA can be isolated from the resorbed embryos.
Embryo tissue could not be dissected freely from maternal tissue.
To determine the time point at which PINCH1−/− embryos died, we set up timed matings and analyzed embryos by a PCR-based protocol. No homozygous mutant embryos were found between E7.5 and E9.5. However, at E7.5 and E8.5, severely developmentally retarded and resorbed embryos were found, with the percentage of resorption being as high as 25%. At E6.5, 25% of all embryos appeared small and misshapen at a gross morphological level (Fig. 2A). We genotyped four abnormal embryos at E6.5 by PCR and found that they were indeed PINCH1−/− mutants (Fig. 2B).
FIG. 2.
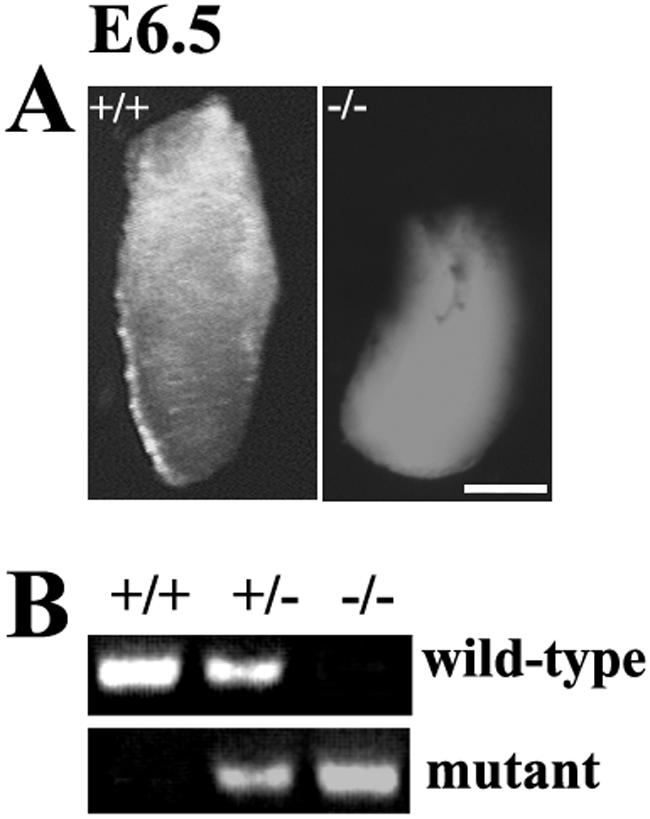
Phenotype of PINCH1−/− embryos at the gross morphological level. (A) At the gross morphological level, a PINCH1−/− embryo of E6.5 appears smaller than its wild-type littermate and degenerated. Bar, 50 μm. (B) The genotypes of embryos were identified by PCR.
To investigate the time course of lethality, we examined serial sections of embryos in utero between E5.5 and E6.5. To genotype embryos at the peri-implantation stage, uterine decidua at proximately E5.5 was immunostained with antibodies specific for PINCH1. As shown in Fig. 3A and C, both wild-type and PINCH1+/− embryos at approximately E5.5 and E6.5 developed into egg cylinders of normal size with a layer of visceral endoderm, which exhibited organized structures with a proximal-distal polarity. In addition, a proamniotic cavity was present in the embryonic region in control embryos at E5.5 (Fig. 3A). In contrast, PINCH1−/− embryos at E5.5 that failed to positively stain for PINCH1 antibody appeared significantly smaller and showed abnormal structures, being oval in shape with disorganized inner and outer layers (Fig. 3B). In addition, the proamniotic cavity was missing in most PINCH1−/− embryos. At E6.5, about 25% of uterine decidua appeared degenerated and partially resorbed, with no organized cellular structure (Fig. 3D). Extensive resorption as high as 25% was observed in uterine decidua at E7.5 (data not shown). These results indicate that PINCH1 homozygous mutant mice die shortly after implantation.
FIG. 3.
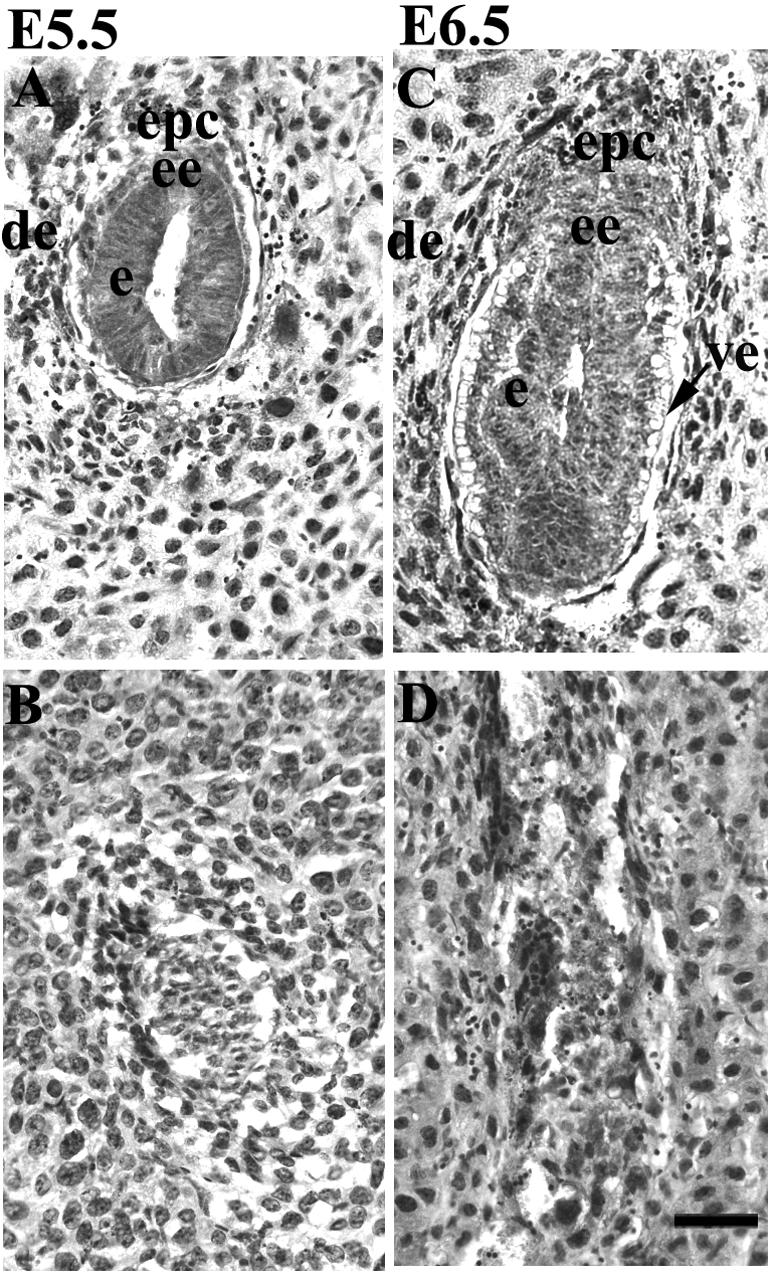
Histological analysis of embryos generated from PINCH1 heterozygous intercrosses by hematoxylin and eosin staining. (A and B) Sagittal sections of E5.5 control (A) and mutant (B) embryos. (C and D) Sagittal sections of E6.5 control (C) and mutant (D) embryos. de, decidua; epc, ectoplacental cone; ee, extraembryonic ectoderm; e, ectoderm; ve, visceral endoderm. Bar, 50 μm.
PINCH1 is expressed in embryos during early developmental stages.
Early embryonic lethality resulting from the deletion of the PINCH1 gene led us to determine the temporal and spatial patterns of PINCH1 expression during peri-implantation stages. To determine the expression profile of PINCH1, we performed both RT-PCR analysis of blastocysts and embryos at E6.5 and E7.5 and whole-mount in situ hybridization of embryos at E6.5 and E7.5. RT-PCR analysis showed that expression of PINCH1 is already detectable in blastocysts around E3.5 (Fig. 4A). Whole-mount in situ hybridization demonstrated that PINCH1 is highly expressed in the neuroectoderm at E6.5 and E7.5, with more diffuse staining in the remainder of the embryo (Fig. 4B). These data imply the importance of PINCH1 for early mouse development.
FIG. 4.
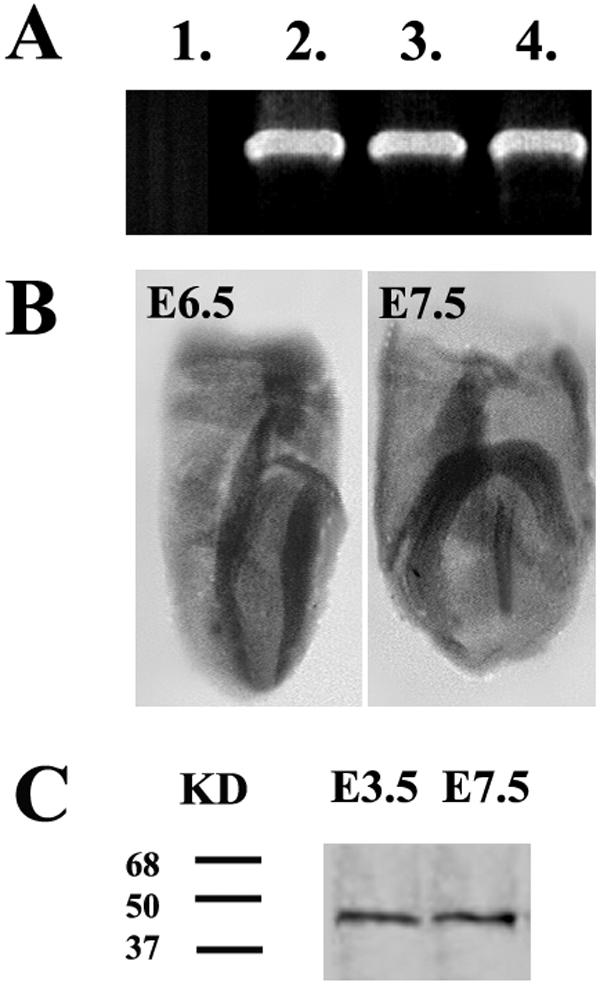
PINCH1 expression in mouse embryos at early stages. (A) RT-PCR shows that PINCH1 expression is already detectable in blastocysts at E3.5. Total RNA was isolated from blastocysts of E3.5 and embryos at E6.5 and E7.5 and analyzed by RT-PCR with specific primers for PINCH1. Lane 1, negative control; lane 2, E3.5; lane 3, E6.5; lane 4, E7.5. (B) Whole-mount in situ hybridization with PINCH1 probe demonstrates that PINCH1 is highly expressed in the neuroectoderm at E6.5 and E7.5, with more diffuse staining in the remainder of the embryo. (C) Western blot analysis shows that PINCH1 is detectable in E3.5 blastocysts. Protein was isolated from E3.5 blastocysts and E7.5 embryos and analyzed with a specific antibody to PINCH1.
Proliferation and apoptosis in PINCH1−/− embryos.
At E5.5, PINCH1−/− embryos appear smaller than control littermates. Accordingly, we examined the rate of proliferation using BrdU incorporation and the extent of apoptosis by performing TUNEL assays in PINCH1 mutant and control littermates at E5.5. To genotype embryos, uterine deciduae were immunostained with antibodies specific for PINCH1 (Fig. 5A and B).
FIG. 5.

BrdU labeling and TUNEL analysis of PINCH1 mutants and control littermates at E5.5. (A and B) Immunostaining with PINCH1 antibody identified control (A) and PINCH1 mutant (B) embryos. (C and D) BrdU labeling showed proliferation indices in control littermates (C) and PINCH1 mutants (D), showing lower proliferative activity in PINCH1 mutants than in control littermates. (E to H) TUNEL analysis performed on sections from control littermates (E) and PINCH1 mutants (F); DAPI staining was used to visualize nuclei of control littermates (G) and mutants (H). Bar, 50 μm.
BrdU labeling showed that the rate of proliferation in PINCH1−/− embryos was significantly reduced in comparison to that of control littermates (Fig. 5C and D). Approximately 85% of cells in visceral endoderm and epiblast of control embryos exhibited BrdU-positive staining, whereas less than 5% of cells in mutants were labeled. As shown by TUNEL staining in Fig. 5E, few apoptotic cells were observed in control embryos where the lumen would be forming. In contrast, excessive TUNEL-positive cells were scattered throughout PINCH1−/− embryos at E5.5 (Fig. 5F). Therefore, it is likely that the growth deficit of PINCH1 homozygous embryos resulted from both decreased cell proliferation and increased cell death.
Mice in which PINCH1 is specifically deleted in ventricular cardiomyocytes exhibit no basal phenotype with regard to mouse survival, cardiac histology, or cardiac function.
It has been shown that mice which have β1 integrin specifically deleted in ventricular cardiomyocytes display myocardial fibrosis and depressed left ventricular contractility and relaxation (40). In addition, a recent study demonstrated that PINCH1 forms a functional complex with β4 thymosin and ILK. This complex was suggested to play an essential role in promoting cardiomyocyte migration and survival (2). To determine the functional role of PINCH1 in cardiomyocytes, we generated mice in which PINCH1 has been specifically deleted in ventricular cardiomyocytes by utilizing MLC2v-Cre mice. These mice have been shown to mediate DNA recombination specifically in ventricular cardiomyocytes starting from embryonic day 8.5 (5). Pups in which PINCH1 has been specifically deleted in ventricular cardiomyocytes (PINCH1flox/flox Cre+/−) were born normally, were externally indistinguishable from littermates of other genotypes, were recovered at Mendelian frequency (for MLC2v wild-type mice, 19 offspring were PINCHflox/flox and 18 were PINCHflox/+; for MLC2v-Cre/+ mice, 18 offspring were PINCHflox/flox and 23 were PINCHflox/+), and grew to adulthood without signs of cardiac malfunction.
DNA analysis of adult animals (1 to 2 months old) confirmed PINCH1 gene excision only in ventricular tissue derived from PINCH1flox/flox Cre+/− mice, with an efficiency of Cre-mediated recombination of approximately 85% (data not shown), which is in agreement with other studies with MLC2v-Cre-mediated excision (5, 12, 20, 22, 40).
To determine whether the PINCH1flox/flox Cre+/− mice display any histological changes, we performed histological studies. Paraffin sections (10 μm) of heart from PINCH1flox/flox Cre+/− and PINCH1flox/flox littermate controls (6 months old) were stained with hematoxylin and eosin. All specimens appeared normal, exhibiting no sign of hypertrophy, myocardial disarray, infarction, necrosis, fibrosis, calcification, or fat infiltration (data not shown). Cardiac function was evaluated noninvasively by echocardiography at 4 to 6 months of age. There were no significant differences in all of the echocardiographic parameters assessed between wild-type and mutant groups (Table 2).
TABLE 2.
Echocardiographic measurements under basal conditionsa
| Measurement | PINCH1flox/flox (n = 7) | PINCH1flox/flox Cre+/− (n = 9) |
|---|---|---|
| BW (g) | 28.1 ± 4.38 | 30.0 ± 5.27 |
| HR (bpm) | 511.6 ± 46.32 | 471.8 ± 101.7 |
| LVEDD (mm) | 3.48 ± 0.38 | 3.71 ± 0.75 |
| LVESD (mm) | 2.00 ± 0.42 | 2.25 ± 0.65 |
| PWth (mm) | 0.63 ± 0.05 | 0.60 ± 0.03 |
| IVSth (mm) | 0.64 ± 0.05 | 0.61 ± 0.04 |
| LV%FS | 43.05 ± 6.31 | 40.01 ± 6.76 |
| Vcf (circ/s) | 8.98 ± 1.70 | 8.42 ± 2.14 |
All data are presented as means ± standard errors. BW, body weight; HR, heart rate; LVEDD, end-diastolic left ventricular dimension; LVESD, end-systolic left ventricular dimension; PWth, left ventricular posterior wall thickness; IVSth, interventricular wall thickness; LV%FS, left ventricular percent fractional shortening; Vcf, velocity of circumferential fiber shortening. bpm, beats per minute; circ, circumference.
DISCUSSION
In the present study, we have investigated the functional role of PINCH1 in mouse embryonic development by generating PINCH1-deficient mice and analyzing PINCH1 expression during early embryogenesis. PINCH1−/− mice failed to form organized egg cylinders or to cavitate and subsequently died at the peri-implantation period. These results demonstrate a pivotal role for PINCH1 in early mouse embryogenesis. PINCH1−/− embryos exhibited decreased cell proliferation and excessive cell death. However, whether these are the primary effects of lack of PINCH1 or secondary to the overall poor embryonic development remains to be determined.
Morphogenesis of peri-implantation mouse embryos involves extensive cell-cell and cell matrix interactions. During the peri-implantation period, the inner cell mass (ICM) of the blastocyst develops into the primitive endoderm and the epiblast, which form the embryo proper (42). The primitive endoderm forms the surface of the ICM of blastocyst and deposits a basement membrane. The basement membrane is required for adjacent ICM cells to polarize and establish the columnar epiblast (8). The importance of integrin-ILK-mediated cell-cell and cell-matrix interactions during early embryonic development is highlighted by genetic studies in mouse models (1, 13, 29, 38, 41). In β1-integrin-deficient embryos, the primitive endoderm fails to produce laminin α1 and therefore no basement membrane is formed (1, 29). In mouse embryos lacking ILK, the primitive endoderm differentiates and produces a basement membrane but the epiblast fails to polarize or cavitate, and mutants die at the peri-implantation stage (38).
Biochemical studies have demonstrated that PINCH1 functions as an important mediator of integrin- and ILK-dependent signaling pathways (14, 19). Here, we show that PINCH1-null mutant mice, like β1-integrin- and ILK-null mutant mice, die at the peri-implantation stage, providing in vivo evidence that PINCH1 is a critical component of the integrin-ILK pathway in vertebrates.
Our data on PINCH1 germ line-knockout mice are consistent with genetic studies in C. elegans and Drosophila (7, 24). Deletion of PINCH in C. elegans results in an embryonic-lethal phenotype called PAT (24), resembling that of β-integrin/PAT-3 (15) or ILK/PAT-4 (31). PINCH-deficient flies exhibit muscle detachment, similar to the phenotypes of ILK and PS-integrin (7, 28, 32, 50). All these data suggest that the β1-integrin-ILK-PINCH complex is a functional complex that is highly conserved from invertebrates to mammals.
To our surprise, mice in which PINCH1 has been specifically deleted in ventricular cardiomyocytes exhibit no basal phenotype with regard to mouse survival, cardiac histology, or cardiac function as measured by echocardiography. Although this is a negative result, we think it is a very significant one, for reasons discussed below.
So far all data from Drosophila, C. elegans, mammalian cells, and our studies with mouse embryos have indicated that PINCH1 is indispensable for β1-integrin function (see discussion above). Mice which have β1-integrin specifically deleted in ventricular cardiomyocytes, with the use of the same MLC2v-Cre mouse that we used in our studies, display myocardial fibrosis, depressed left ventricular contractility and relaxation, and development of heart failure by 6 months of age (40). Our data demonstrate that PINCH1 is dispensable for β1-integrin function in ventricular cardiomyocytes.
Additionally, a recent study demonstrated that PINCH1 forms a functional complex with β4 thymosin and ILK. This complex was suggested to play an essential role in promoting cardiomyocyte migration and survival (2). Our data suggest that, if this complex does play an essential role in cardiomyocyte migration and survival, PINCH1 is certainly dispensable for the complex.
Two highly homologous proteins, PINCH1 and PINCH2, are encoded by two distinct genes (48) and have both been shown to interact with the ANK domain of ILK (48). Both proteins are widely expressed, which raised the possibility that they could be functionally redundant (48). Our data demonstrate that PINCH1 is essential for early murine embryonic development and that PINCH2 cannot compensate for the loss of PINCH1 during early embryonic development. However, it is possible that the lack of phenotype in mice in which PINCH1 is specifically deleted in ventricular cardiomyocytes is due to a redundant role of PINCH2 in cardiomyocytes.
Acknowledgments
We thank Ann Rearden for providing us PINCH1 antibody and Sylvia Evans, Marie-Louise Bang, and Farah Sheikh for critical reading of the manuscript.
This work was supported by a grant from NIH (J. Chen).
REFERENCES
- 1.Aumailley, M., M. Pesch, L. Tunggal, F. Gaill, and R. Fassler. 2000. Altered synthesis of laminin 1 and absence of basement membrane component deposition in β1 integrin-deficient embryoid bodies. J. Cell Sci. 113:259-268. [DOI] [PubMed] [Google Scholar]
- 2.Bock-Marquette, I., A. Saxena, M. D. White, J. M. Dimaio, and D. Srivastava. 2004. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 432:466-472. [DOI] [PubMed] [Google Scholar]
- 3.Brakebusch, C., D. Bouvard, F. Stanchi, T. Sakai, and R. Fassler. 2002. Integrins in invasive growth. J. Clin. Investig. 109:999-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braun, A., R. Bordoy, F. Stanchi, M. Moser, G. G. Kostka, E. Ehler, O. Brandau, and R. Fassler. 2003. PINCH2 is a new five LIM domain protein, homologous to PINCH and localized to focal adhesions. Exp. Cell Res. 284:239-250. [DOI] [PubMed] [Google Scholar]
- 5.Chen, J., S. W. Kubalak, and K. R. Chien. 1998. Ventricular muscle-restricted targeting of the RXRα gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125:1943-1949. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J., S. W. Kubalak, S. Minamisawa, R. L. Price, K. D. Becker, R. Hickey, J. Ross, Jr., and K. R. Chien. 1998. Selective requirement of myosin light chain 2v in embryonic heart function. J. Biol. Chem. 273:1252-1256. [DOI] [PubMed] [Google Scholar]
- 7.Clark, K. A., M. McGrail, and M. C. Beckerle. 2003. Analysis of PINCH function in Drosophila demonstrates its requirement in integrin-dependent cellular processes. Development 130:2611-2621. [DOI] [PubMed] [Google Scholar]
- 8.Coucouvanis, E., and G. R. Martin. 1995. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 83:279-287. [DOI] [PubMed] [Google Scholar]
- 9.Dedhar, S. 2000. Cell-substrate interactions and signaling through ILK. Curr. Opin. Cell Biol. 12:250-256. [DOI] [PubMed] [Google Scholar]
- 10.Dedhar, S. 1999. Integrins and signal transduction. Curr. Opin. Hematol. 6:37-43. [DOI] [PubMed] [Google Scholar]
- 11.Delcommenne, M., C. Tan, V. Gray, L. Rue, J. Woodgett, and S. Dedhar. 1998. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 95:11211-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding, J. H., X. Xu, D. Yang, P. H. Chu, N. D. Dalton, Z. Ye, J. M. Yeakley, H. Cheng, R. P. Xiao, J. Ross, J. Chen, and X. D. Fu. 2004. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J. 23:885-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fassler, R., and M. Meyer. 1995. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 9:1896-1908. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda, T., K. Chen, X. Shi, and C. Wu. 2003. PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J. Biol. Chem. 278:51324-51333. [DOI] [PubMed] [Google Scholar]
- 15.Gettner, S. N., C. Kenyon, and L. F. Reichardt. 1995. Characterization of beta pat-3 heterodimers, a family of essential integrin receptors in C. elegans. J. Cell Biol. 129:1127-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giancotti, F. G., and E. Ruoslahti. 1999. Integrin signaling. Science 285:1028-1032. [DOI] [PubMed] [Google Scholar]
- 17.Giancotti, F. G., and G. Tarone. 2003. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu. Rev. Cell Dev. Biol. 19:173-206. [DOI] [PubMed] [Google Scholar]
- 18.Gu, H., Y. R. Zou, and K. Rajewsky. 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73:1155-1164. [DOI] [PubMed] [Google Scholar]
- 19.Guo, L., and C. Wu. 2002. Regulation of fibronectin matrix deposition and cell proliferation by the PINCH-ILK-CH-ILKBP complex. FASEB J. 16:1298-1300. [DOI] [PubMed] [Google Scholar]
- 20.Gutstein, D. E., G. E. Morley, H. Tamaddon, D. Vaidya, M. D. Schneider, J. Chen, K. R. Chien, H. Stuhlmann, and G. I. Fishman. 2001. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88:333-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hannigan, G. E., C. Leung-Hagesteijn, L. Fitz-Gibbon, M. G. Coppolino, G. Radeva, J. Filmus, J. C. Bell, and S. Dedhar. 1996. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379:91-96. [DOI] [PubMed] [Google Scholar]
- 22.Hirota, H., J. Chen, U. A. Betz, K. Rajewsky, Y. Gu, J. Ross, Jr., W. Müller, and K. R. Chien. 1999. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97:189-198. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch, E., L. Barberis, M. Brancaccio, O. Azzolino, D. Xu, J. M. Kyriakis, L. Silengo, F. G. Giancotti, G. Tarone, R. Fassler, and F. Altruda. 2002. Defective Rac-mediated proliferation and survival after targeted mutation of the beta1 integrin cytodomain. J. Cell Biol. 157:481-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hobert, O., D. G. Moerman, K. A. Clark, M. C. Beckerle, and G. Ruvkun. 1999. A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. J. Cell Biol. 144:45-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes, P. E., and M. Pfaff. 1998. Integrin affinity modulation. Trends Cell Biol. 8:359-364. [DOI] [PubMed] [Google Scholar]
- 26.Hynes, R. O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell 110:673-687. [DOI] [PubMed] [Google Scholar]
- 27.Hynes, R. O., and A. D. Lander. 1992. Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons. Cell 68:303-322. [DOI] [PubMed] [Google Scholar]
- 28.Leptin, M., T. Bogaert, R. Lehmann, and M. Wilcox. 1989. The function of PS integrins during Drosophila embryogenesis. Cell 56:401-408. [DOI] [PubMed] [Google Scholar]
- 29.Li, S., D. Harrison, S. Carbonetto, R. Fassler, N. Smyth, D. Edgar, and P. D. Yurchenco. 2002. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J. Cell Biol. 157:1279-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liddington, R. C., and M. H. Ginsberg. 2002. Integrin activation takes shape. J. Cell Biol. 158:833-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackinnon, A. C., H. Qadota, K. R. Norman, D. G. Moerman, and B. D. Williams. 2002. C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12:787-797. [DOI] [PubMed] [Google Scholar]
- 32.MacKrell, A. J., B. Blumberg, S. R. Haynes, and J. H. Fessler. 1988. The lethal myospheroid gene of Drosophila encodes a membrane protein homologous to vertebrate integrin beta subunits. Proc. Natl. Acad. Sci. USA 85:2633-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelsen, J. W., K. L. Schmeichel, M. C. Beckerle, and D. R. Winge. 1993. The LIM motif defines a specific zinc-binding protein domain. Proc. Natl. Acad. Sci. USA 90:4404-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moens, C. B., B. R. Stanton, L. F. Parada, and J. Rossant. 1993. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development 119:485-499. [DOI] [PubMed] [Google Scholar]
- 35.O'Gorman, S., N. A. Dagenais, M. Qian, and Y. Marchuk. 1997. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA 94:14602-14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rearden, A. 1994. A new LIM protein containing an autoepitope homologous to “senescent cell antigen.” Biochem. Biophys. Res. Commun. 201:1124-1131. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez, C. I., F. Buchholz, J. Galloway, R. Sequerra, J. Kasper, R. Ayala, A. F. Stewart, and S. M. Dymecki. 2000. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25:139-140. [DOI] [PubMed] [Google Scholar]
- 38.Sakai, T., S. Li, D. Docheva, C. Grashoff, K. Sakai, G. Kostka, A. Braun, A. Pfeifer, P. D. Yurchenco, and R. Fassler. 2003. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 17:926-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmeichel, K. L., and M. C. Beckerle. 1994. The LIM domain is a modular protein-binding interface. Cell 79:211-219. [DOI] [PubMed] [Google Scholar]
- 40.Shai, S. Y., A. E. Harpf, C. J. Babbitt, M. C. Jordan, M. C. Fishbein, J. Chen, M. Omura, T. A. Leil, K. D. Becker, M. Jiang, D. J. Smith, S. R. Cherry, J. C. Loftus, and R. S. Ross. 2002. Cardiac myocyte-specific excision of the β1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 90:458-464. [DOI] [PubMed] [Google Scholar]
- 41.Stephens, L. E., A. E. Sutherland, I. V. Klimanskaya, A. Andrieux, J. Meneses, R. A. Pedersen, and C. H. Damsky. 1995. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 9:1883-1895. [DOI] [PubMed] [Google Scholar]
- 42.Tam, P. P., and R. S. Beddington. 1992. Establishment and organization of germ layers in the gastrulating mouse embryo. Ciba Found. Symp. 165:27-41. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka, N., N. Dalton, L. Mao, H. A. Rockman, K. L. Peterson, K. R. Gottshall, J. J. Hunter, K. R. Chien, and J. Ross, Jr. 1996. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation 94:1109-1117. [DOI] [PubMed] [Google Scholar]
- 44.Tu, Y., F. Li, S. Goicoechea, and C. Wu. 1999. The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol. 19:2425-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tu, Y., F. Li, and C. Wu. 1998. Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase-signaling pathways. Mol. Biol. Cell 9:3367-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkinson, D. G. 1992. In situ hybridization: a practical approach. Oxford University Press, New York, N.Y.
- 47.Wu, C. 1999. Integrin-linked kinase and PINCH: partners in regulation of cell-extracellular matrix interaction and signal transduction. J. Cell Sci. 112:4485-4489. [DOI] [PubMed] [Google Scholar]
- 48.Wu, C. 2004. The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim. Biophys. Acta 1692:55-62. [DOI] [PubMed] [Google Scholar]
- 49.Wu, C., and S. Dedhar. 2001. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 155:505-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zervas, C. G., S. L. Gregory, and N. H. Brown. 2001. Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J. Cell Biol. 152:1007-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang, Y., K. Chen, L. Guo, and C. Wu. 2002. Characterization of PINCH-2, a new focal adhesion protein that regulates the PINCH-1-ILK interaction, cell spreading, and migration. J. Biol. Chem. 277:38328-38338. [DOI] [PubMed] [Google Scholar]