

Summary
Given the importance of the kinin B1 receptor in insulin and leptin hormonal regulation, which in turn is crucial in maternal adaptations to ensure nutrient supply to the fetus, we investigated the role of this receptor in maternal metabolism and fetoplacental development. Wild-type and kinin B1 receptor-deficient (B1KO) female mice were mated with male mice of the opposite genotype. Consequently, the entire litter was heterozygous for kinin B1 receptor, ensuring that there would be no influence of offspring genotype on the maternal phenotype. Maternal kinin B1 receptor blockade reduces adiponectin secretion by adipose tissue ex vivo, consistent with lower adiponectin levels in pregnant B1KO mice. Furthermore, fasting insulinemia also increased, which was associated with placental insulin resistance, reduced placental glycogen accumulation, and heavier offspring. Therefore, we propose the combination of chronic hyperinsulinemia and reduced adiponectin secretion in B1KO female mice create a maternal obesogenic environment that results in heavier pups.
Subject areas: Pregnancy, Physiology, Molecular biology
Graphical abstract
Highlights
-
•
B1R controls adiponectin secretion by parametrial adipose tissue
-
•
B1KO triggers decreased maternal levels of HMW adiponectin and hyperinsulinemia
-
•
Maternal B1R deficiency results in placental insulin resistance
-
•
B1KO results in an obesogenic intrauterine environment and heavier pups
Pregnancy; Physiology; Molecular biology
Introduction
Hormonal adaptations during pregnancy are crucial for metabolic adaptation, prioritizing the passage of nutrients to the embryo/fetus to the detriment of their use by the mother.1,2 These endocrine-metabolic adaptations fluctuate during pregnancy according to the stage of fetal-placental development, causing the same hormone to be present at different levels throughout pregnancy.
Adiponectin, insulin, and leptin are key hormones playing a role in gestational metabolic adaptation. In both humans and rodents, adiponectin displays lower levels in the second half of pregnancy, while insulin and leptin present higher levels.3,4,5,6 High levels of insulin and leptin associated with reduced levels of adiponectin at the end of pregnancy are associated with maternal resistance to insulin and leptin, which ensure proper mobilization of nutrients to the fetus.2,6 These adjustments in maternal metabolism need to be orchestrated for adequate fetal-placental development. Metabolic dysfunction during diabetes, obesity, and malnutrition affect fetal and placental growth, increasing the risk of cardiovascular and metabolic diseases in the offspring.7,8,9
The kallikrein-kinin system (KKS), widely known for its role in inflammation,10 nociception,11 and vascular permeability,12 is also involved in the regulation of hepatic glycemic metabolism13,14 as well as adipose tissue homeostasis.15,16 KKS stimulates the increase of glucose uptake in adipocytes and myoblasts, as well as the reduction of hepatic gluconeogenesis, by promoting the insulin-induced translocation of glucose transporter type 4 (GLUT4) to the plasma membrane and the increase in insulin sensitivity,13,15,16,17,18 demonstrating that kinins play a crucial role in the pathogenesis of several metabolic diseases, such as obesity and diabetes.
The actions of the KKS are mediated through the kinin B1 (B1R) and B2 (B2R) receptors. In the absence of B1R, serum insulin and leptin levels are lower, while peripheral insulin sensitivity and central leptin sensitivity are higher.19,20,21 B1R is involved in pancreatic vascularization and insulin secretion, and the lack of B1R reduced the number of pancreatic islets and insulin content in male mice, reducing plasma insulin levels after 8 h of fasting, at the same time that increased insulin sensitivity.19
Besides the regulation of insulin homeostasis by B1R, this receptor modulates leptin homeostasis in an insulin-dependent manner. In normoinsulinemic male animals, the pharmacological activation of B1R by des-arg9-bradykinin (DBK) stimulates leptin secretion by adipose tissue, leading to increased serum leptin levels, which does not occur in the absence of insulin or elevated insulin levels (hyperinsulinemia), for example.21 B1R is involved in the expansion of adipose tissue and predisposition to obesity induced by a high-fat diet.16,22 The lack of B1R reduces serum levels of leptin as well as the adiposity index, culminating in resistance to diet-induced obesity in B1R-deficient mice (B1KO).16,20,22 Furthermore, recently, the participation of B1R in the change of energy source during the metabolic stress caused by fasting was also demonstrated, with a preference for a more glycolytic metabolism in the absence of this receptor.23
Despite the consolidated body of evidence linking the B1R with endocrine-metabolic modulation, little is known about the role of B1R in maternal metabolism. In addition, the function of maternal B1R in the intrauterine environment remains unknown. Here we hypothesized that the B1R plays a role in this context, affecting the maternal microenvironment due to reduced insulin and leptin levels, leading to fetuses being small for gestational age. Thus, we aimed to evaluate the role of this receptor in maternal metabolism, fetal-placental development, and offspring status.
Results
The lack of B1R increased blood glucose and insulin levels in female mice
While the role of B1R in glycemic metabolism and insulin homeostasis has been already well-characterized in B1KO males, no data on females were available. Therefore, we investigated glucose and insulin tolerance in virgin B1KO females. Biochemical parameters such as insulinemia, glycemia, ketonemia, and cholesterol and triglyceride levels were also evaluated. Although basal glycemia (random-fed state) and fasting insulinemia were increased in B1KO females (Figures 1A and 1B), there was no difference in fasting glycemia (Figure 1A), glucose tolerance (Figure 1C), or insulin levels 30 min after glucose injection (Figure 1D). There were also no changes in insulin tolerance (Figure 1E). Fasting ketonemia was also increased in B1KO females (Figure 1F), indicating increased ketogenesis even in the presence of higher insulin levels, suggesting hepatic resistance to this hormone. HMG-CoA lyase, an enzyme suppressed by high insulin levels, is required for ketogenesis.24,25 However, according to the pyruvate tolerance test, the hyperinsulinemia repressed the gluconeogenesis in the absence of B1R (Figure 1G). At the same time, high insulin levels also have increased the levels of cholesterol in B1KO females (Figure 1H), with no change in triglycerides (Figure 1I).
Figure 1.

The lack of B1R increased blood glucose and insulin levels in female mice
Effects of B1R deficiency on feeding and fasting glycemia (A); serum fasting insulin level (B); glucose tolerance test (C); serum insulin level 30 min after glucose injection test (D); insulin tolerance (E); feeding and fasting β-ketone levels (F); pyruvate tolerance test (G); and serum cholesterol (H) and triglycerides (I) levels. The lack of B1R increases feeding glycemia (p < 0.0001), fasting insulin (p = 0.004), β-ketone (p = 0.001), and cholesterol (p = 0.003) levels and reduces the pyruvate tolerance (genotype as the source of variation: p = 0.032). Interaction between genotype and feed state was found for glycemia (p = 0.0001) and ketone levels (p = 0.001); and between genotype and time for pyruvate tolerance test (p = 0.001). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Data were compared by two-way ANOVA with Sidak’s multiple comparison test (A, C, D, F, and G), Mann-Whitney (B and I), and Student’s unpaired t test (D and H) at GraphPad Prism 7. Values expressed as mean ± SD of 3–7 mice per group. AUC – area under the curve. Lilac circle = virgin WT female; Yellow square = virgin B1KO female.
B1KO females are hyperphagic and gain more weight at the end of pregnancy
Consistent with kinin B1 receptor playing a role in maternal metabolism, we observed higher weight gain at the end of gestation in B1KO females accompanied by hyperphagia in the second gestational week compared to wild-type (WT) females (Figures 2A and 2B), without any change in litter size (Figure 2C), resulting by 11.4% heavier pregnant mice in the absence of B1R (Table 1). Placental and fetal mass did not contribute to the difference in gestational weight gain since they did not differ between WT and B1KO mothers (Figures 2D and 2E). Furthermore, the fetal mass/placental mass ratio was analyzed as a parameter of placental efficiency, which showed no difference between WT and B1KO mothers (Figure 2F). Interestingly, even before pregnancy (virgin females), the lack of B1R increased by 7.9% body mass, as well as increased liver and muscle mass by 24% and 16.5%, respectively, while reducing parametrial adipose tissue by 39% (Table 1). As expected, the liver mass doubled in females from both genotypes at the end of pregnancy.26,27 By contrast, the reduction of parametrial adipose tissue during pregnancy did not occur in B1KO females (Table 1), suggesting an impairment in fat metabolization in the absence of B1R.
Figure 2.

B1KO females are hyperphagic and gain more weight at the end of pregnancy body
Influence of B1R deficiency on weight gain (A) and food intake (B) during pregnancy; litter size (C), placental mass (D), litter weight (E), and placental efficiency mean per dam (F). The B1R absence increases the gestational body weight gain (genotype as the source of variation: p = 0.032) and food intake (p = 0.018), with no impact on litter size, placental weight, litter weight, and placental efficiency mean. Interaction between genotype and time was observed for gestational body weight gain (p = 0.036). ∗p < 0.05, ∗∗p < 0.01. Data were compared by two-way ANOVA with Sidak’s multiple comparison test (A), Mann-Whitney (C), and Student’s unpaired t test (B, D, E, and F) at GraphPad Prism 7. Values expressed as mean ± SD of 8–11 mice per group. AUC – area under the curve. Blue = WT dam; Red = B1KO dam. See also Figure S2.
Table 1.
Body composition and tissue weight at virgin females and at the end of pregnancy
| Body weight (g) |
Parametrial adipose tissue (mg) |
Liver (mg) |
Muscle (mg) |
|||||
|---|---|---|---|---|---|---|---|---|
| Virgin | Pregnant | Virgin | Pregnant | Virgin | Pregnant | Virgin | Pregnant | |
| WT | 19.59 ± 0.603 | 34.85 ± 2.643 | 358 ± 70.59 | 229.1 ± 98.46 | 805.8 ± 41.91 | 1,683 ± 169.3 | 93.67 ± 7.35 | 108.2 ± 19.09 |
| B1KO | 21.13 ± 1.391∗ | 38.82 ± 3.096∗∗ | 217.2 ± 40.84∗ | 231.4 ± 82.02 | 1,002 ± 76.56∗ | 1,923 ± 241.9∗ | 109.2 ± 4.57∗ | 120.5 ± 24.86 |
∗p < 0.05 to WT, ∗∗p < 0.01 to WT. Values expressed as mean ± SD of 6–7 virgin female mice per group and 10–11 pregnant mice per group.
B1KO females have higher fasting insulinemia at the end of pregnancy
Given the importance of insulin in coordinating maternal metabolic adaptations and nutrient delivery to the fetus during pregnancy, we measured insulin levels and glucose tolerance in females at gestational day 16.5 (GD16.5), when there is a reported peak of insulin secretion in rodents.28,29 Fasting glucose was unchanged (Figure 3A), as were oral glucose tolerance (Figure 3B) and serum insulin levels after 30 min of glucose administration (Figure 3C). However, fasting insulin levels at the end of pregnancy (GD18.5) were increased (Figure 3D), although there was no change in glycemia (Figure 3E). Similarly, there were no differences in cholesterol and triglyceride levels (Figures 3F and 2G), although fasting ketonemia tended to be lower (Figure 3H). Therefore, our data showed B1KO dams are hyperinsulinemic, despite normal oral glucose tolerance test (OGTT) and insulin levels 30 min after glucose-stimulation, suggesting insulin resistance with unchanged glucose tolerance and insulin production in B1KO females. Moreover, Bdkrb2 expression remained unchanged in maternal metabolic tissues such as the liver, muscle, and adipose tissue, reaffirming that the observed effects are due to B1R absence instead of B2R increase (Figure S1).
Figure 3.

B1KO females have higher fasting insulinemia at the end of pregnancy
Consequence of B1R deficiency on gestational metabolism at gestational day 16.5 (GD16.5) and 18.5 (GD18.5): fasting glycemia (A) and glucose tolerance test at GD16.5 (B), serum insulin level 30 min after glucose gavage (C); fasting insulin (D), blood glucose (E), cholesterol (F), triglycerides (G), and ketone bodies (H) levels at GD18.5. Maternal B1R deficiency increases fasting insulin levels at GD18.5 (p = 0.016). ∗p < 0.05. Data were compared by two-way ANOVA with Sidak’s multiple comparison test (B), Mann-Whitney (A, C, D, F, and G), and Student’s unpaired t test (E and H) at GraphPad Prism 7. Values expressed as mean ± SD of 3–11 mice per group. AUC – area under the curve. Blue = WT dam; Red = B1KO dam. See also Figure S1.
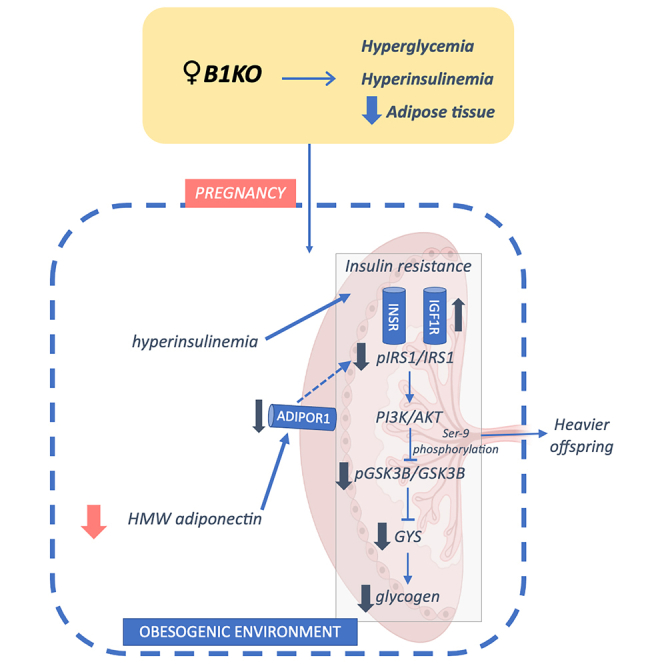
B1KO mothers have lower levels of high molecular weight adiponectin and lower expression of leptin receptors and ADIPOR1 in the placenta
Leptin and adiponectin are adipokines that play a crucial role in maternal adaptations to pregnancy as well as fetal and placental development. Their maternal levels are also related to disease predisposition in the offspring. Therefore, we evaluated the maternal levels of leptin and adiponectin and the expression of their receptors in the placenta. Additionally, some inflammatory mediators have been linked to adiponectin production,30 suggesting that B1R could also modulate adiponectin secretion. Thus, we evaluated the influence of B1R in adipose tissue ex vivo adiponectin secretion.
Maternal serum levels of leptin or placental production of leptin was not altered in B1KO (Figures 4A and 4B). However, gene expression of the two main isoforms of Lepr, isoforms a and b, were reduced in the placentas of B1KO mothers (Figures 4C and 4D). Although there was no difference in total maternal serum adiponectin in B1KO mothers (Figure 5A), its high molecular weight and most metabolically relevant form was reduced (Figure 5B). The same was found for ADIPOR1 levels in the placenta (Figure 5C), whereas ADIPOR2 did not differ between maternal genotypes (Figure 5D). Ex vivo experiments confirmed that inhibition of B1R by the specific antagonist R715 reduced adiponectin secretion from parametrial fat tissue (Figure 5E), demonstrating that adipose tissue B1R controls adiponectin secretion without regulating adiponectin mRNA expression (Figure 5F).
Figure 4.

B1KO mothers have lower expression of leptin receptors in the placenta
Effect of the lack of maternal B1R on serum and placental leptin levels and the gene expression of their receptors in the placenta: serum (A) and placental (B) leptin concentration; isoform a (C) and isoform b (D) of Lepr gene expression. Maternal B1R deficiency reduces leptin receptor expression in heterozygous placenta (isoform a: p = 0.005; isoform b: p = 0.032) with no changes in serum and placental leptin levels. ∗p < 0.05, ∗∗p < 0.01. Data were compared by Mann-Whitney (A) and unpaired t test (B, C, and D) at GraphPad Prism 7. Values expressed as mean ± SD of 4–10 mice per group for serum and placental leptin, or 8–10 placentas per group (2 placentas per dam). Lepr – leptin receptor gene. Blue = WT dam; Red = B1KO dam. See also Figures S3 and S4).
Figure 5.

B1KO mothers have lower levels of high molecular weight adiponectin and lower expression of ADIPOR1 in the placenta
Influence of B1R absence on adiponectin secretion and signaling: serum total (A) and high molecular weight (HMW) (B) adiponectin levels; placental ADIPOR1 (C) and ADIPOR2 (D) protein content normalized by ACTB, and their respective representative western blotting images; adiponectin secretion ex vivo by parametrial adipose tissue explants (E) and its respective gene expression (F). The lack of maternal B1R represses ADIPOR1 protein content (p = 0.012) in the heterozygous placenta with no change in ADIPOR2 protein content. Full-length blots are presented in Figures S5C and S5D. Further to the reduction of placental ADIPOR1 expression, maternal HMW adiponectin (p = 0.027) is also reduced. Moreover, the B1R blockage by its antagonist R715 inhibited the adiponectin secretion ex vivo by parametrial adipose tissue explants (p = 0.0007) (treatment as the source of variation: p = 0.001) without modulating mRNA level. Interaction between treatment and time was found for adiponectin secretion ex vivo (p = 0.02). ∗p < 0.05, ∗∗∗p < 0.001. Data were compared by Mann-Whitney (C and D), unpaired t test (A, B, E, and F), and two-way ANOVA with Sidak’s multiple comparison test (G) at GraphPad Prism 7. Values expressed as mean ± SD of 6 and 10 mice per group for total and HMW adiponectin, respectively, or 6–10 placentas per group (2 placentas per dam), or 4–5 mice per group for adiponectin secretion ex vivo. ADIPOR1 – adiponectin receptor 1; ADIPOR2 – adiponectin receptor 2; AUC – area under the curve. Blue = WT dam; Red = B1KO dam; Black Circle = control; Maroon square = R715. See also Figure S6.
Maternal B1R deficiency represses gene expression of large neutral amino acid transporter 1
After observing metabolic changes in B1KO mothers, even without differences in fetal-placental mass and efficiency (Figures 2D and 2F), we evaluated placental markers related to the passage of nutrients to the fetus. First, we verified whether maternal B1R deficiency would modulate the expression of kinin receptors in the placenta (Figure S2), which remained unchanged.
After verifying the expression pattern of kinin receptors in heterozygous placentas, we analyzed the gene expression of amino acid, fatty acid, and glucose transporters. The transport of amino acids across the placenta to the fetus plays a crucial role in fetal growth and development. Furthermore, it is proposed in the literature the relationship between leptin and the transport of amino acids, especially the sodium-coupled neutral amino acid transporters (SNAT), also called System A amino acid transporters.6,31 However, despite repressed leptin receptors expression (Figures 4C and 4D), no alteration was observed in SNAT mRNA expression (Figures S3A–S3C).
Among the large neutral amino acid transporters (LAT), responsible for essential aromatic amino acids transport to the fetus, only Slc7a5 (LAT1) had diminished expression in placentas from B1KO mothers (Figure S3D). Moreover, Slc3a2 expression was also reduced (Figure S3F)—Slc3a2 is the gene encoding the 4F2hc protein, which heterodimerizes with LAT and promotes the transport of amino acids.32
Regarding fatty acids, in spite of a tendency to increase gene expression of lipoprotein lipase in the placentas of B1KO mothers, there was no alteration of the gene expression of the fatty acid transport proteins studied here (FATPs, CD36, and FABP-pm) (Figure S4). Moreover, there were no changes in the gene expression of GLUT 1 and 3, the main isoforms responsible for the transport by facilitated diffusion of glucose to the fetus (insulin-independent glucose transport) (Figure S5). Therefore, our data indicated that maternal B1R absence did not modulate most macronutrient transporters except for an apparent reduction in certain amino acid transporters. However, it is worth noting the limitation of the study to measure transplacental nutrient transport, with suggestions based on mRNA levels, which are susceptible to translational modulations.
Maternal B1R deficiency reduces glycogen synthesis in the heterozygous placenta
Considering that fasting insulin levels of B1KO mothers were increased, after analyzing the gene expression of nutrient transporters in the placenta, we also evaluated the gene expression of the placental lactogen hormone 2 (Prl3b1), a crucial modulator of maternal insulin secretion by pancreatic β-cells.28 However, Prl3b1 showed no difference in placentas of WT vs. B1KO mothers (Figure 6A).
Figure 6.

Maternal B1R deficiency reduces glycogen synthesis in the heterozygous placenta
Effect of maternal B1R deficiency on placental glycogenesis: Prl3b1 (PL-II) (A), Insr (B), Igf1r (C), and Gys1 (G) gene expression; phosphorylated and total IRS1 and GSK3B content by western blotting and their respective representative images (D and E); placental glycogenesis in B1KO dam representation (F); placental glycogen content (H). Full-length blots are presented in Figures S5A and S5B. Maternal B1R deficiency increases Igf1r expression (p = 0.040) as well as reduces phosphorylated-to-total IRS1 ratio (p = 0.045), phosphorylated and total GSK3B ratio (p = 0.032), Gys1 expression (p = 0.002) and glycogen content (p = 0.038) in the heterozygous placenta. ∗p < 0.05, ∗∗p < 0.01. Data were compared by unpaired t test at GraphPad Prism 7. Values expressed as mean ± SD of 5–10 placentas per group (1 or 2 placentas per dam). AKT – protein kinase B; GSK3B – glycogen synthase kinase 3 beta; Gys1 – glycogen synthase 1 gene expression; Igf1r – insulin-like growth factor 1 receptor gene expression; Insr – insulin receptor gene expression; IRS1 – insulin receptor substrate 1; PI3K – phosphoinositide 3-kinase; PL-II – mouse placental lactogen II. Blue = WT dam; Red = B1KO dam. See also Figures S5 and S6.
Additionally, we investigated the placental insulin signaling pathway. Insulin acts via phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway triggered by insulin receptor substrate 1 and 2 (IRS1/2). Further insulin receptor (INSR), insulin binds with low affinity to insulin-like growth factor 1 receptor (IGF1R).33 Despite no difference in Insr expression (Figure 6B), we observed an increase in Igf1r expression (Figure 6C) and a reduced degree of phosphorylation of IRS1 at tyrosine (Figure 6D) in the placentas of B1KO mothers, suggesting a less active insulin signaling.
Considering the relationship between placental glycogen storage and fetal development, as well as that the insulin stimulates glycogenesis via inhibition of glycogen synthase kinase 3 beta (GSK3B) via PI3K/AKT signaling pathway, we analyzed the placental glycogen storage and the activation of the GSK3B-glycogen synthase 1 (GYS1) pathway, responsible for glycogen synthesis. We found increased total GSK3B in the placentas of B1KO mothers, although the degree of phosphorylation of GSK3B at serine-9 was lower (Figure 6E). We hypothesized that a reduction in the ratio between phosphorylated GSK3B and total GSK3B would lead to inhibition of GYS, as a consequence of decreased IRS1 phosphorylation by INSR (Figure 6F). Consistently, Gys1 expression was repressed (Figure 6G) and placental glycogen content was lower (Figure 6H) in the placentas of B1KO mothers, indicating a reduction in glycogenesis in these placentas.
The absence of maternal B1R increases offspring mass until the thirtieth day of life
After analyzing maternal metabolism and fetal-placental development due to maternal B1R deletion, we evaluated the impact of the absence of this receptor on the weight progression of the offspring obtained by vaginal delivery. B1KO mothers' offspring presented greater mass during the first 30 days of life (Figure 7).
Figure 7.

The absence of maternal B1R increases offspring mass until the thirtieth day of life
Maternal B1R deficiency increases heterozygous offspring body weight at 10 (p < 0.0001), 21 (p < 0.0001), and 30 (p = 0.002) days after birth. ∗∗∗∗p < 0.0001, ∗∗p < 0.01. Data were compared by Mann-Whitney (P30) and unpaired t test (P1, P10, and P30) at GraphPad Prism 7. Values expressed as mean ± SD of 35 and 56 mice on WT dam and B1KO dam group respectively at P1, or 26 mice on WT dam group and 35 mice on B1KO dam group at P10, P21, and P30. Blue = WT dam; Red = B1KO dam.
Discussion
The present work reveals that B1R controls maternal metabolism, which in turn affects offspring weight. It is worth noting the scarcity of studies that aimed to investigate B1R and maternal metabolism; since the studies that showed the role of B1KO on metabolism were carried out in males. This is somewhat surprising, given the existing literature, which we corroborate in the present study, demonstrating the expression of kallikrein and kinin receptors in the placenta and the other reproductive tissues.34,35,36,37,38,39,40 Moreover, some authors have shown that high molecular weight kininogen (HMWK) and its activated form (HKa) are essential for the maintenance of pregnancy homeostasis. HKa and HMWK cleavage-derived peptides, such as bradykinin, increase in the course of pregnancy, which sheds light on their potential consideration as biomarkers during pregnancy-associated disorders.41
KKS disbalances have already been related to gestational loss and preeclampsia.42,43 Bradykinin via B2R has promoted the migration and invasion in the first-trimester trophoblastic cell line HTR-8/SVneo through modifications of the actin cytoskeleton that assist in cell migration,44 important phenotypes during the blastocyst implantation process. In addition to the importance of B2R in the invasion phase of the uterine endometrium during the implantation process, it participates in the regulation of placental blood flow through the spiral arteries remodeling and blood pressure control, participating in the etiology of preeclampsia, for example.45,46,47,48,49,50
Most of the studies have focused on B2R in preeclampsia etiology or trophoblast migration and invasion. As aforementioned, few studies have focused on B1R during pregnancy. In spite of that, it is already known that B1R has no effects on trophoblast migration and invasion, but it participates in angiogenesis modulation.44,46,51 Additionally, Hoegh et al. have described a reduction of placental B1R expression in preeclampsia, suggesting the involvement of B1R in pregnancy disorders.43 As cited previously, the studies on kinin receptors and fetal-placental development are all focused on the inflammatory and angiogenic potential of the KKS.43,44,49,52 This gap reinforces the importance of evaluating the role of kinin receptors in maternal and fetal-placental metabolism.
During pregnancy, the female metabolism passes through modifications that guarantee adequate fetal-placental development. Pregnancy can be divided into two phases, anabolic and catabolic. The anabolic phase comprehends a lipogenic stage, with maternal fat accumulation to be used during the catabolic phase. The catabolism is present at the end of gestation, characterized by a lipolytic metabolism, with increased maternal adipose tissue lipolysis, hepatic gluconeogenesis, and ketogenesis to guarantee fetal glucose availability.53,54 Insulin and leptin resistance plays a pivotal role in maternal adaptations. However, disturbances in maternal metabolism adaptations result in pregnancy disorders such as gestational diabetes and preeclampsia, predisposing the offspring to cardiometabolic diseases.2,8,55,56,57,58
B1KO male mice are insulin and leptin-sensitive with lower levels of these essential gestational hormones.19,20 In this way, our initial hypothesis was that maternal deficiency of B1R could disturb the insulin and leptin resistance necessary for good placental and fetal growth and development, leading to small for gestational age fetuses. Nevertheless, although we did not investigate the central responsiveness to leptin in B1KO females, we observed that they did not present lower leptinemia as virgins or at the end of pregnancy, contrary to what has been seen in males.20
Similarly to the males, virgin B1KO females presented reduced adipose tissue. Conversely, they are heavier, a characteristic that is pronounced with pregnancy. At the end of gestation, the maternal fat accumulated during the anabolic stage decreases by lipolysis of adipose tissue, as observed by less adipose tissue content in WT females at GD18.5. Different gestational complications are linked to adipocyte dysfunction as preeclampsia and gestational diabetes mellitus.59 In preeclampsia, for instance, the expansion of subcutaneous adipose tissue is limited by the impairment of adipocyte maturation caused by maternal insulin resistance.59,60 Our data indicate that B1R is essential for maternal adipose tissue homeostasis since pregnancy did not alter adipose tissue mass in B1KO females. B1R has already been demonstrated as crucial for adipose tissue expansion.22 Although we have not analyzed the maternal adiposity in the first half of pregnancy, we suggest B1KO females were not able to perform adipogenesis and expand the adipose tissue, similarly to the already observed in preeclampsia. The defective fat accumulation could culminate in a metabolic shift, as an increase in ketone body and cholesterol consumption, contributing to the reduction of ketone body and no raising of cholesterol levels at the end of pregnancy.
In addition to the differences between the genders, while B1KO males have lower pancreatic insulin content, reduced glycemia, and higher insulin responsiveness,19,23 B1KO females are hyperglycemic and hyperinsulinemic. These characteristics suggest that B1KO females have increased hepatic insulin resistance and, perhaps, beta cell hyperplasia, which leads to an increase in the synthesis and secretion of insulin in females. Since the absence of B1R has different repercussions in males and females, our results in virgin females substantially modified our hypothesis, reaffirming how important it is to consider sexual dimorphism in metabolic analyses.
Despite the suggestion of increased hepatic insulin resistance, when challenged with pyruvate, B1KO females showed lower blood glucose elevation, suggesting that high insulin levels repressed hepatic gluconeogenesis. The decreased pyruvate sensitivity in B1KO females discarded the hypothesis of hepatic insulin resistance. Although there was no difference in glucose and insulin tolerance, it has previously been shown that insulin signaling can vary among different tissues. Male B1KO mice, although more responsive to insulin, show insulin resistance in adipose tissue,16 which indicates that females could have a similar phenotype.
Moreover, although insulin signaling has not been investigated in different tissues of B1KO females, it has already been demonstrated that B1R participates in the translocation of GLUT4 to the plasma membrane in adipocytes, both basal and induced by insulin.16 Based on this, we suggest that the observed hyperglycemia may be due to impaired glucose uptake in some tissues, such as adipose tissue. Additionally, B1KO females were hyperphagic during pregnancy, suggesting that instead of the lower central sensitivity to leptin, as initially thought, B1KO females must have increased resistance to leptin. In line with the absence of lower leptinemia, we found reduced levels of the mRNAs encoding the two main isoforms of the leptin receptor, isoforms a and b, in the placentas of B1KO mothers, suggestive of peripheral leptin resistance.
Additionally, B1KO mothers were hyperinsulinemic and had reduced HMW adiponectin levels. Our ex vivo experiments also demonstrated that B1R blockage in parametrial adipose tissue reduced adiponectin secretion, indicating a direct modulation of adiponectin secretion by B1R. Adiponectin acts through ADIPOR1 and ADIPOR2, which binds to the adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), responsible for adiponectin signal transduction. APPL1 activated by ADIPOR1 induces IRS1 phosphorylation, promoting insulin sensitivity.61,62 Associated with the maternal reduced HMW adiponectin, placental ADIPOR1 is decreased in B1KO mothers, suggesting a possible contribution to the lower IRS1 induction presented in B1KO dam placentas. On the other hand, some authors have already shown that maternal adiponectin causes placental insulin resistance instead of the sensitivity caused in maternal tissues. In this way, from this perspective, the low adiponectin levels observed in the B1KO dam could lead to placental insulin sensitivity instead of placental insulin resistance.63,64 Hypoadiponectinemia is frequently associated with maternal metabolic disorders such as gestational diabetes mellitus, maternal obesity, and insulin resistance. Furthermore, it has already been described its potential as a predictor of offspring predisposition to obesity.59,65,66,67,68,69
Despite the higher maternal serum insulin levels, the phosphorylated-to-total IRS1 ratio was reduced in the placenta of B1KO females. Luo et al. demonstrated that the p85 regulatory subunit of PI3K downregulates IRS1 signaling via the formation of a sequestration complex composed by IGF1R-induced p85 monomer -IRS1 dimer, recruiting IRS1 to the cytosol and decreasing its activation.70 In this way, we suggest that maternal B1R deficiency increased Igf1r, which induced the dimerization between IRS1 and p85 monomer, resulting in the sequestration of IRS1 from the INSR. Reduced IRS1 bound to insulin receptors could be responsible for the reduced tyrosine-phosphorilated-to-total IRS1, downregulating insulin signaling, which results in placental insulin resistance. Therefore, we suggest decreased IRS1 induction by INSR culminated in a reduced phosphorylated-to-total GSK3B ratio observed in the placenta of B1KO females.
Activation of GYS by inhibiting GSK3B through insulin signaling via PI3K/AKT has already been reported in tissues such as the liver and muscle,71,72 suggesting, therefore, that placental insulin resistance is involved in the reduction of glycogen synthesis by increased activation of GSK3B.73,74 Although placental insulin resistance may account for the reduced glycogenesis in the placenta, some studies suggest that maternal glucose availability is the main determinant of placental glycogen synthesis.75 Glucose levels were not statistically different between WT and B1KO mothers, and neither was the expression of genes encoding glucose transporters in the placenta, strengthening the notion that insulin resistance might be responsible for the reduced placental glycogen levels found in B1KO mothers.
Although it is unclear how placental glycogen storage contributes to fetal development, the presence of the glycogenolysis pathway in the placenta and the fact that it is controlled by insulin suggests that placental glycogen may contribute with an additional source of glucose to account for the high energy demand of the fetus at the end of pregnancy.76,77 Hence, the combination of less placental glycogen and increased peripheral insulin resistance in the mother may increase the flow of glucose to the fetus. The increased use of glucose by the fetus is consistent with the fact that B1KO mothers are hyperinsulinemic despite having normal glycemia and oral glucose tolerance. It is also compatible with the suggestive defect in fat accumulation and lipolysis by B1KO mothers, which could culminate in increased use of other substrates, such as glucose, ketone bodies, and cholesterol, characterizing a maternal metabolic disorder in the absence of B1R. It is worth noting the disbalances observed are caused neither by the deficiency of B1R nor by the compensation by B2R. Although it has already been described the upregulation of B2R in the absence of B1R in some contexts, no differences were observed in B2R expression in maternal tissues after genetic deletion of Bdkrb1.
Adiponectin has also been shown to control glycogen accumulation. Adiponectin induces Gys1 and glycogen synthesis via PI3K in endometrial stromal cells in vitro.78 Further, as aforementioned, adiponectin also induces IRS1, which in turn contributes to promoting insulin sensitivity.79,80 Hence, reduced adiponectin levels in B1KO mothers may further contribute to the reduction of placental insulin signaling and glycogen content.
Several studies have correlated low levels of adiponectin and ADIPOR1 with increased fetal growth as well as changes in placental glycogen content with several fetal programming models.65,67,81,82 Although pups from B1KO mothers showed no difference in fetal mass at GD18.5, there was a trend toward higher birth weight and increased body mass up to 30 days after birth. The lower maternal adiponectin levels and insulin resistance can induce offspring obesity programming and can be the reason for higher body weight observed in B1KO dam offspring.65,69,83,84 In conclusion, our data suggest that B1R controls adiponectin secretion and maternal adipose tissue homeostasis, leading to maternal metabolic disorder and demonstrating its potential as a pregnancy biomarker. At the same time, B1R seems to modulate placental insulin signaling by inhibiting the IRS1-p85 complex induced by IGF1R, which could contribute to placental insulin resistance and glycogen synthesis reduction, which plays a role in fetal programming. Once the B1R deficiency triggers hyperphagia, decreased levels of high-molecular-weight adiponectin, and insulin resistance in mothers during pregnancy, likely contributing to reduced placental glycogen content and heavier offspring.
Limitations of the study
It is already known that placental metabolism could differ between female and male fetuses in some aspects. However, this study neglects, for the purpose of simplicity, the sexual dimorphic metabolism of the placenta. Although we did not separate placentas from female and male fetuses, different randomly collected placentas have been used for distinct analyses. Furthermore, we have used two separate placentas per mother for each assay in an attempt to minimize the possible sexual disbalance.
Moreover, this study did not measure the real transplacental nutrient transport; the modulation of nutrient transport by B1R was inferred by mRNA levels since we do not have the necessary facilities for radiolabeling the nutrients. The measurement of mRNA expression of nutrient transporters may not be informative. Furthermore, the scarcity of studies carried out on humans that investigated the B1R in pregnancy impacts the discussion of the data in a translational context.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AdipoR1 goat polyclonal antibody | Santa Cruz Biotechnology | Cat#sc46749; RRID:AB_2242218 |
| AdipoR2 rabbit polyclonal antibody | Santa Cruz Biotechnology | Cat#sc99184; RRID:AB_2273552 |
| β-actin (D6A8) rabbit monoclonal antibody | Cell Signaling Technology | Cat#8457S; RRID:AB_10950489 |
| GSK-3β (D5C5Z) rabbit monoclonal antibody | Cell Signaling Technology | Cat#12456S; RRID:AB_2636978 |
| Phospho-GSK-3β (Ser9) (D85E12) rabbit monoclonal antibody | Cell Signaling Technology | Cat#5558S; RRID:AB_10013750 |
| IRS1 (D23G12) rabbit monoclonal antibody | Cell Signaling Technology | Cat#3407S; RRID:AB_2127860 |
| Phospho-IRS1 (Tyr612) rabbit polyclonal antibody | Thermo Fisher Scientific | Cat#44-816G; RRID:AB_2533768 |
| Biological samples | ||
| C57BL/6J mouse | CEDEME - UNIFESP | Ref. C57BL/6J |
| B1KO mouse | Federal University of São Paulo (UNIFESP) animal house | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Ketamine - Cetamin 10% | Syntec | N/A |
| Xilazyne - Anazedan 2% | Ceva | N/A |
| R-715 TFA salt | Sigma-Aldrich | R9032 |
| TRIzol | Thermo Scientific, Waltham, MA, USA | Cat#15596026 |
| M-MLV Reverse Transcriptase | Promega, Madison, WI, USA | Cat#M1705 |
| Ribonuclease Inhibitor RNase-Free | EURx Sp. zo.o., Gdansk, Poland | Cat#E4210-02 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | Cat#78440 |
| dNTPs | Promega, Madison, WI, USA | Cat#U1515 |
| GoTaq® qPCR Master Mix, ROX | Promega, Madison, WI, USA | Cat#A6002 |
| Critical commercial assays | ||
| Mouse Leptin Quantikine ELISA | R&D Systems | Cat#MOB00; RRID:AB_2732075 |
| Mouse Adiponectin/Acrp30 Duo-Set ELISA | R&D Systems | Cat#DY1119 |
| Ultra Sensitive Mouse Insulin ELISA Ki | Crystal Chem | Cat#90080; RRID:AB_2783626 |
| Mouse Adiponectin ELISA Kit | Crystal Chem | Cat#80569 |
| Mouse HMW & Total Adiponectin ELISA | Alpco Diagnostics | Cat#80-ADPHU-E01; RRID:AB_2892778 |
| Cholesterol Liquiform | Labtest | Ref. 76 |
| Triglycerides Liquiform | Labtest | Ref. 87 |
| Experimental models: Organisms/strains | ||
| Mouse:C57BL/6 | CEDEME - UNIFESP | Ref. C57BL/6J |
| Mouse:B1KO | UNIFESP animal house | N/A |
| Oligonucleotides | ||
| Primers for RT-qPCR see Table S1. | This paper | N/A |
| Software and algorithms | ||
| Graphpad Prism V.7 | Graphpad | RRID:SCR_002798 |
| Image Lab Software 6.0.1 | Bio-Rad Laboratories, Inc | RRID:SCR_014210 |
| Image Studio Lite Software, version 5.2.5 | LI-COR, Inc | RRID:SCR_013715 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ronaldo Carvalho Araujo (araujorona@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Original western blot images have been incorporated into Supplemental Information and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
12-14-week-old C57BL/6 (Centro de Desenvolvimento de Modelos Experimentais para Medicina e Biologia (CEDEME – UNIFESP), São Paulo, Brazil) wild-type male and female mice; and 12-14-week-old Bdkrb1 knockout (B1KO)85 male and female mice obtained from the Federal University of São Paulo (UNIFESP) animal house were used in this study and housed in 2 females per cage. Pregnant mice were placed in individual cages.
The experimental design was divided into two phases. First, since all previous literature has been based on males, we have characterized the B1KO female mice (Figure S7A). For this, we evaluated virgin 12-week-old WT and B1KO female mice. Considering the role of B1R in insulin and leptin sensitivity observed in male mice, we have measured the glycemia, ketone bodies, and tolerance to glucose and insulin. The animals were euthanized 6 hours after fasting for hormonal and biochemical analyses and body composition analysis. All female mice were at the estrus stage of the estrous cycle when euthanized to ensure there was no influence of hormonal cyclicity on the measurements.
In the second phase, considering the alterations observed in virgin female mice, we evaluated the role of B1R in maternal-placental metabolism and offspring development (Figure S7B). For placental and maternal metabolism evaluation, females were mated with C57BL/6 males, B1KO females mated with WT males, and WT females with B1KO males, thus ensuring that all fetuses were heterozygous for the B1R (B1+/-) and that there would be no influence of offspring genotype (Figure S7B). After the breeding period, the presence of a plug was considered indicative of pregnancy (gestational day (GD) 0.5).
At GD16.5, the pancreatic function was assessed by oral glucose tolerance test (OGTT) after 12h fasting. The cesarean section was performed at GD18.5, also 12h after fasting, to obtain the placenta and maternal tissues (Figure S7B). Some females were submitted to natural delivery for the offspring development evaluation by weight assessment until 90 days after birth (Figure S7B).
The protocols used in this work followed the guidelines of CONCEA. They were approved by the Ethics Committee in the Use of Animals (CEUA Permit number 3410111117) and by the Internal Biosafety Committee (CIBio Permit number 2017/41) of UNIFESP, following the ARRIVE guidelines.
Method details
Glycemic analyses
In the virgin females, the fasting blood glucose was measured at 8 a.m. after 12 hours of fasting, initiating the glucose tolerance test (GTT). While the blood glucose was measured, the beta ketone levels (ketonemia) were also assessed. On the third day after GTT, the blood glucose and ketonemia were evaluated again but in the random-fed state, beginning the insulin tolerance test (ITT) with the injection of 0.75IU/Kg of regular insulin, i.p. (Figure S7A). In the pregnant females, OGTT was performed at GD16.5, after a 12-hour fasting period, and the fasting glycemia and ketonemia were estimated at the moment of cesarean section (GD18.5) (Figure S7B). Glycemia was measured using an Accu-Chek Active glucometer (Roche Diabetes Care, Indianopolis, Indiana, USA) at 0, 15, 30, 60, and 120 minutes after glucose administration. For the OGTT, 1g of glucose per kg of body weight was administered orally in pregnant females.
Pyruvate tolerance test
WT and B1KO virgin females were submitted to a pyruvate tolerance test. For the test, blood glucose was measured after 6 hours of fasting (zero point), as well as in the GTT and ITT, and then 1g/kg of pyruvate was injected i.p., and blood glucose was measured at 15, 30, 45, and 60 minutes after pyruvate injection.
Material collection
The animals were euthanized by exsanguination after anesthesia with 30% ketamine (Cetamin, Syntec, Tamboré, SP, Brazil) (150 mg/Kg) and xylazine (ANASEDAN® PEC, Ceva, Paulínia, SP, Brazil) (10 mg/Kg) solution. The anesthesia was applied intraperitoneally or subcutaneously (pregnant females). Blood was collected for hormonal measurements by cardiac puncture. Furthermore, all fetuses were decapitated. Placenta and maternal tissues were collected and stored at -80°C.
RNA extraction and RT-qPCR
RNA extraction was performed with 50 to 100mg of tissue in 1mL of TRIzol (#15596026, Thermo Scientific, Waltham, MA, USA), using a tissue homogenizer with magnetic beads. To the homogenate was added 200μL of chloroform and mixed by vortexing for approximately 15 seconds. Subsequently, the samples were incubated at room temperature for 10 minutes and centrifugated at 12,000 rpm for 10 minutes at 4°C for phase separation. The upper translucid phase (500μL) was collected and transferred to a 1.5mL tube with 500μL of cold isopropanol. The aqueous phase and the isopropanol were mixed by inversion and incubated at 4°C for 10 minutes for RNA precipitation. After centrifugation at 12,000 rpm for 10 minutes at 4°C, the maximum of isopropanol was removed and the precipitated was washed two times with 1mL of 75% ethanol for better RNA purification by centrifugation and remotion of ethanol. Later, the pellet air-dryed for 15 to 20 minutes and was resuspended with 20 to 50μL of nuclease-free water, followed by incubation at 55°C for 10 minutes. The RNA quality and integrity were assessed by observation of the 260/280nm absorbance ratio detected by nucleic acid spectrophotometer (NanoDrop 1000, Thermo Scientific, USA). Only intact samples were used for the production of complementary DNA (cDNA).
After DNase treatment, cDNA synthesis was carried out with M-MLV Reverse Transcriptase (#M1705, Promega, Madison, WI, USA) using random primers (#C118A, Promega, Madison, WI, USA), dNTPs (#U1515, Promega, Madison, WI, USA), and Ribonuclease Inhibitor RNase-Free (#E4210-02, EURx Sp. zo.o., Gdansk, Poland). First, 0.5μg of random primers were added to 1μg of DNase-treated RNA in a volume of 12.2μL and incubated at 70°C for 5 minutes, followed by 5 minutes cooling on ice. Later, 200U of M-MLV Reverse Transcriptase, 5mM of dNTP, 15U of Ribonuclease Inhibitor RNAse-Free, and the M-MLV Reverse Transcriptase Incubation Buffer (5x) were added to the reaction and submitted to 25°C for 15 minutes, 37°C for 60 minutes, and 70°C for 15 minutes for cDNA synthesis. RT-qPCR was performed using 20ng of cDNA with 10μM of the primers described in Table 1 and GoTaq® qPCR Master Mix, ROX (#A6002, Promega, Madison, WI, USA), 10 μL reaction volume in QuantStudio5 equipment (Applied Biosystems). Relative gene expression was quantified by the 2-ΔΔCt method using Rn18s as a reference gene. The primer sequences used in this study are listed in Table S1.
Protein extraction and western blotting
Protein extraction was performed with half placenta (about 50 mg) in 800μL of lysis buffer (Halt Protease and Phosphatase Inhibitor Cocktail, ThermoFisher Scientific), using a tissue homogenizer with magnetic beads. The homogenate was centrifuged at 14000 rpm for 40 minutes at 4°C. Protein concentration was measured from the supernatant using the Bradford method in plate spectrophotometry. The samples, 15 to 40 μg of placental protein extract, were submitted to a 10% polyacrylamide gel electrophoresis (SDS-PAGE) and transferred into a nitrocellulose membrane by Trans-Blot Turbo (Bio-Rad Laboratories, Inc).
Membranes were blocked with 5% BSA overnight to avoid possible unspecific binding of the primary antibody. For the detection of target proteins, primary antibodies to ADIPOR1 (1:500, Santa Cruz Biotechnology Cat# sc-46749, RRID:AB_2242218), ADIPOR2 (1:300, Santa Cruz Biotechnology Cat# sc-99184, RRID:AB_2273552), GSK3B (1:1000, Cell Signaling Technology Cat# 12456, RRID:AB_2636978), pGSK3B (Ser-9) (1:1000, Cell Signaling Technology Cat# 5558, RRID:AB_10013750), IRS1 (1:1000, Cell Signaling Technology Cat# 3407, RRID:AB_2127860) and pIRS1 (Tyr-612) (1:1000, Thermo Fisher Scientific Cat# 44-816G, RRID:AB_2533768) were incubated for 2h at room temperature or overnight at 4°C. Subsequently, the membranes were incubated with an appropriate secondary antibody: horseradish peroxidase-conjugated anti-rabbit and/or anti-goat IgG for one and a half hours. In order to quantify the density of the bands obtained from the chemiluminescent or fluorescent membranes, the Image Lab Software 6.0.1 (Bio-Rad Laboratories, Inc, RRID:SCR_014210) and the Image Studio Lite Software (version 5.2.5, LI-COR, Inc, RRID:SCR_013715), respectively, were utilized. For the normalization of the target proteins, β-actin (ACTB, 1:2000, Cell Signaling Technology Cat# 8457, RRID:AB_10950489) was used as a reference protein.
Biochemical analysis
Cholesterol and triglyceride levels were measured by enzymatic tests in virgin and pregnant female serum (Cholesterol Liquiform Ref. 76 e Triglycerides Liquiform Ref. 87, Labtest, Lagoa Santa, MG, Brazil). Both enzymatic tests have similar protocols. First, 1mL of Reagent 1, composed of the necessary enzymes for the colorimetric compost formation, was added to a 1.5mL tube. As a second step, 10μL of the standard or the sample was added to the respective tube. Finally, after incubation at 37°C for 10 minutes, 100μL of the samples, standards, and the blank (reagent 1) were pipetted in a 96-well plate and their absorbance read in a plate spectrophotometer at 500nm for cholesterol and 505nm for triglycerides. The cholesterol and triglyceride concentration was determined by the multiplication between the samples' absorbance values and the calibration factor. The calibration factor was calculated by dividing 200 by the standard absorbance value. The sample and standard absorbance values were corrected by subtracting the blank absorbance before any calculation.
Placental glycogen content
The measurement of placental glycogen content was performed in approximately half a placenta (about 50 mg) by the phenol-sulfuric method described by Lo and Taylor86 adapted for 96-well plates. For glycogen extraction, samples were digested in 200μL of 30% w/v KOH (Merck) at 99°C for 20 to 30 minutes. Subsequently, the samples were cooled on ice and precipitated from the alkaline digest with 1.2 volumes (300μL) of 100% v/v ethanol (Synth). The precipitate was obtained by centrifugation at 4500rpm for 15 minutes after 30 minutes on ice and resuspended in 200μL of distilled water. Later, 30μL of 5% v/v phenol was added to 30μL of the resuspended sample diluted in distilled water (proportion of 2:5) or standard solution. Last, 150μL of 98% v/v H2SO4 was rapidly added (10-20 seconds) to the center of the mixture. Finally, the samples were pipetted in triplicate, and their absorbance was read in a plate spectrophotometer at 490nm. The blank (distilled water) absorbance was discounted from samples and standard absorbances. For the glycogen concentration calculation, a standard curve was plotted by glycogen standard solutions absorbance versus glycogen amount in μg (2,5; 5; 10; 20; 30; 40), and the slope of the curve was obtained. The glycogen concentration was calculated as demonstrated below:
| g of glycogen/100g of placenta =A490k×Vv×10−4w, |
where V = sample glycogen solution total volume; v = volume of sample aliquot for colorimetric reaction; A490 = absorbance at 490 nm; w = placenta tissue mass weight in grams; κ = standard curve slope; unity = 1 per microgram of glycogen.
ELISA
Leptin, insulin, adiponectin, and high molecular weight adiponectin levels were measured by Mouse Leptin Quantikine ELISA R&D Systems (R and D Systems Cat# MOB00, RRID:AB_2732075), Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem Cat# 90080, RRID:AB_2783626), Mouse Adiponectin ELISA Kit (Crystal Chem Cat# 80569) or Mouse Adiponectin Duo-Set ELISA R&D Systems (R and D Systems Cat# DY1119), and Mouse HMW & Total Adiponectin ELISA ALPCO kit (Alpco Diagnostics Cat# 80-ADPHU-E01, RRID:AB_2892778), respectively. The analyses were performed by sandwich ELISA technique following the manufacturers’ instructions with some adaptations at the incubation durations in the protocol. Briefly, the samples were incubated in capture antibody-sensitized plates, followed by consecutive washes to remove the unbound sample and incubation with detection antibody conjugated to biotin. After another round of consecutive washes to remove the unbound biotinylated antibody, the samples were incubated with streptavidin-HRP, which reacted with the substrate solution protected from light after the remotion of excessive streptavidin-HRP by consecutive washes. Finally, the reaction was stopped with a sulfuric acid solution and the optical density read at 450 nm in a microplate spectrophotometer.
For the detection of leptin levels in pregnant serum, we have activated leptin to the immunoreactive form by the mixture of 10μL of serum with 20μL of 2.5N Acetic Acid/ 8M Urea and incubation for 10 minutes at room temperature, followed by the neutralization with 20μL of 2.7N NaOH/ 1M HEPES. Prior to the assay, the activated serum samples were 60-fold diluted while the placental samples were 12-fold diluted. The samples, controls, and standards, in both assays as well as in insulin assays, were incubated overnight at room temperature. Furthermore, the time of incubation with the detection antibody was of 2 hours and the incubations with streptavidin-HRP and substrate solution were from 30 minutes each.
The sample dilution for adiponectin detection differed according to the kit utilized. For total adiponectin detection in serum from pregnant mice using Mouse Adiponectin ELISA Kit, the samples were 10,000-fold diluted while in supernatant from adipose tissue explant using Mouse Adiponectin Duo-Set ELISA R&D Systems, the samples were 100-fold diluted. For HMW adiponectin detection using Mouse HMW & Total Adiponectin ELISA ALPCO kit, the samples underwent an enzyme treatment with an incubation at 37°C for 30 minutes. Moreover, they were 8181-fold diluted prior to the assay.
Modulation of adiponectin secretion ex vivo
Adiponectin secretion ex vivo by parametrial adipose tissue was measured similarly as described by Mori et al.21 Parametrial depots of white adipose tissue were collected and minced from overnight fasted female mice. Small pieces of parametrial adipose tissue from 5 mice were incubated in a culture medium (DMEM high-glucose containing 1mg/mL insulin, 20 mM HEPES, 1% bovine serum albumin, 100 μU penicillin/streptomycin, and 10% fetal bovine serum, pH 7.4) in a proportion of 100mg of tissue per mL of medium at 37°C, 5%CO2. After 3h, the culture medium was changed to a fresh culture medium containing 1μM R715 (R9032, Sigma-Aldrich) (B1R antagonist) or not. Aliquots of 2% of the total volume of the conditioned medium were collected at 3h, 6h, 12h, and 24h after changing the culture medium for quantification of adiponectin. At the end of incubation, the adipose tissue depots were stored at -80°C to quantify of adiponectin mRNA expression.
Quantification and statistical analysis
All females used in the experiments were nulliparous, and the maximum mating period of 3 weeks. Females that showed no sign of pregnancy within 3 weeks of mating were excluded from the experiment as well as the pregnant females that delivered less than 5 fetuses. The clustering of observations by litter was considered to represent the experimental unit (N) of fetal and placental weight. The quantitative values were presented as the mean ± standard deviation of the mean (SDM). The difference between the groups was evaluated by Student’s unpaired t-test or non-parametrical Mann-Whitney test, depending on the data normality distribution. The data normality was evaluated by the Kolmogorov-Smirnov test, if p-value > 0.05, the data are treated as parametrical variables. Statistical significance was assigned at p<0.05. Statistical analysis was performed by using GraphPad Prism software, version 7.0 (GraphPad Software Inc., San Diego, CA).
Acknowledgments
We gratefully acknowledge the grants from FAPESP (grants 2015/20082-7 to R.C.A. and 2021/08354-2 to M.A.M.), CAPES/DAAD (grant number PROBRAL 427/2015 to M.B. and R.C.A., subgrant to T.A.-S.), CNPq (grant 307962/2021-0 to L.M.O.) and, as well as the FAPESP Doctoral fellowship to Thaís Alves da Silva (2017/21368-7).
Author contributions
T.A.-S.: designing and drafting the work; sample collection; experiments performance; statistical analysis; interpretation of data; writing. T.G.R.H.: sample collection; interpretation of data and writing review. L.C.F.-L.: sample collection and writing review. G.M.A.: Ex vivo experiment and writing review. A.C.A.: oral glucose tolerance test. R.B.S.: experiment performance. L.M.O.: interpretation of data; funding acquisition; writing review. M.A.S.M.: interpretation of data and writing review. M.B.: designing and drafting the work; interpretation of data; supervision; writing review; funding acquisition. R.C.A.: contributions to the conception; designing and drafting of the work; statistical analysis; interpretation of data; supervision; writing review; funding acquisition. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108409.
Supplemental information
References
- 1.Jansson N., Nilsfelt A., Gellerstedt M., Wennergren M., Rossander-Hulthén L., Powell T.L., Jansson T. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am. J. Clin. Nutr. 2008;87:1743–1749. doi: 10.1093/ajcn/87.6.1743. [DOI] [PubMed] [Google Scholar]
- 2.Ladyman S.R., Augustine R.A., Grattan D.R. Hormone Interactions Regulating Energy Balance During Pregnancy. J. Neuroendocrinol. 2010;22:805–817. doi: 10.1111/j.1365-2826.2010.02017.x. [DOI] [PubMed] [Google Scholar]
- 3.Tomimatsu T., Yamaguchi M., Murakami T., Ogura K., Sakata M., Mitsuda N., Kanzaki T., Kurachi H., Irahara M., Miyake A., et al. Increase of mouse leptin production by adipose tissue after midpregnancy: gestational profile of serum leptin concentration. Biochem. Biophys. Res. Commun. 1997;240:213–215. doi: 10.1006/bbrc.1997.7638. [DOI] [PubMed] [Google Scholar]
- 4.Dahlgren J. Pregnancy and insulin resistance. Metab. Syndr. Relat. Disord. 2006;4:149–152. doi: 10.1089/met.2006.4.149. [DOI] [PubMed] [Google Scholar]
- 5.Marcinkevage J.A., Narayan K.M.V. Gestational diabetes mellitus: taking it to heart. Prim. Care Diabetes. 2011;5:81–88. doi: 10.1016/j.pcd.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Dos Santos E., Duval F., Vialard F., Dieudonné M.-N. The roles of leptin and adiponectin at the fetal-maternal interface in humans. Horm. Mol. Biol. Clin. Invest. 2015;24:47–63. doi: 10.1515/hmbci-2015-0031. [DOI] [PubMed] [Google Scholar]
- 7.Ravelli G.-P., Stein Z.A., Susser M.W. Obesity in Young Men after Famine Exposure in Utero and Early Infancy. N. Engl. J. Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 8.Barker D.J., Hales C.N., Fall C.H., Osmond C., Phipps K., Clark P.M. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 9.Langley-Evans S.C., Phillips G.J., Jackson A.A. In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clin. Nutr. 1994;13:319–324. doi: 10.1016/0261-5614(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 10.Proud D., Kaplan A.P. Kinin formation: mechanisms and role in inflammatory disorders. Annu. Rev. Immunol. 1988;6:49–83. doi: 10.1146/annurev.iy.06.040188.000405. [DOI] [PubMed] [Google Scholar]
- 11.Corrêa C.R., Calixto J.B. Evidence for participation of B1 and B2 kinin receptors in formalin-induced nociceptive response in the mouse. Br. J. Pharmacol. 1993;110:193–198. doi: 10.1111/j.1476-5381.1993.tb13791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whalley E.T. Receptors mediating the increase in vascular permeability to kinins: comparative studies in rat, guinea-pig and rabbit. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1987;336:99–104. doi: 10.1007/BF00177758. [DOI] [PubMed] [Google Scholar]
- 13.Barros C.C., Haro A., Russo F.J., Schadock I., Almeida S.S., Reis F.C., Moraes M.R., Haidar A., Hirata A.E., Mori M., et al. Bradykinin inhibits hepatic gluconeogenesis in obese mice. Lab. Invest. 2012;92:1419–1427. doi: 10.1038/labinvest.2012.105. [DOI] [PubMed] [Google Scholar]
- 14.Morais R.L., Silva E.D., Sales V.M., Filippelli-Silva R., Mori M.A., Bader M., Pesquero J.B. Kinin B1 and B2 receptor deficiency protects against obesity induced by a high-fat diet and improves glucose tolerance in mice. Diabetes Metab. Syndr. Obes. 2015;8:399–407. doi: 10.2147/DMSO.S87635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beard K.M., Lu H., Ho K., Fantus I.G. Bradykinin augments insulin-stimulated glucose transport in rat adipocytes via endothelial nitric oxide synthase-mediated inhibition of Jun NH2-terminal kinase. Diabetes. 2006;55:2678–2687. doi: 10.2337/db05-1538. [DOI] [PubMed] [Google Scholar]
- 16.Mori M.A., Sales V.M., Motta F.L., Fonseca R.G., Alenina N., Guadagnini D., Schadock I., Silva E.D., Torres H.A.M., dos Santos E.L., et al. Kinin B1 receptor in adipocytes regulates glucose tolerance and predisposition to obesity. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isami S., Kishikawa H., Araki E., Uehara M., Kaneko K., Shirotani T., Todaka M., Ura S., Motoyoshi S., Matsumoto K., et al. Bradykinin enhances GLUT4 translocation through the increase of insulin receptor tyrosine kinase in primary adipocytes: evidence that bradykinin stimulates the insulin signalling pathway. Diabetologia. 1996;39:412–420. doi: 10.1007/BF00400672. [DOI] [PubMed] [Google Scholar]
- 18.Miyata T., Taguchi T., Uehara M., Isami S., Kishikawa H., Kaneko K., Araki E., Shichiri M. Bradykinin potentiates insulin-stimulated glucose uptake and enhances insulin signal through the bradykinin B2 receptor in dog skeletal muscle and rat L6 myoblasts. Eur. J. Endocrinol. 1998;138:344–352. doi: 10.1530/eje.0.1380344. [DOI] [PubMed] [Google Scholar]
- 19.Araújo R.C., Mori M.A., Merino V.F., Bascands J.L., Schanstra J.P., Zollner R.L., Villela C.A., Nakaie C.R., Paiva A.C.M., Pesquero J.L., et al. Role of the kinin B1 receptor in insulin homeostasis and pancreatic islet function. Biol. Chem. 2006;387:431–436. doi: 10.1515/BC.2006.057. [DOI] [PubMed] [Google Scholar]
- 20.Mori M.A., Araújo R.C., Reis F.C.G., Sgai D.G., Fonseca R.G., Barros C.C., Merino V.F., Passadore M., Barbosa A.M., Ferrari B., et al. Kinin B1 receptor deficiency leads to leptin hypersensitivity and resistance to obesity. Diabetes. 2008;57:1491–1500. doi: 10.2337/db07-1508. [DOI] [PubMed] [Google Scholar]
- 21.Mori M.A., Araújo R.C., Pesquero J.B. Kinin B1 receptor stimulation modulates leptin homeostasis. Evidence for an insulin-dependent mechanism. Int. Immunopharm. 2008;8:242–246. doi: 10.1016/j.intimp.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 22.Sales V.M., Gonçalves-Zillo T., Castoldi A., Burgos M., Branquinho J., Batista C., Oliveira V., Silva E., Castro C.H.M., Câmara N., et al. Kinin B1 Receptor Acts in Adipose Tissue to Control Fat Distribution in a Cell-Nonautonomous Manner. Diabetes. 2019;68:1614–1623. doi: 10.2337/db18-1150. [DOI] [PubMed] [Google Scholar]
- 23.Freitas-Lima L.C., Budu A., Estrela G.R., Alves-Silva T., Perilhão M.S., Arruda A.C., Carvalho Araujo R. Metabolic fasting stress is ameliorated in Kinin B1 receptor-deficient mice. Life Sci. 2022;294 doi: 10.1016/j.lfs.2021.120007. [DOI] [PubMed] [Google Scholar]
- 24.Fukao T., Lopaschuk G.D., Mitchell G.A. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fatty Acids. 2004;70:243–251. doi: 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Møller N. Ketone Body, 3-Hydroxybutyrate: Minor Metabolite - Major Medical Manifestations. J. Clin. Endocrinol. Metab. 2020;105:2884–2892. doi: 10.1210/clinem/dgaa370. [DOI] [PubMed] [Google Scholar]
- 26.Dai G., Bustamante J.J., Zou Y., Myronovych A., Bao Q., Kumar S., Soares M.J. Maternal hepatic growth response to pregnancy in the mouse. Exp. Biol. Med. 2011;236:1322–1332. doi: 10.1258/ebm.2011.011076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milona A., Owen B.M., van Mil S., Dormann D., Mataki C., Boudjelal M., Cairns W., Schoonjans K., Milligan S., Parker M., et al. The normal mechanisms of pregnancy-induced liver growth are not maintained in mice lacking the bile acid sensor Fxr. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;298:G151–G158. doi: 10.1152/ajpgi.00336.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsons J.A., Brelje T.C., Sorenson R.L. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee R.R., Cyphert H.A., Walker E.M., Chakravarthy H., Peiris H., Gu X., Liu Y., Conrad E., Goodrich L., Stein R.W., Kim S.K. Gestational Diabetes Mellitus From Inactivation of Prolactin Receptor and MafB in Islet β-Cells. Diabetes. 2016;65:2331–2341. doi: 10.2337/db15-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajri T., Tao H., Wattacheril J., Marks-Shulman P., Abumrad N.N. Regulation of adiponectin production by insulin: interactions with tumor necrosis factor-α and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 2011;300:E350–E360. doi: 10.1152/ajpendo.00307.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansson N., Greenwood S.L., Johansson B.R., Powell T.L., Jansson T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J. Clin. Endocrinol. Metab. 2003;88:1205–1211. doi: 10.1210/jc.2002-021332. [DOI] [PubMed] [Google Scholar]
- 32.Jansson T. Amino acid transporters in the human placenta. Pediatr. Res. 2001;49:141–147. doi: 10.1203/00006450-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Sciacca L., Le Moli R., Vigneri R. Insulin analogs and cancer. Front. Endocrinol. 2012;3:21. doi: 10.3389/fendo.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X., Greenbaum L.M., Mahesh V.B., Brann D.W. Characterization of the kinin system in the ovary during ovulation in the rat. Biol. Reprod. 1992;47:945–951. doi: 10.1095/biolreprod47.6.945. [DOI] [PubMed] [Google Scholar]
- 35.Smith C., Perks A.M. The kinin system and ovulation: changes in plasma kininogens, and in kinin-forming enzymes in the ovaries and blood of rats with 4-day estrous cycles. Can. J. Physiol. Pharmacol. 1983;61:736–742. doi: 10.1139/y83-114. [DOI] [PubMed] [Google Scholar]
- 36.Miatello R., Lama M., González S., Damiani T., Nolly H. Biochemical evidence of a kallikrein-like activity in rat reproductive tissues. Hypertension. 1994;23:I193–I197. doi: 10.1161/01.hyp.23.1_suppl.i193. [DOI] [PubMed] [Google Scholar]
- 37.Valdés G., Figueroa C.D., Corthorn J. Temporospatial changes of kallikrein-like enzymes during the estrous cycle and pregnancy in the rat uterus. Biol. Reprod. 1996;55:236–245. doi: 10.1095/biolreprod55.2.236. [DOI] [PubMed] [Google Scholar]
- 38.Figueroa C.D., Chacón C., Corthorn J., Ehrenfeld P., Müller-Esterl W., Valdés G. Temporospatial changes of kinin b2 receptors during the estrous cycle and pregnancy in the rat uterus. Biol. Reprod. 2001;64:1590–1599. doi: 10.1095/biolreprod64.6.1590. [DOI] [PubMed] [Google Scholar]
- 39.Valdés G., Germain A.M., Corthorn J., Chacón C., Figueroa C.D., Müller-Esterl W. Tissue kallikrein and bradykinin B2 receptor in human uterus in luteal phase and in early and late gestation. Endocrine. 2001;16:207–215. doi: 10.1385/ENDO:16:3:207. [DOI] [PubMed] [Google Scholar]
- 40.Murone C., Chai S.Y., Müller-Esterl W., Mendelsohn F.A., Clements J. Localization of bradykinin B2 receptors in the endometrium and myometrium of rat uterus and the effects of estrogen and progesterone. Endocrinology. 1999;140:3372–3382. doi: 10.1210/endo.140.7.6871. [DOI] [PubMed] [Google Scholar]
- 41.Droll S.H., Hsu Y.M.S., Drake S.K., Kim A., Wang W., Calvo K.R., Cao Z., Hu T.Y., Zhao Z. Differential processing of high-molecular-weight kininogen during normal pregnancy. Rapid Commun. Mass Spectrom. 2020;34(Suppl 1) doi: 10.1002/rcm.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugi T., Makino T. Factor XII, kininogen and plasma prekallikrein in abnormal pregnancies. Curr. Drug Targets. 2005;6:551–557. doi: 10.2174/1389450054546060. [DOI] [PubMed] [Google Scholar]
- 43.Hoegh A.M., Borup R., Nielsen F.C., Sørensen S., Hviid T.V.F. Gene Expression Profiling of Placentas Affected by Pre-Eclampsia. J. Biomed. Biotechnol. 2010;2010:787545. doi: 10.1155/2010/787545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erices R., Corthorn J., Lisboa F., Valdés G. Bradykinin promotes migration and invasion of human immortalized trophoblasts. Reprod. Biol. Endocrinol. 2011;9:97. doi: 10.1186/1477-7827-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdés G., Acuña S., Schneider D., Ortíz R., Padilla O. Bradykinin Exerts Independent Effects on Trophoblast Invasion and Blood Pressure in Pregnant Guinea Pigs. Reprod. Sci. 2020;27:1648–1655. doi: 10.1007/s43032-020-00195-6. [DOI] [PubMed] [Google Scholar]
- 46.Valdés G., Corthorn J. Review: The angiogenic and vasodilatory utero-placental network. Placenta. 2011;32:S170–S175. doi: 10.1016/j.placenta.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Thuringer D., Maulon L., Frelin C. Rapid transactivation of the vascular endothelial growth factor receptor KDR/Flk-1 by the bradykinin B2 receptor contributes to endothelial nitric-oxide synthase activation in cardiac capillary endothelial cells. J. Biol. Chem. 2002;277:2028–2032. doi: 10.1074/jbc.M109493200. [DOI] [PubMed] [Google Scholar]
- 48.Miura S., Matsuo Y., Saku K. Transactivation of KDR/Flk-1 by the B2 receptor induces tube formation in human coronary endothelial cells. Hypertension. 2003;41:1118–1123. doi: 10.1161/01.HYP.0000064345.33807.57. [DOI] [PubMed] [Google Scholar]
- 49.Quitterer U., Fu X., Pohl A., Bayoumy K.M., Langer A., AbdAlla S. Beta-Arrestin1 Prevents Preeclampsia by Downregulation of Mechanosensitive AT1-B2 Receptor Heteromers. Cell. 2019;176:318–333.e19. doi: 10.1016/j.cell.2018.10.050. [DOI] [PubMed] [Google Scholar]
- 50.AbdAlla S., Lother H., el Massiery A., Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 51.Morbidelli L., Parenti A., Giovannelli L., Granger H.J., Ledda F., Ziche M. B1 receptor involvement in the effect of bradykinin on venular endothelial cell proliferation and potentiation of FGF-2 effects. Br. J. Pharmacol. 1998;124:1286–1292. doi: 10.1038/sj.bjp.0701943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Húngaro T.G.R., Gregnani M.F., Alves-Silva T., Herse F., Alenina N., Bader M., Araújo R.C. Cortisol Dose-Dependently Impairs Migration and Tube-like Formation in a Trophoblast Cell Line and Modulates Inflammatory and Angiogenic Genes. Biomedicines. 2021;9 doi: 10.3390/biomedicines9080980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrera E. Lipid metabolism in pregnancy and its consequences in the fetus and newborn. Endocrine. 2002;19:43–55. doi: 10.1385/ENDO:19:1:43. [DOI] [PubMed] [Google Scholar]
- 54.Zeng Z., Liu F., Li S. Metabolic Adaptations in Pregnancy: A Review. Ann. Nutr. Metab. 2017;70:59–65. doi: 10.1159/000459633. [DOI] [PubMed] [Google Scholar]
- 55.Ladyman S.R., Brooks V.L. Central actions of insulin during pregnancy and lactation. J. Neuroendocrinol. 2021;33 doi: 10.1111/jne.12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Augustine R.A., Ladyman S.R., Grattan D.R. From feeding one to feeding many: hormone-induced changes in bodyweight homeostasis during pregnancy. J. Physiol. 2008;586:387–397. doi: 10.1113/jphysiol.2007.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.King J.C., Butte N.F., Bronstein M.N., Kopp L.E., Lindquist S.A. Energy metabolism during pregnancy: influence of maternal energy status. Am. J. Clin. Nutr. 1994;59:439S–445S. doi: 10.1093/ajcn/59.2.439S. [DOI] [PubMed] [Google Scholar]
- 58.Barker D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007;261:412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 59.Trivett C., Lees Z.J., Freeman D.J. Adipose tissue function in healthy pregnancy, gestational diabetes mellitus and pre-eclampsia. Eur. J. Clin. Nutr. 2021;75:1745–1756. doi: 10.1038/s41430-021-00948-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huda S.S., Forrest R., Paterson N., Jordan F., Sattar N., Freeman D.J. In preeclampsia, maternal third trimester subcutaneous adipocyte lipolysis is more resistant to suppression by insulin than in healthy pregnancy. Hypertension. 2014;63:1094–1101. doi: 10.1161/HYPERTENSIONAHA.113.01824. [DOI] [PubMed] [Google Scholar]
- 61.Achari A.E., Jain S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu Y., Luo N., Klein R.L., Garvey W.T. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J. Lipid Res. 2005;46:1369–1379. doi: 10.1194/jlr.M400373-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Aye I.L.M.H., Gao X., Weintraub S.T., Jansson T., Powell T.L. Adiponectin inhibits insulin function in primary trophoblasts by PPARα-mediated ceramide synthesis. Mol. Endocrinol. 2014;28:512–524. doi: 10.1210/me.2013-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jones H.N., Jansson T., Powell T.L. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino Acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–1170. doi: 10.2337/db09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aye I.L.M.H., Rosario F.J., Powell T.L., Jansson T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc. Natl. Acad. Sci. USA. 2015;112:12858–12863. doi: 10.1073/pnas.1515484112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paulsen M.E., Rosario F.J., Wesolowski S.R., Powell T.L., Jansson T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. Faseb. J. 2019;33:2899–2909. doi: 10.1096/fj.201801015R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiao L., Wattez J.S., Lee S., Nguyen A., Schaack J., Hay W.W., Shao J. Adiponectin Deficiency Impairs Maternal Metabolic Adaptation to Pregnancy in Mice. Diabetes. 2017;66:1126–1135. doi: 10.2337/db16-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meyer B.J., Stewart F.M., Brown E.A., Cooney J., Nilsson S., Olivecrona G., Ramsay J.E., Griffin B.A., Caslake M.J., Freeman D.J. Maternal obesity is associated with the formation of small dense LDL and hypoadiponectinemia in the third trimester. J. Clin. Endocrinol. Metab. 2013;98:643–652. doi: 10.1210/jc.2012-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pheiffer C., Dias S., Jack B., Malaza N., Adam S. Adiponectin as a Potential Biomarker for Pregnancy Disorders. Int. J. Mol. Sci. 2021;22:1326. doi: 10.3390/ijms22031326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo J., Field S.J., Lee J.Y., Engelman J.A., Cantley L.C. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol. 2005;170:455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cohen P., Frame S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 72.Lizcano J.M., Alessi D.R. The insulin signalling pathway. Curr. Biol. 2002;12:R236–R238. doi: 10.1016/s0960-9822(02)00777-7. [DOI] [PubMed] [Google Scholar]
- 73.Lee J., Kim M.S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 2007;77(Suppl 1):S49–S57. doi: 10.1016/j.diabres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 74.Eldar-Finkelman H., Schreyer S.A., Shinohara M.M., Leboeuf R.C., Krebs E.G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes. 1999;48:1662–1666. doi: 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- 75.Shafrir E., Barash V. Placental glycogen metabolism in diabetic pregnancy. Isr. J. Med. Sci. 1991;27:449–461. [PubMed] [Google Scholar]
- 76.Barash V., Shafrir E. Mobilization of placental glycogen in diabetic rats. Placenta. 1990;11:515–521. doi: 10.1016/s0143-4004(05)80197-3. [DOI] [PubMed] [Google Scholar]
- 77.Roberts G.A.G., Tunster S.J. Characterising the dynamics of placental glycogen stores in the mouse. Placenta. 2020;99:131–140. doi: 10.1016/j.placenta.2020.07.010. [DOI] [PubMed] [Google Scholar]
- 78.Duval F., Dos Santos E., Maury B., Serazin V., Fathallah K., Vialard F., Dieudonné M.N. Adiponectin regulates glycogen metabolism at the human fetal–maternal interface. J. Mol. Endocrinol. 2018;61:139–152. doi: 10.1530/JME-18-0013. [DOI] [PubMed] [Google Scholar]
- 79.Berg A.H., Combs T.P., Du X., Brownlee M., Scherer P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 80.Yamauchi T., Kamon J., Waki H., Terauchi Y., Kubota N., Hara K., Mori Y., Ide T., Murakami K., Tsuboyama-Kasaoka N., et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 81.Qiao L., Wattez J.S., Lee S., Guo Z., Schaack J., Hay W.W., Zita M.M., Parast M., Shao J. Knockout maternal adiponectin increases fetal growth in mice: potential role for trophoblast IGFBP-1. Diabetologia. 2016;59:2417–2425. doi: 10.1007/s00125-016-4061-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akison L.K., Nitert M.D., Clifton V.L., Moritz K.M., Simmons D.G. Review: Alterations in placental glycogen deposition in complicated pregnancies: Current preclinical and clinical evidence. Placenta. 2017;54:52–58. doi: 10.1016/j.placenta.2017.01.114. [DOI] [PubMed] [Google Scholar]
- 83.Aye I.L.M.H., Rosario F.J., Kramer A., Kristiansen O., Michelsen T.M., Powell T.L., Jansson T. Insulin Increases Adipose Adiponectin in Pregnancy by Inhibiting Ubiquitination and Degradation: Impact of Obesity. J. Clin. Endocrinol. Metab. 2022;107:53–66. doi: 10.1210/clinem/dgab680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nicholas L.M., Morrison J.L., Rattanatray L., Zhang S., Ozanne S.E., McMillen I.C. The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int. J. Obes. 2016;40:229–238. doi: 10.1038/ijo.2015.178. [DOI] [PubMed] [Google Scholar]
- 85.Pesquero J.B., Araujo R.C., Heppenstall P.A., Stucky C.L., Silva J.A., Walther T., Oliveira S.M., Pesquero J.L., Paiva A.C., Calixto J.B., et al. Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc. Natl. Acad. Sci. USA. 2000;97:8140–8145. doi: 10.1073/pnas.120035997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lo, S., Russell, J.C., and Taylor, A.W. (1970). Determination of Glycogen in Small Tissue Samples. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original western blot images have been incorporated into Supplemental Information and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.