

Abstract

Ovothiol A and ergothioneine are thiol-histidine derivatives with sulfur substitutions at the δ-carbon or ε-carbon of the l-histidine imidazole ring, respectively. Both ovothiol A and ergothioneine have protective effects on many aging-related diseases, and the sulfur substitution plays a key role in determining their chemical and biological properties, while factors governing sulfur incorporation regioselectivities in ovothiol and ergothioneine biosynthesis in the corresponding enzymes (OvoA, Egt1, or EgtB) are not yet known. In this study, we have successfully obtained the first OvoA crystal structure, which provides critical information to explain their C–S bond formation regioselectivity. Furthermore, OvoATh2 exhibits several additional activities: (1) ergothioneine sulfoxide synthase activity akin to Egt1 in ergothioneine biosynthesis; (2) cysteine dioxygenase activity using l-cysteine and l-histidine analogues as substrates; (3) cysteine dioxygenase activity upon mutation of an active site tyrosine residue (Y406). The structural insights and diverse chemistries demonstrated by OvoATh2 pave the way for future comprehensive structure–function correlation studies.
Keywords: nonheme iron enzyme, ovothiol, ergothioneine, X-ray structure, regioselectivity, sulfur-containing natural products
Introduction
Ovothiol A (1) and ergothioneine (2) are naturally occurring thiol-histidine derivatives, featuring a sulfur substitution at the δ-carbon or the ε-carbon of their imidazole ring (Scheme 1),1,2 respectively. The locations of the sulfur atom within the imidazole side-chain of ovothiol A 1 and ergothioneine 2 resulted in different physical and biological properties.2 The pKa of ovothiol’s sulfur (∼1.4) is significantly more acidic than that of other natural thiols (usually falling within a range of 7.0 to 9.0).2−6 Ovothiol A inhibits cell proliferation due to its activation of an autophagic process in human hepatocarcinoma cell lines, Hep-G2,7 which indicates its potential anticancer activities. In contrast, ergothioneine is found primarily in its thiolate form under physiological conditions.8,9 Ergothioneine’s reduction potential (E0′ = −0.06 V)6,10,11 is significantly higher than that of glutathione (E0′ = −0.25 V).12 Ergothioneine can efficiently eradicate reactive oxygen species (ROS) and reactive nitrogen species (RNS).13−15 Moreover, ergothioneine possesses protective effects in several human diseases, such as rheumatoid arthritis,16,17 Crohn’s disease,18,19 neurodegenerative diseases,20−23 cardiovascular disorders,24 diabetes,25 and fatty liver disease.26,27 Ergothioneine may also cross the blood-brain barrier,20−22 to protect against neurodegenerative diseases. Notably, Ames recently hypothesized that ergothioneine might be a longevity vitamin.28
Scheme 1. Biosynthetic Pathways of Ergothioneine and Ovothiol A.
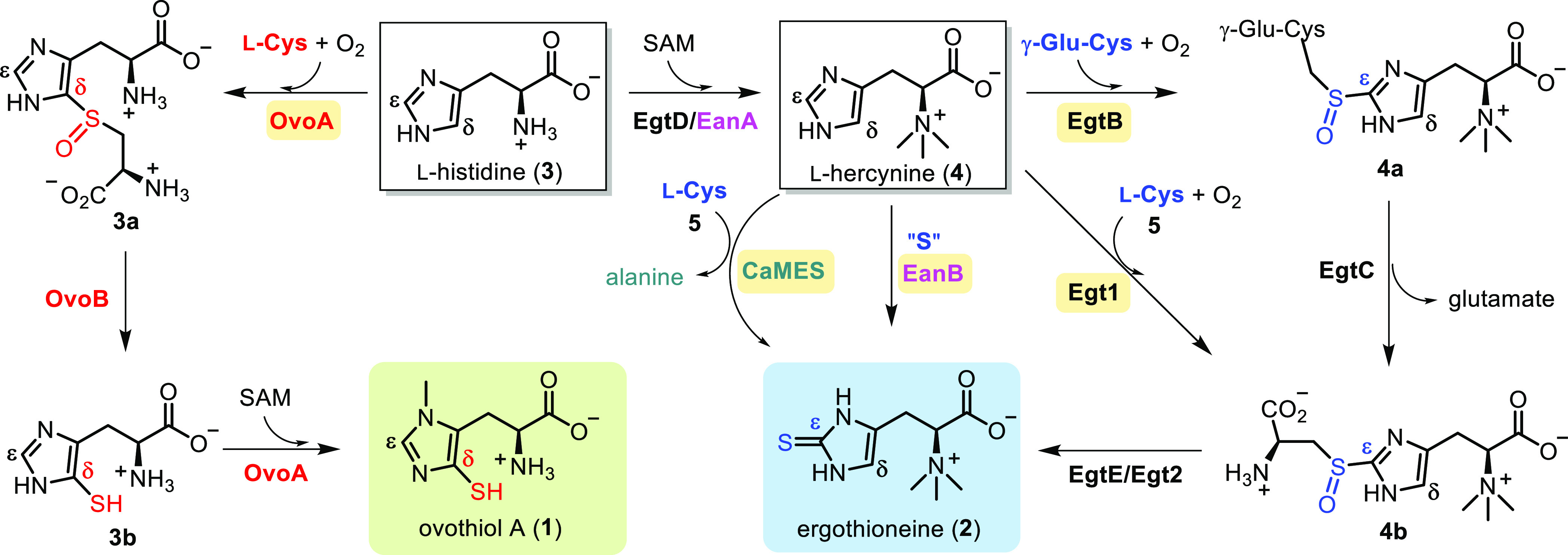
The four ergothioneine biosynthetic routes are depicted on the right, while the aerobic ovothiol A biosynthetic pathway is illustrated on the left. Crucial C–S bond construction steps are catalyzed by EgtB, Egt1, EanB, CaMES, and OvoA, respectively.
Besides their biological activities, the biosynthetic and mechanistic characterization of sulfur-containing natural products has sparked new chemistries and enzymology.29−32 While l-cysteine or methionine commonly serves as sulfur sources in sulfur-containing peptides, the realm of sulfur incorporation chemistry extends far beyond peptide-based natural products. Exemplifying this versatility is ovothiol A and ergothioneine biosynthesis, which present elegant instances across various facets,1,30 encompassing sulfur sources, aerobic vs anaerobic biosynthetic routes, exploration of diverse catalytic strategies, meticulous control over sulfur-transfer regioselectivity, and notably, the shared and distinct features between sulfur and selenium chemistries.33−36 Over the past decade, four biosynthetic pathways for ergothioneine and one for ovothiol A have been elucidated. In the two aerobic pathways leading to ergothioneine, namely the mycobacterial pathway (4 → 4a → 4b → 2 transformations catalyzed by EgtB, EgtC, and EgtE enzymes, Scheme 1)37,38 and the fungal Neurospora crassa pathway (4 → 4b → 2 transformations catalyzed by Egt1 & Egt2/EgtE enzymes, Scheme 1),39,40 pivotal C–S bond formation steps are catalyzed by mononuclear nonheme iron enzymes. In particular, 4 → 4a is catalyzed by EgtB, and 4 → 4b is catalyzed by Egt1, using γ-glutamyl-cysteine (γ-GC)37 or l-cysteine39 as the sulfur sources, respectively. Subsequently, a PLP-dependent C–S lyase orchestrates the 4b → 2 transformation catalyzed by either EgtE or Egt2 (Scheme 1), finalizing the sulfur transfer from l-cysteine to the l-histidine side-chain.38,40 Recently, two anaerobic ergothioneine biosynthetic pathways have come to light. In the pathway of the green sulfur bacterium Chlorobium limicola (3 → 4 → 2 transformations catalyzed by EanA-EanB catalysis, Scheme 1), the sulfur transfer is mediated by a rhodanese-domain-containing protein, EanB, utilizing polysulfide as the direct sulfur source and l-cysteine persulfide as an intermediate.41,42 In the Caldithrix abyssi ergothioneine biosynthetic pathway of involving 3 → 4 → 2 transformations (Scheme 1), a molybdenum-containing enzyme, CaMES,43 is responsible for the vital sulfur transfer step. While l-cysteine has been suggested as the sulfur source, the mechanistic intricacies of CaMES catalysis are yet to be thoroughly investigated. Currently, a solitary ovothiol A biosynthetic pathway has been characterized (3 → 3a → 3b → 1 transformations catalyzed by OvoA and OvoB enzymes, Scheme 1).44,45 In this ovothiol biosynthetic pathway, OvoA is a bifunctional enzyme encompassing sulfoxide synthase activity (3 → 3a, Scheme 1) and methyltransferase activity (3b → 1 transformations catalyzed by OvoA enzyme, Scheme 1).44 Concurrently, OvoB serves as a PLP-dependent C–S lyase.45
Over the past decade, a pivotal query in the realm of ergothioneine and ovothiol biosynthetic research involves unraveling the factors that dictate the regioselectivity of these nonheme iron enzymes (EgtB/Egt1 vs OvoA).46 Recently, two X-ray crystal structures of ergothioneine sulfoxide synthases, namely EgtBMthr47 and EgtBCth,48,49 have emerged. However, no crystal structure of the OvoA has been documented thus far. This report addresses this gap in knowledge by presenting the biochemical and structural insights of an ovothiol sulfoxide synthase, OvoATh2, derived from Hydrogenimonas thermophila. This is the first crystal structure of an ovothiol sulfoxide synthase.
Results and Discussion
Identification of the Ovothiol Biosynthetic Enzymes Derived from a Thermophilic Organism, H. thermophila
Due to the sulfur atom’s importance in determining the chemical and biological properties of ovothiol A and ergothioneine, there has been extensive interest in uncovering the factors governing the sulfoxide synthase regioselectivities, i.e., OvoA vs EgtB/Egt1 selectivity.46 The biochemically characterized OvoA isolated from Erwinia tasmaniensis (OvoAEta),44,46,50,51 is very labile, and all crystallization efforts failed. Using OvoAEta as the query sequence, we searched for the homologues in thermophiles or mesophilic microorganisms from UniProt and NCBI databases.52 In total, 230 homologues were obtained. Using a protein sequence similarity network analysis approach, at an E-value cutoff of 10–60, seventy-five representative sequences were categorized into two clusters (the EgtB branch and the OvoA branch), as well as a separate enzyme EgtBCth carrying both Egt1 and EgtB activities (Figure 1A).48,53 Among those sequences found within the OvoA node is the H. thermophila (WP_092912839) sequence was intriguing due to the optimal growth temperature of the microbe of approximately 55 °C.54 This protein was subsequently named OvoATh2. Analysis of the protein domain organization of OvoATh2 suggests that similar to that of OvoAEta, OvoATh2 has both sulfoxide synthase and methyltransferase domains (Figure S1). Due to thermostability concerns in OvoA crystallization, after overexpression and purification of OvoATh2 (Figure S2) using a protocol similar to those employed for OvoAEta and OvoAMtht characterization,46,52 we assessed the thermostability of OvoATh2 using a nanoDSF assay (Figure 1B).55,56 The thermal unfolding curves of the peptide of OvoAMtht are included for comparison purposes. Our analysis demonstrated that the TM of OvoATh2 is approximately 21 °C higher than that of OvoAMtht (Figure 1B).
Figure 1.

OvoATh2 characteristics. (A) Sequence similarity network analysis with an E-value of 10–60, highlighting EgtB and OvoA nodes; (B) assessment of thermostability for OvoAMtht and OvoATh2 via NanoDSF; (C) OvoATh2-catalyzed reactions using l-histidine and l-cysteine/[β-13C]-l-cysteine substrates, obtained through 1H NMR and 13C NMR analysis.
Domain analysis of OvoATh2 suggested that it possesses an N-terminal formylglycine-generating enzyme (FGE) sulfatase domain as well as a C-terminal methyltransferase domain (Figure S1). OvoAEta contains a similar domain organization, with our biochemical characterization supporting the bifunctionality of OvoAEta: ovothiol sulfoxide synthase and methyltransferase (Scheme 1).45 OvoATh2 catalysis was analyzed using three different assays to provide evidence to support the activity of OvoATh2 as an ovothiol sulfoxide synthase (Figures 1C and S3): (a) 1H NMR to examine the l-histidine imidazole side-chain for ovothiol sulfoxide synthase regioselectivity; (b) 13C NMR assay for l-cysteine activities using [β-13C]-l-cysteine as a substrate; (c) oxygen consumption rate analysis utilizing a Neo-Foxy oxygen electrode to assess the overall O2 consumption rate.
In the 1H NMR spectra, based on previously reported results pertaining to Egt1/EgtB/OvoA,39,46,52 the ∼7.60 and 6.80 ppm peaks were assigned to l-histidine imidazole Hε and Hδ, respectively. In the OvoATh2 catalyzed reaction, when l-histidine (3) and l-cysteine (5) were utilized as substrates, the product 1H NMR signal was found to be ∼7.92 ppm (trace i, Figures 1C and S3), assigned to Hε based on prior studies on OvoAEta,46 which suggests that OvoATh2 catalyzes sulfoxide formation at the δ-position, the ovothiol class of sulfoxide regioselectivity. In the 13C NMR spectrum of the reaction mixture from the l-histidine and [β-13C]-l-cysteine reaction, two signals at 58.0 and 53.2 ppm were observed (trace ii, Figure 1C). The 53.2 ppm signal is consistent with [β-13C]-sulfoxide product 3a′ as reported in prior OvoAEta studies.52,57 The signal at 58.0 ppm is consistent with l-cysteine sulfinic acid,57 while the level of [β-13C]-l-cysteine sulfinic acid 6′ represents approximately ∼10% of the product mixture. Therefore, OvoATh2 indeed acts as an ovothiol sulfoxide synthase. The oxygen consumption assay using a NeoFoxy electrode indicated that the kinetic parameters of OvoATh2 using l-histidine and l-cysteine at 23 °C are kcat, O2 = 589.0 ± 3.5 min–1; KM, l-His = 585.1 ± 28.0 μM, and KM, l-cys = 278.6 ± 9.0 μM (Table 1 and Figure S4).
Table 1. Kinetic Parameters of Wild-Type OvoATh2 and Its Mutants under Different Conditions.
| enzyme | substrates | kcat, O2 [min–1] | KM, l-His or l-His derivatives [μM] | KM, l-Cys [μM] | kcat/KM, l-His or l-His derivatives or l-Cys [min–1μM–1] | % of coupling product from NMR analysis |
|---|---|---|---|---|---|---|
| wild-type | l-His + l-Cys | 589.0 ± 3.5 | 585.1 ± 28.0 | 278.6 ± 9.0 | 2.10 ± 0.09 | ∼90% |
| wild-type | l-Her + l-Cys | 127.2 ± 1.5 | 61.9 ± 3.5 | (1.86 ± 0.04) E3 | 66.9 ± 2.3 | ∼50% |
| wild-type | l-Cys | 26.7 ± 0.4 | (2.6 ± 0.2) E3 | below the detection limit | ||
| wild-type | 3-Me-l-His + l-Cys | 131.2 ± 2.7 | 1.9 ± 0.1 | 252.2 ± 14.5 | 0.52 ± 0.04 | below the detection limit |
| wild-type | 1-Me-l-His + l-Cys | 20.5 ± 0.3 | (3.9 ± 0.1) E3 | (5.3 ± 0.1) E-3 | below the detection limit |
| enzyme | substrates | kcat, O2 [min–1] | KM, l-His [μM] | KM, l-Cys [μM] | kcat/KM, l-Cys [min–1 μM–1] | % of coupling product from NMR analysis |
|---|---|---|---|---|---|---|
| Y406F | l-His + l-Cys | 361.2 ± 5.9 | 38.2 ± 2.6 | 575.2 ± 22.0 | 0.63 ± 0.09 | ∼20% |
| Y68F | l-His + l-Cys | 23.4 ± 0.2 | 50.9 ± 2.5 | (61.9 ± 1.5) E3 | (0.4 ± 0.1) E-3 | ∼30% |
| Y68F/Y406F | l-His + l-Cys | 14.8 ± 0.3 | 31.3 ± 4.0 | (125.7 ± 11.0) E3 | (1.2 ± 0.1) E-4 | ∼10% |
Overall Structure of OvoATh2
After demonstrating that OvoATh2 is the ovothiol sulfoxide synthase for ovothiol biosynthesis, we sought to crystallize it. Because OvoATh2’s thermal unfolding temperature TM is ∼21 °C higher than that of OvoAMtht (Figure 1B), our crystallization efforts on OvoATh2 indeed were successful, obtaining the OvoATh2 crystal structure at a resolution of 2.7 Å in complex with its two substrates, l-histidine and l-cysteine (Figure 2, OvoA·CoII·l-Cys·l-His, PDB ID: 8KHQ). OvoATh2 contains two functional domains: a sulfatase domain at the N-terminus (7–455) and a SAM-dependent methyltransferase domain at the C-terminus (466–707). Each domain forms a distinctive globular region, and these two domains are linked via a linker loop (456–465). The shaft angle between the mass centers of the two domains and the center of the linker is 106.6° (Figure 2A). Two OvoATh2 monomers dimerize through a 2-fold symmetry in which the two C-terminal methyl transferase domains are at the center, flanked by the two N-terminal domains on either side. Such an arrangement results in a flat, rhomboid-shaped structure with three nodes (dimer of C-terminal domains in the middle, two N-terminal domains on each side) (Figure 2B). Based on calculations using the PISA server,58 the dimer interface of OvoATh2 is 1,951 Å2, occupying approximately 6.5% of the total surface accessible area. Two OvoATh2 regions are responsible for the dimerization. The first region is a β-strand resulting from residues T690-Q700 at the C-terminus. The β-strands from the two monomers stack in an antiparallel manner. Moreover, salt bridges were observed to link R692 of one monomer to D682′ of the other monomer. The H696 residue forms a hydrogen bond with the same residue H696′ on the other monomer while interacting with F486′ through π stacking, which may help to orient the C-terminal domains (Figure 2C). The second monomer interface is primarily driven by the C-terminal domain of one monomer with the N-terminal domain of the other monomer. Residues G653-K662 form a short β-hairpin, which slots into the cavity built from N78′ to S100′ and I405′ to D408′ from the N-terminal domain of the other monomer (Figure 2D). The residues flanking this β-hairpin, (i.e., Y638–W650 and S663–D665) also participate in dimerization through interactions with the β-hairpin formed by T560′-L564′ in the other monomer. Apart from hydrophobic interactions, intensive polar interactions also occur, including salt bridging between K647:E563′, K655:D407′, K655:D408′, and E659:K79′, as well as hydrogen bonding between D657:S96′, S663:E561′, and G653:E563′, G653:L564′ (Figures 2D and S5).
Figure 2.

Crystal structure of OvoATh2. (A) Domain arrangement of OvoATh2. The OvoA·CoII·l-Cys·l-His complex (PDB ID: 8KHQ) is shown in cartoon mode with yellow and light blue colors to present the N-terminal and C-terminal domains with a green linker, respectively. (B) Overall architecture of the OvoATh2 homodimer. The two monomers are colored green and cyan. (C and D) Dimer interfaces of OvoATh2. Each monomer was colored green and cyan, and the interacting residues were labeled with polar interactions highlighted in black dashes. (E) The active site of the OvoA·CoII·l-Cys·l-His ternary complex. The residues around the active site in the N-terminal domain were shown as green sticks, while the metal cobalt as a pink sphere. (F) Active site of EgtB·MnII·γ-GC·N,N-dimethyl-l-His ternary complex (PDB ID: 4 × 8D).47 The residues around the active site were shown as gray sticks, while the metal manganese as a sphere. (G) Superimposition between EgtB·MnII·γ-GC·N,N-dimethyl-l-His complex and OvoA·CoII·l-Cys·l-His complex. The Cδ of the l-histidine imidazole ring was shown in sphere.
Active Site Analysis of the OvoATh2·CoII·l-Cys·l-His Ternary Complex
Upon cocrystallization of OvoATh2 with its substrates, l-cysteine and l-histidine, we mixed apo OvoATh2, CoII, l-Cys, and l-His in a molar ratio of 1:3:3:3 and successfully solved the structure of OvoATh2·CoII·l-Cys·l-His complex, which was crystallized anaerobically in an anaerobic Coy-chamber to avoid transformations during the crystallization process (Figures 2E and S6). The metallo-center is coordinated by three l-histidine residues (H71, H162, and H166). The l-histidine substrate coordinates to CoIItrans to H71 using the imidazole N1 atom, and the l-cysteine substrate is trans to H162. The sixth coordination site is occupied by a water molecule (outlined in chain B), which is a potential oxygen-binding site. A conserved tyrosine residue (Y406) forms a hydrogen bond with the coordinating water at a distance of 3.1 Å (visible in chain B, Figure 2E). The l-cysteine substrate is also involved in a hydrogen bonding network involving R62, Y68, the backbone of V102, and the carboxyl group of the l-histidine substrate. Apart from interactions with the l-cysteine substrate, the l-histidine substrate also interacts with the F409 backbone by hydrogen bonds, and a portion of l-histidine is also close to Y406 as well as the nonpolar portion of T413 through nonpolar contacts (Figure 2E).
To examine structural factors crucial for the sulfoxide formation regioselectivity in OvoATh2 relative to ergothioneine sulfoxide synthase (Egt1 or EgtB), we focused on the OvoATh2N-terminal domain as it is the sulfoxide synthase domain. EgtB from Mycobacterium thermophilum (EgtBMthr, PDB ID: 4 × 8D)47,49 possesses the highest structural similarity to OvoATh2, with an RMSD of 2.3 Å. EgtBMthr uses N,N,N-trimethyl-histidine (l-hercynine) and γ-glutamyl-cysteine (γ-GC) as substrates (Scheme 1).37,47,59 Both ergothioneine and ovothiol sulfoxide metallo-centers are coordinated by four imidazoles of l-histidine/hercynine residues (three l-histidine residues from the protein and one from the l-histidine/hercynine substrate) and one l-cysteine, leaving an additional site unoccupied for O2 binding and activation. Seebeck and co-workers successfully solved the crystal structure of EgtBMthr·MnII·γ-GC·N,N-dimethyl-l-histidine complexes (Figure 2F).47 In the OvoATh2·CoII·l-Cys·l-His and EgtBMthr·MnII·γ-GC·N,N-dimethyl-l-histidine complexes (Figure 2G), the l-cysteine portions possess a similar orientation. Interestingly, the imidazole of the N,N-dimethyl-l-histidine in the EgtBMthr·MnII·γ-GC·N,N-dimethyl-l-histidine complexes flips approximately 180° relative to the l-histidine in the OvoATh2·CoII·l-Cys·l-His complex. Such differences expose the l-histidine Cδ to the l-cysteine sulfur atom in the OvoATh2·CoII·l-Cys·l-His complex, while in the EgtBMthr·MnII·γ-GC·N,N-dimethyl-l-histidine complex, the Cε position in N,N-dimethyl-l-histidine is closer to the cysteine sulfur atom in γ-GC (Figure 2G). In our prior biochemical characterizations, we have demonstrated that the use of N,N-dimethyl-l-histidine and l-hercynine as OvoAEta substrates result in the same sulfoxide synthase regioselectivity, the ergothioneine type.46 Therefore, this comparative structural analysis suggests that the relative orientation of the l-histidine imidazole side-chain in the enzyme active site determines the ovothiol and ergothioneine sulfoxide synthase regioselectivities.
The largest difference between the active site of OvoATh2 and EgtBMthr is a loop region (residues 98 to 117 in OvoATh2 vs residues 80–95 in EgtBMthr), acting as a lid to cover the subpocket for the l-cysteine substrate. In EgtBMthr, this loop region flips toward the solvent (Figure S7). We proposed that this loop region modulates the substrate preference in this family of sulfoxide synthases, where the relatively smaller subpocket in OvoA covered by this loop could accommodate only l-cysteine as the sulfur donor. At the same time, the more spacious pocket in EgtBMthr could accommodate γ-GC as the sulfur donor. Similar conclusions can also be drawn from the comparison of the crystal structure of EgtBCth from Chloracidobacterium thermophilum to OvoATh2.48,49 Residues 91 to 111 in EgtBCth represent the loop serving as the lid for covering the subpocket.
In EgtBMthr, a tyrosine residue (Y377) has been proposed to be critical to the sulfoxide synthetase activity.47 In EgtBCth,48,49 two tyrosine residues (Y92 and Y93) are catalytically important. In OvoATh2, there is a conserved tyrosine residue (Y406) in the same position relative to Y337 in EgtBMthr. Given the similarity between OvoATh2 and EgtBMthr, Y406 in OvoATh2 may be critical for ovothiol sulfoxide synthase activity.
Functional Diversities of OvoATh2 Derived from H. thermophila
In two OvoA enzymes reported in the literature (OvoAEta46,57,60 and OvoAMtht52), outside of the ovothiol sulfoxide synthase activities, under some conditions, these enzymes produce l-cysteine sulfinic acid as a dominant product, displaying cysteine dioxygenase (CDO) activity. The partitioning between CDO and ovothiol sulfoxide synthase activities can be tuned through metallo-center ligands or by the secondary coordination shell residues. Critically, both OvoAEta and OvoAMtht have ergothioneine sulfoxide synthase activity when using l-hercynine and l-cysteine as substrates.46,52 In the previous section, we summarized the crystal structure data from OvoATh2. The next key point to be addressed is whether the unique biochemical properties reported in the literature for OvoAEta and OvoAMtht are common to OvoATh2. If OvoATh2 possesses the same properties, the structural information reported for OvoATh2 will be a solid foundation for future structure–function correlation studies.61 To answer this question, we systematically characterized OvoATh2 reactivity under various conditions (Figure 3 and Table 1): (1) ovothiol vs ergothioneine sulfoxide synthase activity; (2) cysteine dioxygenase activity; (3) roles of active site tyrosine residues.
Figure 3.

Characterization of OvoATh2 and its variants under a few different conditions. (A) Results of wild-type OvoATh2-catalyzed reactions using [β-13C]-l-cysteine (5′) and l-histidine (3) or derivatives (l-hercynine (4), 3-methyl-l-histidine (7), and 1-methyl-l-histidine (8)) as substrates or using 5′ as the only substrate. (B) Evaluation of OvoATh2 variants OvoATh2/Y406F and OvoATh2/Y68F and its double mutant OvoATh2/Y68F/Y406F for their relative sulfoxide and CDO-like synthase activity using 5′ and 3 as substrates. (C) 13C NMR analysis of reactions: wild-type OvoATh2 using 5′ and 3 as substrates (i), OvoATh2 using 5′ and 4 as substrates (ii), OvoATh2 using 5′ as the only substrate (iii), OvoATh2 using 5′ and 7 as substrates (iv), OvoATh2 using 5′ and 8 as substrates (v), OvoATh2/Y406F using 5′ and 3 as substrates (vi), OvoATh2/Y68F using 5′ and 3 as substrates (vii), and OvoATh2/Y68F/Y406F using 5′ and 3 as substrates (viii).
OvoATh2 is an ovothiol sulfoxide synthase (Figure 1). When [β-13C]-cysteine was used to replace cysteine, it generated [β-13C]-sulfoxide 3a′ as a major product and [β-13C]-l-cysteine sulfinic acid 6′ as a minor product (∼10% of the reaction mixture). Interestingly, OvoATh2 possesses ergothioneine sulfoxide synthase activity (Figure S8). Upon swapping in l-hercynine for l-histidine, 1H NMR clearly indicated that OvoATh2 is an ergothioneine sulfoxide synthase, as demonstrated by the signal at 7.23 ppm in the 1H NMR spectrum, which is the signal for the imidazole δ-position hydrogen atom (Figure S8). This result suggests sulfoxide formation at the l-hercynine ε-carbon. This conclusion was further supported by 13C NMR analysis of the reaction using l-hercynine and [β-13C]-l-cysteine as substrates. In the 13C NMR spectrum, the signal at 55.0 ppm was derived from [β-13C]-sulfoxide 4b′, and the signal at 58.0 ppm was derived from [β-13C]-l-cysteine sulfinic acid 6′. The ratio between 4b′ and 6′ is approximately 1:1 (trace ii, Figure 3). Kinetic analysis using an O2 consumption assay demonstrated that the kinetic parameters for this reaction at 23 °C are kcat, O2 = 127.2 ± 1.5 min–1 ; KM, L-Her = 61.9 ± 3.5 μM, and KM, l-cys = 1.86 ± 0.04 mM (Figure S9, Table 1). Clearly, upon changing l-histidine to l-hercynine, OvoATh2 makes a corresponding alteration in activity from an ovothiol sulfoxide synthase to an ergothioneine sulfoxide synthase. We measured the CDO activity of the molecule of OvoATh2 using l-cysteine as the substrate. The signal at 58.0 ppm in the 13C NMR experiments did support the CDO activity when l-cysteine was the only substrate (trace iii, Figures 3and S10). The steady-state kinetics parameters are KM, l-Cys = 2.6 ± 0.2 mM, and a much lower kcat, O2 = 26.7 ± 0.4 min–1 (Figure S11, Table 1).
The alterations in activity upon the replacement of l-histidine with l-hercynine clearly suggest that the catalysis of OvoATh2 is sensitive to subtle modifications to the enzyme’s active site. We examined two additional histidine analogs, 3-methyl-l-histidine (7) and 1-methyl-l-histidine (8), which have the two imidazole nitrogen atoms methylated, respectively. Using 3-methyl-l-histidine (7) and l-cysteine as substrates, only [β-13C]-l-cysteine sulfinic acid 6′ (δC 58.0) was observed as a product (trace iv, Figures 3 and S12–S13, Table 1) with the kcat, O2 = 131.2 ± 2.7 min–1. Similar results were also observed when 1-methyl-l-histidine (8) and l-cysteine were used as substrates (trace v, Figures 3 and S14, Table 1), producing only [β-13C]-l-cysteine sulfinic acid 6′ with kcat, O2 = 20.5 ± 0.3 min–1 (Figure S15).
Results from the above experiments demonstrated that the activities of the OvoATh2 can be tuned across ovothiol sulfoxide synthase, ergothioneine sulfoxide synthase, and CDO activities. When l-histidine and l-cysteine were substrates, ∼90% of the product in the reaction mixture was found to be sulfoxide 3a. However, in the case of l-hercynine and l-cysteine reactions, ergothioneine sulfoxide 4b and l-cysteine sulfinic acid 6 are produced at comparable levels. In the cases of both 3-methyl-l-histidine and 1-methyl-l-histidine, OvoATh2 displays CDO activity, with reduced kcat values by ∼4.5 and 28.7-fold, respectively (Table 1).
In the structure of the OvoATh2·CoII·l-Cys·l-His complex, two tyrosine residues, Y68 and Y406, are close to the metallo-center and substrate binding site. It has been identified that active site tyrosine residues are critical to ovothiol and ergothioneine sulfoxide synthase activity.48−52,60 We, therefore, characterized the phenylalanine mutants of these two tyrosine residues. Three mutants have been biochemically and kinetically analyzed, including Y68F and Y406F single mutants and the Y68F/Y406F double mutant of OvoATh2. The kinetic parameters of these mutants are summarized in Table 1.
In the Y406F mutant, the kinetic parameters were measured through an O2 consumption assay at 23 °C (Table 1 and Figures S16–S17). In comparison to the parameters obtained for wild-type OvoATh2, OvoATh2/Y406F has a KM for the l-histidine substrate decreased by 15-fold from 585.1 ± 28.0 μM in wild-type OvoATh2 to 38.2 ± 2.6 μM, while their kcats are comparable. The product analysis of the Y406F reaction determined that the dominant product (>80%) of the Y406F mutant reaction is [β-13C]-l-cysteine sulfinic acid 6′, with a small amount of [β-13C]-sulfoxide 3a′ representing ∼20% of the product mixture (trace vi, Figure 3). In the Y68F mutant, the results differ from both the wild-type and Y406F mutant (Table 1 and Figures S18–S19). Compared to the parameters of the wild-type OvoATh2, the KM for the l-cysteine substrate increases by nearly 200-fold, and the kcat, O2 decreased by approximately 25-fold for OvoATh2/Y68F. As a result, in the OvoATh2 Y68F mutant, the catalytic efficiency (kcat/KM, l-Cys) decreases by nearly 5.5 × 103-fold. 1H NMR and 13C NMR analysis of the Y68F mutant reaction demonstrated that the product ratio of [β-13C]-sulfoxide 3a′, [β-13C]-l-cysteine sulfinic acid 6′, and [β-13C]-l-cystine 9′ is approximately 5:3:2 (trace vii, Figures 3 and S18). Based on the crystal structure depicted in Figure 2, Y68 directly interacts with the carboxylate of the l-cysteine substrate through hydrogen bonding. In the double mutant variant Y68F/Y406F, the kinetic parameters are comparable to those of the Y68F mutant. However, 1H NMR and 13C NMR analysis suggested that the double mutant variant produces l-cysteine sulfinic acid as the dominant product, while sulfoxide and cystine are below the detection levels (Table 1, trace viii in Figures 3 and S20–21). Therefore, with the use of 3-methyl-l-histidine and 1-methyl-l-histidine as the substrate or by using the Y68F/Y406F mutant, OvoATh2 exhibits predominantly the CDO activity rather than an ovothiol sulfoxide synthase activity. Such flexibility in the OvoATh2 activities suggests it is an excellent platform for future structure–function relationship studies.
Conclusions
Sulfur is one of the most abundant elements in the natural world. In recent years, an interdisciplinary approach has been employed to characterize sulfur incorporation chemistry,30 encompassing biosynthetic sulfur sources, intermediates, and, notably, mechanistic insights into new strategies for sulfur-related biosynthetic pathways. With the exponential expansion of microbial genomes accessible through public databases,62 new biosynthetic and mechanistic knowledge has been harnessed in genome mining initiatives to systematically identify related natural products and their underlying biosynthetic routes. In the biosynthesis of sulfur-containing natural products, l-cysteine, or its derivatives (e.g., glutathione and mycothiol), have conventionally served as direct sulfur sources. Recent instances include the use of thiocysteine in leinamycin biosynthesis63 and the utilization of polysulfide in a specific anaerobic ergothioneine biosynthetic pathway.41,42 In numerous other cases, the precise sulfur sources remain unidentified and are simply denoted as “S”.1,29,30,61
Mechanistically, sulfur transfer chemistry can be accomplished through nucleophilic substitution or radical mechanisms. For nucleophilic substitution chemistry, the two common intermediates are thiocarboxylate or l-cysteine persulfide on carrier proteins, as evidenced in biosynthetic studies of BE-7585A.64 For sulfur carrier proteins generating thiocarboxylate intermediates, they terminate with a C-terminal Gly-Gly. Some exceptions to this general structural motif have been reported, in which the C-terminal Gly-Gly motif was released from the carrier protein after proteolysis through the removal of the C-terminal residues by a dedicated protease.65,66 Recently, persulfides and thiocarboxylates have been implicated as key intermediates in the biosynthesis of thioamides, functional groups in sulfur-modified tRNA, and many small molecular natural products.31 Similar to thiazoline/thiazole in peptide-based natural products,67 thioamide generation is generally an ATP-dependent process involving either phosphorylation or adenosylation activation32 and l-cysteine, protein-bound thiocarboxylates, or l-cysteine persulfides have been suggested as sulfur sources, while in many cases, the sulfur sources are unknown.31
In addition to these novel sulfur natural product biosynthetic examples, ergothioneine and ovothiol A biosynthesis represent exceptional examples (Scheme 1). Their important biological properties are consistent with the use of several different strategies to produce these two compounds.28 Both aerobic and anaerobic biosynthetic pathways involving sulfur’s nucleophilic chemistries41−43 and radical chemistries1 have been reported for ergothioneine biosynthesis. For the aerobic ergothioneine and ovothiol A biosynthetic pathways, the crucial transformation is the nonheme iron enzyme-catalyzed sulfoxide formation reactions (EgtB, Egt1, and OvoA). One of the most important mechanistic questions is how these enzymes modulate the sulfoxide formation regioselectivities (Scheme 1). Over the past decade, two X-ray crystal structures of ergothioneine sulfoxide synthases, namely EgtBMthr47 and EgtBCth,48,49 have been reported. However, due to thermostability issues faced in the OvoA studies, it has been difficult to obtain the ovothiol sulfoxide synthase structure. In this work, using enzymes with significantly improved thermostability (∼21 °C increase in thermostability for OvoATh2 relative to OvoAMtht), we have obtained the first ovothiol sulfoxide synthase crystal structure. This paves the way for more detailed structure–function relationship studies.61 More importantly, in spite of the low sequence homology (47.6% identity relative to OvoAMtht) and a significant increase in thermostability, OvoATh2 shares nearly all biochemical properties with OvoAMtht.52 First, OvoATh2 possesses both ovothiol and ergothioneine sulfoxide synthase activities, and such tuning-in activities are controlled by the orientation of the imidazole side chain in the enzyme active site (Figure 2). Second, OvoATh2 has CDO activity, and the activities between CDO and sulfoxide synthase can be modulated using either substrate analogs (e.g., 3-methyl-l-histidine or 1-methyl-l-histidine) or by mutations in active site tyrosine residues (e.g., Y68F/Y406F double mutant). The structural information reported in this study and the rich chemistries displayed by OvoATh2 have laid a solid foundation for future structure–function correlation studies. This structural information may also assist future engineering efforts in obtaining enzymes that are more suitable for the production of ergothioneine and ovothiol in an industrial setting.68
Acknowledgments
The authors thank the staff members at the BL02U1 and BL19U1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF), China, for their assistance in X-ray crystal data collection.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c04026.
Fermentation and compound purification conditions; enzymatic reactions under various combinations and under different conditions; NMR data for substrates, substrate analogs, and products from the enzymatic reactions; and additional crystal structural information (PDF)
Author Contributions
# X.W., S.H., J.W., and T.Z. contributed equally to this work. P.L., X.L., W.Y., and L.Z. designed the study. S.H., T.Z., A.W., and X.W. performed biochemical experiments. W.Y., J.W., X.W., and K.Y. performed crystallization and structural determination. P.L., X.L., W.Y., L.Z., G.Z., and A.V. analyzed data and wrote the manuscript with input from all authors. All authors have given approval for the final version of the manuscript.
This work was supported by the National Key Research and Development Program of China (2019YFA0906201, 2020YFA0907800, 2020YFA090032, 2022YFC2105400), the National Natural Science Foundation of China (32121005, 21977029, 81903529, 32101008, 22307037), the 111 Project (B18022), the Open Project Funding of the State Key Laboratory of Bioreactor Engineering, and the Fundamental Research Funds for the Central Universities. This work is supported in part by the National Institutes of Health (GM140040 to P.L.) and the National Science Foundation (CHE-2004109 to P.L.).
The authors declare no competing financial interest.
Notes
The structure of the OvoATh2·CoII·l-Cys·l-His ternary complex (PDB ID: 8KHQ) can be accessed at https://www.pdbus.org/.
Supplementary Material
References
- Naowarojna N.; Cheng R.; Chen L.; Quill M.; Xu M.; Zhao C.; Liu P. Mini-Review: Ergothioneine and Ovothiol Biosyntheses, an Unprecedented Trans-Sulfur Strategy in Natural Product Biosynthesis. Biochemistry 2018, 57 (24), 3309–3325. 10.1021/acs.biochem.8b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano I.; Seebeck F. P. On ovothiol biosynthesis and biological roles: from life in the ocean to therapeutic potential. Nat. Prod. Rep. 2018, 35 (12), 1241–1250. 10.1039/C8NP00045J. [DOI] [PubMed] [Google Scholar]
- Marjanovic B.; Simic M. G.; Jovanovic S. V. Heterocyclic thiols as antioxidants: why ovothiol C is a better antioxidant than ergothioneine. Free Radic. Biol. Med. 1995, 18 (4), 679–685. 10.1016/0891-5849(94)00186-N. [DOI] [PubMed] [Google Scholar]
- Mirzahosseini A.; Orgován G.; Tóth G.; Hosztafi S.; Noszál B. The complete microspeciation of ovothiol A disulfide: a hexabasic symmetric biomolecule. J. Pharm. Biomed. Anal. 2015, 107, 209–216. 10.1016/j.jpba.2014.12.029. [DOI] [PubMed] [Google Scholar]
- Holler T. P.; Hopkins P. B. Ovothiols as biological antioxidants. The thiol groups of ovothiol and glutathione are chemically distinct. J. Am. Chem. Soc. 1988, 110 (14), 4837–4838. 10.1021/ja00222a057. [DOI] [Google Scholar]
- Hand C. E.; Honek J. F. Biological chemistry of naturally occurring thiols of microbial and marine origin. J. Nat. Prod. 2005, 68 (2), 293–308. 10.1021/np049685x. [DOI] [PubMed] [Google Scholar]
- Russo G. L.; Russo M.; Castellano I.; Napolitano A.; Palumbo A. Ovothiol isolated from sea urchin oocytes induces autophagy in the Hep-G2 cell line. Mar. Drugs 2014, 12 (7), 4069–4085. 10.3390/md12074069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler T. P.; Hopkins P. B. Ovothiols as free-radical scavengers and the mechanism of ovothiol-promoted NAD(P)H-O2 oxidoreductase activity. Biochemistry 1990, 29 (7), 1953–1961. 10.1021/bi00459a042. [DOI] [PubMed] [Google Scholar]
- Turner E.; Klevit R.; Hager L. J.; Shapiro B. M. Ovothiols, a family of redox-active mercaptohistidine compounds from marine invertebrate eggs. Biochemistry 1987, 26 (13), 4028–4036. 10.1021/bi00387a043. [DOI] [PubMed] [Google Scholar]
- Hartman P. E. Ergothioneine as antioxidant. Methods Enzymol. 1990, 186, 310–318. 10.1016/0076-6879(90)86124-E. [DOI] [PubMed] [Google Scholar]
- Fahey R. C. Novel thiols of prokaryotes. Annu. Rev. Microbiol. 2001, 55, 333–356. 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- Scott E. M.; Duncan I. W.; Ekstrand V. Purification and properties of glutathione reductase of human erythrocytes. J. Biol. Chem. 1963, 238 (12), 3928–3933. 10.1016/S0021-9258(18)51808-1. [DOI] [PubMed] [Google Scholar]
- Aruoma O. I.; Whiteman M.; England T. G.; Halliwell B. Antioxidant action of ergothioneine: assessment of its ability to scavenge peroxynitrite. Biochem. Biophys. Res. Commun. 1997, 231 (2), 389–391. 10.1006/bbrc.1997.6109. [DOI] [PubMed] [Google Scholar]
- Franzoni F.; Colognato R.; Galetta F.; Laurenza I.; Barsotti M.; Di Stefano R.; Bocchetti R.; Regoli F.; Carpi A.; Balbarini A. An in vitro study on the free radical scavenging capacity of ergothioneine: comparison with reduced glutathione, uric acid and trolox. Biomed. Pharmacother. 2006, 60 (8), 453–457. 10.1016/j.biopha.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Jang J.-H.; Aruoma O. I.; Jen L.-S.; Chung H. Y.; Surh Y.-J. Ergothioneine rescues PC12 cells from β-amyloid-induced apoptotic death. Free Radic. Biol. Med. 2004, 36 (3), 288–299. 10.1016/j.freeradbiomed.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Taubert D.; Lazar A.; Grimberg G.; Jung N.; Rubbert A.; Delank K.-S.; Perniok A.; Erdmann E.; Schömig E. Association of rheumatoid arthritis with ergothioneine levels in red blood cells: a case control study. J. Rheumatol. 2006, 33 (11), 2139–2145. [PubMed] [Google Scholar]
- Tokuhiro S.; Yamada R.; Chang X.; Suzuki A.; Kochi Y.; Sawada T.; Suzuki M.; Nagasaki M.; Ohtsuki M.; Ono M.; Furukawa H.; Nagashima M.; Yoshino S.; Mabuchi A.; Sekine A.; Saito S.; Takahashi A.; Tsunoda T.; Nakamura Y.; Yamamoto K. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet. 2003, 35 (4), 341–348. 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- Peltekova V. D.; Wintle R. F.; Rubin L. A.; Amos C. I.; Huang Q.; Gu X.; Newman B.; Van Oene M.; Cescon D.; Greenberg G.; Griffiths A. M.; St George-Hyslop P. H.; Siminovitch K. A. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004, 36 (5), 471–475. 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- Leung E.; Hong J.; Fraser A. G.; Merriman T. R.; Vishnu P.; Krissansen G. W. Polymorphisms in the organic cation transporter genes SLC22A4 and SLC22A5 and Crohn’s disease in a New Zealand Caucasian cohort. Immunol. Cell Biol. 2006, 84 (2), 233–236. 10.1111/j.1440-1711.2006.01423.x. [DOI] [PubMed] [Google Scholar]
- Kaneko I.; Takeuchi Y.; Yamaoka Y.; Tanaka Y.; Fukuda T.; Fukumori Y.; Mayumi T.; Hama T. Quantitative determination of ergothioneine in plasma and tissues by TLC-densitometry. Chem. Pharm. Bull. 1980, 28 (10), 3093–3097. 10.1248/cpb.28.3093. [DOI] [PubMed] [Google Scholar]
- Briggs I. Ergothioneine in the central nervous system. J. Neurobiochem. 1972, 19 (1), 27–35. 10.1111/j.1471-4159.1972.tb01250.x. [DOI] [PubMed] [Google Scholar]
- Crossland J.; Mitchell J.; Woodruff G. N. The presence of ergothioneine in the central nervous system and its probable identity with the cerebellar factor. J. Physiol. 1966, 182 (2), 427–438. 10.1113/jphysiol.1966.sp007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncaster J. A.; Walsh D. T.; Gentleman S. M.; Jen L.-S.; Aruoma O. I. Ergothioneine treatment protects neurons against N-methyl-D-aspartate excitotoxicity in an in vivo rat retinal model. Neurosci. Lett. 2002, 328 (1), 55–59. 10.1016/S0304-3940(02)00427-5. [DOI] [PubMed] [Google Scholar]
- Libby P.; Ridker P. M.; Hansson G. K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473 (7347), 317–325. 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Bastard J.-P.; Maachi M.; Van Nhieu J. T.; Jardel C.; Bruckert E.; Grimaldi A.; Robert J.-J.; Capeau J.; Hainque B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87 (5), 2084–2089. 10.1210/jcem.87.5.8450. [DOI] [PubMed] [Google Scholar]
- Cheah I. K.; Halliwell B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta 2012, 1822 (5), 784–793. 10.1016/j.bbadis.2011.09.017. [DOI] [PubMed] [Google Scholar]
- Cheah I. K.; Halliwell B. Could ergothioneine aid in the treatment of coronavirus patients?. Antioxidants (Basel) 2020, 9 (7), 595. 10.3390/antiox9070595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames B. N. Prolonging healthy aging: longevity vitamins and proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (43), 10836–10844. 10.1073/pnas.1809045115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele A. D.; Kiefer A. F.; Shen B. The many facets of sulfur incorporation in natural product biosynthesis. Curr. Opin. Chem. Biol. 2023, 76, 102366 10.1016/j.cbpa.2023.102366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar K. L.; Scharf D. H.; Litomska A.; Hertweck C. Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem. Rev. 2017, 117 (8), 5521–5577. 10.1021/acs.chemrev.6b00697. [DOI] [PubMed] [Google Scholar]
- Mahanta N.; Szantai-Kis D. M.; Petersson E. J.; Mitchell D. A. Biosynthesis and chemical applications of thioamides. ACS Chem. Biol. 2019, 14 (2), 142–163. 10.1021/acschembio.8b01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J.; Schwalen C. J.; Mann G.; Naismith J. H.; Mitchell D. A. YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem. Rev. 2017, 117 (8), 5389–5456. 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich H. J.; Hondal R. J. Why Nature chose selenium. ACS Chem. Biol. 2016, 11 (4), 821–841. 10.1021/acschembio.6b00031. [DOI] [PubMed] [Google Scholar]
- Kayrouz C. M.; Huang J.; Hauser N.; Seyedsayamdost M. R. Biosynthesis of selenium-containing small molecules in diverse microorganisms. Nature 2022, 610 (7930), 199–204. 10.1038/s41586-022-05174-2. [DOI] [PubMed] [Google Scholar]
- Cheng R.; Lai R.; Peng C.; Lopez J.; Li Z.; Naowarojna N.; Li K.; Wong C.; Lee N.; Whelan S. A.; Qiao L.; Grinstaff M. W.; Wang J.; Cui Q.; Liu P. Implications for an imidazol-2-yl carbene intermediate in the rhodanase-catalyzed C-S bond formation reaction of anaerobic ergothioneine biosynthesis. ACS Catal. 2021, 11 (6), 3319–3334. 10.1021/acscatal.0c04886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Li B. How nature incorporates sulfur and selenium into bioactive natural products. Curr. Opin. Chem. Biol. 2023, 76, 102377 10.1016/j.cbpa.2023.102377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebeck F. P. In vitro reconstitution of Mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc. 2010, 132 (19), 6632–6633. 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- Song H.; Hu W.; Naowarojna N.; Her A. S.; Wang S.; Desai R.; Qin L.; Chen X.; Liu P. Mechanistic studies of a novel C-S lyase in ergothioneine biosynthesis: the involvement of a sulfenic acid intermediate. Sci. Rep. 2015, 5, 11870. 10.1038/srep11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Song H.; Sae Her A.; Bak D. W.; Naowarojna N.; Elliott S. J.; Qin L.; Chen X.; Liu P. Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus Neurospora crassa ergothioneine biosynthetic pathway. Org. Lett. 2014, 16 (20), 5382–5385. 10.1021/ol502596z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani S.; Naowarojna N.; Tang Y.; Kathuria K. R.; Wang S.; Dhembi A.; Lee N.; Yan W.; Lyu H.; Costello C. E.; Liu P.; Zhang Y. J. Snapshots of C-S cleavage in Egt2 reveals substrate specificity and reaction mechanism. Cell Chem. Biol. 2018, 25 (5), 519–529.e4. 10.1016/j.chembiol.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn R.; Misson L.; Meury M.; Seebeck F. P. Anaerobic origin of ergothioneine. Angew. Chem., Int. Ed. 2017, 56 (41), 12508–12511. 10.1002/anie.201705932. [DOI] [PubMed] [Google Scholar]
- Cheng R.; Wu L.; Lai R.; Peng C.; Naowarojna N.; Hu W.; Li X.; Whelan S. A.; Lee N.; Lopez J.; Zhao C.; Yong Y.; Xue J.; Jiang X.; Grinstaff M. W.; Deng Z.; Chen J.; Cui Q.; Zhou J.; Liu P. Single-step replacement of an unreactive C-H bond by a C-S bond using polysulfide as the direct sulfur source in anaerobic ergothioneine biosynthesis. ACS Catal. 2020, 10 (16), 8981–8994. 10.1021/acscatal.0c01809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliaeva M. A.; Seebeck F. P. Discovery and characterization of the metallopterin-dependent ergothioneine synthase from Caldithrix abyssi. JACS Au 2022, 2 (9), 2098–2107. 10.1021/jacsau.2c00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunshausen A.; Seebeck F. P. Identification and characterization of the first ovothiol biosynthetic enzyme. J. Am. Chem. Soc. 2011, 133 (6), 1757–1759. 10.1021/ja109378e. [DOI] [PubMed] [Google Scholar]
- Naowarojna N.; Huang P.; Cai Y.; Song H.; Wu L.; Cheng R.; Li Y.; Wang S.; Lyu H.; Zhang L.; Zhou J.; Liu P. In vitro reconstitution of the remaining steps in ovothiol A biosynthesis: C–S lyase and methyltransferase reactions. Org. Lett. 2018, 20 (17), 5427–5430. 10.1021/acs.orglett.8b02332. [DOI] [PubMed] [Google Scholar]
- Song H.; Leninger M.; Lee N.; Liu P. Regioselectivity of the oxidative C-S bond formation in ergothioneine and ovothiol biosyntheses. Org. Lett. 2013, 15 (18), 4854–4857. 10.1021/ol402275t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharenko K. V.; Vit A.; Blankenfeldt W.; Seebeck F. P. Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway. Angew. Chem., Int. Ed. 2015, 54 (9), 2821–2824. 10.1002/anie.201410045. [DOI] [PubMed] [Google Scholar]
- Naowarojna N.; Irani S.; Hu W.; Cheng R.; Zhang L.; Li X.; Chen J.; Zhang Y. J.; Liu P. Crystal Structure of the ergothioneine sulfoxide synthase from Candidatus chloracidobacterium thermophilum and structure-guided engineering to modulate its substrate selectivity. ACS Catal. 2019, 9 (8), 6955–6961. 10.1021/acscatal.9b02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfli A. R.; Goncharenko K. V.; Meury M.; Dubey B. N.; Schirmer T.; Seebeck F. P. An alternative active site architecture for O2 activation in the ergothioneine biosynthetic EgtB from Chloracidobacterium thermophilum. J. Am. Chem. Soc. 2019, 141 (13), 5275–5285. 10.1021/jacs.8b13023. [DOI] [PubMed] [Google Scholar]
- Chen L.; Naowarojna N.; Song H.; Wang S.; Wang J.; Deng Z.; Zhao C.; Liu P. Use of a tyrosine analogue to modulate the two activities of a nonheme iron enzyme OvoA in ovothiol biosynthesis, cysteine oxidation versus oxidative C-S bond formation. J. Am. Chem. Soc. 2018, 140 (13), 4604–4612. 10.1021/jacs.7b13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Naowarojna N.; Chen B.; Xu M.; Quill M.; Wang J.; Deng Z.; Zhao C.; Liu P. Mechanistic studies of a nonheme iron enzyme OvoA in ovothiol biosynthesis using a tyrosine analogue, 2-amino-3-(4-hydroxy-3-(methoxyl) phenyl) propanoic acid (MeOTyr). ACS Catal. 2019, 9, 253–258. 10.1021/acscatal.8b03903. [DOI] [Google Scholar]
- Cheng R.; Weitz A. C.; Paris J.; Tang Y.; Zhang J.; Song H.; Naowarojna N.; Li K.; Qiao L.; Lopez J.; Grinstaff M. W.; Zhang L.; Guo Y.; Elliott S.; Liu P. OvoA(Mtht) from Methyloversatilis thermotolerans ovothiol biosynthesis is a bifunction enzyme: thiol oxygenase and sulfoxide synthase activities. Chem. Sci. 2022, 13 (12), 3589–3598. 10.1039/D1SC05479A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13 (11), 2498–504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai K.; Nealson K. H.; Horikoshi K. Hydrogenimonas thermophila gen. nov., sp. nov., a novel thermophilic, hydrogen-oxidizing chemolithoautotroph within the epsilon-Proteobacteria, isolated from a black smoker in a Central Indian Ridge hydrothermal field. Int. J. Syst. Evol. Microbiol. 2004, 54 (Pt 1), 25–32. 10.1099/ijs.0.02787-0. [DOI] [PubMed] [Google Scholar]
- Wen J.; Lord H.; Knutson N.; Wikstrom M. Nano differential scanning fluorimetry for comparability studies of therapeutic proteins. Anal. Biochem. 2020, 593, 113581 10.1016/j.ab.2020.113581. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Yoo H. J.; Park E. J.; Na D. H. Nano differential scanning fluorimetry-based thermal stability screening and optimal buffer selection for immunoglobulin G. Pharmaceuticals (Basel) 2022, 15 (1), 29. 10.3390/ph15010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Her A. S.; Raso F.; Zhen Z.; Huo Y.; Liu P. Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Org. Lett. 2014, 16 (8), 2122–2125. 10.1021/ol5005438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372 (3), 774–97. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Gao S. S.; Naowarojna N.; Cheng R.; Liu X.; Liu P. Recent examples of alpha-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosyntheses. Nat. Prod. Rep. 2018, 35 (8), 792–837. 10.1039/C7NP00067G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharenko K. V.; Seebeck F. P. Conversion of a non-heme iron-dependent sulfoxide synthase into a thiol dioxygenase by a single point mutation. Chem. Commun. 2016, 52 (9), 1945–1948. 10.1039/C5CC07772A. [DOI] [PubMed] [Google Scholar]
- Paris J. C.; Hu S.; Wen A.; Weitz A. C.; Cheng R.; Gee L. B.; Tang Y.; Kim H.; Vegas A.; Chang W. C.; Elliott S. J.; Liu P.; Guo Y. An S = 1 Iron(IV) intermediate revealed in a non-heme iron enzyme-catalyzed oxidative C-S bond formation. Angew. Chem., Int. Ed. 2023, 62, e202309362 10.1002/anie.202309362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemert N.; Alanjary M.; Weber T. The evolution of genome mining in microbes - a review. Nat. Prod. Rep. 2016, 33 (8), 988–1005. 10.1039/C6NP00025H. [DOI] [PubMed] [Google Scholar]
- Pan G.; Xu Z.; Guo Z.; Hindra; Ma M.; Yang D.; Zhou H.; Gansemans Y.; Zhu X.; Huang Y.; Zhao L. X.; Jiang Y.; Cheng J.; Van Nieuwerburgh F.; Suh J. W.; Duan Y.; Shen B. Discovery of the leinamycin family of natural products by mining actinobacterial genomes. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (52), E11131–E11140. 10.1073/pnas.1716245115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki E.; Zhang X.; Sun H. G.; Lu M. Y.; Liu T. L.; Ou A.; Li J. Y.; Chen Y. H.; Ealick S. E.; Liu H. W. Co-opting sulphur-carrier proteins from primary metabolic pathways for 2-thiosugar biosynthesis. Nature 2014, 510 (7505), 427–431. 10.1038/nature13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Xu X.; You C.; Yang C.; Guo J.; Sang M.; Geng C.; Cheng F.; Du L.; Shen Y.; Wang S.; Lan H.; Yang F.; Li Y.; Tang Y. J.; Zhang Y.; Bian X.; Li S.; Zhang W. Biosynthesis of chuangxinmycin featuring a deubiquitinase-like sulfurtransferase. Angew. Chem., Int. Ed. 2021, 60 (46), 24418–24423. 10.1002/anie.202107745. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy K.; Begley T. P. Protein thiocarboxylate-dependent methionine biosynthesis in Wolinella succinogenes. J. Am. Chem. Soc. 2011, 133 (2), 379–386. 10.1021/ja107424t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya R.; Dahiya S.; Fuloria N. K.; Kumar S.; Mourya R.; Chennupati S. V.; Jankie S.; Gautam H.; Singh S.; Karan S. K.; Maharaj S.; Fuloria S.; Shrivastava J.; Agarwal A.; Singh S.; Kishor A.; Jadon G.; Sharma A. Natural bioactive thiazole-based peptides from marine resources: structural and pharmacological aspects. Mar. Drugs 2020, 18 (6), 329. 10.3390/md18060329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N.; Kawano Y.; Satoh Y.; Dairi T.; Ohtsu I. Gram-scale fermentative production of ergothioneine driven by overproduction of cysteine in Escherichia coli. Sci. Rep. 2019, 9 (1), 1895. 10.1038/s41598-018-38382-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.