

Summary
Persistent DNA double-strand breaks (DSBs) in neurons are an early pathological hallmark of neurodegenerative diseases including Alzheimer’s Disease (AD), with the potential to disrupt genome integrity. We used single-nucleus RNA-seq in human post-mortem prefrontal cortex samples and found that excitatory neurons in AD were enriched for somatic mosaic gene fusions. Gene fusions were particularly enriched in excitatory neurons with DNA damage repair and senescence gene signatures. In addition, somatic genome structural variations and gene fusions were enriched in neurons burdened with DSBs in the CK-p25 mouse model of neurodegeneration. Neurons enriched for DSBs also had elevated levels of cohesin along with progressive multiscale disruption of the 3D genome organization aligned with transcriptional changes in synaptic, neuronal development, and histone genes. Overall, this study demonstrates the disruption of genome stability and the 3D genome organization by DSBs in neurons as pathological steps in the progression of neurodegenerative diseases.
Keywords: Neurodegeneration, DNA double-strand breaks, Alzheimer’s Disease, Structural Variations, 3D genome organization, Genomic Mosaicism, Somatic Mosaicism, Epigenome, Transcriptome, Senescence
eTOC
Disruption of genome stability and 3D genome organization by DNA double-strand breaks in neurons are pathological steps in the progression of neurodegeneration.
Graphical Abstract
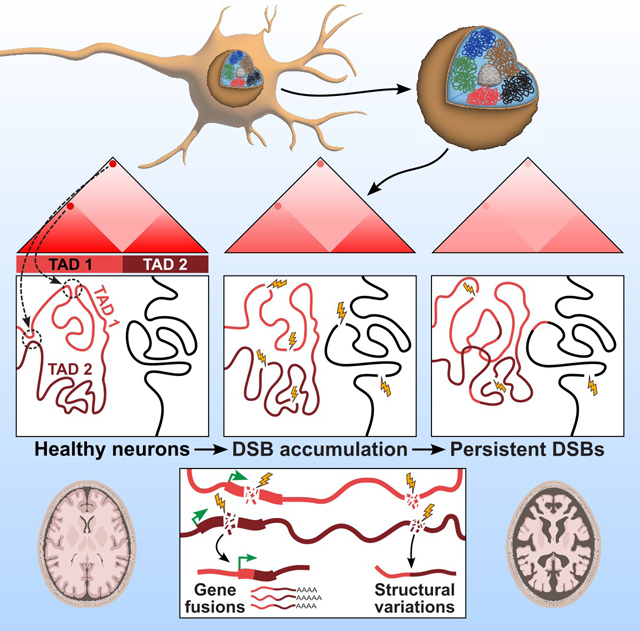
Introduction
DNA damage and defective DNA repair are hallmarks of aging and several neurodegenerative disorders including Alzheimer’s Disease (AD)1–3. Neurons are generally post-mitotic cells that can last the life span of an organism making them especially vulnerable to the accumulation of DNA repair defects. Studies using post-mortem human brain samples show that neurons, astrocytes, and oligodendrocytes accumulate DNA double-strand breaks (DSBs) early in the progression of AD2,4–7. In addition to pathological sources of DNA breaks, neurons also actively induce both DSBs and single-stranded breaks as part of their normal physiology8–11. Further, extensive active demethylation following neuronal activity through the Tet-TDG pathway-associated base excision repair can also be a source of DNA breaks8,12,13. While several studies have focused on understanding DNA repair mechanisms in AD, only a few studies have attempted to understand the epigenetic consequences of DSBs in AD2,14,15.
Persistent DSBs are drivers of genomic instability16. The major repair pathway that resolves DSBs in neurons is the non-homologous end-joining (NHEJ)17. However, NEHJ is error-prone and can cause genome structural variations, potentially leading to gene fusions with pathological gain or loss of function18–20. These pathological effects of structural variations are well demonstrated in various cancers21,22. In addition, structural variations have the potential to rewire genes with ectopic regulatory elements23–25. Therefore, the observation that DSBs accumulate in neurons in the early stages of AD begs the question of whether these DSBs lead to genome structural variations in neurons26.
The genome is hierarchically organized at different scales within the nucleus. Chromatin interactions including chromatin loops form self-interacting topologically-associating domains (TADs) that spatially segregate in the nucleus to give rise to the transcriptionally active and repressive compartments. This organization of the genome is closely associated with several fundamental biological paradigms including gene expression, DNA replication, genome stability, DNA repair, development, cell cycle, antibody repertoire generation by V(D)J recombination, etc. to name a few among the growing list of processes regulated by the 3D folding of the genome27–35. There is now emerging evidence that the 3D genome is actively regulated during learning and memory formation 36–41. Further, the disruption of 3D genome organization has been demonstrated in neurodevelopmental and neurological neurodegenerative disorders such as Huntington’s disease, Fragile X syndrome, Down syndrome, etc. 42,43. Chromatin accessibility has also been shown to be disrupted in the AD brain 44,45. Other studies have demonstrated how disease-associated SNPs in non-coding areas of the genome influence genes associated with diseases including Alzheimer’s Disease (AD) 46–50. DSBs have the potential to directly affect the 3D genome organization 51–54. Persistent DSBs in neurons could also indirectly impact the 3D genome organization through genome structural variations55,56.
To explore the genomic consequences of DSBs in Alzheimer’s Disease, we used both human postmortem brain samples and a mouse model of neurodegeneration that recapitulate the pre-symptomatic accumulation of DSBs. We show increased gene fusions, genome structural variations, and changes in 3D genome organization in neurons harboring DSBs. Our results demonstrate the disruption of genome stability and the 3D genome organization mediated by DSBs as pathological steps in the progression of neurodegenerative diseases.
Results
1. DSB-bearing neurons have mosaic genome structural variations in neurodegeneration
We obtained post-mortem human prefrontal cortex brain samples from 47 Religious Orders Study and Rush Memory and Aging Project (ROSMAP) participants and classified them into 23 non-pathologic AD individuals (NIA-Reagan 3–4) and 24 individuals with pathologic AD (NIA-Reagan 1–2). We used well-based single-nucleus RNA-seq full-transcript profiles (snRNA-seq, Smart-Seq2 v2.032) across all 47 individuals and obtained 6,180 cells (5,421 after QC) (Figure 1A, S1A, B, Table S1, page 1)57. We used known marker gene expression to annotate 2,121 oligodendrocytes (39.1%), 1,170 excitatory neurons (21.6%), 255 microglia (4.7%), 221 inhibitory neurons (4.1%), 220 astrocytes (4.1%), 94 OPCs (1.7%), and 40 endothelial cells (0.7%) (Table S1, page 2). We also identified a cluster of 1,300 cells (24%) that we refer to here as “unidentified/dying” with reduced transcription, and loss of cell-type-identity markers, positioned between neurons and oligodendrocytes in the two-dimensional t-SNE cell embedding. This cluster also had a lower number of reads and genes per cell, indicating that they are unlikely to be doublets (Figure S1C).
Figure 1: DSBs in neurons lead to mosaic genome structural variations. See also Figure S1, S2 and Table S1, pages 1–6.
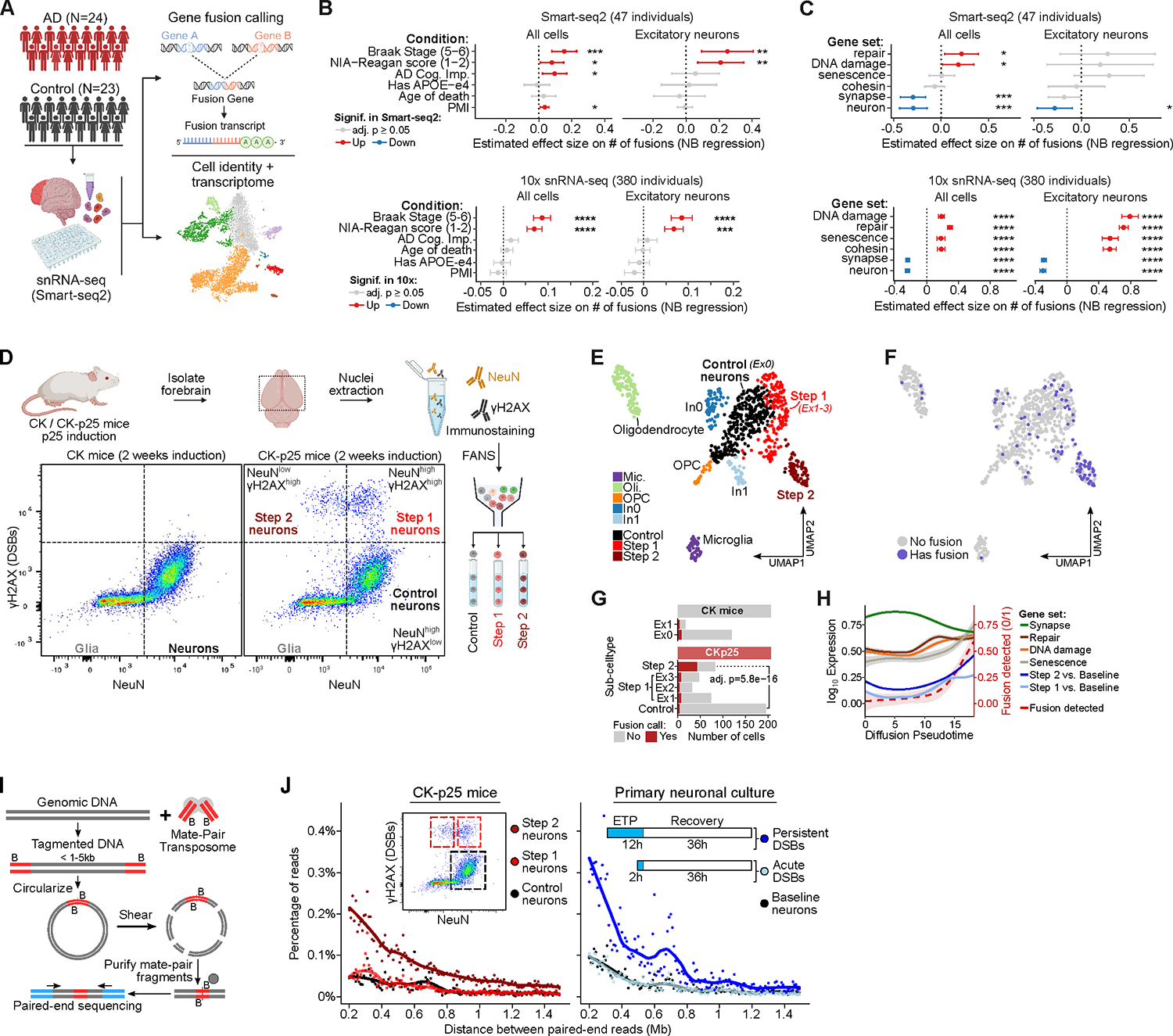
A) Schematic of snRNA-seq (Smart-seq2) analysis in human post-mortem brains.
B) The effect size of the number of gene fusions using human snRNA-seq as a function of human sample covariates. Significantly increased (red) or decreased (blue) gene fusions shown for adjusted p-value ≤ 0.05. Human sample covariates include AD cognitive impairment score (AD Cog, Imp) and post-mortem interval (PMI).
C) The effect size of the number of gene fusions as a function of gene sets associated with different Gene Ontology (GO) terms, similar to panel B.
D) Representative FANS dot-plot of CK/CK-p25 forebrain after 2 weeks induction using NeuN (neuron) and γH2AX (DSB) marker.
E) UMAP embedding of CK/CK-p25 snRNA-seq after 1 or 2 weeks of induction. Colors indicate cell-type annotations. Control neurons (Ex0) and Step 1 neurons are represented by clusters Ex1, Ex2, and Ex3.
F) Overlay of cells containing at least 1 gene fusion event (blue) on the snRNA-seq UMAP embedding.
G) Quantification of the number of cells with at least one gene fusion and the total number of cells for Control, Step 1 (Ex1, Ex2, Ex3 clusters), and Step 2. Significance from Fisher’s exact test between Control and Step 2 in CKp25 mice.
H) Trajectory analysis of Control, Step 1, and Step 2. Lines indicate the fraction of cells with gene fusions (dotted red lines) or smoothed expression of gene sets along the pseudo-time trajectory. Step 1 and Step 2 gene sets are defined as significantly upregulated genes from Step 1 vs. Control and Step 2 vs. Control comparisons from bulk RNA-seq (Welch et al.,2022) respectively (adjusted p-value<0.05, log2 fold-change≥1.0).
I) Schematic of mate-pair sequencing. Genomic DNA is tagmented to sizes between 1 – 5 kb with Tn5 transposase carrying biotinylated adaptors. The fragments are circularized and then sheared into 200–300 bp fragments. The ligation junctions are then enriched through biotin pull-down and paired-end sequenced. Mapped read pairs will be separated by 1– 5 kb and large deviations from the expected separation indicates genome structural variation events.
J) Plots of mate-pair sequencing quantifying the percentage of read pairs separated by large genomic distances greater than 200 kb that indicate genome structural variation events.
We then called mosaic gene fusion events in each cell independently, using STAR-fusion to detect chimeric snRNA-seq reads mapping between two genes as a proxy for genome structural variations (Table S1, page 3)58. As expected, the majority of cells did not have any gene fusions, and 95% of the cells with gene fusions had between 1–4 fusion events (Figure S1D). Only 3.3% of pairs of genes involved in intra-chromosomal fusions were adjacent to each other, making gene fusions due to RNA polymerase run-through unlikely as a major source of false positives (Figure S1E, F). We quantified enrichments of putative fusion events across multiple conditions using a negative binomial regression framework controlling for cell quality and individual-level covariates including post-mortem interval, sex, and age of death. Strikingly, we found that gene fusions were significantly enriched in cells from individuals with AD pathology as ascertained by the NIA-Reagan score (pathology diagnosis, AD=1–2 and non-AD=3–4) relative to cells from non-AD individuals (adj. p-value=0.046). We also found that gene fusions were significantly enriched in cells from late-AD individuals, as defined by the presence of neurofibrillary tangles (NFTs) in their prefrontal cortex (Braak stages 5–6 versus non/early-AD stages 1–4, adj. p-value=0.00031) (Figure 1B). Stratifying the gene fusion enrichment by cell type revealed that excitatory neurons in particular were enriched for gene fusions and that they showed a higher effect size compared to all other cell types in late AD individuals (Figure 1B, S1G). To validate these results in a second cohort of individuals, we used 10x snRNA-seq data for over 2 million cells from 380 individuals (profiled in the accompanying Mathys et al. study59). We re-aligned and called fusions on the 10x snRNA-seq reads to detect gene fusion junctions involving the 3’ end of the 10x cDNA molecule. Repeating the analysis done in the Smart-seq2 cohort, we observed significantly increased gene fusion events (adjusted p-value<0.001) in cells from AD individuals compared to non-AD individuals as ascertained by NIA-Reagan and Braak stage (late-AD versus non/early-AD) (Figure 1B, Table S1, page 4).
Since genome structural variations can result from errors in DNA repair and as DNA repair pathways can be up-regulated in the presence of DNA damage, we asked whether cells with increased activity of specific damage-associated and normal neuronal genes showed different gene fusions burden60. We scored all cells in the Smart-seq2 and 10x datasets by their expression of genes involved in functional pathways (GO) related to DNA damage, repair, senescence, neuronal, and synaptic functions. Excitatory neurons with higher expression of DNA damage, repair, and senescence-associated gene sets (Table S2, page 5) showed a significant increase in their gene fusion burden in the 10x dataset (Figure 1C). On the other hand, excitatory neurons with higher expression of genes associated with neuronal identity and synaptic function were depleted for gene fusions in both datasets (Figure 1C). Analyzing all cells jointly showed significant effects in the same directions but with reduced effect sizes (in the 10x data) for the enrichment of fusions in repair, DNA damage, and senescence-associated cells (Figure 1C). The increased mosaic gene fusion burden in cells with these expression profiles suggests that DNA breaks in neurons could potentially lead to a senescence-like state with increased genome structural variations. DNA damage-related gene expression in excitatory neurons was associated with several key measures of AD pathology in our 10x data, in line with previous studies that found a link between DNA damage and Alzheimer’s Disease (AD) (Figure S1H)2,4–7.
To investigate the genomic impact of DSBs in neurons, we utilized the CK-p25 mouse model of inducible and synchronous neurodegeneration which recapitulates the pre-symptomatic accumulation of DSBs and expression of senescence-associated genes in excitatory neurons61. By using this model, we were able to isolate neurons at different states of DSB accumulation, enabling us to more accurately assess the effects of DSBs on genome integrity. In these mice, the expression of p25, an activator of cyclin-dependent kinase 5 (Cdk5), is under the control of the CamKII promoter and can be switched on by withdrawing doxycycline from the animal’s diet through doxycycline (dox)-off system62. In CK-p25, DSB accumulation starts at 1 week, with maximum levels of DSB at 2 weeks after the removal of doxycycline63. Thereafter, intracellular amyloid-beta accumulation occurs as early as 2–3 weeks after induction64. Neuronal loss, learning deficits, and tau hyperphosphorylation are also observed between 4 to 12 weeks after doxycycline removal62,64. Using fluorescence-activated nuclei sorting (FANS), we can isolate three distinct populations of neuronal nuclei using pan-nuclear neuronal marker NeuN and DSBs marker γH2AX. After 2 weeks of p25 induction in CK-p25 mice, we isolated 1) neurons with high levels of NeuN and baseline levels of γH2AX (Control neurons), 2) neurons with high levels of NeuN and γH2AX (Step 1 neurons), 3) cells with low levels of NeuN and high levels of γH2AX (Step 2 neurons) (Figure 1D). We previously showed that Step 2 neurons are indeed neurons that enter a later step of neurodegeneration that is characterized by the expression of senescence and immune genes61.
We used the previously published snRNA-seq in CK-p25 mice to test if neurons enriched for γH2AX have increased levels of gene fusions as a proxy for increased genome structural variation. Single-nucleus RNA-seq (Smart-seq2) was performed on all four FANS gates (Control neurons - NeuNhigh γH2AXlow, Glia - NeuNlow γH2AXlow, Step 1 neurons - NeuNhigh γH2AXhigh, and Step 2 neurons - NeuNlow γH2AXhigh ) from both CK and CK-p25 mice induced for 1 or 2 weeks. Step 2 neurons from CK-p25 mice formed a distinct expression cluster, whereas Step 1 was distributed into three clusters (Ex1, Ex2, and Ex3) between the Control (Ex0) and Step 2 clusters on a UMAP representation of the cells (Figure 1E). Step 2 neurons were significantly enriched for gene fusion calls (p-value=5.8e−16) whereas Step 1 neurons showed a modest increase in gene fusions (p-value > 0.05) (Figures 1F, G, S2 A–C, Table S1, page 6). We used Monocle 3 to order Control, Step 1, and Step 2 neuron nuclei along a gene expression pseudotime trajectory65. As we previously described, signatures of the Step 1 state (from comparing Step 1 vs Control bulk RNA-seq) increased from Control to Step 1 and then plateaued, whereas the signatures of Step 2 (from comparing Step 2 vs Control bulk RNA-seq) neurons continue to increase into Step 2 neurons as a function of pseudo time61. Remarkably, gene fusion frequency increased as a function of pseudo time, along with genes associated with senescence, similar to the Step 2 signature. On the other hand, genes associated with DNA damage and repair increased and plateaued similar to the Step 1 signature (Figures 1H, S2D, E). These results show that neurons with increased DSB burden (Step 1) progress to a senescence-like state that is characterized by increased gene fusions (Step 2).
We wondered whether increased gene fusions are the result of a general increase in genome-wide genome structural variations. To test this, we performed long-insert mate-pair sequencing to detect structural variations genome-wide. Here, genomic DNA was fragmented by Tn5 transposase carrying biotinylated adaptors. The fragments were circularized and then sheared into 200–300 bp fragments. The ligation junctions were then enriched through biotin pull-down and paired-end sequenced (Figure 1I). We first performed mate-pair sequencing in neurons with baseline levels of yH2AX and in yH2AXhigh neurons. Since the insert size was centered around 1–5 kb, pairs of reads that are separated by much larger distances would indicate genome structural variations. As expected, the vast majority of the read pairs (95.4 ± 0.9%) were separated by a distance of less than 10 kb. To measure structural variations with high confidence, we examined the percentage of reads in each sample separated by distances of 0.2 to 1.5 Mb. We saw a modest increase (22% increase) in the frequency of structural variations in yH2AXhigh neurons compared to neurons with baseline levels of yH2AX (Figure S2F). Remarkably, mate-pair sequencing in yH2AXhigh neurons partitioned into Step 1 and Step 2 revealed that Step 2 neurons had a much higher frequency of structural variation events (241% increase), whereas the structural variation frequency of Step 1 neurons was similar to that of Control neurons (Figure 1J). Structural variation frequencies follow chromosome spatial organization and tend to decay exponentially with the increasing genomic distance between the translocated loci30,66,67. Consistent with this, structural variation frequencies in all samples decreased as a function of distance (Figure 1J). These results suggest that the persistent DSBs and delayed repair in Step 1 neurons may increase the chance of repair errors and cause them to transition into Step 2 neurons with increased genome structural variations.
Next, we tested if inducing DSBs through an alternate mechanism would induce genome structural variations in neurons. We treated primary cortical neurons with Etoposide (5 μM), a Topoisomerase IIb poison that induces widespread DSBs. To model the acute vs persistent DSBs, we treated mouse primary cortical neurons with etoposide for either 2 hours (acute DSBs) or 12 hours (persistent DSBs) and allowed the neurons to recover in a basal etoposide-free media for 36 hours. Mate-pair sequencing revealed that neurons with acute DSBs had similar levels of structural variations compared to DMSO-treated baseline neurons, whereas neurons with persistent DSBs had a markedly higher frequency of structural variation (201% increase) similar to that of the increase observed in Step 2 neurons (Figure 1J). The 2 hours of treatment with Etoposide was sufficient to induce DSBs, as measured by yH2AX staining (Figure S2G), but not sufficient to accumulate structural variation (Figure 1J). Together, these results show that delayed repair due to persistent DSBs in excitatory neurons leads to the accumulation of mosaic genome structural variation.
2. Highly expressed genes and long genes are vulnerable sites of genome structural variations.
We found that fusion genes from Control, Step 1, and Step 2 neurons had significantly higher expression (from bulk RNA-seq) than expressed genes in neurons, suggesting that increased transcription is associated with increased genome structural variation (Figure 2A)61. This is consistent with the finding that genome structural variation induced by Top2B DNA breakage is dependent on transcription68. Next, we mapped DSBs directly using Break Labeling in situ and Sequencing (BLISS) in Control, Step 1, and Step 2 neurons and found that higher gene expression was strongly associated with increased DSBs across Control, Step 1, and Step 2 neurons (Figures 2B, S3A)69. Surprisingly, the level of enrichment of DSBs at highly transcribed genes was lower in Step 1 and Step 2 neurons compared to Control. This suggested that, while highly transcribed genes were hotspots for DSBs, the increased DSBs in Step 1 are distributed stochastically across the genome. BLISS uses Unique Molecular Identifiers (UMIs) to differentiate between independent DSB events and PCR duplicates. Therefore, conserved DSB locations across cells (hotspots) will be associated with multiple UMIs, whereas stochastic DSBs will be supported by fewer UMIs. Consistent with increased stochastic DSBs, Step 1 DSBs have fewer UMIs per genomic location. Interestingly, Step 2 neurons had a UMI distribution closer to Control suggesting partial repair of stochastic DSBs gained in Step 1 leading to possible mosaic structural variations (Figure S3B). Finally, we observed enrichment of DSBs at the gene fusion junctions with the highest enrichment in Control cells, indicating gene-fusion junctions are frequent DSB locations (Figure 2C). Example top gene fusion calls (Inka2—Atp5f1 and Cdip1—Ubald1), based on the number of fusion junction spanning fragments, show increased gene expression and DNA breakpoints at the fusion junctions (Figure 2D, Figure S3C). Interestingly, Inka2 and Cdip1 are targets of p53, an important regulator of senescence, and their transcriptional disruption through genome structural variation may promote a senescent-like state70–72.
Figure 2: Highly expressed genes and long genes are vulnerable sites for genome structural variations in neurons. See also Figure S3, Table S1, pages 1–4, 6, 7, and 17.
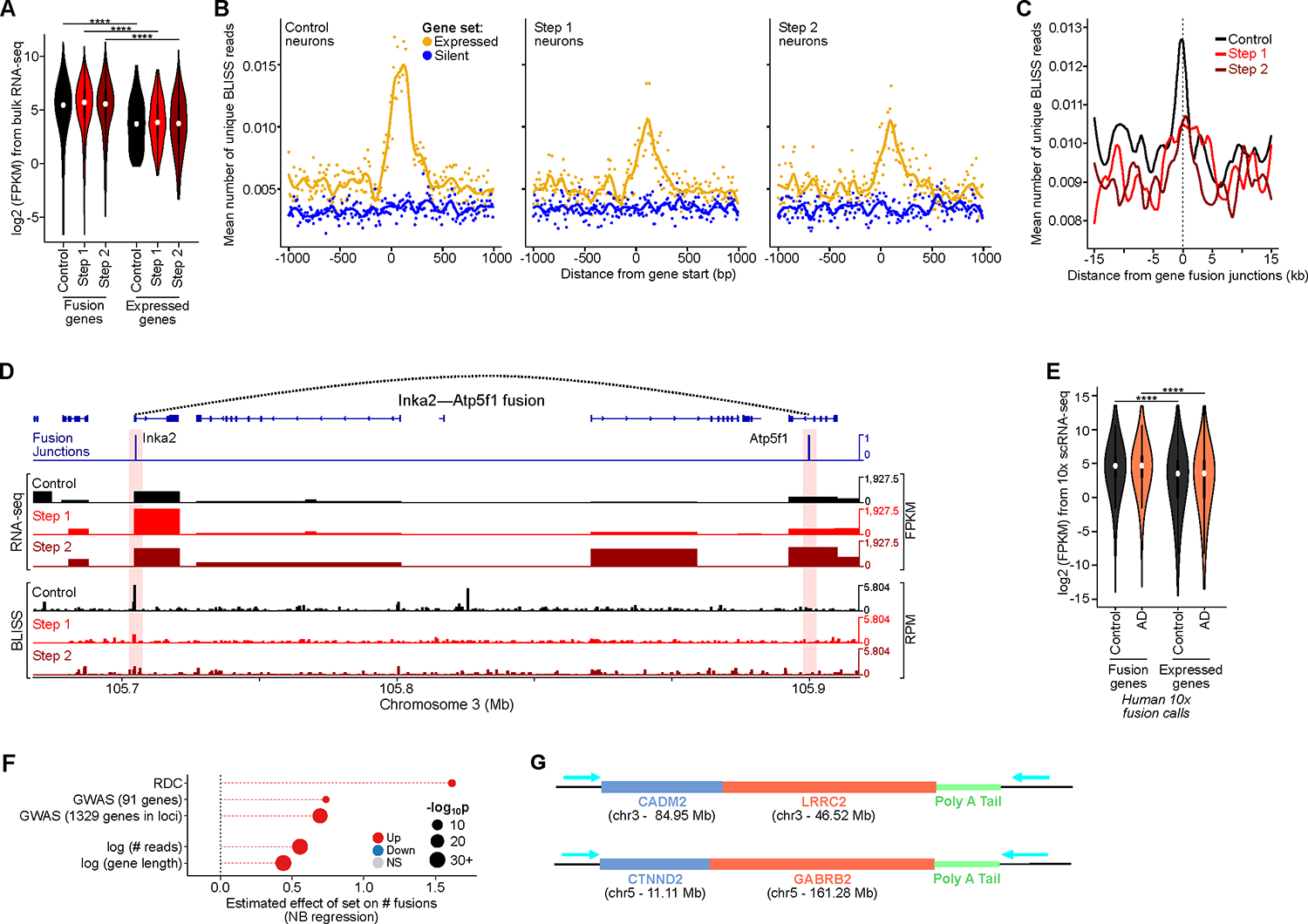
A) Expression level of genes involved in CK-p25 gene fusions (Wilcoxon test with Benjamini–Hochberg correction).
B) Aggregate plots of BLISS reads centered at gene starts for the top 2500 expressed (yellow) and the bottom 2500 silent genes (blue), for Control, Step 1, and Step 2 neurons.
C) Aggregate plots of BLISS reads centered at gene fusion junctions called by snRNA-seq in CK-p25 neurons.
D) Genome browser snapshot of example gene fusion in CK-p25 mice Step 2 neurons. Panels show fusion junctions (top), bulk RNA-seq signal (panels 2–4), and BLISS in the three populations (panels 5–7).
E) Expression level of genes involved in human gene fusions (10x snRNA-seq) vs all expressed genes (Wilcoxon test with BH correction).
F) Effect size of the number of gene fusions as a function of: genes identified as RDC gene, AD GWAS genes, gene expression level (reads), and gene length (bp).
G) Schematics of example gene fusions identified through ONT long-read cDNA sequencing.
Similar to our observations from BLISS in CK-p25, human fusion genes in excitatory neurons also tended to have increased gene expression in both AD and control individuals (Figure 2E). We saw a significant overlap between genes involved in fusions and a set of actively transcribed, long neural genes that are important for synaptic function and cell-cell adhesion and are known to harbor recurrent DSB clusters (RDC) in neural precursor cells (Figure 2F)73. We found 25 of the 27 recurrent DSB cluster (RDC) genes in human excitatory gene fusion calls, including CTNND2, LSAMP, and NRXN1 (Table S1, page 7). The length and active transcription of these RDC genes likely drive their increased propensity for DSBs and gene fusions. Across all genes, both higher transcription levels and increased gene length were significantly associated with increased gene fusions (Figure 2F). The association between gene length and gene fusions was observed in mouse neurons and is consistent with increased DSBs at highly transcribed genes and long genes (Figure 2A, S3D, E). Interestingly, genes linked to AD by genome-wide association studies (GWAS) were significantly longer than expected (Figure S3F) and were significantly enriched within fusion genes (Figure 2F, Table S1, page 7)74. We also found a significant association between fusion genes and a larger list of genes linked to AD by GWAS and polygenic risk scores (Figure 2F) (NHGRI-EBI GWAS Catalog, ID: MONDO_0004975)75.
Finally, to validate fusion events observed by short-read sequencing, we performed Oxford nanopore technologies (ONT) long-read sequencing of cDNA from two AD post-mortem samples. We identified several gene fusions which also included RDC genes (e.g. CADM2, CTNND2) (Figure 2G, Figure S3G). Taken together, these results show that increased DNA breaks associated with long genes and highly transcribed genes make them vulnerable to structural variation and gene fusion.
3. Neurons burdened with DSBs are associated with the disruption of 3D genome organization
In parallel, we identified a significant elevation in cohesin complex expression along with a coordinated increase in DNA damage response in excitatory neurons from subjects with high AD pathology in the large-scale snRNA-seq across 427 AD and control individuals from our companion manuscript59. The cohesin complex is an important architectural protein that organizes the 3D genome and is also important in DSB repair 32,76–78. In particular, cohesin has been shown to protect against genome structural variations 79,80. Further, loop extrusion mediated by cohesin and CTCF mediates the formation of chromatin loops and self-interacting Topologically Associating Domains (TADs)77,81–86. To directly investigate the levels of the cohesin complex in excitatory neurons with DSBs, we stained the CK-p25 hippocampus for the cohesin subunit RAD21 after 2 weeks of induction. RAD21 was significantly enriched in neurons with DSBs compared to those with baseline DSBs (Figure 3A). Interestingly, snRNA-seq revealed increased gene expression of another nuclear architectural protein Lamin B1 (Lmnb1) in Step 1 neurons and a subsequent decrease in expression from Step 1 to Step 2 (Figure S2E). Lamin B1 plays a crucial role in the 3D genome organization by tethering heterochromatin to the nuclear periphery87. Additionally, Lamin B1 has been shown to control the release of DSB repair protein 53BP1 and overexpression of LaminB1 impeded DSB repair leading to persistent DSBs88. Immunohistochemistry of Lamin B1 in CK-p25 mice induced for 2 weeks revealed increased Lamin B1 and abnormalities in the structure of Lamin B1 in neurons bearing DSBs. Thereafter, Lamin B1 intensity decreased after 6 weeks of induction, consistent with the snRNA-seq (Figure S4 A–C). Indeed, we show decreased Lamin B1 in late-stage human AD associated with high epigenomic erosion in our companion manuscript (Xiong et al.89).
Figure 3: Neurons burdened with DSBs exhibit global disruption of the 3D genome organization at multiple scales. See also Figure S4, Table S1, pages 8–10 and 16.
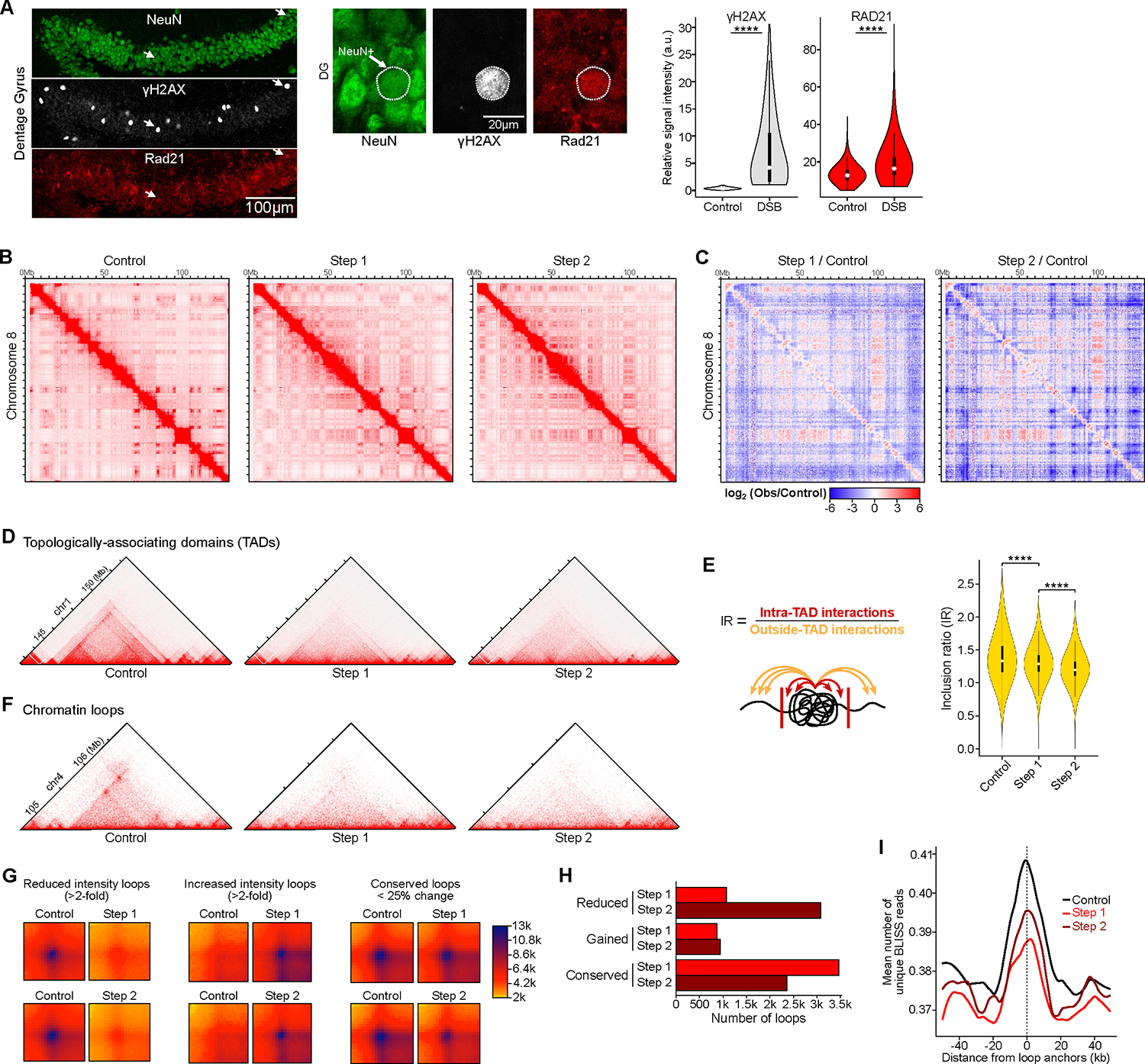
A) Representative immunohistochemistry images and quantification of RAD21 (cohesin subunit) in 2-week induced CK-p25 mice, comparing neurons with baseline DSBs to neurons enriched for DSBs. Representative images show dentate gyrus (left) and higher-magnification images (right). Violin plots quantify RAD21 levels between neuronal nuclei with baseline DSBs (ϒH2AX relative intensity < 1) and neurons enriched for DSBs (ϒH2AX relative intensity > 1) (Wilcoxon test). The mean relative intensity of ϒH2AX (gray) and RAD21 (red) was measured within NeuN surfaces. Control (n=5,316 cells), DSB (n=725 cells).
B) Full chromosome (chr8) Hi-C chromatin interaction heatmaps.
C) Differential heatmaps comparing Step 1/Control and Step 2/Control, colored by increased (red) or decreased (blue) interactions over control.
D) Representative Hi-C interaction plots showing the disruption of TADs. Hi-C heatmaps were rotated 45 degrees and only the upper triangle is shown.
E) Quantification of TAD disruption through Inclusion Ratio (IR). IR is the ratio of intra-TAD interactions to outside-TAD interactions (Wilcoxon test with BH correction).
F) Representative Hi-C interaction plots showing the disruption of chromatin loops.
G) Aggregate heatmaps of reduced intensity (>2-fold), increased intensity (>2-fold), and conserved (<25% change) chromatin loops for Step 1 vs. Control and Step 2 vs. Control.
H) Bar plot indicating quantification of chromatin loop disruption in Step 1 (red) and Step 2 (dark red).
I) Aggregate plots of BLISS reads centered at loop anchors in CK-p25 neurons.
To investigate if DSBs and the alterations in chromatin structural proteins can impact the 3D genome organization, we performed in situ Hi-C90 in Control, Step 1 and Step 2 neurons. Hi-C revealed genome-wide alterations in the 3D genome organization that involved both gain and loss of long-range interactions (Figure 3B, C). Overall, there was a net decrease in the intensity of long-range interactions above 1 Mb, in Step 1 neurons. This was even more pronounced in Step 2 neurons with the intensity of long-range interactions decreasing from 0.5 Mb (Figure S4E). Chromosomes are spatially segregated into generally active A compartment and repressive B compartment91. While the A compartment is largely localized to the nuclear interior, the B compartment is tethered to the nuclear periphery by Lamin B192. Surprisingly, despite the disruption of Lamin B1, the nuclear A to B compartmentalization derived from Hi-C was largely intact. However, approximately 8.5% of the genome shifted compartments and these changes were generally progressive from Step 1 to Step 2 (Figure S4 F, G, Table S1, page 8).
Since chromatin is organized into loops and self-interacting units called TADs by cohesin and CTCF, we wondered how DSBs and the misregulation of cohesin would affect loop and TAD organization. We used a metric called inclusion ratio that measures the ratio between intra-TAD interactions to outside-TAD interactions to quantify changes in TAD definition genome-wide93. This analysis revealed an overall significant decrease in the TAD definitions in Step 1 that was exacerbated in Step 2 (Figure 3 D, E, Table S1, page 9). Remarkably, the accompanying manuscript by Xiong et al. describes widespread changes in cell-type specific chromatin accessibility within excitatory neurons in human AD samples which they term “epigenome erosion”89. This highlights the deterioration of chromatin structure and organization as a general pathological step in AD progression.
We also observed the disruption of chromatin loops in Step 1 and Step 2 neurons compared to control neurons (Figure 3F, Table S1, page 10). Aggregate plots of loops show genome-wide disruption of loops involving both reduction and gain of loop intensity (Figure 3G). Overall, more loops showed a reduction than a gain in intensity, and loop loss was much more pronounced in Step 2 than in Step 1 neurons (Figure 3H). Given the overall reduction in loop intensities, we wondered if loop boundaries are generally hotspots of DSBs in neurons. We found an enrichment of DSBs at loop boundaries in Control, Step 1, and Step 2 (Figure 3I). This aligns well with the observation that loop boundaries are hotspots for TOP2 B-mediated DNA breaks in T-cells and B-cells 28. The enrichment of DSBs at loop boundaries was lowest in Step 1, followed by Step 2, which is consistent with the stochastic increase in DSB burden in Step 1 and partial repair in Step 2 (Figures 3I, S3B).
4. Induction of DSBs in neurons is sufficient to disrupt the 3D genome organization
Next, we sought to test if the induction of DSBs by an alternate mechanism independent of the CK-p25 model would recapitulate the DSB-mediated effects on the 3D genome organization. We observed increased genome structural variations in cultured primary neurons where persistent DSBs were induced by etoposide treatment (Figure 1J). Therefore, we wondered if the induction of DSBs by Etoposide would also elicit changes in the 3D genome organization. To test this, we treated primary mouse cortical neurons with either 5 μM etoposide for 48 hours or DMSO. We FANS sorted NeuN+ve neurons with high γH2AX from the etoposide treatment and NeuN+ve neurons with baseline γH2AX from the DMSO treatment (Figure 4A). Next, we performed in situ Hi-C, but at a lower read depth. We observed a reduction in TAD integrity and loop intensities similar to CK-p25 (Figure S5A). Quantifying TAD integrity using inclusion ratio revealed a significant reduction in the etoposide-treated primary neuronal cultures similar to that of in-vivo neurons from CK-p25 mice with high DSB burden (Figure 4B, Table S1, page 11). The level of induction of DSBs, as measured by yH2AX levels in FANS, was dependent on etoposide concentration (Figure S5B), and treating primary neurons with a lower concentration of etoposide (1 μM, 48 hours) also produced a significant reduction in the inclusion ratio (Figure S5C).
Figure 4: DSBs in neurons are sufficient to disrupt the 3D genome organization. See also Figures S5, Table S1, page 11 and 16.
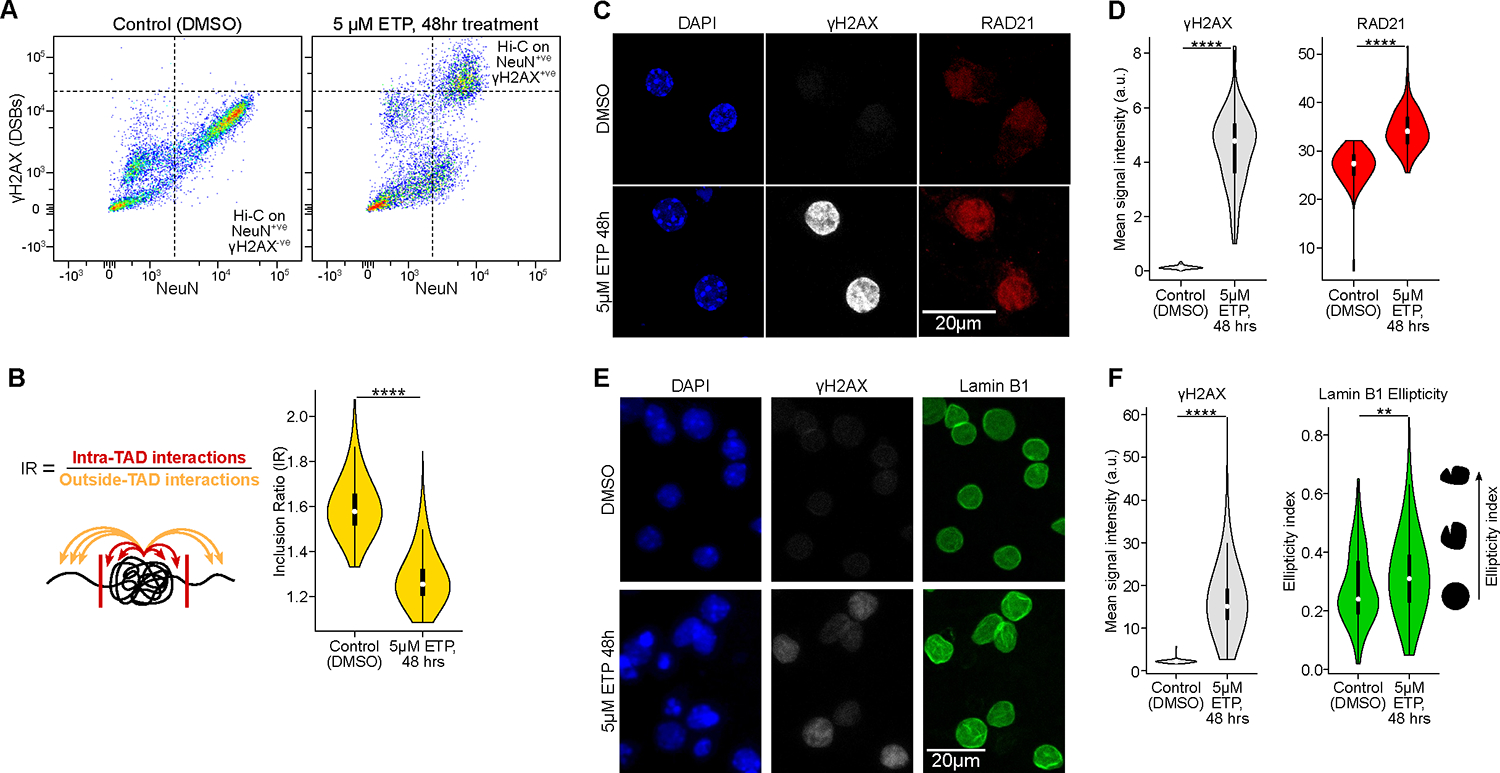
A) FANS dot-plots of NeuN versus ϒH2AX immunoreactivity after etoposide (5μM for 48 hours) or vehicle (DMSO) treatment in primary neuronal culture.
B) Quantification of TAD disruption (IR, Wilcoxon test).
C) Representative images and quantification of RAD21 (cohesin subunit) IHC in primary culture neurons (DIV 12) after Etoposide (5μM for 48 hours) or vehicle treatment (DMSO).
D) Violin plots quantifying the images for ϒH2AX levels (gray) and RAD21 levels (red) in control neurons and etoposide-treated neurons (Wilcoxon test). The mean relative intensity of ϒH2AX and RAD21 was measured within NeuN surfaces. Control (n=111 cells), ETP (n=112 cells).
E) Representative images of Lamin B1 IHC in primary culture neurons (DIV 12) after Etoposide (5μM for 48 hours) or vehicle treatment (DMSO).
F) Violin plots quantifying the images for ϒH2AX levels (red) and Lamin B1 ellipticity (green) in control neurons and etoposide-treated neurons (Wilcoxon test). The mean relative intensity of ϒH2AX was measured within Lamin B1 surfaces that were positive for NeuN. Control (n=78), ETP (n=809).
CK-p25 neurons burdened with DSBs exhibited disruption of RAD21 and Lamin B1, two structural proteins involved in DSB repair and 3D genome organization (Figures 3A, S4A–D). To measure the effect of etoposide-induced DSBs on levels of cohesin, we stained for RAD21 in primary cortical neurons treated with 5 μM etoposide for 48 hours compared to DMSO-treated control. Like CK-p25 neurons with high DSB burden, RAD21 levels were significantly elevated in neurons harboring etoposide-mediated DSBs (Figure 4C, D). Next, we stained for Lamin B1 in primary neurons treated with etoposide and observed the disruption of Lamin B1 at the nuclear envelope similar to the CK-p25 mouse model (Figure 4E, F, S4D). Etoposide-treated neurons also showed increased senescence-like and immune gene expression signatures indicating the similar molecular mechanisms underlying the disruption of genome organization and stability between etoposide-treated neurons and the CK-p25 mouse model (Figure S5D).
To further explore the association between DSBs and the 3D genome disruption in neurons, we turned to the Tau P301S model of tauopathy, an additional mouse model of neurodegeneration94. While not as pronounced as the CK-p25 model, where a distinct subset of neurons harbors high levels of DSBs, neurons in Tau P301S mice show a pan-neuronal increase in DSBs95. We performed in situ Hi-C on FANS-sorted NeuN+ve neuronal nuclei from the Tau P301S forebrain and WT litter mates. Consistent with the observations in CK-p25 mice and etoposide-treated neurons, Hi-C in P301S neurons showed a decreased number of chromatin loops and a significant decrease in TAD integrity as measured by inclusion ratio (Figure S5E, F, Table S1, pages 12, 13). Together, these results demonstrate that DSBs in neurons are sufficient to disrupt the 3D genome organization.
5. Disruption of the 3D genome organization by DSBs is associated with differential gene regulation.
The 3D genome plays an important role in neuronal gene expression and function36,38–40,96,97. Chromatin loops enable interactions between enhancers and promoters enabling fine control of gene expression, in addition to the regulation mediated by transcription factors and other epigenetic modifications27,98. To investigate if the chromatin loop dysregulation in Step 1 and Step 2 neurons is associated with gene expression changes, we stratified differential loops at increasing fold-change thresholds and calculated the percentage of differential up and down-regulated genes (fold-change > 2, p-value<0.05) near these loops (+/− 5 kb from anchors) using bulk RNA-seq from Control, Step 1, and Step 2 neurons (Figure 5A)61. Reduction in loop intensity was strongly associated with higher percentages of down-regulated genes in both Step 1 and Step 2 neurons and with lower percentages of up-regulated genes (Figure 5B). Down-regulated genes associated with loop loss were significantly enriched for synaptic, cell adhesion, and neuronal development genes (Figure 5C, Table S1, page 14). On the other hand, loops with increased intensity did not have a consistent relationship with differential gene expression, suggesting that the association between loop gain and gene expression is context-dependent (Figure S6A)99.
Figure 5: DSB-associated dysregulation of the 3D genome aligns with the differential gene expression. See also Figure S6, Table S1, page 10, 14 and 16.
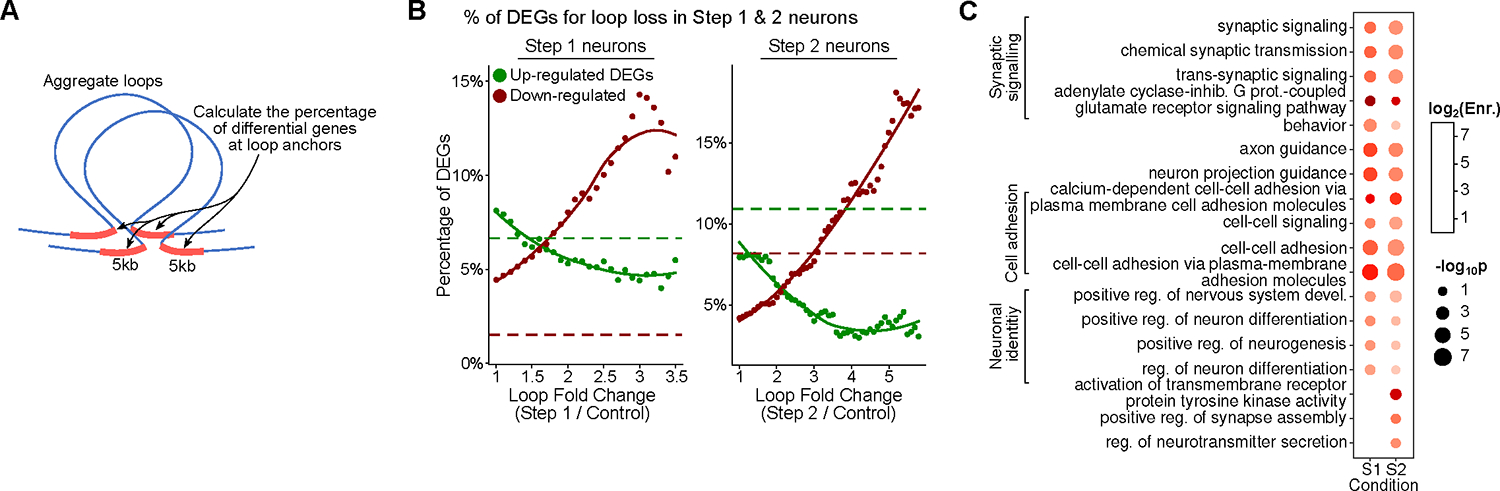
A) Schematic for comparison of differential loops to differential gene expression in neurons with DSBs. Differential loops were identified for increasing fold change thresholds of loop score. For each differential loop, genes within +/− 5 kb of the loop base were identified and the percentage of differentially expressed genes (DEGs) was calculated. Percentage DEGs were then plotted against the thresholds used for calling the differential loops.
B) Percentage of up (green) and down (red) DEGs vs loop fold change for loop loss in both Step 1 and 2. Red and green dotted lines are the baseline percentage of up and down DEGs genome-wide, respectively.
C) Gene ontology analysis (GORILLA) of loop loss-associated down-regulated genes.
Among the differential genes in Step 1 and Step 2 neurons, we found that cohesin complex genes were up-regulated alongside several previously reported DNA damage, senescence, and immune response genes (Figure 6A)61. Cohesin complex genes were consistently and significantly up-regulated in excitatory neurons from subjects with high AD pathology in the 10x snRNA-seq data across 427 individuals (Figure 6B)59. The increase in cohesin complex gene expression is also consistent with the increased levels of RAD21 in both CK-p25 neurons and etoposide-treated primary neuronal cultures (Figure 3A, 4C). Finally, we used immunohistochemistry to measure the levels of cohesin subunit RAD21 in human post-mortem tissue and found significantly increased levels of RAD21 in neurons from subjects with AD pathology (Figure 6C, D).
Figure 6: Up-regulation of cohesin and histone genes in Alzheimer’s Disease. See also Figure S6 and Table S1, page 5, 15, and 16.
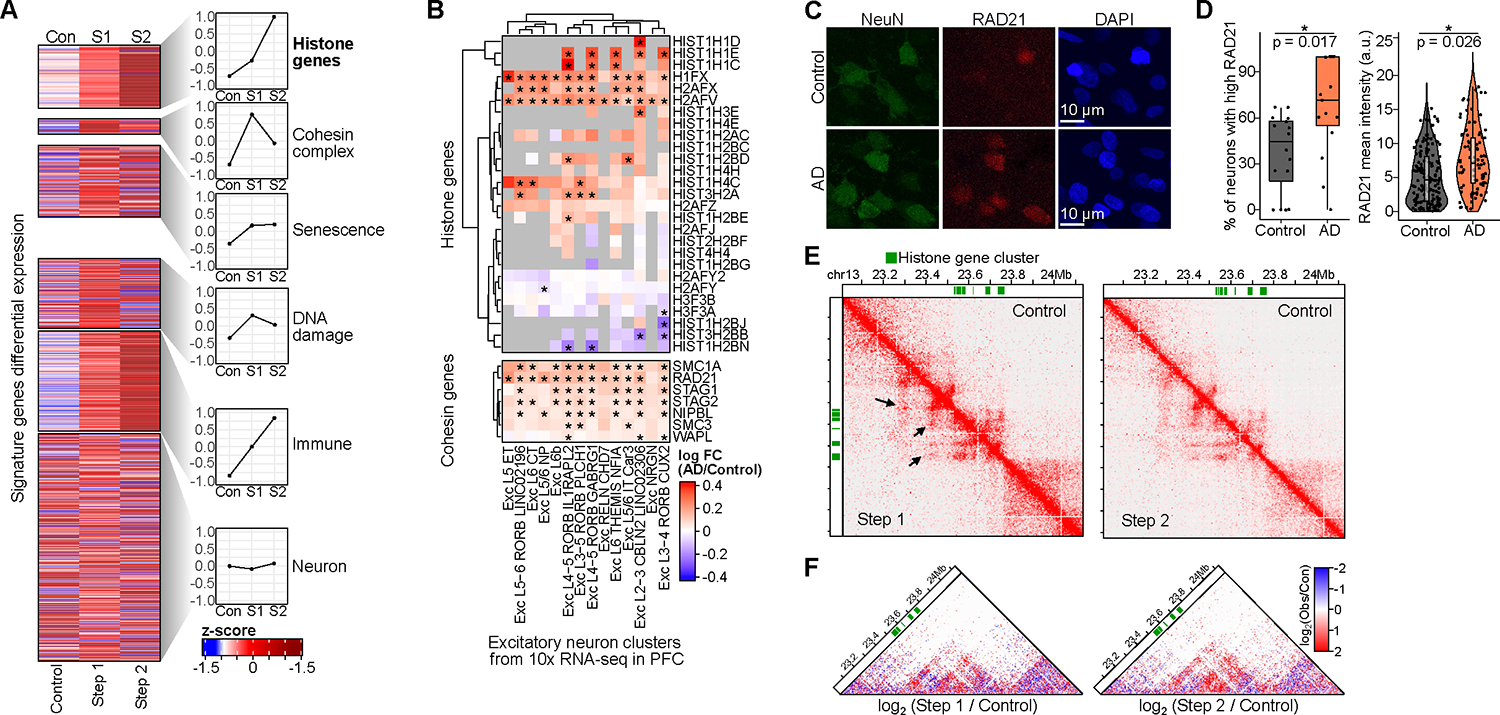
A) Heatmap of differential gene expression as z-scores from bulk RNA-seq in Control, Step 1, and Step 2 for signature gene sets. The right panels show average values for each set.
B) Heatmap of differential expression of histone and cohesin genes across excitatory subtypes in 10x snRNA-seq from Mathys et al. (*differential).
C) Representative immunostaining images of RAD21 in AD and Control human post-mortem tissue.
D) RAD21 staining was quantified as the percentage of neurons with high RAD21 (mean intensity > 50th percentile) per image (left) or using RAD21 mean intensity (right). Significance was calculated using a linear mixed-model approach across 3 AD and 3 Control individuals (Table S15).
E) Hi-C interaction matrix for histone gene cluster (green blocks, chr13). The lower diagonal is Step 1 or 2, upper is Control.
F) Differential Hi-C heatmaps comparing Step 1/Control and Step 2/Control, colored by increased (red) or decreased (blue) interactions over control. Hi-C heatmaps were rotated 45 degrees and only the upper triangle is shown.
Interestingly, we noticed histone genes were consistently and progressively upregulated in Step 1 and Step 2 neurons (Figure 6A). Their differential changes were significantly correlated with increased histone gene expression in etoposide-treated primary neuronal cultures, suggesting that DSBs play a key role in their dysregulation (Figure S6B). To measure the regulation of histone genes in human AD pathology, we leveraged snRNA-seq across 427 individuals from Mathys et al.59 Indeed, we found that several histones were upregulated in human AD in excitatory neurons along with the upregulation of the cohesin complex genes (Figure 6B). Strikingly, the strongly up-regulated histone genes included H2AFX, which encodes the histone H2AX, whose phosphorylation marks DNA damage. Another upregulated histone gene, H2AFV, codes for H2A.Z, whose increase is associated with persistent DNA damage, brain aging, and is negatively associated with memory formation100–102. A large number of histone genes are located as clusters in a 1 Mb region on mouse chromosome 13103. Remarkably, along with the increased gene expression of histone genes, the histone gene clusters on chromosome 13 gained de novo loops in Step 1 and Step 2 neurons (Figures 6E, F, S6C, D). This highlights the 3D genome level regulation underlying the upregulation of histone gene clusters. Together these results demonstrate the association between DSB-mediated 3D genome changes and gene regulation programs underlying neurodegeneration.
Discussion
Neurons’ long lifespan and the lack of sister chromatids for homologous recombination repair leave them vulnerable to DSBs104. Recent studies have shown neuronal activity-dependent DSBs and associated repair mechanisms9,11,105. In addition, extensive neuronal activity-induced demethylation could be a source of DNA single-stranded breaks through the Tet-TDG pathway-mediated base excision repair12,13. Neuronal genomes have a non-random distribution of DNA damage106, notably, neuronal enhancers are hotspots of single-stranded breaks presumably through multiple cycles of enhancer cytosine methylation and demethylation8,10. These processes compound the vulnerability of neurons to DNA damage compared to other brain cell types.
Neurons start accumulating DSBs early in the progression of AD2,4–7. We found a significant association between the expression of DNA repair pathway genes and AD pathology in single-nucleus profiling of AD brains (Mathys et al. co-submitted)59. In addition, mutations in DNA repair pathway components lead to neurodevelopment and neurodegenerative diseases107,108. Interestingly, we found a modest but not significant enrichment of protein-coding genes related to DNA damage repair in the published AD GWAS catalog (NHGRI-EBI, OR= 1.22, 62 GWAS/DNA damage repair genes)75, including crucial DSB repair genes such as ATM, HMGA2, and EXO1. This apparent lack of statistical significance may be because certain mutations in DNA repair genes predispose individuals to premature death before AD onset through accelerated aging and other severe diseases such as cancer and diabetes, leading to their underrepresentation in GWAS109,110. Consistent with this hypothesis, DNA damage repair pathways were found to be significantly enriched in Huntington’s disease GWAS analysis, a neurodegenerative disease with a much earlier age of disease onset111.
DSBs are repaired via the classical non-homologous end joining (NHEJ) pathway in post-mitotic cells and can lead to genome structural variations18–20. Further, genome structural variations can lead to gene fusions and are often drivers of cancer112, and are detectable from bulk RNA-seq58,113–115. Gene fusions are an attractive proxy for comparing the rate of genome structural variations in AD and non-AD individuals using single-cell RNA-seq, but detection and validation of independent mosaic fusions or structural variations in single cells remain challenging in the absence of clonality116–120. Instead, in this study we report an increased burden of mosaic gene fusions in excitatory neurons in individuals with AD pathology by calling fusion events in single-cell RNA-seq in human post-mortem samples in each cell independently. Gene fusions are also particularly enriched in excitatory neurons with DNA damage, senescence, and cohesin gene signatures. Indeed, accumulation of DNA damage can lead to a senescence-like state in neurons and has been implicated in the progression of AD.121–123.
In the CK-p25 model of neurodegeneration, neurons progress through an initial step with increased DSBs (Step 1) to a later step enriched for gene fusions and structural variation and marked by senescence genes (Step 2)61. We hypothesize that persistent DSBs in neurons lead to genome structural variations, consistent with DSB induction by etoposide, where persistent but not acute DSB induction leads to increased structural variations. Gene fusion junctions were enriched for DSBs and fusions were enriched for genes with higher expression and length, consistent with previous observations in non-neuronal cells and neural precursor cells68,73. Surprisingly, Step 1 showed lower DSB enrichments for highly transcribed and long genes, and DSBs were supported by fewer UMIs, suggesting that Step 1 DSBs are largely stochastic. By contrast, UMIs in Step 2 were distributed similarly to controls, suggesting increased repair of DSBs at the price of structural variations. We propose that the increased stochastic DSB burden strains the DNA repair resources, leading to defective DNA repair and genome structural variations that disproportionately affect hotspots of DNA breaks such as highly expressed genes, long genes, and loop anchors. Future studies that map DSBs directly in post-mortem tissue could reveal additional hotspots associated with structural variations in AD.
Previous studies have shown increased neuronal genomic mosaicism (including SNVs, copy number variations, and cDNA insertions in neurodegeneration)124–128. Recently, a study identified an increase in mosaicism due to recombination between Alu and L1 elements in Parkinson’s and Alzheimer’s disease120. Here we report gene fusions and genome structural variations mediated by DSBs in neurons as a class of mosaic somatic genomic change exacerbated in neurodegeneration. Genome structural variations can bring ectopic regulatory elements next to genes causing pathological up or down-regulation of genes. Interestingly, genome structural variations have been shown to cause pathological TAD fusions and create neo-TADs in several cancers. For instance, a neo-TAD caused by structural variation was associated with higher expression of the oncogene MYC in neuroblastomas23. Therefore, it’s likely that structural variations in neurons could impact the expression of important neuronal genes in AD. Gene fusions at important neural genes can result in a pathological gain or loss of their function and result in defects in neuronal function. Indeed, we find that long-neural genes with synaptic and cell adhesion genes associated with the recurrent DNA double-strand breaks and AD GWAS genes are enriched for gene fusions 73. We also demonstrate a link between genome structural variations and the senescence-like state in neurons. Indeed, genomic mosaicism by retro transposition of LINE-1 elements has been associated with the neuronal genome and the derepression of LINE-1 elements has been associated with senescence129–131. These observations combined with our results suggest that genomic mosaicism is an important driver of senescence in neurons and could contribute to the pathogenesis of neurodegenerative disease.
In addition to the disruptions in the linear genome, we show that the DSB-mediated senescence-like state in neurons leads to the disruption of the 3D genome in neurodegeneration. We observed a general loss of TAD definition and loop intensities especially in Step 2 senescence-like neurons along with differential regulation of several genes associated with neuronal identity. This is strikingly similar to epigenome erosion, particularly in excitatory neurons and oligodendrocytes, characterized by the loss of cell-type specific chromatin accessibility profile reported in the companion manuscript89. Interestingly, erosion of epigenetic information, TAD insulation, and cell identity was recently reported in a mouse model with inducible DNA damage (ICE mouse model) accompanied by brain aging and cognitive decline132. A recent study proposed that DSBs can act as roadblocks to loop extrusion32. It’s conceivable that adjacent DSBs may cause premature loop extrusion block on both sides, thereby disrupting chromatin loops and larger domains. Surprisingly, X-ray radiation-induced DSBs increased the segregation of TAD fibroblasts54. Future studies should explore how acute DSBs formed by irradiation vs. persistent DSBs impact genome organization, and additionally how post-mitotic cells and dividing cells differ in their response to DSBs. We also observed DSB-mediated changes in the levels of nuclear structural proteins, cohesin, and Lamin B1, in neurons enriched for DSBs, similar to previous observations in AD neurons and also in senescent astrocytes133,134. Despite the disruption of Lamin B1, the A/B compartmentalization of the genome remained relatively unaffected which is in concordance with the observations in retinal rod neurons, where despite the loss of heterochromatin interactions with the nuclear lamina, phase separation drives the formation of A/B compartments135.
Changes in the 3D genome aligned with transcriptional changes overall, with reduced loop intensities having a striking correlation with the down-regulation of genes. Interestingly, we observed the de-novo establishment of loops at histone gene clusters that aligned with the upregulation of several histone genes. Previous studies have shown the importance of histones in DNA damage response, senescence, brain aging, neuronal identity, and memory formation100–102,136–139. These results highlight a link between the 3D genome organization in neurons and transcriptional programs associated with the neurodegenerative state. Indeed, while the gain of chromatin loops is associated with histone gene up-regulation, other independent mechanisms of gene regulation may underlie the up-regulation of genes in degenerating neurons. Additionally, studies have shown senescence-associated 3D genome reorganization in dividing cell types42,140–143. Our results show that the 3D genome organization changes in neurons in a senescence-like state may mediate genome dysfunction in neurodegeneration.
Overall, our work demonstrates that DNA double-strand breaks lead to mosaic genome structural variations and the disruption of 3D genome organization in neurons. We observe increased mosaic gene fusions caused by genome structural variations in excitatory neurons associated with increased cohesin, DNA damage, and senescence-like gene expression in Alzheimer’s disease. These data link neuronal genomic integrity and 3D genome organization to cellular pathologies underlying neurodegeneration.
Limitations of the Study
Gene fusions arise from a subset of genome structural variations and future technologies coupling single-cell RNA sequencing with long-read single-cell genome sequencing will be required to resolve the full extent of DNA damage-dependent genome structural variations in neurodegeneration. Single-cell RNA seq provides a sparse readout of gene expression and is likely to generally underestimate the number of gene fusions in each cell. While relatively uncommon, chimeric molecules arising through PCR artifacts and random ligations during library preparation are difficult to distinguish from chimeric molecules arising from true mosaic gene fusions. We make the assumption that AD vs. Control classification and various gene expression pattern enrichments explored in this do not modify PCR artifacts and random ligations during library preparation. The disruption in the 3D genome organization could be caused both directly by unresolved DSBs and by genome structural variations. In the current study, we are unable to resolve the precise role of each of these components. Future studies that perform targeted Hi-C at unresolved DSBs locations vs. post-repair locations will be better positioned to answer these questions.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Li-Huei Tsai (lhtsai@mit.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
Single-nucleus RNA-seq data will be available through the AD Knowledge Portal on Synapse upon publication of this manuscript. The data will be available here: https://www.synapse.org/#!Synapse:syn52293426. The data are available under controlled use conditions set by human privacy regulations. To access the data, a data use agreement is needed. This registration is in place solely to ensure the anonymity of the ROSMAP study participants. A data use agreement can be agreed with either Rush University Medical Center (RUMC) or with SAGE, which maintains Synapse, and can be downloaded from their websites (https://adknowledgeportal.synapse.org/)
All mouse data generated in this study, including raw and processed files, are available at GSE227445.
We have established a web portal for this paper at http://compbio.mit.edu/ad_dna_damage/ to better describe and provide access to our datasets and code, which is also available at https://doi.org/10.5281/zenodo.8290268.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human subjects
We selected 427 individuals from the Religious Orders Study and Rush Memory and Aging Project (ROSMAP), both ongoing longitudinal clinical-pathologic cohort studies of aging and dementia, in which all participants are brain donors. The studies include clinical data collected annually, detailed postmortem pathological evaluations, and extensive genetic, epigenomic, transcriptomic, proteomic, and metabolomic bulk-tissue profiling144. Individuals were balanced between sexes (male:female ratio 212:215). Informed consent was obtained from each subject, and the Religious Orders Study and Rush Memory and Aging Project were each approved by an Institutional Review Board (IRB) of Rush University Medical Center. Participants also signed an Anatomic Gift Act, and a repository consent to allow their data to be repurposed. Participants enrolled without known dementia and agreed to annual clinical evaluation and organ donation.
For single-nucleus Smart-seq2, we selected a subset of 47 individuals comprised of 24 and 23 age and sex-matched cases with AD pathology (NIA-Reagan 1–2) and no pathologic AD (NIA-Reagan 3–4). For the gene fusion burden analysis, we additionally split the samples independent of NIA-Reagan by:
Animal models
All the experiments were approved by the Committee for Animal Care of the Division of Comparative Medicine at the Massachusetts Institute of Technology (MIT) and carried out at MIT. Tg(Camk2a-tTA) and Tg(Prnp-MAPT*P301S)PS19 were obtained from the Jackson laboratory. Tg(tetO-CDK5R1/GFP) was generated in our lab. All the transgenic mice were bred and maintained in our animal facility. We used male TauP301S mice at 3 months and 11–12 months of age. CK-p25 mice are generated by breeding CaMKIIα promoter-tTA mice (CK controls) with tetO-CDK5R1/GFP mice and raised on doxycycline-containing food to repress p25 expression. CKp-p25 mice were induced for 1, 2, or 6 weeks depending on the experiment by replacing the doxycycline diet with a normal rodent diet. We used adult CK-p25 mice (4–8 months old) for all experiments. Both male and female mice were used for Hi-C, and female mice were used for all other experiments.
Primary Neuron Culture
Cortices were dissected from E15 Swiss-Webster embryos in ice-cold HBSS (Thermo Fisher Scientific, 14175103) and dissociated with papain (Worthington Biochemical Corp, LS003126) and DNase I (Roche, 10104159001). Cells were resuspended in plating media (Neurobasal media (Thermo Fisher Scientific, 21103049), 1% Penicillin/Streptomycin Solution (Gemini Bio-Products, 400-109), 10% FBS) and filtered through a 100 μM cell strainer (VWR, 21008-950). Cell density was quantified using a Countess II Automated Cell Counter (Thermo Fisher Scientific, AMQAX1000), then plated on poly-D-Lysine-coated 12-well culture dishes at 0.5×106 cells or in 100mm culture dishes at 5×106 cells. Cultures were maintained in 5% CO2 at 37 °C in a cell culture incubator. After allowing four hours for the cells to adhere to the plate, the media was replaced and maintained with neurobasal media supplemented with B-27 (Invitrogen, 17504-044), 1% Penicillin/Streptomycin, and 1% GlutaMAX Supplement (Thermo Fisher Scientific, 35050–079).
MADRC brain tissue samples
Immunohistochemistry was performed using fresh frozen postmortem PFC brain samples which were generously provided by the Massachusetts Alzheimer’s Disease Research Center (MADRC). Sample selection was based on the Braak staging. Braak stage147, where stages 1–4 indicate the absence of NFTs in the prefrontal cortex (PFC), and stages 5–6 indicate the presence of NFTs in the PFC. Three of the samples identified as AD had a Braak score above 5. Meanwhile, the remaining three non-AD samples had Braak scores less than or equal to 3. For more information on the samples, the sample metadata can be found in Supplementary Table S1, page 15.
METHOD DETAILS
Single-nucleus Smart-seq2
We adapted the protocol for the isolation of nuclei from frozen postmortem brain tissue from Swiech et al. 2015148. All procedures were carried out on ice or at 4 °C. Briefly, postmortem brain tissue was homogenized in 2 ml Homogenization Buffer (320 mM Sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 10 mM Tris-HCl pH 7.8, 0.1 mM EDTA pH 8.0, 0.1% IGEPAL CA-630, 1 mM b-mercaptoethanol, 0.4 U/microliter Recombinant RNase Inhibitor (Takara, 2313A) using a Wheaton Dounce Tissue Grinder (10 strokes with the loose pestle). 3 ml of Homogenization Buffer was added (final volume 5 ml) and the homogenized tissue was incubated on ice for 5 minutes. Then the homogenized tissue was filtered through a 40 μm cell strainer, mixed with an equal volume of Working Solution (83% OptiPrep™ Density Gradient Medium (Sigma-Aldrich), 5 mM CaCl2, 3 mM Mg(Ac)2, 10 mM Tris HCl pH 7.8, 0.1 mM EDTA pH 8.0, 1 mM β-mercaptoethanol) and loaded on top of an OptiPrep density gradient (10 ml 29% OptiPrep solution (29% OptiPrep™ Density Gradient Medium, 134 mM Sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 10 mM Tris-HCl pH 7.8, 0.1 mM EDTA pH 8.0, 1 mM β-mercaptoethanol, 0.04 % IGEPAL CA-630, 0.17 U/microliter Recombinant RNase Inhibitor) on top of 5 ml 35 % OptiPrep solution (35% OptiPrep™ Density Gradient Medium, 96 mM Sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 10 mM Tris-HCl pH 7.8, 0.1 mM EDTA pH 8.0, 1 mM β-mercaptoethanol, 0.03 % IGEPAL CA-630, 0.12 U/microliter Recombinant RNase Inhibitor)). The nuclei were separated by ultracentrifugation using an SW32 rotor (20 minutes, 9000 rpm, 4 °C). 3 ml of nuclei were collected from the 29%/35% interphase and washed with 15 ml ice-cold PBS containing 0.5% BSA and 2 mM EDTA. The nuclei were centrifuged at 100 g for 5 minutes (4 °C) and resuspended in 1 ml PBS containing 0.5% BSA and 2 mM EDTA. Then the nuclei were stained by adding two drops of NucBlue Live ReadyProbes Reagent (ThermoFisher Scientific, R37605) and passed through a 40 μm cell strainer (Falcon Cell Strainers, Sterile, Corning, 352340). Single Hoechst-positive nuclei were sorted into 96-well plates (Eppendorf twin.tec PCR Plate 96, 951020401) containing 5 microliters of Buffer TCL (QIAGEN, 1031576) per well containing 1% beta-mercaptoethanol (Sigma-Aldrich, M6250), snap frozen on dry ice, and then stored at −80 °C before whole transcriptome amplification, library preparation, and sequencing.
Single-nucleus RNA sequencing libraries were generated based on the Smart-Seq2 protocol57 with the following modifications. RNA from single cells was first purified with Agencourt RNAClean XP beads (Beckman Coulter, A63987) before oligo-dT primed reverse transcription with Maxima H Minus Reverse Transcriptase (Thermo Fisher, EP0753), which was followed by 21 cycle PCR amplification using KAPA HiFi HotStart ReadyMix (KAPA Biosystems, KK2602) with subsequent Agencourt AMPure XP bead (Beckman Coulter, A63881) purification. Libraries were tagmented using the Nextera XT DNA Library Preparation Kit (Illumina, FC-131–1096) and the Nextera XT Index Kit v2 Sets A, B, C, and D according to the manufacturer’s instructions with minor modifications. Specifically, reactions were run at one-fourth the recommended volume, the tagmentation step was extended to 10 minutes, and the extension time during the PCR step was increased from 30 seconds to 60 seconds. Libraries from 192 cells with unique barcodes were combined and sequenced on the Illumina HiSeq 2000 platform at the MIT BioMicro Center.
Oxford Nanopore Technology (ONT) sequencing and analysis
We selected two AD subjects (Female, age=83, NIA-Regan=2 and Male, age=76, NIA-Regan=1) from the Mathys et al.59 cohort to perform Oxford Nanopore Long-read sequencing. cDNA (2–10 ng) from the 10x snRNA-seq pipeline was amplified using custom universal primers NNNCTACACGACGCTCTTCCGATCT and NNNAAGCAGTGGTATCAACGCAGAGTACAT using Q5® High-Fidelity 2X Master Mix (New England Biolabs, M0492S) for 10 cycles. Amplified cDNA libraries were purified with 0.65x SPRISelect and ONT sequencing libraries were prepared with the Ligation Sequencing Kit (ONT, SQK-LSK109 ) (PCR-free) and barcode using Native Barcoding Kit 24 V14 (ONT, SQK-NBD114.24) according to the manufacturer’s instructions149.
Fluorescence-activated nuclei sorting.
Frozen forebrains were disrupted with a loose dounce homogenizer (30 strokes) in ice-cold PBS with protease inhibitors (Roche, 11836170001) and RNase inhibitors (Thermo Fisher Scientific, EO0382). Samples were fixed with 1% formaldehyde for 10 minutes at room temperature, then quenched with 2.5M glycine for 5 minutes. Homogenized tissue was centrifuged at 1000g at 4°C for 5 minutes and the pellet was suspended in lysis buffer (Sigma, Nuclei PURE Prep, NUC201–1KT) followed by dounce-homogenization using the tight pestle (30 strokes) followed by filtration with a 70 μM cell strainer (VWR, 21008–952). Nuclei were isolated through sucrose gradient centrifugation (Ultra-Clear Centrifuge Tubes, 1 × 3.5 in. (25 × 89 mm), Beckman, 344058) at 30,000×g for 45 minutes at 4°C as per manufacturer’s instructions for Sigma Nuclei PURE Prep. Primary neuronal cultures were processed as described above, except the cells were fixed in the dish and scraped using a cell scraper. The following antibodies were used to immunostain nuclei: anti-H2A.X-Phosphorylated (Ser139) antibody conjugated to APC (BioLegend, 613416), anti-NeuN antibody conjugated to Alexa Fluor 488 (EMD Millipore, MAB377X). Antibodies were incubated with nuclei in 1% BSA/PBS at 4°C for one hour or overnight. Samples were strained through a 40um filter (VWR, 21008–949) and stained with DAPI (Sigma Aldrich, D9542) before sorting. Sorting was performed on a FACSAria at the Koch Institute Flow Cytometry Core (BD Biosciences, US).
In situ Hi-C
In situ Hi-C was performed as previously described in Rao et al., 201490. 80–120k FANS isolated nuclei were used for CK-p25. Nuclei were permeabilized. DNA was digested with 100 units of MboI, and the ends of restriction fragments were labeled using biotinylated nucleotides and ligated in a small volume. After reversal of crosslinks, ligated DNA was purified and sheared to a length of ~400 bp, at which point ligation junctions were pulled down with streptavidin beads and prepped for Illumina sequencing. Illumina libraries were sequenced in 38 bp paired-end mode. Three replicates were performed, where each replicate was a pool of 3 CK-p25 mice forebrains (Table S1, page 16). P301S Tau mice and primary neuronal culture Hi-C were performed using 120–170k FANS isolated nuclei with the Dovetail Hi-C kit (SKU: 21004) according to the manufacturer’s instructions.
Break-Labeling in situ and Sequencing (BLISS)
BLISS was performed as previously described with minor modifications69. Sorted neurons were attached to a MatTek 35 mm dish, No. 1.0 Coverslip, 14 mm Glass Diameter, Poly-D-Lysine Coated (P35GC-1.0–14-C) by adding the cells to the center coverslip in 4% formaldehyde in PBS and spinning at 2000×g for 2 minutes. The attached nuclei were then end-repaired using the NEBNext End Repair Module (New England Biolabs, E6050S) and A-tailed using the NEBNext dA-Tailing Module (New England Biolabs, E6053S), with washes using cold PBS and equilibrations using 1X CutSmart Buffer (New England Biolabs, B7204S) performed between the reactions. BLISS adaptors with unique barcodes were ligated to respective samples overnight at 16°C using the T4 DNA Ligase (2,000,000 units/ml) (New England Biolabs, M0202M). The excess BLISS adaptors were removed using a High Salt Buffer wash according to the original protocol. Nuclei were then scraped off the coverslip using a mini scraper and DNA extraction buffer with proteinase K. Samples were reverse cross-linked overnight at 55°C, and genomic DNA was isolated using ethanol precipitation, adding 1 μl GlycoBlue™ Coprecipitant (Ambion by Life Technologies, AM9515), 0.1 volume 3M NaoAc, and 2.5 volumes of 100% ethanol, followed by overnight incubation at −80°C and ethanol wash. The purified DNA was sonicated to an average size of 500 bp and was then in-vitro transcribed using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, E2040S) for 14 hours. Following this, template DNA was removed by incubating the in-vitro transcription (IVT) products with 1 μl of DNaseI (RNase-Free) (New England Biolabs, M0303S) for 15 min, and the RNA was purified using RNAXP clean beads. The RA3 adapter (/5rApp/TGG AAT TCT CGG GTG CCA AGG /3SpC3/) was ligated onto the RNA using T4 RNA Ligase 2, truncated (New England Biolabs, M0242L). The RNA was then reverse transcribed using the reverse primers RTP (5′-GCCTTGGCACCCGAGAATTCCA-3′) and the SuperScript™ IV First-Strand Synthesis System (Thermo Fisher Scientific, 18091050). Lastly, the DNA was amplified using the Q5® High-Fidelity Master Mix, 2X (New England Biolabs, M0492S) using RP1 and RPI primers for 16–17 cycles. The libraries were purified using 0.8X volume AMPure beads and sequenced using single-end 75 bp reads on an Illumina NextSeq 500.
Mate-pair sequencing
Mate-pair sequencing was performed using Nextera Mate Pair Library Prep Kit (Illumina, FC-132–1001). Briefly, formaldehyde-fixed FANS sorted nuclei were reverse crosslinked, and genomic DNA was purified. Genomic DNA was tagmented to a size range of 1–5 kb using mate pair transposome containing biotinylated adaptors followed by strand displacement to repair single-stranded gaps. Tagmented genomic DNA was circularized and then fragmented using sonication. The junctions were purified using streptavidin beads. Sequencing libraries were prepared using TruSeq DNA LT Sample Prep (Illumina, 15027084) provided with the mate pair kit or NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, E7645S) and NEBNext Multiplex Oligos for Illumina (NEB, E6440S). Illumina libraries were sequenced in 75bp or 40bp paired-end mode using Nextseq500.
Minor modifications were made to the manufacturer’s instructions to account for the lower amount of genomic DNA from FANS isolated nuclei used for the assay as described below. A minimum of 100k nuclei obtained by FANS was used for the experiments. Samples were reverse-crosslinked in a 300 μl reaction with 0.2M NaCl, 0.5% SDS, and 4U of Proteinase K (NEB, P8107S) and heating at 95°C for 30 minutes with shaking at 900 rpm. An additional 4U of Proteinase K was added and incubated at 55°C for 30 minutes. DNA was purified by ethanol precipitation by adding 1 μl GlycoBlue™ Coprecipitant (Ambion by Life Technologies, AM9515), 0.1 volume 3M NaoAc, and 2.5 volumes of 100% ethanol, followed by overnight incubation at −80°C and ethanol wash. Genomic DNA was resuspended in Tris-EDTA buffer and quantified using Qubit dsDNA HS Assay Kit (Invitrogen, Q32851). A maximum of 250 ng of DNA was used to proceed with mate-pair sequencing protocol as described per the manufacturer’s instructions. All reactions were performed at 0.5X volume unless otherwise noted. Tagmentation was performed using 0.25X Tn5 enzyme for 30 minutes. Circulation was performed in 124 μl instead of 300 μl and all reagents were scaled down accordingly. Circularized DNA was sheared using Covaris E220 using microTUBE AFA Fiber Pre-Slit Snap-Cap 6×16mm (Covaris, 520045) to an average size of 500 bp. Purification of biotinylated DNA was performed using 0.5x the recommended amount of Dynabeads™ M-280 Streptavidin (Thermo Fisher Scientific, 11205D). The final PCR amplification was done in 50 μl according to the kit instructions. Illumina libraries were sequenced in 75 bp paired-end mode.
Immunofluorescence microscopy
Mice were transcardially perfused with ice-cold PBS, then fixed with ice-cold 4% paraformaldehyde in PBS. Dissected mouse brains or human brain tissue were drop-fixed overnight in 4% paraformaldehyde in PBS at 4°C. Brain tissue or entire forebrains (mice) were sectioned with a vibrating microtome (Leica BioSystems, Wetzlar Germany) to generate 40 μM coronal slices. Slices were blocked for two hours at room temperature in blocking buffer (10% NGS, 0.3% Triton X-100, PBS), then incubated with primary antibody overnight at 4°C. Slices were washed 3 × 10 minutes with PBS, and Alexa Fluor Secondary antibodies (Thermo Fisher Scientific) were added at a 1:1000 dilution for 1 hour at room temperature. Slices were washed again 3 × 10 minutes with PBS, then stained with DAPI (Millipore Sigma, D9542) and mounted onto Fisherbrand™ Superfrost™ Plus Microscope Slides (Thermo Fisher Scientific, 12–550-15) with Fluoromount-G™ Slide Mounting Medium (VWR, 100502–406). Primary neurons cultured on cover glass were washed once with PBS, then fixed with 4% paraformaldehyde/PBS for 10 minutes at room temperature. Immunostaining proceeded as described.
Primary neurons cultured on poly-D-Lysine-coated coverslips (VWR, 194310012A) were washed once with PBS, then fixed with 4% paraformaldehyde/PBS for 10 minutes at room temperature. Immunostaining proceeded as described.
We stained for RAD21 with Anti-RAD21 antibody (Millipore, 05–908, 1:500 dilution), Lamin B1 with Anti-Lamin B1 antibody (Abcam, ab16048, 1:500 dilution), NeuN with Anti-NeuN antibody (Synaptic Systems, 266 004, 1:500 dilution), and ϒH2AX with Anti-phospho-Histone H2A.X antibody (Millipore, 05–636, 1:100 dilution) or anti-H2A.X-Phosphorylated (Ser139) antibody conjugated to APC (BioLegend, 613416). Mounted samples were imaged with Zeiss LSM 710 and 880 confocal microscopes. Images were quantified using ImageJ (NIH Image Analysis) and Imaris (Oxford Instruments). At least two coronal slices were used for each mouse for image quantification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Smart-seq2 cell identities.
For each of the 6,180 cells in the smart-seq2 dataset, we used HTSeq-count150 on its filtered and recalibrated bam file to compute the cell’s transcriptomic coverage over each gene’s exons in GENCODE gene annotation (v28 lifted to b37). We used SCANPY151 to process and cluster the expression profiles and infer cell identities. We kept only 19,765 protein-coding genes detected in at least 3 cells and filtered out 37 cells with less than 100 expressed genes. We identified and filtered out 322 additional cells showing very strong individual-specific batch effects, leaving 5,821 cells over 47 individuals. We used the filtered dataset to calculate the low dimensional embedding of the cells (t-Stochastic Neighbor Embedding: t-SNE) (default parameters, perplexity=50), built a nearest neighbors graph (n=10), and clustered it with the Louvain clustering (resolution=2), giving 24 preliminary clusters. We then manually assigned clusters based on the following 2–3 major marker genes per class: Neuronal: GRIN1, SNAP25, SYT1; Excitatory neurons: CAMK2A, NRGN, SLC17A7; Inhibitory neurons: GAD1, GAD2; Astrocytes: AQP4, GFAP; Microglia: C3, CD74, CSF1R; Oligodendrocytes: MBP, MOBP, PLP1; Oligodendrocyte progenitor cells (OPCs): PDGFRA, VCAN; Endothelial: FLT1 and CLDN5. We merged clusters sharing marker genes to obtain 9 final clusters, defining two broad neuronal subtypes (1,170 excitatory and 221 inhibitory cells), four glial clusters (220 astrocytes, 255 microglia, 2121 oligodendrocytes, and 94 OPCs), 40 endothelial cells, 400 cells with strong individual-specific batch effect and cancer signatures (DNMT3A, COL6A3), and 1,300 senescent cells, marked by CARD8, FAM126A, IRX2, ALK, SENP7, and GMFB and lower overall transcription (Table S1, page 2).
For the 10x dataset, we collected 2,026,710 cells across 380 unique individuals in the ROSMAP cohort from Mathys et al. (co-submitted). We removed the ‘SM_171013Tsa’ batch from the original dataset due to markedly lower quality, number of reads, and number of genes detected per cell. These batch effects were because this was the oldest batch in the collection and had been collected using 10x v2 chemistry as opposed to v3. The cell annotation was performed as described in Mathys et al. (co-submitted) and the resulting 2M cells contained 902k excitatory neurons (44.5% of cells). Differential expression analysis was performed in each excitatory neuron subtype separately at the pseudo-bulk level using Muscat for differential testing 152.
Gene fusions calling
For the human PFC Smart-seq2 and 10x datasets, we ran STAR-Fusion v1.10.1 on the GRCh37 genome and GENCODE v19 Mar012021 CTAT library with STAR version v2.7.3 but with the --max-sensitivity flag to collect all high-quality chimeric read alignments in each cell separately 114. For 10x we removed any non-uniquely mapping reads, assigned each read to its corresponding barcode, and kept only those barcodes with cell assignments within the final set of 2M cells. We further filtered the human fusion calls by keeping unique breakpoints across all fusion calls and removing ribosomal and mitochondrial genes, immunoglobulin locus, pseudogenes, and MALAT1 (Table S1, page 3). Mouse Smart-seq2 data QC, quantification, cell type annotation, and pseudotime trajectory analysis is previously described in Welch et al., 2022. To re-align, call, and quantify gene fusions for each cell in this dataset separately, we ran STAR-Fusion v1.10.1 on the mm10 genome and GENCODE M24 Apr062020 CTAT library, with STAR version v2.7.3 and default parameters 58. We further filtered the mouse fusion calls by removing fusions containing promiscuously chimeric lncRNA Snhg14 (Table S1, page 4).
Gene fusions quantification
Quantification of fusions in the mouse Smart-seq2 analysis was performed both at the mouse and cell level, grouping all close fusions (<1Mb) and inter-chromosomal events. For human Smart-seq2 and human 10x data, we evaluated the relative contribution to detected fusion counts of the following individual-level variables: AD status (NIA-Reagan score 1–2 vs. 3–4, Braak stage 5–6 vs. 1–4, and AD cognitive impairment diagnosis 1–3 vs. 4–5), APOE e4 genotype, post-mortem interval (PMI, in units of 10 hours), age of death (in units of 10 years). We quantified the enrichment of putative fusion events at the cell level across conditions using a negative binomial generalized linear model where we model the number of fusions in each cell with an offset term for the number of reads captured for that cell (log(n_counts)). We include as cell-level covariates the cell type (if not evaluating on a single cell type), the number of captured genes (log(n_genes)), and the number of reads per gene. As individual-level variables, we include sex, age of death (rescaled by dividing by 10), and PMI (also rescaled by a factor of 10). We adjust the resulting p-values using the Benjamini-Hochberg method (BH in p.adjust in R). Regression coefficient plots show regression estimates plus or minus 2*SE on either side.
Gene set signatures were built by taking all unique genes in GO terms containing the relevant keywords (senescence, repair, DNA damage, synapse, neuron) from MGI (mouse, 2022–08-06) and GO (human, 2019–11-02). In CK/CK-p25 mouse analysis, Step 2 and Step 1 signatures are as previously described 61. Gene set signatures in each cell were computed as the sum of expression of all constituent genes (TP10k), log1p transformed, z-scored, and cells stratified into high (z-score > 1) and low (z-score <= 1) signature expression for comparison with fusions. For the cell-level stratified gene signature scores we performed the same regression analysis as for the human individual-level variables and included the same cell and individual-level covariates.
For gene-level tests, we aggregated putative fusions in each gene across all individuals in the 10x cohort. We tested the contribution of various gene-level labels on the number of fusions in each gene by a negative binomial generalized linear model. We tested whether the gene was in the RDC gene set, in the AD GWAS gene set, or whether their length or expression (log-transformed), contributed to the number of fusions. We tested the GWAS association using both a reduced set of 91 prioritized genes and an expanded set of 1329 genes covering the majority of genes in GWAS loci74.
Mate-pair seq analysis
Mate-pair paired-end fastq reads were trimmed using cutadpt to remove any Illumina adaptors. The paired reads were aligned separately to the mm10 genome using bowtie2 with a quality score above 10153. Mapped reads were converted to bam files using samtoolsView in samtools. The bam files were filtered for PCR duplicates using samtools Rmdup and converted to bed files using bamtobed in bedtools. Bed files from corresponding paired fastq files were merged to make pair files for each sample.
BLISS data analysis
BLISS fastq reads were demultiplexed into individual sample fastq reads based on sample-specific barcodes. The reads were filtered based on Unique Molecular Index (UMI) to remove PCR duplicates. The resulting fastq files were trimmed to remove any sample barcodes using cutadapt (https://cutadapt.readthedocs.io/en/stable/). The reads were aligned to the mm10 genome using bowtie2 with a quality score above 10153. Mapped reads were converted to bam files using samtoolsView in samtools, then aggregated into bed files using bamtobed in bedtools154. These steps were performed using R wrapper scripts from the R travis package (https://github.com/dvera/travis). Bed files binned into 0.5 kb windows were used for the visualization of data using Integrative Genomics Viewer (IGV).
Oxford Nanopore Technology (ONT) sequencing analysis
We filtered ONT reads for complete, non-chimeric 10x cDNA products with appropriate flanking primers following Lebrigand et al., 2020149. We mapped reads to the hg38 genome using minimap2 (v2.22-r1101) with parameters “-ax splice -uf --MD --sam-hit-only -t 20 --junc-bed” and junction bed file built from the GENCODE v28 release with additional scaffolds 155. We added overlapping gene and sequence tags and extracted reads with clipped alignments over genic regions. We remapped these with “-ax splice -k 13 -t 20” and the same junction file. To scan for fusion events, we filtered the resulting alignments with sam flags “-F 1792 -f 2048” to keep reads whose primary alignment passed QC and was tagged with supplementary alignments. We further filtered to reads with alignments over coding regions and removed any malformed or chimeric reads remaining. We sorted the alignments by alignment quality and alignment edit distance to visualize examples, which we further confirmed by running Blast on the original reads.
Hi-C analysis
Hi-C was analyzed using the HOMER Hi-C analysis pipeline (http://homer.ucsd.edu/homer/interactions2/index.html)93 and Juicer90,156. A/B compartment scores and changes in compartment scores were computed using HOMER default settings with a resolution of 25 kb. For Control and Step 1 neurons, all three bio-replicates were used for the calculation of statistical significance, and for Step 2 neurons bio-replicates 2 and 3 were used. Bio-replicate 1 of Step 2 neurons was excluded from the analysis of differential A/B compartment statistical significance owing to its lower read depth. Compartment bins were marked as differentially regulated if there was a magnitude change of greater than 0.5 along with a sign change and an adjusted p-value < 0.05. TADs and Loops were called using HOMER default settings with -res 3000 -window 15000 and -res 10000 -window 25000 and merged. (Table S1 pages 9, 10). For low-resolution Hi-C from primary neuronal cultures and P301S Tau mice, TADs were called using -res 25000 -window 50000, and loops were called using HOMER setting -res 10000 -window 25000 (Table S1, pages 11–13), and Juicer tools were used to visualize Hi-C heatmaps and create aggregate plots of loops.
Gene Ontology analysis
Gene ontology analysis in Figure 5C was performed using GOrilla157 with two unranked lists of genes, downregulated genes (adj. p-value < 0.05, log2 (fold change) > 1), and all expressed genes. Only GO terms with adj. p-value < 0.01 in at least Step 1 or Step 2 were kept.
Human RAD21 staining analysis
To assess the differences in RAD21 values between control and Alzheimer’s disease (AD) individuals, we employed a linear mixed-effects model using the lme4 package in R158. The dataset consisted of Rad21 mean intensity values measured in each neuron and the percentage of neurons with high RAD21 (> 50th percentile) per image from both control and AD individuals. We fitted a linear mixed-effects model with the percentage of high RAD21 neurons or mean intensity of RAD21 as the response variable, group (Control or AD) as the fixed effect, and individual (ID) as the random effect. To test the significance of the group effect, we performed a likelihood ratio test comparing the full model (with the group effect) against a null model excluding the group effect.
Supplementary Material
Figure S1. Smart-seq2 and 10X snRNA-seq gene fusion calling in human post-mortem brain samples. (Related to Main Figure 1)
A) UMAP for Smart-seq2 from post-mortem human PFC, colored by cell-type.
B) Expression of cell-type-specific markers (top). Heatmap of cell-type-specific markers in each cluster (bottom).
C) Boxplots of QC metrics in each cell cluster in Smart-seq2.
D) Histogram of the number of chimeric reads identified per cell.
E) Histogram of the number of intervening genes between pairs of fused genes.
F) Histogram of the distance between pairs of fused genes.
G) Heatmap of gene fusions (effect size) for human subject covariates against cell types (*adjusted p-value<0.05). Covariates include AD cognitive impairment and post-mortem interval (PMI).
H) Enrichment of excitatory neuron up-regulated DEGs for various AD ascertainment variables in DNA damage gene ontology terms (hypergeometric test, terms with p < 0.05 and with more than one associated diagnosis shown).
Figure S2. Step 2 neurons with senescence signature have increased gene fusions and genome structural variations. (Related to Main Figure 1)
A) Fraction and number of cells with or without at least one gene fusion in each cell type in snRNA-seq in CK mice.
B) Same as panel A, for CK-p25 mice.
C) Percent of cells with at least one gene fusion at the mouse level in Control, Step 1, and Step 2 neurons (t-test). Each dot represents one animal.
D) Expression (z-scored) of genes in gene ontology terms with specific keywords (senescence, DNA damage, repair, neuron, synapse) across all single cells from Control, Step 1, and Step 2 neurons. Cells with at least one gene fusion detected are marked by a black line.
E) Overlay of Cdkn1a gene expression, Lmnb1 gene expression, senescence gene set z-score, and DNA damage gene set z-score on UMAP.
F) Percentage of mate-pair sequencing read pairs separated by large genomic distances (>200kb), indicating genome structural variation events.
G) FANS dot-plot of neuronal culture after vehicle (DMSO), etoposide (2hr), and etoposide recovery (2hr ETP + 36hr recovery) using NeuN (neuron) and γH2AX (DSB) marker.
Figure S3. Long and highly expressed genes have increased DSBs and a higher propensity for gene fusions (Related to Main Figure 2)
A) Aggregate plots of BLISS reads centered at gene starts for the top 2500 expressed (yellow) and the bottom 2500 silent genes (blue), for Control, Step 1, and Step 2 neurons in both replicates.
B) Number of DSBs (log10, normalized) for each number of supporting UMIs (BLISS data).
C) Genome browser snapshot of example gene fusion in CK-p25 mice Step 2 neurons. Panels show fusion junctions (top), bulk RNA-seq signal (panels 2–4), and BLISS in the three populations (panels 5–7).
D) Gene lengths of fusion genes called in CK-p25 excitatory neurons versus all mouse genes (Wilcoxon test)
E) Aggregate plots of BLISS reads centered at gene starts for the top 2500 longest genes (yellow) and the bottom 2500 shortest genes (blue), for Control neurons.
F) Gene lengths of GWAS genes compared to all human genes (Wilcoxon test).
G) Complete long-read sequences of fusion gene examples from Figure 2G.
Figure S4. 3D genome organization is disrupted in neurons harboring DSBs. (Related to Main Figure 3)
A) Representative immunohistochemistry images and quantification of Lamin B1 in 2-weeks and 6-weeks induced CK-p25 mice in the dentate gyrus.
B) Representative higher magnification images comparing neurons with baseline DSBs to neurons enriched for DSBs
C) Violin plots quantifying Lamin B1 mean intensity in neurons enriched for 𝛾H2AX and neurons with baseline 𝛾H2AX in CK/CK-p25 mice (Wilcoxon test with Benjamini–Hochberg correction).
D) Violin plots quantifying Lamin B1 ellipticity in neurons from control CK mice and neurons enriched for 𝛾H2AX from CK-p25 2-week induction (Wilcoxon test).
E) Distance decay of chromatin interaction frequency measured by Hi-C as a function of the distance between the interacting loci for Control, Step 1, and Step 2 neurons. Log2 fold change and significance between Step 1 and 2 vs Control neurons are shown in the top two panels.
F) Representative plot showing A/B compartmentalization (principle component 1) measured by Hi-C in Control, Step 1, and Step 2 neurons.
G) K-means clustering of A/B compartmentalization (PC1) across Control, Step 1, and Step 2 neurons.
Figure S5. DSBs are sufficient to disrupt the 3D genome organization in neurons. (Related to Main Figure 4, Table S1, pages 12 and 13)
A) Representative Hi-C heatmaps comparing the disruption of 3D genome organization in etoposide-treated primary neurons to CK-p25 neurons.
B) FANS dot-plot of neuronal culture after etoposide (different concentrations) or vehicle (DMSO) treatment using NeuN (neuron) and γH2AX (DSB) marker.
C) Quantification of the TAD disruption through Inclusion Ratio (IR).
D) Correlation of differentially expressed senescence and immune-related genes between Etoposide treatment (25μM for 6 hours) and CK-p25 (Welch et al. 202261, RNA-seq).
E) Number of chromatin loops from Hi-C in P301S Tau model in both young (3 months) and old (11–12 months) animals.
F) TAD disruption (IR) in the P301S Tau model in both young (3 months) and old (11–12 months) animals (Wilcoxon test).
Figure S6. Re-organization of the 3D genome aligns with gene expression changes. (Related to Main Figure 5, 6)
A) Percentage of up and down DEGs vs. loop fold change for loop gain. Colored dotted lines represent baselines (genome-wide percentage of DEGs).
B) Correlation of differentially expressed histone genes between Etoposide treatment (25μM for 6 hours) and CK-p25 (Welch et al. 2022, RNA-seq).
C) Hi-C interaction matrix for second histone gene cluster (green blocks, chr13). The lower diagonal is Step 1 or 2, upper is Control.
D) Differential Hi-C heatmaps comparing Step 1/Control and Step 2/Control, colored by increased (red) or decreased (blue) interactions over control. Hi-C heatmaps were rotated 45 degrees and only the upper triangle is shown.
Table S1: Table S1: Metadata, gene fusions, gene lists, CK-p25-hiC compartment analysis and TADs, Chromatin loops identified, GO terms, Hi-C sequencing information, and BLISS adaptors and primer sequences (Related to Figures 1– 6).
Highlights.
Increased somatic mosaic gene fusions in excitatory neurons in Alzheimer’s Disease
DNA double-strand breaks lead to mosaic gene fusions and genome structural variations
Changes in 3D genome organization in neurons enriched for DNA double-strand breaks
3D genome changes align with differential gene expression in neurodegeneration
Acknowledgments
This work was supported in part by NIH grants NS102730, NS051874, AG054012, Cure Alzheimer’s Fund, JPB Foundation, and the Glenn Foundation for Aging Research to L.-H.T. NIH U01 NS110453 and NIH R01 AG058002 for M.K and LHT. V.D. was supported by NIH Pathway to Independence Award K99 AG073466-01 and Alzheimer’s Association fellowship AARF-19-618751. C.A.B was supported by NIH training grant GM087237. H.M. was supported by an Early Postdoc Mobility fellowship from the Swiss National Science Foundation (P2BSP3_151885). ROSMAP is supported by P30AG10161, P30AG72975, R01AG15819, R01AG17917. U01AG46152, U01AG61356 to D.A.B. ROSMAP resources can be requested at https://www.radc.rush.edu. We thank Dr. Ping-Chieh Pao, Dr. Jay Penny, Dr. Matheus Victor, Dr. William Ralvenius, the members of Tsai Lab, Dr. Maria Kousi, the members of Kellis Lab, and Dr. Vladimir Seplyarskiy for all the constructive discussions and feedback on the manuscript. We would also like to thank Ying Zhou and Erica McNamara of the Tsai Lab, Glenn Paradis and the team at Koch Institute Flow Cytometry Core, as well as Dr. Stuart Levine and the team at the MIT BioMicro Center.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests
L-HT is a member of the Scientific Advisory Board of Cognito Therapeutics, 4M Therapeutics, Cell Signaling Technology, and Souvien Therapeutics, which has no association to the work described in this manuscript.
ADDITIONAL RESOURCES
We have established a web portal for this paper at http://compbio.mit.edu/ad_dna_damage/ to better describe and provide access to our datasets and code.
References
- 1.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, and Yankner BA (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 2.Madabhushi R, Pan L, and Tsai LH (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Provasek VE, Mitra J, Malojirao VH, and Hegde ML (2022). DNA Double-Strand Breaks as Pathogenic Lesions in Neurological Disorders. Int. J. Mol. Sci. 23. 10.3390/ijms23094653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamec E, Vonsattel JP, and Nixon RA (1999). DNA strand breaks in Alzheimer’s disease. Brain Res. 849, 67–77. 10.1016/S0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- 5.Thadathil N, Delotterie DF, Xiao J, Hori R, McDonald MP, and Khan MM (2020). DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol, 1–14. 10.1007/s12035-020-02109-8. [DOI] [PubMed] [Google Scholar]
- 6.Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A, Rissman RA, Masliah E, and Mucke L (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7, 1–18. 10.1186/s40478-019-0723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tse KH, Cheng A, Ma F, and Herrup K (2018). DNA damage-associated oligodendrocyte degeneration precedes amyloid pathology and contributes to Alzheimer’s disease and dementia. Alzheimer’s Dement. 14. 10.1016/j.jalz.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu W, Hill SE, Nathan WJ, Paiano J, Callen E, Wang D, Shinoda K, van Wietmarschen N, Colón-Mercado JM, Zong D, et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444. 10.1038/s41586-021-03468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao P-C, et al. (2015). Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605. 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reid DA, Reed PJ, Schlachetzki JCMM, Nitulescu II, Chou G, Tsui EC, Jones JR, Chandran S, Lu AT, McClain CA, et al. (2021). Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science (80-. ). 372, 91–94. 10.1126/science.abb9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, and Mucke L (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 16, 613–621. 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, et al. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351. 10.1038/NN.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeNizio JE, Dow BJ, Serrano JC, Ghanty U, Drohat AC, and Kohli RM (2021). TET-TDG active DNA demethylation at CpG and non-CpG sites. J. Mol. Biol. 433, 166877. 10.1016/J.JMB.2021.166877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H, Harrison FE, and Xia F (2018). Altered DNA repair; An early pathogenic pathway in Alzheimer’s disease and obesity. Sci. Rep. 8, 5600. 10.1038/s41598-018-23644-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suberbielle E, Djukic B, Evans M, Kim DH, Taneja P, Wang X, Finucane M, Knox J, Ho K, Devidze N, et al. (2015). DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6, 1–14. 10.1038/ncomms9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aguilera A, and García-Muse T (2013). Causes of genome instability. Annu. Rev. Genet. 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 17.Iyama T, and Wilson DM (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 12, 620. 10.1016/J.DNAREP.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson C, and Jasin M (2000). Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 405, 697–700. 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson DO, and Alt FW (2001). DNA double strand break repair and chromosomal translocation: Lessons from animal models. Oncogene 20, 5572–5579. 10.1038/sj.onc.1204767. [DOI] [PubMed] [Google Scholar]
- 20.Lin C, Yang L, Tanasa B, Hutt K, Ju B. gun, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, et al. (2009). Nuclear Receptor-Induced Chromosomal Proximity and DNA Breaks Underlie Specific Translocations in Cancer. Cell 139, 1069–1083. 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabbitts TH (1994). Chromosomal translocations in human cancer. Nat. 1994 3726502 372, 143–149. 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, et al. (2020). Patterns of somatic structural variation in human cancer genomes. Nature 578. 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardımcı GG, Chakraborty A, Bann DV, Wang Y, et al. (2018). Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 10.1038/s41588-018-0195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Song F, Lyu H, Kobayashi M, Zhang B, Zhao Z, Hou Y, Wang X, Luan Y, Jia B, et al. (2022). Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 611. 10.1038/s41586-022-05365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Botten GA, Zhang Y, Dudnyk K, Kim YJ, Liu X, Sanders JT, Imanci A, Droin N, Cao H, Kaphle P, et al. (2023). Structural variation cooperates with permissive chromatin to control enhancer hijacking–mediated oncogenic transcription. Blood. 10.1182/blood.2022017555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caldecott KW, Ward ME, and Nussenzweig A (2022). The threat of programmed DNA damage to neuronal genome integrity and plasticity. Nat. Genet. 2022 542 54, 115–120. 10.1038/s41588-021-01001-y. [DOI] [PubMed] [Google Scholar]
- 27.Schoenfelder S, and Fraser P (2019). Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- 28.Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon K-R, Pekowska A, Zhang H, Rao SSP, et al. (2017). Genome Organization Drives Chromosome Fragility. Cell 170, 507–521.e18. 10.1016/j.cell.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Emerson DJ, Gilgenast TG, Titus KR, Lan Y, Huang P, Zhang D, Wang H, Keller CA, Giardine B, et al. (2019). Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature. 10.1038/s41586-019-1778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, and Dekker J (2012). Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921. 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ba Z, Lou J, Ye AY, Dai HQ, Dring EW, Lin SG, Jain S, Kyritsis N, Kieffer-Kwon KR, Casellas R, et al. (2020). CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature. 10.1038/s41586-020-2578-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnould C, Rocher V, Finoux AL, Clouaire T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, et al. (2021). Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, et al. (2018). A pathway for mitotic chromosome formation. Science (80-. ). 359. 10.1126/science.aao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dileep V, Ay F, Sima J, Vera DL, Noble WS, and Gilbert DM (2015). Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 10.1101/gr.183699.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera-Mulia JC, Trevilla-Garcia C, et al. (2019). Identifying cis Elements for Spatiotemporal Control of Mammalian DNA Replication. Cell 176. 10.1016/j.cell.2018.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada T, Yang Y, Valnegri P, Juric I, Abnousi A, Markwalter KH, Guthrie AN, Godec A, Oldenborg A, Hu M, et al. (2019). Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature. 10.1038/s41586-019-1190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Albert J, Lipinski M, Lopez-Cascales MT, Rowley MJ, Martin-Gonzalez AM, del Blanco B, Corces VG, and Barco A (2019). Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci. 10.1038/s41593-019-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, and Phillips-Cremins JE (2020). Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci. 10.1038/s41593-020-0634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marco A, Meharena HS, Dileep V, Raju RM, Davila-Velderrain J, Zhang AL, Adaikkan C, Young JZ, Gao F, Kellis M, et al. (2020). Mapping the epigenomic and transcriptomic interplay during memory formation and recall in the hippocampal engram ensemble. Nat. Neurosci, 1–12. 10.1038/s41593-020-00717-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dileep V, and Tsai LH (2021). Three-dimensional chromatin organization in brain function and dysfunction. Curr. Opin. Neurobiol. 69, 214–221. 10.1016/J.CONB.2021.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Marco A (2022). Activity-dependent remodeling of genome architecture in engram cells facilitates memory formation and recall. Neural Regen. Res. 17. 10.4103/1673-5374.324834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meharena HS, Marco A, Dileep V, Lockshin ER, Akatsu GY, Mullahoo J, Watson LA, Ko T, Guerin LN, Abdurrob F, et al. (2022). Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 29, 116–130.e7. 10.1016/J.STEM.2021.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, et al. (2018). Disease-Associated Short Tandem Repeats Colocalize with Chromatin Domain Boundaries. Cell. 10.1016/j.cell.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis PN, Lukiw WJ, De Boni U, and Crapper McLachlan DR (1981). Changes in Chromatin Structure Associated with Alzheimer’s Disease. J. Neurochem. 10.1111/j.1471-4159.1981.tb04670.x. [DOI] [PubMed] [Google Scholar]
- 45.Bendl J, Hauberg ME, Girdhar K, Im E, Vicari JM, Rahman S, Fernando MB, Townsley KG, Dong P, Misir R, et al. (2022). The three-dimensional landscape of cortical chromatin accessibility in Alzheimer’s disease. Nat. Neurosci. 25. 10.1038/s41593-022-01166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Won H, De La Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casiño C, Powell S, Yashaswini C, LaMarca EA, Kassim B, et al. (2018). Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science (80-. ). 10.1126/science.aat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science (80-. ). 366, 1134–1139. 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikuchi M, Hara N, Hasegawa M, Miyashita A, Kuwano R, Ikeuchi T, and Nakaya A (2019). Enhancer variants associated with Alzheimer’s disease affect gene expression via chromatin looping. BMC Med. Genomics. 10.1186/s12920-019-0574-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javierre BM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, Freire-Pritchett P, Spivakov M, Fraser P, Burren OS, et al. (2016). Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell. 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aymard F, and Legube G (2016). A TAD closer to ATM. Mol. Cell. Oncol. 3, e1134411. 10.1080/23723556.2015.1134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M, et al. (2017). Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 24, 353–361. 10.1038/nsmb.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aten JA, Stap J, Krawczyk PM, Van Oven CH, Hoebe RA, Essers J, and Kanaar R (2004). Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science (80-. ). 303, 92–95. 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 54.Sanders JT, Freeman TF, Xu Y, Golloshi R, Stallard MA, Hill AM, San Martin R, Balajee AS, and McCord RP (2020). Radiation-induced DNA damage and repair effects on 3D genome organization. Nat. Commun. 2020 111 11, 1–14. 10.1038/s41467-020-20047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Luan Y, and Yue F (2022). EagleC: A deep-learning framework for detecting a full range of structural variations from bulk and single-cell contact maps. Sci. Adv. 8. 10.1126/sciadv.abn9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chakraborty A, and Ay F (2018). Identification of copy number variations and translocations in cancer cells from Hi-C data. Bioinformatics 34. 10.1093/bioinformatics/btx664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2013 91 9, 171–181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 58.Haas BJ, Dobin A, Li B, Stransky N, Pochet N, and Regev A (2019). Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 20, 1–16. 10.1186/S13059-019-1842-9/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathys H, Boix C, Peng Z, Victor MB, Leary N, Babu S, Abdelhady G, Jiang X, Ng AP, Ghafari K, et al. (2023). Single-cell atlas of the aged human brain reveals cellular and molecular correlates of high cognitive function, dementia, and resilience to Alzheimer’s disease pathology. Cell 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silva E, and Ideker T (2019). Transcriptional responses to DNA damage. DNA Repair (Amst). 79. 10.1016/j.dnarep.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welch GM, Boix CA, Schmauch E, Davila-Velderrain J, Victor MB, Dileep V, Bozzelli PL, Su Q, Cheng JD, Lee A, et al. (2022). Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 8, eabo4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cruz JC, Tseng HC, Goldman JA, Shih H, and Tsai LH (2003). Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483. 10.1016/S0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 63.Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, et al. (2008). Deregulation of HDAC1 by p25/Cdk5 in Neurotoxicity. Neuron 60, 803–817. 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, and Tsai LH (2006). p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 26, 10536–10541. 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014 324 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramsden DA, and Nussenzweig A (2021). Mechanisms driving chromosomal translocations: lost in time and space. Oncogene 40, 4263–4270. 10.1038/S41388-021-01856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roukos V, Voss TC, Schmidt CK, Lee S, Wangsa D, and Misteli T (2013). Spatial dynamics of chromosome translocations in living cells. 341, 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Canela A, Maman Y, Huang S. yin N., Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters JM, et al. (2019). Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 75, 252–266.e8. 10.1016/J.MOLCEL.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L, et al. (2017). BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 8. 10.1038/ncomms15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu YY, Tanikawa C, Ueda K, and Matsuda K (2019). INKA2, a novel p53 target that interacts with the serine/threonine kinase PAK4. Int. J. Oncol. 54, 1907–1920. 10.3892/IJO.2019.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lai CH, Barik P, Hsieh DJY, Day CH, Ho TJ, Chen RJ, Kuo WW, Padma VV, Shibu MA, and Huang CY (2020). Inhibition of cell death-inducing p53 target 1 through miR-210-3p overexpression attenuates reactive oxygen species and apoptosis in rat adipose-derived stem cells challenged with Angiotensin II. Biochem. Biophys. Res. Commun. 532, 347–354. 10.1016/J.BBRC.2020.07.052. [DOI] [PubMed] [Google Scholar]
- 72.Mijit M, Caracciolo V, Melillo A, Amicarelli F, and Giordano A (2020). Role of p53 in the Regulation of Cellular Senescence. Biomolecules 10. 10.3390/BIOM10030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, and Schwer B (2016). Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 164, 644–655. 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022 544 54, 412–436. 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, Groza T, Güneş O, Hall P, Hayhurst J, et al. (2023). The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51. 10.1093/nar/gkac1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell. 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caron P, Aymard F, Iacovoni JS, Briois S, Canitrot Y, Bugler B, Massip L, Losada A, and Legube G (2012). Cohesin Protects Genes against γH2AX Induced by DNA Double-Strand Breaks. PLOS Genet. 8, e1002460. 10.1371/JOURNAL.PGEN.1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Covo S, Westmoreland JW, Gordenin DA, and Resnick MA (2010). Cohesin Is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet. 6, 1–16. 10.1371/JOURNAL.PGEN.1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gelot C, Guirouilh-Barbat J, Le Guen T, Dardillac E, Chailleux C, Canitrot Y, and Lopez BS (2016). The Cohesin Complex Prevents the End Joining of Distant DNA Double-Strand Ends. Mol. Cell 61, 15–26. 10.1016/J.MOLCEL.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 81.Hansen AS, Cattoglio C, Darzacq X, and Tjian R (2018). Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20. 10.1080/19491034.2017.1389365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters JM (2019). DNA loop extrusion by human cohesin. Science (80-. ). 366, 1338–1345. 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 83.Kim Y, Shi Z, Zhang H, Finkelstein IJ, and Yu H (2019). Human cohesin compacts DNA by loop extrusion. Science 366, 1345–1349. 10.1126/SCIENCE.AAZ4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 485, 376. 10.1038/NATURE11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, Van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nat. 2012 4857398 485, 381–385. 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 88.Etourneaud L, Moussa A, Rass E, Genet D, Willaume S, Chabance-Okumura C, Wanschoor P, Picotto J, Thézé B, Dépagne J, et al. (2021). Lamin B1 sequesters 53BP1 to control its recruitment to DNA damage. Sci. Adv. 7, 3799–3826. 10.1126/SCIADV.ABB3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xiong X, James BT, Boix CA, Park Y, Galani K, Victor MB, Sun N, Hou L, Dileep V, Ho L-L, et al. (2023). Single-cell epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion. Cell 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science 326, 289. 10.1126/SCIENCE.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression (NIH Public Access) 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed]
- 93.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576. 10.1016/J.MOLCEL.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TCC, Maeda J, Suhara T, Trojanowski JQ, and Lee VMY (2007). Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 53. 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 95.Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al. (2013). SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. 16, 1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-PP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu B, Won H, Mah W, Park RB, Kassim B, Spiess K, Kozlenkov A, Crowley CA, Pochareddy S, Ashley-Koch AE, et al. (2021). Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021 121 12, 1–13. 10.1038/s41467-021-24243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim JH, Rege M, Valeri J, Dunagin MC, Metzger A, Titus KR, Gilgenast TG, Gong W, Beagan JA, Raj A, et al. (2019). LADL: light-activated dynamic looping for endogenous gene expression control. Nat. Methods. 10.1038/s41592-019-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, and Furlong EEM (2014). Enhancer loops appear stable during development and are associated with paused polymerase. Nat. 2014 5127512 512, 96–100. 10.1038/nature13417. [DOI] [PubMed] [Google Scholar]
- 100.Kalocsay M, Hiller NJ, and Jentsch S (2009). Chromosome-wide Rad51 Spreading and SUMO-H2A.Z-Dependent Chromosome Fixation in Response to a Persistent DNA Double-Strand Break. Mol. Cell 33, 335–343. 10.1016/J.MOLCEL.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 101.Stefanelli G, Azam AB, Walters BJ, Brimble MA, Gettens CP, Bouchard-Cannon P, Cheng HYM, Davidoff AM, Narkaj K, Day JJ, et al. (2018). Learning and Age-Related Changes in Genome-wide H2A.Z Binding in the Mouse Hippocampus. Cell Rep. 22, 1124–1131. 10.1016/J.CELREP.2018.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM, and Sweatt JD (2014). Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nat. 2014 5157528 515, 582–586. 10.1038/nature13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang ZF, Krasikov T, Frey MR, Wang J, Matera AG, and Marzluff WF (1996). Characterization of the mouse histone gene cluster on chromosome 13: 45 histone genes in three patches spread over 1Mb. Genome Res. 6, 688–701. 10.1101/GR.6.8.688. [DOI] [PubMed] [Google Scholar]
- 104.Mckinnon PJ (2013). Maintaining Genome Stability in the Nervous System. Nat. Neurosci. 16, 1523. 10.1038/NN.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pollina EA, Gilliam DT, Landau AT, Lin C, Pajarillo N, Davis CP, Harmin DA, Yap EL, Vogel IR, Griffith EC, et al. (2023). A NPAS4–NuA4 complex couples synaptic activity to DNA repair. Nature 614. 10.1038/s41586-023-05711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhu Q, Niu Y, Gundry M, and Zong C (2021). Single-cell damagenome profiling unveils vulnerable genes and functional pathways in human genome toward DNA damage. Sci. Adv. 7. 10.1126/sciadv.abf3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chow HM, and Herrup K (2015). Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 10.1038/nrn4020. [DOI] [PubMed] [Google Scholar]
- 108.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JAM, Rathbun GA, Swat W, Wang J, et al. (1998). A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95, 891–902. 10.1016/S0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 109.Shimizu I, Yoshida Y, Suda M, and Minamino T (2014). DNA damage response and metabolic disease. Cell Metab. 20. 10.1016/j.cmet.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 110.Schumacher B, Pothof J, Vijg J, and Hoeijmakers JHJ (2021). The central role of DNA damage in the ageing process. Nature 592. 10.1038/s41586-021-03307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee JM, Wheeler VC, Chao MJ, Vonsattel JPG, Pinto RM, Lucente D, Abu-Elneel K, Ramos EM, Mysore JS, Gillis T, et al. (2015). Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 162. 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Edwards PAW (2010). Fusion genes and chromosome translocations in the common epithelial cancers. J. Pathol. 220, 244–254. 10.1002/PATH.2632. [DOI] [PubMed] [Google Scholar]
- 113.Uhrig S, Ellermann J, Walther T, Burkhardt P, Fröhlich M, Hutter B, Toprak UH, Neumann O, Stenzinger A, Scholl C, et al. (2021). Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 31. 10.1101/GR.257246.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Davidson NM, Majewski IJ, and Oshlack A (2015). JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 7. 10.1186/s13073-015-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jin Z, Huang W, Shen N, Li J, Wang X, Dong J, Park PJ, and Xi R (2022). Single-cell gene fusion detection by scFusion. Nat. Commun. 13. 10.1038/s41467-022-28661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gao T, Soldatov R, Sarkar H, Kurkiewicz A, Biederstedt E, Loh PR, and Kharchenko PV (2023). Haplotype-aware analysis of somatic copy number variations from single -cell transcriptomes. Nat. Biotechnol. 41. 10.1038/s41587-022-01468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fan J, Lee HO, Lee S, Ryu DE, Lee S, Xue C, Kim SJ, Kim K, Barkas N, Park PJ, et al. (2018). Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 28. 10.1101/gr.228080.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80-. ). 344. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pascarella G, Hon CC, Hashimoto K, Busch A, Luginbühl J, Parr C, Hin Yip W, Abe K, Kratz A, Bonetti A, et al. (2022). Recombination of repeat elements generates somatic complexity in human genomes. Cell 185, 3025–3040.e6. 10.1016/J.CELL.2022.06.032. [DOI] [PubMed] [Google Scholar]
- 121.Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Jill Saffrey M, Cameron K, et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996. 10.1111/J.1474-9726.2012.00870.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saez-Atienzar S, and Masliah E (2020). Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat. Rev. Neurosci. 2020 218 21, 433–444. 10.1038/s41583-020-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Welch G, and Tsai L (2022). Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 23. 10.15252/EMBR.202154217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, et al. (2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science (80-. ). 359, 555–559. 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miller MB, Yue Huang A, Kim J, Zhou Z, Kirkham SL, Maury EA, Ziegenfuss JS, Reed HC, Neil JE, Rento L, et al. (2022). Somatic genomic changes in single Alzheimer’s disease neurons. | Nat. | 604. 10.1038/s41586-022-04640-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, et al. (2018). Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature. 10.1038/s41586-018-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kousi M, Boix C, Park YP, Mathys H, Sledzieski S, Peng Z, Bennett DA, Tsai L-H, and Kellis M (2022). Single-cell mosaicism analysis reveals cell-type-specific somatic mutational burden in Alzheimer’s Dementia. bioRxiv, 2022.04.21.489103. 10.1101/2022.04.21.489103. [DOI] [Google Scholar]
- 128.McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, et al. (2017). Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science (80-. ). 356. 10.1126/science.aal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bedrosian TA, Quayle C, Novaresi N, and Gage FH (2018). Early life experience drives structural variation of neural genomes in mice. Science (80-. ). 10.1126/science.aah3378. [DOI] [PubMed] [Google Scholar]
- 130.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. (2019). LINE-1 derepression in senescent cells triggers interferon and inflammaging. Nature 566, 73. 10.1038/S41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valle F. Della, Reddy P, Yamamoto M, Liu P, Saera-Vila A, Bensaddek D, Zhang H, Martinez JP, Abassi L, Celii M, et al. (2022). LINE-1 RNA causes heterochromatin erosion and is a target for amelioration of senescent phenotypes in progeroid syndromes. Sci. Transl. Med. 14. 10.1126/scitranslmed.abl6057. [DOI] [PubMed] [Google Scholar]
- 132.Yang JH, Hayano M, Griffin PT, Amorim JA, Bonkowski MS, Apostolides JK, Salfati EL, Blanchette M, Munding EM, Bhakta M, et al. (2023). Loss of epigenetic information as a cause of mammalian aging. Cell 186. 10.1016/j.cell.2022.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Frost B, Bardai FH, and Feany MB (2016). Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 10.1016/j.cub.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matias I, Diniz LP, Damico IV, Araujo APB, Neves L. da S., Vargas G, Leite REP, Suemoto CK, Nitrini R, Jacob-Filho W, et al. (2022). Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 21. 10.1111/ACEL.13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, et al. (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. 10.1038/s41586-019-1275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Isermann A, Mann C, and Rübe CE (2020). Histone Variant H2A.J Marks Persistent DNA Damage and Triggers the Secretory Phenotype in Radiation-Induced Senescence. Int. J. Mol. Sci. 21, 1–20. 10.3390/IJMS21239130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux PF, Bischof O, Courbeyrette R, Carvalho C, Thuret JY, Ma Z, et al. (2017). Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017 81 8, 1–18. 10.1038/ncomms14995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rübe CE, Bäumert C, Schuler N, Isermann A, Schmal Z, Glanemann M, Mann C, and Scherthan H (2021). Human skin aging is associated with increased expression of the histone variant H2A.J in the epidermis. npj Aging Mech. Dis. 2021 71 7, 1–11. 10.1038/s41514-021-00060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Funk OH, Qalieh Y, Doyle DZ, Lam MM, and Kwan KY (2022). Postmitotic accumulation of histone variant H3.3 in new cortical neurons establishes neuronal chromatin, transcriptome, and identity. Proc. Natl. Acad. Sci. U. S. A. 119. 10.1073/pnas.2116956119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret JY, Andrews S, Fraser P, and Reik W (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471–483. 10.1016/J.CELREP.2014.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Criscione SW, Cecco M. De, Siranosian B, Zhang Y, Kreiling JA, Sedivy JM, and Neretti N (2016). Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2. 10.1126/SCIADV.1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Iwasaki O, Tanizawa H, Kim KD, Kossenkov A, Nacarelli T, Tashiro S, Majumdar S, Showe LC, Zhang R, and Noma K ichi (2019). Involvement of condensin in cellular senescence through gene regulation and compartmental reorganization. Nat. Commun. 2019 101 10, 1–20. 10.1038/s41467-019-13604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zirkel A, Nikolic M, Sofiadis K, Mallm JP, Brackley CA, Gothe H, Drechsel O, Becker C, Altmüller J, Josipovic N, et al. (2018). HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol. Cell 70, 730–744.e6. 10.1016/j.molcel.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 144.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, and Schneider JA (2018). Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers. Dis. 64, S161–S189. 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, and Bach J (2002). Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205. 10.1212/WNL.59.2.198. [DOI] [PubMed] [Google Scholar]
- 146.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, and Wilson RS (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. 10.1212/01.WNL.0000219668.47116.E6. [DOI] [PubMed] [Google Scholar]
- 147.Bennett DA, Schneider JA, Bienias JL, Evans DA, and Wilson RS (2005). Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 64, 834–841. 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 148.Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, and Zhang F (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102–106. 10.1038/NBT.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lebrigand K, Magnone V, Barbry P, and Waldmann R (2020). High throughput error corrected Nanopore single cell transcriptome sequencing. Nat. Commun. 2020 111 11, 1–8. 10.1038/s41467-020-17800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/BIOINFORMATICS/BTU638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5. 10.1186/S13059-017-1382-0/FIGURES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Crowell HL, Soneson C, Germain PL, Calini D, Collin L, Raposo C, Malhotra D, and Robinson MD (2020). muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 11. 10.1038/S41467-02019894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li H (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 1–7. 10.1186/1471-2105-10-48/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bates D, Mächler M, Bolker BM, and Walker SC (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67. 10.18637/jss.v067.i01. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Smart-seq2 and 10X snRNA-seq gene fusion calling in human post-mortem brain samples. (Related to Main Figure 1)
A) UMAP for Smart-seq2 from post-mortem human PFC, colored by cell-type.
B) Expression of cell-type-specific markers (top). Heatmap of cell-type-specific markers in each cluster (bottom).
C) Boxplots of QC metrics in each cell cluster in Smart-seq2.
D) Histogram of the number of chimeric reads identified per cell.
E) Histogram of the number of intervening genes between pairs of fused genes.
F) Histogram of the distance between pairs of fused genes.
G) Heatmap of gene fusions (effect size) for human subject covariates against cell types (*adjusted p-value<0.05). Covariates include AD cognitive impairment and post-mortem interval (PMI).
H) Enrichment of excitatory neuron up-regulated DEGs for various AD ascertainment variables in DNA damage gene ontology terms (hypergeometric test, terms with p < 0.05 and with more than one associated diagnosis shown).
Figure S2. Step 2 neurons with senescence signature have increased gene fusions and genome structural variations. (Related to Main Figure 1)
A) Fraction and number of cells with or without at least one gene fusion in each cell type in snRNA-seq in CK mice.
B) Same as panel A, for CK-p25 mice.
C) Percent of cells with at least one gene fusion at the mouse level in Control, Step 1, and Step 2 neurons (t-test). Each dot represents one animal.
D) Expression (z-scored) of genes in gene ontology terms with specific keywords (senescence, DNA damage, repair, neuron, synapse) across all single cells from Control, Step 1, and Step 2 neurons. Cells with at least one gene fusion detected are marked by a black line.
E) Overlay of Cdkn1a gene expression, Lmnb1 gene expression, senescence gene set z-score, and DNA damage gene set z-score on UMAP.
F) Percentage of mate-pair sequencing read pairs separated by large genomic distances (>200kb), indicating genome structural variation events.
G) FANS dot-plot of neuronal culture after vehicle (DMSO), etoposide (2hr), and etoposide recovery (2hr ETP + 36hr recovery) using NeuN (neuron) and γH2AX (DSB) marker.
Figure S3. Long and highly expressed genes have increased DSBs and a higher propensity for gene fusions (Related to Main Figure 2)
A) Aggregate plots of BLISS reads centered at gene starts for the top 2500 expressed (yellow) and the bottom 2500 silent genes (blue), for Control, Step 1, and Step 2 neurons in both replicates.
B) Number of DSBs (log10, normalized) for each number of supporting UMIs (BLISS data).
C) Genome browser snapshot of example gene fusion in CK-p25 mice Step 2 neurons. Panels show fusion junctions (top), bulk RNA-seq signal (panels 2–4), and BLISS in the three populations (panels 5–7).
D) Gene lengths of fusion genes called in CK-p25 excitatory neurons versus all mouse genes (Wilcoxon test)
E) Aggregate plots of BLISS reads centered at gene starts for the top 2500 longest genes (yellow) and the bottom 2500 shortest genes (blue), for Control neurons.
F) Gene lengths of GWAS genes compared to all human genes (Wilcoxon test).
G) Complete long-read sequences of fusion gene examples from Figure 2G.
Figure S4. 3D genome organization is disrupted in neurons harboring DSBs. (Related to Main Figure 3)
A) Representative immunohistochemistry images and quantification of Lamin B1 in 2-weeks and 6-weeks induced CK-p25 mice in the dentate gyrus.
B) Representative higher magnification images comparing neurons with baseline DSBs to neurons enriched for DSBs
C) Violin plots quantifying Lamin B1 mean intensity in neurons enriched for 𝛾H2AX and neurons with baseline 𝛾H2AX in CK/CK-p25 mice (Wilcoxon test with Benjamini–Hochberg correction).
D) Violin plots quantifying Lamin B1 ellipticity in neurons from control CK mice and neurons enriched for 𝛾H2AX from CK-p25 2-week induction (Wilcoxon test).
E) Distance decay of chromatin interaction frequency measured by Hi-C as a function of the distance between the interacting loci for Control, Step 1, and Step 2 neurons. Log2 fold change and significance between Step 1 and 2 vs Control neurons are shown in the top two panels.
F) Representative plot showing A/B compartmentalization (principle component 1) measured by Hi-C in Control, Step 1, and Step 2 neurons.
G) K-means clustering of A/B compartmentalization (PC1) across Control, Step 1, and Step 2 neurons.
Figure S5. DSBs are sufficient to disrupt the 3D genome organization in neurons. (Related to Main Figure 4, Table S1, pages 12 and 13)
A) Representative Hi-C heatmaps comparing the disruption of 3D genome organization in etoposide-treated primary neurons to CK-p25 neurons.
B) FANS dot-plot of neuronal culture after etoposide (different concentrations) or vehicle (DMSO) treatment using NeuN (neuron) and γH2AX (DSB) marker.
C) Quantification of the TAD disruption through Inclusion Ratio (IR).
D) Correlation of differentially expressed senescence and immune-related genes between Etoposide treatment (25μM for 6 hours) and CK-p25 (Welch et al. 202261, RNA-seq).
E) Number of chromatin loops from Hi-C in P301S Tau model in both young (3 months) and old (11–12 months) animals.
F) TAD disruption (IR) in the P301S Tau model in both young (3 months) and old (11–12 months) animals (Wilcoxon test).
Figure S6. Re-organization of the 3D genome aligns with gene expression changes. (Related to Main Figure 5, 6)
A) Percentage of up and down DEGs vs. loop fold change for loop gain. Colored dotted lines represent baselines (genome-wide percentage of DEGs).
B) Correlation of differentially expressed histone genes between Etoposide treatment (25μM for 6 hours) and CK-p25 (Welch et al. 2022, RNA-seq).
C) Hi-C interaction matrix for second histone gene cluster (green blocks, chr13). The lower diagonal is Step 1 or 2, upper is Control.
D) Differential Hi-C heatmaps comparing Step 1/Control and Step 2/Control, colored by increased (red) or decreased (blue) interactions over control. Hi-C heatmaps were rotated 45 degrees and only the upper triangle is shown.
Table S1: Table S1: Metadata, gene fusions, gene lists, CK-p25-hiC compartment analysis and TADs, Chromatin loops identified, GO terms, Hi-C sequencing information, and BLISS adaptors and primer sequences (Related to Figures 1– 6).
Data Availability Statement
Single-nucleus RNA-seq data will be available through the AD Knowledge Portal on Synapse upon publication of this manuscript. The data will be available here: https://www.synapse.org/#!Synapse:syn52293426. The data are available under controlled use conditions set by human privacy regulations. To access the data, a data use agreement is needed. This registration is in place solely to ensure the anonymity of the ROSMAP study participants. A data use agreement can be agreed with either Rush University Medical Center (RUMC) or with SAGE, which maintains Synapse, and can be downloaded from their websites (https://adknowledgeportal.synapse.org/)
All mouse data generated in this study, including raw and processed files, are available at GSE227445.
We have established a web portal for this paper at http://compbio.mit.edu/ad_dna_damage/ to better describe and provide access to our datasets and code, which is also available at https://doi.org/10.5281/zenodo.8290268.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request