

Abstract
With the rapid advancement of medical technologies in genomic and molecular medicine, the number of treatable neurometabolic diseases is quickly expanding. Spastic paraplegia 56 (SPG56), one of the severe autosomal recessive forms of neurodegenerative disorders caused by pathogenic variants in the CYP2U1 gene, has no reported specific targeted treatment yet. Here we report 2 Chinese brothers with CYP2U1 bi-allelic pathogenic variants with cerebral folate deficiency who were treated for over a decade with folinic acid supplement. Patients have remained stable under therapy.
Keywords: CYP2U1, Cerebral folate deficiency, Cerebral folate, Folinic acid, Spastic paraplegia 56, 5-methyltetrahydrofolate, 5-MTHF
1. Introduction
Hereditary spastic paraplegia (HSP) is one of the most heterogeneous groups of inherited neurodegenerative disorders, both clinically and genetically. They are mainly characterized by slowly progressive lower limb spasticity as a consequence of corticospinal tract degeneration. Affected patients are classified as uncomplicated (or pure) HSP or complicated (or complex) HSP according to the presence of additional neurological or extra-neurological signs including cerebellar ataxia, extrapyramidal signs, chorioretinal dystrophy, peripheral neuropathy and intellectual disability [[1], [2], [3], [4], [5]]. Complicated HSP most often present with autosomal recessive inheritance [5]. With the expansion of next generation sequencing, there are >80 different disease loci or genes being identified [5].
Spastic paraplegia 56 (SPG56) (OMIM#615030), one of the severe autosomal recessive forms of HSP is caused by pathogenic variants in CYP2U1 [4]. So far, loss of function variants in this gene have been detected in 32 affected individuals who developed early-onset spastic paraplegia with a wide spectrum of clinical signs, including upper limb involvement, dystonia, cognitive impairment, and heterogeneous neuroimaging features with brain calcification and hypomyelination [6]. Variants in this gene have also been recently implicated in a neurocutaneous syndrome with pseudoxanthoma elasticum and pigmentary degenerative maculopathy [7,8]. However, there is no specific treatment except multidisciplinary management of symptoms.
Cerebral folate deficiency (CFD), on the other hand, is a potentially treatable condition that prompts recognition and initiation of appropriate therapy [9]. It was defined by Ramaekers and Blau as any neurological syndrome associated with a low 5-methyltetrahydrofolate (5-MTHF) in the cerebrospinal fluid (CSF), in the presence of normal folate metabolism outside the nervous system [10]. However, it is often used to describe conditions where CSF 5-MTHF is low, regardless of peripheral folate status. Oral treatment with 5-formyltetrahydrofolate (folinic acid) is recommended to normalize CSF 5-MTHF values and often resulted in a favorable clinical response. A list of causes of cerebral folate deficiency has been described including primary and secondary CFD [5,9]. However, there has been no reported association between CYP2U1-related SPG56 and CFD.
Here we report 2 Chinese brothers with SPG56 due to CYP2U1 homozygous pathogenic variants, who were previously diagnosed as CFD and consequently treated with folinic acid supplement for over a decade. This treatment was associated with remarkable clinical stability over years.
2. Clinical history
2.1. Elder brother
A 19-year-old boy was born from non-consanguineous Chinese parents with uneventful antenatal, perinatal and postnatal courses. There was no relevant family history. He enjoyed normal growth and development till around 2 years old. At the age of 2, he could run well, walk upstairs using 1 ft per step unaided and downstairs 2 ft per step, feed himself with a spoon without spillage, speak in complete sentences and was able to sing a song with clear articulations. Subsequently, he was noted to have frequent falls which worsened over a year with unsteadiness in walking, difficulty in managing stairs, clumsiness in feeding, frequent drooling of saliva and slurring of speech. No seizures were reported. Physical examination showed mixed spasticity and dystonia over four limbs, more prominent in lower limbs, with brisk deep tendon reflexes and bilateral sustained ankle clonus. Vision and hearing were normal. Magnetic resonance imaging (MRI) of the brain at 3 years of age was normal as well as baseline neurometabolic work up – liver and renal function, blood lactate, ammonia, glucose, lipid profile, vitamin B12, folate, copper, ceruloplasmin, amino acid, transferrin isoelectric focusing pattern, carnitine profile, homocysteine, very long chain fatty acids, cholestanol and urine organic acids. Griffiths Mental Developmental Scale at the chronological age of 44.2 months showed a mental age of 29 months (general quotient 66).
A trial of L-dopa up to 8.7 mg per kg body weight per day for possible dopa-responsive dystonia showed no clinical response. CSF studies including neurotransmitters and folate measurements were performed at 3 year and 11 months. These showed normal cell count, glucose, lactate and amino acids but low 5-MTHF, 38.2 (Ref: 63–111 nmol/L for age) with normal 5-hydroxyindoleacetic acid (5-HIAA), homovanillic acid (HVA) and pterins (Table 1). Folinic acid (1 mg/kg/day) was started at 4 years of age with the diagnosis of CFD. CSF 5-MTHF was normalized 6 months later with on and off elevation of CSF neopterin ranging from 20 (Ref: 9–20 nmol/L for age) to 41.5 (Ref: 8–30 nmol/L for age) and biopterin 32 (Ref: 10–30 nmol/L) (Table 1). Currently, the patient has been on regular folinic acid for over 15 years (1.9 mg/kg/day). The latest CSF exam at the age of 19 years 2 months showed low CSF 5-HIAA of 40 (Ref: 66–141 nmol/L) and HVA of 97 (Ref: 115–488 nmol/L) (Table 1). Surprisingly, this patient showed no further cognitive and motor deterioration upon supplementation with folinic acid (Table 1). At the time of preparing this manuscript, this young gentleman (20 years old) was still ambulatory with limited intelligence – he has been receiving special education all along – but independent for activities of daily living. Latest eye examination showed early features of macula telangiectasia but no pigmentary retinopathy. Clinically there was no visual or hearing concern.
Table 1.
Folinic acid dosages, CSF folates and pterins in the 2 brothers.
| Patient | Age at CSF sampling# | Folinic acid (mg/kg/day) | 5-MTHF (nmol/L) | Neopterin (nmol/L) | Biopterin (nmol/L) | 5HIAA (nmol/L) | HVA (nmol/L) | Significant clinical progress |
|---|---|---|---|---|---|---|---|---|
| Elder brother | 3y11m | Baseline (started at 4y1m) | ↓38.2 (63–111) | 26.4 (8–30) | 17.5 (10−30) | 150 (105–299) | 552 (211–871) | Spasticity and dystonia over four limbs, more prominent over lower limbs with brisk jerks and sustained bilateral clonus, oromotor dysfunction with significant drooling; GMDS: mental age 29 m at 44.2 m (general quotient 66) |
| 4yr6m | 1 | 59.1 (41–117) | ↑41.5 (8–30) | 15.1 (10–30) | 120 (105–299) | 392 (211–871) | More alert, improvement in speech, drooling and cognitive function, no further deterioration in neurological signs | |
| 5y2m | 1.3 | 87 (41–117) | 19.2 (9–20) | 19.6 (10–30) | 146 (88–178) | 418 (144–801) | GMDS: age equivalent 39 m at 65.5 m (general quotient 60) | |
| 6y4m | 1.6 | 84 (41–117) | 17.2 (9–20) | 12.3 (10–30) | 215 (88–178) | 469 (144–801) | ||
| 7y5m | 1.6 | 92 (41–117) | ↑25 (9–20) | 21 (10–30) | 201 (88–178) | 451 (144–801) | ||
| 8y9m | 1.6 | 92 (41–117) | 20 (9–20) | 20 (10–30) | 103 (88–178) | 354 (144–801) | ||
| 10y10m | 1.5 | 87 (41–117) | ↑21 (9–20) | ND | 141 (88–178) | 354 (144–801) | ||
| 14y10m | 1.7 | 100 (41–117) | 3 (< 18) | ND | 61 (74–163) | 167 (133–551) | ||
| 16y10m | 1.5 | 61 (41–117) | 2.8 (<18) | ND | 72 (66–141) | 172 (115–488) | WISC-IV (HK) at 15y4m: IQ 75 | |
| 19y2m | 1.9 | 109 | 2.7 (<18) | ND | ↓40 (66–141) | ↓97 (115–488) | At preparation of manuscript, still community walker and can manage stairs by himself; WAIS-IV (HK) at 20y4m: IQ 72 | |
| Younger brother | 3y1m | Baseline (started at | ↓19.9 (63–111) | 13 (9–30) | 13.5 (10–30) | 126 (105–2996) | 600 (211–871) | GMDS: mental age 25.2 m at 27.8 m (general quotient 91); no focal neurological sign |
| 3y5m | 1 | 93.3 (63–111) | 11.5 (9–30) | 11.5 (10–30) | 115 (105–299) | 491 (211–871) | ||
| 4y1m | 1 | 83 (63–111) | 31.3 (9–30) | 18.4 (10–30) | 171 (105–299) | 558 (211–871) | ||
| 4y5m | 1.8 | 102 (63–111) | 47.5 (9–30) | 14.5 (10–30) | 197 (105–299) | 538 (211–871) | ||
| 5y6m | 1.8 | 93 (41–117) | 40 (9–20) | 23 (10–30) | 238 (88–178) | 567 (144–801) | ||
| 6y10m | 1.2 | 97 (41–117) | 17 (9–20) | 26 (10–30) | 179 (88–178) | 709 (144–801) | WISC-IV (HK): IQ 80–89* | |
| 13y 10 m | 1.7 | 72 (41–117) | 2.8 (<18) | ND | 95 (74–163) | 390 (133–551) | ||
| 17y6m | 1.7 | 85 (41–117) | 2.2 (<18) | ND | 81 (66–141) | 236 (115–488) | At preparation of manuscript, subtle clumsiness in tandem walking and studying in mainstream school |
Keys: y: years; m: months; (bracket): reference range for age; GMDS: Griffiths Mental Developmental Scale; WISC-IV (HK): Wechsler Intelligence Scale for Children-Fourth Edition (Hong Kong); WAIS-IV (HK): Wechsler Adult Intelligence Scale-Fourth Edition (Hong Kong); IQ: intelligent quotient; ND: not done; ↓: Values below reference range; ↑:Values above reference range; #CSF analyses were performed in 2 laboratories throughout the clinical course: Division of Clinical Chemistry and Biochemistry, University Children's Hospital Zurich (2005–2007) then in Division of Clinical Biochemistry, Queen Mary Hospital, Hong Kong.
2.2. Younger brother
The younger brother was seen at 2 years of age when his elder brother was diagnosed with CFD. At that time, he had normal growth and development without any focal neurological signs. However, the family was worried that he might be affected too. CSF for neurotransmitter studies was then performed and confirmed low 5-MTHF 19.9 (Ref: 63–111 nmol/L for age). He then started supplementation with folinic acid (1 mg/kg/day). The Griffiths Mental Developmental Scale revealed a mental age of 25.2 months at 27.8 months and a repeated cognitive assessment at the age of 6 showed a low average intellectual quotient. Serial CSF neurotransmitter studies showed normalization of 5-MTHF.
At the time of preparing the manuscript, he was 19 years old without significant hearing, visual, cognitive or motor concern and enjoyed normal school. Latest eye examination showed early features of macula telangiectasia. Notably, his compliance to folinic acid varied during adolescence. In the past 2 years, he was noted to have very subtle clumsiness in tandem walking without any hard neurological signs.
2.3. Other significant biochemical findings
In the past 15 years, low complement C3 (C3) at 61–68 (Ref: 76–150 mg/dL) was documented periodically in the elder brother with low normal complement C4 (C4) at 12–15 (Ref: 9-35mg/dL) while the younger brother also had occasionally low C3 at 68-85 mg/dL and low C4 at 0.12 g/L (Ref: 0.13–0.38 g/L). Extensive immunological work up was unrevealing.
Low serum copper 9.9–12 (Ref: 10.0–18.0 umol/L) and ceruloplasmin at 0.16–0.24 (Ref: 0.18–0.35 g/L) were also measured in the elder brother, which also reached the low normal range over time (11 umol/L and 0.17 g/L respectively). Serum copper (13.6–16 Ref: 11.5–23.4 umol/L) and ceruloplasmin (0.17–0.27 Ref: 0.18–0.35 g/L) were in the low normal range in the younger brother as well. Both of them had vitamin D deficiency and were supplemented. Interestingly, the elder brother also had an isolated episode of acute-onset iron deficiency anemia that required blood transfusion. His serum folate was normal all along, and no other etiology was identified.
2.4. Brain imaging
EIder brother's MRI in 2005 (aged 3 y) and 2017 (aged 16 y) showed no parenchymal abnormality. However, susceptibility weighted sequences, a technique most sensitive to presence of minerals, were not performed. CT in 2015 (aged 13 y) demonstrated tiny hyperdensities in left globus pallidus and bilateral frontal subcortical white matter compatible with calcifications (Fig. 1).
Fig. 1.
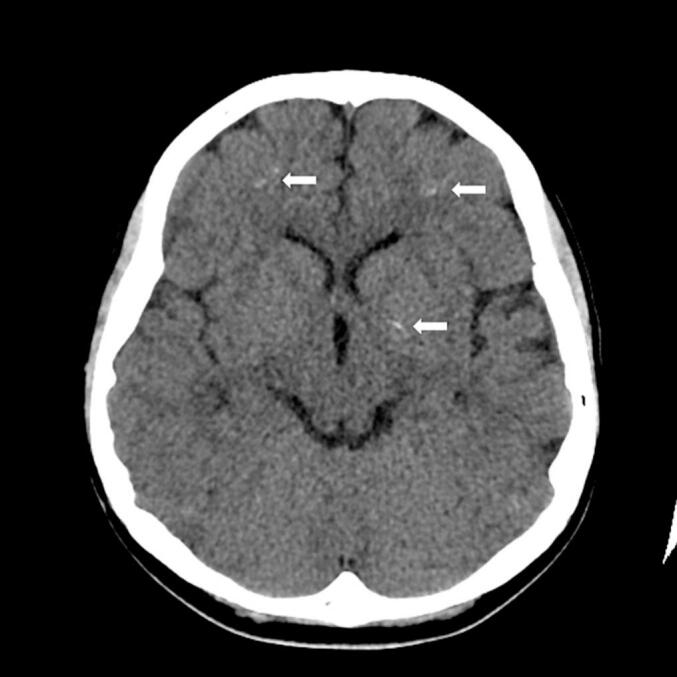
Non-contrast CT in 2015 (aged 13) demonstrated in left globus pallidus and frontal subcortical white matter calcifications (arrows).
Younger brother's MRI in 2017 (aged 13 y) showed susceptibility artefacts in globus pallidus and subcortical white matter of the bilateral frontal and left parietal lobes, suggestive of mineral deposits such as calcium (Fig. 2). Subsequent CT in 2018 (aged 14 y) showed hyperdensities in bilateral globus pallidi and frontal and parietal subcortical white matter in keeping with calcifications (Fig. 2B).
Fig. 2.

A: MRI using susceptibility weight sequence showed blooming artefacts in globus pallidus and subcortical white matter of the bilateral frontal and left parietal lobes (arrows), suggesting presence of mineral deposits.
B: Non-contrast CT in 2018 demonstrated bilateral frontal and parietal subcortical white matter hyperdensities in keeping with calcifications (arrowheads).
2.5. Genetic studies
In view of the decreased cerebral 5-MTHF in both brothers, targeted gene analysis of FOLR1 for primary CFD was performed but negative. Work up for possible underlying mitochondrial disorders as a secondary cause for CFD including mitochondrial DNA common point mutations and whole exome sequencing were all negative. Recently, a quad whole exome sequencing analysis was performed using the Roche KAPA HyperExome assay and in-house bioinformatics analysis based on Burrow-Wheeler Aligner and GATK HaplotypeCaller. The mean sequencing depth was around 120× and over 99% of the target regions reached a sequencing depth of 20×. This analysis found a homozygous1-bp deletion NM_183075.3(CYP2U1):c.471del; p.(Ile158SerfsTer2) in both affected boys with both parents being heterozygous carriers. c.471del is a frameshift variant located in CYP2U1 exon 1, which encodes part of the cytochrome P450 functional domain. It creates a premature stop codon and is predicted to cause loss of normal protein function by nonsense-mediated mRNA decay. This variant is present at very low frequency in the gnomAD (v2.1.1). It has been reported once in ClinVar as pathogenic (accession: VCV000655971.1).
3. Discussion
Spastic paraplegia-56 (OMIM#615030), one of the recently described severe autosomal recessive forms of HSP caused by pathogenic variants in CYP2U1 was first reported in 2012 by Tesson et al. In 2021, from Pujol et al.'s study on the clinical summaries of all published SPG56 patients, it showed that most of them had disease onset in early childhood [6]. Pujol et al. also performed CSF studies from 3 patients with biallelic CYP2U1 variants which showed elevated neopterin and IFN-α levels and considered that likely to be markers of folate deficiency [6]. Both neopterin and IFN-α have been reported elevated during the activation of the immune system [11]. However, this hypothesis had not yet been validated in human studies. Here, we report 2 Chinese brothers with elevated CSF neopterin, low CSF 5-MTHF and cerebral calcifications, consistent with the conclusion from Pujol et al [6] Furthermore, Pujol et al. suggested that early folate supplement by in utero sodium formate supplementation may prevent the neurodegenerative process in CYP2U1 −/− mouse model [6]. With over 15 years of folinic acid supplement and maintenance of a normal CSF 5-MTHF level, we successfully prevented the 2 brothers from deteriorating from this devastating neurodegenerative disease. Taking together Pujol's recent mouse model study results and our clinical evidence, we propose CYP2U1-related SPG56 as a novel potentially treatable neurometabolic disease. To have a better understanding of this postulation, further studies investigating the relationship of the loss of function in CYP2U1 and cerebral folate deficiency should be carried out.
The CSF findings of transient elevated CSF neopterin in the elder brother together with low complement C3 in both brothers during late childhood that subsequently normalized during adolescent period require further studies. It may relate to the stable clinical course of our patients, or potentially to an underlying immunological process. No definite temporal relationship was noted between the level of neopterin and dosages of folinic acid treatment in the elder brother. These parameters may also serve as future treatment or disease biomarkers.
Folate, in the form of folic acid, folinic acid and 5-MTHF are the 3 currently available formulations that have different properties [12]. Folic acid is not an active drug. For the treatment of CFD, most reporting studies used folinic acid with good results as the brain has a limited capacity to reduce folic acid into metabolically active forms. For 5-MTHF, it is well absorbed in the intestine and is also thought to be the most efficient way to restore CSF 5-MTHF concentrations [9]. Further clinical studies on the efficiency of folinic acid verses 5-MTHF should be considered. We suggest checking CSF for neurotransmitters including 5-MTHF and neopterin levels in other patients with CYP2U1 pathogenic variants. In the future, there is imminent need to carry out a prospective natural history study including the cerebral folate deficiency in CYP2U1-related spastic paraplegia 56 and a formal clinical trial using folinic acid supplementation in these patients with a low or borderline low CSF 5-MTHF.
4. Conclusion
This case report is the first human report supporting preclinical findings from Pujol et al. Likewise, CYP2U1-related SPG56 may be associated with cerebral folate deficiency and early folate supplement can be associated with clinical stabilization.
Authors contributions
Dr. Sheila Suet-Na Wong contributed to the study by creating the original design, writing the manuscript and overseeing the study as a whole. Dr. Liz Yuet-Ping Yuen played a role in molecular diagnosis, manuscript writing and proofreading. Dr. Elaine Kan contributed to radiological diagnosis, manuscript writing and proofreading. Dr. Nenad Blau and Dr. Richard Rodenburg contributed to biochemical diagnosis, manuscript writing and proofreading. Prof Virginia Chun-Nei Wong, Dr. Fanny Mochel, and Dr. Ron Wevers contributed to the original design of the study and proofreading of the manuscript. Dr. Cheuk-Wing Fung contributed to the study by creating the original design, writing the manuscript and overseeing the study as a whole.
Funding information
The biochemical and molecular analyses from overseas laboratories of the patients are generously supported by The Society for the Relief of Disabled Children (SRDC). The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Ethics statement
This study has approved by the Central Institutional Review Board, Hospital Authority (Ref no.: PAED-2023-054). Written consent was obtained from the patients for the publication of this case report.
Declaration of Competing Interest
All authors declare that they have no conflict of interest.
Data availability
The data that support the findings of this study are available on request from the corresponding author. These data are not publicly available due to privacy or ethical restrictions.
References
- 1.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Tallaksen C.M., Durr A., Brice A. Recent advances in hereditary spastic paraplegia. Curr. Opin. Neurol. 2001;14(4):457–463. doi: 10.1097/00019052-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Fink J.K. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003;184(Suppl. 1):S106–S110. doi: 10.1016/j.expneurol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Tesson C., Nawara M., Salih M.A., Rossignol R., Zaki M.S., Al Balwi M., et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91(6):1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murala S., Nagarajan E., Bollu P.C. Hereditary spastic paraplegia. Neurol. Sci. 2021;42(3):883–894. doi: 10.1007/s10072-020-04981-7. [DOI] [PubMed] [Google Scholar]
- 6.Pujol C., Legrand A., Parodi L., Thomas P., Mochel F., Saracino D., et al. Implication of folate deficiency in CYP2U1 loss of function. J. Exp. Med. 2021;218(11) doi: 10.1084/jem.20210846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Legrand A., Pujol C., Durand C.M., Mesnil A., Rubera I., Duranton C., et al. Pseudoxanthoma elasticum overlaps hereditary spastic paraplegia type 56. J. Intern. Med. 2021;289(5):709–725. doi: 10.1111/joim.13193. [DOI] [PubMed] [Google Scholar]
- 8.Leonardi L., Ziccardi L., Marcotulli C., Rubegni A., Longobardi A., Serrao M., et al. Pigmentary degenerative maculopathy as prominent phenotype in an Italian SPG56/CYP2U1 family. J. Neurol. 2016;263(4):781–783. doi: 10.1007/s00415-016-8066-7. [DOI] [PubMed] [Google Scholar]
- 9.Pope S., Artuch R., Heales S., Rahman S. Cerebral folate deficiency: analytical tests and differential diagnosis. J. Inherit. Metab. Dis. 2019;41(4):655–672. doi: 10.1002/jimd.12092. [DOI] [PubMed] [Google Scholar]
- 10.Ramaekers V.T., Blau N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2004;46(12):843–851. doi: 10.3390/nu14153096. [DOI] [PubMed] [Google Scholar]
- 11.Dale R.C., Brilot F., Fagan E., Earl J. Cerebrospinal fluid neopterin in paediatric neurology: a marker of active central nervous system inflammation. Dev. Med. Child Neurol. 2009;51(4):317–323. doi: 10.1111/j.1469-8749.2008.03225.x. [DOI] [PubMed] [Google Scholar]
- 12.Scaglione F., Panzavolta G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica. 2014;44(5):480–488. doi: 10.3109/00498254.2013.845705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. These data are not publicly available due to privacy or ethical restrictions.