

Abstract
Background and Purpose:
The μ-opioid receptor (μ receptor) is the primary target for opioid analgesics. The 7-transmembrane (TM) and 6TM μ receptor isoforms mediate inhibitory and excitatory cellular effects. Here, we developed compounds selective for 6TM- or 7TM-μ receptors to further our understanding of the pharmacodynamic properties of μ receptors.
Experimental Approach:
We performed virtual screening of the ZINC Drug Now library of compounds using in silico 7TM- and 6TM-μ receptor structural models and identified potential compounds that are selective for 6TM- and/or 7TM-μ receptors. Subsequently, we characterized the most promising candidate compounds in functional in vitro studies using Be2C neuroblastoma transfected cells, behavioural in vivo pain assays using various knockout mice and in ex vivo electrophysiology studies.
Key Results:
Our virtual screen identified 30 potential candidate compounds. Subsequent functional in vitro cellular assays shortlisted four compounds (#5, 10, 11 and 25) that demonstrated 6TM- or 7TM-μ receptor-dependent NO release. In in vivo pain assays these compounds also produced dose-dependent hyperalgesic responses. Studies using mice that lack specific opioid receptors further established the μ receptor-dependent nature of identified novel ligands. Ex vivo electrophysiological studies on spontaneous excitatory postsynaptic currents in isolated spinal cord slices also validated the hyperalgesic properties of the most potent 6TM- (#10) and 7TM-μ receptor (#5) ligands.
Conclusion and Implications:
Our novel compounds represent a new class of ligands for μ receptors and will serve as valuable research tools to facilitate the development of opioids with significant analgesic efficacy and fewer side-effects.
Keywords: 6TM-μ receptor, 7TM-μ receptor, opioid, opioid receptor isoform, pain
1 |. INTRODUCTION
Opioid analgesics, primarily μ-opioid receptor agonists, are the most widely prescribed and clinically utilized drugs to treat moderate-tosevere pain (Busse et al., 2018). The analgesic mechanisms of action of opioids involve μ receptor-mediated activation of Gαi/o proteins, resulting in the inhibitory cellular effects mediated by reduction of adenylyl cyclase activity, inhibition of voltage-gated Ca2+ channels and an increase in cellular K+ conductance (Waldhoer et al., 2004). These molecular mechanisms ultimately lead to inhibition of synaptic transmission, neurotransmitter release, and antinociceptive processes (Serohijos et al., 2011; Waldhoer et al., 2004). Despite clear analgesic efficacy, at least in acute settings, the clinical utility of opioid analgesics has been limited by various adverse effects, including respiratory depression, constipation, physical dependence and addiction liability (Ballantyne & Shin, 2008; Busse et al., 2018). Prolonged use of opioids can also lead to clinically significant problems of tolerance and opioid-induced hyperalgesia (OIH) (Lee et al., 2011; Noble et al., 2010).
It is now well established that the μ receptor gene (Oprm1) undergoes extensive alternative pre-mRNA splicing and generates many splice variants (Shabalina et al., 2009). To date, over 50 splice variants of Oprm1, consisting of the full length 7-transmembrane (TM), truncated 6-TM and single TM isoforms, have been identified in rodents and humans and are available in Ensembl, NCBI Gene and UCSC Browser databases (Convertino, Samoshkin, Gauthier, et al., 2015; Pasternak & Pan, 2013). The 6TM-μ receptor isoform lacks the extracellular N-terminus and first cytoplasmic domain but possesses a cytoplasmic N-terminus followed by six transmembrane domains and the C-terminus of 7TM-μ receptors. The 6TM-μ receptor then retains the ligand binding pocket that is distributed across the conserved transmembrane helical (TMH)-2, TMH-3 and TMH-7 domains and is capable of binding μ receptor agonists (Samoshkin et al., 2015). Although the stimulation of the canonical 7TM-μ receptor has been associated with classical inhibitory cellular effects through the Gαi/o binding, by inhibiting AC and voltage-gated Ca2+ channels and by activating K+ channels, there is also much evidence that 6TM-μ receptors produce excitatory cellular effects of opioids mediated by increased levels of intracellular Ca2+ and NO release (Convertino, Samoshkin, Gauthier, et al., 2015; Convertino, Samoshkin, Viet, et al., 2015; Gris et al., 2010; Samoshkin et al., 2015; Shabalina et al., 2009). The absence of the extracellular N-terminus in the 6TM-μ receptor isoform leads to impaired translocation to the cell membrane when expressed alone, and this isoform is retained intracellularly following heterologous expression (Yang et al., 1998). However, a few dimerization partners have been proposed and demonstrated to facilitate 6TM-μ receptor function and translocation to membrane (Majumdar et al., 2011; Marrone, Grinnell, et al., 2016; Zhang et al., 2020).
Recently, it has been demonstrated that the heterodimerization of 6TM-μ receptors and β2-adrenoceptor underlies the development of OIH in mice (Samoshkin et al., 2015). Previous studies have also shown that the knockdown of the 1K splice variant of the 6TM-μ receptor, via chronic intrathecal siRNA administration not only prevents the development of OIH but also unmasks latent morphine analgesia (Oladosu et al., 2015). Finally, morphine analgesia occurs, following acute administration, in exon 11 KO mice that express 7TM-μ receptor variants but not in exon 1 KO mice which express 6TM-μ receptor variants or in triple KO mice that lack the 7TM-isoform of μ-, δ- and κ- opioid receptors but express 6TM-μ receptor variants (Juni et al., 2007; Pan et al., 2009; Schuller et al., 1999). These findings, along with the observation that OIH develops following continuous infusion of morphine in triple 7TM KO mice (Juni et al., 2007) suggest that 6TM-, but not 7TM-μ receptor isoforms, in large part, underlie the cellular excitatory effects of opioids.
In apparent contradiction to this view, a recently synthesized 6TM-μ receptor ligand, 3′-iodobenzoyl-6β-naltrexamide (IBNtxA), produces an opioid receptor-mediated analgesic response with a vastly improved side-effect profile (Grinnell et al., 2014; Majumdar et al., 2011; Wieskopf et al., 2014). These findings are contrary to human studies where individuals who harbour the minor T allele of rs563649 show reduced morphine analgesia (Shabalina et al., 2009). Of note, this allele codes for higher expression of the 1K variant of 6TM-μ receptors, which produces cellular excitatory responses to morphine or IBNtxA exposure (Samoshkin et al., 2015). Studies that increase our understanding of the biological and cellular mechanisms mediated by the 6TM- and 7TM-μ receptor isoforms will facilitate the development of novel opioid compounds with a high degree of analgesic efficacy and fewer side-effects.
In the present study, we have performed a virtual screening by docking compounds of the ZINC Drug Now library using both 6TM- and 7TM-μ receptor structural models. We identified novel candidate compounds that are potentially selective for 6TM- and/or 7TM-μ receptors. We then characterized the most promising candidate compounds in functional in vitro studies using Be2C neuroblastoma transfected cells, behavioural in vivo pain assays using a range of knockout mice, and in ex vivo electrophysiology studies.
2 |. METHODS
2.1 |. In silico modelling
2.1.1 |. 7TM- and 6TM-μ receptor structural models
We generated the structural models of 7TM- and 6TM-μ receptors, by applying minor changes to the crystallographic coordinates, at 2.80 Å resolution, of the 7TM-μ receptor chimera (PDB-ID: 4DKL; Manglik et al., 2012), as described previously (Convertino, Samoshkin, Viet, et al., 2015; Samoshkin et al., 2015). Briefly, we modelled the 7TM-μ receptor by removing the coordinates of the co-crystallized antagonist β-funaltrexamide, as well as the T4-lysozyme, which is co-crystallized between helices 5 and 6 of the opioid receptor. To generate the structural model of the 6TM-μ receptor, we removed the receptor’s first trans-membrane helix. For both 7TM- and 6TM-μ receptor isoforms, we manually built the amino acids constituting the i3 loop, and explored its conformational space using discrete molecular dynamics simulations (Dokholyan et al., 1998; Shirvanyants et al., 2012). No critical issues were found in the structural models of the two isoforms of opioid receptor, as assessed by Gaia (http://chiron.dokhlab.org; RRID:SCR_009182), an in-house developed software, which compares the intrinsic structural properties of the generated models to high-resolution crystallographic structures (Kota et al., 2011; Ramachandran et al., 2011).
2.1.2 |. Molecular library preparation
As described previously (Politi et al., 2016), we took advantage of the chemical space redundancy of the ZINC library by optimizing only representative compounds of different chemical classes included in the ZINC-12 Drug Now subset (Irwin et al., 2012), which contains ~11 million entries. Compounds in the library subset were characterized by (i) net charge between −3 and +3, (ii) H-bond donors ≤5, (iii) H-bond acceptors ≤10, (iv) cLogP between −6 and 5, (v) MW between 150 and 500 Da, (vi) PSA between 0 and 150, and (vii) rotatable bonds ≤7. To perform clustering reduction of the database entries, ligands were sorted by increasing MW. The SUBSET 1.0 algorithm was then used to progressively select compounds that differed from those previously selected by at least the Tanimoto cut-off. Representatives were selected via Tanimoto similarity-based clustering approach (Voigt et al., 2001), which reduces the chemical space to about 350,000 distinct chemical entities (Tanimoto coefficients’ mode of post-screening top-ranked compounds equals 0.3; Figure S1A, B). The resulting representatives had two properties: (i) each representative differed from all the others by at least the Tanimoto cut-off, and (ii) all the molecules in the subset were within the Tanimoto cut-off of at least one representative. Thus, the representatives can be said to “cover” the chemical space of the subset at a given Tanimoto level. All of the entries were further optimized using the LigPrep application (RRID:SCR_016746), as available via the Schrödinger Suite, using the MMFFs force field (Halgren, 1996), and titratable groups are protonated at pH = 7.0. Compounds’ chirality was retained as specified in the ZINC library.
2.1.3 |. In silico docking calculations
Docking calculations were performed using MedusaDock (Ding et al., 2010; J. Wang & Dokholyan, 2019), an in-house developed software that simultaneously models the flexibility of both ligand and receptor; while docking poses were evaluated using MedusaScore (Yin et al., 2008), a scoring function based on the physics-based force field accounting for the protein-ligand interaction energy. By properly modelling the induced-fit phenomenon upon ligand binding (Dokholyan, 2016), MedusaDock is not sensitive to the starting conformations of amino acids in both 7TM- and 6TM-binding sites. Thus, the docking results were not biased by the starting conformations of the 7TM-μ receptor in complex with β-funaltrexamide. Starting from a low number of independent docking attempts, we isolated an ensemble of candidates that have the lowest binding energies to each receptor’s binding site (i.e. ‘best classes’ of compounds). The isolated ligands underwent two additional and more exhaustive tiers of independent docking calculations, which were focused on the identification of putative hit compounds.
The maximum number of independent docking calculations (i.e. 500 in this study) was chosen after analysing the convergence of docking poses energies as described in (Convertino & Dokholyan, 2016). The estimated binding energies for all the docking pose of any docked compound follows a normal distribution. Therefore, according to the central limit theorem, only the docking poses, for which the Z-score value was lower than −2, were retrieved as statistically significant solutions, which can pass the tier-based filters. In this study, is defined as:
where was the estimated binding energy of a specific docking poses of a given compounds and and were the mean and the SD of the binding energies in the population of binding poses for all of docked molecules, respectively.
2.1.4 |. In silico targets prediction
We employed a in-house-developed software DRIFT (http://drift.dokhlab.org; https://dokhlab.med.psu.edu/drift) to predict potential targets of the screened compounds. DRIFT generates 2D FP2 fingerprints and 3D pharmacophores for the query compound and performs chemical similarity search against large curated datasets of bioactive molecules with drug-like properties [ChEMBL (RRID:SCR_014042), ZINC (RRID:SCR_008596), HMDB (RRID:SCR_007712) and BindingDB (RRID:SCR_000390)]. The identified compounds were then used as baits to obtain their associated targets and were subsequently ranked and listed based on the extent of similarity with the query structure.
2.2 |. In vitro pharmacology
2.2.1 |. Cell culture
The human Be2C neuroblastoma cells (RRID:CVCL_0529) or HEK293 cells (RRID:CVCL_0045; American Tissue Culture Collection [ATTC]) were transiently transfected with human 6TM-, 7TM-μ receptor, β2-adrenoceptor, 6TM-μ receptor / β2-adrenoceptor or empty vector (EV) constructs and cultured, as described previously (Gris et al., 2010; Samoshkin et al., 2015). Briefly, the cells were grown in T75 flasks in DMEM/F12 (Be2C) or DMEM (HEK293) medium supplemented with 10% FBS and penicillin/streptomycin at 37°C. When confluent, the cells were seeded on 6 cm cell culture dishes and transfected next day using 3 μg of plasmid per dish, and XtremeGENE transfection reagent (Roche) as per manufacturer instructions.
2.2.2 |. NO assay
NO release is a cellular phenotype we have shown previously to be characteristic of the activation of the 6TM-μ receptor isoform (Gris et al., 2010; Samoshkin et al., 2015). The NO release from the cells was measured using NO-specific amperometric probe (Innovative Instruments, Tampa, FL), as described previously (Gris et al., 2010). Briefly, NO release was measured 48 h post-transfection in PBS to prevent clogging of the probe pores by medium. The probe was lowered just above the cells in the 6 cm dishes and allowed to reach a stable baseline before adding any compounds by gentle pipetting. Initial experiments involved treating transfected cells with all experimental compounds at a 10 μM concentration. The majority of the compounds did not produce any NO signal, serving as an internal negative control. A separate cohort of cells were then treated with a logarithmic concentration range of compounds 5, 10, 11 and 25.
For studies involving co-administration of a non-selective opioid antagonist (naloxone; 10 μM) and a selective β2-adrenoceptor antagonist (ICI 118,551; 10 μM), the transfected cells were first incubated for 30 min with the antagonists at 37°C before placing the probe in the plate and adding the experimental compounds (10 μM). Each experiment was repeated at least three times along with appropriate control cell lines. The antagonistic properties of compounds were examined in 6TM-μ receptor-transfected cells by testing their ability to block morphine-dependent NO release. The transfected cells were pretreated with 10 μM of test compounds, for 30 min, before morphine treatment (10 μM).
2.2.3 |. cAMP assay
Production of cAMP, stimulated by isoprenaline, was determined using the luciferase biosensor assay (GloSensor™ cAMP Assay; Promega), as described previously (Martin et al., 2015). Briefly, the cells were transfected in 6 cm cell culture dishes as described above, and co-transfected with a plasmid coding for GloSensor-22F vector. Following 24 h post-transfection, the cells were seeded onto a white tissue culture 384-well plate coated with poly-L-lysine (20,000 cells per well) with a clear bottom. The next day, the well plates were incubated with the GloSensor™ cAMP reagent for 2 h at room temperature. Cells were then treated with a logarithmic concentration range of compounds for 30 min before adding 100 nM isoprenaline to stimulate cAMP production. We also tested the cAMP-stimulating properties of the compounds without isoproterenol treatment. The antagonistic properties of compounds were examined in 7TM-MOR transfected cells by testing their ability to block morphine-induced cAMP inhibition. The transfected cells were pretreated with 10 μM of test compounds 30 min before morphine was added at log-concentrations. To determine cAMP levels in each experiment, the luminescence was measured after 20 min of stimulating treatment using the PHERAStar microplate reader (BMG Labtech). Each experiment was repeated at least three times along with appropriate control cell lines.
2.3 |. In vivo pharmacology
2.3.1 |. Animals
All animal care and experimental procedures were approved by McGill University’s Animal Care Committee and were performed in accordance with the guidelines of the International Association for the Study of Pain. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
All experiments were performed on naïve adult (8–12 weeks old) male and female mice. C57BL/6 (Stock# 000664; RRID: IMSR_JAX:000664) mice were purchased from The Jackson Laboratory. Homozygous breeders of Adrb1tm1Bkk Adrb2tm1Bkk/J (Stock# 003810; RRID:IMSR_JAX:003810) and B6.129S2-Oprm1tm1Kff/J (Stock# 007559; RRID:IMSR_JAX:007559) were obtained from The Jackson Laboratory and were bred in-house to generate Adrb1/2−/− and Oprm1−/− mice. Null mutant mice lacking expression of opioid receptor delta 1 gene (Oprd1−/−; RRID:IMSR_JAX:007557) and opioid receptor kappa 1 gene (Oprk1−/−; RRID:IMSR_JAX:007558) were bred in-house from homozygous breeders obtained from Dr. Brigitte L. Kieffer (McGill University). Mice were housed with their same-sex littermates in standard shoebox cages, maintained in a temperature controlled (20 ± 1°C) environment (12:12 h light/dark cycle; lights on at 07:00 h) and received standard rodent food and water ad libitum.
2.3.2 |. Tail-withdrawal assay
Tail-withdrawal latency (TWL) values were determined as previously described (Mogil et al., 2006). Briefly, adult naïve male and female C57BL/6 (wild-type), Oprm1−/−, Oprd1−/−, Oprk1−/− and Adrb1/2−/− mice were lightly restrained in a cloth and cardboard holder. The distal half of the mouse’s tail was dipped into a water bath thermostatically maintained at 47°C. The latency of response to the heat stimuli by vigorous tail flexion was measured. The baseline TWL values are the average of four latency determinations collected at least 30 min apart. All behavioural experiments were undertaken in a blinded manner.
2.3.3 |. Administration of test compounds
Solutions of the test compounds in the desired concentrations were prepared by one person and were assigned codes independently by a second person. The coded test compound solutions were administered to mice randomly, and the tail withdrawal assay was undertaken in a blinded manner. The test compounds 5, 10, 11, 22, 25 and 28 were dissolved in 20% polyethylene glycol 200 (PEG) and administered s.c. ICI 118,551 and naloxone hydrochloride were dissolved in physiological saline and administered i.p.
2.3.4 |. In vivo pharmacological characterization
Following baseline TWL determinations, groups of adult naïve male and female C57BL/6 mice received single s.c. bolus doses of compounds 5 (10, 20 and 40 mgkg−1), 10 (10, 20 and 40 mgkg−1), 11 (10, 20 and 40 mgkg−1), 22 (20 mgkg−1), 25 (10, 20 and 40 mgkg−1), 28 (20 mgkg−1) or vehicle (20% PEG). The TWL values were determined at 15-, 30-, 60-, 90- and/or 120- min post-administration of compounds.
Separate groups of naïve male and female C57BL/6 mice received an ~ED75 dose of compounds 5 (20 mgkg−1, s.c.), 10 (20 mgkg−1, s.c.), 11 (40 mgkg−1, s.c.) and 25 (20 mgkg−1, s.c.). Subsequently, these mice received an i.p. bolus dose of ICI 118,551 (5 mgkg−1), naloxone hydrochloride (1 mgkg−1 e.q.) or vehicle (saline), and the TWL values were determined at 30- and 60-min post-dosing time points. Given the 10–15-fold higher affinity of naloxone for the μ receptor over the δ- and κ-opioid receptors (Tam, 1985), a low dose of naloxone was used to try to ensure only μ receptors were blocked.
Finally, groups of adult male and female naïve Oprm1−/−, Oprd1−/−, Oprk1−/− and Adrb1/2−/− mice received an ~ED75 single s.c. bolus doses of compounds 5 (20 mgkg−1), 10 (20 mgkg−1), 11 (40 mgkg−1) and 25 (20 mgkg−1) or vehicle (20% PEG), and the TWL values were assessed at 30-, 60- and 120-min post-dosing time points.
The TWL values are presented as differences from their baselines (ΔTWL). The extent and duration of hyperalgesia (area over the ΔTWL vs. time curves) for test compounds or vehicle in individual mouse were determined using trapezoidal integration. Dose–response curves were generated, and non-linear regression method was used to estimate effective dose 75% (ED75) values.
2.4 |. Ex vivo electrophysiology
2.4.1 |. Spinal cord slice preparation
Adult male mice (5–7 weeks old; n = 7–8 mice per compound) were anaesthetized using urethane (1.5–2.0 gkg−1, i.p.). The lumbosacral spinal cord was carefully removed and submerged in ice-cold sucrose-artificial CSF (ACSF, in mM): 240 sucrose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2 and 3.5 MgCl2, which was saturated with 95% O2 and 5% CO2 at room temperature. After extraction and still under anaesthesia, animals were killed by decapitation. Transverse slices (300–400 μm) were cut using a vibrating microslicer (Leica-VT1200S). The slices were incubated at 32°C for at least 30 min in regular ACSF (in mM) (NaCl 126, KCl 3, MgCl2 1.3, CaCl2 2.5, NaHCO3 26, NaH2PO4 1.25 and glucose 11) equilibrated with 95% O2 and 5% CO2.
2.4.2 |. Electrophysiological recording
Patch clamp recordings were conducted previously reported (Wang et al., 2020). The spinal cord slice placed in the recording chamber was completely submerged and superfused at a rate of 2–4 mlmin−1 with ACSF. Lamina II neurons in lumbar segments were identified as a translucent band under a microscope (BX51WIF; Olympus) with light transmitted from below. Whole-cell voltage-clamp recordings were made from lamina II neurons by using patch-pipettes. Patch-pipette solution used to record excitatory postsynaptic currents (EPSCs) contained (in mM): K-gluconate 135, KCl 5, CaCl2 0.5, MgCl2 2, EGTA 5, HEPES 5, Mg-ATP 5 (pH 7.3 adjusted with KOH, 300 mOsm). The tip of patch-pipettes had a resistance of 8–10 M. The spontaneous EPSCs (sEPSC) recordings were made at a holding potential (VH) of −70 mV in the presence of 10 μM picrotoxin and 2 μM strychnine. Signals were acquired using an Axopatch 700B amplifier. The data were stored and analysed with a personal computer using pCLAMP 10.3 software (RRID:SCR_011323). sEPSC events were detected and analysed using Mini Analysis Program ver. 6.0.3 (RRID:SCR_002184).
2.5 |. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. In line with BJP’s recommendations, statistical analysis was not undertaken for our in vitro data as the ‘n’ were less than 5 for some of the experimental groups. All in vivo studies involved mice of n > 5 per experimental group, except for the vehicle group mice (n = 4) shown in Figure 5e. The differences in the ‘n’ number of mice used per experimental group in our studies shown in Figures 5–7 were due to random allocation of mice to the treatment groups. Our in vivo studies were also undertaken in mice of both sexes, and the distribution of male and female mice in each experimental group was approximately equal. For behavioural analysis, the group sizes reported are the number of independent mice studied. In electrophysiological experiments, the ‘n’ refers to the number of the neurons studied. Statistical analyses were performed using GraphPad Prism v.8.4 (RRID:SCR_002798) and statistical comparisons were performed using Student’s t-test or two-way ANOVA (with Bonferroni), as indicated. The post hoc tests were conducted only if F in ANOVA achieved P < .05, and there were no significant variance inhomogeneity. All data are presented as mean ± SEM, and the level of significance was in all cases set to P < .05.
FIGURE 5.
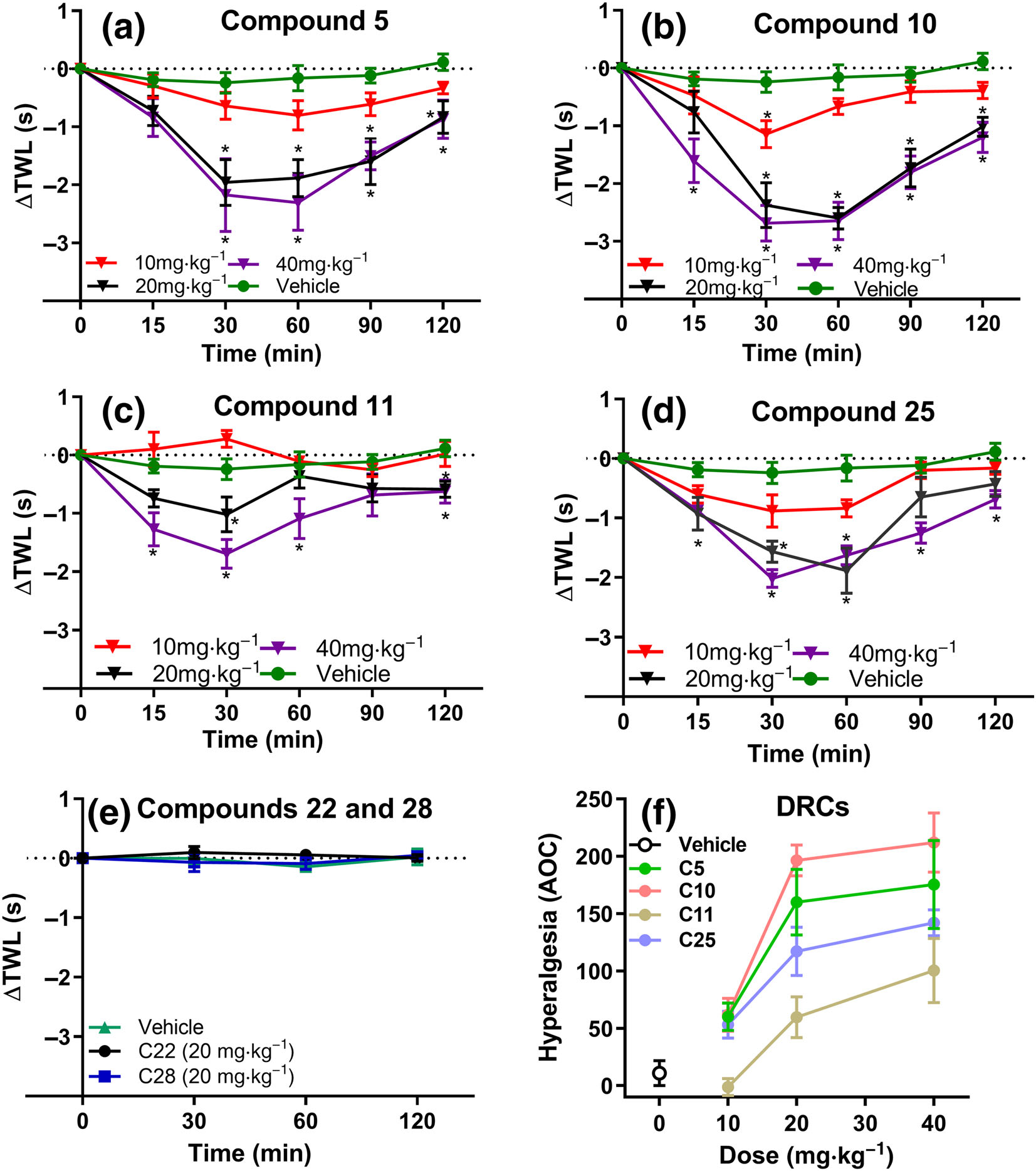
Temporal changes in the mean ± SEM ΔTWL values assessed using the tail-withdrawal assay (47°C). Subcutaneous administration of compound (a) 5 (n = 7, 8 and 7 respectively for 10, 20 and 40 mgkg−1) (b) 10 (n = 7, 8 and 7, respectively for 10, 20 and 40 mgkg−1) (c) 11 (n = 7, 10 and 7, respectively for 10, 20 and 40 mgkg−1) and (d) 25 (n = 7, 10 and 7, respectively for 10, 20 and 40 mgkg−1) but not vehicle (n = 10), produced significant dose-dependent temporal changes in the thermal hyperalgesia of naïve adult C57BL/6 mice. In contrast, administration of compounds (e) 22 and 28 (n = 7 per compound) did not elicit any temporal changes in the ΔTWL values, relative to vehicle (n = 4). (f) Dose response curves (DRCs) for each of the compounds tested in the tail-withdrawal nociceptive assay. Data presented as mean ± SEM. *P < 0.05, significantly different from vehicle (20% PEG); two-way ANOVA, with post hoc Bonferroni test
FIGURE 7.
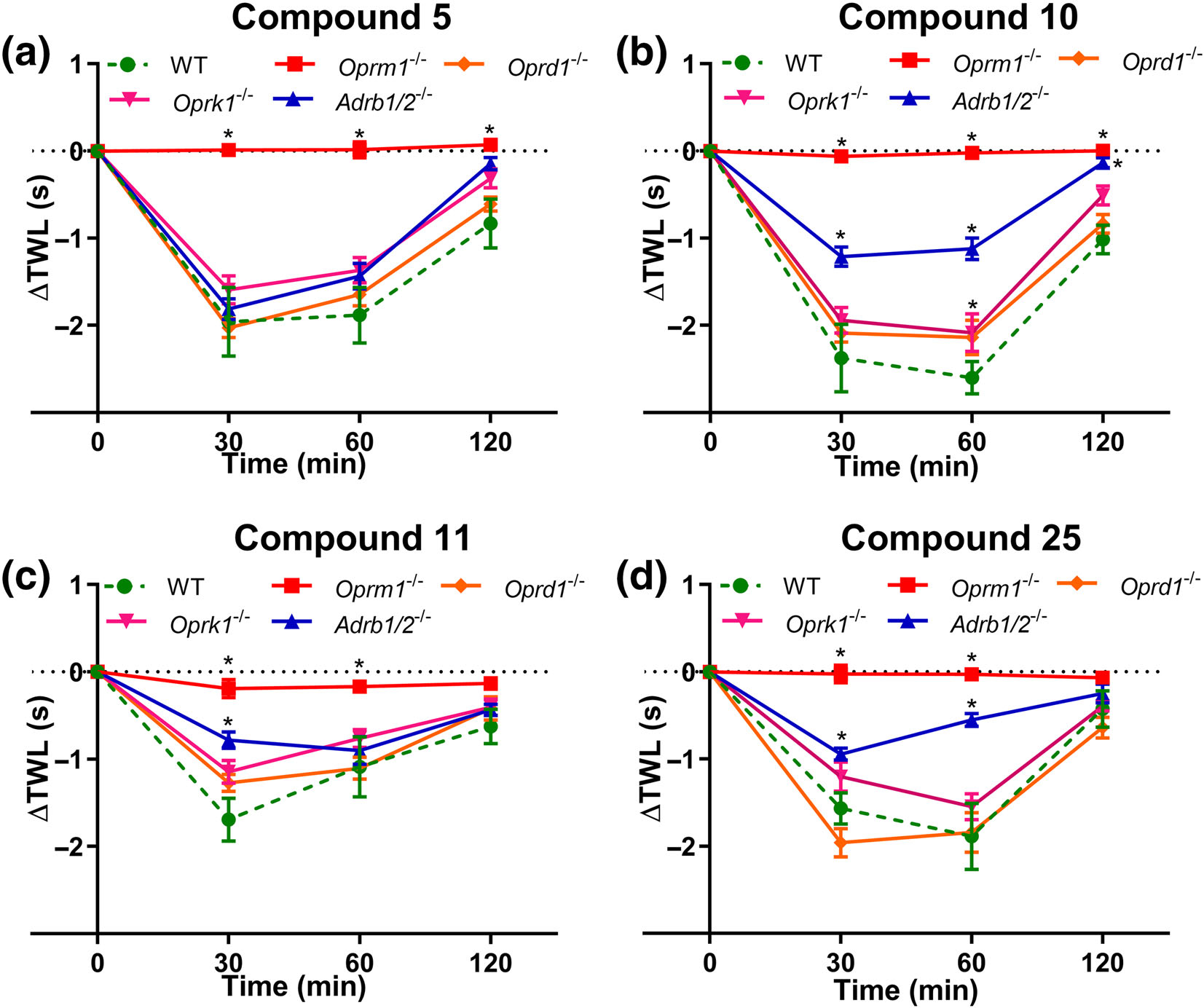
Pharmacological characterization of compounds (a) 5, (b) 10, (c) 11 and (d) 25 in adult male and female naïve Oprm1−/− (n = 8, 8, 6 and 6 for compounds 5, 10, 11 and 25, respectively), Oprd1−/− (n = 7, 7, 6 and 7 for compounds 5, 10, 11 and 25, respectively), Oprk1−/− (n = 9, 9, 6 and 9 for compounds 5, 10, 11 and 25, respectively), Adrb1/2−/− (n = 8, 8, 6 and 8 for compounds 5, 10, 11 and 25, respectively) and C57BL/6J mice (n = 8, 8, 7 and 10 for compounds 5, 10, 11 and 25, respectively). Homozygous ablation of Oprd1 or Oprk1 did not significantly affect the thermal hyperalgesia induced by ~ED75 doses of compounds 5, 10, 11 and 25 compared with wild-type C57BL/6 mice. In contrast, subcutaneous administration of an ~ED75 dose of all compounds - 5, 10, 11 and 25 - failed to produce thermal hyperalgesia in Oprm1−/− mice lacking both 6- and 7-TM MOR. Homozygous ablation of Adrb1 and Adrb2 only partly restored the thermal hypersensitivity induced by compounds 10, 11 and 25, but not the hyperalgesic effects of compound 5. Data presented as mean ± SEM. *P < 0.05, significantly different from WT C57BL/6 mice; two-way ANOVA with post hoc Bonferroni test
2.6 |. Materials
ICI 118,551, isoprenaline and naloxone hydrochloride were supplied by Sigma Aldrich (Oakville, Canada); picrotoxin and strychnine were supplied by Sigma Aldrich (St Louis, MO, USA); IBNtxA was supplied by SRI Int. (Menlo Park, CA, USA); morphine was supplied by Medisca Pharmaceutique Inc, (Quebec, Canada). The compounds 5, 10, 11 and 25, and 22 and 28 were supplied by ChemBridge, San Diego, CA, USA.
2.7 |. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Christopoulos, et al., 2019; Alexander, Fabbro et al., 2019; Alexander, Mathie et al., 2019).
3 |. RESULTS
3.1 |. In silico modelling
We performed the virtual screening process involving a multitier-based docking strategy (Figure 1), with MedusaDock software (Ding et al., 2010) and the MedusaScore scoring function (Yin et al., 2008), using structural models of both 7TM- and 6TM-μ receptors. From 350,000 compounds present in the ZINC Drug Now library, we identified a short list of 56 ligands with selectivity for 7TM-μ receptors, 38 ligands with selectivity for 6TM-μ receptors and 103 compounds with putative binding activities for both opioid isoforms (non-selective). For each of the three classes of molecules (i.e. 7TM- and 6TM-selective, and non-selective compounds), we chose the first 10 top-ranked compounds (Table S1). We were able to obtain 22 out of the top-ranked 30 compounds, and subsequently tested them for 6TM- and 7TM-μ receptor-mediated cellular responses.
FIGURE 1.
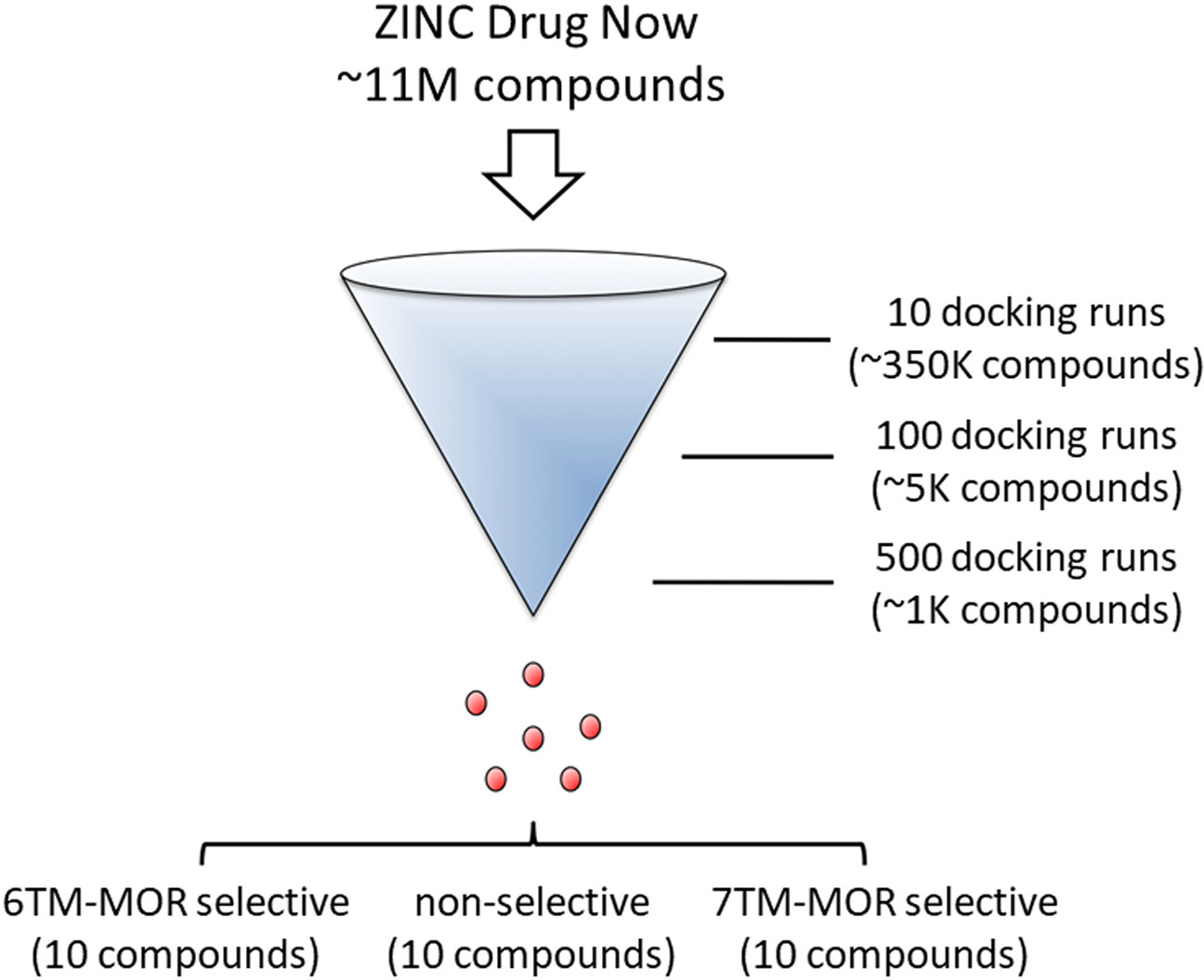
Tier-based strategy for virtual drug screening. We initiated the screening campaign by exploring the ability of 350,000 representative entries of the ZINC Drug Now repository (containing ~11 million commercially available compounds) to bind to the 6TM-μ receptor (6-TM-MOR) and 7TM-μ receptor (7-TM-MOR). The initial low number (i.e. 10) of independent docking attempts allowed the identification of best classes of putative binders. The isolated entries underwent two more exhaustive tiers of independent docking calculations (i.e. 100 and 500 attempts) that were designed to filter out putative hits with favourable binding energy towards μ receptor isoforms (additional details in the Methods section). We were able to purchase a total of 22 compounds that were retrieved from the top lists of (i) 6TM-, (ii) 7TM-selective, and (iii) non-isoform-selective docking solutions
3.2 |. In vitro pharmacological characterization of candidate compounds
3.2.1 |. Test compound responses in a NO release assay using cells expressing μ receptors
Morphine-dependent release of NO has been previously shown to reduce opioid analgesia and to increase analgesic tolerance and OIH in animal models (Toda et al., 2009). Our previous work has also demonstrated NO release to be characteristic of activation of 6TM-μ receptors (Gris et al., 2010; Samoshkin et al., 2015). Hence, we first tested each of the 22 compounds (n = 3 per transfected cell line) for their ability to modulate NO release. We used 10 μM for each of the test compounds on Be2C human neuroblastoma cell lines transfected with 6TM-μ receptors, 7TM-μ receptors or an empty vector (EV), using morphine as a positive control (Figure S2A–C). Although Be2C cells endogenously express both 6TM- and 7TM-μ receptors (Gris et al., 2010), we additionally transfected the cells with the corresponding gene to enhance the cellular response. We also tested IBNtxA, which is a ligand that acts in a 6TM-μ receptor-dependent manner (Grinnell et al., 2014; Majumdar et al., 2011; Wieskopf et al., 2014).
Morphine (n = 7, 5 and 6 in 6TM-μ receptor, 7TM-μ receptor and EV transfected cell line, respectively) released NO in EV and this effect was augmented in 6TM-μ receptor transfected cells (Figure S2A), in line with previous findings (Gris et al., 2010). Most of the compounds, including IBNtxA (n = 3 per transfected cell line), did not lead to NO release from the cells (Figure S2A–C). In contrast, compounds 5, 10, 11, and 25 (Figure 2) increased NO release (n = 3 per transfected cell line; Figure S2A). Compounds 10, 11, and 25 had a similar profile, releasing high levels of NO in 6TM-μ receptor transfected cells and low levels in EV and 7TM-μ receptor transfected cells (Figure S2A). In contrast, compound 5 had virtually no effect on 6TM-μ receptor cells but released NO in EV and 7TM-μ receptor cells in a similar manner (Figure S2A). The difference of NO release levels by different compounds in EV-transfected cells probably reflects the selectivity of a test compound between 6TM- or 7TM-μ receptor- dependent excitatory effects, as endogenous expression of 7TM-μ receptors is much higher in Be2C cells than that of 6TM-μ receptors. Of note, our virtual drug screening predicted compound 25 as a 6TM-μ receptor selective molecule, and compounds 5, 10 and 11 as non-selective μ receptor ligands. All four compounds have little similarity to known inhibitors, as predicted by the DRIFT algorithm, suggesting poor off-target activity (Figure S3A–D).
FIGURE 2.
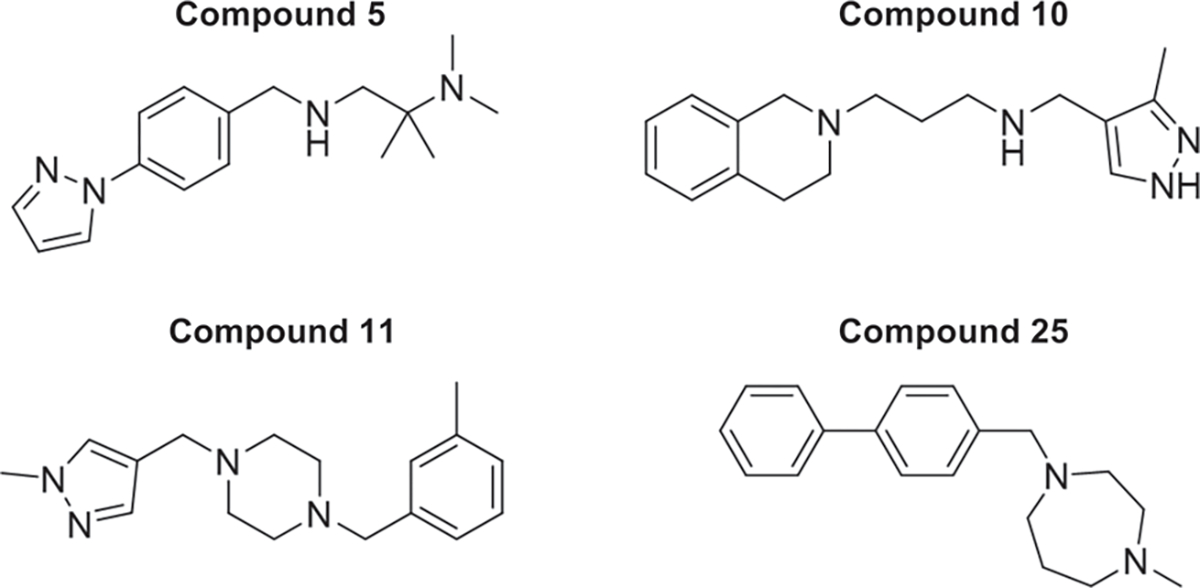
Chemical structures of compounds 5, 10, 11 and 25
The effects of compounds 5, 10, 11 and 25 were further characterized in subsequent dose–response studies, where the compounds were applied to 6TM- or 7TM-μ receptor-transfected cells at 10 nM to 50 or 100 μM concentrations (Figure 3a–d). Compounds 10 (Figure 3b), 11 (Figure 3c) and 25 (Figure 3d) induced a dose-dependent release of NO in cells transfected with 6TM-μ receptors. In these experiments, we also co-transfected Be2C cells with 6TM-μ receptors with β2-adrenoceptors because our previous experiments showed that heterodimerization of these receptors significantly increases the plasma membrane expression of and the functionality of 6TM-μ receptors (Samoshkin et al., 2015). In line with these findings, we observed a pronounced dose-dependent NO release with compounds 10 (n = 3, 4, 3 and 4 per dose tested in 7TM-μ receptor, 6TM-μ receptor, β2-adrenoceptor and 6TM-μ receptor/β2-adrenoceptor transfected cell lines respectively), 11 (n = 3, 4, 3 and 4 per dose tested in 7TM-μ receptor, 6TM-μ receptor, β2-adrenoceptor and 6TM-μ receptor/β2-adrenoceptor transfected cell line, respectively), and 25 (n = 3 for log doses −8.0, −7.0 and −6.0 per transfected cell line and n = 3, 5, 3 and 5 in 7TM-μ receptor, 6TM-μ receptor, β2-adrenoceptor and 6TM-μ receptor/β2-adrenoceptor transfected cell lines, respectively, for log doses −5.0, −4.3 and −4.0) in cells co-transfected with β2-adrenoceptors in comparison with their effect on cells transfected with 6TM-μ receptor alone. The compounds had no effect in cells transfected only with β2-adrenoceptors. In contrast, compound 5 released NO dose-dependently in 7TM-μ receptor-transfected cells (n = 4 per dose tested; Figure 3a) as well as EV transfected cells to a similar level (data not shown). Importantly, compound 5 had no effect in cells transfected with either β2-adrenoceptors alone (n = 3 per dose tested) or 6TM-μ receptors /β2-adrenoceptors (n = 4 per dose tested; Figure 3a).
FIGURE 3.
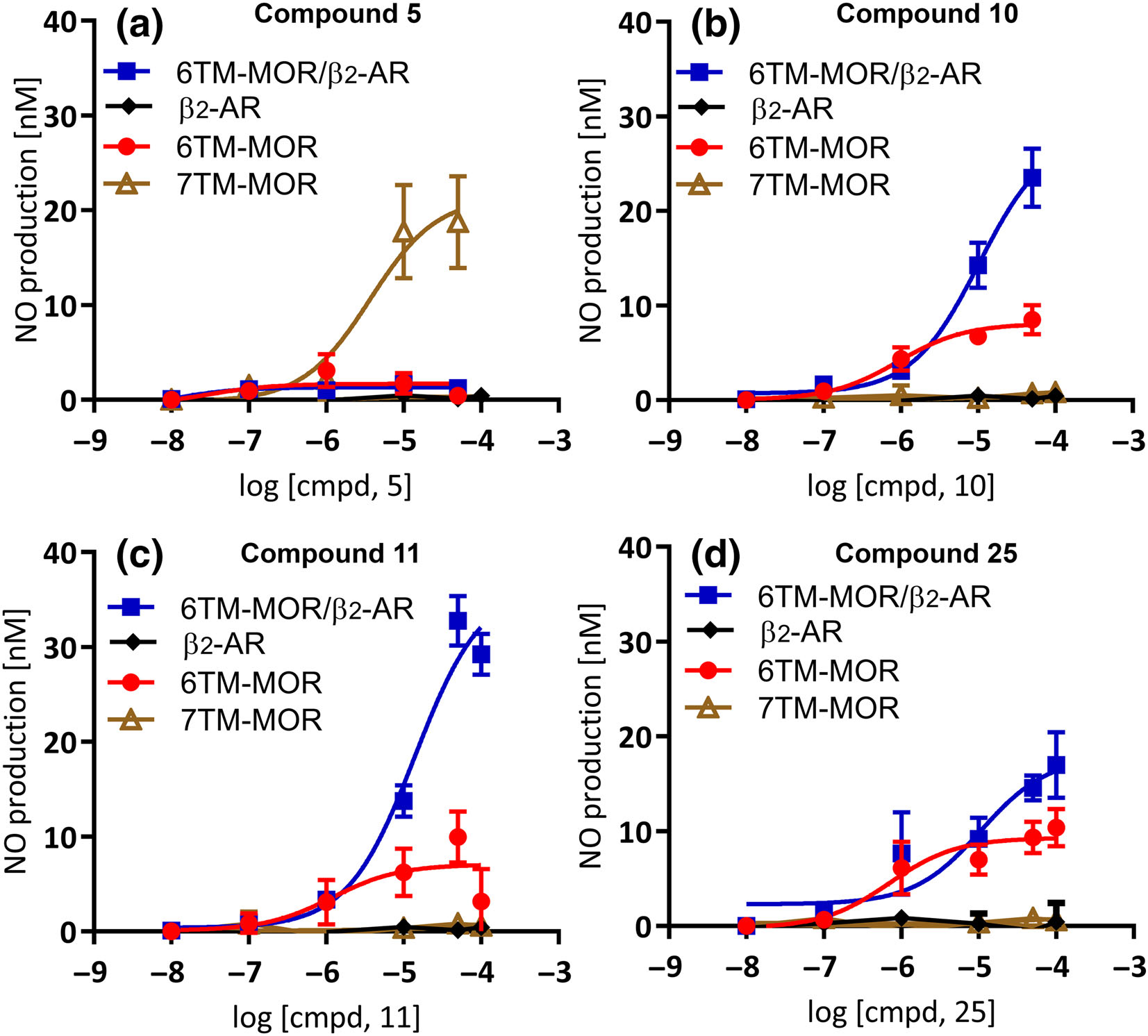
Dose-dependent NO release for (a–d) compounds 5, 10, 11 and 25 in Be2C cells transfected with 7TM-μ receptors (7TM-MOR), 6TM-μ receptors (6TM-MOR), β2-adrenoceptors (β2-AR) or 6TM-MOR+β2-AR. (a) Compound 5 (n = 4, 4, 3 and 4 per dose tested in 7TM-MOR, 6TM-MOR, β2-AR and 6TM-MOR/β2-AR transfected cell line, respectively) induced a dose-dependent release of NO in 7TM-MOR transfected cells only. Compounds (b) 10 (n = 3, 4, 3 and 4 per dose tested in 7TM-MOR, 6TM-MOR, β2-AR and 6TM-MOR/β2-AR transfected cell line, respectively), (c) 11 (n = 3, 4, 3 and 4 per dose tested in 7TM-MOR, 6TM-MOR, β2-AR and 6TM-MOR/β2-AR transfected cell line, respectively) and (d) 25 (n = 3 for log doses −8.0, −7.0 and −6.0 per transfected cell line and n = 3, 5, 3 and 5 in 7TM-MOR, 6TM-MOR, β2-AR and 6TM-MOR/β2-AR transfected cell lines, respectively, for log doses −5.0, −4.3 and −4.0) induced a dose-dependent release of NO in 6TM-MOR transfected cells, and this effect was potentiated by co-transfection with β2-AR. None of the compounds released NO when the cells were transfected only with β2-AR. Data expressed as mean ± SEM
We also screened all compounds for antagonistic properties by testing their ability to block morphine-dependent NO release in 6TM-μ receptor transfected cells. However, none of the compounds showed effects on morphine-dependent NO release (data not shown).
3.2.2 |. Characterization of μ receptor-dependent NO release by compounds 5, 10, 11 and 25 with naloxone and ICI 118,551
Next, we pretreated cells with a non-selective opioid antagonist, naloxone or a selective β2-adrenoceptor antagonist, ICI 118,551 to block opioid receptors or β2-adrenoceptors respectively, before application of test compounds inducing NO release (Figure 4a–d). The NO release induced by compound 5 was blocked by naloxone (10 μM), but not by ICI 118,551 (10 μM) in 7TM-μ receptor-(Figure 4a) and EV-transfected cells (data not shown), supporting the binding of compound 5 to 7TM-μ receptors (n = 5, 3 and 4 for vehicle, naloxone and ICI 118,551, respectively). Conversely, the ability of ICI 118,551 (10 μM), but not naloxone (10 μM), to inhibit the NO-release induced by compounds 10 (Figure 4b), 11 (Figure 4c) and 25 (Figure 4d) (10 μM), support binding of compounds 10, 11 and 25 to the 6TM-isoform (n = 5, 4 and 4 for vehicle, naloxone and ICI 118,551 per transfected cell line per compound). Note that NO-release was inhibited by ICI 118,551 in both 6TM-μ receptor and 6TM-μ receptor /β2-adrenoceptor transfected cells, due to endogenous expression of β2-adrenoceptors in Be2C cells, although the amplitude of this inhibition was greatly enhanced in 6TM-μ receptor /β2-adrenoceptor-transfected cells. The subsequent experiments were designed to determine if the test compounds show MOR-dependent activity.
FIGURE 4.
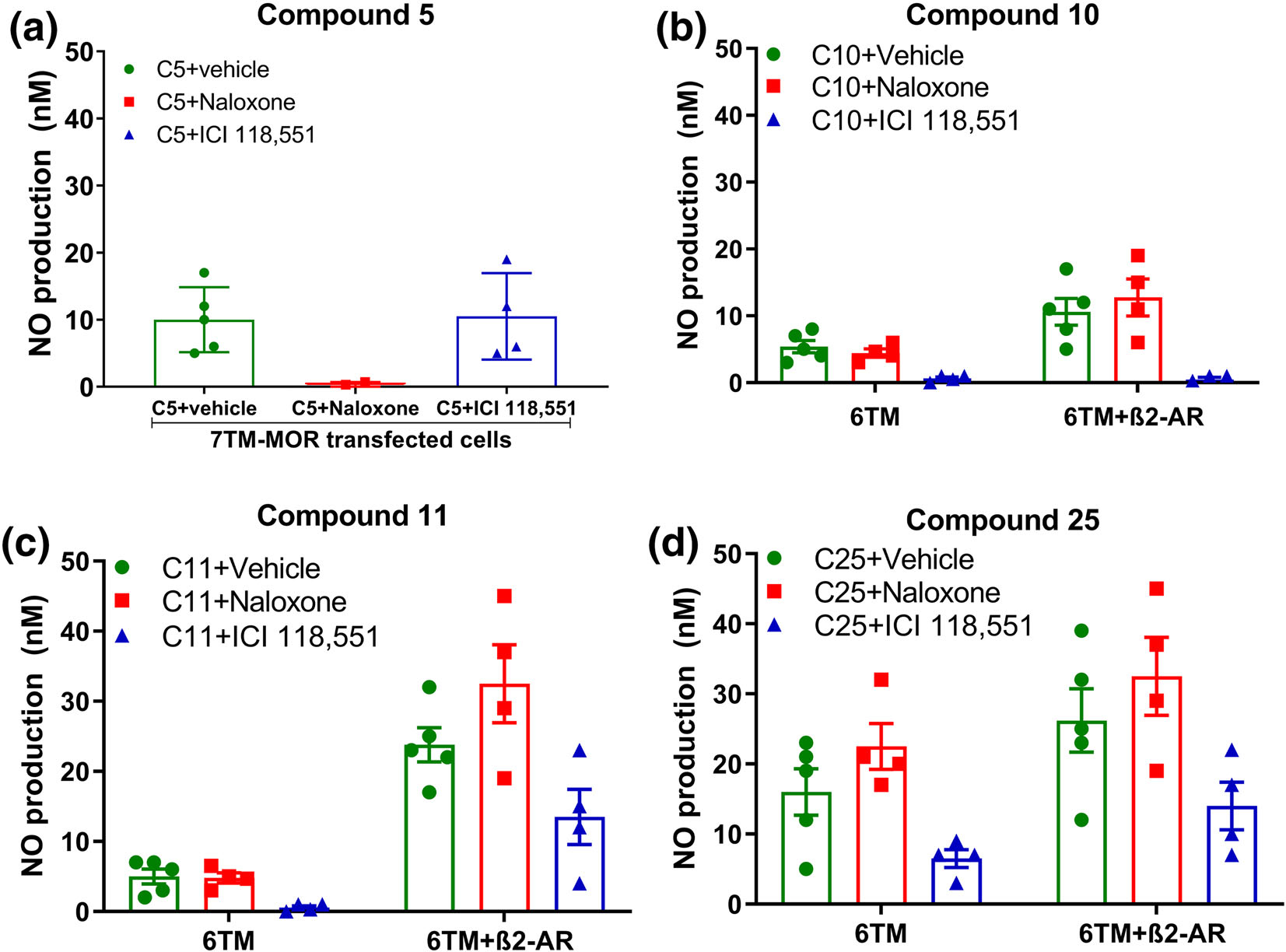
Antagonistic effects of the non-selective opioid receptor antagonist, naloxone, and the selective β2-adrenoceptor (β2-AR) antagonist, ICI 118,551, on NO-release induced by compounds 5, 10, 11 and 25. (a) NO release induced by 10 μM compound 5 (n = 5, 3 and 4 for vehicle, naloxone and ICI 118,551, respectively) in 7TM-MOR transfected cells was abolished by pretreatment with 10 μM naloxone but not by ICI 118,551. (b–d) NO release induced by 10 μM compounds (b) 10, (c) 11 or (d) 25 (n = 5, 4 and 4 for vehicle, naloxone and ICI 118,551 per transfected cell line per compound) was blocked by pretreatment with 10 μM ICI 118,551 in cells transfected with 6TM-MOR alone or together with β2-AR. Data expressed as mean ± SEM
3.2.3 |. Screening of compounds in a cAMP production assay
The inhibition of AC is characteristic of 7TM-μ receptor-activation and has been implicated in the suppression of neuronal activity (Crain & Shen, 2000; Waldhoer et al., 2004). Thus, we next tested the effects of the 22 compounds on cAMP production in 7TM-μ receptor- and EV-transfected HEK293 cells, using a cAMP-sensitive luciferase reporter assay. Because the 6TM-isoform has been shown to produce excitatory cellular responses (Convertino, Samoshkin, Viet, et al., 2015; Gris et al., 2010; Samoshkin et al., 2015; Shabalina et al., 2009), we screened 6TM-μ receptor-transfected cells (data not shown) in both conditions of stimulated cAMP levels with 100 nM of isoprenaline (to detect ligand-dependent inhibition of cAMP levels), and in unstimulated cells (to detect ligand-dependent stimulation of cAMP levels). Morphine was used as positive control, and, as expected, it induced a dose-dependent reduction (IC50 ~ 3.6 × 10−8 M) in cAMP production induced by 100 nM isoprenaline in 7TM-μ receptor-transfected cells (Figure S4).
Generally, the experimental compounds, including compound 5 (Figure S4A), did not appear to have any effect on cAMP production in 7TM- or 6TM-μ receptor transfected cells, neither stimulatory nor inhibitory (data not shown for all compounds or transfected cell lines). We observed a slight reduction in isoprenaline-stimulated cAMP in both 7TM-μ receptor- and EV transfected cells at 10 μM for compounds 10 and 11 (Figure S4B,C). Compound 25 partially reduced cAMP production dose-dependently, with the highest concentrations reaching ~60% reduction (Figure S4D). Furthermore, for all three compounds—10, 11, and 25—this inhibition appeared to be non-specific (Figure S4B–D). None of the compounds were able to antagonize the cAMP production-lowering effect of morphine in 7TM-μ receptor-transfected cells (data not shown).
3.3 |. In vivo pharmacological characterization of candidate 6TM- and/or 7TM-μ receptor agonists
3.3.1 |. Compounds 5, 10, 11 and 25 produced dose-dependent thermal hyperalgesia in naïve adult C57BL/6 mice
The compounds 5, 10, 11 and 25 that elicited an increased NO production in the transfected human Be2C neuroblastoma cell lines were pharmacologically characterized in vivo in naïve adult C57BL/6 mice. For comparative purposes, two candidate compounds (compounds 22 and 28) that did not elicit NO were included as negative controls.
Subcutaneous administration of compound 5 (Figure 5a; n = 7, 8 and 7, respectively for 10, 20 and 40 mgkg−1), compound 10 (Figure 5b; n = 7, 8 and 7, respectively for 10, 20 and 40 mgkg−1), compound 11 (Figure 5c; n = 7, 10 and 7, respectively for 10, 20 and 40 mgkg−1) and compound 25 (Figure 5d; n = 7, 10 and 7, respectively for 10, 20 and 40 mgkg−1), but not vehicle (20% PEG; n = 10), to naïve C57BL/6 mice produced dose-dependent hyperalgesia in the tail-withdrawal nociceptive assay. Administration of compounds 22 and 28 (n = 7 per compound; Figure 5e), which failed to evoke NO release, did not produce any changes in the tail-withdrawal nociceptive assay. Figure 5f shows the dose-hyperalgesic response curves of each of the compounds tested.
3.3.2 |. Effect of naloxone and ICI 118,551 on thermal hyperalgesia induced by compounds 5, 10, 11 and 25 in naïve adult C57BL/6 mice
To further characterize the compounds, we tested the effect of low-dose naloxone, and a selective β2-adrenoceptor antagonist, ICI 118,551, following administration of an ~ED75 dose of compounds 5, 10, 11 and 25 in naïve adult C57BL/6 mice (Figure 6a–d). Thermal hyperalgesia induced by Compound 5 was significantly reversed by i.p. administration of low-dose naloxone (1 mgkg−1; n = 7), but not ICI 118,551 (5 mgkg−1; n = 7) or vehicle (saline; n = 8) (Figure 6a). In contrast, thermal hyperalgesia induced by s.c. administration of compounds 10 (Figure 6b), 11 (Figure 6c) and 25 (Figure 6d) were reversed by i.p. administration of ICI 118,551 (n = 7 per test compound), but not by low-dose naloxone (n = 7 per test compound) or vehicle (n = 7). These results are consistent with our findings from in vitro cellular assays.
FIGURE 6.
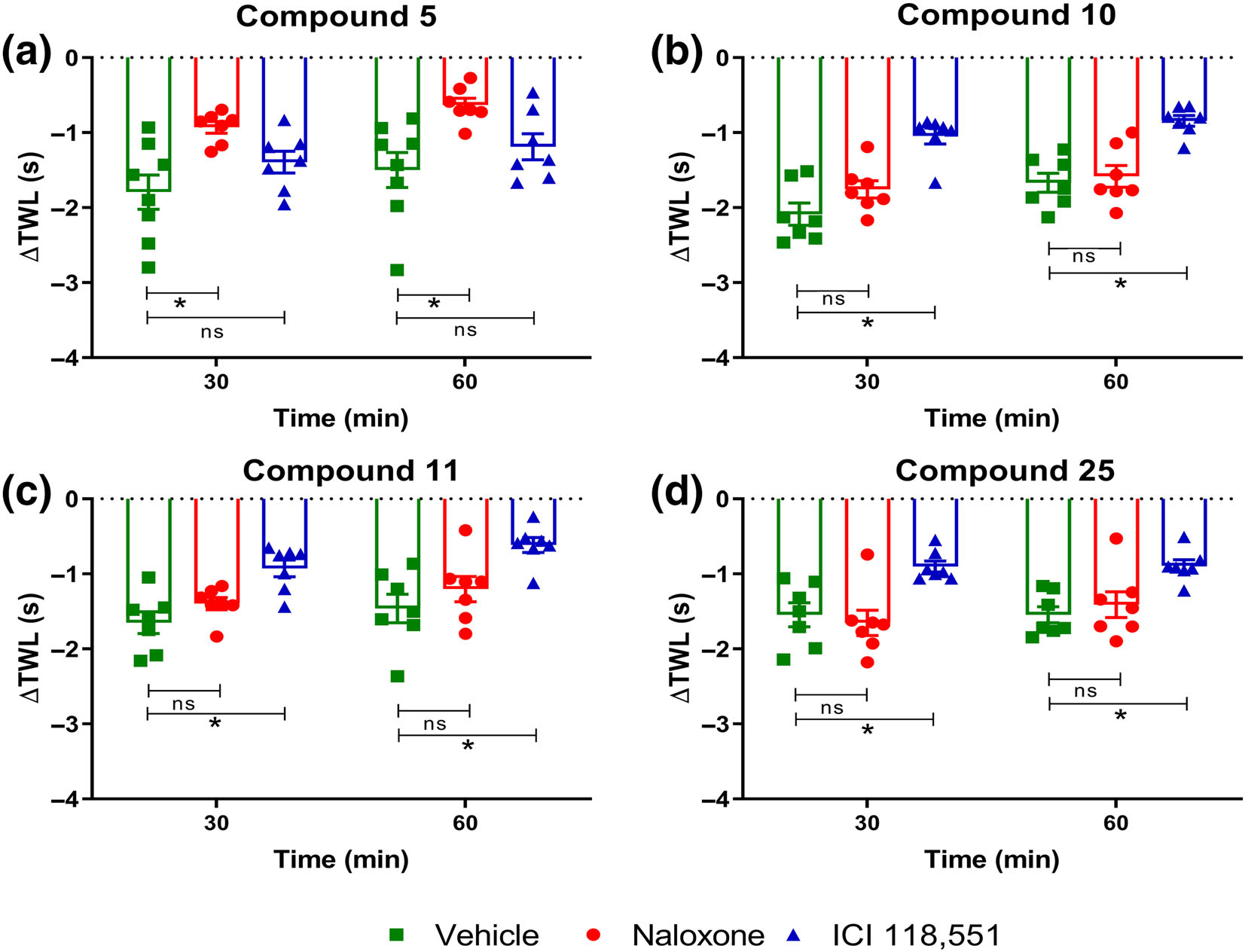
Effect of low-dose naloxone and ICI 118,551 on thermal hyperalgesia induced by compounds (a) 5, (b) 10, (c) 11 and (d) 25 in naïve adult C57BL/6 mice. The temporal changes in the ΔTWL values induced by s.c. administration of ~ED75 dose of (a) compound 5 (n = 8, 7 and 7 for vehicle, naloxone and ICI 118,551, respectively) was significantly reversed by i.p. administration of low dose naloxone (1 mgkg−1), but not ICI 118,551 (5 mgkg−1). In contrast, thermal hyperalgesia induced by s.c. administration of ~ED75 doses of compounds (b) 10, (c) 11 and (d) 25 were significantly attenuated by i.p. administration of ICI 118,551 (5 mgkg−1; n = 7 per test compound), but not the non-selective opioid antagonist, naloxone (1 mgkg−1; n = 7 per test compound), relative to vehicle (n = 7 per test compound). Data presented as mean ± SEM. *P < 0.05, significantly different, ns, non-significantly different, as indicated; two-way ANOVA with post hoc Bonferroni test
3.3.3 |. Effects of compounds 5, 10, 11 and 25 in mice lacking opioid or β-adrenoceptors
To assess the opioid receptor specificity of the candidate compounds, we pharmacologically characterized compounds 5, 10, 11 and 25 in adult male and female naïve Oprm1−/− (n = 8, 8, 6 and 6 for compounds 5, 10, 11 and 25, respectively), Oprd1−/− (n = 7, 7, 6 and 7 for compounds 5, 10, 11 and 25, respectively), Oprk1−/− (n = 9, 9, 6 and 9 for compounds 5, 10, 11 and 25, respectively) knockout mice, relative to C57BL/6J mice (n = 8, 8, 7 and 10 for compounds 5, 10, 11 and 25, respectively). because the 6TM-μ receptor isoform specifically heterodimerizes with β2-adrenoceptor, and that this dimerization largely underlies a molecular mechanism for cellular signalling by the 6TM isoforms (Samoshkin et al., 2015), we also pharmacologically characterized these compounds in Adrb1/2−/− double knockout mice (n = 8, 8, 6 and 8 for compounds 5, 10, 11 and 25, respectively).
Homozygous ablation of Oprd1 or Oprk1 did not significantly affect the thermal hyperalgesia induced by ~ED75 doses of compounds 5 (Figure 7a), 10 (Figure 7b), 11 (Figure 7c) and 25 (Figure 7d), indicating that these compounds do not mediate the observed effects through either δ- or κ-opioid receptors. In contrast, the inability of all compounds assessed herein to produce thermal hyperalgesia in Oprm1−/− mice implies a requirement of μ-opioid receptors in the observed effects on nociception (Figure 7a–d). Importantly, homozygous ablation of Adrb1 and Adrb2 partly reversed the thermal hyperalgesia induced by ~ED75 doses of compounds 10, 11 and 25 (Figure 7b–d) but not the hyperalgesic effects of compound 5 (Figure 7a).
3.4 |. Compounds 10 and 5 enhanced spontaneous excitatory transmission in spinal lamina II neurons
To further determine the excitatory effects of these compounds, we examined effects on sEPSC in isolated spinal cord slices. Patch clamp recordings were conducted in spinal cord lamina II interneurons in outer lamina II (IIo), which are predominantly excitatory neurons implicated in nociceptive transmission. In vivo spinal cord slice preparations were used to assess sEPSCs generation using the 7TM-μ receptor (compound 5) and the 6TM-μ receptor ligands (compound 10). For sEPSC recordings, each spinal cord slice was used only once. All recorded neurons tested had a resting membrane potential lower than −60 mV and exhibited sEPSC at a holding potential of −70 mV, near the reversal potential for inhibitory postsynaptic currents.
Compound 10 (20 μM) produced an enhancement of sEPSC in seven out of 11 recorded neurons, as shown in the upper trace of Figure 8a. A similar increase was found by superfusing Compound 5 at 20 μM in six out of nine recorded neurons (Figure 8b). The sEPSC frequency increased gradually over time, peaking around 2 min after compound 10 application, without any change in sEPSC amplitude (Figure 8c). Experiments with compound 5 showed that the sEPSC frequency increased similarly, peaking around 2 min after compound 5 application, without an alteration in its amplitude (Figure 8d). Additionally, compound 5 produced a slight inward current in four neurons (four out of nine recorded neurons), and the average current was −4.5 ± 0.29 pA (data not shown).
FIGURE 8.
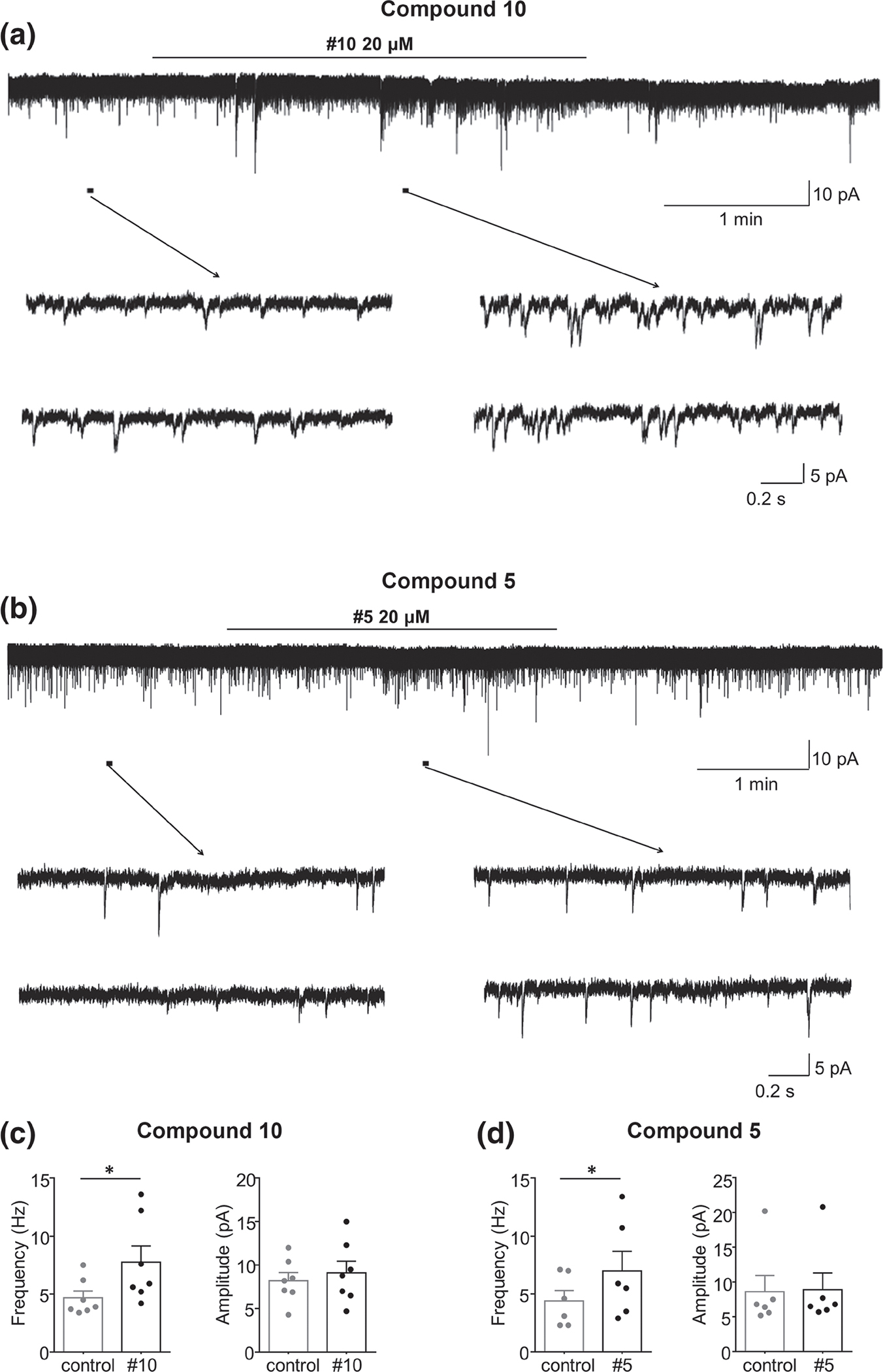
Compounds 10 and 5 enhanced spontaneous excitatory transmission in spinal lamina II neurons of adult male mice (n = 6–7 neurons per group; individual values are shown). (a) Recordings of sEPSCs in the absence and presence of compound 10. (b) Recordings of sEPSCs in the absence and presence of compound 5. (c and d) Mean ± SEM changes in the sEPSCs frequency (left) and amplitude (right) before and under the action of compound (c) 10 or (d) 5. The duration of drug superfusion is shown by a horizontal bar above the chart recording, and two consecutive traces of sEPSCs for a period indicated by a short bar below the chart recording are shown in an expanded time scale. VH = −70 mV. *P < 0.05, significantly different as indicated; Student’s t-test
4 |. DISCUSSION
In this work, our systematic investigation identified three novel agonist ligands for the 6TM-μ receptor (compounds 10, 11, and 25) and one novel agonist for the 7TM-μ receptor (compound 5) that produces dose-dependent NO-release and hyperalgesia in mice.
Recent research evidence suggests that the full-length 7TM- and truncated 6TM-μ receptor isoforms may mediate different cellular effects, namely, the classical inhibitory effects and novel excitatory cellular effects (Convertino, Samoshkin, Gauthier, et al., 2015; Pasternak & Pan, 2013). The lack of availability of selective ligands has hindered our advances in understanding the mechanism of action and the pharmacodynamic properties of 7TM- and 6TM-μ receptor isoforms. To address this knowledge gap, we first used a multitier-based strategy combined with the use of the most accurate docking techniques available (Convertino & Dokholyan, 2016; Ding et al., 2010) to rapidly survey the chemical space in the structural models of 6TM- and 7TM-μ receptors and identified potential 7TM-selective, 6TM-selective and non-selective compounds.
Next, we assessed the NO release (Gris et al., 2010; Samoshkin et al., 2015) and cAMP production (Crain & Shen, 2000; Waldhoer et al., 2004), which are characteristic of 6TM- and 7TM-μ receptor-mediated cellular responses respectively, of the top-ranked compounds. Our functional in vitro cellular assays in transfected human Be2C neuroblastoma cells suggested potential agonistic properties for compounds 5, 10, 11 and 25 with respect to NO release. In line with our previous findings (Gris et al., 2010), morphine induced NO release in EV transfected cells, a phenomenon that can be attributed to the endogenous expression of μ receptors, as well as other human opioid receptors, in human Be2C cells (Gris et al., 2010). Interestingly, IBNTxA, a ligand suggested to mediate 6TM-μ receptor-dependent cellular responses (Grinnell et al., 2014; Majumdar et al., 2011; Wieskopf et al., 2014), did not evoke NO release. Although IBNtxA has been shown to increase intracellular Ca2+ levels in a 6TM-μ receptor-dependent manner (Samoshkin et al., 2015), there are no published reports that IBNtxA produces NO release. It is possible that the IBNtxA’s biased signalling pathways does not include an activation effect on NO release. However, this remains to be investigated as the direction of cellular effects of the IBNtxA continues to be elusive despite its documented binding properties (Majumdar et al., 2011).
Subsequent characterization of the μ receptor-dependent NO release of our compounds using the non-specific opioid antagonist, naloxone, and the selective β2-adrenoceptor antagonist, ICI 118,551, supported the 7TM-μ receptor-binding properties for compound 5, and 6TM-μ receptor binding properties for compounds 10, 11 and 25. Importantly, our present findings showing inhibition of NO release stimulated by compound 5, but not for compounds 10, 11 and 25, by naloxone is unique. This is because our current data as well as our previous findings with the canonical 7TM-μ receptor-agonist, morphine (Gris et al., 2010), have not revealed a 7TM-μ receptor-mediated increase in NO. The observation that compound 5 produces NO via a 7TM-μ receptor process can be explained by the possibility that compound 5 engages intracellular excitatory processes of 7TM-μ receptors that are coupled to a NO release pathway. Importantly, an absence of a morphine-like response for cAMP production in 7TM-μ receptor-transfected cells for compound 5 (Figure S4A) was expected, taking into account that compound 5 produces hyperalgesia, and opioid-dependent cAMP inhibition is associated with analgesia (Gris et al., 2010).
In line with our functional in vitro data, compounds 5, 10, 11 and 25 produced dose-dependent hyperalgesia in the in vivo tail-withdrawal nociceptive assay. In addition, our data showing inhibition of compound 5-mediated hyperalgesia by low dose naloxone and ICI 118,551-mediated inhibition of hyperalgesia produced by compounds 10, 11 and 25 indicate potentially different molecular pharmacological mechanisms of our novel compounds. Consistent with this notion, the hyperalgesic properties of compounds 10, 11 and 25 were partly reversed in Adrb1/2 KO mice, and hyperalgesic properties all four compounds were abolished in μ receptor-KO mice, but not in mice lacking other opioid receptors. These data highlight that the hyperalgesic effects of compounds 10, 11 and 25 are likely to be mediated via 6TM-μ receptor / β2-adrenoceptor dimers and that of compound 5 is mediated by 7TM-μ receptor. These data also emphasize the potential clinical utility of β2-adrenoceptor antagonists to inhibit the unwanted hyperalgesic effects of opioid agonists. It is also worth noting that the Oprm1−/− mice used here lack exon 2, and thus does not express either 6TMor 7TM-μ receptors. We used these mice instead of the exon-11 Oprm1−/− knockout mice, which have been proposed to completely lack 6TM-μ receptor isoforms (Juni et al., 2007; Pan et al., 2009; Schuller et al., 1999). However, this may not be the case because it is now well known that multiple 6TM-μ receptor isoforms in rodents and humans are initiated from at least three different promoters on Oprm1 (as seen in Ensembl, NCBI Gene and UCSC Browser; (Pasternak & Pan, 2013)). Finally, we would like to point that the in vitro NO release assays showed relatively weak on target activity while the in vivo potency is moderate to high (10–40 mgkg−1), suggesting that the compounds should have high brain penetration. Off-target, non-opioid, effects are unlikely due to the combination of the absence of ligand-dependent behavioural phenotypes in μ receptor KO mice and the original design of our computation modelling to identify selective 6TM/ 7TM-μ receptor ligands.
Finally, we examined the effects of the novel 7TM-μ receptor ligand, compound 5, and the most potent novel 6TM-μ receptor ligand, compound 10, on sEPSCs in spinal cord lamina IIo neurons. The sEPSCs in lamina IIo neurons of spinal cord slices are mediated by glutamate AMPA receptors, as it can be completely blocked by CNQX, a selective antagonist of AMPA receptors (Park et al., 2011; Yang et al., 1998). Application of morphine to these cells has also previously been shown to produce an opioid-receptor mediated reduction in sEPSCs (Braz et al., 2014; Todd, 2010; Z. Wang et al., 2020). Because compounds 5 and 10 increased the frequency, but not amplitude of sEPSCs, and μ receptors are expressed by primary afferent neurons projecting to the lamina II (Ji et al., 1995; Scherrer et al., 2009), these two compounds are likely to act on presynaptic μ receptors in the lamina II, releasing excitatory substances from primary afferents. Our ex vivo electrophysiology data further validates the hyperalgesic properties of compounds 5 and 10 by enhancing glutamate releases from primary afferents. Additionally, as compound 5 induced inward currents in some neurons, this compound may also act on postsynaptic neurons to produce excitatory actions.
Our results are in line with the originally proposed hyperalgesic function of the 6TM-μ receptor isoform based on the genetic association study (Shabalina et al., 2009), identified excitatory molecular mechanisms for 6TM-μ receptor cellular signalling (Gris et al., 2010; Samoshkin et al., 2015), and heterodimerization of 6TM-μ receptors with β2-adrenoceptors contributing to an opioid-induced hyperalgesia (Samoshkin et al., 2015). The results from the Pasternak group also demonstrated that morphine hyperalgesia is restricted to 6TM-μ receptor mechanisms (Marrone et al., 2017), although both analgetic and hyperalgesic behavioural effects have been reported for 6TM-μ receptor-dependent IBNtxA responses (Majumdar et al., 2011, 2012; Samoshkin et al., 2015). Analgesia produced by other opioid compounds have also been shown to be significantly attenuated or completely absent in 6TM-μ receptor KO mice (Majumdar et al., 2011; Marrone, Lu, et al., 2016; Pan et al., 2009). Recently demonstrated G protein signalling bias at μ receptors for IBNtxA provides one explanation for such diverse responses (Che et al., 2018). Together, our new and earlier results suggest that the analgesic and hyperalgesic functions may be intrinsic for both 6TM- and 7TM-μ receptors, with a hyperalgesic function being more prominent for the 6TM-μ receptor, and manifestation of either function depends on the ligand biased properties of the test ligands and relative strength of the binding to either isoform. Furthermore, these functions can be further modulated by the presence of binding partners such as β2-adrenoceptors.
In conclusion, this is the first time that 6TM- or 7TM-μ receptor ligands have been identified that produce hyperalgesic responses. The hyperalgesic function also correlates with cellular NO release. Importantly, our work herein has addressed a crucial knowledge gap that existed in the field of opioid research, the lack of selective 6TM-μ receptor ligands, as the only known 6TM-μ receptor ligand, IBNtxA, is not selective for 6TM-μ receptor but also binds to the 7TM isoform and some other opioid receptors (Majumdar et al., 2011). Further investigation of our novel 6TM-μ receptor ligands using radioligand binding studies is warranted, which is currently not practically feasible due to the lack of commercially available radio-labelled compound that would bind selectively to the 6TM-μ receptors. Furthermore, functional characterization of leads at 6TM-and 7TM-μ receptors, and β2-adrenoceptors should be carried out. However, overall, the novel compounds identified in the study represent a new class of μ receptor-dependent ligands that can serve as a valuable research tools and will enable the development of new approaches, such as the use of 7TM-μ receptor agonists together with a 6TM-μ receptor antagonist, or specific μ receptor antagonists for anti-hyperalgesia properties.
Supplementary Material
What is already known
7TM- and 6TM-μ receptor isoforms mediate both the classical inhibitory and novel excitatory cellular effects.
Lack of selective 6TM-μ receptor ligands hinders our understanding of the pharmacodynamics of this receptor.
What does this study add
Four novel compounds that represent a new class of 6TM- or 7TM-μ receptor ligands.
Elucidation of the hyperalgesic properties of μ receptor ligands, along with their analgesic properties.
What is the clinical significance
These novel μ receptor ligands will help to develop new and better μ receptor antagonists.
ACKNOWLEDGEMENTS
A.M. was supported by the Ronald Melzack Fellowship in Chronic Pain Research awarded by the Louise and Alan Edwards Foundation. L.D., W.M. and N.V.D. were supported by National Institute on Drug Abuse STTR 1R41DA032293 grant. N.V.D. also acknowledge support from the National Institutes for Health grants 1R35 GM134864 and UL1 TR002014, and the Passan Foundation. J.S.M. was supported by grants from the Canadian Institutes for Health Research and the Natural Sciences and Engineering Council of Canada. L.D. was supported by a Pfizer Canada Professorship in Pain Research and the Canadian Excellence Research Chairs Program (CERC9).
Abbreviations:
- EV
empty vector
- OIH
opioid-induced hyperalgesia
- TM
transmembrane
- TWL
tail withdrawal latency
Footnotes
CONFLICT OF INTEREST
Authors have no competing interests to disclose.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
REFERENCES
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & Collaborators C (2019). The concise guide to pharmacology 2019/20: G protein-coupled receptors. British Journal of Pharmacology, 176(Suppl 1), S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & CGTP Collaborators. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA, Veale EL, Striessnig J, Kelly E, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & CGTP Collaborators. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176, S142–S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne JC, & Shin NS (2008). Efficacy of opioids for chronic pain: A review of the evidence. Clinical Journal of Pain, 24(6), 469–478. 10.1097/AJP.0b013e31816b2f26 [DOI] [PubMed] [Google Scholar]
- Braz J, Solorzano C, Wang X, & Basbaum AI (2014). Transmitting pain and itch messages: A contemporary view of the spinal cord circuits that generate gate control. Neuron, 82(3), 522–536. 10.1016/j.neuron.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse JW, Wang L, Kamaleldin M, Craigie S, Riva JJ, Montoya L, Mulla SM, Lopes LC, Vogel N, Chen E, Kirmayr K, de Oliveira K, Olivieri L, Kaushal A, Chaparro LE, Oyberman I, Agarwal A, Couban R, Tsoi L, … Guyatt GH (2018). Opioids for chronic noncancer pain: A systematic review and meta-analysis. JAMA, 320(23), 2448–2460. 10.1001/jama.2018.18472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee MY, Pardon E, Steyaert J, Huang XP, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, … Roth BL (2018). Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell, 172(1–2), 55–67e15. 10.1016/j.cell.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertino M, & Dokholyan NV (2016). Computational modeling of small molecule ligand binding interactions and affinities. Computational Design of Ligand Binding Proteins, 1414, 23–32. 10.1007/978-1-4939-3569-7_2 [DOI] [PubMed] [Google Scholar]
- Convertino M, Samoshkin A, Gauthier J, Gold MS, Maixner W, Dokholyan NV, & Diatchenko L (2015). Mu-opioid receptor 6-transmembrane isoform: A potential therapeutic target for new effective opioids. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 62, 61–67. 10.1016/j.pnpbp.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertino M, Samoshkin A, Viet CT, Gauthier J, Li Fraine SP, Sharif-Naeini R, Schmidt BL, Maixner W, Diatchenko L, & Dokholyan NV (2015). Differential regulation of 6- and 7-transmembrane helix variants of mu-opioid receptor in response to morphine stimulation. PLoS One, 10(11), e0142826. 10.1371/journal.pone.0142826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain SM, & Shen KF (2000). Antagonists of excitatory opioid receptor functions enhance morphine’s analgesic potency and attenuate opioid tolerance/dependence liability. Pain, 84(2–3), 121–131. 10.1016/s0304-3959(99)00223-7 [DOI] [PubMed] [Google Scholar]
- Ding F, Yin SY, & Dokholyan NV (2010). Rapid flexible docking using a stochastic Rotamer library of ligands. Journal of Chemical Information and Modeling, 50(9), 1623–1632. 10.1021/ci100218t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan NV (2016). Controlling allosteric networks in proteins. Chemical Reviews, 116(11), 6463–6487. 10.1021/acs.chemrev.5b00544 [DOI] [PubMed] [Google Scholar]
- Dokholyan NV, Buldyrev SV, Stanley HE, & Shakhnovich EI (1998). Discrete molecular dynamics studies of the folding of a protein-like model. Folding & Design, 3(6), 577–587. 10.1016/S1359-0278(98)00072-8 [DOI] [PubMed] [Google Scholar]
- Grinnell SG, Majumdar S, Narayan A, Le Rouzic V, Ansonoff M, Pintar JE, & Pasternak GW (2014). Pharmacologic characterization in the rat of a potent analgesic lacking respiratory depression, IBNtxA. The Journal of Pharmacology and Experimental Therapeutics, 350(3), 710–718. 10.1124/jpet.114.213199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris P, Gauthier J, Cheng P, Gibson DG, Gris D, Laur O, Pierson J, Wentworth S, Nackley AG, Maixner W, & Diatchenko L (2010). A novel alternatively spliced isoform of the mu-opioid receptor: Functional antagonism. Molecular Pain, 6, 33. 10.1186/1744-8069-6-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA (1996). Merck molecular force field.1. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry, 17(5–6), 490–519. [DOI] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, & Coleman RG (2012). ZINC: A free tool to discover chemistry for biology. Journal of Chemical Information and Modeling, 52(7), 1757–1768. 10.1021/ci3001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Law PY, Low HH, Elde R, & Hokfelt T (1995). Expression of mu-, delta-, and kappa-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. The Journal of Neuroscience, 15(12), 8156–8166. 10.1523/JNEUROSCI.15-12-08156.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juni A, Klein G, Pintar JE, & Kest B (2007). Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience, 147(2), 439–444. 10.1016/j.neuroscience.2007.04.030 [DOI] [PubMed] [Google Scholar]
- Kota P, Ding F, Ramachandran S, & Dokholyan NV (2011). Gaia: Automated quality assessment of protein structure models. Bioinformatics, 27(16), 2209–2215. 10.1093/bioinformatics/btr374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB, & Manchikanti L (2011). A comprehensive review of opioid-induced hyperalgesia. Pain Physician, 14(2), 145–161. [PubMed] [Google Scholar]
- Lilley E, Stanford SC, Kendall DE, Alexander SP, Cirino G, Docherty JR, George CH, Insel PA, Izzo AA, Ji Y, Panettieri RA, Sobey CG, Stefanska B, Stephens G, Teixeira M, & Ahluwalia A (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan Y-X, & Pasternak GW (2011). Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proceedings of the National Academy of Sciences of the United States of America, 108(49), 19778–19783. 10.1073/pnas.1115231108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Subrath J, Le Rouzic V, Polikar L, Burgman M, Nagakura K, Ocampo J, Haselton N, Pasternak AR, Grinnell S, Pan Y-X, & Pasternak GW (2012). Synthesis and evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated mu opioid receptor (MOR-1) splice variants. Journal of Medicinal Chemistry, 55(14), 6352–6362. 10.1021/jm300305c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, & Granier S (2012). Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature, 485(7398), 321–326. 10.1038/nature10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone GF, Grinnell SG, Lu Z, Rossi GC, Le Rouzic V, Xu J, Majumdar S, Pan Y-X, & Pasternak GW (2016). Truncated mu opioid GPCR variant involvement in opioid-dependent and opioid-independent pain modulatory systems within the CNS. Proceedings of the National Academy of Sciences of the United States of America, 113(13), 3663–3668. 10.1073/pnas.1523894113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone GF, Le Rouzic V, Varadi A, Xu J, Rajadhyaksha AM, Majumdar S, Pan YX, & Pasternak GW (2017). Genetic dissociation of morphine analgesia from hyperalgesia in mice. Psychopharmacology, 234(12), 1891–1900. 10.1007/s00213-017-4600-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone GF, Lu Z, Rossi G, Narayan A, Hunkele A, Marx S, Xu J, Pintar J, Majumdar S, Pan YX, & Pasternak GW (2016). Tetrapeptide Endomorphin analogs require both full length and truncated splice variants of the mu opioid receptor gene Oprm1 for analgesia. ACS Chemical Neuroscience, 7(12), 1717–1727. 10.1021/acschemneuro.6b00240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Piltonen MH, Gauthier J, Convertino M, Acland EL, Dokholyan NV, Mogil JS, Diatchenko L, & Maixner W (2015). Differences in the Antinociceptive effects and binding properties of propranolol and bupranolol enantiomers. The Journal of Pain, 16(12), 1321–1333. 10.1016/j.jpain.2015.09.004 [DOI] [PubMed] [Google Scholar]
- Mogil JS, Ritchie J, Sotocinal SG, Smith SB, Croteau S, Levitin DJ, & Naumova AK (2006). Screening for pain phenotypes: Analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain, 126(1–3), 24–34. 10.1016/j.pain.2006.06.004 [DOI] [PubMed] [Google Scholar]
- Noble M, Treadwell JR, Tregear SJ, Coates VH, Wiffen PJ, Akafomo C, & Schoelles KM (2010). Long-term opioid management for chronic noncancer pain. Cochrane Database of Systematic Reviews, 1, CD006605. 10.1002/14651858.CD006605.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladosu FA, Conrad MS, O’Buckley SC, Rashid NU, Slade GD, & Nackley AG (2015). Mu opioid splice variant MOR-1K contributes to the development of opioid-induced hyperalgesia. PLoS One, 10(8), e0135711. 10.1371/journal.pone.0135711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, & Pasternak GW (2009). Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proceedings of the National Academy of Sciences of the United States of America, 106 (12), 4917–4922. 10.1073/pnas.0811586106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, & Ji RR (2011). Resolving TRPV1- and TNF-a-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. The Journal of Neuroscience, 31(42), 15072–15085.31/42/15072[pii]. 10.1523/JNEUROSCI.2443-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, & Pan YX (2013). Mu opioids and their receptors: Evolution of a concept. Pharmacological Reviews, 65(4), 1257–1317. 10.1124/pr.112.007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, … Würbel H (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi R, Convertino M, Popov K, Dokholyan NV, & Tropsha A (2016). Docking and scoring with target-specific pose classifier succeeds in native-like pose identification but not binding affinity prediction in the CSAR 2014 benchmark exercise. Journal of Chemical Information and Modeling, 56(6), 1032–1041. 10.1021/acs.jcim.5b00751 [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Kota P, Ding F, & Dokholyan NV (2011). Automated minimization of steric clashes in protein structures. Proteins, 79 (1), 261–270. 10.1002/prot.22879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoshkin A, Convertino M, Viet CT, Wieskopf JS, Kambur O, Marcovitz J, Patel P, Stone LS, Kalso E, Mogil JS, Schmidt BL, Maixner W, Dokholyan NV, & Diatchenko L (2015). Structural and functional interactions between six-transmembrane mu-opioid receptors and beta2-adrenoreceptors modulate opioid signaling. Scientific Reports, 5, 18198. 10.1038/srep18198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, Kieffer BL, & Basbaum AI (2009). Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell, 137(6), 1148–1159doi: S0092–8674(09)00444–9 [pii]. 10.1016/j.cell.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, & Pintar JE (1999). Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nature Neuroscience, 2 (2), 151–156. 10.1038/5706 [DOI] [PubMed] [Google Scholar]
- Serohijos AW, Yin S, Ding F, Gauthier J, Gibson DG, Maixner W, Dokholyan NV, & Diatchenko L (2011). Structural basis for mu-opioid receptor binding and activation. Structure, 19(11), 1683–1690. 10.1016/j.str.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Zaykin DV, Gris P, Ogurtsov AY, Gauthier J, Shibata K, Tchivileva IE, Belfer I, Mishra B, Kiselycznyk C, Wallace MR, Staud R, Spiridonov NA, Max MB, Goldman D, Fillingim RB, Maixner W, & Diatchenko L (2009). Expansion of the human mu-opioid receptor gene architecture: Novel functional variants. Human Molecular Genetics, 18(6), 1037–1051. 10.1093/hmg/ddn439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirvanyants D, Ding F, Tsao D, Ramachandran S, & Dokholyan NV (2012). Discrete molecular dynamics: An efficient and versatile simulation method for fine protein characterization. Journal of Physical Chemistry B, 116(29), 8375–8382. 10.1021/jp2114576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SW (1985). (+)-[3H]SKF 10,047, (+)-[3H]ethylketocyclazocine, mu, kappa, delta and phencyclidine binding sites in guinea pig brain membranes. European Journal of Pharmacology, 109(1), 33–41. 10.1016/0014-2999(85)90536-9 [DOI] [PubMed] [Google Scholar]
- Toda N, Kishioka S, Hatano Y, & Toda H (2009). Modulation of opioid actions by nitric oxide signaling. Anesthesiology, 110(1), 166–181. 10.1097/ALN.0b013e31819146a9 [DOI] [PubMed] [Google Scholar]
- Todd AJ (2010). Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews Neuroscience, 11(12), 823–836doi:nrn2947 [pii]. 10.1038/nrn2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt JH, Bienfait B, Wang SM, & Nicklaus MC (2001). Comparison of the NCI open database with seven large chemical structural databases. Journal of Chemical Information and Computer Sciences, 41(3), 702–712. 10.1021/Ci000150t [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Bartlett SE, & Whistler JL (2004). Opioid receptors. Annual Review of Biochemistry, 73, 953–990. 10.1146/annurev.biochem.73.011303.073940 [DOI] [PubMed] [Google Scholar]
- Wang J, & Dokholyan NV (2019). MedusaDock 2.0: Efficient and accurate protein-ligand docking with constraints. Journal of Chemical Information and Modeling, 59(6), 2509–2515. 10.1021/acs.jcim.8b00905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang C, He Q, Matsuda M, Han Q, Wang K, Bang S, Ding H, Ko M-C, & Ji RR (2020). Anti-PD-1 treatment impairs opioid antinociception in rodents and nonhuman primates. Science Translational Medicine, 12(531), eaaw6471. 10.1126/scitranslmed.aaw6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieskopf JS, Pan YX, Marcovitz J, Tuttle AH, Majumdar S, Pidakala J, Pasternak GW, & Mogil JS (2014). Broad-spectrum analgesic efficacy of IBNtxA is mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain, 155(10), 2063–2070. 10.1016/j.pain.2014.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, & Yoshimura M (1998). Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neuroscience Letters, 255(3), 135–138. 10.1016/s0304-3940(98)00730-7 [DOI] [PubMed] [Google Scholar]
- Yin S, Biedermannova L, Vondrasek J, & Dokholyan NV (2008). MedusaScore: An accurate force field-based scoring function for virtual drug screening. Journal of Chemical Information and Modeling, 48(8), 1656–1662. 10.1021/ci8001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Xu J, & Pan YX (2020). A truncated six transmembrane splice variant MOR-1G enhances expression of the full-length seven transmembrane mu-opioid receptor through Heterodimerization. Molecular Pharmacology, 98(4), 518–527. 10.1124/mol.120.119453 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.