

Summary
The lacZ gene of Escherichia coli encodes β-galactosidase (β-gal), a lactose metabolism enzyme of the lactose operon. Previous chemical modification or site-directed mutagenesis experiments have identified 21 amino acids that are essential for β-gal catalytic activity. We have assembled over 10,000 lacZ mutations from published studies that were collected using a positive selection assay to identify mutations in lacZ that disrupted β-gal function. We analyzed 6,465 independent lacZ mutations that resulted in 2,732 missense mutations that impaired β-gal function. Those mutations affected 492 of the 1,023 lacZ codons, including most of the 21 previously known residues critical for catalytic activity. Most missense mutations occurred near the catalytic site and in regions important for subunit tetramerization. Overall, our work provides a comprehensive and detailed map of the amino acid residues affecting the structure and catalytic activity of the β-gal enzyme.
Subject areas: Genetics, Molecular genetics
Graphical abstract
Highlights
-
•
Characterized 2,732 missense mutations in lacZ gene that impaired β-gal function
-
•
Missense mutations affected 492 (48%) of the 1,023 lacZ codons
-
•
Enhanced understanding of structural features affecting catalytic activity of β-gal
Genetics; Molecular genetics
Introduction
The lactose (lac) operon and the lacZ gene of Escherichia coli have a long history in the field of molecular biology, dating back to the discovery of gene regulation in the pioneering studies of Jacob and Monod that examined the production of β-galactosidase (β-gal) from lacZ in the presence of inducing substrates.1 The lacZ gene has played an important role in recombinant DNA technology through colorimetric selection of recombinant clones via the α-complementation phenomenon.2,3,4,5 Today, β-gal is extensively used in the food industry and in a range of other industries with broader implications across medical and biotechnological applications.6 The structure and function of β-gal has been studied extensively as a model for understanding glycosidase mechanisms.7
The E. coli lacZ coding sequence comprises 3,075 nucleotides (GenBank: V00296.1) generating a β-gal subunit consisting of 1,023 amino acid residues. The protein is divided into an α-peptide chain, four domain regions, and an inter-domain region between domains 3 and 4. The domains consist of the sugar binding (49-219; PF02837), β-galactosidase (221-334; PF00703), TIM barrel (336-630; PF02836), and the β-galactosidase small chain (749-1022; PF02929) domains. The β-gal enzyme is a homotetramer, and tetramerization is dependent on the complementation region (first 50 residues).8,9 Structural studies using site-directed mutagenesis have identified and characterized 21 amino acid residues with functional significance for the β-gal protein, most of which are present in the TIM Barrel Domain (14/21; Table 1). Substitutions of those amino acid residues can impair β-gal function through different means, including alterations to substrate binding, alteration to the catalytic site, transition state destabilization, and active site loop destabilization.10 However, many key structural features important for β-gal function are yet unidentified because site-directed mutagenesis is low-throughput and rarely investigates the role of mutations beyond the active site.
Table 1.
Characteristics of the 21 previously identified codons demonstrated to be important for β-gal function through site-directed mutagenesis or protein structure analyses
| Domain | Nucleotide range | Codon | NGS missense mutationsa | Sanger missense mutationsb | Total missense | Sequence context | Reported mutation | Reported mutation an SNV? (Y or N)c | Brief Description of (proposed) mutational effects | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 304–306 | Asn102 | 2 | 0 | 2 | accAACgtg | Asn → Ala | N | Alters binding affinity to substrate | Wheatley et al.10 |
| 1 | 601–603 | Asp201 | 28 | 8 | 36 | cagGATatg | Asp → Glu, Asn, Phe | Y | Alters binding affinity to substrate | Xu et al.69 |
| 3 | 1069–1071 | His357 | 4 | 1 | 5 | cgtCACgag | His → Asp, Phe, Leu, Asn | Y | Destabilizes transition state | Roth et al.66 |
| 3 | 1171–1173 | His391 | 17 | 6 | 23 | tcgCATtat | His → Glu, Phe, Lys | N | Destabilizes transition state | Huber et al.68 |
| 3 | 1246–1248 | Glu416 | 10 | 7 | 17 | attGAAacc | Glu → Gln, Val | Y | Alters binding affinity to substrate | Roth et al.64 |
| 3 | 1252–1254 | His418 | 8 | 3 | 11 | accCACggc | His → Asn, Glu, Phe | Y | Distorts Gly794, alters binding affinity to substrate | Wheatley et al.7, Wheatley et al.10, Roth et al.60, Juers et al.70 |
| 3 | 1378–1380 | Asn460 | 12 | 6 | 18 | gggAATgaa | Asn → Asp, Thr, Ser | Y | Destabilizes transition state | Wheatley et al.73 |
| 3 | 1381–1383 | Glu461 | 13 | 4 | 17 | aatGAAtca | Glu → His, Asp, Gly, Gln, His, Lys | Y | Destabilizes transition state; alters binding affinity to substrate | Cupples et al.58, Edwards et al.59, Martinez-Bilbao et al.62, Richard et al.63 |
| 3 | 1507–1509 | Tyr503 | 24 | 2 | 26 | atgTACgcg | Tyr → Phe, His, Cys, Lys | Y | Reduction in enzyme catalytic activity | Ring et al.34, Ring et al.57, Edwards et al.59, Penner et al.67 |
| 3 | 1549–1551 | Lys517 | 0 | 0 | 0 | ccgAAAtgg | Lys → Ala | N | Alters binding affinity to substrate | Wheatley et al.10 |
| 3 | 1609–1611 | Glu537 | 37 | 52 | 89 | tgcGAAtac | Glu → Gln, Asp, Val | Y | Inhibits covalent binding to galactosyl moiety of substrate; reduction in enzyme catalytic activity | Juers et al.32, Yuan et al.61 |
| 3 | 1618–1620 | His540 | 59 | 8 | 67 | gccCACgcg | His → Glu, Phe, Asn | Y | Alters binding affinity to substrate, destabilizes transition state | Roth et al.65 |
| 3 | 1624–1626 | Met542 | 2 | 1 | 3 | gcgATGggt | Met → Ala | N | Alters stability of 794–804 loop that open and closes active site | Dugdale et al.71 |
| 3 | 1702–1704 | Trp568 | 47 | 7 | 54 | gtcTGGgac | – | – | Distorts Gly794 | Wheatley et al.7 |
| 3 | 1795–1797 | Arg599 | 3 | 0 | 3 | gatCGCcag | Arg → Ala | N | Alters stability of 794–804 loop that open and closes active site | Dugdale et al.72 |
| 3 | 1801–1803 | Phe601 | 0 | 0 | 0 | cagTTCtgt | Phe → Ala | N | Alters stability of 794–804 loop that open and closes active site | Juers et al.32 |
| 4 | 2380–2382 | Gly794 | 0 | 0 | 0 | attGGCgta | Gly → Ala | Y | Alters binding affinity to substrate | Wheatley et al.10, Juers et al.80 |
| 4 | 2386–2388 | Ser796 | 0 | 1 | 1 | gtaAGTgaa | Ser → Ala, Thr, Asp | N | Alters binding affinity to substrate | Wheatley et al.10, Jancewicz et al.81 |
| 4 | 2389–2391 | Glu797 | 0 | 1 | 1 | agtGAAgcg | Glu → Ala, Leu | Y | Alters binding affinity to substrate | Wheatley et al.10, Sutendra et al.82 |
| 4 | 2422–2424 | Glu808 | 0 | 0 | 0 | gtcGAAcgc | – | – | Alters stability of 794–804 loop that open and closes active site | Jancewicz et al.81 |
| 4 | 2995–2997 | Trp999 | 1 | 0 | 1 | tccTGGagc | Trp → Leu, Phe, Gly, Tyr | Y | Alters binding affinity to substrate | Wheatley et al.10, Huber et al.83 |
Beal et al. 2020.
Historical data curated in this study.
Y designates that an SNV can lead to the reported mutation in the lacZ sequence, and N designates that it takes multiple mutations to get the amino acid change. For example, Lys → Ala requires a sequence change of AAA to GCA.
Computational approaches have advanced substantially to enable predictions of protein structure using the amino acid sequence11,12 and evaluate the impact of missense mutations.13 When coupled with deep mutational scanning14,15,16 or genetic interaction mapping,17,18 computational approaches allow an unparalleled understanding of structural features affecting protein function. Furthermore, a high-throughput method to assess the functional impact of mutations on the catalytic activity of a bacterial enzyme has recently been published.19 Application of these methods shows that a full understanding of the functional features of an enzyme requires exploration of how amino acid changes outside of the active site affect its catalytic activity and/or interaction with other proteins.
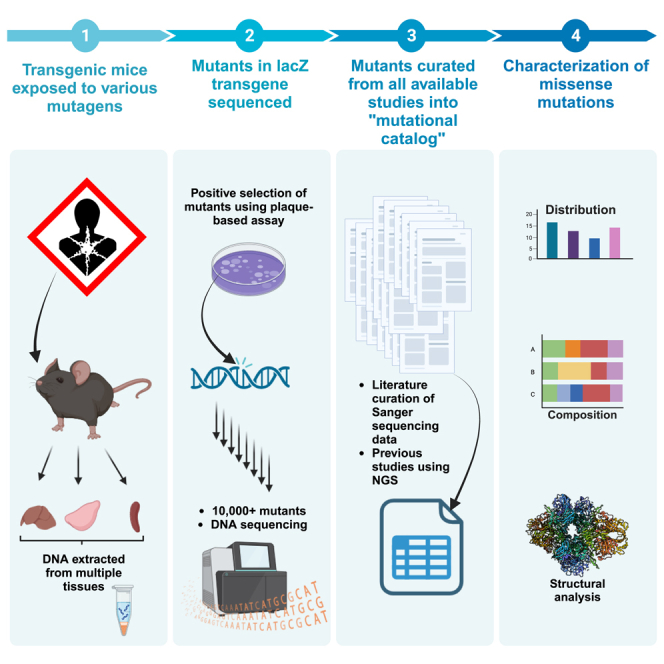
Here, we curated data from published in vivo mutagenesis studies with transgenic rodent models using either Sanger or next-generation sequencing (NGS) to characterize mutations throughout the lacZ sequence that affect β-gal function and to further define its use as a target for mutagenesis studies. Transgenic mouse models carrying bacterial reporter genes, such as lacZ, have been developed to provide an efficient method for detecting in vivo mutations.20,21,22 In particular, MutaMouse and LacZ plasmid mouse models have been genetically modified to carry multiple copies of lacZ shuttle vectors in their genomes and use positive selection methods in bacteria to detect loss-of-function mutants.23 Specifically, the lacZ shuttle vectors are excised from high-molecular-weight mouse genomic DNA and packaged into lambda phages that are used to infect E. coli C (lacZ− galE−) grown on a medium containing phenyl-β-D-galactopyranoside (P-gal). Under these conditions, P-gal becomes toxic to E. coli C infected by a phage with a functional lacZ gene.24 Thus, only phages containing a lacZ gene carrying a mutation that inactivates β-gal will be able to complete the lytic cycle and form plaques. Sequencing of mutations arising in these transgenic rodents has been used for decades to provide insight into mutagenic mechanisms associated with chemical exposures.25 However, sequencing results can also be harnessed to identify amino acids that have important roles in the structure and function of β-gal. For that latter purpose, the transgenic rodent model is particularly powerful because the transgene is not expressed in rodent tissues and DNA lesions are therefore not subject to processes such as transcription-coupled repair that might influence the mutant yield and mutation location. Thus, we produced and investigated a large catalog of lacZ mutations to identify amino acid residues of functional significance in β-gal.
Results
Curation of the lacZ mutant database
We have previously used NGS26 to sequence a total of 10,417 lacZ mutants (8,434 single nucleotide variations [SNVs]; 1,475 deletions; 508 insertions) arising spontaneously in MutaMouse animals or following exposure to four mutagens (Figure 1; Table S1). When the same mutation was observed multiple times within a sample, it was considered to be the result of clonal expansion of a single mutation and counted as one mutation (henceforth referred to as “independent mutations”); however, when the same mutation was observed in different samples, it was considered to have originated from independent events. Analysis of the 10,417 mutants identified a total of 4,805 independent mutations (3,748 SNVs; 867 deletions; and 190 insertions; Tables S2, S3, S4, S5, S6, and S7, respectively). Furthermore, as previously summarized,25 data were collected from 17 other studies with eight additional mutagens that used Sanger sequencing to characterize a separate set of 1,660 independent mutants (1,399 SNVs; 213 deletions; 28 insertions; and 20 complex mutations; Table S8), although mutants characterized by Sanger sequencing were often limited to the complementation regions.27 Overall, we collected a set of 6,465 independent mutations including 2,724 missense mutations that identified many previously uncharacterized amino acid residues that are relevant for the structural and catalytic function of β-gal (Tables S9, S10, S11, S12, S13, and S14).
Figure 1.

Composition of the complete dataset used in this study
We examined a total of 6,465 independent mutations in the lacZ gene to establish the importance of different amino acid residues in the function of β-gal. A wide variety of sequenced mutants induced by chemical or radiation exposure, different genetic backgrounds, and spontaneous mutants were compiled into the single dataset used in this study. Data from Beal et al. 2020.
Characterization of lacZ mutants
Missense mutations represented ∼42% of all independent mutations (2,732 out of 6,465) and 33% of all unique mutations (895 unique out of 2,732) detected (Table S14). We identified 492 (48% of total) codons, including all 21 previously characterized codons, with mutations that disrupted β-gal activity. This is very similar to results reported in the lacI repressor protein; specifically, more than 4,000 substitutions across residues 2–329 found that 192/328 sites (41%) are generally intolerant to substitutions.28,29 Of 492 codons with functional mutations, 384 were identified by NGS and 266 by Sanger sequencing (Figure 2; 158 codons were identified by both methods). Spontaneous missense mutations identified 204 of those codons (Figure S1) compared with 439 in mutagen-exposed samples (Figure S2; 151 codons had a missense mutation in both mutagen-exposed and non-exposed samples). Including mutations induced by exogenous sources greatly increases the chances of observing a more complete set from the full range of possible mutations, both because of the increased numbers of mutants recovered and because of the differing mutational profiles of induced compared with spontaneous mutations. The majority of the 492 codons (263; 53.5%) were identified by at least two independent missense mutations, i.e., these mutations originated in different animals and/or tissues, providing strong support for their functional role, as the number of times that a mutation is observed in each codon strengthens the evidence that the amino acid residue is important for β-gal function. Silent mutations represented only 3.5% of all independent mutations (Figure 1). These likely represent “passenger” mutations that happened to occur on the same molecule containing a functional mutation. Using the assumption that the false-positive rate is equal to the percentage of silent mutations detected by our approach, the confidence that a given mutation has a functional impact on β-gal would be equal to 1–0.035n, where n is the number of times a mutation occurs across samples, i.e., the chance of a mutation observed once, twice, or three times to be a false positive is 3.5%, 0.12%, and. 0.004%, respectively. Therefore, even the 229 codons that were identified by a single mutational event likely have functional importance rather than representing passenger mutations associated with lacZ functional mutations. Overall, our study shows that about 50% of the lacZ amino acid sequence is essential for β-gal function.
Figure 2.

Locations of lacZ codons with sequenced mutations that disrupted β-gal function
Mutations from historical data identified by Sanger sequencing are shown in the bottom panel; mutations identified by NGS in the present study are shown in the middle panel; the top panel shows all missense mutations from both datasets combined. The domains of the protein are labeled. Mutations were observed within all domains as well as outside the domain regions. The y axis shows the number of independent mutations that were observed.
Next, we evaluated whether missense, nonsense, and silent mutations were associated with specific mutation types. We found that all mutation types are associated with missense mutations in equal proportions; however, there are biases for the recovery of silent and nonsense mutations (Figure S3). Specifically, silent mutations are most likely a result of G:C → A:T transitions and are less likely caused by A:T transversions. Nonsense mutations are most commonly caused by G:C → A:T or T:A SNVs. It is not possible for A:T → G:C mutations to produce new stop codons, such as UAG or UGA, because UAA is also a stop codon. At the 24 positions where a nonsense mutation was not observed, the reference nucleotides were 11 T, 5 G, 4 C, and 4 A. The most common nonsense mutation that was absent (9/24) was of the type A:T → C:G, which is the rarest mutation in this study. Hence, A:T → G:C mutations are less likely to be recovered by this assay compared with other mutation types. Nevertheless, the data show that all mutation types contributed to the induction of missense mutations.
Distribution of mutations along the lacZ gene
Mutations affecting β-gal function occur every 2.4 codons on average throughout the length of the lacZ gene (Figure 3). The longest gap without an observed missense mutation is a stretch of 15 codons in the TIM barrel domain at residues 574–588. The percentages of codons with missense mutations occurring in the α-peptide region and domains 1 to 4 are 1%, 13%, 6%, 48%, and 29%, respectively. As is the case in site-directed mutagenesis studies, domains 3 and 4 have the highest number of amino acids that are important for function. Conversely, the 2,206 nonsense mutations, 1,298 indels, and 227 silent mutations (Table S1; Figure S4) in our study are distributed throughout the lacZ sequence, irrespective of domain region.
Figure 3.

Distribution of mutation types along the lacZ gene
Nonsense mutations were distributed throughout the gene, whereas some missense mutations were observed repeatedly at locations of functionally important codons. Both deletions and insertions are distributed evenly throughout lacZ, with enrichment near repetitive sequences. The y axis represents the number of independent mutations.
We used Swiss-PdbViewer30 and PyMOL31 to visualize the location of the 263 amino acids whose functional role was supported by at least two independent mutations. As shown in Figure 4, the 21 previously characterized amino acids and the majority of amino acids with the highest numbers of independent mutations in our dataset (see below) are near the binding site for the substrate (i.e., allolactose for this experimental structure) in the β-gal homotetramer (PDB ID 1JZ8).32 However, the rest of the newly identified amino acids are found throughout the β-gal subunit, suggesting that they may have a role in protein folding, stability, and subunit interactions.
Figure 4.

Visualization of functional missense mutations within the β-Gal homotetramer
Far left image shows the complete homotetramer (subunits colored blue, yellow, green, and red). Second image zooms in on one subunit. Third image only shows the 263 amino acids with at least two independent mutations. The last image on the right only shows the active site and the 33 most highly substituted amino acids from this study (which are high-confidence mutations in codons presumed to be functionally important). Previously characterized amino acids known to be important for β-Gal function are aqua, and highly mutated residues from this study are yellow. The allolactose substrate is colored in blue. Image was created using PyMOL.
Identification of SNV and codon hotspots
Nucleotide positions were considered hotspots for SNV formation if the position had a mutation count that was one standard deviation above the mean number of mutations per nucleotide position. The hotspot with the most mutations was at position 1,169 with 112 independent SNVs. This corresponds to the second position of the Ser390 codon (TCG), which had not been previously identified as functionally important, but is adjacent to His391, one of the 21 previously identified residues critical for function (the mutation of this residue is known to destabilize the transition state; Table 1). Nucleotide 2,356 was also frequently mutated, with 106 independent mutations identified at this position. This corresponds to the first position of the Arg786 codon (wild-type sequence of CGT). Mutations at that position accounted for 74% of the total SNVs in that codon, whereas the second and third positions in the codon accounted for 25% and <1% of mutations, respectively. The low proportion of mutations at the third position of the Arg786 codon is expected, as not all SNVs will change the coded amino acid. The trinucleotide motifs of the wild-type sequence for the first and second position of Arg786 are CCG (nucleotides 2,355–2,357) and ACG (nucleotides 2,356–2,358; complementary strand of CGT), respectively. In total, there were 74 sites with 14 or more independent mutations that accounted for 2,069 of the total 5,147 SNVs (40.2%) identified by NGS and Sanger sequencing (Figure 1).
As suggested by the trinucleotide context of the two most frequently mutated codons, CpG sites were common among the hotspots, with 45 of 74 (60.8%) of mutation hotspots occurring at the motif NCG, where N is any nucleotide. The three positions with the most indels also occurred at CpG dinucleotide repeats (2 or 4 repeats per site). Among all the lacZ mutations we curated, 84% of the SNVs occurred at C or G bases, whereas the G/C content of the lacZ reference sequence is 56%. This is likely because the majority of mutations were induced by chemicals that preferentially target guanines. Thus, our conclusion is that guanine residues and/or CpG sites have been well sampled in our data, whereas A/T sites are sampled to a lower depth (Figure S5).
As for SNVs, codons were considered hotspots for mutations if they had a missense count that was one standard deviation above the mean number of mutations per amino acid position. These 33 codons are shown in Table 2. The Ser390 codon with 100 separate observations of missense mutations was the most frequently mutated. Six of the 21 previously characterized sites (Asp201, His391, Tyr503, Glu537, His540, Trp568) were among these highly mutated codons.
Table 2.
Highly mutated codons with strong evidence of importance for β-gal function identified by sequencing lacZ mutants
| Domain 1 Sugar binding (PF02837) |
Domain 2 β-Galactosidase (PF00703) |
Domain 3 TIM barrel (PF02836) |
Domain 4 β-Galactosidase small chain (PF02929) |
||||
|---|---|---|---|---|---|---|---|
| Codona | Mutations | Codon | Mutations | Codon | Mutations | Codon | Mutations |
| Gly207 | 56 | Glu304 | 32 | Ser390 | 100 | Arg786 | 142 |
| Trp203 | 37 | Trp301 | 21 | Glu537 | 89 | Arg909 | 91 |
| Asp201 | 36 | Glu358 | 72 | Gly899 | 67 | ||
| His540 | 67 | Arg881 | 53 | ||||
| Gly605 | 59 | Asp792 | 49 | ||||
| Trp568 | 54 | Thr941 | 23 | ||||
| Pro393 | 39 | Asp908 | 23 | ||||
| Arg439 | 33 | Asn791 | 20 | ||||
| Gly547 | 30 | Gly901 | 19 | ||||
| Gly406 | 29 | ||||||
| Gly564 | 27 | ||||||
| Tyr503 | 26 | ||||||
| Gly459 | 25 | ||||||
| Gly565 | 24 | ||||||
| Gly353 | 23 | ||||||
| His391 | 23 | ||||||
| Pro501 | 23 | ||||||
| Met502 | 20 | ||||||
| Ser545 | 19 | ||||||
Codons were included if the codon had more mutations (i.e., 19) than one standard deviation of the mean number of mutations per codon (mean 5.5 mutations per codon plus one standard deviation, 13.1; excluding codons where no mutations were detected).
Examination of protein structural features and their relationship to the types of substitutions observed
We examined the distribution, and types, of amino acid changes in the context of structural features of the β-gal enzyme. Our dataset contains 895 unique missense SNVs that resulted in 861 different amino acid changes at 492 codons. We used Grantham distance to estimate the impact of a given amino acid substitution. We found that the degree of conservation of amino acid change may be correlated with the type of secondary structure in which the substitution occurred. The π-helix had a significant positive coefficient in our model, suggesting that radical substitutions within this secondary structure is associated with a mutant phenotype (Table S15; Figure S6). Solvent accessibility appears to be weakly negatively associated with the Grantham distance (t-statistic −3.37, p = 0.0008; Table S16), indicating that conservative changes in solvent-exposed residues are sufficient to disrupt enzyme function. However, based on examination of the relationship between solvent accessibility and Grantham distance, the effect size of this association appears to be small (Figure S7). When comparing the impact on change in side chain volume and change in hydropathy for substitutions, we found that turns in the protein secondary structure were prone to reductions, rather than increases, in both side chain volume and hydropathy (i.e., a higher ratio of change for mutant:wild type; Figure S8).
We further examined the specific types of amino acid substitutions observed in our dataset to determine whether some substitutions were differentially represented between types of secondary structures. First, we observed that the most frequently substituted wild-type residue was glycine (Figure S9). Furthermore, the most frequently observed mutant residue was arginine, and, indeed, the overall most frequent substitutions were G > V (32 instances) and G > R (25 instances). Glycine was the wild-type target in four of the top five most frequent substitutions. However, the distribution of the observed mutations in secondary structures did not differ drastically from the proportion of residues in the GenBank reference sequence (Figure S10). We then used our logistic regression model to test whether specific amino acid substitutions vary in their distribution between types of secondary structures (Table S17). As shown in Figure 5, several patterns are visible (e.g., the apparent over-representation of substitutions at a glycine residue). Overall, we found that there were 13 substitutions (Figure S11) that were significantly different in proportion between the coil regions and regions of secondary structure.
Figure 5.

The wild-type and mutant residues for each of the 862 unique amino acid changes observed in this study
Some of the specific substitutions are present in different proportions based on the type of secondary structure. Glycine (G) is the most commonly mutated wild-type residue (i.e., within columns); conversely, arginine (R) is the most common mutant residue observed (i.e., within rows), although this pattern is not as visually striking, because the majority are G > V.
Modeling the maximum number of functional amino acids
We used a mathematical approach to explore whether some functional amino acids were not identified in our study (see STAR Methods: theoretical maximum number of functional targets). The enrichment of nonsense mutations (45%) and missense mutations compared with the theoretical proportions of the different mutation types (4% nonsense, 23% silent, 73% missense; Figure S3) indicates that the assay favors mutations that result in knocking out β-gal function by producing a truncated form of the protein. Thus, the proportion of observed nonsense to total possible nonsense mutations may serve as a good proxy for the theoretical maximum number of functional amino acids. In total, there were 332 unique nonsense mutations characterized out of the 356 possible mutations (93.3%) or 266 codons with nonsense mutations out of a possible 290 (91.7%). The rarefaction curve provides supportive evidence that nonsense mutations are near saturation (Figure S12). Interestingly, the last three possible nonsense mutations at nucleotide positions 3,063 (Cys TGT > Stop TGA); 3,064 (Gln CAA > Stop TAA); and 3,067 (Lys AAA > Stop TAA) were not recovered. In contrast, there were 10 independent nonsense mutations recovered at position 3,060 (Trp TGG > Stop TGA), possibly indicating a highly precise cut-off location at the end of the lacZ gene where truncation does not affect enzyme function. Thus, our recovery of possible nonsense events with a functional impact is 92.7% (260/287). Assuming that we recovered a comparable proportion of functional nonsynonymous events, this suggests that there are approximately 39 additional amino acid substitutions ((492/0.927)-492) detrimental to protein function that may have not been detected in our data.
Discussion
We identified a set of 2,732 independent missense mutations in the lacZ gene that provided a comprehensive and detailed map of the amino acid residues affecting the structure and catalytic activity of the β-gal enzyme. Prior to this study, little was known about the distribution of functional amino acid residues along the lacZ gene product. Our analysis shows that missense mutations are found at multiple sites in each of the functional domains. Contrary to lacI, another common reporter gene for mutation studies, where the majority of mutations occur in the negative complementing domain region alone,33 our results show that functional amino acid residues occur throughout the length β-gal enzyme and that the entire lacZ gene represents a relatively unbiased target for mutagenesis studies with transgenic rodent models.
Our results reaffirm the importance of the 21 amino acids previously identified by site-directed mutagenesis, as loss-of-function mutations were detected within each of these residues. Most importantly, sequencing revealed hundreds of additional residues that have a functional role in β-gal. Since most of these codons were mutated across multiple biological replicates, we have high confidence that they represent amino acids residues that are critical for β-gal function. Here, we highlight 33 codons with the most missense mutations observed in our data. For simplicity, those codons are referred to as highly mutated codons or amino acid residues. As expected, the 3-dimensional structure of these sites revealed that the previously characterized amino acid residues (Table 1; Figure 4) are in the active site in close proximity to the substrate. Similarly, most of the highly mutated functional amino acid residues (Table 2; Figure 4) are in close proximity to the substrate on the second coordination shell around the active site. For example, Met502 is one of the highly substituted amino acid residues closest to the substrate. Previous studies using chemical modifications have indicated that Met502 is not required for catalytic activity.34 Therefore, it may be its interaction with Tyr503 that is important for β-gal function. This is supported by additional missense mutations detected by NGS in Tyr503 and neighboring codons Thr494, Ala495, Thr496, Asp497, Ile498, Ile499, Cys500, Pro501, Met502, Ala504, and Arg505. Other substituted amino acid residues that are in close proximity to the active site in 3-dimensional space are located proximal to previously characterized amino acid residues involved in substrate coordination (e.g., Glu358, Ser390, Glu412, Gly459, Pro501, Ala541, Ser545, Gly605, Asn791, and Asp792), and several other highly substituted amino acid residues are located proximal to those 10 amino acid residues. Thus, those amino acid residues in close proximity to the active site likely form the backbone of each monomer and play a role in active site stability through different mechanisms (non-covalent or covalent bonding, pKa perturbation, etc.). The rest of the highly substituted amino acid residues are located far from the active site and are likely involved in protein folding, stability, and subunit interactions.
A notable example of one of the 33 highly substituted amino acid residues is Arg786 in the β-gal small chain domain, which had a total of 143 independent mutations (142 missense; 1 silent). There is currently no known function for Arg786 in β-gal, even though it occurs in a region that is relatively conserved among five β-gal homologues.35,36 Visual inspection of Arg786 in the homotetramer led us to predict that it participates in multiple hydrogen bonds with four nearby residues (Figure S13). Hydrogen bonds are formed with Tyr816, Met991, Gly992, and Asp792 (identified in this study as an amino acid residue likely important for β-gal function; Table 2). As expected, the six types of missense mutations that were detected in Arg786 would cause major alterations in those interactions. Specifically, substitutions to Cys, Ser, Leu, Pro, and His would result in the loss of four hydrogen bonds, retaining only a bond to Tyr816; substitution to Gly would result in complete loss of hydrogen bonds. Arg786Pro also would cause steric hindrance of the neighboring amino acid residue Thr785. Arg786His would also result in the formation of a new hydrogen bond and affect the stability of intramolecular reactions with Asp792. Although those amino acid residues have not previously been shown to be critical for β-gal function, the loss of interactions with Arg786 may modify the orientation of side chain and backbone that they interact with, affecting residues more proximal to the active site. Thus, although Arg786 may not be involved in substate binding or hydrolysis directly, it contributes to the structural integrity of the protein.
There are some differences in the distribution of mutations that we observed when compared with previously characterized amino acid substitutions that affect function and those identified by our study. For example, we found comparatively few missense mutations in the active site loop (codons 794–804), despite the fact that this region is known to be important for enzymatic function. This suggests that the individual amino acids in the loop are not as important as the loop as a whole. Indeed, Wheatley et al.10 pointed out that most of the studied mutations to the active site loop do not have much effect on kcat/Km of hydrolysis. These authors argue that the loop is essential for allolactose synthesis by promoting transgalactosylation. The data support the conclusion that the loop is not critical for the hydrolytic reaction, and changes in transgalactosylation would not be detected by our assay. In addition, some of the mutations investigated by site-directed mutagenesis require more than one SNV to obtain the reported amino acid substitution. For example, Lys517Ala alters binding affinity to substrates,10 but this substitution requires a mutation from AAA to GCA, which would be much rarer than a single-base substitution. Nevertheless, our approach effectively identified previously characterized and many novel highly mutated protein sites relevant for β-gal function.
We found that the magnitude of the Grantham distance for a given substitution was not strongly determined by the region of secondary structure in which it occurred, although we did identify a potential weak correlation with π-helix, β-strand, and β-bridge features, in which a higher Grantham distance was observed. All secondary structures, when compared with the coil, had positive coefficients in our linear modeling (although not all were significant or large effect sizes); this indicates that the coil regions were most likely to contain conservative amino acid substitutions, whereas regions of secondary structure in β-gal require more radical substitutions to disrupt function. Similarly, the magnitude of the Grantham distance is weakly negatively correlated with solvent accessibility. A possible conclusion is that residues with a high solvent accessibility do not require a large Grantham distance to be detected in the functional assay, suggesting that they may be less tolerant of conservative substitutions. Overall, the degree of conservation for substitutions does not have a striking correlation with any structural features we could identify, suggesting that substitutions are mostly randomly distributed.
In our data, turns in secondary structure are correlated with an increase in side chain volume and hydropathy for substitutions. Furthermore, glycine was the most frequently substituted residue. These observations are consistent because glycine is a common residue at turns37 and has a small volume; thus, substitutions at glycine usually increase side chain volume. Therefore, we expect that this observation is driven largely by substitutions at glycine residues. Aside from glycine, we identified 15 specific substitution types that were more likely to occur in regions of secondary structure compared with coils. These substitutions may be of interest as targets for future structural studies in β-gal.
An objective of this work was to determine the proportion of the 3,096 bases in the lacZ gene that were functional targets for mutagenesis. Rarefaction curves (Figures S5 and S12) indicated that nonsense mutations are closer to saturation than missense mutations and suggested that there could potentially be more functional mutations that were not recovered in our dataset. Furthermore, examining the different types of base substitutions separately shows that SNVs at G:C nucleotides are the closest to saturation, as the majority of mutations recovered were at these positions (Figure S5). In contrast, mutations at A:T nucleotides are more likely to have been missed. Previous work exploring mutations in lacZ has demonstrated that mutations are more likely to occur at specific nucleotide positions over others due to the interaction between mutagens and DNA, the action of the repair and replication machineries, and sequence context.38 Thus, it is possible that the mutagenic specificity of chemicals used for mutation induction in the available studies contributed to the undersampling of mutations at A:T nucleotides. Further studies using chemicals that induce A:T → C:G mutations would improve the mutation saturation of lacZ gene and identify additional functional mutations.
In summary, our study has assembled the largest number of sequenced lacZ mutations to identify functional amino acid residues throughout β-gal. Analysis of 6,465 mutations recovered from the lacZ mutant selection assay showed that 48% (492/1,023) of the residues of lacZ had missense mutations and that these mutations were distributed throughout the lacZ gene. These results greatly expand our understanding of the structural features important for the catalytic site of β-gal and also identify novel amino acid residues important for homotetramerization, which also plays a critical role in enzymatic function. Overall, we provide comprehensive foundational data to define the amino acid residues that determine the catalytic and structural activity of this important enzyme.
Limitations of the study
A limitation of this study is that we did not undertake a biochemical characterization of the mutants recovered here. By virtue of the positive selection assay for the mutant lacZ gene, it is understood that any plaques we collected for sequencing originated from a phage that had an impaired β-gal function; in fact, we recovered all previously known single-base mutations where a biochemical impact on enzymatic function was observed. However, we did not verify this for all new mutations that were identified here. Indeed, a limitation of the methodology is that in order to obtain high numbers of sequenced mutants, they must first be pooled, so characterizing the individual plaques further would require first creating the mutant using site-directed mutagenesis and then performing more downstream assays. These limitations, conversely, also speak to the opportunities for practical application of the data. By presenting the catalog of mutations we categorized in this study, researchers could pose numerous hypotheses that could be tested using these mutants as a starting point to explore functional residues of the β-gal protein. However, as a final caveat to using these data in future studies, it should be noted that the positive selection assay using P-gal may not always recapitulate the catalytic activity of β-gal under different conditions, and using different substrates. Thus, researchers are cautioned that the substitutions we have identified herein may have residual catalytic activity when tested under different experimental conditions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Raw sequencing reads of lacZ mutants obtained from Ion Torrent sequencing of collected plaques | NCBI BioProject | PRJNA640660 |
| Curated complete list of lacZ mutants used as input for analysis | This paper; Mendeley Data | 10.17632/4n3bsmyskj.1 |
| Software and algorithms | ||
| RMarkdown script used to reproduce all the analyses, numbers, models and figures used in the manuscript | GitHub | https://github.com/EHSRB-BSRSE-Bioinformatics/Beal_et_al_2023_lacZ |
| R | https://www.r-project.org/ | 4.3.1 |
| bio3d | CRAN (R 4.3.1) | 2.4-4 |
| Biostrings | Bioconductor | 2.68.1 |
| doBy | CRAN (R 4.3.1) | 4.6.18 |
| flextable | CRAN (R 4.3.1) | 0.9.2 |
| GenomicRanges | Bioconductor | 1.52.0 |
| GenVisR | Bioconductor | 1.31.1 |
| ggh4x | CRAN (R 4.3.1) | 0.2.5 |
| ggplot2 | CRAN (R 4.3.1) | 3.4.3 |
| grantham | CRAN (R 4.3.1) | 0.1.1 |
| Gviz | Bioconductor | 1.44.0 |
| msa | Bioconductor | 1.32.0 |
| oddsratio | CRAN (R 4.3.1) | 2.0.1 |
| plyranges | Bioconductor | 1.20.0 |
| Pviz | Bioconductor | 1.34.0 |
| reutils | CRAN (R 4.2.2) | 0.2.3 |
| tidyverse | CRAN (R 4.3.1) | 2.0.0 |
| vtree | CRAN (R 4.3.1) | 5.6.5 |
Resource availability
Lead contact
Further information and request for resources should be directed to and will be fulfilled by the Lead Contact, Francesco Marchetti (francesco.marchetti@hc-sc.gc.ca).
Materials availability
The present study used published data obtained using the MutaMouse or LacZ plasmid mouse models.20,22,25,26,27,35,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55 No new animal work was conducted and no new unique reagents were generated.
Data and code availability
-
•
All data regarding the mutations reported in study are shown in supplementary material and available in Mendeley Data (see key resources table).
-
•
RMarkdown scripts are available at GitHub (see key resources table).
Experimental model and study participant details
No new laboratory data were generated, and all previously published sequencing data are listed in the key resources table.
Method details
Experimental design and data collection
We curated thousands of lacZ mutations from published studies that were collected using a positive selection assay20 and NGS data from our laboratory.25,26,39,40 In addition, we collected data on lacZ mutation distribution from studies in which lacZ transgene mutants from MutaMouse or LacZ plasmid animals had been sequenced using Sanger sequencing.22,27,35,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55 All mutants considered in this study are shown in Table S1. Only data from studies that reported the position of each mutation were included. It is important to note that some of the studies reported the position of each mutation differently. Specifically, some studies reported the coding sequence position while others reported the position within the plasmid construct. Therefore, the mutation positions were adjusted to reflect the position within the lacZ coding sequence for consistency. The lacZ reference sequence used in this study matched the coding sequence in MutaMouse because that is where the majority of the mutations were observed. The MutaMouse lacZ coding sequence has three SNVs relative to that of E. coli (GenBank: V00296.1)56 including two silent mutations and one missense mutation (Phe1007Leu). In addition, there is a 15 bp insertion into codon 852 (referred to here as codons 8a-f to prevent disruption of the downstream codon number). Thus, the total sequence length in studies conducted on MutaMouse is 3,096 bp. The final position of each mutation was adjusted to match the reference sequence. Finally, data regarding protein sites relevant for β-gal function were obtained from site-directed mutagenesis and crystallographic protein structure studies.7,10,32,34,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73
Protein structure examination
Swiss PDB Viewer30 and PyMOL31 were used to visualize β-gal (1JZ832) and predict the impact of uncharacterized functional sites. The mutate feature in Swiss PDB Viewer was used to determine the effects of different amino acid substitutions.
Examination of protein structural features and their relationship to the types of substitutions observed
We obtained the secondary structure and degree of solvent exposure at each residue from the PDB file in R using the bio3d package (version 2.4.4)76 and DSSP (version 3.0.0).77 Then, we used the Grantham R package (version 0.1.1) to annotate each substitution in our data with Grantham’s using the default parameters. We classified each change into conservative substitutions (Grantham distance less than 50) or radical substitutions (Grantham distance greater than 50). Additionally, we annotated the hydropathy (as calculated by78) of each amino acid residue (wild type and mutant), using the idpr R package (version 1.9.15) and calculated the change in hydropathy for each substitution by subtraction. For each wild type and mutant residue, we also annotated the volume of side chains, chemical subclass, charge, hydrogen donor or acceptor atoms class, and polarity, based on the physicochemical classes data provided by IGMT.79
We built regression models in R (using the glm() function from the stats package, version 4.3.1) to test if the degree to which conservative amino acid substitutions are tolerated (or not) may be related to whether a residue is solvent exposed, or it is found in a region of secondary structure. We built two models using the formulas: ‘Grantham distance ∼ class of secondary structure, and ‘Grantham distance ∼ solvent accessible amino acid’. We used the quasipoisson distribution to account for overdispersion.
To test whether specific amino acid changes were over- or under-represented in regions of secondary structure, we performed a logistic regression using the binomial distribution and the formula ‘class of secondary structure ∼0 + amino acid change’, where the secondary structure variable was a factor specifying the region of secondary structure for a given substitution, and the amino acid change variable was the specific observed substitution (e.g., R > C). The odds ratio was calculated as the exponent of the estimated coefficient of the model.
Theoretical maximum number of functional targets
There are 9,288 possible SNVs that can be observed in the lacZ gene (3,096 positions × 3 SNVs/position), and 7,162 of these have the potential to affect function (missense, nonsense, or loss of start mutations). Our study identified 895 unique missense mutations out of 2,732 characterized. Extending the analysis to all SNV types (including nonsense and silent mutations) shows that there were 1,399 unique SNVs out of 5,147 total. To explore how many functional lacZ codons were not identified in our sequencing data we simulated all possible SNVs that could have occurred in lacZ and their associated consequences in β-gal function (code available at https://github.com/MarcBeal/HC-MSD/tree/master/lacZ_Saturation; Figure S6).
Quantification and statistical analysis
All statistical analyses and simulations were done using the R programming language.74 Rarefaction analysis (Figures S5 and S12) was done using the R library “vegan”.75 Regression models for Tables S15 and S16 were built in Rusing the glm() function from the stats package (version 4.3.1) using the formulas: ‘Grantham distance ∼ class of secondary structure, and ‘Grantham distance ∼ solvent accessible amino acid’. Logistic regression for Table S17 was done using the binomial distribution and the formula ‘class of secondary structure ∼0 + amino acid change’, where the secondary structure variable was a factor specifying the region of secondary structure for a given substitution, and the amino acid change variable was the specific observed substitution. All the code to reproduce the statistical tests is available at https://github.com/EHSRB-BSRSE-Bioinformatics/Beal_et_al_2023_lacZ. The versions of software (i.e., R packages) used within the code are listed in the key resources table.
Additional resources
There are no additional resources.
Acknowledgments
We would like to thank Drs Clotilde Maurice and Jason O’Brien and Beverly Allan, John Gingerich, Marianela Rosales, and Lynda Soper for all their help with lacZ analyses and mutation collection and Rémi Gagné for his contributions to NGS analyses. Work funded by the Health Canada’s Chemicals Management Plan and Genomics Research and Development Initiative to FM. CLY gratefully acknowledges the Canada Research Chairs Program.
Author contributions
Conceptualization: F.M., C.Y.L., I.L.B.; Software: M.A.B., M.J.M.; Formal Analysis: M.A.B., M.J.M., F.M., C.Y.L., I.L.B.; Investigation: M.A.B., M.J.M., A.D.; Resources: F.M., C.Y.L.; Data Curation: M.A.B., M.J.M.; Writing—Original Draft: M.A.B., M.J.M., F.M.; Writing—Review & Editing; M.A.B., M.J.M., A.D., I.B.L., C.Y.L., F.M.; Visualization: M.A.B., M.J.M.; Supervision: F.M., C.L.Y.; Funding Acquisition: F.M.
Declaration of interests
The authors declare no competing interests.
Published: November 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108407.
Supplemental information
References
- 1.Jacob F., Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 2.Ullmann A., Jacob F., Monod J. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the beta-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 1967;24:339–343. doi: 10.1016/0022-2836(67)90341-5. [DOI] [PubMed] [Google Scholar]
- 3.Langley K.E., Villarejo M.R., Fowler A.V., Zamenhof P.J., Zabin I. Molecular basis of beta-galactosidase alpha-complementation. Proc. Natl. Acad. Sci. USA. 1975;72:1254–1257. doi: 10.1073/pnas.72.4.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messing J., Gronenborn B., Müller-Hill B., Hans Hopschneider P. Filamentous coliphage M13 as a cloning vehicle: insertion of a HindII fragment of the lac regulatory region in M13 replicative form in vitro. Proc. Natl. Acad. Sci. USA. 1977;74:3642–3646. doi: 10.1073/pnas.74.9.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vieira J., Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 6.Husain Q. Beta galactosidases and their potential applications: a review. Crit. Rev. Biotechnol. 2010;30:41–62. doi: 10.3109/07388550903330497. [DOI] [PubMed] [Google Scholar]
- 7.Wheatley R.W., Huber R.E. An allolactose trapped at the lacZ beta-galactosidase active site with its galactosyl moiety in a (4)H3 conformation provides insights into the formation, conformation, and stabilization of the transition state. Biochem. Cell. Biol. 2015;93:531–540. doi: 10.1139/bcb-2015-0037. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson R.H., Zhang X.J., DuBose R.F., Matthews B.W. Three-dimensional structure of beta-galactosidase from E. coli. Nature. 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- 9.Juers D.H., Matthews B.W., Huber R.E. LacZ beta-galactosidase: structure and function of an enzyme of historical and molecular biological importance. Protein Sci. 2012;21:1792–1807. doi: 10.1002/pro.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheatley R.W., Lo S., Jancewicz L.J., Dugdale M.L., Huber R.E. Structural explanation for allolactose (lac operon inducer) synthesis by lacZ beta-galactosidase and the evolutionary relationship between allolactose synthesis and the lac repressor. J. Biol. Chem. 2013;288:12993–13005. doi: 10.1074/jbc.M113.455436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A., et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596:590–596. doi: 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng J., Novati G., Pan J., Bycroft C., Žemgulytė A., Applebaum T., Pritzel A., Wong L.H., Zielinski M., Sargeant T., et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381:eadg7492. doi: 10.1126/science.adg7492. [DOI] [PubMed] [Google Scholar]
- 14.Livesey B.J., Marsh J.A. Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 2020;16:e9380. doi: 10.15252/msb.20199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rollins N.J., Brock K.P., Poelwijk F.J., Stiffler M.A., Gauthier N.P., Sander C., Marks D.S. Inferring protein 3D structure from deep mutation scans. Nat. Genet. 2019;51:1170–1176. doi: 10.1038/s41588-019-0432-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braberg H., Echeverria I., Bohn S., Cimermancic P., Shiver A., Alexander R., Xu J., Shales M., Dronamraju R., Jiang S., et al. Genetic interaction mapping informs integrative structure determination of protein complexes. Science. 2020;370 doi: 10.1126/science.aaz4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmiedel J.M., Lehner B. Determining protein structures using deep mutagenesis. Nat. Genet. 2019;51:1177–1186. doi: 10.1038/s41588-019-0431-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markin C.J., Mokhtari D.A., Sunden F., Appel M.J., Akiva E., Longwell S.A., Sabatti C., Herschlag D., Fordyce P.M. Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Science. 2021;373 doi: 10.1126/science.abf8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert I.B., Singer T.M., Boucher S.E., Douglas G.R. Detailed review of transgenic rodent mutation assays. Mutat. Res. 2005;590:1–280. doi: 10.1016/j.mrrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Ono T., Ikehata H., Nakamura S., Saito Y., Hosoi Y., Takai Y., Yamada S., Onodera J., Yamamoto K. Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutat. Res. 2000;447:165–177. doi: 10.1016/s0027-5107(99)00200-6. [DOI] [PubMed] [Google Scholar]
- 22.Dollé M.E.T., Snyder W.K., Dunson D.B., Vijg J. Mutational fingerprints of aging. Nucleic Acids Res. 2002;30:545–549. doi: 10.1093/nar/30.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gossen J.A., Vijg J. A selective system for lacZ- phage using a galactose-sensitive E. coli host. Biotechniques. 1993;14 326, 330. [PubMed] [Google Scholar]
- 24.Gossen J.A., Molijn A.C., Douglas G.R., Vijg J. Application of galactose-sensitive E. coli strains as selective hosts for LacZ- plasmids. Nucleic Acids Res. 1992;20:3254. doi: 10.1093/nar/20.12.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beal M.A., Meier M.J., LeBlanc D.P., Maurice C., O’Brien J.M., Yauk C.L., Marchetti F. Chemically induced mutations in a MutaMouse reporter gene inform mechanisms underlying human cancer mutational signatures. Commun. Biol. 2020;3:438. doi: 10.1038/s42003-020-01174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beal M.A., Gagne R., Williams A., Marchetti F., Yauk C.L. Characterizing Benzo[a]pyrene-induced lacZ mutation spectrum in transgenic mice using next-generation sequencing. BMC Genom. 2015;16 doi: 10.1186/s12864-015-2004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglas G.R., Gingerich J.D., Gossen J.A., Bartlett S.A. Sequence spectra of spontaneous lacZ gene mutations in transgenic mouse somatic and germline tissues. Mutagenesis. 1994;9:451–458. doi: 10.1093/mutage/9.5.451. [DOI] [PubMed] [Google Scholar]
- 28.Markiewicz P., Kleina L.G., Cruz C., Ehret S., Miller J.H. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as "spacers" which do not require a specific sequence. J. Mol. Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 29.Pace H.C., Kercher M.A., Lu P., Markiewicz P., Miller J.H., Chang G., Lewis M. Lac repressor genetic map in real space. Trends Biochem. Sci. 1997;22:334–339. doi: 10.1016/s0968-0004(97)01104-3. [DOI] [PubMed] [Google Scholar]
- 30.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 31.PyMOL . Schrödinger, LLC; 2022. The PyMOL Molecular Graphics System, Version 2.0. [Google Scholar]
- 32.Juers D.H., Heightman T.D., Vasella A., McCarter J.D., Mackenzie L., Withers S.G., Matthews B.W. A structural view of the action of Escherichia coli (lacZ) beta-galactosidase. Biochemistry. 2001;40:14781–14794. doi: 10.1021/bi011727i. [DOI] [PubMed] [Google Scholar]
- 33.Gu M., Ahmed A., Wei C., Gorelick N., Glickman B.W. Development of a lambda-based complementation assay for the preliminary localization of lacI mutants from the Big Blue mouse: implications for a DNA-sequencing strategy. Mutat. Res. 1994;307:533–540. doi: 10.1016/0027-5107(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 34.Ring M., Huber R.E. Multiple replacements establish the importance of tyrosine-503 in beta-galactosidase (Escherichia coli) Arch. Biochem. Biophys. 1990;283:342–350. doi: 10.1016/0003-9861(90)90652-f. [DOI] [PubMed] [Google Scholar]
- 35.Ikehata H., Takatsu M., Saito Y., Ono T. Distribution of spontaneous CpG-associated G: C--> A: T mutations in the lacZ gene of Muta mice: effects of CpG methylation, the sequence context of CpG sites, and severity of mutations on the activity of the lacZ gene product. Environ. Mol. Mutagen. 2000;36:301–311. doi: 10.1002/1098-2280(2000)36:4<301::aid-em6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 36.Juers D.H., Jacobson R.H., Wigley D., Zhang X.J., Huber R.E., Tronrud D.E., Matthews B.W. High resolution refinement of beta-galactosidase in a new crystal form reveals multiple metal-binding sites and provides a structural basis for alpha-complementation. Protein Sci. 2000;9:1685–1699. doi: 10.1110/ps.9.9.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trevino S.R., Schaefer S., Scholtz J.M., Pace C.N. Increasing protein conformational stability by optimizing beta-turn sequence. J. Mol. Biol. 2007;373:211–218. doi: 10.1016/j.jmb.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogozin I.B., Pavlov Y.I. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat. Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 39.O'Brien J.M., Beal M.A., Yauk C.L., Marchetti F. Next generation sequencing of benzo(a)pyrene-induced lacZ mutants identifies a germ cell-specific mutation spectrum. Sci. Rep. 2016;6 doi: 10.1038/srep36743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meier M.J., O'Brien J.M., Beal M.A., Allan B., Yauk C.L., Marchetti F. Utero Exposure to Benzo[a]Pyrene Increases Mutation Burden in the Soma and Sperm of Adult Mice. Environ. Health Perspect. 2017;125:82–88. doi: 10.1289/EHP211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Douglas G.R., Jiao J., Gingerich J.D., Gossen J.A., Soper L.M. Temporal and molecular characteristics of mutations induced by ethylnitrosourea in germ cells isolated from seminiferous tubules and in spermatozoa of lacZ transgenic mice. Proc. Natl. Acad. Sci. USA. 1995;92:7485–7489. doi: 10.1073/pnas.92.16.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douglas G.R., Jiao J., Gingerich J.D., Soper L.M., Gossen J.A. Temporal and molecular characteristics of lacZ mutations in somatic tissues of transgenic mice. Environ. Mol. Mutagen. 1996;28:317–324. doi: 10.1002/(SICI)1098-2280(1996)28:4<317::AID-EM4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 43.Frijhoff A.F., Rebel H., Mientjes E.J., Kelders M.C., Steenwinkel M.J., Baan R.A., van Zeeland A.A., Roza L. UVB-induced mutagenesis in hairless lambda lacZ-transgenic mice. Environ. Mol. Mutagen. 1997;29:136–142. doi: 10.1002/(sici)1098-2280(1997)29:2<136::aid-em4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 44.Dollé M.E., Martus H.J., Novak M., van Orsouw N.J., Vijg J. Characterization of color mutants in lacZ plasmid-based transgenic mice, as detected by positive selection. Mutagenesis. 1999;14:287–293. doi: 10.1093/mutage/14.3.287. [DOI] [PubMed] [Google Scholar]
- 45.Ikehata H., Masuda T., Sakata H., Ono T. Analysis of mutation spectra in UVB-exposed mouse skin epidermis and dermis: frequent occurrence of C-->T transition at methylated CpG-associated dipyrimidine sites. Environ. Mol. Mutagen. 2003;41:280–292. doi: 10.1002/em.10153. [DOI] [PubMed] [Google Scholar]
- 46.Ikehata H., Nakamura S., Asamura T., Ono T. Mutation spectrum in sunlight-exposed mouse skin epidermis: small but appreciable contribution of oxidative stress-mediated mutagenesis. Mutat. Res. 2004;556:11–24. doi: 10.1016/j.mrfmmm.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 47.Jiao J., Douglas G.R., Gingerich J.D., Soper L.M. Analysis of tissue-specific lacZ mutations induced by N-nitrosodibenzylamine in transgenic mice. Carcinogenesis. 1997;18:2239–2245. doi: 10.1093/carcin/18.11.2239. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki T., Hayashi M., Wang X., Yamamoto K., Ono T., Myhr B.C., Sofuni T. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta Mouse) Mutat. Res. 1997;395:75–82. doi: 10.1016/s1383-5718(97)00144-7. [DOI] [PubMed] [Google Scholar]
- 49.Mientjes E.J., Luiten-Schuite A., van der Wolf E., Borsboom Y., Bergmans A., Berends F., Lohman P.H., Baan R.A., van Delft J.H. DNA adducts, mutant frequencies, and mutation spectra in various organs of lambda lacZ mice exposed to ethylating agents. Environ. Mol. Mutagen. 1998;31:18–31. [PubMed] [Google Scholar]
- 50.Souliotis V.L., van Delft J.H., Steenwinkel M.J., Baan R.A., Kyrtopoulos S.A. DNA adducts, mutant frequencies and mutation spectra in lambda lacZ transgenic mice treated with N-nitrosodimethylamine. Carcinogenesis. 1998;19:731–739. doi: 10.1093/carcin/19.5.731. [DOI] [PubMed] [Google Scholar]
- 51.Ono T., Ikehata H., Nakamura S., Saito Y., Komura J., Hosoi Y., Yamamoto K. Molecular nature of mutations induced by a high dose of x-rays in spleen, liver, and brain of the lacZ-transgenic mouse. Environ. Mol. Mutagen. 1999;34:97–105. [PubMed] [Google Scholar]
- 52.Hakura A., Tsutsui Y., Sonoda J., Tsukidate K., Mikami T., Sagami F. Comparison of the mutational spectra of the lacZ transgene in four organs of the MutaMouse treated with benzo[a]pyrene: target organ specificity. Mutat. Res. 2000;447:239–247. doi: 10.1016/s0027-5107(99)00213-4. [DOI] [PubMed] [Google Scholar]
- 53.Ono T., Ikehata H., Vishnu Priya P., Uehara Y. Molecular nature of mutations induced by irradiation with repeated low doses of X-rays in spleen, liver, brain and testis of lacZ-transgenic mice. Int. J. Radiat. Biol. 2003;79:635–641. doi: 10.1080/09553000310001596931. [DOI] [PubMed] [Google Scholar]
- 54.Staedtler F., Suter W., Martus H.J. Induction of A: T to G: C transition mutations by 5-(2-chloroethyl)-2'-deoxyuridine (CEDU), an antiviral pyrimidine nucleoside analogue, in the bone marrow of Muta Mouse. Mutat. Res. 2004;568:211–220. doi: 10.1016/j.mrfmmm.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 55.Dollé M.E.T., Busuttil R.A., Garcia A.M., Wijnhoven S., van Drunen E., Niedernhofer L.J., van der Horst G., Hoeijmakers J.H.J., van Steeg H., Vijg J. Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat. Res. 2006;596:22–35. doi: 10.1016/j.mrfmmm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 56.Kalnins A., Otto K., Rüther U., Müller-Hill B. Sequence of the lacZ gene of Escherichia coli. EMBO J. 1983;2:593–597. doi: 10.1002/j.1460-2075.1983.tb01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ring M., Armitage I.M., Huber R.E. m-Fluorotyrosine substitution in beta-galactosidase; evidence for the existence of a catalytically active tyrosine. Biochem. Biophys. Res. Commun. 1985;131:675–680. doi: 10.1016/0006-291x(85)91290-2. [DOI] [PubMed] [Google Scholar]
- 58.Cupples C.G., Miller J.H., Huber R.E. Determination of the roles of Glu-461 in beta-galactosidase (Escherichia coli) using site-specific mutagenesis. J. Biol. Chem. 1990;265:5512–5518. [PubMed] [Google Scholar]
- 59.Edwards R.A., Cupples C.G., Huber R.E. Site specific mutants of beta-galactosidase show that Tyr-503 is unimportant in Mg2+ binding but that Glu-461 is very important and may be a ligand to Mg2+ Biochem. Biophys. Res. Commun. 1990;171:33–37. doi: 10.1016/0006-291x(90)91352-s. [DOI] [PubMed] [Google Scholar]
- 60.Roth N.J., Huber R.E. Site directed substitutions suggest that His-418 of beta-galactosidase (E. coli) is a ligand to Mg2+ Biochem. Biophys. Res. Commun. 1994;201:866–870. doi: 10.1006/bbrc.1994.1781. [DOI] [PubMed] [Google Scholar]
- 61.Yuan J., Martinez-Bilbao M., Huber R.E. Substitutions for Glu-537 of beta-galactosidase from Escherichia coli cause large decreases in catalytic activity. Biochem. J. 1994;299(Pt 2):527–531. doi: 10.1042/bj2990527. 10.1042/bj2990527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez-Bilbao M., Gaunt M.T., Huber R.E. E461H-beta-galactosidase (Escherichia coli): altered divalent metal specificity and slow but reversible metal inactivation. Biochemistry. 1995;34:13437–13442. doi: 10.1021/bi00041a022. [DOI] [PubMed] [Google Scholar]
- 63.Richard J.P., Huber R.E., Lin S., Heo C., Amyes T.L. Structure-reactivity relationships for beta-galactosidase (Escherichia coli, lac Z). 3. Evidence that Glu-461 participates in Bronsted acid-base catalysis of beta-D-galactopyranosyl group transfer. Biochemistry. 1996;35:12377–12386. doi: 10.1021/bi961028j. [DOI] [PubMed] [Google Scholar]
- 64.Roth N.J., Huber R.E. Glu-416 of beta-galactosidase (Escherichia coli) is a Mg2+ ligand and beta-galactosidases with substitutions for Glu-416 are inactivated, rather than activated, by MG2+ Biochem. Biophys. Res. Commun. 1996;219:111–115. doi: 10.1006/bbrc.1996.0190. [DOI] [PubMed] [Google Scholar]
- 65.Roth N.J., Huber R.E. The beta-galactosidase (Escherichia coli) reaction is partly facilitated by interactions of His-540 with the C6 hydroxyl of galactose. J. Biol. Chem. 1996;271:14296–14301. doi: 10.1074/jbc.271.24.14296. [DOI] [PubMed] [Google Scholar]
- 66.Roth N.J., Rob B., Huber R.E. His-357 of beta-galactosidase (Escherichia coli) interacts with the C3 hydroxyl in the transition state and helps to mediate catalysis. Biochemistry. 1998;37:10099–10107. doi: 10.1021/bi972796t. [DOI] [PubMed] [Google Scholar]
- 67.Penner R.M., Roth N.J., Rob B., Lay H., Huber R.E. Tyr-503 of beta-galactosidase (Escherichia coli) plays an important role in degalactosylation. Biochem. Cell. Biol. 1999;77:229–236. doi: 10.1139/o99-042. [DOI] [PubMed] [Google Scholar]
- 68.Huber R.E., Hlede I.Y., Roth N.J., McKenzie K.C., Ghumman K.K. His-391 of beta-galactosidase (Escherichia coli) promotes catalyses by strong interactions with the transition state. Biochem. Cell. Biol. 2001;79:183–193. doi: 10.1139/o00-101. [DOI] [PubMed] [Google Scholar]
- 69.Xu J., McRae M.A.A., Harron S., Rob B., Huber R.E. A study of the relationships of interactions between Asp-201, Na+ or K+, and galactosyl C6 hydroxyl and their effects on binding and reactivity of beta-galactosidase. Biochem. Cell. Biol. 2004;82:275–284. doi: 10.1139/o04-004. [DOI] [PubMed] [Google Scholar]
- 70.Juers D.H., Rob B., Dugdale M.L., Rahimzadeh N., Giang C., Lee M., Matthews B.W., Huber R.E. Direct and indirect roles of His-418 in metal binding and in the activity of beta-galactosidase (E. coli) Protein Sci. 2009;18:1281–1292. doi: 10.1002/pro.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dugdale M.L., Dymianiw D.L., Minhas B.K., D'Angelo I., Huber R.E. Role of Met-542 as a guide for the conformational changes of Phe-601 that occur during the reaction of β-galactosidase (Escherichia coli) Biochem. Cell. Biol. 2010;88:861–869. doi: 10.1139/O10-009. [DOI] [PubMed] [Google Scholar]
- 72.Dugdale M.L., Vance M.L., Wheatley R.W., Driedger M.R., Nibber A., Tran A., Huber R.E. Importance of Arg-599 of beta-galactosidase (Escherichia coli) as an anchor for the open conformations of Phe-601 and the active-site loop. Biochem. Cell. Biol. 2010;88:969–979. doi: 10.1139/O10-144. [DOI] [PubMed] [Google Scholar]
- 73.Wheatley R.W., Kappelhoff J.C., Hahn J.N., Dugdale M.L., Dutkoski M.J., Tamman S.D., Fraser M.E., Huber R.E. Substitution for Asn460 cripples beta-galactosidase (Escherichia coli) by increasing substrate affinity and decreasing transition state stability. Arch. Biochem. Biophys. 2012;521:51–61. doi: 10.1016/j.abb.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 74.R Core Team . 2016. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 75.Oksanen J., Kindt R., Legendre P., O’Hara B., Stevens M.H.H. The vegan package. Community Ecology Package. 2007;10:631–637. [Google Scholar]
- 76.Grant B.J., Rodrigues A.P.C., ElSawy K.M., McCammon J.A., Caves L.S.D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 77.Touw W.G., Baakman C., Black J., te Beek T.A., Krieger E., Joosten R.P., Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015;43:D364–D368. doi: 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 79.Pommié C., Levadoux S., Sabatier R., Lefranc G., Lefranc M.P. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recogn. 2004;17:17–32. doi: 10.1002/jmr.647. [DOI] [PubMed] [Google Scholar]
- 80.Juers D.H., Hakda S., Matthews B.W., Huber R.E. Structural basis for the altered activity of Gly794 variants of Escherichia coli beta-galactosidase. Biochemistry. 2003;42:13505–13511. doi: 10.1021/bi035506j. [DOI] [PubMed] [Google Scholar]
- 81.Jancewicz L.J., Wheatley R.W., Sutendra G., Lee M., Fraser M.E., Huber R.E. Ser-796 of beta-galactosidase (Escherichia coli) plays a key role in maintaining a balance between the opened and closed conformations of the catalytically important active site loop. Arch. Biochem. Biophys. 2012;517:111–122. doi: 10.1016/j.abb.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 82.Sutendra G., Wong S., Fraser M.E., Huber R.E. Beta-galactosidase (Escherichia coli) has a second catalytically important Mg2+ site. Biochem. Biophys. Res. Commun. 2007;352:566–570. doi: 10.1016/j.bbrc.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 83.Huber R.E., Hakda S., Cheng C., Cupples C.G., Edwards R.A. Trp-999 of beta-galactosidase (Escherichia coli) is a key residue for binding, catalysis, and synthesis of allolactose, the natural lac operon inducer. Biochemistry. 2003;42:1796–1803. doi: 10.1021/bi0270642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data regarding the mutations reported in study are shown in supplementary material and available in Mendeley Data (see key resources table).
-
•
RMarkdown scripts are available at GitHub (see key resources table).