

Abstract
Post-traumatic stress disorder (PTSD) is a debilitating neuropsychiatric disease that is highly comorbid with major depressive disorder (MDD) and bipolar disorder (BD). The overlap in symptoms is hypothesized to stem from partially shared genetics and underlying neurobiological mechanisms. To delineate conservation between transcriptional patterns within and across disorders we performed RNA-sequencing in the postmortem brain of two prefrontal cortex regions and two amygdala regions from neurotypical donors (N=109) as well as donors diagnosed with PTSD (N=107) or MDD (N=109). We identified a limited number of differentially expressed genes (DEGs) specific to PTSD, with nearly all mapping to cortical versus amygdala regions. PTSD-specific DEGs were enriched in gene sets associated with immune-related pathways and microglia, and with sub-populations of GABAergic inhibitory neurons. While we identified a greater number of DEGs associated with MDD, most overlapped with PTSD, and only a few were MDD-specific. We used weighted gene co-expression network analysis (WGCNA) as an orthogonal approach to confirm the observed cellular and molecular associations. These findings provide supporting evidence for involvement of immune signaling and neuroinflammation in MDD and PTSD pathophysiology, and extend evidence that GABAergic neurons have functional significance in PTSD.
Introduction
Post-traumatic stress disorder (PTSD) is a debilitating disorder that develops in a subset of individuals following trauma exposure. PTSD is highly comorbid with other mental health disorders (1–3); for example >50% of individuals with PTSD also have a major depressive disorder (MDD) diagnosis (4–6), and PTSD prevalence among individuals with a bipolar disorder (BD) diagnosis is 2–3 times the general population (7, 8). PTSD is characterized by a unique set of clinical phenotypes, but shares some diagnostic symptoms with depressive disorders. Comorbidity may arise from shared mechanistic underpinnings, including overlapping genetic heritability and common environmental risk factors such as chronic stress and trauma exposure (4). However, the cellular and molecular mechanisms unique to PTSD versus shared with MDD are not well-understood. Here we examined transcriptional patterns within and across PTSD and MDD in the human cortex and amygdala.
Aberrant activity in neural circuits that link amygdala and prefrontal cortical regions has been identified in individuals with PTSD as well as in animal models relevant for PTSD (9–11). The amygdala and prefrontal cortex are critical for emotional regulation, including the expression and extinction of fear, behavioral functions that are dysregulated in PTSD. Coordinated patterns of neural activity in cortico-amygdala circuits underlie functional connectivity between these regions, which controls fear and anxiety (12–15). In accordance with human neuroimaging studies showing aberrant cortico-amygdala activity in PTSD (12, 15–17), animal studies demonstrate that function in these circuits is strongly impacted by exposure to trauma, and experimentally manipulating neuronal activity or key cell signaling pathways in cortico-amygdala circuits impacts fear processing and anxiety (18–21).
At the cellular level, deficits in inhibitory neurotransmission in cortico-amygdala circuits are associated with PTSD and depressive disorders (22, 23). Chronic stress and trauma exposure are hypothesized to impair inhibitory neuron function, impacting excitation-inhibition balance in cortico-amygdala circuits (24, 25). This is important because GABAergic inhibitory neurons in these circuits control neural activity and synaptic plasticity to regulate fear-related behaviors in animal models (26). However, how the molecular sequelae following chronic stress and trauma exposure impacts inhibitory neuron function is not well understood. In addition to GABAergic inhibition, inflammation and immune signaling have emerged as potential contributors to PTSD and depressive disorders (27–30). While inflammatory markers and genes related to immune signaling are altered in PTSD (31–33), whether observed changes result from central versus peripheral immune signaling pathways, and whether they reflect increased risk or epiphenomenon related to the pathophysiological sequelae of PTSD is not clear.
Conducting the largest RNA-sequencing study of PTSD in the human brain to date, we identified down-regulation of microglial-related transcripts and immune-related co-expression modules in both the cortex and amygdala. We identified notable reductions in specific transcripts encoding neuromodulators that are associated with GABAergic neuron function, but there was also evidence for increased expression of transcripts associated with both excitatory and inhibitory neurons. Collectively, the findings contribute evidence supporting the involvement of immune signaling, neuroinflammation and inhibitory neuron function in MDD and PTSD.
Methods
Detailed methods are available in the Supplement.
Postmortem human brains were donated through US medical examiners’ offices at the time of autopsy and a retrospective clinical diagnostic review was conducted on every brain to diagnose each donor into one of the three diagnosis groups (control, PTSD, MDD). Tissue was dissected from two subregions of the frontal cortex (dorsolateral prefrontal cortex; dlPFC and dorsal anterior cingulate cortex; dACC), and two subregions of the amygdala (basolateral amygdala; BLA, and medial amygdala; MeA) under visual guidance. RNA was extracted and sequenced using Ribo-Zero Gold ribosomal RNA depletion on an Illumina HiSeq 3000. Raw sequencing reads were processed as previously described (34) to obtain gene counts relative to GENCODE release 25 (GRCh38.p7). Quality control - including sequencing quality and sample identity checks - resulted in 1285 post-QC samples across 325 unique donors and 4 brain regions. We performed differential expression analyses within and across brain subregions using limma voom (35), adjusting for clinical and technical covariates, as well as quality variables (qSVs) (36). These models account for donors from all three diagnosis groups to jointly estimate the effects of PTSD versus control, MDD versus control, and PTSD versus MDD. We performed RNAscope to validate cell type specificity of candidate DEGs. We defined sets of marginally significant (at P < 0.005) genes, with and without enforcing directionality of effects (i.e. higher versus lower expression in PTSD versus control), and performed gene set and cell type enrichment analyses using the hypergeometric test. We lastly performed WGCNA (37) to assign genes to modules and assess the role of diagnosis on co-expressed gene sets.
Results
We generated deep bulk/homogenate RNA-sequencing (RNA-seq) data from postmortem human tissue in two subregions of the frontal cortex (dorsolateral prefrontal cortex; dlPFC and dorsal anterior cingulate cortex; dACC), and two subregions of the amygdala (basolateral amygdala; BLA, and medial amygdala; MeA; Table S1, Table S2, Table S3) from neurotypical donors as well as donors with singular diagnosis of PTSD or MDD, or PTSD comorbid with MDD or BD (Results S1, Table S2). After extensive and rigorous quality control of RNA-seq data (see Methods, Figure S1, Table S4), we performed differential expression and network analyses using 1285 samples from 325 unique donors (Table 1) and across 26,020 jointly expressed genes (Results S2).
Table 1:
demographic and RNA quality information for the subjects and associated brain tissue in this study.
| Group | Control | MDD | PTSD | P-value | |
|---|---|---|---|---|---|
| N | 109 | 109 | 107 | PTSD vs Control | PTSD vs MDD |
| Sex (%M) | 78.9 | 55 | 50.5 | 1.62E-05 | 0.586 |
| Race (%Cauc) | 69.7 | 80.7 | 87.9 | 3.01E-03 | 0.285 |
| Age: Mean | 49.6 | 45.9 | 40.8 | 1.93E-06 | 4.04E-03 |
| Age: SD | 15.1 | 14.5 | 11.3 | ||
| RIN: Mean | 7.4 | 7.3 | 7.3 | 0.239 | 0.957 |
| RIN: SD | 0.8 | 0.9 | 0.9 | ||
| PMI (hr): Mean | 29.4 | 26.7 | 29.1 | 0.822 | 0.0596 |
| PMI (hr): SD | 11.1 | 7.71 | 10.7 | ||
| Smoke (%) | 16.5 | 65.1 | 73.6 | 8.16E-18 | 0.187 |
| Opioids (%) | 6.42 | 62.4 | 66.4 | 1.18E-21 | 0.572 |
| Manner of Death (%Suicide) | NA | 22.9 | 24.3 | NA | 0.873 |
| Drug Related Death (%) | NA | 45.9 | 69.2 | NA | 5.97E-04 |
Expression differences related to PTSD diagnosis
We first explored the gene expression effects of PTSD diagnosis versus neurotypical control donors. We identified 41 PTSD differentially expressed genes (DEGs) in cortex (Figure 1A) and 1 PTSD DEG in amygdala (Figure 1B) at genome-wide significance (FDR < 0.05), while a more liberal threshold of FDR < 0.1 identified an additional 78 genes in cortex (with no additional genes in amygdala). We highlight several representative DEGs in PTSD versus neurotypical control donors in cortex including decreased expression of CORT, which is expressed in a subpopulation of GABAergic inhibitory neurons (38) (Figure 1C); increased expression of the histone deacetylase HDAC4 (Figure 1D); and increased expression of SPRED1, which encodes a protein involved in the Ras/MAPK signaling pathway (Figure 1E). In amygdala, a single gene was consistently downregulated in PTSD versus neurotypical controls across both subregions - CRHBP (Figure 1F), the gene encoding corticotropin-releasing hormone binding protein, which is an antagonist of the stress hormone corticotropin-releasing hormone (CRH) (39). Overall, cortical regions showed more association with PTSD than amygdala subregions, and the observed expression differences were largely consistent across subregions of the cortex, with only 5 genes showing marginal interaction (at p<0.01) between PTSD diagnosis and cortical subregion (NRSN1, PHF20L1, RP11–505E24.2, OXLD1, CARD8-AS1), and CRHBP only showing modest interaction between PTSD and amygdala subregions (p=0.037).
Figure 1:
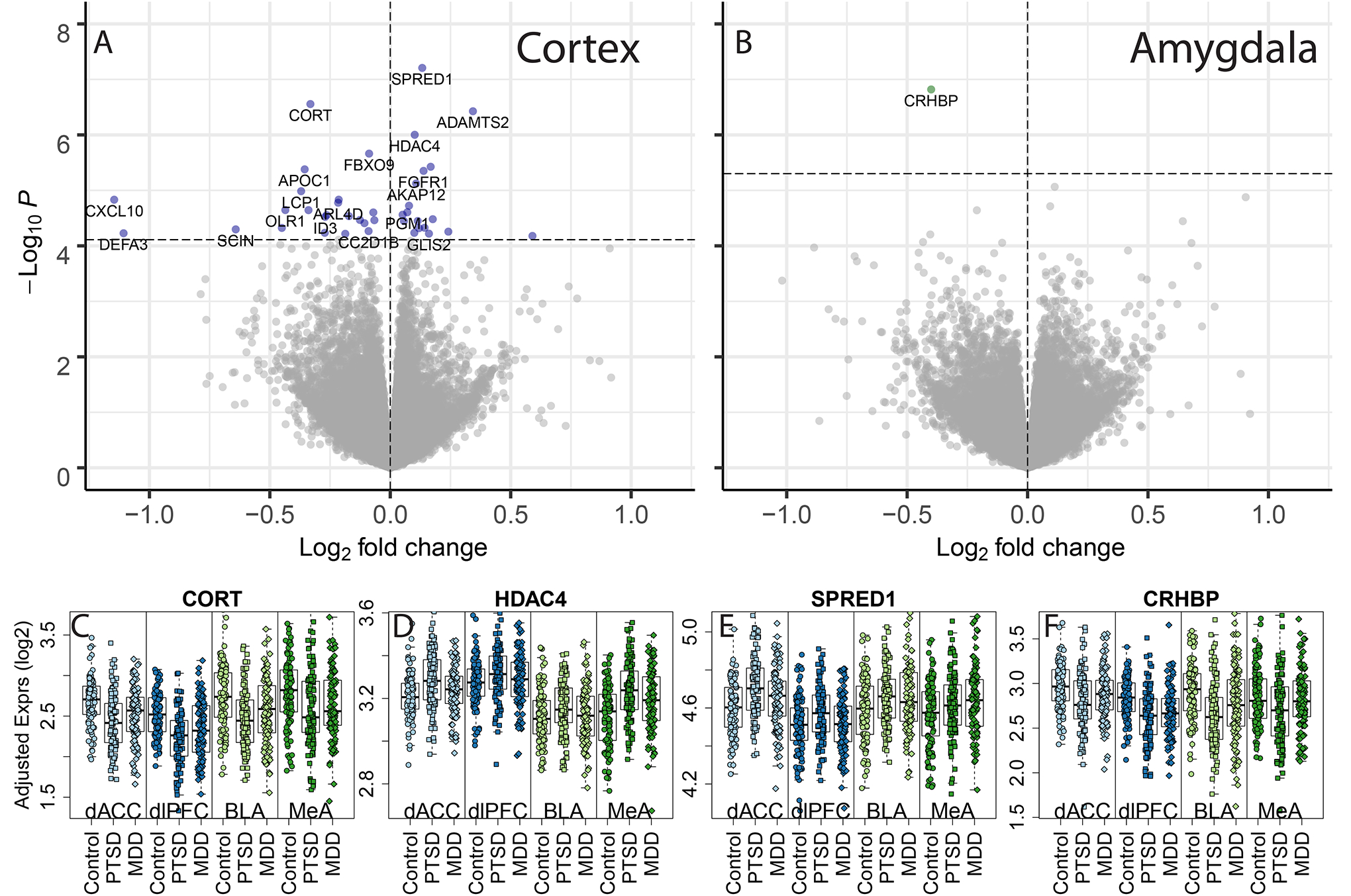
Differential gene expression associated with PTSD diagnosis, compared to neurotypical controls. Volcano plots for (A) cortex and (B) amygdala subregion-combined dataset. P-values were calculated using linear mixed effects modeling and the horizontal dashed line indicates the p-value that controls a false discovery rate (FDR) < 0.05. Positive log2 fold changes indicated higher expression in PTSD versus neurotypical subjects and negative log2 fold changes indicated lower expression in the PTSD group. Example differentially expressed genes include (C) CORT (D) HDAC4, (E) SPRED1, and (F) CRHBP, with “Adjusted” expression on the y-axis (regressing out unwanted technical and clinical confounders, preserving group and region effects, see Methods).
We next performed secondary analyses within each of the four subregions (dlPFC, dACC, BLA and MeA) to identify additional DEGs associated with PTSD diagnosis. Differential expression statistics were highly correlated with the combined subregion analyses, with the cortical associations driven predominantly by dACC, and the amygdala associations driven primarily by BLA (Figure S2). The cortical subregions again showed more PTSD DEGs, with 16 genes in the dACC (Figure S3A) and 1 gene in the dlPFC (Figure S3B) (and no genes in amygdala subregions) at genome-wide significance (FDR < 0.05). Using a more liberal cutoff of FDR < 0.1, we identified 74 unique genes across the cortical subregions (dACC: 72, dlPFC: 3, with one gene shared: AC124804.1, a novel transcript, antisense to SDK2) and 18 unique genes across amygdala subregions (BLA: 3, MeA: 16 with one gene: CORT, shared). Joint analysis of all data identified 117 genes with consistent PTSD versus control effects across all 4 subregions (at FDR < 0.05, with 276 genes at FDR < 0.1, Figure S4) further highlighting the similar effects of PTSD across multiple brain regions. Interestingly, these cross-region results were best represented by the amygdala (predominantly BLA), and not cortex, even though the cortex had more DEGs when considered alone (Figure S2). A comprehensive list of all differential expression statistics for all expressed genes and all statistical models is presented in Data S1.
We next used a series of sensitivity analysis to determine the robustness of our differential expression model by specifically interrogating the role of potential confounders and risk factors. Specifically, we tested a series of additional potential variables (including antidepressant treatment, and presence of opioids via toxicology) for attenuating the DEGs identified above in each brain region. Overall, subsequently adjusting our models for these variables had minimal effects on differential expression signals across all expressed genes, including those identified as DEGs (Figure S5). We further examined the role of sex on our identified DEGs using sex-specific analyses and found that subsets of DEGs were more strongly explained by effects within a single sex (Figure S6, Figure S7, Table S5). The identified DEGs showed significant confirmation with a recent manuscript that used a cohort of partially overlapping subjects with upwards of 80% of expressed genes being directionally consistent (Results S2, Figure S7A) (40). We lastly assessed the effects of combat, comparing the 25 combat-exposed donors with PTSD to the 82 PTSD donors without combat exposure within each brain region and found DEGs exclusively in the MeA (3 genes at FDR < 0.05, 29 at FDR <0.1 and 116 at FDR < 0.2, Table S6).
Taken together, by performing analysis on the largest postmortem brain dataset of PTSD to date, these analyses identified robust sets of differentially expressed genes associated with PTSD that are not a result of association with substance abuse or mood disorder diagnoses.
Gene sets and cell types associated with PTSD
We next performed gene set and pathway enrichment analyses to identify biological and molecular functions associated with PTSD within and across subregions. To facilitate these analyses we used more liberal significance thresholds to define PTSD DEGs (marginal p < 0.005 rather than FDR control) and directionality, and tested for enrichment among DEGs more highly and more lowly expressed in PTSD cases versus neurotypical controls. Overall, genes associated with PTSD showed the strongest enrichment for immune-related gene sets and pathways in both the cortex and amygdala (Figure 2A, Table S7), largely driven by decreased expression of genes in donors with PTSD compared to controls. Interrogating PTSD differences within subregions further identified unique molecular associations. For example, the MeA and dlPFC each showed decreased expression of genes associated with receptor ligand activity (that were further marginally significant in other regions). Interestingly, dlPFC associations were driven by eight genes (CORT/CSF1/SST/OSTN/CXCL10/CXCL11/GDF9/CCL3) and MeA associations by ten genes (CORT/TNFSF10/CXCL11/SFRP2/OSGIN2/OGN/IGF2/CTF1/CCL5/TTR) with only two genes in common (CORT and CXCL11), highlighting the convergence of molecular functions across brain regions.
Figure 2:
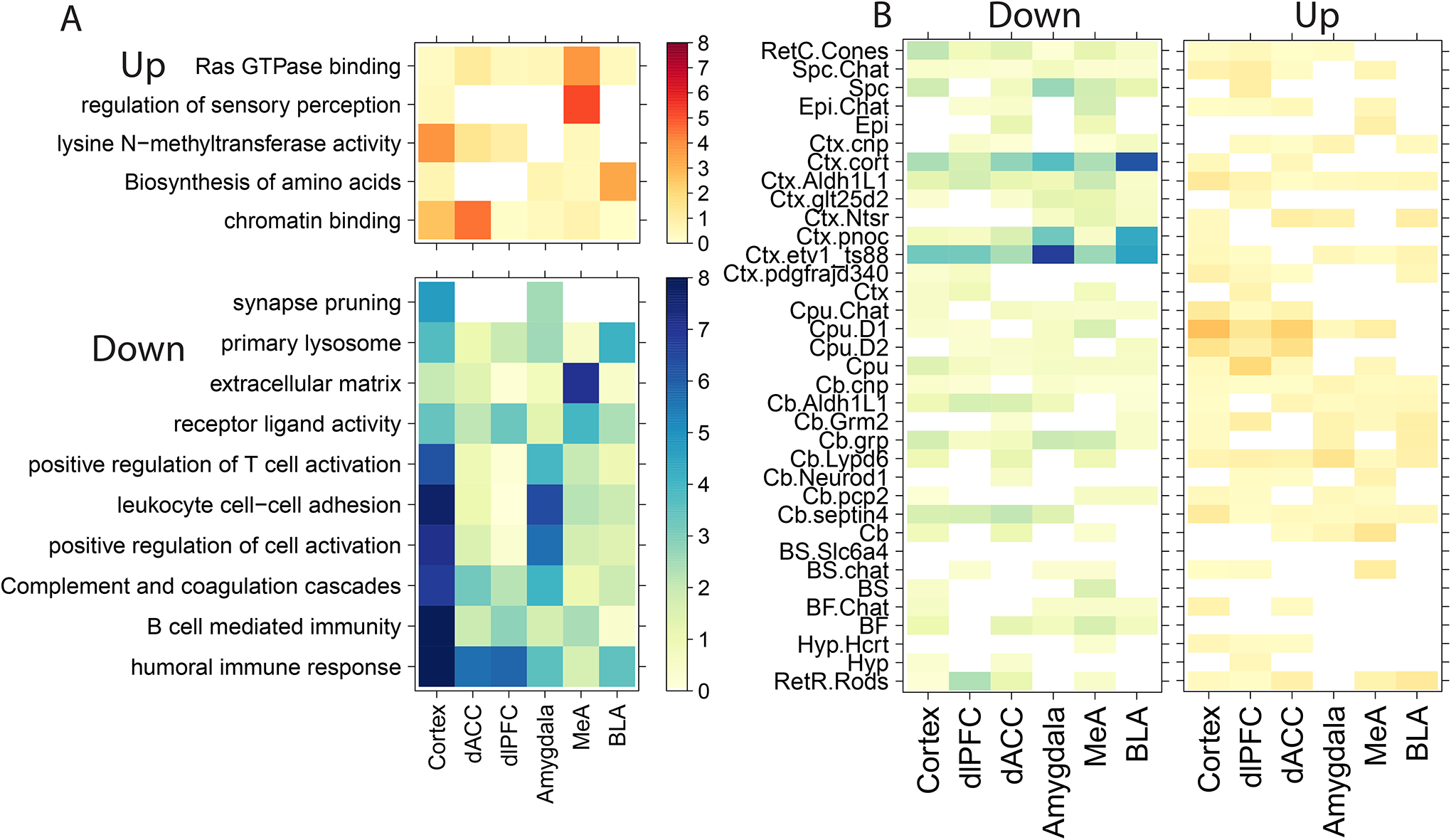
Molecular and cellular enrichments for genes associated with PTSD versus neurotypical controls. A) Gene set enrichment and (B) cell specific enrichment analyses for genes more highly expressed in PTSD (“Up”) or more lowly expressed (“Down”) compared to neurotypical donors. Color indicates −log10(p-values).
We next used cell type-specific enrichment analyses (CSEA) (41) to identify cell types that preferentially express these sets of differentially expressed genes. We found consistent enrichment of cortistatin-expressing GABAergic inhibitory neurons (“Ctx.cort”) and immune cells (“Ctx.etv1_ts88”) among genes where expression was decreased in donors with PTSD compared to neurotypical controls. Stronger enrichments were observed in the amygdala, particularly the BLA, compared to the cortex (Figure 2B, Table S8). For example, using a specificity threshold of pSI < 0.01 and the BLA, immune cell enrichments were driven by decreased expression of FERMT3, CRHBP, FOLR2, PTGS1, SLCO1C1, P2RY13 and GLT8D2 (odds ratio, OR= 8.9, p=2.94e-5) and cortistatin-positive inhibitory neuron enrichments were driven by decreased expression of NPY, CORT, CRHBP, DLL3, NXPH2, and SST (OR=23.7, p=5.9e-7).
We further confirmed enrichment of PTSD DEGs related to immune signaling and inhibitory neurons using snRNA-seq data generated in the human brain from amygdala and dlPFC (42) (Table S9). For amygdala DEGs where expression was lower in individuals with PTSD compared to neurotypical controls, we found strong enrichment within microglial populations identified in human amygdala (42). These enrichments for DEGs with lower expression in PTSD were strongest in a combined subregion analysis (OR=7.1, p=1.8e-23), but results were driven by the BLA (OR=3.0, p=9.7e-7) with no significant enrichment in the MeA (p=0.17). These more lowly expressed DEGs were also enriched in T-cells at the subregion level (p=2.7e-5), with these results driven by the MeA (p=8.6e-3). Unlike CSEA, we found evidence for inhibitory neuron enrichments among DEGs with higher expression in PTSD, particularly in the MeA (Inhib_C: OR=2.6, p = 2e-5; Inhib_F: OR=3.6, p=1.8e-9). However, this discrepancy could arise due to low expression levels in the snRNA-seq data of some genes that drove enrichments in the CSEA analysis. Analogous enrichment analyses using snRNA-seq data on cortical cell types in human brain similarly showed strong enrichment with PTSD DEGs. Using our snRNA-seq data from dlPFC (42), we found similar strong microglial cell enrichments among DEGs with decreased expression in PTSD (Microglia p=7.7e-22; Macrophage p=1.8e-15), whereas as DEGs with increased expression in PTSD were enriched in neuronal populations (Excit_A p = 7.1e-6; Excit_E p = 3.1e-7; Inhib_B p = 1.1e-6; Inhib_D=1.1e-6) (42). Using snRNA-seq data from a second study of human prefrontal and cingulate cortices (43), we identified that DEGs with increased expression in PTSD were most enriched in a somatostatin (SST)-expressing inhibitory neuron population (IN-SST p=5.3e-5), whereas DEGs decreased in PTSD were enriched for microglia p=4.5e-24 and endothelial p = 7.1e-6 populations. Finally, other snRNA-seq data from human prefrontal cortex (44) showed that DEGs with increased expression in PTSD were associated with Ast0, Ex12, In0, In1, In6, In7, In9 populations, while DEGs with decreased expression in PTSD were associated with Ast2, End1, Mic0, Mic1, Mic2, Mic3 populations (Table S9). Given the limitations of snRNA-seq for detecting relatively rare cell populations, we used an RNAscope single molecule fluorescence in situ hybridization (smFISH, see Methods) approach in BLA and dlPFC tissue derived from independent neurotypical donors to better understand co-expression of PTSD DEGs associated with inhibitory neurons. For RNAscope analysis we targeted expression of PTSD DEGs: CORT, SST, and CRHBP, Data S1), as well as GAD2 as a cell marker of inhibitory GABAergic cells (Figures 3B). We compared expression levels of these genes across nuclei, and found high correlations (Figures 3B), with the highest between CORT and SST (ρ = 0.72,p=8.7e-84) to the lowest between GAD2 and CRHBP (ρ = 0.166, p=2.1e-13). Almost all SST+ neurons co-expressed CORT whereas less than half of CORT+ neurons co-expressed SST. The top amygdala DEG - CRHBP - showed co-expression with both CORT and GAD2 across many ROIs in both brain regions (45, 46).
Figure 3:
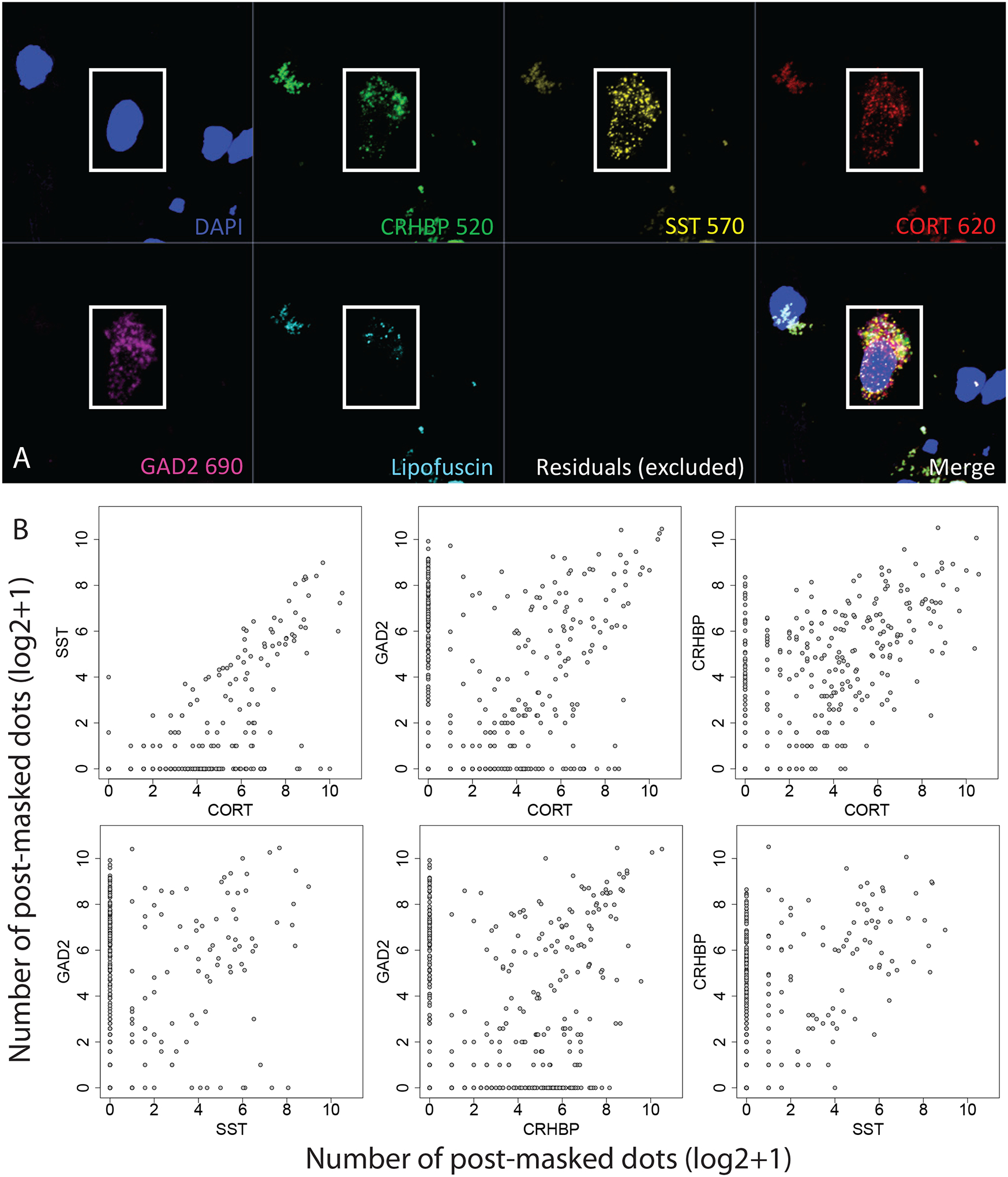
Single molecule fluorescence in situ hybridization (smFISH) validation of inhibitory neuron co-expression. (A) Representative image of co-expressing region-of-interest (ROI)/nucleus across multi-channel image. (B) Pairwise co-expression plots among four target genes, where axes indicate the number of post lipofuscin-masked segmented transcript dots (on the log2 scale)
Gene expression comparisons between PTSD and MDD
We next incorporated existing bulk RNA-seq data from MDD donors to better understand the gene expression differences unique to PTSD. We first compared donors with MDD to neurotypical controls among the broader cortical and amygdala brain regions, and again identified a larger number of differentially expressed genes in the cortex (182 genes at FDR < 0.05, 352 at FDR < 0.1, Table 2) compared to amygdala (0 genes at FDR < 0.05, 1 at FDR < 0.1). These differences were driven by the dACC (249 genes at FDR <0.1) compared to dlPFC (2 genes at FDR<0.1), similar to PTSD effects. There were similarly increased MDD differences in the MeA (16 genes at FDR < 0.05, 32 at FDR < 0.1) and no differences in BLA when stratifying the amygdala into subregions. Genes with decreased expression in MDD donors compared to neurotypical controls showed analogous enrichment of immune-related processes in the cortex using both gene set enrichment analysis (Table S10) and CSEA (“Ctx.etv1_ts88” cell type, Table S11). CSEA results related to cortistatin-positive neurons were attenuated compared to PTSD, particularly in the amygdala (best p-value = 0.01)
Table 2:
Number of differentially expressed genes in each dataset/brain region at two false discovery rate (FDR) cutoffs.
| PTSD vs CONT | MDD vs CONT | |||
|---|---|---|---|---|
| Dataset | FDR < 0.05 | FDR < 0.1 | FDR < 0.05 | FDR < 0.1 |
| Cortex | 41 | 119 | 182 | 352 |
| dACC | 16 | 74 | 67 | 249 |
| dlPFC | 1 | 3 | 1 | 2 |
| Amygdala | 1 | 1 | 0 | 1 |
| BLA | 0 | 3 | 0 | 0 |
| MeA | 0 | 16 | 16 | 34 |
| Joint | 117 | 276 | 55 | 192 |
Globally, there was high concordance between PTSD and MDD effects on gene expression (Figure S10, ρ range=0.647–0.695), with highly overlapping DEGs at marginal significance in each brain region or subregion (all Fisher’s p-value < 1.72e-46). While global effects were correlated and significant genes were overlapping, there was nevertheless variation among significantly differentially expressed genes across the two disorders. For example, among the genes marginally associated with MDD in each subregion, only a quarter were significantly differentially expressed comparing PTSD to controls (each at p < 0.005), and among those genes marginally associated with PTSD, only a third of genes in cortical regions and a quarter of genes in amygdala regions showed similar marginal association in MDD.
We therefore directly compared expression between PTSD and MDD donors to better partition these differences across diagnoses (see Methods), and identified only a limited number of differentially expressed genes (at FDR< 0.1). Specifically, we saw increased expression of KCNC1, FAM234B and RASD2 and decreased expression of CH507–513H4.4 in PTSD versus MDD in cortex, decreased expression of LMCD1 in PTSD in MeA and decreased expression of DNAH11 in PTSD in dlPFC. In cortex, marginally significant genes that were more highly expressed in PTSD versus MDD (at p < 0.005) were associated with neuronal processes and synapses (both inhibitory and excitatory), whereas marginally significant genes with decreased expression in PTSD versus MDD in amygdala were associated with neuronal migration and PI3K signaling (Table S12). There were no enrichments for the immune-related gene sets for these disorder-specific contrasts, suggesting decreased expression of immune processes and/or microglia involvement were shared across both disorders, relative to neurotypical individuals (Table S13). These results together suggest largely similar transcriptomic changes in PTSD and MDD compared to neurotypical donors.
We then used RNA deconvolution to better determine if microglia or neurons were more or less prevalent in PTSD and MDD donors (see Methods) (47). While the proportion of microglia and neuron RNAs differed by brain region (Figures S11A, S11B), there were no differences between diagnosis for either cell type (microglia: PTSD p=0.84 and MDD p=0.59; neurons PTSD p=0.632 and MDD p=0.649). The RNA fractions across all evaluated cell types also strongly associated with the qSVs used to control for latent heterogeneity - in line with our previous work (34) - suggesting our DEGs were not confounded by tissue composition (Figure S11C).
We lastly performed weighted gene co-expression analyses (WGCNA) to better understand network-level gene expression differences between PTSD and MDD (Results S4). This analysis identified a total of 156 modules across six WGCNA runs (regions: cortex, amygdala; subregions: dACC, dlPFC, MeA, BLA; Table S14), of which 35 were enriched for PTSD (N=22) or MDD (N=22; 9 overlapping) DEGs (Table 3, Table S15). In the cortex and its subregions, the strongest disorder-related module (Cortex.ME7) related to regulation of cell activation, a broad category encompassing many immune processes, associated with both PTSD (p=1.6e-25) and MDD (p=3.3e-126) DEGs, with its eigengene further associated with these diagnoses at the subject-level (PTSD p=2.9e-4, MDD p=8.4e-6). The strongest disorder-related module in the amygdala (Amygdala.ME2) was specifically enriched with PTSD DEGs (p=2.97e-23) with its eigengene further associated with PTSD compared to controls (p=0.005). Sensitivity analyses for other potential confounders including combat, childhood maltreatment, and toxicology-determined smoking, SSRI antidepressant use, and opioid use showed minor effects on the WGCNA eigengene associations to PTSD or MDD diagnosis. These variables themselves had weak associations to only a few eigengenes (Results S4), These analyses further highlight biological processes associated with PTSD and MDD using convergent approaches to traditional gene set enrichments of DEGs.
Table 3:
Module-level associations to PTSD and MDD. Fisher’s exact test enrichment for DEGs (at p < 0.005) in PTSD (PTSD_Pval) and MDD (MDD_Pval) among module gene membership. Eigengene subject-level associations to PTSD vs control (PTSD_p) and MDD vs control (MDD_p). The top gene ontology biological process is shown for module gene membership (GOBP_Description) with corresponding p-value (GOBP_pvalue).
| Module_Name | numGenes | DEG Enrichment | Eigengene Assoc. | Gene Ontology (BP) | Cellular Enrichment | ||||
|---|---|---|---|---|---|---|---|---|---|
| PTSD | MDD | PTSD | MDD | Description | P-value | Class | P-value | ||
| Cortex_ME2 | 1360 | 4.40E-03 | 4.66E-11 | 1.69E-01 | 2.58E-05 | synapse organization | 3.49E-14 | Excit_A | 5.7E-192 |
| Cortex_ME7 | 620 | 1.57E-25 | 3.29E-126 | 2.85E-04 | 8.45E-06 | regulation of cell activation | 1.68E-42 | Micro | <1E-300 |
| Cortex_ME18 | 125 | 3.61E-01 | 2.22E-03 | 3.03E-01 | 5.81E-04 | modulation of chemical synaptic transmission | 6.21E-07 | Excit_B | 2.1E-52 |
| Cortex_ME30 | 56 | 7.94E-03 | 6.65E-01 | 3.86E-01 | 2.14E-01 | regulation of neurotransmitter receptor activity | 8.82E-05 | Inhib_A | 6.4E-14 |
| Cortex_ME31 | 55 | 1.70E-10 | 1.88E-01 | 3.22E-02 | 2.42E-01 | learning or memory | 1.37E-04 | Inhib_B | 2.7E-03 |
| Cortex_ME34 | 35 | 7.27E-03 | 1.00E+00 | 8.39E-02 | 3.82E-01 | response to cAMP | 1.55E-08 | Astro | 1.4E-08 |
| dlPFC_ME3 | 1104 | 3.53E-01 | 1.12E-03 | 1.53E-01 | 2.78E-03 | regulation of synaptic plasticity | 6.77E-12 | Excit_E | 4.7E-67 |
| dlPFC_ME6 | 502 | 7.77E-05 | 8.26E-02 | 2.35E-03 | 6.92E-03 | regulation of leukocyte activation | 5.16E-41 | Micro | <1E-300 |
| dlPFC_ME11 | 299 | 6.12E-01 | 1.89E-06 | 1.93E-02 | 6.93E-04 | meiotic chromosome separation | 1.87E-04 | Inhib_E | 4.9E-07 |
| dlPFC_ME20 | 117 | 2.71E-15 | 4.44E-02 | 9.14E-03 | 3.23E-01 | UDP-N-acetylglucosamine metabolic process | 1.36E-04 | Tcell | 2.4E-03 |
| dlPFC_ME22 | 98 | 2.37E-03 | 6.32E-01 | 1.38E-01 | 8.25E-01 | response to estradiol | 4.15E-04 | Excit_B | 1.4E-25 |
| dlPFC_ME24 | 84 | 1.61E-04 | 2.69E-03 | 5.41E-02 | 7.92E-02 | membrane depolarization during action potential | 7.82E-07 | Inhib_D | 1.1E-21 |
| dACC_ME3 | 1393 | 1.55E-03 | 1.61E-23 | 1.01E-02 | 1.84E-08 | modulation of chemical synaptic transmission | 2.95E-22 | Excit_A | 6.9E-263 |
| dACC_ME5 | 747 | 3.60E-03 | 2.70E-06 | 2.20E-02 | 1.10E-03 | forebrain development | 6.78E-10 | Excit_B | 3.6E-172 |
| dACC_ME7 | 702 | 2.66E-03 | 3.28E-84 | 1.26E-03 | 2.03E-05 | lymphocyte activation | 3.57E-42 | Micro | <1E-300 |
| dACC_ME9 | 599 | 1.42E-08 | 6.02E-01 | 4.79E-01 | 4.68E-02 | modulation of chemical synaptic transmission | 2.01E-09 | Excit_B | 3.1E-42 |
| dACC_ME12 | 197 | 1.03E-01 | 6.02E-04 | 8.96E-01 | 1.11E-01 | heart development | 1.48E-06 | Astro | 4.7E-121 |
| dACC_ME13 | 177 | 6.56E-03 | 1.00E+00 | 6.39E-01 | 8.99E-02 | negative regulation of translation | 9.03E-05 | Tcell | 5.8E-07 |
| Amygdala_ME1 | 1035 | 1.90E-05 | 3.66E-04 | 2.49E-01 | 3.99E-02 | myelination | 2.73E-13 | Oligo | <1E-300 |
| Amygdala_ME2 | 904 | 2.97E-23 | 2.59E-03 | 4.70E-03 | 1.15E-01 | lymphocyte activation | 1.70E-38 | Micro | <1E-300 |
| Amygdala_ME4 | 696 | 1.90E-01 | 3.67E-05 | 2.56E-01 | 1.12E-01 | modulation of chemical synaptic transmission | 5.27E-26 | Excit_B | 5.2E-169 |
| Amygdala_ME19 | 47 | 9.15E-02 | 7.72E-03 | 7.88E-03 | 5.43E-03 | extracellular matrix constituent secretion | 3.05E-04 | Inhib_D | 7.3E-06 |
| Amygdala_ME21 | 39 | 8.55E-03 | 2.86E-01 | 1.49E-02 | 1.15E-02 | regulation of system process | 3.77E-03 | Inhib_B | 3.8E-06 |
| Amygdala_ME24 | 32 | 2.94E-01 | 2.60E-03 | 3.17E-01 | 1.09E-01 | formation of quadruple SL/U4/U5/U6 snRNP | 4.84E-07 | Astro_A | 1.7E-01 |
| MeA_ME3 | 431 | 9.88E-12 | 5.72E-01 | 4.09E-02 | 5.13E-01 | modulation of chemical synaptic transmission | 7.17E-18 | Inhib_D | 8.3E-147 |
| MeA_ME4 | 375 | 3.63E-01 | 3.22E-04 | 2.60E-01 | 4.32E-02 | modulation of chemical synaptic transmission | 1.71E-28 | Excit_A | 1.1E-159 |
| MeA_ME5 | 281 | 1.11E-01 | 3.82E-03 | 6.05E-02 | 5.99E-03 | regulation of ion transmembrane transport | 1.21E-07 | Inhib_C | 2.9E-69 |
| MeA_ME7 | 153 | 5.00E-03 | 2.78E-06 | 1.53E-01 | 7.61E-02 | extracellular structure organization | 2.98E-10 | Astro_B | 1.2E-90 |
| MeA_ME9 | 86 | 6.34E-01 | 7.33E-04 | 4.35E-01 | 2.20E-03 | ameboidal-type cell migration | 1.45E-04 | Excit_A | 3.9E-31 |
| MeA_ME10 | 85 | 3.21E-01 | 1.65E-05 | 1.01E-01 | 4.65E-04 | regulation of membrane potential | 7.35E-06 | Inhib_F | 8.3E-27 |
| MeA_ME13 | 63 | 7.22E-21 | 2.34E-26 | 5.51E-03 | 7.62E-04 | extracellular matrix organization | 1.36E-18 | Mural | 3.0E-36 |
| BLA_ME2 | 1300 | 3.25E-09 | 5.09E-01 | 1.32E-02 | 9.98E-02 | lymphocyte activation | 1.81E-30 | Micro | <1E-300 |
| BLA_ME12 | 199 | 4.08E-03 | 6.60E-02 | 2.52E-02 | 1.37E-01 | homophilic cell adhesion via plasma membrane adhesion molecules | 4.41E-06 | Astro_A | 3.1E-92 |
| BLA_ME13 | 176 | 2.36E-07 | 6.47E-01 | 3.85E-03 | 5.21E-01 | locomotory behavior | 1.98E-06 | Inhib_D | 2.6E-68 |
| BLA_ME20 | 37 | 5.31E-02 | 1.13E-08 | 6.37E-02 | 1.17E-02 | spliceosomal tri-snRNP complex assembly | 9.98E-08 | Astro_A | 1.1E-01 |
Discussion
The goal of this study was to identify shared versus divergent in transcriptional patterns within and across PTSD and MDD in the prefrontal cortex and amygdala. We identified a limited number of DEGs specific to PTSD, with nearly all mapping to cortex versus amygdala. PTSD-specific DEGs were enriched in gene sets associated with immune-related pathways and microglia, and with sub-populations of GABAergic inhibitory neurons. While we identified a greater number of DEGs associated with MDD, most overlapped with PTSD, and only a few were MDD-specific. These findings provide supporting evidence for involvement of immune signaling and neuroinflammation in MDD and PTSD pathophysiology, and extend evidence that GABAergic neurons have functional significance in PTSD.
Decreased expression of genes included in immune-related Gene Ontology sets were associated with PTSD diagnosis in both cortical and amygdala brain regions (Figure 3A). CSEA using mouse cell-specific markers and snRNA-seq data from human brain demonstrated enrichment of these DEGs with decreased expression in PTSD among microglia profiles (41, 48). Genes with decreased expression in MDD donors compared to neurotypical controls showed analogous enrichment of immune-related processes using both gene set enrichment analysis and CSEA, and there were no enrichments for the immune and microglia-related genes when contrasting PTSD and MDD, suggesting that decreased expression of immune processes and microglia involvement are not specific to PTSD. The downward direction of dysregulation was somewhat surprising considering that higher levels of pre-trauma C-reactive protein (a marker of blood inflammation) has been reported to predict elevated PTSD symptoms after trauma (49). Furthermore elevated levels of selected markers of low-grade blood inflammation have been reported in a meta-analysis of PTSD studies (50). However, over time, and with repeated exposure to chronic stress and trauma, immune function may become dysregulated in a myriad of ways, with neuronal, glial and peripheral systems attempting to compensate for immune activation and increased inflammation (33, 51–53). While general decreased expression of the microglial immune transcriptome and/or reductions in microglial cell ratios are possible explanations for the present data, we identified that a number of the genes included in the affected immune Gene Ontology sets that encode proteins with known immunosuppressive activity, which could also explain the somewhat paradoxical finding of decreased expression of immune-related genes. For example, in the immune-related regulation of cell activity category, we identified 13 member PTSD DEGs - and of these 13, 7 (bolded) have potential immunosuppressive activity (IL1RL2/DPP4/IGFBP2/TGFBR2/TAC1/MDK/CD4/PTPN6/TESPA1/IGF1/ITGAM/TYROBP/ITGB2) (54). These observations do not support a high level of microglial immune activation in chronic PTSD or MDD in cortex or amygdala, but do suggest dysregulation or possibly a compensatory response to stress.
We observed down-regulation of CORT mRNA across all four subregions in individuals with PTSD. CORT encodes the secreted neuropeptide cortistatin, which is expressed in the cerebral cortex, hippocampus and amygdala in a subset of GABAergic neurons (55), (38). Loss of cortistatin cells in mice causes spontaneous seizures, demonstrating that these cells provide strong inhibitory control (56, 57). In the rodent cells expressing cortistatin constitute a subset of SST-expressing neurons (56), and in human brain we confirmed co-expression of CORT with GABAergic inhibitory neuron markers (GAD2, SST and CRHBP). Decreased CORT and SST expression were previously reported in amygdala of female postmortem human brain donors with MDD (58), and our CSEA analyses showed enrichment of genes differentially expressed in PTSD in cortistatin expressing cells. We also identified enrichment of PTSD DEGs with specific inhibitory neuron clusters from snRNA-seq data in human amygdala that have been associated with anxiety and HPA axis function (42, 59). Weighted gene coexpression network analyses (WGCNA) further implicated inhibitory neuron function in line with the gene set enrichment results applied directly to PTSD DEGs. Decreased expression of CORT, SST and CRHBP mRNA provides additional support for the hypothesis that GABAergic neuron dysfunction is mechanistically associated with PTSD (22). Strong evidence implicates GABAergic neurons in controlling fear-related behaviors in preclinical animal models relevant for PTSD and other trauma-related disorders by controlling neural activity and synaptic plasticity in cortico-amygdala circuits (23). For example, firing of excitatory cells that project from the BLA to the frontal cortex is under tight regulation by local GABAergic inhibitory neurons (25), which provides negative feedback regulation of the BLA to control both the expression and extinction of fear (26, 60). Strong evidence links somatostatin signaling and SST+ cells in cortico-amygdala circuits with threat perception and fear memory processing (61–65). CRHBP, which we showed to be co-expressed with CORT and SST in GABAergic neurons in the human brain, was the only gene consistently downregulated in PTSD versus neurotypical controls across both subregions of the amygdala. CRHBP encodes corticotropin-releasing hormone binding protein, which sequesters and antagonizes CRH signaling (39). The robust DEG signal for CRHBP is interesting given many studies implicating the stress hormones corticotrophin-releasing hormone (CRH) and cortisol in PTSD (66–69). CRH activates the release of adrenocorticotropic releasing hormone (ACTH), which stimulates production of cortisol to control the body’s response to stress and trauma, which is important given that stress is a leading risk factor for the development of PTSD (70). Additional genetic support for a critical role of the stress axis in PTSD comes from recent GWAS associations of CRHR1 (71), which encodes the primary CRH receptor - CRF1, and CRH binds to CRF1 to mediate the behavioral and endocrine responses to stress exposure.
We further ran a number of analyses to better identify gene expression differences that were selectively associated with PTSD, but not MDD. In general, we identified more DEGs for MDD than PTSD, particularly in the cortex (which were primarily driven by the dACC). However, gene expression differences were highly concordant between the two diagnoses, with most highly significant DEGs showing the same directionality of effects (i.e. log2 fold changes) in both diagnoses. Marginally selective between-diagnoses DEG gene sets included more highly-expressed glutamatergic synapse-related DEGs in PTSD cortex (driven by the dACC) and more highly-expressed neuronal activity-related DEGs in MDD amygdala (driven by the BLA). Differences between the two diagnoses were more prominent in WGCNA analyses, where seven potentially overlapping modules showed PTSD-specific enrichment (Cortex_ME31, dlPFC_ME20, dACC_ME9, Amygdala_ME2, MeA_ME3, BLA_ME2, BLA_ME13) and five modules showed MDD-specific enrichment (dlPFC_ME11, dACC_ME3, dACC_ME7, MeA_ME9, BLA_ME20).
DEGs from a recent RNA-seq study of human postmortem PTSD tissue (40), which used a partially overlapping set of donors (see below) provides support for top DEGs identified here. For example, within our combined cortical PTSD analyses (see Figure 1), 6 of the 7 most robustly affected transcripts comparing PTSD versus controls (CORT, HDAC4, CRHBP, ADAMTS2, FBXO9, APOC1) were directionally consistent and at least marginally significant in this previous dataset. These genes further showed decreased expression in MDD versus controls in the present study, with at least marginal significance, suggesting that these particular findings may be related to shared pathophysiological changes accompanying PTSD and MDD. A key up-regulated gene identified in Girgenti et al. (40), ELK1, was significantly up-regulated in both cortical regions in the present study, and SST, identified as robustly down-regulated in several regions of the cortex in Girgenti et al., was in the top ten of all down-regulated transcripts in both dlPFC and dACC here. ADAMTS2, the second highest upregulated DEG in the combined cortical sample, was the top up-regulated gene in the dACC and the third most up-regulated in the dlPFC in Girgenti et al., 2020 (40). HDAC4, a top DEG, has been associated previously with both human PTSD and rodent models of PTSD (72, 73). The Girgenti study (40) also identified enrichment of down-regulated PTSD-associated DEGs using CSEA that were related to GABAergic neurons and their molecular functions.
In addition to these common elements, the present results extend previous findings from Girgenti et al. in several key areas (40). First, this study extended the search for differential gene expression beyond the cortex and into the amygdala, a relatively under-studied brain area in postmortem human brain research with high relevance to PTSD. Second, we provide compelling evidence implicating decreased expression of immune-related genes and associated processes in PTSD and MDD compared to neurotypical controls. This is an important observation because it runs counter to most expectations for immune response directionality. We further refined these cellular enrichment in GABAergic neurons more specifically to CORT-positive interneurons, which we subsequently validated with RNAscope. The cell type analyses in the present study provide direct evidence of these enrichments by interrogating DEGs directly against cell type-specific genes from both human and mouse studies, complementing the indirect strategy taken by Girgenti et al of first identifying genes in discrete co-expressed modules, and then associating those genes with both PTSD DEGs and cell type-specific genes separately (such that different genes captured the cell type versus PTSD signal in the same module). Third, we believe our >2-fold increased sample size in all diagnostic groups (total N=325 versus N=143) - obtained from a single postmortem brain collection under identical sample ascertainment and inclusion criteria - refined several of the clinical associations identified by Girgenti et al. We identified more similarities than differences between PTSD and MDD and replicated this finding across both amygdala and cortical subregions, with far less sex-specific diagnosis-associated signal than discussed in this previous study. Contributing to both the similarities and differences between the two studies was the fact that 77 donors were shared across both studies, although the two studies used different hemispheres, independent dissections and RNA extractions, as well as different data analysis pipelines. Over half (53.8%) of the donors in Girgenti et al. overlapped with a quarter (23.7%) of the donors in the present study (40).
Therefore, it might seem counter-intuitive that we identified many fewer DEGs in this much larger study, particularly with overlapping donors. We believe these differences can be accounted for by our more conservative statistical analyses - including modeling both diagnostic groups in a single statistical model against the neurotypical group that further accounted for robust observed and latent confounders. The differential expression models in Girgenti et al only adjusted for age, RIN, PMI, and race, and lack of accounting for sequencing-derived RNA quality metrics and other latent confounders, which can greatly increase false positive rates in human postmortem brain gene expression studies (36). For example, this less comprehensive statistical model applied to our larger dataset resulted in 1,243 DEGs in DLPFC, 1,719 DEGs in dACC, 1,813 DEGs in BLA and 10,283 DEGs in MeA for PTSD at FDR < 0.05, which is many more genes than obtained with our more conservative approach. There has been some debate regarding the optimal methods of latent variable correction in these types of postmortem studies, including the potential for “over-correction” (74). A major analytic element of the present investigation was the use of quality surrogate variable (qSV) analysis to identify and correct for expressed sequences that are particularly prone to degradation in human post-mortem brain (36). The qSVs utilized here were defined from the top 1000 degradation-susceptible expressed regions generated from independent time course experiments. Dropping the qSVs from our main analyses resulted in 209 DEGs in DLPFC, 43 DEGs in dACC, 62 DEGs in BLA and 1,054 DEGs in MeA (at FDR < 0.05) for PTSD in the present dataset. It is however possible that if sex or disease-associated interactions related to gene transcript degradation exist, use of qSV may have limited the emergence of these genes as DEGs and contributed to the differences between the present findings and those discussed in Girgenti et al (40). Similarly, in MDD, the most prominent previous report of differential gene expression actually identified no DEGs when correcting for multiple testing via the FDR (from the supplementary tables included in that manuscript) (75) making it difficult to assess replication of our DEGs using previously-published datasets. While these issues may seem rather nuanced, they nevertheless have important consequences on identifying DEGs in human postmortem RNA-seq datasets, and require careful consideration in past and future work.
Limitations of this work include potential under-representation of female donors in the neurotypical control group (21.1% female) compared to cases (~50% female). While we adjusted for sex in differential expression analyses, secondary analyses did suggest some potential differences in diagnosis effects across sex. Our cohort included donors with only a PTSD or only an MDD diagnosis, as well as PTSD donors with comorbid MDD or comorbid BD diagnosis. However, we did not have subjects with only a BD diagnosis. Furthermore, as is common in most psychiatric post-mortem human studies, psychotropic medications, substance use, smoking and suicide were more common in the MDD and PTSD groups, and further work will be needed to investigate their potential influence on brain gene expression patterns. In summary, these analyses of the largest postmortem brain cohort of patients with PTSD and MDD to date highlight microglia and other immune cell types as having potential functional significance in PTSD, and provide additional evidence for dysregulated neuroinflammation and neuroimmune signaling in MDD and PTSD pathophysiology.
Supplementary Material
Figure 4:
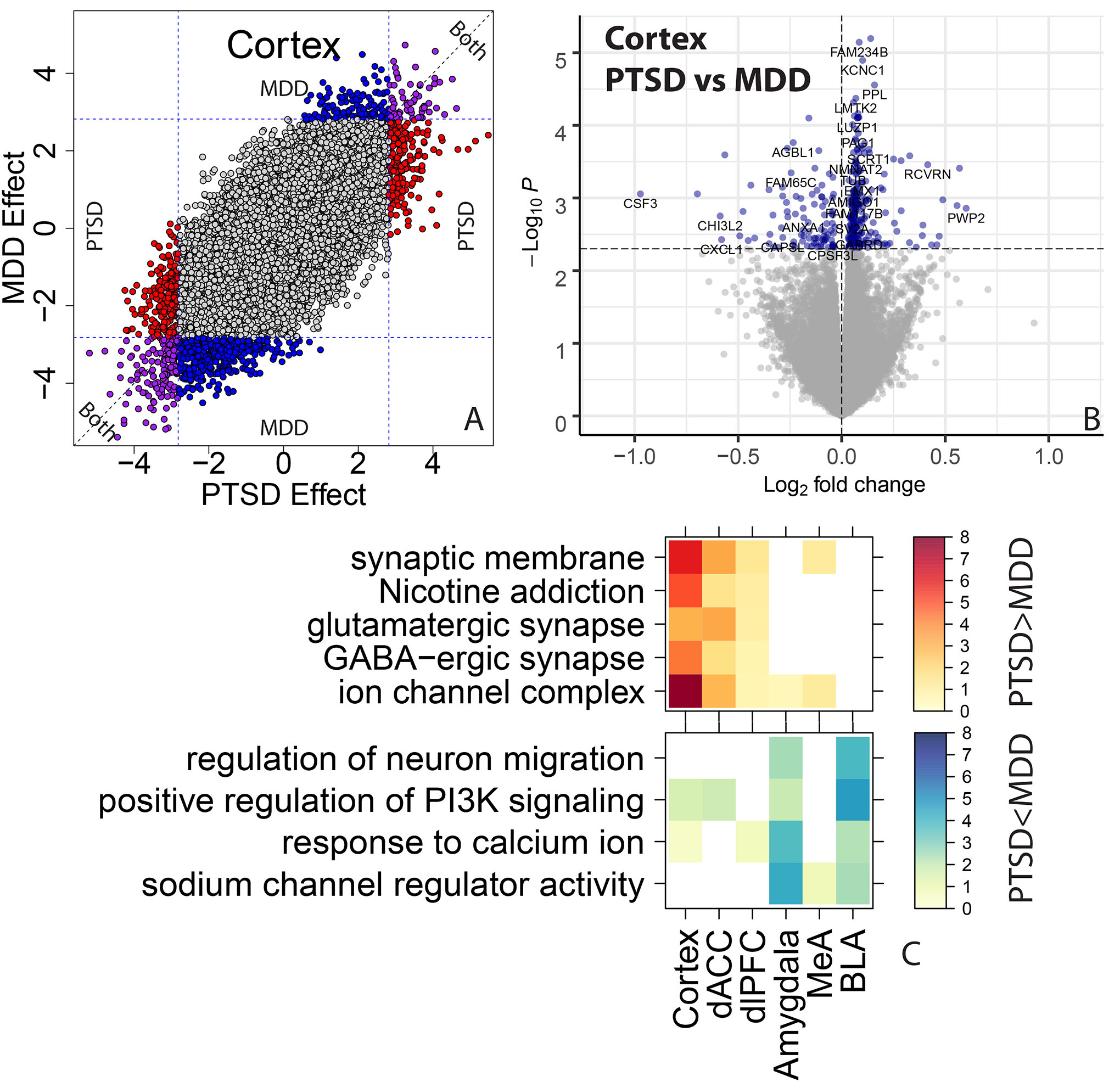
Contrasting PTSD and MDD effects on gene expression. (A) Scatterplot comparing the t-statistics for MDD versus control differential expression effects (y-axis) against PTSD versus control effects (x-axis). Colors indicate marginal significance at p < 0.005 for PTSD (red), MDD (blue), or both (purple). (B) Volcano plot directly comparing PTSD and MDD groups to each other. Horizontal line indicates marginal P < 0.005. (C) Gene set enrichment analyses for genes differentially expressed between PTSD and MDD, stratified by directionality.
Acknowledgements
The authors express their gratitude to our colleagues whose tireless efforts have led to the donation of postmortem tissue to advance these studies: the Office of the Chief Medical Examiner of the District of Columbia, the Office of the Chief Medical Examiner for Northern Virginia, Fairfax Virginia, the Office of the Chief Medical Examiner of the State of Maryland, Baltimore Maryland, the Office of the Chief Medical Examiner for Kalamazoo County, Kalamazoo Michigan, the University of North Dakota School of Medicine Department of Pathology, Forensic Pathology Center, Grand Forks North Dakota, and the Santa Clara County Office of the Chief Medical Examiner, San Jose California. This work was supported with resources and use of facilities at the VA Connecticut Health Care System, West Haven, CT, Central Texas Veterans Health Care System, Temple, TX, Durham VA Healthcare System, Durham NC, VA San Diego Healthcare System, La Jolla, CA, VA Boston Healthcare System, Boston, MA, USA and the National Center for PTSD, U.S. Department of Veterans Affairs. The views expressed here are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs (VA) or the U.S. government. We acknowledge the contributions of Amy Deep-Soboslay and Llewellyn B. Bigelow, MD for their diagnostic expertise, and Dr. Daniel R. Weinberger for providing constructive commentary and editing of the manuscript. Finally, we are indebted to the generosity of the families of the decedents, who donated the brain tissue used in these studies.
Funding:
This work was supported by the Lieber Institute for Brain Development and Contract #(VA-241-17-C-0138) from the US Department of Veterans Affairs.
Footnotes
Disclosures: Andrew E. Jaffe is now employed by Neumora Therapeutics, a for-profit biotechnology company, which is unrelated to the contents of this manuscript. No other authors have financial relationships with commercial interests.
Data availability
All code and figures associated with this manuscript are available through GitHub: https://github.com/LieberInstitute/LIBD_VA_PTSD_RNAseq_4Region. All raw and processed data may be requested via the PTSD Brain Bank Resource Request process described on the “For Investigators” tab of the following page: https://www.research.va.gov/programs/tissue_banking/ptsd/default.cfm
Bibliography
- 1.Armenta RF, Walter KH, Geronimo-Hara TR, et al. : Longitudinal trajectories of comorbid PTSD and depression symptoms among U.S. service members and veterans. BMC Psychiatry 2019; 19:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter KH, Levine JA, Highfill-McRoy RM, et al. : Prevalence of Posttraumatic Stress Disorder and Psychological Comorbidities Among U.S. Active Duty Service Members, 2006–2013. J Trauma Stress 2018; 31:837–844 [DOI] [PubMed] [Google Scholar]
- 3.Kang B, Xu H, McConnell ES: Neurocognitive and psychiatric comorbidities of posttraumatic stress disorder among older veterans: A systematic review. Int J Geriatr Psychiatry 2019; 34:522–538 [DOI] [PubMed] [Google Scholar]
- 4.Flory JD, Yehuda R: Comorbidity between post-traumatic stress disorder and major depressive disorder: alternative explanations and treatment considerations. Dialogues Clin Neurosci 2015; 17:141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessler RC, Sonnega A, Bromet E, et al. : Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 1995; 52:1048–1060 [DOI] [PubMed] [Google Scholar]
- 6.Rytwinski NK, Scur MD, Feeny NC, et al. : The co-occurrence of major depressive disorder among individuals with posttraumatic stress disorder: a meta-analysis. J Trauma Stress 2013; 26:299–309 [DOI] [PubMed] [Google Scholar]
- 7.Otto MW, Perlman CA, Wernicke R, et al. : Posttraumatic stress disorder in patients with bipolar disorder: a review of prevalence, correlates, and treatment strategies. Bipolar Disord 2004; 6:470–479 [DOI] [PubMed] [Google Scholar]
- 8.Neria Y, Olfson M, Gameroff MJ, et al. : Trauma exposure and posttraumatic stress disorder among primary care patients with bipolar spectrum disorder. Bipolar Disord 2008; 10:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenster RJ, Lebois LAM, Ressler KJ, et al. : Brain circuit dysfunction in post-traumatic stress disorder: from mouse to man. Nat Rev Neurosci 2018; 19:535–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berretta S: Cortico-amygdala circuits: role in the conditioned stress response. Stress 2005; 8:221–232 [DOI] [PubMed] [Google Scholar]
- 11.Lobo I, de Oliveira L, David IA, et al. : The neurobiology of posttraumatic stress disorder: Dysfunction in the prefrontal-amygdala circuit? Psychol Neurosci 2011; 4:191–203 [Google Scholar]
- 12.Brown VM, LaBar KS, Haswell CC, et al. : Altered resting-state functional connectivity of basolateral and centromedial amygdala complexes in posttraumatic stress disorder. Neuropsychopharmacology 2014; 39:351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu X, Helpman L, Papini S, et al. : Altered resting state functional connectivity of fear and reward circuitry in comorbid PTSD and major depression. Depress Anxiety 2017; 34:641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen AC, Etkin A: Hippocampal network connectivity and activation differentiates post-traumatic stress disorder from generalized anxiety disorder. Neuropsychopharmacology 2013; 38:1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rauch SL, Shin LM, Phelps EA: Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research--past, present, and future. Biol Psychiatry 2006; 60:376–382 [DOI] [PubMed] [Google Scholar]
- 16.Stevens JS, Jovanovic T, Fani N, et al. : Disrupted amygdala-prefrontal functional connectivity in civilian women with posttraumatic stress disorder. J Psychiatr Res 2013; 47:1469–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun D, Gold AL, Swanson CA, et al. : Threat-induced anxiety during goal pursuit disrupts amygdala-prefrontal cortex connectivity in posttraumatic stress disorder. Transl Psychiatry 2020; 10:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Likhtik E, Stujenske JM, Topiwala MA, et al. : Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nat Neurosci 2014; 17:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S, Hultman R, Hughes D, et al. : Prefrontal cortex reactivity underlies trait vulnerability to chronic social defeat stress. Nat Commun 2014; 5:4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukalo O, Pinard CR, Silverstein S, et al. : Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv 2015; 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, et al. : Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron 2012; 76:804–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fogaça MV, Duman RS: Cortical gabaergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci 2019; 13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang Q, Li Z, Huang G-D, et al. : Traumatic stress produces distinct activations of gabaergic and glutamatergic neurons in amygdala. Front Neurosci 2018; 12:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tovote P, Fadok JP, Lüthi A: Neuronal circuits for fear and anxiety. Nat Rev Neurosci 2015; 16:317–331 [DOI] [PubMed] [Google Scholar]
- 25.Ghosal S, Hare B, Duman RS: Prefrontal cortex gabaergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr Opin Behav Sci 2017; 14:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krabbe S, Gründemann J, Lüthi A: Amygdala inhibitory circuits regulate associative fear conditioning. Biol Psychiatry 2018; 83:800–809 [DOI] [PubMed] [Google Scholar]
- 27.Yu B, Becnel J, Zerfaoui M, et al. : Serotonin 5-hydroxytryptamine(2A) receptor activation suppresses tumor necrosis factor-alpha-induced inflammation with extraordinary potency. J Pharmacol Exp Ther 2008; 327:316–323 [DOI] [PubMed] [Google Scholar]
- 28.Lori A, Maddox SA, Sharma S, et al. : Dynamic Patterns of Threat-Associated Gene Expression in the Amygdala and Blood. Front Psychiatry 2018; 9:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Règue M, Poilbout C, Martin V, et al. : Increased 5-HT2C receptor editing predisposes to PTSD-like behaviors and alters BDNF and cytokines signaling. Transl Psychiatry 2019; 9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young MB, Howell LL, Hopkins L, et al. : A peripheral immune response to remembering trauma contributes to the maintenance of fear memory in mice. Psychoneuroendocrinology 2018; 94:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatt S, Hillmer AT, Girgenti MJ, et al. : PTSD is associated with neuroimmune suppression: evidence from PET imaging and postmortem transcriptomic studies. Nat Commun 2020; 11:2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breen MS, Tylee DS, Maihofer AX, et al. : PTSD Blood Transcriptome Mega-Analysis: Shared Inflammatory Pathways across Biological Sex and Modes of Trauma. Neuropsychopharmacology 2018; 43:469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michopoulos V, Powers A, Gillespie CF, et al. : Inflammation in Fear- and Anxiety-Based Disorders: PTSD, GAD, and Beyond. Neuropsychopharmacology 2017; 42:254–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collado-Torres L, Burke EE, Peterson A, et al. : Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron 2019; 103:203–216.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law CW, Chen Y, Shi W, et al. : voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaffe AE, Tao R, Norris AL, et al. : qSVA framework for RNA quality correction in differential expression analysis. Proc Natl Acad Sci USA 2017; 114:7130–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langfelder P, Horvath S: WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Lecea L, del Rio JA, Criado JR, et al. : Cortistatin is expressed in a distinct subset of cortical interneurons. J Neurosci 1997; 17:5868–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laryea G, Arnett MG, Muglia LJ: Behavioral Studies and Genetic Alterations in Corticotropin-Releasing Hormone (CRH) Neurocircuitry: Insights into Human Psychiatric Disorders. Behav Sci (Basel) 2012; 2:135–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Girgenti MJ, Wang J, Ji D, et al. : Transcriptomic organization of the human brain in post-traumatic stress disorder. Nat Neurosci 2021; 24:24–33 [DOI] [PubMed] [Google Scholar]
- 41.Xu X, Wells AB, O’Brien DR, et al. : Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci 2014; 34:1420–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran MN, Maynard KR, Spangler A, et al. : Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 2021; 109:3088–3103.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velmeshev D, Schirmer L, Jung D, et al. : Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019; 364:685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathys H, Davila-Velderrain J, Peng Z, et al. : Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019; 570:332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ketchesin KD, Huang NS, Seasholtz AF: Cell Type-Specific Expression of Corticotropin-Releasing Hormone-Binding Protein in GABAergic Interneurons in the Prefrontal Cortex. Front Neuroanat 2017; 11:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calakos KC, Blackman D, Schulz AM, et al. : Distribution of type I corticotropin-releasing factor (CRF1) receptors on GABAergic neurons within the basolateral amygdala. Synapse 2017; 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burke EE, Chenoweth JG, Shin JH, et al. : Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat Commun 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran MN, Maynard KR, Spangler A, et al. : Single-nucleus transcriptome analysis reveals cell type-specific molecular signatures across reward circuitry in the human brain. BioRxiv 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eraly SA, Nievergelt CM, Maihofer AX, et al. : Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry 2014; 71:423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Passos IC, Vasconcelos-Moreno MP, Costa LG, et al. : Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2015; 2:1002–1012 [DOI] [PubMed] [Google Scholar]
- 51.Haroon E, Raison CL, Miller AH: Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012; 37:137–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ménard C, Pfau ML, Hodes GE, et al. : Immune and neuroendocrine mechanisms of stress vulnerability and resilience. Neuropsychopharmacology 2017; 42:62–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deslauriers J, Powell S, Risbrough VB: Immune signaling mechanisms of PTSD risk and symptom development: insights from animal models. Curr Opin Behav Sci 2017; 14:123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, He M, Wang D, et al. : HisgAtlas 1.0: a human immunosuppression gene database. Database 2017; 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Lecea L: Cortistatin--functions in the central nervous system. Mol Cell Endocrinol 2008; 286:88–95 [DOI] [PubMed] [Google Scholar]
- 56.Hill JL, Jimenez DV, Mai Y, et al. : Cortistatin-expressing interneurons require TrkB signaling to suppress neural hyper-excitability. Brain Struct Funct 2019; 224:471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maynard KR, Kardian A, Hill JL, et al. : TrkB Signaling Influences Gene Expression in Cortistatin-Expressing Interneurons. eNeuro 2020; 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guilloux JP, Douillard-Guilloux G, Kota R, et al. : Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry 2012; 17:1130–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Babaev O, Piletti Chatain C, Krueger-Burg D: Inhibition in the amygdala anxiety circuitry. Exp Mol Med 2018; 50:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janak PH, Tye KM: From circuits to behaviour in the amygdala. Nature 2015; 517:284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cummings KA, Clem RL: Prefrontal somatostatin interneurons encode fear memory. Nat Neurosci 2020; 23:61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koppensteiner P, Von Itter R, Melani R, et al. : Diminished Fear Extinction in Adolescents Is Associated With an Altered Somatostatin Interneuron-Mediated Inhibition in the Infralimbic Cortex. Biol Psychiatry 2019; 86:682–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu H, Liu L, Tian Y, et al. : A disinhibitory microcircuit mediates conditioned social fear in the prefrontal cortex. Neuron 2019; 102:668–682.e5 [DOI] [PubMed] [Google Scholar]
- 64.Sun Y, Qian L, Xu L, et al. : Somatostatin neurons in the central amygdala mediate anxiety by disinhibition of the central sublenticular extended amygdala. Mol Psychiatry 2020; [DOI] [PubMed] [Google Scholar]
- 65.Yu K, Garcia da Silva P, Albeanu DF, et al. : Central amygdala somatostatin neurons gate passive and active defensive behaviors. J Neurosci 2016; 36:6488–6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyrka AR, Price LH, Gelernter J, et al. : Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol Psychiatry 2009; 66:681–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dedic N, Kühne C, Jakovcevski M, et al. : Chronic CRH depletion from GABAergic, long-range projection neurons in the extended amygdala reduces dopamine release and increases anxiety. Nat Neurosci 2018; 21:803–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Risbrough VB, Stein MB: Role of corticotropin releasing factor in anxiety disorders: a translational research perspective. Horm Behav 2006; 50:550–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toth M, Flandreau EI, Deslauriers J, et al. : Overexpression of forebrain CRH during early life increases trauma susceptibility in adulthood. Neuropsychopharmacology 2016; 41:1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gradus J: Epidemiology of PTSD-PTSD: National Center for PTSD. US Department of Veterans Affairs; 2016; [Google Scholar]
- 71.Gelernter J, Sun N, Polimanti R, et al. : Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nat Neurosci 2019; 22:1394–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maddox SA, Kilaru V, Shin J, et al. : Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD. Mol Psychiatry 2018; 23:658–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chaby LE, Sadik N, Burson NA, et al. : Repeated stress exposure in mid-adolescence attenuates behavioral, noradrenergic, and epigenetic effects of trauma-like stress in early adult male rats. Sci Rep 2020; 10:17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gandal MJ, Zhang P, Hadjimichael E, et al. : Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018; 362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Labonté B, Engmann O, Purushothaman I, et al. : Sex-specific transcriptional signatures in human depression. Nat Med 2017; 23:1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code and figures associated with this manuscript are available through GitHub: https://github.com/LieberInstitute/LIBD_VA_PTSD_RNAseq_4Region. All raw and processed data may be requested via the PTSD Brain Bank Resource Request process described on the “For Investigators” tab of the following page: https://www.research.va.gov/programs/tissue_banking/ptsd/default.cfm