

Abstract
Prostate cancer (PCa) is an increasingly prevalent health problem in the developed world. Effective treatment options exist for localized PCa, but metastatic PCa has fewer treatment options and shorter patient survival. PCa and bone health are strongly entwined, as PCa commonly metastasizes to the skeleton. Since androgen receptor signaling drives PCa growth, androgen-deprivation therapy whose sequelae reduce bone strength constitutes the foundation of advanced PCa treatment. The homeostatic process of bone remodeling – produced by concerted actions of bone-building osteoblasts, bone-resorbing osteoclasts, and regulatory osteocytes — may also be subverted by PCa to promote metastatic growth. Mechanisms driving skeletal development and homeostasis, such as regional hypoxia or matrix-embedded growth factors, may be subjugated by bone metastatic PCa. In this way, the biology that sustains bone is integrated into adaptive mechanisms for the growth and survival of PCa in bone. Skeletally metastatic PCa is difficult to investigate due to the entwined nature of bone biology and cancer biology. Herein, we survey PCa from origin, presentation, and clinical treatment to bone composition and structure and molecular mediators of PCa metastasis to bone. Our intent is to quickly yet effectively reduce barriers to team science across multiple disciplines that focuses on PCa and metastatic bone disease. We also introduce concepts of tissue engineering as a novel perspective to model, capture, and study complex cancer-microenvironment interactions.
Keywords: prostatecancer, androgenreceptor, bone, hypoxia, tissueengineering, metastasis
Introduction
Prostate cancer (PCa) is a highly prevalent health problem in the developed world. As an age-related disease that is exacerbated by Western diets and lifestyles, the global burden of PCa will only continue to rise as lifespans increase and diets converge. Localized PCa can be treated through combinations of surgery, radiotherapy, and systemic therapies to yield a 97% 5-year survival rate (Siegel et al. 2023). In contrast, advanced PCa that escapes its local environment and travels to distant sites results in markedly increased morbidity and mortality (Siegel et al. 2023, Ottewell et al. 2014, Sweeney et al. 2015, Hu et al. 2017a). While PCa can metastasize to lung, liver, and lymph nodes, metastasis to bone dominates clinically. Of nearly 3600 patients presenting with metastatic PCa from 1991 to 2009, the vast majority (>90%) demonstrated metastatic bone disease and markedly lower incidence of metastasis to lymph nodes (8.7%) or liver (4.5%) (Gandaglia et al. 2015). Site-specific metastasis associates with survival: men with liver metastasis have shorter median overall survival (13.5 months) compared to men with bone-only metastases (21.3 months) (Halabi et al. 2016). Thus, the fundamental nature, composition, and anatomy of the skeletal microenvironment present as an area of mechanistic and clinical interest for PCa metastasis, treatment, and prevention.
Advances in therapeutic approaches for preventing or treating skeletally metastatic PCa may be achieved through engaging the diverse perspectives of a team of researchers from multiple disciplines. Insight from new perspectives may elucidate previously unconsidered mechanisms or offer different techniques to interrogate scientific questions. However, cross-disciplinary collaborations present challenges in communicating nuances and pathways key to cancer or bone biology. Consequently, this review synthesizes the knowledge and perspectives on PCa of several disciplines, beginning with bone structure and function, clinical presentation, and molecular mechanisms of cancer biology. We then introduce a final perspective – that of biomedical engineering – to highlight opportunities to better model, capture, and study complex metastatic cancer–bone interactions in a 3D context that captures native in vivo geometry. Through this review, we aim to harmonize the language and perspectives of different disciplines of biology to promote engagement in collaborative research, effective communication across disciplines, and minimize barriers to team science.
The bone microenvironment
The skeleton is the most common metastatic site for PCa. The structure and composition of bone, and the cells responsible for building, remodeling, and maintaining bone, all provide putative mechanisms wherein the bone is a permissive soil for disseminated tumor cells. Thus, by introducing the bone microenvironment, we may uncover mechanisms of PCa metastatic organotropism.
Bone function in homeostasis
Bone is a multifunctional organ (Moreira et al. 2000, Robling et al. 2006, Clarke 2008). The structural and protective roles of the skeleton are readily apparent, providing attachment sites for muscles and ligaments and shielding organs from damage. The skeleton also houses bone marrow (BM), providing the site for blood cell production and maturation, and resorbs and rebuilds itself to maintain mineral homeostasis.
Bones are composites of proteins, minerals, and water. Bone is grossly arranged into cortical or trabecular microarchitecture depending on structure and location. Cortical (or compact) bone forms the dense, protective outer shell, whereas trabecular (or cancellous or spongy) bone is a network of struts and rods inside the ends of long bones and throughout the interior of flat and irregular bones (Fig. 1A). Cortical and trabecular bone work in concert to provide biomechanical strength and flexibility to withstand applied loads without breaking. Bone strength originates from its arrangement and composite architecture. Type I collagen fibrils within bone are intercalated with the mineral hydroxyapatite (Ca10 (PO4)6 (OH)2) (often abbreviated HA) which provides resistance to compression and confers biomechanical strength to the skeleton. The careful arrangement of collagen fibrils and HA into lamellar sheets further increases strength. Acidification of bone by bone-resorbing osteoclasts (OCLs) promotes HA hydrolysis into calcium and phosphate, which maintains serum calcium and phosphate levels within a narrow physiologic range.
Figure 1.
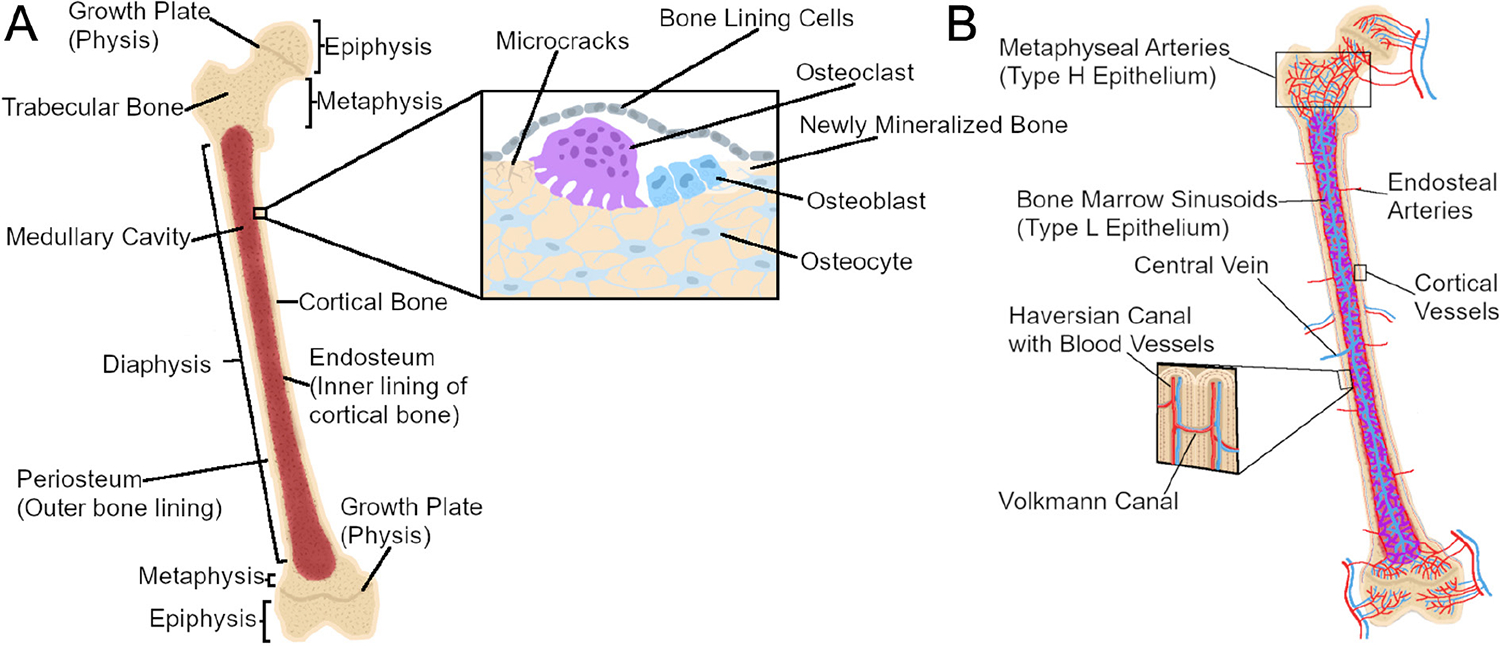
Long bone anatomy and cellular composition. (A) Long bones are hollow, marrow-filled shafts with rounded, sealed ends. The diaphysis is the middle, tubular shaft of bone in of the bone which encases the medullary cavity and the bone marrow. Proximal and distal to the diaphysis are the conical epiphyses, whose rounded ends provide attachment sites for connective tissue. In growing individuals, the epiphysis and diaphysis are separated by the metaphysis, whose growth plates (physes) allow for longitudinal growth before ultimately fusing with the epiphysis in adulthood. Bone tissue is composed of spongy trabecular bone found inside bone, primarily within the epiphysis, while compact cortical bone forms the thick outer shell of the bone. The cellular composition of bone involves bone-resorbing osteoclasts, bone-forming osteoblasts, and matrix-entrapped osteocytes that coordinate the activity of osteoblasts and osteoclasts. Bone-lining cells cover bone surfaces in the absence of formation or resorption; they can be mobilized to active osteoblasts during remodeling. (B) Bones are intricately vascularized to meet high metabolic demands. Cortical bone is supplied through large arteries which enter the bone in the diaphysis. Cortical bone vasculature is then organized into Haversian and Volkmann canals which supply blood longitudinally and sagittally, respectively. In contrast, the trabecular vasculature enters near the metaphysis to provide oxygen-rich blood (red) to the growth plate before draining into the medullary cavity to provide less oxygen-rich blood (purple) to the bone marrow. Oxygen-poor blood in the medullary cavity (blue) then drains through a central vein which exits the bone through the diaphysis.
The central cavity of long bones is termed the medullary cavity (Fig. 1A). BM and stromal cells lie within the medullary cavity to support and protect hematopoietic stem cells (HSCs) that give rise to red blood cells and leukocytes. Some blood cells such as B cells remain and mature in the BM, egressing once fully differentiated. Other cells such as neutrophils and monocytes can be mobilized from the BM quickly in response to physiological challenges such as infection (Swirski et al. 2009).
Remodeling and the role of skeletal effector and regulatory cells
Bone strength and resistance to fracture paradoxically require that microscopic packets of bone tissue are routinely degraded and replaced. This process, termed remodeling, removes specific regions of the mineralized matrix that has accumulated damage; thus, remodeling improves bone strength and reduces fracture risk. OCLs resorb areas of matrix damage, which is followed by the deposition of new matrix and minerals by osteoblasts (OBs). The activity of OBs and OCLs is regulated by osteocytes (OCYs), which perceive mechanical stimuli and drive OB and OCL activity via secreted factors or direct cell–cell engagement to generate suitable bone for the mechanical environment (Fig. 1A) (Regard et al. 2012, Hart et al. 2020).
Remodeling is a cyclical process employed to maintain bone strength throughout the lifespan. Bone remodeling is induced by myriad factors, including traumatic fracture, hormone signaling, or tissue-level strain. In fracture repair, disorganized woven bone is deposited quickly and irregularly to provide immediate strength; over time, woven bone is remodeled into organized, biomechanically superior lamellar bone. Hormones such as parathyroid hormone (PTH) or sex steroids influence remodeling by activating or inhibiting OBs and OCLs, causing net gain or loss of bone based on the dose and duration of the hormone signal (Dobnig & Turner 1995, Dempster et al. 2001, Jilka 2007, Jilka et al. 2009). Bone experiences tissue-level strain continuously from daily activity: exercise, walking, and even standing all exert force on bone tissue. Tissue strains are converted into biophysical stimuli like fluid shear stress or matrix deformation at the cellular level, which then drive adaptive or reparative processes. Repetitive activities damage the mineralized matrix which accumulates in the form of microcracks (Martin 2003); these must be remodeled before coalescing into tissue-level damage. Thus, remodeling allows for continuous, piecewise repair of bone by coupling osteoclastic resorption of the damaged bone to osteoblastic deposition of new bone. The cyclical nature of remodeling allows for tissue repair without the need for the individual to discontinue the daily activity. Instead, the newly synthesized, undamaged bone matrix provides biomechanical strength to the tissue while damaged and biomechanically inferior tissue is resorbed and replaced. Thus, remodeling is critical to bone health and is driven by the proper coupling of OB and OCL activity, which is regulated through OCYs.
Osteoclastic bone resorption removes bone and liberates mineral from the matrix
OCLs resorb segments of bone. OCLs develop from the differentiation and fusion of macrophages into a single multinucleated cell which dissolves the mineralized HA matrix by generating a low pH environment directly onto bone surfaces; the organic matrix is dissolved through OCL-secreted enzymes and proteases like tartrate-resistant acid phosphatase, collagenases, and matrix metalloproteinases (MMPs). Liberated minerals are transcytosed from the apical membrane and secreted at the basolateral membrane to maintain serum calcium and phosphate levels. In contrast, degraded organic components remain in the vicinity of bone resorption and are released to the local environment upon cessation of resorption (Bruzzaniti & Baron 2006). OCL formation and activity are tightly regulated by local cytokine presentation, often from bone-forming cells. OBs, OCYs, and BM cells produce the cytokine Receptor Activator of NF-κB Ligand (RANKL; Tnfsf11) which binds to the cognate receptor RANK on macrophages and thereby drive OCL formation or activity. Similarly, OBs and OCYs produce a decoy receptor, osteoprotegerin (OPG; Tnfrsf11b), which sequesters RANKL to limit RANKL-RANK interactions. Thus, the relative availability of RANKL to OPG directs OCL formation and localization to specific bone segments.
Osteoblasts build bone by depositing and mineralizing the extracellular matrix
The counterpart to the OCL is the OB, the cell responsible for building bone (Robling 2012). Derived from mesenchymal stem cells, cuboidal OBs synthesize and secrete proteins that make extracellular matrix, such as collagen type 1; further, OBs secrete HA which intercalates in type 1 collagen fibrils to mineralize and thus strengthen bone matrix. OB differentiation and activity – and thus bone deposition – is regulated by myriad growth factors and cytokines, including bone morphogenetic proteins (BMPs) and Wnts. Canonical Wnt/β-catenin signaling drives bone deposition through Wnt ligands binding to membrane-bound Frizzled receptors and Lrp co-receptors on OBs. Restraint of Wnt signaling occurs through the secretion of direct antagonists of Wnt receptors, like Sclerostin and Dkk1, or secreted decoy receptors like secreted frizzled-related proteins. The robust contribution of Wnt signaling in skeletal anabolism has led to the development of monoclonal antibodies such as romosozumab (Evenity™; anti-sclerostin antibody) which neutralize Wnt signaling inhibitors to stimulate bone deposition (Sharifi et al. 2015, Reid 2022).
Osteocytes maintain skeletal and mineral homeostasis through mechanosensation and diverse signaling
As OBs generate new bone matrix, a subset of OBs become entrapped in the matrix and differentiate into a new cell, the OCY (Donahue et al. 2020, Buettmann et al. 2022, Delgado-Calle & Bellido 2022). Within the calcified matrix, OCYs have a distinct stellate morphology: dendritic processes extend from the cell body and thread throughout the mineralized matrix in the lacuno-canalicular system. These processes allow for extensive crosstalk with neighboring OCYs or other cells in bone through gap junctions or Notch signaling or indirect means such as secretion of cytokines or exosomal vesicles. OCYs have a limited capacity to directly resorb or deposit bone and instead direct the formation and activity of OBs and OCLs in response to hormonal signals or mechanical loads. Historically, systemic calcium and phosphate demands were considered primary drivers of bone mass; postnatal changes in bone mass or diameter or length were attributed to variations in hormonal milieu or serum ion concentration, rather than as adaptive responses to biophysical forces (Frost 2000). Yet, observations such as trabecular alignment in the femoral neck and estimated principal stress suggested a third mechanism – applied forces – in controlling the bone size and strength (Martin et al. 1998). As the most abundant cell type in bone and due to their network of direct and indirect engagement with other cell types, OCYs are recognized as orchestrators of bone remodeling. For example, genetically engineered mice that lack OCYs fail to alter bone mass under disuse conditions, demonstrating their obligate role as mechanosensory cells (Tatsumi et al. 2007). Distinct and convergent signaling pathways are activated in OCYs in response to mechanical loads, signals which are transduced to OBs and OCLs or their progenitors. Mechanical loading occurs with locomotion. OCYs promote OB activity by reducing OCY secretion of Wnt antagonists like Sclerostin and Dkk1 while promoting OCL formation by increasing RANKL production. Thus, OBs, OCLs, and OCYs collectively and concertedly work to develop and maintain bone tissue through tightly coordinated activity.
OCY contribution to tissue and organ homeostasis exceeds skeletal impact. OCYs interact with myriad tissues and organs including the kidney, BM, blood vessels, nerves, and muscles via both direct and paracrine signaling. The first description of the OCY as a paracrine cell stemmed from the observation that OCY-derived Fgf23 regulates phosphate homeostasis in the kidney (Feng et al. 2006). This observation shifted the view on OCYs as local orchestrators of remodeling to the bone as an endocrine organ. Although osteocalcin was once proposed to be another bone-derived hormone that established a role for OCYs in regulating energy metabolism, male fertility and cognition, such findings are under scrutiny (Lee et al. 2007, Oury et al. 2011, Diegel et al. 2020, Dasgupta et al. 2021).
Bone-lining cells in the bone marrow microenvironment and remodeling
Though OBs, OCLs, and OCYs are prioritized in the literature for their roles in remodeling, other cells in bone participate in this process. The periosteal and endosteal surfaces of cortical bone are also covered by fibroblasts, osteoprogenitors, and bone-lining cells (BLCs). BLCs represent a different fate for OBs, a fate in which cell size and metabolism decrease and shapes convert from cuboidal to squamous. BLCs reside on the periosteal and endosteal bone surfaces that undergo neither modeling nor remodeling and, in doing so, maintain ionic differences between the bone fluid compartment and interstitial fluid (Talmage 1970). Recent advances reveal that endosteal fibroblasts and BLCs maintain the BM microenvironment and actively participate in remodeling, respectively. Fibroblasts aid OBs in maintaining the HSC niche through CXCL12, which directs HSC homing to the BM (Sugiyama et al. 2006). When bone is remodeled, BLCs form a canopy which separates the bone remodeling unit from BM and non-remodeling (Hauge et al. 2001). Further, BLCs can de-differentiate in response to mechanical loading (Pead et al. 1988), PTH (Dobnig & Turner 1995), or romosozumab (Kim et al. 2017) to directly participate in bone formation.
Bone marrow
BM fills the medullary cavity. BM contains heterogeneous populations of cells involved in myelopoiesis, hematopoiesis, mesenchymal progenitors, and supportive reticular cells (Dolgalev & Tikhonova 2021). BM adipose tissue (BMAT) exerts endocrine and energy storage functions and, in doing so, participates in central and local metabolism, osteogenesis, and hematopoiesis (Zhang et al. 2021a). BMAT burden reflects chronologic age, diet, and treatment decisions (Hotte & Saad 2010, Martin et al. 2017). BMAT volume inversely relates to bone mass and skeletal health (Devlin et al. 2010, Cohen et al. 2013, Li et al. 2022), signaled through the release of adipokines and pro-inflammatory cytokines which contribute to senescence and low-grade systemic inflammation (Aaron et al. 2022).
Bone vascularization
Bone is highly metabolic and requires efficient oxygen transport, sources of cellular energy, and removal of metabolic waste. Correspondingly, it is intricately and thoroughly vascularized. The necessity of skeletal vascularity often indicates the functional coupling of angiogenesis with osteogenesis. For example, vascular endothelial cells (ECs) promote bone growth through Notch-mediated angiocrine signaling (Ramasamy et al. 2014) and secretion of the osteoinductive factor bone morphogenetic protein-2 (BMP-2) (Willette et al. 1999). Ultimately, the rich vascular network on and within bone that is necessary for its survival and function may also enrich metastasis, perhaps through changes in fluid flow and local oxygen tensions. Here, we focus on the vascular network supplying the medullary cavity and BM sinusoids, as bone metastases are most often found within the medulla as opposed to the periosteal surface (Roudier et al. 2008).
Nutrient arteries supplying the cortical bone enter the cortex near the diaphysis, then branch proximally and distally along the long axis of the bone through holes known as Haversian canals (Fig. 1B). Haversian blood vessels are bridged via perpendicular canals known as Volkmann canals (Bilezikian et al. 2019). Trabecular bone employs a different vascular organization, with nutrient arteries passing through the cortical bone near the ends of the bone supplying the growth plate with highly oxygenated blood, next supplying the medullary cavity with less oxygenated blood, and finally exiting the bone through a large central vein in the diaphysis (Ramasamy et al. 2016) (Fig. 1B).
Trabecular bone vascularity is well described in the long bones, where arteries pass through the cortex near the metaphysis (Kusumbe et al. 2014, Ramasamy et al. 2016). Arteries feed oxygen-rich blood to capillaries near the metaphysis of the bone where longitudinal growth takes place in youth. Metaphyseal capillaries are columnar in shape and experience a high shear force (Ramasamy et al. 2016). They also contain specialized ECs, termed ‘type H’ due to high expression of Pecam1 and endomucin (Kusumbe et al. 2014). Blood flows directly from the type H vessels in the metaphysis or from arteries along the endosteum to the capillaries in the medullary cavity (Fig. 1B). The capillaries in the medullary cavity are sinusoidal and highly branched and thus experience a rapid reduction in blood velocity (Ramasamy et al. 2016), the endothelium of which are termed ‘type L’ ECs due to low expression of Pecam1 and endomucin (Kusumbe et al. 2014). Blood subsequently drains from type L endothelium to a central vein in the diaphysis (Ramasamy et al. 2016). Capillary specialization impacts blood flow through and oxygenation in the skeleton: cells near type H ECs in the metaphysis experience the highest oxygen tension while cells near the type L ECs in the marrow are hypoxic as the blood reaching the marrow is already oxygen poor (Kusumbe et al. 2014, Ramasamy et al. 2014, 2016, Spencer et al. 2014). Specialization of the capillary endothelium and relative oxygen tension is also important for stem and progenitor cell maintenance (Dobnig & Turner 1995, DaSilva et al. 2009). Osteoprogenitors preferentially associate with type H endothelium, which expresses higher levels of growth factors associated with osteoprogenitor survival and proliferation (Kusumbe et al. 2014, Langen et al. 2017). Type L endothelium and the vessel-associated mesenchymal cells experience low oxygen tensions and secrete signals that maintain HSC populations (Sugiyama et al. 2006, Ding et al. 2012). Thus, regional differences in oxygen tensions and endothelium type play an important role in the progenitor cell niche.
Oxygen tension is spatially heterogeneous in the skeleton: the highest tension is observed periosteally and decreases in cortical bone (Spencer et al. 2014). Intracortical oxygen tension heterogeneity is revealed by the expression of hypoxia-sensitive transcripts in OCYs, wherein OCYs deep within cortical bone express markers of glycolysis and the oxygen-regulated protein ORP150, which do not occur in OCYs closer to the bone surface (Frikha-Benayed et al. 2016). Similarly, oxygen tension is lower within BM than in the endosteum. The BM has no direct arterial blood supply. Instead, blood supplying oxygen to the BM first donates oxygen to the metaphysis or the endosteum – leaving less oxygen in the blood to supply the BM with – ultimately contributing to hypoxia within the marrow (Ramasamy et al. 2016). Hypoxia within the marrow may also be attributed to the higher metabolic demand of marrow stromal or HSCs (Spencer et al. 2014).
Long bone vascular structure changes as a function of age. The proportion of type L ECs increases throughout the lifespan while type H ECs decline with age (Kusumbe et al. 2014, Langen et al. 2017). This age-related shift has important implications for metastasis, as it has been proposed that bone metastasis is likely mediated through sluggish blood flow through type L ECs (Peng et al. 2020). However, it is yet unclear if the same blood vessel organization occurs in the flat bones, which is where bone metastasis occurs more frequently, for example, the pelvis and ribs. To date, the reported microvascular networks in the pelvis are like long bones; however, type H and type L endothelium have yet to be identified in these bones (Pannarale et al. 1997). As such, further interrogation of blood vessel organization in these bones is warranted.
Sex steroids and bone
Bone remodeling is driven, in part, by sex hormone signaling; this is often appreciated in post-menopausal osteoporosis, wherein bone mineral density rapidly declines following cessation of estrogen production. Estrogen influences both and OBs, restraining osteoclastic bone resorption via suppression of RANKL while promoting OB survival and therein maintaining the ideal coupling of the remodeling cycle of resorption followed by formation (Hughes et al. 1996, Khosla et al. 2012). Without estrogen, remodeling is uncoupled to increase bone resorption relative to formation and cause net bone loss with each remodeling cycle (Khosla et al. 2012). Estrogen signaling impacts both females and males. In females, selective estrogen receptor α (ERα) deletion from OCLs (Ctsk-cre;ERaf/f) increases OCL number and trabecular bone resorption (Nakamura et al. 2007, Martin-Millan et al. 2010), while Osx-, Col1-, Bglap-, or Dmp1-cre-driven deletion of ERα in OBs or OCYs reduces bone mass and OB number (Almeida et al. 2013, Määttä et al. 2013a, Windahl et al. 2013, Kondoh et al. 2014, Melville et al. 2014). In males, ERα deletion exerts transient or sustained impacts on bone mass when deleted in pre-OBs (Osx-cre) (Ucer et al. 2015) vs mature OBs and OCYs (Col1- or Dmp1-cre) (Windahl et al. 2013, Ucer et al. 2015). The influence of sex steroids on the skeleton is not restricted to estrogen, as progesterone and androgens also drive skeletal development and maintain skeletal homeostasis.
Androgen receptors (ARs) are expressed in epiphyseal chondrocytes and in growth plate cartilage cells – therefore affecting longitudinal bone growth in early puberty. Androgens were found to regulate proliferation as well as differentiation of epiphyseal chondrocytes and growth hormone secretion during puberty (Clarke & Khosla 2009). The AR consists of an N-terminal domain, followed by a DNA-binding domain, a hinge region, and a C-terminal ligand-binding domain where the ligands bind (Wang et al. 2007). In the inactive state, the AR is bound to a complex regulated by the heat shock protein HSP90 that maintains it in the cytoplasm, but ligand binding releases this binding – allowing the AR to translocate to the nucleus, where it binds to the promoters of target genes at sequences called the androgen response element (ARE). Both testosterone and dihydrotestosterone (DHT) stimulate OB precursors and may also promote OB differentiation. By upregulating transforming growth factor-beta (TGF-β) and insulin-like growth factor (IGF) 1/2 – which stimulate bone formation – and downregulating interleukin (IL)-6 which stimulates osteoclastogenesis, they ensure that bone density is maintained at a high level. In OBs, androgens promote IL-1β and fibroblast growth factor (FGF), while suppressing PGE2 (Prostaglandin E2) and cAMP production by IL-1 and PTH (Wiren et al. 1997). DHT has also been shown to prevent osteoclastogenesis by reducing OPG levels. Many of these genes are direct targets of the AR and were found to contain an ARE in their proximal promoter. For example, IL-10, which inhibits bone resorption, IL-1, and IL-6 all are direct transcriptional targets of the AR. Taken together, the AR regulates bone density by increasing OB proliferation and preventing osteoclastogenesis.
Androgen control of bone has a greater impact in males than females. AR deletion from OBs or OCYs in male mice results in reduced bone mass, reminiscent of the low bone mass of men on androgen deprivation therapy (ADT) (Yeh et al. 2002, Notini et al. 2007, Lauretani et al. 2008, Chiang et al. 2009, Sinnesael et al. 2012, Määttä et al. 2013b); similarly, AR deletion from OBs and OCYs reduces bone mass in female mice, though less so than in males (Määttä et al. 2013b). These studies highlight that bone is highly responsive to sex hormone signaling whose contributions are sex-, age-, and compartment-dependent. Because sex hormones are a target of cancer therapies, treatment strategies that alter hormone levels or function must consider the skeletal sequelae of these choices.
Prostate cancer clinical course and treatment
PCa remains the most common cancer diagnosed and the second most common cause of cancer death in men in the United States (Siegel et al. 2023). Though many treatments exist for PCa, research continues to further improve treatment to improve patient outcomes. Introducing the disease course and common strategies to treat PCa will highlight current molecular mechanisms and may identify clinical needs which need to be met.
Screening, diagnosis, and cancer treatment
Current American Cancer Society guidelines recommend screening with serum prostate-specific antigen (PSA) in men ages 40–50 depending on their risk; however, frequency of screening is often controversial based on the recommending organization (Smith et al. 2019, Desai et al. 2022). Serum PSA is a common marker used in screening for PCa, although changes in PSA levels lack both specificity and sensitivity for early disease. PSA screens thus can lead to high rates of overdiagnosis and overtreatment; instead, screening is most effective when patient risk and symptoms are also considered. The most common presenting symptoms for patients with PCa are lower urinary tract symptoms including urinary frequency and hesitancy and poor urinary stream. Unfortunately, such symptoms are often common with those for benign prostatic hyperplasia and prostatitis, therein highlighting the need for more specific and less sensitive diagnostic biomarkers to correctly identify patients with early PCa before symptom onset (McNally et al. 2020). Localized PCa is diagnosed by digital rectal examination and prostate biopsies which identify the Gleason grade (Sauter et al. 2016).
PCa treatment depends on the stage, risk stratification, and extent of progression (Fig. 2). In the US, depending on risk stratification, common treatments for localized PCa can involve radiation therapy, prostatectomy, or active surveillance for disease progression (Brawley et al. 2018). These treatments are curative in most patients with PCa. However, after the United States Preventative Services Task Force recommended against annual PSA screening in 2012, there has been a noted increase in the incidence of metastatic PCa (mPCa) in the National Cancer Institute’s Surveillence, Epidemiology and End Results data despite an overall decrease in PCa diagnosis. The increase could also result from improved diagnostic imaging techniques which identify higher rates of low-volume metastases and high-volume mPCa (Desai et al. 2022). Common sites of metastasis for PCa include bone, lymph node, brain, liver, and lung. Most patients with mPCa are initially castration-sensitive (metastatic castration-sensitive prostate cancer (mCSPC)) and respond to ADT. ADT treatment can be achieved through luteinizing hormone-releasing hormone agonists such as leuprolide acetate or goserelin, luteinizing hormone-releasing hormone antagonists like degarelix or relugolix, or surgical castration via orchiectomy (Sun et al. 2016). Currently, the addition of docetaxel, a chemotherapy agent, or abiraterone, a CYP17 inhibitor, to ADT is the first treatment of choice for patients with mCSPC (James et al. 2017, Kyriakopoulos et al. 2018). In practice, it seems abiraterone with ADT may have more benefits for patients with low-volume disease whereas docetaxel plus ADT confers survival benefits in high-volume PCa (Hahn et al. 2018). Additional agents more recently approved for mCSPC in conjunction with ADT include antiandrogens apalutamide and enzalutamide (Sayegh et al. 2022).
Figure 2.
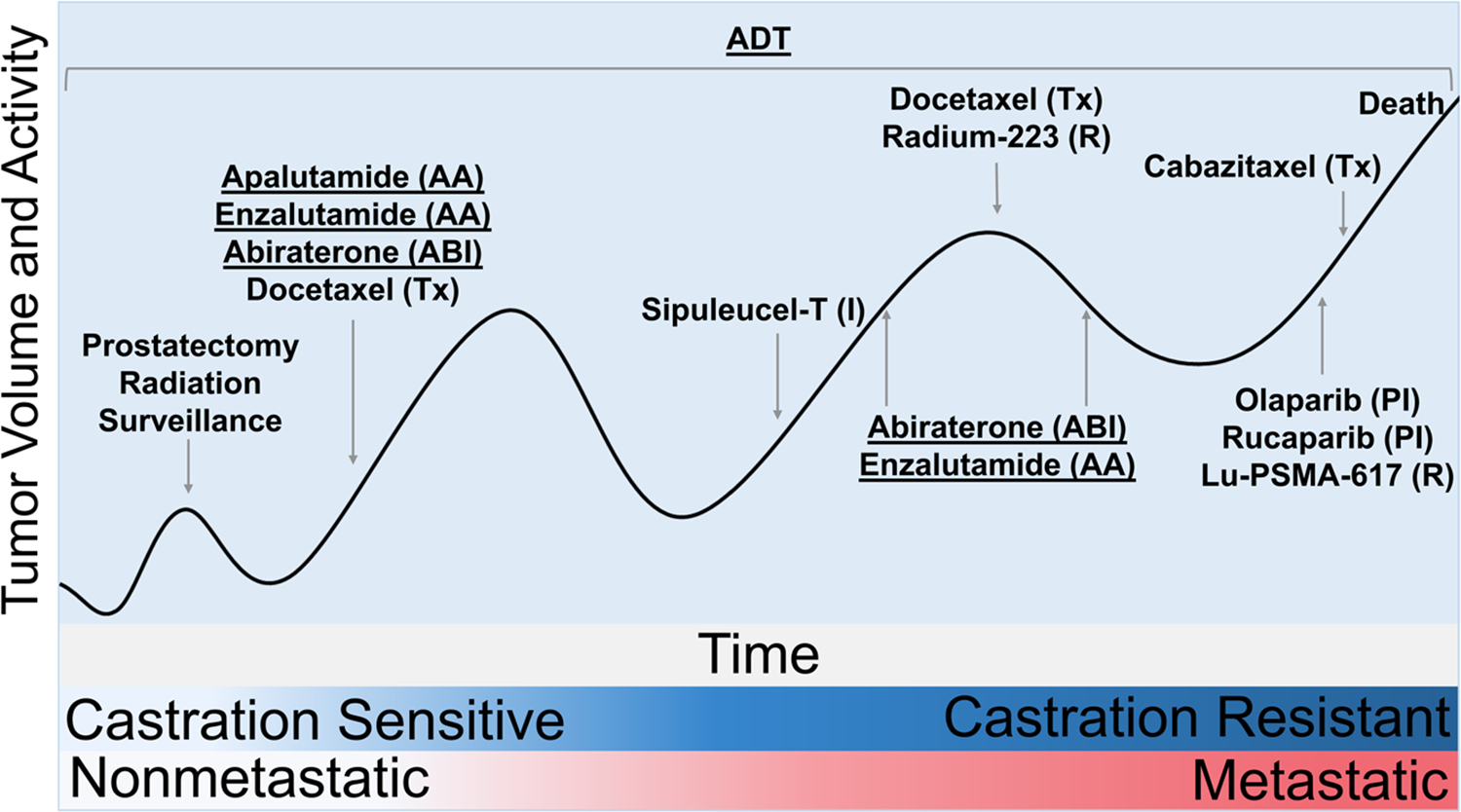
Treatment of prostate cancer over its disease course. The initial stage of prostate cancer involves prostate-confined disease, referred to as local or nonmetastatic disease; local disease is treated with surgery or radiation and is surveilled for recurrence. Recurrent or metastatic disease if caught early is normally castration-sensitive and is thus treated with surgical or chemical androgen deprivation therapy (ADT). ADT is the foundation of prostate cancer treatment throughout the disease course. Prostate cancer eventually becomes castration-resistant when the disease no longer responds to ADT (denoted by blue gradient). Castration-resistant prostate cancer is often subsequently treated with androgen biosynthesis inhibitors (ABI), antiandrogens (AA), or taxanes (Tx). New therapies for metastatic castration-resistant prostate cancer include immunotherapy (I), radiopharmaceuticals (R), and PARP inhibitors (PI). Prostate cancer can metastasize during either the castration-resistant or castration-sensitive phase (denoted by the pink gradient). Metastasis becomes more likely as the disease progresses and alters the treatment of the cancer. Therapies impacting bone mass (denoted by underlining) are given throughout the disease.
Patients with mCSPC eventually develop castration resistance (metastatic castration-resistant prostate cancer (mCRPC)). Once mCSPC progresses to mCRPC, the disease becomes much more aggressive with poorer prognosis, yet a variety of adjunctive therapies can be used (Sayegh et al. 2022). In patients with no previous chemotherapy exposure, docetaxel is the preferred agent though cabazitaxel is another chemotherapeutic agent approved for mCRPC. In practice, it is often used after progression on abiraterone acetate or enzalutamide; both agents also provide a survival benefit in chemonaïve or docetaxel-pretreated mCRPC patients (Sayegh et al. 2022). Radium-223 – a radioactive radium isotope which acts as a calcium mimetic incorporated during osteogenesis – is a therapy with proven benefit, for patients with primarily osseous metastatic disease and symptoms from bony metastases. Immunotherapy with sipuleucel-T, a dendritic cell vaccine created from patients’ own cells has demonstrated survival benefits in patients with asymptomatic mCRPC. Patients with select mutations benefit from treatment with PARP inhibitors olaparib or rucaparib which interfere with DNA repair. A radiopharmaceutical ligand, Lu-PSMA-617, was also recently approved for patients with mCRPC who have progressed through antiandrogen, androgen biosynthesis inhibition, and taxane therapies (Sayegh et al. 2022). Many factors, including disease burden, presence of bone or visceral metastases and symptoms, influence sequencing decisions and treatment selection, especially when patients with mCRPC experience disease progression.
Treatment-associated bone loss
Crucially, patients who develop skeletal complications (termed skeletal-related events (SREs)) like hypercalcemia, bone pain, or pathologic fractures have markedly worse prognoses and survival outcomes than those without SREs (Coleman 1997): 5-year survival rates are 56% in PCa patients without bone metastasis, but plummet to <1% with metastasis and a SRE such as fracture (Nørgaard et al. 2010). Thus, there is a critical link between bone health and patient survival, which necessitates investigation of the crosstalk between bone and metastatic PCa. Indeed, PCa and its treatment associates with or causes skeletal comorbidities. For example, bone mass is reduced in PCa patients, particularly in patients with bone metastasis: 25 and 9% of patients are osteopenic or osteoporotic, respectively (Kwon et al. 2014). Many treatments for PCa such as ADT and enzalutamide can also cause osteoporosis and osteopenia, illustrating the entwined nature of bone health in PCa patients. ADT significantly associates with reduced bone mineral density in the first three years of treatment (Kim et al. 2019). Indeed, ADT – either surgical or chemical – causes rapid onset of osteoporosis and bone loss at trabecular and cortical sites in the skeleton (Stoch et al. 2001, Hörnberg et al. 2011). In men undergoing ADT, the mean trabecular bone mineral density of the lumbar spine decreased by 8.5 ± 1.8% (Smith et al. 2001). Further, PCa patients on ADT experienced significantly more fractures compared to patients not on ADT (Shahinian et al. 2005). The AR antagonist enzalutamide associates with higher fracture rates than the placebo in the ARCHES trial (Armstrong et al. 2019). Pre-clinical studies in mice demonstrate that enzalutamide reduces bone mass in the axial but not appendicular skeleton (Wu et al. 2016). Similar to enzalutamide, in patients with non-metastatic CRPC, treatment with apalutamide significantly increased fracture risk compared to placebo (11.7 vs 6.5%) (Smith et al. 2018). In vitro, abiraterone acetate inhibits OCL differentiation and activity while promoting OB differentiation, activity, and bone deposition (Iuliani et al. 2015). An important consideration for abiraterone acetate is that it is commonly administered with prednisone which reduces bone strength via upregulation of OCL function, reduction in OB function and dysregulated Wnt signaling from OCYs (Yao et al. 2008, 2013). Thus, the relationship between abiraterone acetate plus prednisone on bone health is likely complex.
The use of advanced AR antagonists, including abiraterone and the antiandrogens, gives rise to alternately spliced AR variants that lack the ligand-binding domain while maintaining the N-terminal domain and the DNA-binding domain, such as AR-V7 and AR-567 (Messner et al. 2020a). AR variants transcribe genes not regulated by the full-length receptor while many genes that were previously regulated are no longer maintained. For example, the GDF15 promoter contains an ARE that is activated by AR splice variants; once expressed, GDF15 promotes RANKL activation resulting in bone metastasis (Siddiqui et al. 2022a). AR-regulated transcription involves multiple co-regulators that bind to the AR–ARE complex. Many of these co-regulators bind at the ligand-binding domain, and therefore, it is assumed that loss of these co-regulators alters the transcriptional pattern of the AR.
Bone-sparing therapeutics in prostate cancer
Current therapy options for improving bone health in patients undergoing PCa treatment focus on preventing bone resorption, but their administration is low in real-world studies. Zoledronic acid, a bisphosphonate, and denosumab, a RANKL inhibitor, are the primary agents that delay the onset of SREs and bone metastasis-related pain (Roviello et al. 2022). Co-treatment with bone-targeted agents in mCRPC patients significantly reduces fracture risk. Radiation therapy is also an effective palliative option for bone metastasis-related pain (Roviello et al. 2022). Though osteoanabolic therapies such as romosozumab (an anti-sclerostin antibody) and Forteo (recombinant human PTH (1–84)) are FDA-approved for post-menopausal osteoporosis, neither have been considered in PCa patients. However, inhibition of the Wnt antagonist DKK1 promotes tumor progression and metastasis in PCa (Thudi et al. 2011, Zhang et al. 2014), which may dampen enthusiasm for anti-sclerostin therapy in PCa. Similarly, PTH-related protein drives tumor-associated bone destruction in breast cancer (BCa); given the common targets and mechanisms of action between PTHrP and PTH, the involvement of PTHrP may generate reluctance toward a clinical trial for Forteo (Guise et al. 1996). Thus, a thorough investigation of the effects of these osteoanabolic drugs on bone metastases is needed before clinical trials can begin.
Upon metastasis to bone, PCa can present as osteoblastic (bone-forming), osteolytic (bone-resorbing), or mixed lesions in the skeleton (Fig. 3). Bone resorption and formation are coupled with tumor progression (Thalmann et al. 2000, Yi et al. 2002, Yin et al. 2003), evidence for which is provided by increased levels of biochemical markers of OB activity, increased cellular activity and metastasis (measured by technetium diphosphonate at bone metastatic sites) (Cachin et al. 2006), and elevated serum markers of bone resorption (Percival et al. 1987, Pelger et al. 1998, Garnero et al. 2000). Histologically, PCa cells localize to OB-rich regions of the bone hence bone metastases are primarily osteoblastic in PCa (Charhon et al. 1983, Roudier et al. 2008). The presence of OBs adjacent to the tumor itself a priori suggests that PCa drives OB activity, which creates osteosclerotic woven bone physically adjacent to the metastatic tumor (Charhon et al. 1983). OB presence near metastases is not universal, as this is not observed in bone metastases from breast, lung, or kidney cancers (Logothetis & Lin 2005). Further, OBs are generally absent from normal bone that is not undergoing adaptation or remodeling. Combined with increased serum markers of OB activity (Ibrahim et al. 2010), it is apparent that functional engagement between PCa and skeletal cells drives disease burden.
Figure 3.
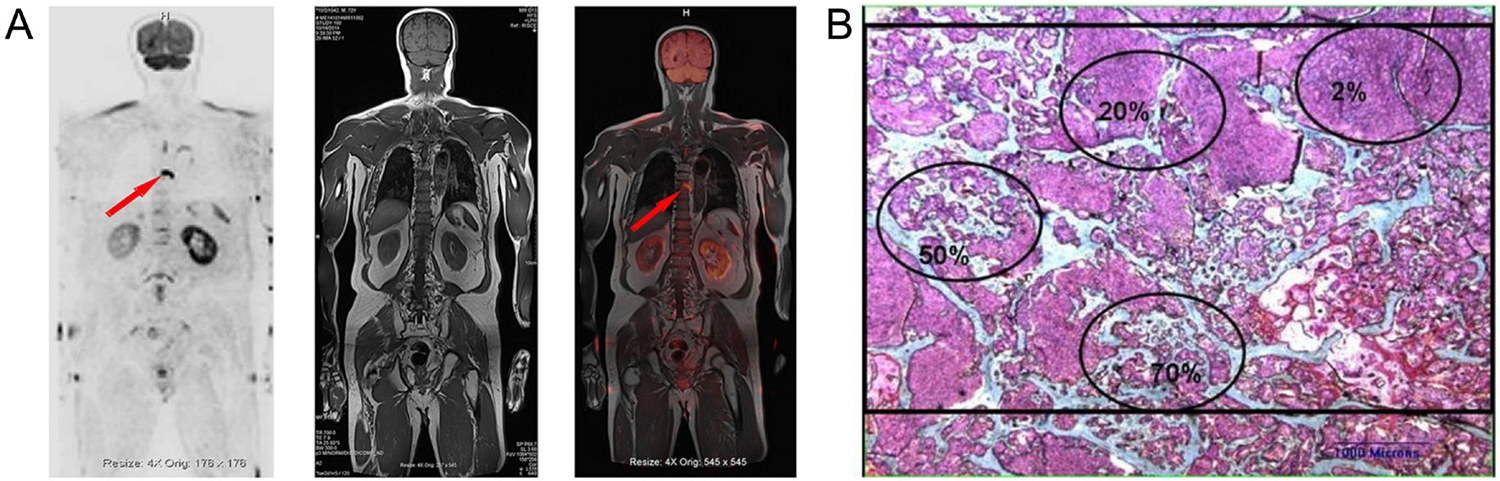
Radiographic detection of prostate cancer metastases and histologic patterns. (A) Whole-body magnetic resonance imaging (MRI) showing suspected metastatic lesion in thoracic vertebra seven; metastasis to axial bones is most common in prostate cancer. Reproduced with permission from (Chen et al. 2021). (B) Metastatic bone biopsy demonstrating lytic, blastic, and mixed lesions within a single biopsy stained with Goldner-Masson trichrome. Green areas indicate bone, pink areas indicate prostate cancer, and red areas indicate osteoid. Ovals indicate differences among approximate bone volumes, including 70, 50, 20, and 2% (clockwise from bottom). Bar indicates 1 mm. Reproduced with permission from (Roudier et al. 2008).
Molecular mechanisms of prostate cancer growth and bone metastasis
Metastatic PCa is often treated with ADT, illustrating the importance of AR signaling in PCa development, growth, and survival. ADT is an imperfect treatment due to side effects and tumor escape from androgen sensitivity. While these therapies cause tumor repression, at least initially, they also affect bone mineral density and associate with falls and fractures. Understanding the mechanisms governing mCSPC vs mCRPC may assist in developing or repurposing existing therapies to better address clinical needs and ultimately improve patient outcomes.
Androgen receptor signaling in prostate cancer and homeostasis
The male gonadal hormones testosterone and DHT elicit their molecular functions by binding to the AR. As a member of the steroid receptor nuclear receptor family, AR exhibits ligand-dependent dimerization, nuclear translocation, and DNA binding (Heemers & Tindall 2007, Wilson et al. 2016, Messner et al. 2020b). The AR is broadly expressed in the body, where its actions impact the development and maintenance of myriad systems including the cardiovascular, immune, reproductive, hematopoietic, and musculoskeletal systems (Davey & Grossmann 2016). AR signaling drives expression of prostatic PSA (Riegman et al. 1991), cytochrome P450 and TGF-β in the liver (Kanda & Yokosuka 2015), and oxytocin in the brain system (Karlsson et al. 2016); further, nongenomic AR signaling activates and interacts with other signaling pathways including Gq/PLC/IP3 (Foradori et al. 2008), MAPK (Foradori et al. 2008), IL-6/STAT3, PI3/Akt (Leung & Sadar 2017), and NF-κB (Hu et al. 2017b). Despite the initial effectiveness of ADT, loss of androgen dependence and rise of a CRPC phenotype invariably occur. CRPC employs multiple molecular strategies to progress, including AR amplification, AR mutations which reduce ligand specificity and thereby enable ligand-independent AR signaling, and alternately spliced AR variants that lack ligand-binding domains (Jathal et al. 2011). The net effect of such molecular adaptations sustains AR signaling even under conditions of markedly reduced androgen synthesis and bioavailability.
The AR is broadly expressed in organs and tissues including the ovary and testis, accessory sex organs, muscle, fat, bone, and myeloid tissue (de Gendt & Verhoeven 2012). Thus, it is unsurprising, if not unexpected, that strategies which mitigate AR signaling – whether ADT, inhibiting androgen synthesis (abiraterone) or AR antagonists (enzalutamide and apalutamide) – also exert systemic sequelae, notably in the skeleton. In bone, androgens restrain OCL formation while maintaining OB and OCY viability (Manolagas et al. 2013, Messner et al. 2020b). Germline deletion of AR produces mice with low bone mass driven by high bone turnover (Kawano et al. 2003); conditional deletion in specific bone cell subtypes elicits muted impacts on bone mass, with limited impact on cortical and trabecular microarchitectural parameters compared to global AR deletion (Manolagas et al. 2013). Thus, the greatest weapon currently available to combat recurrent PCa – AR inhibition – has negative consequences on bone health independent of tumor burden. Consequently, an ideal PCa treatment would retain the efficacy of ADT while improving the specificity of treatment, targeting solely PCa while sparing AR signaling in other tissues.
How prostate cancer metastasizes to bone
PCa metastasis to bone involves several steps: transformation, angiogenesis, invasion, and distal migration from the primary site, followed by endothelial attachment, extravasation, and site-specific establishment of metastases at a secondary site (Clarke et al. 2009) (Fig. 4). As a densely mineralized and highly rigid tissue, the skeleton may present as a hostile environment for tumor cell growth without adaptation (Martin 2002). To accomplish this, tumor cells migrate across the sinusoidal wall to invade and survive within BM stroma prior to migration to endosteal bone surfaces (Suva et al. 2011) (Fig. 4). The endosteal bone surface is covered with lining cells whose motility is stimulated by the newly resident tumor cells (Fig. 4). The cancer cells communicate with local BM stromal cells and release factors that stimulate the motility of the lining cells, activating bone resorption (Suva et al. 2011) (Fig. 5). This provides access to the demineralized bone surface for subsequent tumor cell adhesion and proliferation (Roodman 2004, Suva et al. 2009).
Figure 4.
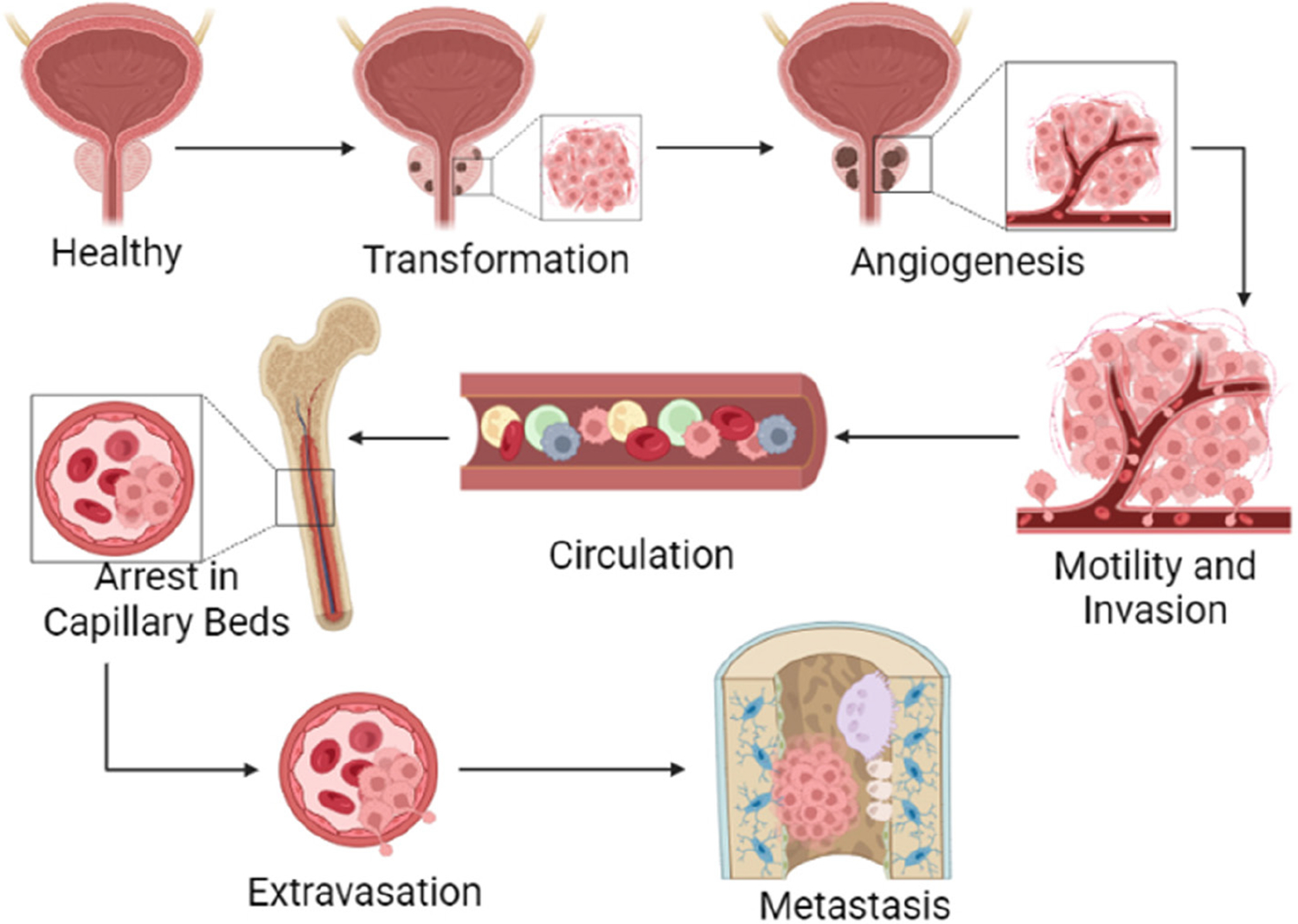
The metastatic cascade. The multi-step metastatic cascade begins with transformation, where healthy tissue develops a tumor. Next, the tumor must stimulate angiogenesis to create its own blood supply and meet its metabolic needs. Some cancer cells will enter the vasculature in a step termed motility and invasion. Invasion into the vasculature enables cancer cell circulation throughout the body until it arrives at a suitable secondary site. Once at a suitable secondary site, cancer arrests in a capillary bed. The cancer cell can then extravasate from the capillary and proliferate in the secondary metastatic site.
Figure 5.
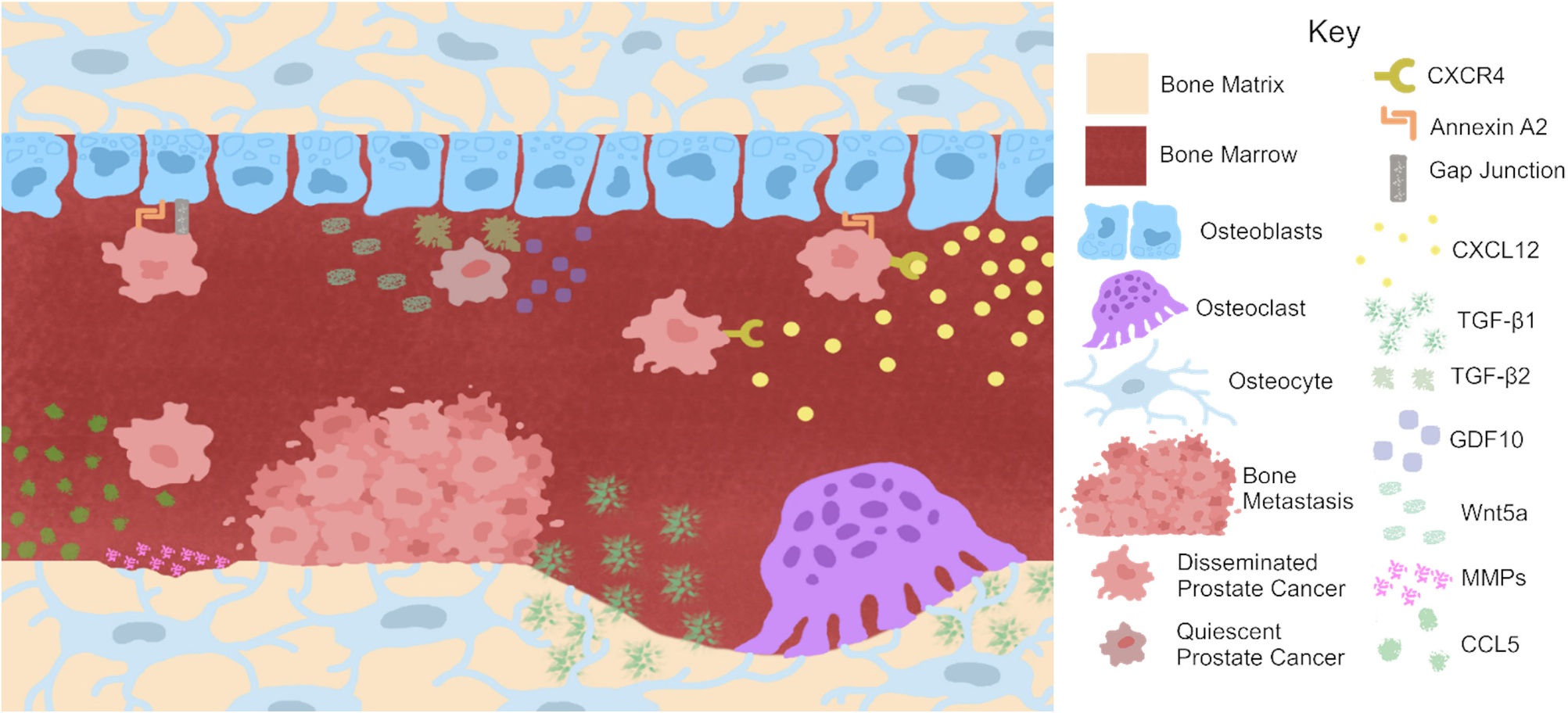
Prostate cancer engages bone cells to promote metastatic growth. Prostate cancer (PCa) recruitment to bone involves myriad pathways including the CXCL12/CXCR4 axis. Osteocytes may also recruit PCa to bone via production of CCL5. Once in bone, PCa binds to the endosteal surface via annexin A2 binding. Growth factors released from the bone matrix by osteoclasts such as TGF-β1 promote PCa growth. Further breakdown of the bone matrix is accomplished by osteocyte production of matrix metalloproteinases (MMPs). Calcium donation through gap junctions between PCa and osteoblasts may also promote PCa growth and metastasis. However, osteoblast production of Wnt5a has been shown to induce dormancy in PCa. Further, the production of GDF10 and TGF-β2 by osteoblasts induces quiescence in PCa cells. Thus, bone cells may also act as sentries against PCa growth.
Chemokines attract prostate cancer and initiate downstream signaling
Concentration gradients of chemokines produced by bone attract cancer cells (Lazennec & Richmond 2010). The CXCR4/CXCL12 binding pair is involved in bone metastases (Fig. 5). CXCL12 that is secreted from cancer-associated fibroblasts and OBs in bone binds to its cognate receptor CXCR4 on expressed in PCa cells, thus recruiting cancer to bone. Chemokine ligand-receptor binding encourages the adhesion of PCa cells to ECs or the extracellular bone matrix α5 and β3 integrins at the metastatic site (Engl et al. 2006); in turn, this initiates a cascade of downstream signals including MAP kinases, PI3K/Akt, Ras, Rho GTPases, and NF-κB, the concerted effect of which is to aggravate inflammation, cancer cell invasion, proliferation, and survival (Vindrieux et al. 2009). Thus, pharmacologic targeting of the CXCR4/CXCL12 binding pair may diminish PCa recruitment to the bone, preventing bone metastases and their resultant sequelae.
Bone marrow microenvironmental cues promote adhesion, migration, and growth
The heterogeneity of the BM microenvironment contributes mightily to PCa metastasis, tumor growth, and tumor dormancy. Endothelial adhesion of metastasis PCa cells to BM endothelium is enriched compared to other endothelial sources (Haq et al. 1992, Lehr & Pienta 1998, Scott et al. 2001). Myriad cell–cell adhesion proteins are implicated in mPCa cell–BM endothelium interactions, including galectin-3 and LFA-1 (Lehr & Pienta 1998), SDF1/CXCL12 and CXCR4 (Taichman et al. 2002), PSA (Romanov et al. 2004), fractalkine-CX3CR1 (Shulby et al. 2004), and E-selectin (Dimitroff et al. 2004), among others (reviewed in Kan et al. 2016). BMAT produces adipokines, such as leptin and adiponectin, that promote cancer cell growth and migration (reviewed in Hardaway et al. 2014, Otley & Sinal 2022).
Hypoxic circuits promote prostate cancer growth and drug resistance
Variances in oxygen bioavailability may influence PCa metastasis and survival in bone. Hypoxic signaling within and from bone prompts the secretion of proteins such as VEGF and shifts metabolism to facilitate cancer cell growth and survival. Hypoxia and androgen signaling create feedback circuits: androgen ablation causes hypoxia, whereas hypoxia, in turn, enhances ligand-independent AR transcriptional activity (Mitani et al. 2011). Thus, hypoxia may enable androgen resistance by allowing for AR signaling without its ligand to drive CRPC growth. This is likely mediated by redirecting glucose flux from the AR-dependent pentose phosphate pathway to the hypoxia-induced glycolysis pathway (Geng et al. 2018), thus triggering AR-independent enzalutamide resistance. Therapeutic resistance may thusly be conferred to PCa by the hypoxic environment of bone. Such interactions between metastatic cancer and its niche highlight the importance of combination therapeutic strategies which target both implicit cancer biology as well as the microenvironmental signals which drive cancer evolution. Though microenvironmental cues may drive therapeutic resistance, these same cues may sensitize PCa to other therapies. Thus, understanding interactions which drive synergy or antagonism between a therapeutic, the cancer, and its microenvironment provides an opportunity to develop more effective therapeutics.
Overlapping signaling pathways in bone homeostasis and prostate cancer
Parathyroid hormone participates in bone remodeling and prostate cancer-associated hypercalcemia
PTH and PTH-related peptide (PTHrP) impact bone mineral density. PTH or PTHrP binds to the receptor PTH1R which is a G protein-coupled receptor; this activates the Gs heterotrimeric G-protein that activates cyclic AMP/PKA pathway, ultimately phosphorylating the OB transcription factor Runx2, activating several genes in these cells. This includes the phosphorylation of the Ras small GTP-binding protein – leading to a kinase cascade cumulating in the phosphorylation and activation of a MAPK – including Erk1/2, JNK/SAPK, and p38MAPK. This pathway also phosphorylates and activates the p85 subunit of PI3K. Class IA PI3K is a dimer of the p110 catalytic subunit and one of five p85 regulatory subunits. Phosphorylation of PI3K activates several downstream targets, including Akt and p70S6 kinase, a downstream target of mTOR. The activation of these pathways increases osteoblastogenesis and OB survival (Jilka 2007). In PCa, PTHrP is a major cause of hypercalcemia. DHT stimulation led to the inhibition of PTHrP, while the latter stabilized the AR by phosphorylation at Y534 (DaSilva et al. 2009). Testosterone stimulation inhibited PTH-stimulated osteoclastogenesis (Chen et al. 2001). Therefore, it is estimated that AR inhibition would reverse these effects, resulting in bone loss.
Insulin-like growth factor, fibroblast growth factor, epidermal growth factor, and other receptor tyrosine kinase activating pathways
Through activation of the PI3K pathway, PTH stimulates insulin-like growth factors IGF-1 and IGF-2 expression to stimulate OB proliferation and differentiation and to regulate OCL activity. The functions of IGF-1/2 are regulated by a family of six secreted transporters that have high affinity to the IGFs – named IGF-binding proteins 1–6 (IGFBP1–6). IGFBPs bind and stabilize IGF – for example, maintaining IGFs in circulation or inactivated in the OBs for prolonged periods of time, thereby preventing homing and metastasis. Upon dissociation from IGFBPs, IGFs can then bind to their receptor and activate the PI3K signaling cascade. IGFBPs can have both stimulatory and inhibitory effects on the IGFs in bone, with IGFBP-3 and -5 being stimulatory and IGFBP-1, 2, -4, and -6 being inhibitory.
The IGF receptors belong to a family of receptor tyrosine kinases (RTKs) that can activate the PI3K cascade, the Ras/MAPK cascade or both. There are 20 different classes of RTKs encoding about 90 RTKs in all. Many of these RTKs are active in the bone and mediate bone metastasis. The FGF receptors also mediate osteoblastogenesis and bone formation. There are 23 FGFs that can bind to four FGFRs. While FGFR1–4 are involved in the proliferation of OB precursor cells, FGFR2 is also involved in OB differentiation (Choi et al. 2008). Mutations in FGFR2 result in increased transcriptional activity of the transcription factor Runx2, which results in increased expression of osteogenic markers. FGFRs also activate the Ras/MAPK and PI3K/Akt pathways.
Another RTK, the epidermal growth factor receptor (EGFR), is a glycoprotein that also activates the ERK and Akt pathways. There are four RTKs in the EGFR family, EGFR/ErbB1, HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4. This family of RTKs regulate the proliferation and differentiation of OBs, OCLs, and chondrocytes, thereby regulating bone formation and cancer metastasis. EGFR stimulation affects OCLs, by inhibition of OPG expression and upregulation of RANKL. Decreased signaling of ErbB RTKs reduces trabecular and cortical bone mass caused by reduced osteoblastogenesis and increased bone resorption (Feng & McDonald 2011). We have previously demonstrated that the ErbB3 RTKs are highly activated in PCa (Chen et al. 2011, Jathal et al. 2019). Overall, these studies indicate that activation of RTKs by their respective growth factors promotes bone mineral density while their loss causes loss of bone mineral density.
Regulation of bone remodeling by Wnt/β-catenin signaling in prostate cancer
Wnts are secreted glycoproteins that comprise a large family of 19 proteins. Wnts activate two major signaling pathways – the canonical β-catenin dependent pathway and the non-canonical pathways. The receptors for the Wnt ligands are called frizzled (Fzd) and consist of 10 members. The Fzd receptors are akin to G protein-coupled receptors and require co-receptors to mediate Wnt signals, such as LRP5/6. In the absence of Wnt activation, β-catenin is cytoplasmic and is ultimately degraded by a complex composed of Axin, APC, PP2A, GSK3, and CK1α (Komiya & Habas 2008). Wnt binding to Fzd and LRP5/6 disrupts this complex and stabilizes β-catenin, which then translocates to the nucleus, where it acts as a transcriptional co-activator to the transcriptional factors LEF/TCF. Non-canonical Wnt signaling pathways include Fzd binding to Dsh which triggers downstream signaling cascades that regulate cell polarity (called the planar cell polarity pathway). Wnt4, Wnt5A, and Wnt 11 activate non-canonical pathways that activate the small GTPases Rho and Rac or activate JNK to modulate the cytoskeleton. A second branch of the non-canonical Wnt signaling pathway stimulates intracellular Ca2+ mediated by Gq. Wnt5A and Wnt11 can activate this pathway (usually using Fzd2) and are mediated by PKC and CamKII. The latter has also been shown to inhibit canonical Wnt signaling.
The canonical Wnt/β-catenin pathway, alongside RUNX2, modulates skeletal development, OB differentiation, and bone formation (Krishnan et al. 2006, Monroe et al. 2012). It promotes mesenchymal commitment to the osteoblastic lineage and OB differentiation during bone formation. Wnt10b signaling increases bone mass by stimulation of osteoblastogenesis and regulates trabecular bone and serum osteocalcin (Bennett et al. 2005). Wnt6a also influences the differentiation of mesenchymal precursors into OBs (Cawthorn et al. 2012). Wnt3A, Wnt4, and Wnt16 regulate RANKL and OPG expression in OBs. In bone cells, non-canonical Wnt signaling was shown to mediate interaction with BMP signaling, the RANK/RANKL/NF-κB pathway, Hippo, Notch, and Hedgehog signaling pathways, mTOR and EGF signaling (Lojk & Marc 2021). Osteoblastic Wnt5A increases RANK expression through JNK signaling that recruits the c-Jun transcription factor to the RANK promoter, thus increasing RANK transcription and the susceptibility of OCL precursor cells to RANKL (Maeda et al. 2012). In contrast, Wnt4 inhibits OCL differentiation of primary BM macrophages, which also inhibit the activation of the canonical Wnt signaling pathway. Wnt3A induces the Warburg effect in differentiating cells, by switching transcription to favor the glycolytic pathway, resulting in OB differentiation. Wnt3A, Wnt7B, and Wnt10B activate the mTOR signaling pathway, increasing OB number and activity, and increasing bone formation. Non-canonical Wnt signaling is also associated with crosstalk with the Hippo pathway as well as the ERK/MAPK cascade. YAP/TAZ, the main effector of the Hippo pathway, interacts with both β-catenin and Dsh. Non-canonical Wnt ligands Wnt5a, Wnt5b, Wnt4, and Wnt3a are also strong activators of YAP/TAZ activity, through activation of the Fzd2 or Fzd5 receptors, and the G-protein Gα12/13, which lead to downstream activation of RhoA and Rac kinases and subsequent inhibition of the YAP/TAZ inhibitors Lats1/2. Both the Wnt and Hippo signaling pathways are required for osteogenic differentiation of mesenchymal cells, but suppression of YAP/TAZ abolished WNT4-induced OB differentiation. Wnt4-induced p38 MAPK pathway has also been shown to be involved in melatonin-induced osteogenic differentiation and suppression of osteoclastogenesis. Together, these results show that while canonical Wnt signaling is primarily associated with osteogenic differentiation and renewal of mesenchymal cells, non-canonical Wnt signaling has been associated with cell survival and the prevention of apoptosis.
PCa is usually characterized by increased levels of β-catenin at the nucleus, the cytoplasm, and the plasma membrane. In the plasma membrane, β-catenin regulates the activation of cadherins – E-cadherin in epithelial cells and N-cadherin in neuronal cells. Activating mutations in β-catenin in treatment-naive PCa is rare but has been observed in 12% of CRPC cases (Beltran et al. 2013). The AR is a crucial partner for β-catenin in PCa. Studies have shown competition of the AR/β-catenin complex with the TCF/β-catenin complex in the nucleus with the AR/β-catenin complex being predominant in PCa. Inactivation of AR by ADT or AR antagonists leads to redirection of β-catenin to a complex with TCF. Dkk1 is an inhibitor of Wnt signaling that is overexpressed in PCa, resulting in bone metastasis and the induction of osteolytic lesions (Thudi et al. 2011). The non-canonical Wnt signaling pathways are also regulated by the AR. Wnt7b, which non-canonically activates PKC and can induce an osteoblastic response, contains an ARE and is a target of various mutated and alternately spliced AR in CRPC. Thus, Wnt signaling plays a very important role in the regulation of bone metastasis as well as its regulation by androgen withdrawal.
Transforming growth fsctor-β and bone marrow morphogenetic protein signaling influence osteoblastogenesis and androgen receptor signaling
The TGF-β and BMP signaling pathways control bone remodeling and maintenance. About 29 factors of this superfamily act through heteromeric receptor complexes (seven type I and five type II) that transduce intracellular signals via the Smad and MAPK pathways. TGF-βs and BMPs have diverse functions in skeletogenesis, including mesenchyme condensation, skeleton morphogenesis, growth plate development, and OB differentiation (Chen et al. 2012). They also regulate the maintenance of postnatal bone and cartilage. TGFβRII phosphorylases TGFβRI, in turn, phosphorylating receptor-activated Smads (R-Smads), Smad2 and 3. R-Smads further activate Smad4 and translocate into the nucleus, where they regulate transcription. TGF-β also activates a non-Smad-dependent pathway, whereby TGF-β activated kinase 1 (TAK1) and TAK1-binding protein 1 activate the MAPK kinase cascade resulting in the activation of ERK, and p38MAPK. Similarly, there are 14 BMPs that work through BMP receptors type I and type II.
When initially synthesized, TGF-β is non-covalently bound to latency-associated protein (LAP) and remains inactive. Osteoclastic bone resorption releases active TGF-β1 from LAP and induces enrichment of osteoprogenitors in the bone resorption lacunae. Loss of TGF-β1 in mice reduces trabecular bone density and OB number on the bone surface. TGF-β1 does not induce osteogenesis in mesenchymal pluripotent cells but increases osteoprogenitors by inducing chemotaxis and proliferation. TGF-β1 is also necessary for survival during the differentiation of OBs into OCYs. TGF-β also blocked OB mineralization. On the other hand, active TGF-βs regulate OCL bone resorption. While TGF-β promotes OCL differentiation at low doses, it can inhibit OCL differentiation at high doses. Similarly, BMP signaling also promotes chondrocyte proliferation and differentiation, via the expression of Sox9, a transcription factor that is essential for chondrogenic commitment and differentiation. BMP-2 conditional knockout mice have frequent fractures that fail to heal and may be important in bone maintenance. Additional studies show that coordination between BMP-2 and BMP-4 is required for osteoblastogenesis as well as chondrocyte proliferation, differentiation, and apoptosis.
The AR interacts with the TGF-β pathway (Song et al. 2008) as well as with BMP ligands (Lee et al. 2013). ADT increased TGF-β, TGFβRI, TGFβRII, and activation of Smads 2 and 3, resulting in apoptosis by a process where the AR interacts with Smad3. In contrast, BMP6 is thought to induce the expression of IL-6, which in turn increases AR levels. Thus, TGF-β may have opposite effects on the AR as compared to BMPs.
Prostate cancer and bone
PCa often metastasizes to the skeleton. Compared to other common sites of PCa metastasis such as the lung, the bone microenvironment more efficiently encourages subsequent metastasis of disseminated tumor cells, likely from genetic selection and epigenetic reprogramming. Indeed, skeletal metastasis augments the ability of disseminated tumor cells to generate secondary metastases in other organs (Zhang et al. 2021b). While AR signaling and its pharmacologic restraint dominate the research and clinical course, PCa growth and metastasis are driven by myriad mechanisms. Elaborating the breadth of molecular pathways subjugated by PCa and their synergistic or antagonistic interactions may identify novel druggable targets. Such therapeutics would prove useful, particularly in treating metastatic CRPC, which has the highest patient mortality rate and is the most difficult to treat. To do so, it is necessary to establish and refine mechanisms of PCa-skeletal engagement.
Bone remodeling and prostate cancer metastasis
PCa subverts homeostatic processes to promote metastasis, colonization, and growth. Bone remodeling – the continuous process by which bone repairs itself – is subjugated by PCa to promote metastasis to bone (Fig. 5). PCa bone metastases are most frequently observed in the spine, ribs, pelvis, and proximal limbs, the sites of active remodeling even throughout adulthood (Imbriaco et al. 1998). Remodeling occurs more frequently during skeletal development, a feature which was exploited in a murine model to elaborate the impact of remodeling and its inhibition on PCa burden. Intracardiac injection of PCa cells into young (7 weeks) or old (52 weeks) mice demonstrated that metastatic burden was higher in young compared to old mice (Kalikin et al. 2003); increased metastatic burden in the young mice was attributed to higher remodeling rates exhibited by the young animals. Similarly, inducing remodeling with PTH also produced more PCa metastasis to bone compared to vehicle-treated animals; conversely, inhibition of bone remodeling with a bisphosphonate reduced the remodeling-induced increase in bone (Schneider et al. 2005). Predilection for PCa to metastasize to remodeling sites is evident within a given bone. PCa cells localize more frequently to the lateral side of the tibia, which exhibits seven-fold higher bone remodeling than the medial side (Wang et al. 2014). The physical contact between PCa cells, OBs, and OCLs develops a cycle of mutually driven growth (Kimura et al. 2017) (Fig. 5). This autocatalytic cycle enhances the growth and spread of the bone metastases and the tumor itself (Logothetis et al. 2018). The richly complex interactions between native cells in bone and invading cancer involve myriad intracellular and extracellular signaling cascades, among them tumor-derived molecules CXCR4 and vascular endothelial growth factor that further recruit metastatic PCa to bone and enable survival therein (Logothetis et al. 2018). Other physical environment factors like extracellular Ca2+, low pH, and hypoxia then activate signaling pathways within these metastatic PCa cells leading to the release of additional factors like PTHrP, IGF, and TGF-β, ultimately perpetuating tumor cell growth and survival within bone (Kingsley et al. 2007, Weilbaecher et al. 2011).
TGF-β drives PCa through myriad conserved pathways. The TGF-β superfamily includes TGF-β, BMPs, growth and differentiation factors (GDFs), and activins. TGF-β members have diverse roles in patterning and cell fate decisions during development (Tabata & Takei 2004, Ozair et al. 2013), homeostasis (Carlson & Conboy 2007), and disease (Yingling et al. 2004, Siddiqui et al. 2022b). TGF-β initially functions as a tumor suppressor that inhibits prostate cell proliferation yet after tumorigenesis acts as a tumor promoter during cancer progression (Bello-DeOcampo & Tindall 2003). TGF-β1 promotes bone metastases through canonical Smad-dependent and Smad-independent pathways; the bone matrix, in turn, is a rich source of TGF-β1 which can be liberated by osteoclastic bone resorption to promote tumor progression (Pfeilschifter & Mundy 1987, Guise et al. 1996). TGF-β1 signaling is highly integrated, making it difficult to target: TGF-β integrates with key regulatory pathways in bone health like Notch and Wnt (Klüppel & Wrana 2005, Hall et al. 2006, Lindsey & Langhans 2014, van den Bosch et al. 2016) and integrates with AR signaling. Ligand-bound AR inhibits TGF-β transcriptional activity (Chipuk et al. 2002) such that androgen ablation via ADT activates TGF-β signaling and gene transcription to promote bone metastasis. Thus, although ADT is a cornerstone of PCa treatment, the complex web of interactions between androgens and other signaling molecules such as TGF-β1 highlights the need for a more cancer-specific therapeutic. Collectively, such discoveries highlight the obligation of bone remodeling toward PCa invasion into bone.
Osteoblasts and prostate cancer
OBs can be found near PCa cells, which is not observed in other skeletally tropic cancers like BCa. Mechanisms wherein OBs may promote PCa metastasis to bone involve their role in maintaining the HSC niche and their role in bone remodeling.
Osteoblast maintenance of the hematopoietic stem cell niche may recruit prostate cancer to bone
The first step in PCa colonization of bone is invasion of the marrow space. There is marked overlap among bones that are destinations for metastatic PCa vs BM distribution, promoting the hypothesis that the BM niche provides a permissive soil or selects for PCa metastasis (Imbriaco et al. 1998). During homeostasis, OBs lining the endosteum maintain the HSC niche through CXCR4/CXCL12 interactions, which regulate HSC homing to the marrow (Taichman et al. 2002, Zhang et al. 2003, Shiozawa et al. 2011). However, PCa competes with HSCs for the BM niche utilizing the same CXCR4/CXCL12 axis (Taichman et al. 2002, Shiozawa et al. 2011) (Fig. 5). PCa cells can be mobilized out of the marrow and into circulation through pharmacologic blockade of CXCR4. Further, more PCa cells can be found in the marrow when the OB number is increased by treatment with PTH (Shiozawa et al. 2011). Another mechanism promoting niche selection in bone involves Annexin A2 (AnxA2), a plasmalemmal calcium-dependent phospholipid (Fig. 5); AnxA2 expression in OBs promotes HSC homing and binding to the endosteal niche of the BM microenvironment (Jung et al. 2007). Metastatic PCa subverts this homeostatic process to engraft in bone via CXCL12 (Jung et al. 2015). Following PCa homing to bone, PCa-OB AnxA2 interactions elicit two further responses: (i) promotes expression of dormancy-regulating receptors Axl, Sky, and Mer in PCa cells and (ii) induces OB expression of Gas6 which binds to PCa cells to prevent PCa proliferation, promote their dormancy, and protect against chemotherapy-induced apoptosis.
Pleiotropic role of osteoblasts on prostate cancer growth
OBs communicate extensively with PCa cells to both promote and prevent metastasis. OBs form gap junctions with breast and PCa cells, thereby providing intracellular Ca2+ which promotes metastasis (Wang et al. 2018a) (Fig. 5). OBs also promote PCa metastasis through secretion of factors like TGF-β (Fig. 5) which increases PCa migration and promotes an aggressive phenotype (Karlsson et al. 2018). The relationship between OB and PCa is complex, as OBs also prevent PCa metastasis and induce PCa dormancy. Wnt5a secreted by OBs reduces PCa proliferation and prevents metastasis in vivo. PCa cells treated with Wnt5a were also resistant to docetaxel, the most common treatment for metastatic PCa, suggesting that Wnt5a induces dormancy in PCa (Ren et al. 2019) (Fig. 5). Similarly, differentiated OBs induce PCa quiescence by secreting TGF-β2 and GDF10 (Yu-Lee et al. 2018) (Fig. 5). Thus, the complexity of OB–PCa engagement reflects context-dependent signaling, wherein cytokines and growth factors exert discrete spatiotemporal impact. This is reflected, for example, by the Wnt antagonist Dkk1, whose expression increases early in PCa development only to decrease during progression from primary tumor to metastasis and, in doing so, functions as a molecular switch transitioning PCa phenotype from osteolytic to osteoblastic (Hall et al. 2008).
Osteoclastic bone resorption has a muted role in prostate cancer bone metastases
OCLs resorb bone by breaking down the extracellular matrix of bone. Bone resorption is necessary for homeostasis as it removes old, damaged, or biomechanically inferior bone for replacement with new bone. During bone resorption, OCL release growth factors such as TGF-β from the bone matrix, which promotes the progression of lytic metastatic cancers like BCa (Pfeilschifter & Mundy 1987, Guise et al. 1996) (Fig. 5). PCa also exhibits lytic and mixed lesions histologically, with OCL found on the border of many PCa bone metastases (Russell et al. 2009, Wang et al. 2018b) (Fig. 3). Thus, it has been proposed that the role of OCL in PCa is the liberation of growth factors from the bone matrix (Russell et al. 2009). Resultingly, preventing OCL activity was once pursued as a target to mitigate cancer metastasis and burden. OCL activity and viability can be dampened pharmacologically through bisphosphonates – an analog of pyrophosphate naturally found in bone – and denosumab – an inhibitor of RANKL (Bekker et al. 2004). However, clinical trials of these drugs in patients with PCa have not been promising. Several clinical trials which incorporated bisphosphonates along with the standard of care for patients with PCa saw no difference in rates of overall survival compared to the control group (Vale et al. 2016, Hayes et al. 2018). Further, treatment with bisphosphonates failed to prevent bone metastases in PCa patients (Wirth et al. 2015). Less data exist on the efficacy of denosumab for the treatment of patients with PCa as only a single trial has been conducted. Patients treated with denosumab saw delayed time to first bone metastasis and increased metastasis-free survival, but survival modestly increased by four months (Smith et al. 2012). Together, the data demonstrate that bisphosphonates do not impact PCa disease progression. Further, the effects seen in other OCL inhibitors such as denosumab are modest. These data suggest that cells other than OCL may play a more prominent role in PCa metastasis to bone.
Osteocyte mechanosensation is disrupted by prostate cancer to promote cancer growth
Our understanding of PCa and OCY interactions is emergent. As OCY are the most abundant and longest-living cell in bone, it is plausible and highly likely that disseminated PCa encounters and engages OCY when growing within the bone. Investigation into the interactions of PCa and OCY is nascent yet growing. Intratibial injection of breast or PCa cells into immunocompetent mice demonstrated direct contact between OCY and tumor cells in BM (Fig. 5), which functionally increased OCY lacunar volume and may impede proper OCY mechanosignaling. Osteosclerotic lesions increased OCY lacuna size – indicative of osteocytic osteolysis and newly embedded OCY – with a concomitant reduction in canalicular connectivity (Hemmatian et al. 2021). Reduced connectivity between OCY in the presence of PCa cells has also been recapitulated in vitro in a 3D tissue-engineered model (Choudhary et al. 2018). This suggests that PCa tumors disrupt the highly organized lacuno-canalicular system in bone, which may impact OCY mechanosensation and signal transduction and could cause bone turnover imbalances characteristic of bone metastases. PCa-driven dysregulated mechanosensation in OCYs has been studied in vitro, wherein pressure generated from tumor growth in the bone induced further tumor growth and invasion. Pressure-induced OCYs produce CCL5 and MMPs which facilitate the migration and invasion of several PCa cell lines (Sottnik et al. 2015) (Fig. 5). CCL5 is a chemokine whose primary role is as a chemoattractant for immune cells, and this observation is consistent with previous reports showing that CCL5 induces migration in prostate, breast, lung, and osteosarcoma tumors (Yaal-Hahoshen et al. 2006, Huang et al. 2009, Wang et al. 2012). MMPs, which degrade the organic aspect of the matrix as occurs in remodeling, drive cancer invasion (Nemeth et al. 2002). Such findings were corroborated in a related study but failed to investigate candidate cytokines (Cui et al. 2016). Further, Cui et al focused on unstimulated OCY-conditioned media, suggesting that the OCY secretome may have latent pro-tumorigenic properties (Cui et al. 2016). Together, these papers establish a role for bidirectional crosstalk between OCYs and PCa, particularly in PCa subjugating or disrupting the normal mechanosensory function of OCYs to promote abnormal remodeling or promote PCa migration and invasion.
Osteocytes and other cancers: lessons to impart?
The relative dearth of information on the relationship between PCa and OCYs limits the field’s understanding of skeletal metastasis, as there is little information regarding crosstalk between PCa and the most abundant cell type in bone. This may impede the development of new drugs to combat skeletal metastasis, ultimately stalling improvements in patient outcomes. In this section, we will discuss mechanisms of communication between OCYs and other cancers where this relationship is better defined. Investigating these mechanisms of communication in the context of OCYs and PCa crosstalk may unveil novel targetable pathways to mitigate PCa skeletal metastasis.
OCYs are obligate mechanosensory cells in bone. The proper sensation of mechanical loading of bone through exercise and signal transduction to the effector cells in bone is crucial to skeletal maintenance; dysregulation of this signaling and subsequent imbalance of OCL/OB activity can lead to osteopenia, osteoporosis, or other skeletal phenotypes. Recent evidence has suggested that mechanically stimulated OCYs promote BCa migration and prevent cancer apoptosis. Thus, BCa may subjugate OCY mechanosignaling to promote cancer progression (Ma et al. 2018). However, OCY mechanosignaling may promote an anti-metastatic phenotype in other bone cells, establishing a pleiotropic role of OCY mechanical signaling in cancer metastasis. Pre-treating OCLs or ECs with conditioned media from mechanically loaded OCYs reduced BCa migration and increased cancer apoptosis from OCL- and endothelial-derived factors (Ma et al. 2019). Thus, OCY mechanical signaling may prevent cancer metastasis by signaling through other cells in closer physical proximity to BCa during the early stages of metastasis. Other groups have reported similar anti-metastatic effects of mechanical loading in vivo. For example, engagement of the Wnt co-receptor Lrp5 in OCYs may underly this anti-metastatic effect (Feng et al. 2021, Liu et al. 2021). Given the contribution of OCY mechanical signaling in BCa metastasis, mechanical signaling may also play a role in PCa metastasis; further, PCa subjugates OCY mechanosensation to promote metastasis through tumor-induced pressure (Sottnik et al. 2015). Consequently, investigation of other mechanical signaling pathways in bone and their contribution to PCa metastasis is warranted.
Multiple myeloma (MM) is a cancer of plasma cells residing within BM whose progression is regulated by OCYs. Reciprocal interactions between MM cells and OCYs involving Notch signaling drive OCY apoptosis and MM cell proliferation; increased expression of RANKL and Sost promote bone resorption and inhibit bone formation, respectively, consistent with osteolytic lesions that are hallmarks of MM (Delgado-Calle et al. 2016). Direct signaling via Notch may also be present in PCa metastases. Previous work has demonstrated direct contact between OCYs and PCa metastases in vivo, though the nature of this contact has not been described. Further, lytic PCa lesions – similar to the lytic lesions seen in MM – show empty OCY lacunae adjacent to the lesion (Hemmatian et al. 2021). Because lytic lesions have similar characteristics such as direct contact between cancer cells and OCYs and signs of OCY apoptosis, Notch signaling may underly this phenomenon in PCa skeletal metastases. Identifying Notch signaling in PCa skeletal metastases would establish new therapeutic targets in this disease. Studies in MM show that treatment with Notch inhibitors reduces tumor burden and osteolytic lesions in a mouse model of MM (Sabol et al. 2021, 2022). If Notch signaling is also present in PCa bone metastases, Notch inhibitors may also be utilized to prevent osteolytic and mixed lesions in PCa.
Engineered models to study prostate cancer and metastasis
Tissue-engineered models provide opportunistic weapons in the fight against cancer. Such models are valuable to improve our understanding of underlying stimuli for tumorigenesis and metastasis, develop better therapeutics and diagnostics, and facilitate the translation of such discoveries to the clinical setting (Sitarski et al. 2018, González Díaz et al. 2019). Many experimental approaches have focused solely on cancer cells in a 2D environment; in contrast, tissue-engineered models provide both tunability and additional complexity, enabling the study of the role of the tumor microenvironment through interrogation of cell–matrix interactions and crosstalk between cancer cells and resident tissue cells. The development of effective tissue-engineered models of bone metastases may accelerate the identification of new druggable targets resulting in improved patient outcomes for those with metastatic bone disease. While a variety of models are available, several aspects of the chosen model can be modified to provide the most useful model for a given study, some of which are highlighted below.
Scaffolds for studying prostate cancer
The development of engineered scaffolds to serve as an artificial environment facilitates the examination of multiple biophysical characteristics of metastasis and growth including composition, stiffness, and elasticity.
Composition
PCa behavior has been studied in an array of biomaterials including natural polymers like collagen (Koutsilieris et al. 1994), Matrigel (Edmondson et al. 2016), alginate (Xu et al. 2019), or synthetic polymers such as poly (ethylene glycol) (PEG) (Sieh et al. 2012, 2014, Taubenberger et al. 2016) and self-assembling peptides (Hainline et al. 2019). While the material selection is guided by the hypothesis under examination, it is imperative that the material embodies physiologically relevant attributes for its effective use as a model system. Numerous biophysical properties can be interrogated based on the selection of material including cell adhesion, degradation, stiffness, fibrous architecture, and extracellular matrix composition (Fig. 6).
Figure 6.
Engineered models to study prostate cancer. Tissue-engineered models provide both tunability and complexity, enabling the study of the role of the tumor microenvironment through interrogation of cell–matrix interactions and crosstalk between cancer cells and resident tissue cells. Common approaches in engineered models aim to model bone composition and biophysical properties, the metastatic potential of cancer cells through biological mediated regulation of substrate degradation, and the incorporation of other contributing cells to understand their influence on tumorigenesis and metastasis.
Such tunability of various biomaterials allows for the interrogation of cell–matrix interactions and the development of superior in vitro models. For example, composite hydrogels of chitosan and chondroitin sulfate – an extracellular matrix protein found in high abundance in PCa metastases – upregulated epithelial-to-mesenchymal transition markers and increased resistance to docetaxel treatment compared to PCa cells grown in 2D culture (Xu et al. 2020). Thus, the culture of PCa cells in tunable hydrogels facilitates a cell phenotype more reminiscent of aggressive metastatic PCa, offering a more biologically relevant platform for candidate drug testing.
Biophysical properties
In PCa, biophysical properties of the tumor microenvironment influence changes in gene and protein expression, alter the response to chemotherapeutics, and exhibit a positive correlation with aggressive cancers (Leight et al. 2017). Tissue stiffness is a biophysical property that correlates with disease state, which can only be evaluated using tissue-engineered platforms (Fig. 6). Stiffness can be tuned using various biomaterials through regulation of molecular weight, degradability, and porosity, as well as deposition of biomimetic extracellular matrix by associated cells. In one example, PC-3 PCa cells were encapsulated with BJ-5ta fibroblasts in a PEG-fibrinogen hydrogel to test the influence of biomechanical cues on the capacity of in vitro models to recapitulate tumors in vivo (Habbit et al. 2022). Through variation of matrix composition, the Young’s moduli of acellular hydrogels were tuned from 50 to 10,000 Pa to mimic clinical PCa stiffness values; gene sets associated with stiffened matrix include epithelial-to-mesenchymal transition, metabolic plasticity, and angiogenesis.
The metabolic activity of cancer cells shifts as they undergo changes in aggressiveness, in part due to diminished nutrient availability associated with cancer cell growth which outpaces vascularization and the deposition of the dense matrix which restricts diffusion of nutrients (Netti et al. 2000). While nutrient availability can be simulated in 2D culture through varying nutrient concentrations, this does not recapitulate interstitial transport or nutrient gradients that would exist within a tissue. Thus, tissue-engineered platforms provide an opportunity to effectively model simple physiological phenomena like nutrient availability. In one study, PCa cells were grown in monolayer overlaid on top of or beneath a disulfide-crosslinked hyaluronic acid hydrogel to simulate high and low nutrient availability, respectively (Tam et al. 2020). Cells grown on top of the hyaluronic acid gels were more viable than cells cultured beneath the gel, suggesting that hyaluronic acid may restrict the transport of nutrients below the gel. ATP content within PCa cells decreased over time and with increasing hydrogel concentration, perhaps through autophagy, which can be used as a survival mechanism under conditions of nutrient deprivation. Collectively, this approach represents an interesting strategy to study the role of interstitial transport on PCa behavior in vitro.
Complexity of co-culture
The study of cancer cells in monolayer culture is a common, inexpensive approach to study molecular mechanisms and evaluate the efficacy of therapeutics. While this reduction in system complexity creates an easy workflow, it reduces biological relevance which is reflected in the generation of many cancer therapeutics with few candidates exceeding a Phase 1 clinical trial. Thus, the incorporation of co-culture and biologically relevant cues through tissue-engineered models may allow for more reliable translation of results from bench to bedside (Fong et al. 2016a) (Fig. 6).
PCa patient-derived xenograft cells do not grow well on tissue culture plastic once isolated from the murine host. To effectively study PCa patient-derived xenograft tissue in a controlled environment, PCa patient-derived xenograft cells were encapsulated in a hyaluronic acid hydrogel with murine MC3T3-E1 pre-OBs to model the bone–PCa interaction (Fong et al. 2016b). PCa cells grew better in the presence of MC3T3-E1s than when maintained in monoculture within the scaffold, while expression of bone-specific markers (e.g. osteocalcin, bone sialoprotein, and alkaline phosphatase) was significantly higher in co-culture than for MC3T3-E1 alone. The engineering of 3D bone tissue models reflects host anatomy and thus predictive value of observed outcomes in vitro. This approach has revealed the direct impact of PCa cells on OCY viability and Wnt signaling which were not captured in 2D culture (Choudhary et al. 2018). Similarly, tunable gelatin methacrylamide models have enabled simultaneous coculture of osteogenic, adipose, and tumor microtissues to model the human bone tumor microenvironment more faithfully (Bessot et al. 2023). Thus, tissue-engineered models may provide essential cues for patient cell survival with a simpler workflow and reduced harm to animals.
Conclusions, challenges, and opportunities for advancement
Despite improvements in patient survival outcomes in recent years, there remain myriad unmet clinical needs. As identified earlier, there is still a need for a PCa-specific biomarker. Though serum PSA is currently used to identify the presence of PCa, PSA is not specific to PCa, nor does it correlate with disease severity. Second, both PCa and its treatment impart burdens on the patient, particularly for the skeleton. Notable sequelae of ADT, enzalutamide, apalutamide, and prednisone include bone loss and increased risk of fracture, together limiting the quality of life for patients on PCa treatment. Though bone loss may appear on the surface to be an acceptable off-target effect of PCa treatment, bone loss may have more dire consequences than previously considered including permitting metastasis to or growth within bone. Thus, treatments for bone health must also be considered alongside PCa treatment to improve patient quality of life and lifespan. Finally, myriad questions remain regarding the relationship between PCa and bone. Understanding of differences in bone tropism between PCa that is metastatic at diagnosis vs metastatic following local treatment may highlight differences in molecular mechanisms between these clinical entities. Further, understanding which cells within the bone – OB, OCL, OCY, or components of the BM – contribute most to PCa metastasis may identify new treatment or preventative strategies to mitigate the metastatic burden.
Funding
The contents reported/presented within do not represent the views of the Department of Veterans Affairs or the United States Government. This work was supported in part by a Biomedical Laboratory Research & Development (BLRD) Merit Award (I01BX004423, PMG) from the Department of Veterans Affairs and by R01CA185509 (PMG) from the National Institutes of Health. This work was supported in part by the Prostate Cancer Research Program Early Investigator Research Award W81XWH2110073 to MKJ. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers R01 AR073772 to DCG, T32 AR079099 to KVW and JKL, R01 AR079211 to JKL. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding bodies. Schematics were created with Biorender.
Footnotes
Declaration of interest
None of the authors have anything to disclose.
References
- Aaron N, Costa S, Rosen CJ & Qiang L 2022. The implications of bone marrow adipose tissue on inflammaging. Frontiers in Endocrinology 13 853765. ( 10.3389/fendo.2022.853765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, Onal M, Xiong J, Weinstein RS, Jilka RL, et al. 2013. Estrogen receptor-α signaling in osteoblast progenitors stimulates cortical bone accrual. Journal of Clinical Investigation 123 394–404. ( 10.1172/JCI65910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Szmulewitz RZ, Petrylak DP, Holzbeierlein J, Villers A, Azad A, Alcaraz A, Alekseev B, Iguchi T, Shore ND, et al. 2019. Arches: a randomized, Phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. Journal of Clinical Oncology 37 2974–2986. ( 10.1200/JCO.19.00799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, Holmes GB, Dunstan CR & DePaoli AM 2004. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. Journal of Bone and Mineral Research 19 1059–1066. ( 10.1359/JBMR.040305) [DOI] [PubMed] [Google Scholar]
- Bello-DeOcampo D & Tindall DJ 2003. TGF-betal/Smad signaling in prostate cancer. Current Drug Targets 4 197–207. ( 10.2174/1389450033491118) [DOI] [PubMed] [Google Scholar]
- Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, et al. 2013. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. European Urology 63 920–926. ( 10.1016/j.eururo.2012.08.053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD & MacDougald OA 2005. Regulation of osteoblastogenesis and bone mass by Wnt10b. PNAS 102 3324–3329. ( 10.1073/pnas.0408742102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessot A, Gunter J, Waugh D, Clements JA, Hutmacher DW, McGovern J & Bock N 2023. GelMA and biomimetic culture allow the engineering of mineralized, adipose, and tumor tissue human microenvironments for the study of advanced prostate cancer in vitro and in vivo. Advanced Healthcare Materials e2201701. ( 10.1002/adhm.202201701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikian J, Raisz L & Rodan G 2019. Principles of Bone Biology. Cambridge, MA, USA: Academic Press. ( 10.1016/C2015-1-01622-2) [DOI] [Google Scholar]
- Brawley S, Mohan R & Nein CD 2018. Localized prostate cancer: treatment options. American Family Physician 97 798–805. [PubMed] [Google Scholar]
- Bruzzaniti A & Baron R 2006. Molecular regulation of osteoclast activity. Reviews in Endocrine and Metabolic Disorders 7 123–139. ( 10.1007/s11154-006-9009-x) [DOI] [PubMed] [Google Scholar]
- Buettmann EG, Goldscheitter GM, Hoppock GA, Friedman MA, Suva LJ & Donahue HJ 2022. Similarities between disuse and age-induced bone loss. Journal of Bone and Mineral Research 37 1417–1434. ( 10.1002/jbmr.4643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachin F, Kelly A & Maublant J 2006. Evaluation of the therapeutic response: role of isotopic imaging. Bulletin du Cancer 93 1191–1199. [PubMed] [Google Scholar]
- Carlson ME & Conboy IM 2007. Regulating the Notch pathway in embryonic, adult and old stem cells. Current Opinion in Pharmacology 7 303–309. ( 10.1016/j.coph.2007.02.004) [DOI] [PubMed] [Google Scholar]
- Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, Martinez-Santibañez G & MacDougald OA 2012. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 50 477–489. ( 10.1016/j.bone.2011.08.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charhon SA, Chapuy MC, Delvin EE, Valentin-Opran A, Edouard CM & Meunier PJ 1983. Histomorphometric analysis of sclerotic bone metastases from prostatic carcinoma special reference to osteomalacia. Cancer 51 918–924 ( 10.1002/1097-0142(19830301)51:5<918::aid-cncr2820510526>3.0.co;2-j) [DOI] [PubMed] [Google Scholar]
- Chen G, Deng C & Li YP 2012. TGF-β and BMP signaling in osteoblast differentiation and bone formation. International Journal of Biological Sciences 8 272–288. ( 10.7150/ijbs.2929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Mooso BA, Jathal MK, Madhav A, Johnson SD, van Spyk E, Mikhailova M, Zierenberg-Ripoll A, Xue L, Vinall RL, et al. 2011. Dual EGFR/HER2 inhibition sensitizes prostate cancer cells to androgen withdrawal by suppressing ErbB3. Clinical Cancer Research 17 6218–6228. ( 10.1158/1078-0432.CCR-11-1548) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kaji H, Sugimoto T & Chihara K 2001. Testosterone inhibits osteoclast formation stimulated by parathyroid hormone through androgen receptor. FEBS Letters 491 91–93. ( 10.1016/S0014-5793(01)02160-3) [DOI] [PubMed] [Google Scholar]
- Chen R, Yang Q, Chen W, Yang Y, Cheng C, Sun Y & Lu J 2021. Whole-body MRI-based multivariate prediction model in the assessment of bone metastasis in prostate cancer. World Journal of Urology 39 2937–2943. ( 10.1007/s00345-020-03571-8) [DOI] [PubMed] [Google Scholar]
- Chiang C, Chiu M, Moore AJ, Anderson PH, Ghasem-Zadeh A, McManus JF, Ma C, Seeman E, Clemens TL, Morris HA, et al. 2009. Mineralization and bone resorption are regulated by the androgen receptor in male mice. Journal of Bone and Mineral Research 24 621–631. ( 10.1359/jbmr.081217) [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Cornelius SC, Pultz NJ, Jorgensen JS, Bonham MJ, Kim SJ & Danielpour D 2002. The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. Journal of Biological Chemistry 277 1240–1248. ( 10.1074/jbc.M108855200) [DOI] [PubMed] [Google Scholar]
- Choi SC, Kim SJ, Choi JH, Park CY, Shim WJ & Lim DS 2008. Fibroblast growth Factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K-Akt and ERK1/2 signaling pathways. Stem Cells and Development 17 725–736. ( 10.1089/scd.2007.0230) [DOI] [PubMed] [Google Scholar]
- Choudhary S, Ramasundaram P, Dziopa E, Mannion C, Kissin Y, Tricoli L, Albanese C, Lee W & Zilberberg J 2018. Human ex vivo 3D bone model recapitulates osteocyte response to metastatic prostate cancer. Scientific Reports 8 17975. ( 10.1038/s41598-018-36424-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke B 2008. Normal bone anatomy and physiology. Clinical Journal of the American Society of Nephrology: CJASN 3 (Supplement 3) S131–S139. ( 10.2215/CJN.04151206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke BL & Khosla S 2009. Androgens and bone. Steroids 74 296–305. ( 10.1016/j.steroids.2008.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke NW, Hart CA & Brown MD 2009. Molecular mechanisms of metastasis in prostate cancer. Asian Journal of Andrology 11 57–67. ( 10.1038/aja.2008.29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Dempster DW, Recker RR, Lappe JM, Zhou H, Zwahlen A, Müller R, Zhao B, Guo X, Lang T, et al. 2013. Abdominal fat is associated with lower bone formation and inferior bone quality in healthy premenopausal women: a transiliac bone biopsy study. Journal of Clinical Endocrinology and Metabolism 98 2562–2572. ( 10.1210/jc.2013-1047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RE 1997. Skeletal complications of malignancy. Cancer 80 1588–1594. ( 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z) [DOI] [PubMed] [Google Scholar]
- Cui YX, Evans BA & Jiang WG 2016. New roles of osteocytes in proliferation, migration and invasion of breast and prostate cancer cells. Anticancer Research 36 1193–1201. [PubMed] [Google Scholar]
- Dasgupta K, Lessard S, Hann S, Fowler ME, Robling AG & Warman ML 2021. Sensitive detection of Cre-mediated recombination using droplet digital PCR reveals Tg (BGLAP-Cre) and Tg (DMP1-Cre) are active in multiple non-skeletal tissues. Bone 142 115674. ( 10.1016/j.bone.2020.115674) [DOI] [PubMed] [Google Scholar]
- DaSilva J, Gioeli D, Weber MJ & Parsons SJ 2009. The neuroendocrine-derived peptide parathyroid hormone–related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Research 69 7402–7411. ( 10.1158/0008-5472.CAN-08-4687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey RA & Grossmann M 2016. Androgen receptor structure, function and biology: from bench to bedside. Clinical Biochemist. Reviews 37 3–15. [PMC free article] [PubMed] [Google Scholar]
- de Gendt K & Verhoeven G 2012. Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Molecular and Cellular Endocrinology 352 13–25. ( 10.1016/j.mce.2011.08.008) [DOI] [PubMed] [Google Scholar]
- Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, Yoneda T, Mohammad KS, Plotkin LI, Roodman GD, et al. 2016. Bidirectional notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Research 76 1089–1100. ( 10.1158/0008-5472.CAN-15-1703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Calle J & Bellido T 2022. The osteocyte as a signaling cell. Physiological Reviews 102 379–410. ( 10.1152/physrev.00043.2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetić K, Müller R, Bilezikian J, et al. 2001. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. Journal of Bone and Mineral Research 16 1846–1853. ( 10.1359/jbmr.2001.16.10.1846) [DOI] [PubMed] [Google Scholar]
- Desai MM, Cacciamani GE, Gill K, Zhang J, Liu L, Abreu A & Gill IS 2022. Trends in incidence of metastatic prostate cancer in the US. JAMA Network Open 5 e222246. ( 10.1001/jamanetworkopen.2022.2246) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, Baron R, Rosen CJ & Bouxsein ML 2010. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. Journal of Bone and Mineral Research 25 2078–2088. ( 10.1002/jbmr.82) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diegel CR, Hann S, Ayturk UM, Hu JCW, Lim KE, Droscha CJ, Madaj ZB, Foxa GE, Izaguirre I, Transgenics Core VVA, et al. 2020. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genetics 16 e1008361. ( 10.1371/journal.pgen.1008361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitroff CJ, Lechpammer M, Long-Woodward D & Kutok JL 2004. Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelium under shear flow is mediated by E-selectin. Cancer Research 64 5261–5269. ( 10.1158/0008-5472.CAN-04-0691) [DOI] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G & Morrison SJ 2012. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481 457–462. ( 10.1038/nature10783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnig H & Turner RT 1995. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 136 3632–3638. ( 10.1210/endo.136.8.7628403) [DOI] [PubMed] [Google Scholar]
- Dolgalev I & Tikhonova AN 2021. Connecting the dots: resolving the bone marrow niche heterogeneity. Frontiers in Cell and Developmental Biology 9 622519. ( 10.3389/fcell.2021.622519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue HJ, Friendman MJ & Genetos D 2020. Osteocyte mechanobiology in aging and disease. In Mechanobiology: From Molecular Sensing to Disease, pp. 1–21. Philadelphia, PA, USA: Elsevier. ( 10.1016/B978-0-12-817931-4.00001-7) [DOI] [Google Scholar]
- Edmondson R, Adcock AF & Yang L 2016. Influence of matrices on 3D-cultured prostate cancer cells’ drug response and expression of drug-action associated proteins. PLoS One 11 e0158116. ( 10.1371/journal.pone.0158116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engl T, Relja B, Marian D, Blumenberg C, Müller I, Beecken WD, Jones J, Ringel EM, Bereiter-Hahn J, Jonas D, et al. 2006. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia (New York, NY) 8 290–301. ( 10.1593/neo.05694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, et al. 2006. Loss of DMP1 causes rickets and osteomalacia and identifies a role of osteocytes in mineral metabolism. Nature Genetics 38 1310–1315. ( 10.1038/ng1905) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X & McDonald JM 2011. Disorders of bone remodeling. Annual Review of Pathology 6 121–145. ( 10.1146/annurev-pathol-011110-130203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Liu S, Zha R, Sun X, Li K, Robling A, Li B & Yokota H 2021. Mechanical loading-driven tumor suppression is mediated by Lrp5-dependent and independent mechanisms. Cancers 13 1–17. ( 10.3390/cancers13020267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong EL, Harrington DA, Farach-Carson MC & Yu H 2016a. Heralding a new paradigm in 3D tumor modeling. Biomaterials 108 197–213. ( 10.1016/j.biomaterials.2016.08.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong EL, Wan X, Yang J, Morgado M, Mikos AG, Harrington DA, Navone NM & Farach-Carson MC 2016b. A 3D in vitro model of patient-derived prostate cancer xenograft for controlled interrogation of in vivo tumor-stromal interactions. Biomaterials 77 164–172. ( 10.1016/j.biomaterials.2015.10.059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foradori CD, Weiser MJ & Handa RJ 2008. Non-genomic actions of androgens. Frontiers in Neuroendocrinology 29 169–181. ( 10.1016/j.yfrne.2007.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikha-Benayed D, Basta-Pljakic J, Majeska RJ & Schaffler MB 2016. Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes. Bone 90 15–22. ( 10.1016/j.bone.2016.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost HM 2000. Muscle Bone and the Utah Paradigm: a 1999 overview. Medicine and Science in Sports and Exercise 32 911–917. ( 10.1097/00005768-200005000-00006) [DOI] [PubMed] [Google Scholar]
- Gandaglia G, Karakiewicz PI, Briganti A, Passoni NM, Schiffmann J, Trudeau V, Graefen M, Montorsi F & Sun M 2015. Impact of the site of metastases on survival in patients with metastatic prostate cancer. European Urology 68 325–334. ( 10.1016/j.eururo.2014.07.020) [DOI] [PubMed] [Google Scholar]
- Garnero P, Buchs N, Zekri J, Rizzoli R, Coleman RE & Delmas PD 2000. Markers of bone turnover for the management of patients with bone metastases from prostate cancer. British Journal of Cancer 82 858–864. ( 10.1054/bjoc.1999.1012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Xue C, Mendonca J, Sun XX, Liu Q, Reardon PN, Chen Y, Qian K, Hua V, Chen A, et al. 2018. Interplay between hypoxia and androgen controls a metabolic switch conferring resistance to androgen/AR-targeted therapy. Nature Communications 9 4972. ( 10.1038/s41467-018-07411-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- González Díaz EC, Sinha S, Avedian RS & Yang F 2019. Tissue-engineered 3D models for elucidating primary and metastatic bone cancer progression. Acta Biomaterialia 99 18–32. ( 10.1016/j.actbio.2019.08.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, Yoneda T & Mundy GR 1996. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. Journal of Clinical Investigation 98 1544–1549. ( 10.1172/JCI118947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbit NL, Anbiah B, Anderson L, Suresh J, Hassani I, Eggert M, Brannen A, Davis J, Tian Y, Prabhakarpandian B, et al. 2022. Tunable three-dimensional engineered prostate cancer tissues for in vitro recapitulation of heterogeneous in vivo prostate tumor stiffness. Acta Biomaterialia 147 73–90. ( 10.1016/j.actbio.2022.05.011) [DOI] [PubMed] [Google Scholar]
- Hahn AW, Higano CS, Taplin ME, Ryan CJ & Agarwal N 2018. Metastatic castration-sensitive prostate cancer: optimizing patient selection and treatment. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting 38 363–371. ( 10.1200/EDBK_200967) [DOI] [PubMed] [Google Scholar]
- Hainline KM, Gu F, Handley JF, Tian YF, Wu Y, de Wet L, vander Griend DJ & Collier JH 2019. Self-assembling peptide gels for 3D prostate cancer spheroid culture. Macromolecular Bioscience 19 e1800249. ( 10.1002/mabi.201800249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, Tangen CM, Rosenthal M, Petrylak DP, Hussain M, et al. 2016. Meta-analysis evaluating the impact of site of metastasis on overall survival in men with castration-resistant prostate cancer. Journal of Clinical Oncology 34 1652–1659. ( 10.1200/JCO.2015.65.7270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CL, Daignault SD, Shah RB, Pienta KJ & Keller ET 2008. Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate 68 1396–1404. ( 10.1002/pros.20805) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CL, Kang S, MacDougald OA & Keller ET 2006. Role of Wnts in prostate cancer bone metastases. Journal of Cellular Biochemistry 97 661–672. ( 10.1002/jcb.20735) [DOI] [PubMed] [Google Scholar]
- Haq M, Goltzman D, Tremblay G & Brodt P 1992. Rat prostate adenocarcinoma cells disseminate to bone and adhere preferentially to bone marrow-derived endothelial cells. Cancer Research 52 4613–4619. [PubMed] [Google Scholar]
- Hardaway AL, Herroon MK, Rajagurubandara E & Podgorski I 2014. Bone marrow fat: linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Reviews 33 527–543. ( 10.1007/s10555-013-9484-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart NH, Newton RU, Tan J, Rantalainen T, Chivers P, Siafarikas A & Nimphius S 2020. Biological basis of bone strength: anatomy, physiology and measurement. Journal of Musculoskeletal and Neuronal Interactions 20 347–371. [PMC free article] [PubMed] [Google Scholar]
- Hauge EM, Qvesel D, Eriksen EF, Mosekilde L & Melsen F 2001. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. Journal of Bone and Mineral Research 16 1575–1582. ( 10.1359/jbmr.2001.16.9.1575) [DOI] [PubMed] [Google Scholar]
- Hayes AR, Brungs D & Pavlakis N 2018. Osteoclast inhibitors to prevent bone metastases in men with high-risk, nonmetastatic prostate cancer: a systematic review and meta-analysis. PLoS One 13 e0191455. ( 10.1371/journal.pone.0191455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemers HV v. & Tindall DJ 2007. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrine Reviews 28 778–808. ( 10.1210/er.2007-0019) [DOI] [PubMed] [Google Scholar]
- Hemmatian H, Conrad S, Furesi G, Mletzko K, Krug J, Faila AV, Kuhlmann JD, Rauner M, Busse B & Jähn-Rickert K 2021. Reorganization of the osteocyte lacuno-canalicular network characteristics in tumor sites of an immunocompetent murine model of osteotropic cancers. Bone 152 116074. ( 10.1016/j.bone.2021.116074) [DOI] [PubMed] [Google Scholar]
- Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A & Wikström P 2011. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 6 e19059. ( 10.1371/journal.pone.0019059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotte SJ & Saad F 2010. Current management of castrate-resistant prostate cancer. Current Oncology 17 S72–S79. ( 10.3747/co.v17i0.718) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Nguyen P, Mao J, Halpern J, Shoag J, Wright JD & Sedrakyan A 2017a. Increase in prostate cancer distant metastases at diagnosis in the United States. JAMA Oncology 3 705–707. ( 10.1001/jamaoncol.2016.5465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Wang G & Sun T 2017b. Dissecting the roles of the androgen receptor in prostate cancer from molecular perspectives. Tumour Biology 39 1010428317692259. ( 10.1177/1010428317692259) [DOI] [PubMed] [Google Scholar]
- Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC & Tang CH 2009. CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochemical Pharmacology 77 794–803. ( 10.1016/j.bcp.2008.11.014) [DOI] [PubMed] [Google Scholar]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR & Boyce BF 1996. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nature Medicine 2 1132–1136. ( 10.1038/nm1096-1132) [DOI] [PubMed] [Google Scholar]
- Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P & Amadori D 2010. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 116 1406–1418. ( 10.1002/cncr.24896) [DOI] [PubMed] [Google Scholar]
- Imbriaco M, Larson SM, Yeung HW, Mawlawi OR, Erdi Y, Venkatraman ES & Scher HI 1998. A new parameter for measuring metastatic bone involvement by prostate cancer: the bone scan index. Clinical Cancer Research 4 1765–1772. [PubMed] [Google Scholar]
- Iuliani M, Pantano F, Buttigliero C, Fioramonti M, Bertaglia V, Vincenzi B, Zoccoli A, Ribelli G, Tucci M, Vignani F, et al. 2015. Biological and clinical effects of abiraterone on anti-resorptive and anabolic activity in bone microenvironment. Oncotarget 6 12520–12528. ( 10.18632/oncotarget.3724) [DOI] [PMC free article] [PubMed] [Google Scholar]
- James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, Ritchie AWS, Amos CL, Gilson C, Jones RJ, et al. 2017. Abiraterone for prostate cancer not previously treated with hormone therapy. New England Journal of Medicine 377 338–351. ( 10.1056/NEJMoa1702900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jathal MK, Chen L, Mudryj M & Ghosh PM 2011. Targeting ErbB3: the new RTK (id) on the prostate cancer block. Immunology, Endocrine and Metabolic Agents in Medicinal Chemistry 11 131–149. ( 10.2174/187152211795495643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jathal MK, Steele TM, Siddiqui S, Mooso BA, D’Abronzo LS, Drake CM, Whang YE & Ghosh PM 2019. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. British Journal of Cancer 121 237–248. ( 10.1038/s41416-019-0496-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka RL 2007. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40 1434–1446. ( 10.1016/j.bone.2007.03.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka RL, O’Brien CA, Ali AA, Roberson PK, Weinstein RS & Manolagas SC 2009. Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone 44 275–286. ( 10.1016/j.bone.2008.10.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Wang J, Lee E, McGee S, Berry JE, Yumoto K, Dai J, Keller ET, Shiozawa Y & Taichman RS 2015. Annexin 2-CXCL12 interactions regulate metastatic cell targeting and growth in the bone marrow. Molecular Cancer Research 13 197–207. ( 10.1158/1541-7786.MCR-14-0118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Wang J, Song J, Shiozawa Y, Wang J, Havens A, Wang Z, Sun YX, Emerson SG, Krebsbach PH, et al. 2007. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 110 82–90. ( 10.1182/blood-2006-05-021352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalikin LM, Schneider A, Thakur MA, Fridman Y, Griffin LB, Dunn RL, Rosol TJ, Shah RB, Rehemtulla A, McCauley LK, et al. 2003. In vivo visualization of metastatic prostate cancer and quantitation of disease progression in immunocompromised mice. Cancer Biology and Therapy 2 656–660. ( 10.4161/cbt.2.6.531) [DOI] [PubMed] [Google Scholar]
- Kan C, Vargas G, Pape FL & Clézardin P 2016. Cancer cell colonisation in the bone microenvironment. International Journal of Molecular Sciences 17 1674. ( 10.3390/ijms17101674) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T & Yokosuka O 2015. The androgen receptor as an emerging target in hepatocellular carcinoma. Journal of Hepatocellular Carcinoma 2 91–99. ( 10.2147/JHC.S48956) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson SA, Studer E, Kettunen P & Westberg L 2016. Neural androgen receptors modulate gene expression and social recognition but not social investigation. Frontiers in Behavioral Neuroscience 10 41. ( 10.3389/fnbeh.2016.00041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson T, Sundar R, Widmark A, Landström M & Persson E 2018. Osteoblast-derived factors promote metastatic potential in human prostate cancer cells, in part via non-canonical transforming growth factor β (TGFβ) signaling. Prostate 78 446–456. ( 10.1002/pros.23489) [DOI] [PubMed] [Google Scholar]
- Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, Nakamura T, Fukuda T, Yoshimura K, Yoshizawa T, et al. 2003. Suppressive function of androgen receptor in bone resorption. Proceedings of the National Academy of Sciences of the United States of America 100 9416–9421. ( 10.1073/pnas.1533500100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, Oursler MJ & Monroe DG 2012. Estrogen and the skeleton. Trends in Endocrinology and Metabolism: TEM 23 576–581. ( 10.1016/j.tem.2012.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Lee JY, Kim KJ, Hong N, Kim JW, Hah YS, Koo KC, Kim JH & Cho KS 2019. Effect of androgen-deprivation therapy on bone mineral density in patients with prostate cancer: a systematic review and meta-analysis. Journal of Clinical Medicine 8. ( 10.3390/jcm8010113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Lu Y, Williams EA, Lai F, Lee JY, Enishi T, Balani DH, Ominsky MS, Ke HZ, Kronenberg HM, et al. 2017. Sclerostin antibody administration converts bone lining cells into active osteoblasts. Journal of Bone and Mineral Research 32 892–901. ( 10.1002/jbmr.3038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Matsugaki A, Sekita A & Nakano T 2017. Alteration of osteoblast arrangement via direct attack by cancer cells: new insights into bone metastasis. Scientific Reports 7 44824. ( 10.1038/srep44824) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley LA, Fournier PGJ, Chirgwin JM & Guise TA 2007. Molecular biology of bone metastasis. Molecular Cancer Therapeutics 6 2609–2617. ( 10.1158/1535-7163.MCT-07-0234) [DOI] [PubMed] [Google Scholar]
- Klüppel M & Wrana JL 2005. Turning it up a Notch: cross-talk between TGF beta and Notch signaling. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology 27 115–118. ( 10.1002/bies.20187) [DOI] [PubMed] [Google Scholar]
- Komiya Y & Habas R 2008. Wnt signal transduction pathways. Organogenesis 4 68–75. ( 10.4161/org.4.2.5851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh S, Inoue K, Igarashi K, Sugizaki H, Shirode-Fukuda Y, Inoue E, Yu T, Takeuchi JK, Kanno J, Bonewald LF, et al. 2014. Estrogen receptor α in osteocytes regulates trabecular bone formation in female mice. Bone 60 68–77. ( 10.1016/j.bone.2013.12.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsilieris M, Sourla A, Pelletier G & Doillon CJ 1994. Three-dimensional type I collagen gel system for the study of osteoblastic metastases produced by metastatic prostate cancer. Journal of Bone and Mineral Research 9 1823–1832. ( 10.1002/jbmr.5650091120) [DOI] [PubMed] [Google Scholar]
- Krishnan V, Bryant HU & Macdougald OA 2006. Regulation of bone mass by Wnt signaling. Journal of Clinical Investigation 116 1202–1209. ( 10.1172/JCI28551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK & Adams RH 2014. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507 323–328. ( 10.1038/nature13145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T, Jeong IG, Park M, You D, Lee J, Kim HK, Hong S, Hong JH, Ahn H & Kim CS 2014. Bone mineral density in prostate cancer: a comparative study of patients with prostate cancer and healthy controls using propensity score matching. Urology 83 385–392. ( 10.1016/j.urology.2013.08.045) [DOI] [PubMed] [Google Scholar]
- Kyriakopoulos CE, Chen YH, Carducci MA, Liu G, Jarrard DF, Hahn NM, Shevrin DH, Dreicer R, Hussain M, Eisenberger M, et al. 2018. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: long-term survival analysis of the randomized Phase III E3805 CHAARTED trial. Journal of Clinical Oncology 36 1080–1087. ( 10.1200/JCO.2017.75.3657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen UH, Pitulescu ME, Kim JM, Enriquez-Gasca R, Sivaraj KK, Kusumbe AP, Singh A, di Russo J, Bixel MG, Zhou B, et al. 2017. 2017 cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nature Cell Biology 19 189–201. ( 10.1038/ncb3476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauretani F, Bandinelli S, Griswold ME, Maggio M, Semba R, Guralnik JM & Ferrucci L 2008. Longitudinal changes in BMD and bone geometry in a population-based study. Journal of Bone and Mineral Research 23 400–408. ( 10.1359/jbmr.071103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazennec G & Richmond A 2010. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends in Molecular Medicine 16 133–144. ( 10.1016/j.molmed.2010.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GT, Jung YS, Ha YS, Kim JH, Kim WJ & Kim IY 2013. Bone morphogenetic protein-6 induces castration resistance in prostate cancer cells through tumor infiltrating macrophages. Cancer Science 104 1027–1032. ( 10.1111/cas.12206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Sowa H, Hinoi E, Ferron M, Deok Ahn JD, Confavreux C, Dacquin R, Mee PJ, Mckee MD, Jung DY, et al. 2007. Endocrine regulation of energy metabolism by the skeleton. Cell 130 456–469. ( 10.1016/j.cell.2007.05.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr JE & Pienta KJ 1998. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. Journal of the National Cancer Institute 90 118–123. ( 10.1093/jnci/90.2.118) [DOI] [PubMed] [Google Scholar]
- Leight JL, Drain AP & Weaver VM 2017. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annual Review of Cancer Biology 1 313–334. ( 10.1146/annurev-cancerbio-050216-034431) [DOI] [Google Scholar]
- Leung JK & Sadar MD 2017. Non-genomic actions of the androgen receptor in prostate cancer. Frontiers in Endocrinology 8 2. ( 10.3389/fendo.2017.00002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bagchi DP, Zhu J, Bowers E, Yu H, Hardij J, Mori H, Granger K, Skjaerlund J, Mandair G, et al. 2022. Constitutive bone marrow adipocytes suppress local bone formation. JCI Insight 7 e160915. ( 10.1172/jci.insight.160915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey S & Langhans SA 2014. Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition. Frontiers in Oncology 4 358. ( 10.3389/fonc.2014.00358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu D, Sun X, Fan Y, Zha R, Jalali A, Feng Y, Li K, Sano T, Vike N, et al. 2021. Overexpression of Lrp5 enhanced the anti-breast cancer effects of osteocytes in bone. Bone Research 9 32. ( 10.1038/s41413-021-00152-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis C, Morris MJ, Den R & Coleman RE 2018. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Reviews 37 189–196. ( 10.1007/s10555-017-9719-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis CJ & Lin SH 2005. Osteoblasts in prostate cancer metastasis to bone. Nature Reviews. Cancer 5 21–28. ( 10.1038/nrc1528) [DOI] [PubMed] [Google Scholar]
- Lojk J & Marc J 2021. Roles of non-canonical Wnt signalling pathways in bone biology. International Journal of Molecular Sciences 22 10840. ( 10.3390/ijms221910840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YV v., Lam C, Dalmia S, Gao P, Young J, Middleton K, Liu C, Xu H & You L 2018. Mechanical regulation of breast cancer migration and apoptosis via direct and indirect osteocyte signaling. Journal of Cellular Biochemistry 119 5665–5675. ( 10.1002/jcb.26745) [DOI] [PubMed] [Google Scholar]
- Ma YV, Xu L, Mei X, Middleton K & You L 2019. Mechanically stimulated osteocytes reduce the bone-metastatic potential of breast cancer cells in vitro by signaling through endothelial cells. Journal of Cellular Biochemistry 120 7590–7601. ( 10.1002/jcb.28034) [DOI] [PubMed] [Google Scholar]
- Määttä JA, Büki KG, Gu G, Alanne MH, Vääräniemi J, Liljenbäck H, Poutanen M, Härkönen P & Väänänen K 2013a. Inactivation of estrogen receptor in bone-forming cells induces bone loss in female mice. FASEB Journal 27 478–488. ( 10.1096/fj.12-213587) [DOI] [PubMed] [Google Scholar]
- Määttä JA, Büki KG, Ivaska KK, Nieminen-Pihala V, Elo TD, Kähkönen T, Poutanen M, Härkönen P & Väänänen K 2013b. Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. BoneKEy Reports 2 440. ( 10.1038/bonekey.2013.174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, et al. 2012. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nature Medicine 18 405–412. ( 10.1038/nm.2653) [DOI] [PubMed] [Google Scholar]
- Manolagas SC, O’Brien CA & Almeida M 2013. The role of estrogen and androgen receptors in bone health and disease. Nature Reviews. Endocrinology 9 699–712. ( 10.1038/nrendo.2013.179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Arm J, Smart J, Palazzi K, Capp A, Ainsworth P & Cowin G 2017. Spinal multiparametric MRI and DEXA changes over time in men with prostate cancer treated with androgen deprivation therapy: a potential imaging biomarker of treatment toxicity. European Radiology 27 995–1003. ( 10.1007/s00330-016-4434-z) [DOI] [PubMed] [Google Scholar]
- Martin RB 2003. Fatigue microdamage as an essential element of bone mechanics and biology. Calcified Tissue International 73 101–107. ( 10.1007/s00223-002-1059-9) [DOI] [PubMed] [Google Scholar]
- Martin RB, Burr DB & Sharkey NA 1998. Mechanical properties of bone. In Skeletal Tissue Mechanics, pp. 127–180. Eds. Martin RB, Burr DB & Sharkey NA. New York, NY, USA: Springer. ( 10.1007/978-1-4757-2968-9_4) [DOI] [Google Scholar]
- Martin TJ 2002. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. Journal of Clinical Investigation 110 1399–1401. ( 10.1172/JCI17124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, Jilka RL, O’Brien CA & Manolagas SC 2010. The estrogen receptor-α in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Molecular Endocrinology 24 323–334. ( 10.1210/me.2009-0354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally CJ, Ruddock MW, Moore T & McKenna DJ 2020. Biomarkers that differentiate benign prostatic hyperplasia from prostate cancer: a literature review. Cancer Management and Research 12 5225–5241. ( 10.2147/CMAR.S250829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville KM, Kelly NH, Khan SA, Schimenti JC, Ross FP, Main RP & Van Der Meulen MCH 2014. Female mice lacking estrogen receptor-alpha in osteoblasts have compromised bone mass and strength. Journal of Bone and Mineral Research 29 370–379. ( 10.1002/jbmr.2082) [DOI] [PubMed] [Google Scholar]
- Messner EA, Steele TM, Tsamouri MM, Hejazi N, Gao AC, Mudryj M & Ghosh PM 2020a. The androgen receptor in prostate cancer: effect of structure, ligands and spliced variants on therapy. Biomedicines 8 422. ( 10.3390/biomedicines8100422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner EA, Steele TM, Tsamouri MM, Hejazi N, Gao AC, Mudryj M & Ghosh PM 2020b. The androgen receptor in prostate cancer: effect of structure, ligands and spliced variants on therapy. Biomedicines 8 1–19. ( 10.3390/biomedicines8100422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani T, Yamaji R, Higashimura Y, Harada N, Nakano Y & Inui H 2011. Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1α in a low androgen environment. Journal of Steroid Biochemistry and Molecular Biology 123 58–64. ( 10.1016/j.jsbmb.2010.10.009) [DOI] [PubMed] [Google Scholar]
- Monroe DG, McGee-Lawrence ME, Oursler MJ & Westendorf JJ 2012. Update on Wnt signaling in bone cell biology and bone disease. Gene 492 1–18. ( 10.1016/j.gene.2011.10.044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira CA, Dempster DW & Baron R 2000. Anatomy and ultrastructure of bone – histogenesis, growth and remodeling. In KR Feingold, et al. Endotext. South Dartmouth, MA, USA: MDText.com. [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, et al. 2007. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130 811–823. ( 10.1016/j.cell.2007.07.025) [DOI] [PubMed] [Google Scholar]
- Nemeth JA, Yousif R, Herzog M, Che M, Upadhyay J, Shekarriz B, Bhagat S, Mullins C, Fridman R & Cher ML 2002. Matrix metalloproteinase activity, bone matrix turnover, and tumor cell proliferation in prostate cancer bone metastasis. Journal of the National Cancer Institute 94 17–25. ( 10.1093/jnci/94.1.17) [DOI] [PubMed] [Google Scholar]
- Netti PA, Berk DA, Swartz MA, Grodzinsky AJ & Jain RK 2000. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research 60 2497–2503. [PubMed] [Google Scholar]
- Nørgaard M, Jensen AØ, Jacobsen JB, Cetin K, Fryzek JP & Sørensen HT 2010. Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark (1999 to 2007). Journal of Urology 184 162–167. ( 10.1016/j.juro.2010.03.034) [DOI] [PubMed] [Google Scholar]
- Notini AJ, McManus JF, Moore A, Bouxsein M, Jimenez M, Chiu WSM, Glatt V, Kream BE, Handelsman DJ, Morris HA, et al. 2007. Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. Journal of Bone and Mineral Research 22 347–356. ( 10.1359/jbmr.061117) [DOI] [PubMed] [Google Scholar]
- Otley MOC & Sinal CJ 2022. Adipocyte-cancer cell interactions in the bone microenvironment. Frontiers in Endocrinology 13 903925. ( 10.3389/fendo.2022.903925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottewell PD, Wang N, Meek J, Fowles CA, Croucher PI, Eaton CL & Holen I 2014. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocrine-Related Cancer 21 769–781. ( 10.1530/ERC-14-0199) [DOI] [PubMed] [Google Scholar]
- Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, Hermo L, Suarez S, Roth BL, Ducy P, et al. 2011. Endocrine regulation of male fertility by the skeleton. Cell 144 796–809. ( 10.1016/j.cell.2011.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozair MZ, Kintner C & Brivanlou AH 2013. Neural induction and early patterning in vertebrates. Wiley Interdisciplinary Reviews. Developmental Biology 2 479–498. ( 10.1002/wdev.90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pead MJ, Skerry TM & Lanyon LE 1988. Direct transformation from quiescence to bone formation in the adult periosteum following a single brief period of bone loading. Journal of Bone and Mineral Research 3 647–656. ( 10.1002/jbmr.5650030610) [DOI] [PubMed] [Google Scholar]
- Pelger RC, Hamdy NA, Zwinderman AH, Lycklama à Nijeholt AA & Papapoulos SE 1998. Effects of the bisphosphonate olpadronate in patients with carcinoma of the prostate metastatic to the skeleton. Bone 22 403–408. ( 10.1016/s8756-3282(97)00289-5) [DOI] [PubMed] [Google Scholar]
- Peng Y, Wu S, Li Y & Crane JL 2020. Type H blood vessels in bone modeling and remodeling. Theranostics 10 426–436. ( 10.7150/thno.34126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival RC, Urwin GH, Harris S, Yates AJ, Williams JL, Beneton M & Kanis JA 1987. Biochemical and histological evidence that carcinoma of the prostate is associated with increased bone resorption. European Journal of Surgical Oncology: the Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology 13 41–49. [PubMed] [Google Scholar]
- Pfeilschifter J & Mundy GR 1987. Modulation of type beta transforming growth factor activity in bone cultures by osteotropic hormones. PNAS 84 2024–2028. ( 10.1073/pnas.84.7.2024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy SK, Kusumbe AP, Schiller M, Zeuschner D, Bixel MG, Milia C, Gamrekelashvili J, Limbourg A, Medvinsky A, Santoro MM, et al. 2016. Blood flow controls bone vascular function and osteogenesis. Nature Communications 7 13601. ( 10.1038/ncomms13601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy SK, Kusumbe AP, Wang L & Adams RH 2014. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507 376–380. ( 10.1038/nature13146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regard JB, Zhong Z, Williams BO & Yang Y 2012. Wnt signaling in bone development and disease: making stronger bone with Wnts. Cold Spring Harbor Perspectives in Biology 4 a007997. ( 10.1101/cshperspect.a007997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid IR 2022. EXTENSIVE EXPERTISE IN ENDOCRINOLOGY: osteoporosis management. European Journal of Endocrinology 187 R65–R80. ( 10.1530/EJE-22-0574) [DOI] [PubMed] [Google Scholar]
- Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, Huang S, Chen X, Lai Y, Du H, et al. 2019. Wnt5a induces and maintains prostate cancer cells dormancy in bone. Journal of Experimental Medicine 216 428–449. ( 10.1084/jem.20180661) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegman PHJ, Vlietstra RJ, van der Korput JAGM, Brinkmann AO & Trapman J 1991. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Molecular Endocrinology 5 1921–1930. ( 10.1210/mend-5-12-1921) [DOI] [PubMed] [Google Scholar]
- Robling AG 2012. The interaction of biological factors with mechanical signals in bone adaptation: recent developments. Current Osteoporosis Reports 10 126–131. ( 10.1007/s11914-012-0099-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robling AG, Castillo AB & Turner CH 2006. Biomechanical and molecular regulation of bone remodeling. Annual Review of Biomedical Engineering 8 455–498. ( 10.1146/annurev.bioeng.8.061505.095721) [DOI] [PubMed] [Google Scholar]
- Romanov VI, Whyard T, Adler HL, Waltzer WC & Zucker S 2004. Prostate cancer cell adhesion to bone marrow endothelium: the role of prostate-specific antigen. Cancer Research 64 2083–2089. ( 10.1158/0008-5472.can-03-3487) [DOI] [PubMed] [Google Scholar]
- Roodman GD 2004. Mechanisms of bone metastasis. New England Journal of Medicine 350 1655–1664. ( 10.1056/NEJMra030831) [DOI] [PubMed] [Google Scholar]
- Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL & Ott SM 2008. Histopathological assessment of prostate cancer bone osteoblastic metastases. Journal of Urology 180 1154–1160. ( 10.1016/j.juro.2008.04.140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roviello G, Catalano M, Ottanelli C, Giorgione R, Rossi V, Gambale E, Casadei C, De Giorgi U & Antonuzzo L 2022. Castration-resistant prostate cancer with bone metastases: toward the best therapeutic choice. Medical Oncology 39 145. ( 10.1007/s12032-022-01739-3) [DOI] [PubMed] [Google Scholar]
- Russell MR, Jamieson WL, Dolloff NG & Fatatis A 2009. The α-receptor for platelet-derived growth factor as a target for antibody-mediated inhibition of skeletal metastases from prostate cancer cells. Oncogene 28 412–421. ( 10.1038/onc.2008.390) [DOI] [PubMed] [Google Scholar]
- Sabol HM, Amorim T, Ashby C, Halladay D, Anderson J, Cregor M, Sweet M, Nookaew I, Kurihara N, Roodman GD, et al. 2022. Notch3 signaling between myeloma cells and osteocytes in the tumor niche promotes tumor growth and bone destruction. Neoplasia 28 100785. ( 10.1016/j.neo.2022.100785) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabol HM, Ferrari AJ, Adhikari M, Amorim T, McAndrews K, Anderson J, Vigolo M, Lehal R, Cregor M, Khan S, et al. 2021. Targeting notch inhibitors to the myeloma bone marrow niche decreases tumor growth and bone destruction without gut toxicity. Cancer Research 81 5102–5114. ( 10.1158/0008-5472.CAN-21-0524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter G, Steurer S, Clauditz TS, Krech T, Wittmer C, Lutz F, Lennartz M, Janssen T, Hakimi N, Simon R, et al. 2016. Clinical utility of quantitative Gleason grading in prostate biopsies and prostatectomy specimens. European Urology 69 592–598. ( 10.1016/j.eururo.2015.10.029) [DOI] [PubMed] [Google Scholar]
- Sayegh N, Swami U & Agarwal N 2022. Recent advances in the management of metastatic prostate cancer. JCO Oncology Practice 18 45–55. ( 10.1200/OP.21.00206) [DOI] [PubMed] [Google Scholar]
- Schneider A, Kalikin LM, Mattos AC, Keller ET, Allen MJ, Pienta KJ & McCauley LK 2005. Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology 146 1727–1736. ( 10.1210/en.2004-1211) [DOI] [PubMed] [Google Scholar]
- Scott LJ, Clarke NW, George NJ, Shanks JH, Testa NG & Lang SH 2001. Interactions of human prostatic epithelial cells with bone marrow endothelium: binding and invasion. British Journal of Cancer 84 1417–1423. ( 10.1054/bjoc.2001.1804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian VB, Kuo YF, Freeman JL & Goodwin JS 2005. Risk of fracture after androgen deprivation for prostate cancer. New England Journal of Medicine 352 154–164. ( 10.1056/NEJMoa041943) [DOI] [PubMed] [Google Scholar]
- Sharifi M, Ereifej L & Lewiecki EM 2015. Sclerostin and skeletal health. Reviews in Endocrine and Metabolic Disorders 16 149–156. ( 10.1007/s11154-015-9311-6) [DOI] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, et al. 2011. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. Journal of Clinical Investigation 121 1298–1312. ( 10.1172/JCI43414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulby SA, Dolloff NG, Stearns ME, Meucci O & Fatatis A 2004. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Research 64 4693–4698. ( 10.1158/0008-5472.CAN-03-3437) [DOI] [PubMed] [Google Scholar]
- Siddiqui JA, Seshacharyulu P, Muniyan S, Pothuraju R, Khan P, Vengoji R, Chaudhary S, Maurya SK, Lele SM, Jain M, et al. 2022a. GDF15 promotes prostate cancer bone metastasis and colonization through osteoblastic CCL2 and RANKL activation. Bone Research 10 6. ( 10.1038/s41413-021-00178-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui JA, Pothuraju R, Khan P, Sharma G, Muniyan S, Seshacharyulu P, Jain M, Nasser MW & Batra SK 2022b. Pathophysiological role of growth differentiation factor 15 (GDF15) in obesity, cancer, and cachexia. Cytokine and Growth Factor Reviews 64 71–83. ( 10.1016/j.cytogfr.2021.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Wagle NS & Jemal A 2023. Cancer statistics, 2023. CA: a Cancer Journal for Clinicians 73 17–48. ( 10.3322/caac.21763) [DOI] [PubMed] [Google Scholar]
- Sieh S, Taubenberger AV v., Lehman ML, Clements JA, Nelson CC & Hutmacher DW 2014. Paracrine interactions between LNCaP prostate cancer cells and bioengineered bone in 3D in vitro culture reflect molecular changes during bone metastasis. Bone 63 121–131. ( 10.1016/j.bone.2014.02.001) [DOI] [PubMed] [Google Scholar]
- Sieh S, Taubenberger AV v., Rizzi SC, Sadowski M, Lehman ML, Rockstroh A, An J, Clements JA, Nelson CC& Hutmacher DW 2012. Phenotypic characterization of prostate cancer LNCaP cells cultured within a bioengineered microenvironment. PLoS One 7 e40217. ( 10.1371/journal.pone.0040217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnesael M, Claessens F, Laurent M, Dubois V, Boonen S, Deboel L & Vanderschueren D 2012. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: evidence from targeted AR disruption in mouse osteocytes. Journal of Bone and Mineral Research 27 2535–2543. ( 10.1002/jbmr.1713) [DOI] [PubMed] [Google Scholar]
- Sitarski AM, Fairfield H, Falank C & Reagan MR 2018. 3D tissue engineered in vitro models of cancer in bone. ACS Biomaterials Science and Engineering 4 324–336. ( 10.1021/acsbiomaterials.7b00097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, McGovern FJ, Zietman AL, Fallon MA, Hayden DL, Schoenfeld DA, Kantoff PW & Finkelstein JS 2001. Pamidronate to prevent bone loss during androgen-deprivation therapy for prostate cancer. New England Journal of Medicine 345 948–955. ( 10.1056/NEJMoa010845) [DOI] [PubMed] [Google Scholar]
- Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, Olmos D, Mainwaring PN, Lee JY, Uemura H, et al. 2018. Apalutamide treatment and metastasis-free survival in prostate cancer. New England Journal of Medicine 378 1408–1418. ( 10.1056/NEJMoa1715546) [DOI] [PubMed] [Google Scholar]
- Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, Miller K, Sieber P, Karsh L, Damião R, et al. 2012. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet 379 39–46. ( 10.1016/S0140-6736(11)61226-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Andrews KS, Brooks D, Fedewa SA, Manassaram-Baptiste D, Saslow D & Wender RC 2019. Cancer screening in the United States, 2019: a review of current American Cancer Society guidelines and current issues in cancer screening. CA: a Cancer Journal for Clinicians 69 184–210. ( 10.3322/caac.21557) [DOI] [PubMed] [Google Scholar]
- Song K, Wang H, Krebs TL, Kim SJ & Danielpour D 2008. Androgenic control of transforming growth factor-β signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-β receptor II. Cancer Research 68 8173–8182. ( 10.1158/0008-5472.CAN-08-2290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottnik JL, Dai J, Zhang H, Campbell B & Keller ET 2015. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Research 75 2151–2158. ( 10.1158/0008-5472.CAN-14-2493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, et al. 2014. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508 269–273. ( 10.1038/nature13034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoch SA, Parker RA, Chen L, Bubley G, Ko YJ, Vincelette A & Greenspan SL 2001. Bone loss in men with prostate cancer treated with gonadotropin-releasing hormone agonists. Journal of Clinical Endocrinology and Metabolism 86 2787–2791. ( 10.1210/jcem.86.6.7558) [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M & Nagasawa T 2006. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25 977–988. ( 10.1016/j.immuni.2006.10.016) [DOI] [PubMed] [Google Scholar]
- Sun M, Choueiri TK, Hamnvik OPR, Preston MA, De Velasco G, Jiang W, Loeb S, Nguyen PL & Trinh QD 2016. Comparison of gonadotropin-releasing hormone agonists and orchiectomy: effects of androgen-deprivation therapy. JAMA Oncology 2 500–507. ( 10.1001/jamaoncol.2015.4917) [DOI] [PubMed] [Google Scholar]
- Suva LJ, Griffin RJ & Makhoul I 2009. Mechanisms of bone metastases of breast cancer. Endocrine-Related Cancer 16 703–713. ( 10.1677/ERC-09-0012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva LJ, Washam C, Nicholas RW & Griffin RJ 2011. Bone metastasis: mechanisms and therapeutic opportunities. Nature Reviews. Endocrinology 7 208–218. ( 10.1038/nrendo.2010.227) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong YN, Hahn N, Kohli M, Cooney MM, et al. 2015. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. New England Journal of Medicine 373 737–746. ( 10.1056/NEJMoa1503747) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. 2009. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325 612–616. ( 10.1126/science.1175202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T & Takei Y 2004. Morphogens, their identification and regulation. Development (Cambridge, England) 131 703–712. ( 10.1242/dev.01043) [DOI] [PubMed] [Google Scholar]
- Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS & Mccauley LK 2002 Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Research 62 1832. –1837. [PubMed] [Google Scholar]
- Talmage RV 1970. Morphological and physiological considerations in a new concept of calcium transport in bone. The American Journal of Anatomy 129 467–476. ( 10.1002/aja.1001290408) [DOI] [PubMed] [Google Scholar]
- Tam NW, Chung D, Baldwin SJ, Simmons JR, Xu L, Rainey JK, Dellaire G & Frampton JP 2020. Material properties of disulfide-crosslinked hyaluronic acid hydrogels influence prostate cancer cell growth and metabolism. Journal of Materials Chemistry. B 8 9718–9733. ( 10.1039/d0tb01570a) [DOI] [PubMed] [Google Scholar]
- Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S & Ikeda K 2007. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metabolism 5 464–475. ( 10.1016/j.cmet.2007.05.001) [DOI] [PubMed] [Google Scholar]
- Taubenberger AV v., Bray LJ, Haller B, Shaposhnykov A, Binner M, Freudenberg U, Guck J & Werner C 2016. 3D extracellular matrix interactions modulate tumour cell growth, invasion and angiogenesis in engineered tumour microenvironments. Acta Biomaterialia 36 73–85. ( 10.1016/j.actbio.2016.03.017) [DOI] [PubMed] [Google Scholar]
- Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, Pathak S & Chung LW 2000. LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate 44 91–103. ( 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l) [DOI] [PubMed] [Google Scholar]
- Thudi NK, Martin CK, Murahari S, Shu ST, Lanigan LG, Werbeck JL, Keller ET, McCauley LK, Pinzone JJ & Rosol TJ 2011. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate 71 615–625. ( 10.1002/pros.21277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucer S, Iyer S, Bartell SM, Martin-Millan M, Han L, Kim HN, Weinstein RS, Jilka RL, O’Brien CA, Almeida M, et al. 2015. The effects of androgens on murine cortical bone do not require AR or ERα signaling in osteoblasts and osteoclasts. Journal of Bone and Mineral Research 30 1138–1149. ( 10.1002/jbmr.2485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale CL, Burdett S, Rydzewska LHM, Albiges L, Clarke NW, Fisher D, Fizazi K, Gravis G, James ND, Mason MD, et al. 2016. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: a systematic review and meta-analyses of aggregate data. Lancet Oncology 17 243–256. ( 10.1016/S1470-2045(15)00489-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch MH, Gleissl TA, Blom AB, van den Berg WB, van Lent PL & van der Kraan PM 2016. Wnts talking with the TGF-β superfamily: WISPers about modulation of osteoarthritis. Rheumatology (Oxford, England) 55 1536–1547. ( 10.1093/rheumatology/kev402) [DOI] [PubMed] [Google Scholar]
- Vindrieux D, Escobar P & Lazennec G 2009. Emerging roles of chemokines in prostate cancer. Endocrine-Related Cancer 16 663–673. ( 10.1677/ERC-09-0109) [DOI] [PubMed] [Google Scholar]
- Wang H, Tian L, Liu J, Goldstein A, Bado I, Zhang W, Arenkiel BR, Li Z, Yang M, Du S, et al. 2018a. The osteogenic niche is a calcium reservoir of bone micrometastases and confers unexpected therapeutic vulnerability. Cancer Cell 34 823–839.e7. ( 10.1016/j.ccell.2018.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KR, Abraham JA, McCue P, Schiewer MJ, Bussard KM, Fatatis A, Languino L, Leiby B, O’Neill R, Li J, et al. 2018b. Characterization of a bone biorepository: comparison of bone metastases from breast, prostate, renal, lung cancers, and myeloma. Journal of Clinical Oncology 36 e24019. ( 10.1200/JCO.2018.36.15_suppl.e24019) [DOI] [Google Scholar]
- Wang N, Docherty FE, Brown HK, Reeves KJ, Fowles ACM, Ottewell PD, Dear TN, Holen I, Croucher PI & Eaton CL 2014. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models. Journal of Bone and Mineral Research 29 2688–2696. ( 10.1002/jbmr.2300) [DOI] [PubMed] [Google Scholar]
- Wang SW, Wu HH, Liu SC, Wang PC, Ou WC, Chou WY, Shen YS & Tang CH 2012. CCL5 and CCR5 interaction promotes cell motility in human osteosarcoma. PLoS One 7 e35101. ( 10.1371/journal.pone.0035101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kreisberg JI & Ghosh PM 2007. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Current Cancer Drug Targets 7 591–604. ( 10.2174/156800907781662248) [DOI] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA & McCauley LK 2011. Cancer to bone: a fatal attraction. Nature Reviews. Cancer 11 411–425. ( 10.1038/nrc3055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willette RN, Gu JL, Lysko PG, Anderson KM, Minehart H & Yue T-L 1999. BMP-2 gene expression and effects on human vascular smooth muscle cells. Journal of Vascular Research 36 120–125. ( 10.1159/000025634) [DOI] [PubMed] [Google Scholar]
- Wilson S, Qi J & Filipp FV v 2016. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Scientific Reports 6 32611. ( 10.1038/srep32611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windahl SH, Börjesson AE, Farman HH, Engdahl C, Movérare-Skrtic S, Sjögrena K, Lagerquist MK, Kindblom JM, Koskela A, Tuukkanen J, et al. 2013. Estrogen receptor-α in osteocytes is important for trabecular bone formation in male mice. PNAS 110 2294–2299. ( 10.1073/pnas.1220811110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiren KM, Zhang X, Chang C, Keenan E & Orwoll ES 1997. Transcriptional up-regulation of the human androgen receptor by androgen in bone cells. Endocrinology 138 2291–2300. ( 10.1210/endo.138.6.5163) [DOI] [PubMed] [Google Scholar]
- Wirth M, Tammela T, Cicalese V, Gomez Veiga F, Delaere K, Miller K, Tubaro A, Schulze M, Debruyne F, Huland H, et al. 2015. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: efficacy and safety results of the Zometa European study (ZEUS) European Urology 67 482–491. ( 10.1016/j.eururo.2014.02.014) [DOI] [PubMed] [Google Scholar]
- Wu J, Movérare-Skrtic S, Börjesson AE, Lagerquist MK, Sjögren K, Windahl SH, Koskela A, Grahnemo L, Islander U, Wilhelmson AS, et al. 2016. Enzalutamide reduces the bone mass in the axial but not the appendicular skeleton in male mice. Endocrinology 157 969–977. ( 10.1210/en.2015-1566) [DOI] [PubMed] [Google Scholar]
- Xu K, Ganapathy K, Andl T, Wang Z, Copland JA, Chakrabarti R & Florczyk SJ 2019. 3D porous chitosan-alginate scaffold stiffness promotes differential responses in prostate cancer cell lines. Biomaterials 217 119311. ( 10.1016/j.biomaterials.2019.119311) [DOI] [PubMed] [Google Scholar]
- Xu K, Wang Z, Copland JA, Chakrabarti R & Florczyk SJ 2020. 3D porous chitosan-chondroitin sulfate scaffolds promote epithelial to mesenchymal transition in prostate cancer cells. Biomaterials 254 120126. ( 10.1016/j.biomaterials.2020.120126) [DOI] [PubMed] [Google Scholar]
- Yaal-Hahoshen N, Shina S, Leider-Trejo L, Barnea I, Shabtai EL, Azenshtein E, Greenberg I, Keydar I & Ben-Baruch A 2006. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clinical Cancer Research 12 4474–4480. ( 10.1158/1078-0432.CCR-06-0074) [DOI] [PubMed] [Google Scholar]
- Yao W, Cheng Z, Busse C, Pham A, Nakamura MC & Lane NE 2008. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis and Rheumatism 58 1674–1686. ( 10.1002/art.23454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W, Dai W, Jiang JX & Lane NE 2013. Glucocorticoids and osteocyte autophagy. Bone 54 279–284. ( 10.1016/j.bone.2013.01.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh S, Tsai M-Y, Xu Q, Mu X-M, Lardy H, Huang K-E, Lin H, Yeh S-D, Altuwaijri S, Zhou X, et al. 2002. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. PNAS 99 13498–13503. ( 10.1073/pnas.212474399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi B, Williams PJ, Niewolna M, Wang Y & Yoneda T 2002. Tumor-derived platelet-derived growth factor-BB plays a critical role in osteosclerotic bone metastasis in an animal model of human breast cancer. Cancer Research 62 917–923. [PubMed] [Google Scholar]
- Yin JJ, Mohammad KS, Käkönen SM, Harris S, Wu-Wong JR, Wessale JL, Padley RJ, Garrett IR, Chirgwin JM & Guise TA 2003. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proceedings of the National Academy of Sciences of the United States of America 100 10954–10959. ( 10.1073/pnas.1830978100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling JM, Blanchard KL & Sawyer JS 2004. Development of TGF-beta signalling inhibitors for cancer therapy. Nature Reviews. Drug Discovery 3 1011–1022. ( 10.1038/nrd1580) [DOI] [PubMed] [Google Scholar]
- Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, Yu KJ, Liu B, Creighton CJ, Rodriguez-Canales J, et al. 2018. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbRIII–p38MAPK–pS249/T252RB pathway. Cancer Research 78 2911–2924. ( 10.1158/0008-5472.CAN-17-1051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yu C, Dai J, Keller JM, Hua A, Sottnik JL, Shelley G, Hall CL, Park SI, Yao Z, et al. 2014. Parathyroid hormone-related protein inhibits DKK1 expression through c-Jun-mediated inhibition of β-catenin activation of the DKK1 promoter in prostate cancer. Oncogene 33 2464–2477. ( 10.1038/onc.2013.203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. 2003. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425 836–841. ( 10.1038/nature02041) [DOI] [PubMed] [Google Scholar]
- Zhang X, Robles H, Magee KL, Lorenz MR, Wang Z, Harris CA, Craft CS & Scheller EL 2021a. A bone-specific adipogenesis pathway in fat-free mice defines key origins and adaptations of bone marrow adipocytes with age and disease. eLife 10 e66275. ( 10.7554/eLife.66275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bado IL, Hu J, Wan YW, Wu L, Wang H, Gao Y, Jeong HH, Xu Z, Hao X, et al. 2021b. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 184 2471–2486. ( 10.1016/j.cell.2021.03.011) [DOI] [PMC free article] [PubMed] [Google Scholar]