

SUMMARY
Monocytes are abundant immune cells that infiltrate inflamed organs. However, the majority of monocyte studies focus on circulating cells, rather than those in tissue. Here, we identify and characterize an intravascular synovial monocyte population resembling circulating non-classical monocytes and an extravascular tissue-resident monocyte-lineage cell (TR-MC) population distinct in surface marker and transcriptional profile from circulating monocytes, dendritic cells, and tissue macrophages that are conserved in rheumatoid arthritis (RA) patients. TR-MCs are independent of NR4A1 and CCR2, long lived, and embryonically derived. TR-MCs undergo increased proliferation and reverse diapedesis dependent on LFA1 in response to arthrogenic stimuli and are required for the development of RA-like disease. Moreover, pathways that are activated in TR-MCs at the peak of arthritis overlap with those that are downregulated in LFA1−/− TR-MCs. These findings show a facet of mononuclear cell biology that could be imperative to understanding tissue-resident myeloid cell function in RA.
Graphical Abstract
In brief
Montgomery et al. identify a population of immune cells (tissue-resident monocytic-lineage cells, TR-MC) that reside in joints, are embryo derived, are functionally distinct from other immune cells, and undergo proliferation in response to inflammatory arthritis. These findings increase our understanding of rheumatoid arthritis.
INTRODUCTION
In recent years, our understanding of the mononuclear phagocyte system has expanded, highlighting previously unknown complexities in cell origin and function. However, to date, few studies have examined the role of monocytes in tissues, instead centering on circulating monocytes or monocyte-derived macrophages. Circulating monocytes exist in three main states, characterized by CCR2, CX3CR1, CD43, and Ly6c in mice: classical (CM) (CCR2+CX3CR1lowCD43−Ly6chi), intermediate (IM) (CCR2±CX3CR1lowCD43±Ly6cint), and non-classical (NCM) (CCR2−CX3CR1hiCD43+Ly6clow).1,2 CMs require CCR2 to exit the bone marrow, while NCMs utilize sphingosine-1-phosphate receptor 5 (S1PR5) and/or CX3CR1.3 Consequently, CCR2−/− mice have reduced numbers of CMs in circulation, while S1PR5−/− and CX3CR1−/− mice have reduced NCMs.3 NCMs also require CEBP/β for transcriptional activation of NR4A1 and CSF1R to maintain survival.4 As such, NR4A1−/− and CEBP/β−/− mice also display markedly reduced numbers of circulating NCMs. While transcriptional studies have exposed critical gene signatures for CMs and NCMs in the bone marrow and circulation, no such studies have examined heterogeneity and function at the tissue level.
In contrast to the well-characterized inflammatory CMs,3 the direct impact of NCMs on steady state and inflammation is unclear. The current dogma for circulating NCMs centers on barrier maintenance due to the ability of NCMs to adhere to and patrol the endothelium.5 In this context, NCMs maintain the endothelium, scavenge debris, and elicit removal of damaged endothelial cells by neutrophils.5 To date, only one study has proposed the existence of an NCM population in tissue.6 The investigators identified a CD64+CD16.2+ subpopulation among extravascular CD45+Ly6clo cells in the lung that are derived from circulating NCMs and require NR4A1. These cells were considered monocytes but were putative precursors of interstitial macrophages. It is not known whether similar populations are found in other tissues.
It is established that recruitment of monocytes to the inflamed synovium is a requisite for sustainment and progression of rheumatoid arthritis (RA).7 Support for a functional role for NCMs in RA comes from murine models. While complete ablation of circulating monocytes using clodronate-laden liposomes (clo-lips) prevents the effector phase of K/BxN serum-transfer-induced arthritis (STIA), pathology is restored exclusively with transfer of NCMs, not CMs.8,9 CX3CR1−/− mice also display a reduction in STIA.10 In contrast, depletion of CMs via anti-CCR2 antibody or deficiency in CCR2 has no effect on arthritis development in STIA,8 TNF-α-transgenic mice,11 or collagen-induced arthritis.12 However, NR4A1−/− mice remain sensitive to STIA and collagen-induced arthritis regardless of reduced numbers of circulating NCMs.13,14 Taken together, these studies present a quandary regarding the role NCMs play in RA.
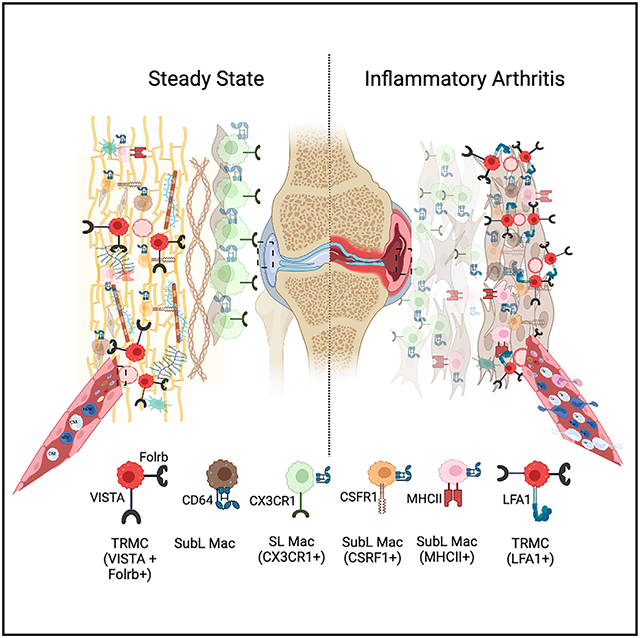
To distinguish functional roles of monocytes, we focused on the heterogeneity of CD64−Ly6c− monocytes in synovial tissue. We uncovered three subpopulations of synovial (Syn) CD64−Ly6c− cells, which lack known markers of macrophages and can be separated by expression of major histocompatibility complex class II (MHCII) and their intra- and extravascular location in the mouse synovium. MHCII+CD64−Ly6c− cells identified as dendritic cells are localized to the extravascular compartment of the synovium. Intravascular CD64−Ly6c− cells retain a phenotype similar to that of circulating NCMs, while extravascular Syn CD64−Ly6c− cells (tissue-resident monocyte-lineage cells [TR-MCs]) are transcriptionally distinct from circulating NCMs, independent of NR4A1 and CCR2, and long lived. Furthermore, TR-MCs are critical for the pathology of inflammatory arthritis, via an LFA-dependent mechanism. These data document an essential role for newly described TR-MC in inflammatory arthritis.
RESULTS
Non-classical monocytes in the synovium are distinct from those in the circulation
We sought to determine the contribution of circulating NCMs to inflammatory arthritis by inducing STIA in NR4A1−/− mice, which are depleted of NCMs in peripheral blood (PB) (Figures 1A, S1A, and S1B). NR4A1−/− mice developed STIA of severity and onset comparable to that of C57BL/6 controls (Figure 1B), in agreement with a previous report.13 Flow cytometry was then performed to identify monocyte populations preserved in the synovium of NR4A1−/− mice, which may explain the sensitivity of these mice to inflammatory arthritis. We identified a Syn myeloid niche defined as CD45+CD11b+Ly6G−SiglecF−CD64−, of which the majority were Ly6c−. Based on this gating strategy Syn macrophages (CD64+) were the most abundant myeloid population in the synovium (Figure S1C), while Ly6c− represented 10% of the Syn mononuclear phagocyte compartment, and Ly6c+ and Ly6cint cells composed less than 1%. Syn Ly6c− cell numbers remained unchanged in NR4A1−/− mice (Figure 1C), even while NCMs were markedly reduced in PB. To confirm that Syn Ly6c− cells were not dependent on CCR2, STIA was induced in CCR2−/− mice lacking CM in PB (Figures 1D and S1B). CCR2−/− mice also showed clinical scores in STIA comparable to those of C57BL/6 controls (Figure 1E), as reported in previous studies,8,10 and their numbers of Syn Ly6c− cells were unchanged compared with controls (Figure 1F). Taken together, these data confirm that neither subtype of circulating monocyte is required for inflammatory arthritis, while Syn Ly6c− cells, independent of NR4A1 and CCR2 and lacking expression of macrophage-associated markers, may play an essential role in STIA.
Figure 1. Synovial Ly6c− cells are distinct from circulating NCMs.

(A–F) Classical (CM), intermediate (IM), and non-classical (NCM) monocytes in the peripheral blood (PB); (B) STIA severity; and (C) Syn Ly6c+, Ly6cint, and Ly6c− in C57BL/6 compared with NR4A1−/− synovium and (D–F) compared with CCR2−/−.
(G and H) Changes in numbers of (G) Syn Ly6c+, Ly6cint, and Ly6c− and (H) PB CMs, IMs, and NCMs during STIA. Data shown are n ≥ 4 ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
(I) Pairwise Pearson’s correlation of global gene expression between replicates of PB CMs, PB NCMs, and Syn Ly6c−.
(J) Fold-change expression of monocyte-associated genes from Mildner et al. compared with PB CMs or NCMs.4
(K) Principal component analysis (PCA) of 10,206 genes expressed by PB CMs, PB NCMs, and Syn Ly6c− from C57BL/6 and NR4A1−/− mice.
(L) k-means clustering of 5,115 differentially expressed genes across PB CMs, PB NCMs, and Syn Ly6c− cells.
(M) Mean expression of representative genes from PB CMs, PB NCMs, Syn Ly6c−, and Syn macrophage (Syn Mac) populations (RNA-seq data: n = 3, error bars indicate SEM).
Cell numbers were measured throughout STIA. Syn Ly6c+ and Ly6c− numbers significantly expanded on STIA day 3 (D3) (p = 0.003, p = 0.006) and D7 (p = 0.003, p = 0.03) compared with D0, while Syn Ly6cint cells were not significantly different at any time point (Figure 1G). By D14, Syn Ly6c+ cells returned to baseline, whereas Syn Ly6c− cells plateaued on D14 and D21 compared with D0 (p = 0.05, p = 0.02). Similarly, PB CMs reached a peak prior to 21 days post-STIA, while PB NCMs continued to increase (Figure 1H).
To determine if Syn Ly6c− cells exhibit a transcriptional state distinct from PB monocytes, we isolated PB CMs and PB NCMs and Syn Ly6c− cells for bulk RNA sequencing (RNA-seq). Given that PB IMs are likely an intermediate state, these cells were excluded. PB CMs, PB NCMs, and Syn Ly6c− cells exhibited distinct transcriptional profiles compared with one another (Figures 1I and S1D). We then compared expression of genes associated with PB CMs vs. PB NCMs as described.4 Expression of monocyte genes in PB CMs and PB NCMs largely aligned with expectations, but Syn Ly6c− cells did not uniformly express PB CM genes—such as Irf7, Ccr2, Ifi30, Mmp8, and Cebpd—or those associated with PB NCM—such as Apoe, Csf1r, Fcgr4, Pparg, Nr4a1, and Cx3cr1 (Figure 1J). Furthermore, loss of NR4A1 had a minimal effect on the transcriptional profile of Syn Ly6c− cells compared with wild type (WT) (Figures 1K and S1E). These data show that Syn Ly6c− cells are transcriptionally distinct from circulating cells and are not NR4A1 dependent.
k-means clustering of 5,116 differentially expressed genes identified three gene clusters preferentially expressed by PB CMs, PB NCMs, or Syn Ly6c− cells (Figure 1L and Table S1A). Compared with others, the Syn Ly6c− cluster (cluster 3) was enriched for genes associated with extracellular matrix organization, hormone secretion, cell division, cell adhesion, and regulation of biological processes (Figure S1F and Table S1B). In addition, genes involved in antigen presentation (H2-Aa, Cd74), immune activation (Cd9, Pf4, Cd36), cell-to-cell interaction (Adam8, Anxa8), and cell differentiation (Cd24a, Crip2) were increased in Syn Ly6c− cells compared with PB CMs and PB NCMs (Figure 1L). On both the individual gene and the global level, Syn Ly6c− cells exhibited a transcriptional profile distinct from each of the four CD64+ Syn macrophage (Syn Mac) populations (Figures 1M and S1G-S1H). Taken together, these data uncover a CD64− Syn Ly6c− population present in the synovium.
Single-cell RNA sequencing identifies synovial Ly6c− population
We utilized single-cell RNA-seq (scRNA-seq) to investigate the heterogeneity of Syn Ly6c− (CD45+CD11b+Ly6G−SiglecF−CD64−) in steady state. We defined six clusters (0–5) of mononuclear subpopulations using unsupervised graph-based clustering of 7,160 cells sequenced from C56BL/6 mice (Figures 2A, S2A, and S2B and Table S2A). The expression of Cd14 and Itgam (CD11b) confirmed mononuclear lineage, while low expression of Fcgr1 (CD64), Timd4 (Tim4), and Mertk confirmed non-macrophage classification (Figures 2B and S2C). Using singleR and the reference population ImmGen dataset, we found that subpopulations 0–3 exhibited a mixture of monocyte, macrophage, and dendritic cell (DC) annotations expected of tissue-resident mononuclear cells, while subpopulations 4 and 5 were primarily assigned to monocytes and DCs, respectively (Figure 2C). Subpopulation 4 also displayed similarity to PB CMs and PB NCMs, suggesting these may be monocytes retained in tissue vasculature (Figures 2D, S2B, and S2D-S2F and Table S2B). Subpopulation 5 had highest expression of genes associated with conventional DCs15 (Figures 2E and S2G). Subpopulation 2 expressed high levels of cell-cycle genes (Figures S2H-S2J), suggesting these cells were actively dividing. Therefore, the formerly labeled Syn Ly6c− compartment contains a heterogeneous population with different identities from monocytes to DCs.
Figure 2. scRNA-seq analysis of joint myeloid niche identifies tissue Syn Ly6c− cells.

(A) Uniform manifold approximation and projection (UMAP) depicting six subpopulations of total Ly6c− (CD45+CD11b+Ly6G−SiglecF−CD64−Ly6c−) cells from scRNA-seq data.
(B) Expression of myeloid markers Cd14 and Itgam.
(C) Percentage of cells assigned to ImmGen cell types by singleR.
(D and E) Module score for scRNA-seq subpopulations representing expression of key genes in (D) PB CMs, PB NCMs, Syn Ly6c−, and (E) conventional dendritic cell (cDCs).
(F) Integration of scRNA-seq data on total Ly6c− (CD45+CD11b+Ly6G−SiglecF−CD64−Ly6c−) cells from CCR2−/− and NR4A1−/− mice with C57BL/6 (subsampled to 2,000 cells).
(G) Proportion of cells annotated as each subpopulation in C57BL/6, CCR2−/−, and NR4A1−/− mice.
(H) Reclassification of cluster 2 as cycling cells, cluster 4 as monocytes, cluster 5 as cDCs, and clusters 0, 1, and 3 as MHCII+ or MHCII− based on expression of H2-eb1.
(I and J) Ridge plots and UMAP visualization of gene expression by MHCII compartment. The p value by Wilcoxon test is indicated.
We then performed scRNA-seq on CD45+CD11b+Ly6G−SiglecF−CD64− Syn cells from NR4A1−/− and CCR2−/− mice at steady state to determine if compositions were affected by these deficiencies (Figure 2F and Table S2C). These datasets were integrated with control (C57BL/6) and subpopulation annotation was superimposed (Figure S2K). The distribution of CCR2−/− and NR4A1−/− cells across the six subpopulations was significantly different from controls (p < 2.2e–16, p = 4.89e–10) (Figure 2G), suggesting that at least some of these cells are derived from circulating monocytes. In particular, the depletion of subpopulation 4 in CCR2−/− mice supports the contribution of CMs.
Although subpopulations 0, 1, and 3 all had high similarity to Syn Ly6c− cells from Figure 1 (Figures 2D and S2D), the variability in gene expression profiles suggested distinct subtypes. Subpopulation 1 exhibited higher expression of genes associated with the DC lineage (Figures 2E and S2B). Since MHCII genes (H2-Eb1, Ab1, Aa, DMb1, DMa) are associated with DCs, we partitioned subpopulations 0, 1, and 3 based on their expression, specifically on H2-Eb1 (Figures 2H, S2L, and S2M and Table S2D). As expected, the MHCII+ compartment exhibited elevated expression of genes associated with DCs, Cd209a, Cd74, and Nr4a316-18 (Figure 2I). The remaining non-DC MHCII− fraction was enriched for genes known to have regulatory functions in inflammation, lipid metabolism, and angiogenesis, including Crip2, Fxyd2, and Rnase419-21 (Figure 2J). Furthermore, there was no significant difference in the proportion of cells annotated as MHCII− vs. MHCII+ in CCR2−/− mice, and MHCII− cells were increased in NR4A1−/− mice (Figure S2N). Collectively, these data demonstrate that MHCII− cells represent a distinct Ly6c− population residing in the synovium of mice that is independent of CCR2 and NR4A1.
We next compared the differentially expressed genes associated with our subpopulations to the top 20 marker genes from single-cell CD45+CD11b+Ly6G− myeloid subpopulations (excluding neutrophils) identified by Culemann et al. on D5 post-STIA22 (Figure S2O and Table S2E). The MHCII− cells displayed a high unique overlap in genes with the CX3CR1+, RELMα+, MHCII+, and CCR2+Arg1+ populations, while MHCII+ cells overlapped genes from MHCII+ DCs as well as CX3CR1+ and MHCII+ populations from the Culemann dataset. The cycling cells and monocyte populations overlapped significantly with STMN1+ proliferating cells and CCR2+IL1b+ mononuclear cells, respectively. These results support the identification of these individual populations, but do not exactly match the Culemann clusters, which would contain large numbers of macrophages outnumbering the tissue-resident Syn Ly6c− cells.22
Synovial Ly6c− cells exist as three distinct populations
To validate our partitioning of cells based on MHCII, we separately sorted Syn CD45+CD11b+Ly6G−SiglecF−CD64− cells into MHCII− and MHCII+ compartments from the same mice for cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) providing both transcriptional (RNA) and surface marker (antibody-derived tags [ADTs]) data. By comparison of transcriptional profiles, we annotated these cells to the populations from Figure 2 (Figures 3A, 3B, S3A, and S3B). As expected, MHCII− cells contained an expanded proportion of the MHCII− population, which we now term TR-MCs, and monocytes with a very small MHCII+ population and no DCs. In contrast, the vast majority of the MHCII+ compartment was annotated as MHCII+ with a very minor MHCII− population; thus, we term these cells generally as DCs (Figures S3C and S3D). In addition, a substantial portion of the MHCII− compartment was annotated as cycling cells and included neutrophils (Figure S2B). The surface marker intensity of CD163, FrB, C5aR, and Vista, as well as gene expression, discriminated the TR-MC populations from the DC and monocytes (Figure 3C and Table S3A). Moreover, we merged the MHCII− CITE-seq data with CD64+ macrophages from the same mice and confirmed that macrophage-associated genes are markedly higher expressed in macrophages compared with TR-MCs or DCs (Figures S3E and S3F). Next, we examined the epigenomic landscape of Syn CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− cells using single-cell assay for transposase-accessible chromatin by sequencing (scATAC-seq) assay. We transferred the transcriptional annotations based on similarity with chromatin accessibility levels at the corresponding genes and obtained similar proportions of the populations (Figure 3D). Using these data to assess transcription factor (TF) activity, we found that MAFB and MYC expression and activity were specific to TR-MCs, while FLI1 and IRF8 were affiliated with the cycling cell and the monocyte populations, respectively (Figures 3E and 3F and Table S3B). Thus, sorting on cell-surface levels of MHCII effectively enriched for TR-MCs. However, the MHCII− compartment still contains both typical monocytes and TR-MCs.
Figure 3. Identification of intra- and extravascular Syn Ly6c− cells by flow cytometry.

(A) Annotation of CITE-seq data on C57BL/6 CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− cells to subpopulations defined in total Syn Ly6c− (CD45+CD11b+Ly6G−SiglecF−CD64−) (Figure 2H).
(B) Expression of TR-MC (Lyve1), DC (CD74), cycling (Top2a), and monocyte (Plac8) subpopulation genes.
(C) Mean ADT intensity of surface markers.
(D) Annotation of scATAC-seq data on C57BL/6 CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− cells to subpopulations defined in total Syn Ly6c− (Figure 2H).
(E and F) (E) Transcription factor (TF) activity and (F) expression of corresponding genes associated with TR-MCs (MafB and MYC), cycling (Fli1), and monocyte (Irf8) subpopulations.
(G) Gating strategy to distinguish intravascular NCMs vs. extravascular DCs and TR-MCs.
(H) NCMs, DCs, and TR-MCs in steady state.
To further distinguish these populations, we investigated whether they differed in anatomical location. We used an established in vivo intravascular labeling system followed by flow cytometry.23 Only intravascular cells were labeled by intravenous (I.V.) administration of a fluorescently conjugated anti-CD45 antibody (αCD45-BUV661-(I.V.)) prior to euthanasia. Over 90% of the circulating leukocytes were labeled 5 min post-I.V. administration (Figure S3G). Thus, immune cells that co-label with αCD45-BUV661-(I.V.) and the ex vivo (E.V.) anti-CD45 antibody (αCD45-AF700-(E.V.)) are considered intravascular, while single-positive αCD45-AF700-(E.V.) cells are extravascular (Figure 3G). Syn macrophages were distinguished using CD64+ (Figures 3G and S3H) and expressed markedly higher levels of F4/80, CD68, and Tim4 compared with DC and TR-MCs. Intra- and extravascular cells were then gated based on CD45+ CD11b+Ly6G−SiglecF−CD64−Ly6c− expression to isolate the Syn Ly6c− cells. Extravascular (e.v.) cells were divided using MHCII to obtain TR-MCs and DCs. We confirmed the identity of the MHCII+ compartment as DC due to expression of the DC TF ZBT46 using zDC-Cre mice crossed with zsGFP reporter mice (zDC-GFP), consistent with the scRNA-seq data (Figures S3I-S3K). Numerically, the DCs had the highest number, while the TR-MCs (e.v. MHCII−Syn. Ly6c− cells) were ~3× less and NCMs (intravascular [i.v.] Syn. Ly6c− cells) were less than 500 per synovium (Figure 3H). These numbers are consistent with the scRNA-seq data in Figures 1 and 2. Using flow cytometry to compare cell-surface levels, we found that FcγRIV and Treml4 discriminated NCMs, while Folrb, VISTA, and Lyve1 were higher in some but not all TR-MCs (Figure S3L).
Intra- and extravascular synovial Ly6c− exhibit different functionalities
To investigate the properties of the two Syn Ly6c− monocyte populations (NCMs and TR-MCs) and contrast them with DCs (Syn Ly6c−MHCII+), we compared the transcriptional profiles of these cells using bulk RNA-seq. Each population was highly reproducible across replicates and characterized by unique transcriptional profiles (Figures 4A and S4A). We further merged the CD45+CD11b+Ly6G−SiglecF−CD64− cells from the MHCII+ and MHCII− compartments (Figure 3) into one dataset and confirmed that the single-cell annotations matched the bulk transcriptional profiles (Figures S4B and S4C). Taken together, these data demonstrate that there are three populations comprising Syn CD64−Ly6c− (CD45+CD11b+Ly6G−SiglecF−CD64−), NCMs, TR-MCs, and DCs, based on three separate scRNA-seq studies as well as bulk RNA-seq.
Figure 4. Extravascular tissue location confers phenotype of TR-MCs.

(A) Pairwise Pearson’s correlation of global gene expression between replicates of NCMs, DCs, and TR-MCs.
(B) Relative expression of representative genes from gene ontology (GO) processes associated with NCMs, DCs, and TR-MCs.
(C) Clustering of RA patient CD45+CD11b+ synovial cells based on gene expression from CITE-seq.
(D) ADT intensity of CD45, CD11b, and CD14 on RA patient synovial cells.
(E and F) TR-MC module score in RA patient data.
(G and H) Expression of Trem2 and Timd4 in RA patient data.
(I–K) (I) ADT intensity of TIM4 in RA patient data. Immunofluorescence staining of synovial tissue from (J) healthy control and (K) RA patient.
We then performed k-means clustering on 5,127 differentially expressed genes to define four clusters: one each with expression specific to NCM cells (1), DCs (2), and TR-MCs (3) and a final cluster (4) preferentially expressed in both DCs and TR-MCs (Figures 4B and S4D-S4H and Table S4A). The NCMs expressed the highest levels of Cx3cr, Spn (CD43), Cebpb, and Nr4a1, genes associated with PB NCMs. Further, the bulk-sorted DC population preferentially expressed DC-associated genes, including Cd74, Cd209a, Zbtb46, and Flt3. Meanwhile, TR-MCs expressed genes observed above in MHCII− cells, such as Alox5, Pf4, Fxyd2, and Rnase4. The cluster specific to NCMs was enriched for genes associated with collagen fibril organization (Col6a1/2, Col4a1/2, Adamts2), blood vessel morphogenesis (Smad4, Pdgfra, Syk), and blood vessel development (Akt1, Notch2, Foxo1) (Table S4B). As expected, the DC-specific cluster was enriched for genes involved in MHCII antigen-presentation molecules (H2-Aa, Cd74), as well as genes involved in myeloid DC differentiation (Irf4, Dcstamp, Btf3) and genes that play a role in T cell differentiation (Ccr7, Stat5a, Tnfsf9) (Table S4C). For the TR-MC-specific cluster, enriched gene ontology (GO) processes included complement receptor-mediated signaling (C1qa/c, C5ar1, Fcna/b) and chemotaxis and cell-migration pathways, including Pf4, Pmp22, and P2ry12 (Table 4SD). In addition, the leukotriene metabolic pathway was enriched in this cluster with genes including Alox5, Ltc4s, and Ncf1. The cluster shared between DCs and TR-MCs included cell cycle (Ccna2, Tubb6, Cdc23), DNA repair (Lig1, Hdac9, Cdca5), and cellular component organization or biogenesis (Rpf2, Ipo4, Lmna) (Table S4E).
Next, we sought to identify the human equivalent of the TR-MC population utilizing single-cell datasets on myeloid cells isolated from the human synovium. Using our merged MHCII− and CD64+ CITE-seq data (Figure S3E), we were able to define the TR-MC module based on the unique gene markers for MHCII − cells (Table S4F). The AMP Consortium presented scRNA-seq data on CD14+ mononuclear cells sorted from human RA synovium split into leukocyte-rich vs. leukocyte-poor, alongside osteoarthritis (OA) patients.24 We identified a subset of these cells that resembled TR-MCs (Figure S4I); OA and leukocyte-poor RA tended to exhibit higher levels of the MHCII module (Figures S4J and S4K). Similarly, these genes were highest in the NUPR1+ and IFN-activated clusters defined by AMP associated with leukocyte-poor RA and OA (Figures S4L and S4M). Next, we performed CITE-seq on sorted CD45+CD11b+ Syn cells from an RA patient collected through the Rheumatoid Arthritis Synovial Tissue Network (REASON).25 We used de novo clustering to divide these 1,614 cells into 11 clusters (Figure 4C). From the ADT data, these cells exhibited myeloid cell-surface markers CD45, CD11b, and CD14 (Figure 4D). Based on expression of TR-MC genes, the orthologous counterpart was narrowed down to cluster 7 (Figures 4E and 4F). These cells exhibited distinct expression of Trem2 at the RNA level (Figure 4G) and TimD4 at both the RNA and the protein level (Figures 4H and 4I). Further, we showed that TR-MCs are CD11b+CD68+TREM2−MERTK−CCR2−MHCII−CD163−Tim4+ using immunofluorescence (IF) on healthy control, and a portion gains CD163 in RA Syn tissue (Figures 4J and 4K). These data are consistent with the Tim4+ population observed in Alivernini et al.26 Overall, these findings demonstrate that Syn CD14+Tim4+ cells represent the human ortholog of TR-MCs.
TR-MCs are long lived
Tissue-resident macrophage populations originate from either the fetal liver or adult hematopoiesis, but the origins of TR-MCs are unknown. We crossed a tamoxifen-inducible CX3CR1-Cre (CX3CR1CreER) with a GFP reporter (zsGFP) to generate a mouse (CX3CR1CreER.zsGFP) in which CX3CR1+ cells express GFP after administration of tamoxifen (TMX). Mice were treated with two subsequent daily doses of TMX to measure the steady-state lifespan of circulating and Syn cells. The vast majority of PB monocytes gained GFP, which was lost by day 14 (Figure 5), in line with the previously reported lifespan of PB NCMs.27 In contrast, 18% ± 0.5% of the TR-MCs were GFP+ at D3, but this level remained stable throughout the 21-day time course (Figure 5B). These data suggest that the TR-MCs are long lived and distinct from short-lived circulating monocytes. Retention of GFP throughout the 21-day time course also suggests these cells did not differentiate into Syn macrophages (Figure 5C).
Figure 5. TR-MCs are fetal liver derived and long lived.

(A–C) GFP expression in (A) PB monocytes, (B) TR-MCs, and (C) CD64+ cells from adult CX3CR1Cre.ER.zsGFP mice treated with 50 mg/kg TMX at D0 analyzed on D1.
(D) Experimental approach for lineage tracing.
(E–G) Labeling of (E) PB monocytes and (F and G) NCMs and TR-MCs in adult CX3CR1Cre.ER.zsGFP without TMX, and offspring of CX3CR1Cre.ER.zsGFP mice treated with TMX on day 15 of gestation (E15).
(H) Total NCMs and TR-MCs in steady state and 24 h post-non-lethal irradiation.
(I) Percentage of TR-MCs in irradiated CD45.1 → CD45.2 bone marrow chimeras.
(J) Experimental approach for shielded chimeras.
(K and L) Percentage of GFP+ NCMs and TR-MCs in (K) CX3CR1Cre.ER.zsGFP → C57BL/6 and (L) C57BL/6 → CX3CR1Cre.ER.zsGFP 7 bone marrow chimeras 28 days after administration of 50 mg/kg TMX.
(M) 5-ethynyl-2′-deoxyuridine (EdU) expression in TR-MCs 14 days post-EdU treatment in CD45.2 → CD45.1 bone marrow chimeras. All graphs are mean n > 4 + SEM; p value was calculated using unpaired t test; *p < 0.05, **p < 0.01, ****p < 0.001.
We then utilized CX3CR1CreER.zsGFP mice for TR-MC fate-mapping studies (Figure 5D). TMX was delivered at E15 to pregnant mothers from the cross of CX3CR1CreER with zsGFP mice, which will allow for identification of embryonically derived cells not derived from yolk sac. TMX-naive CX3CR1CreER.zsGFP mice provided background levels of GFP. PB monocytes from progeny of TMX-treated E15 mice displayed GFP expression similar to background (Figure 5E). Meanwhile, 11% ± 1.5% of TR-MCs were GFP+ in the E15 TMX-treated CX3CR1CreER × zsGFP progeny compared with 0% in NCM cells (Figures 5F and 5G) and neutrophils (Ly6G+) (Figure S5A). Approximately 70% of the CD64+ population were GFP+.
We also subjected C57BL/6 mice to non-lethal gamma-irradiation-induced cell death to further support the tissue residency of TR-MCs. We found that the bulk of TR-MCs, but not NCM cells, were initially refractory toward non-lethal, whole-body gamma-irradiation at 1 day post-irradiation (Figure 5H). However, by 8 weeks post-irradiation and non-shielding, 12% of the host TR-MCs remained (Figure 5I). These data suggest that TR-MCs may be partially derived from embryonic precursors, which has not previously been shown in cells lacking canonical macrophage markers.
Next we examined whether TR-MCs were replenished by PB monocytes over time. C57BL/6 mice were treated with non-lethal gamma-irradiation with hind joints shielded to retain the Syn compartment and received CX3CR1CreERzsGFP bone marrow (CX3CR1CreERzsGFP → C57BL/6) immediately after irradiation. TMX was administered on experimental D0 and D1 at 8 weeks post-irradiation and transfer. GFP was analyzed directly after the second dose of TMX (D1) and at 7 and 28 days post-TMX (Figure 5J). PB monocytes from CX3CR1CreERzsGFP → C57BL/6 chimeric mice displayed a dramatic increase in GFP expression, which eventually disappeared by 28 days post-TMX administration (Figure S5C). Over 12% of the NCMs were GFP+ in the CX3CR1CreERzsGFP → C57BL/6 chimeric mice on D7 post-TMX, which was reduced to undetectable levels by day 28 post-TMX. These data support that NCMs are derived from donor PB monocytes and are short lived (Figures 5K and S5C). In contrast, the expression of GFP was not significantly above background in TR-MCs throughout the 28-day time course, which indicates that the donor monocytes do not contribute to TR-MCs when the niche is not disrupted. Furthermore, a reverse chimera was also generated (C57BL/6 → CX3CR1CreERzsGFP) (Figure 5J). In these chimeric mice, the NCMs were negative for GFP throughout the 28 days post-TMX, further supporting that the NCMs are derived from circulating monocytes. In contrast, only a small fraction of TR-MCs (2.5% ± 0.3%) were GFP+ cells at D7, which increased to 8.5% ± 1.3% by D28 (p = 0.025) (Figure 5L). Since the numbers of GFP+ TR-MCs are reduced in C57BL/6 → CX3CR1CreERzsGFP chimeric mice compared with CX3CR1CreERzsGFP mice at steady state, these data suggest that the TR-MC niche is retained locally. To address whether the increase in GFP+ TR-MCs over time in C57BL/6 → CX3CR1CreERzsGFP chimeric mice supports the self-replenishment of TR-MCs, we continuously administered the nucleotide analog EdU for 14 days to 8 weeks post-irradiation and hind-joint-shielded CD45.2 → CD45.1 bone marrow chimeric mice. Over 17.8% ± 1.4% of CD45.1 (host) TR-MCs were EdU+ after 14 days, whereas <1% of CD45.2 donor TR-MCs were EdU+ (Figure 5M). Collectively, these data suggest the TR-MCs are derived embryonically and radioresistant, but may be capable of self-renewal via circulating monocytes or a Syn precursor when the Syn niche is disrupted.
Rapid expansion of TR-MCs in the synovium is required for the development of inflammatory arthritis
To assess the dynamics of TR-MCs during induction of inflammatory arthritis, we determined the numbers of myeloid cells at 1 and 24 h post-STIA. NCMs and TR-MCs increased by 4-(p = 0.009) and 2-fold (p = 0.008), respectively, at 1 h compared with steady state, but only TR-MCs continued to expand (2-fold) from 1 to 24 h (Figures 6A and S6A). In contrast, neutrophils in the synovium were not significantly changed during the first 24 h post-STIA, consistent with previous studies.8 These results demonstrate that TR-MC expansion precedes neutrophil infiltration.
Figure 6. TR-MCs are the critical population for development of inflammatory arthritis.

(A) NCMs, TR-MCs, and neutrophils at 1 and 24 h post-STIA.
(B and C) NCMs and TR-MCs (B) at steady state and (C) at 1 h post-STIA in C57BL/6, NR4A1−/−, and CCR2−/− mice.
(D) Depletion of NCMs and TR-MCs following 200 μL of clo-lip.
(E) STIA severity induced 24 h after clo-lip treatment in C57BL/6 and NR4A1−/− mice.
(F) Antibody labeling system.
(G) Labeling of PB and NCM via I.V. administration of anti-CD43.
(H and I) Labeling of TR-MCs and DCs with I.V. anti-CD43 after 1 h of steady state.
(J and K) Labeling of NCMs and TR-MCs with anti-CD43 in C57BL/6 and NR4A1−/− (J) after 1 h of steady state and (K) 1 h after STIA.
(L) Surface expression of CD43 and CD11c on I.V. αCD43-BUV395+/− TR-MCs. Graphs are mean n > 4 + SEM; p-value was calculated with unpaired t test; *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
We then examined the TR-MC response in NR4A−/− and CCR2−/− mice to determine whether the expansion of TR-MCs requires contribution from PB NCMs or PB CMs. Under steady-state conditions, NR4A1−/− and CCR2−/− mice have reduced NCMs and levels of TR-MCs comparable to those of C57BL/6 mice (Figure 6B). One h after administration of STIA, NCMs in NR4A1−/− mice remained lower than in C57BL/6 (p = 0.0003) and CCR2−/− (p = 0.0001) mice, which underwent even more significant expansion than C57BL/6 (p = 0.02) (Figures 6C and S6B). Meanwhile TR-MC levels remained comparable across genotypes, having undergone similar levels of expansion 1 h post-STIA (Figures 6C and S6B). To determine if TR-MCs are a critical population in STIA, we examined whether clo-lips would deplete i.v. and TR-MCs. Both NCMs and TR-MCs are markedly reduced in response to clo-lips at 24 h (p = 0.007, p = 0.006) (Figure 6D). As expected clo-lip treatment depleted circulating monocytes but did not affect the numbers of Syn macrophages (Figures S6C and S6D), similar to previous studies.8 Administration of clo-lips prior to STIA completely prevented disease in both C57BL/6 and NR4A1−/− mice (Figure 6E). Given the redundancy of NCMs in NR4A1−/− mice, these data suggest that the TR-MCs are essential for inflammatory arthritis.
Extravascular TR-MCs have access to vasculature
We observed that TR-MCs are sensitive to clo-lips, while Syn macrophages are unaffected, even though both cells are within the e.v. compartment. These results may be explained if TR-MCs have access to the circulation. To investigate this notion, we modified the in vivo labeling system to include a second I.V. antibody (Figure 6F), αCD43-BUV395-(I.V.), which labels 95% ± 1.1% of PB NCMs and 96% ± 1.8% of the Syn NCMs (Figure 6G and S6E). With this system of administering αCD45-BUV661-(I.V.) 1 h prior to αCD43-BUV395-(I.V.), NCMs that are retained in the i.v. space remain dual positive for I.V. αCD45-BUV661 and I.V. αCD43-BUV395, while TR-MCs that access the circulation within the hour will be positive only for αCD43-BUV395-(I.V.) (Figure 6H). Using this strategy, 5% ± 1.2% of TR-MCs were labeled with αCD43-BUV395-(I.V.) during steady state, whereas only 1% ± 0.4% of DCs became labeled, confirming that labeling was due to cell activity and not passive diffusion of antibody into the e.v. space (Figures 6H and 6I). A similar trend was observed in NR4A1−/− mice (Figures 6J and 6K). Further, the addition of STIA as an inflammatory stimulus increased the labeling by 2-fold of TR-MCs with αCD43-BUV395-(I.V.) in C57BL/6 (p = 0.045) and NR4A1−/− mice (p = 0.025) within 1 h post-STIA. Since TR-MCs undergo enhanced labeling response to STIA, these data suggest that depletion of TR-MCs by clo-lips is due to increased contact with vasculature following an inflammatory stimulus.
Increased contact of TR-MCs may be through antigen sampling, which has been demonstrated in CX3CR1+ macrophages.28 Similarly, monocytes in liver are capable of reverse extravasation associated with enhanced expression of CD11c.29 The TR-MCs that were labeled with αCD43-BUV395+-(I.V.) exhibited a significant elevation in CD11c expression (p = 0.007) (Figure 6L) compared with the cells that remained e.v., whereas the level of CD43 remained consistent (Figures 6L and S6F). Together, these data and the clo-lip studies demonstrate that TR-MCs have continued access to the vasculature and these mechanisms may be involved in inflammation.
LFA1 is required for STIA-induced reverse extravasation of TR-MCs
To investigate if contact with vasculature by TR-MCs is required for development of STIA, we used LFA1−/− mice. Previous studies have shown that LFA1 is required for leukocyte extravasation into tissue and for STIA.30 LFA1−/− mice failed to develop inflammatory arthritis (Figure 7A), consistent with reports.30,31 There were no significant differences in the numbers of PB CMs, PB NCMs, and i.v. and TR-MCs between LFA1−/− mice and control mice at steady state (Figures S7A-S7C). However, neither LFA1−/− NCMs or TR-MCs exhibited the expansion in response to arthritogenic serum (Figure 7B) observed in C57BL/6 mice (Figure 6A). In addition, LFA1−/− TR-MCs did not undergo increased labeling with αCD43-BUV395-(I.V.)) 1 h following STIA compared with C57BL/6 mice, suggesting that access to vasculature does not increase in response to inflammatory stimulus in LFA1−/− mice (Figure 7C).
Figure 7. Deletion of LFA1 reduced the pro-inflammatory phenotype of TR-MCs.

(A) STIA clinical score in C57BL/6 and LFA1−/− mice.
(B) NCMs, TR-MCs, and neutrophils 1 h post-STIA in C57BL/6 and LFA1−/− mice.
(C) TR-MCs labeled with I.V. αCD43-BUV395 at steady state and 1 h post-STIA. Graphs are mean n > 4 + SEM; p value was calculated with unpaired t test; *p < 0.05, **p < 0.01.
(D) Ratio of cells annotated as either MHCII− (TR-MCs) or MHCII+ (DCs) in C57BL/6 and LFA1−/− mice.
(E) Selected GO processes associated with differentially expressed genes in the MHCII− compartment (representing TR-MCs) between LFA1−/− and C57BL/6 mice.
(F) Expression of representative genes increased or decreased in MHCII− cells from LFA1−/− compared with C57BL/6 mice.
(G) Merged CITE-seq data on CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− cells from D0 (shown in Figure 3) and D7 STIA.
(H) Expression of Lyve1 (TR-MC marker) in D0 and D7 STIA.
(I) ADT intensity of FrB protein in D0 and D7 STIA.
(J) Annotated subpopulations from merged D0 and D7 STIA CITE-seq data based on de novo clustering and overlap with D0 annotations.
(K) Pairwise Pearson’s correlation between the average expression profile at D7 STIA of annotated subpopulations.
(L) Mean ADT intensity of surface markers at D7 STIA across annotated subpopulations.
(M) Selected GO processes associated with genes that are increased in expression in TR-MCs at D7 STIA compared with D0.
(N–R) Expression of representative genes that are increased in expression in TR-MCs at D7 STIA across clusters stratified by time point.
LFA1 deletion reduces pro-inflammatory transcriptional profile of TR-MCs
To determine how LFA1 deletion affected TR-MCs on the transcriptional level, we performed scRNA-seq on isolated CD45+CD11b+Ly6G−SiglecF−CD64− Syn cells from LFA1−/− mice at steady state. As before, we integrated and superimposed the six subpopulations (0–5) defined from C57BL/6 mice on 3,500 LFA1−/− cells (Figure S7D). LFA1−/− mice displayed an altered distribution of cells across the six subpopulations (p < 2.2e–16) (Figures S7E and S7F and Table S5A) but the ratio of MHCII+ (DCs) to MHCII− cells (TR-MCs) was comparable to that in C57BL/6 mice (Figure 7D). Moreover, LFA1−/− TR-MCs exhibited decreased expression of genes associated with chemotaxis (Ccl17, Itgb2, Ccl9), defense response (Arg1, Mif, Cfp, Itgax), regulation of cell adhesion (Ccr5, Lgals3, Adam8), and stress response (Pycard, Flt1, Prdx5, Vegfa) (Figures 7E and 7F and Tables S5B andS6C). In contrast, genes associated with regulation of cell differentiation (Fos, Cd36, Mef2c, Mafb, Csf1r), regulation of angiogenesis (Tcf4, Pf4, Tgfbr2), and response to wounding (Macf1, Aqp1, Cfh) were increased in expression in LFA1−/− TR-MCs compared with C57BL/6 (Figures 7E and 7F and Tables S5B and S5C). Based on these data, we demonstrate that TR-MCs are associated with chemotaxis of leukocytes such as granulocytes to the synovium during inflammation.
TR-MCs play vital roles in the chemotaxis of the leukocytes during STIA
We next determined the effects of STIA on the TR-MC population. CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− cells were isolated at D7 post-STIA, and 10,131 cells were analyzed by CITE-seq. The CITE-seq data from D0 (Figure 3) was merged with those from the D7 STIA (Figures 7G and S7G) to yield 14 clusters. We defined subpopulations based on previously observed gene expression in the D0 CITE-seq dataset and canonical markers (Figures 7H-7J and S7H-S7K and Table S6A). The D7 STIA data included osteoclast-like cells, fibroblasts, and monocyte subpopulations not observed at D0. Because the last clustered separately from steady-state monocytes and possibly reflect cells in transition to either TR-MCs or macrophages, we simply labeled them STIA monocytes 1–5 (Figure 7K). The cluster labeled as TR-MC had comparable expression of Lyve1, Gas6, Timd4, and Pmepa1 at D0 and D7 with high ADT intensity of TR-MC surface markers, CD163, FrB, CD107a, and C5AR-CD88 (Figures 7L and S7L and Table S6B). Comparison of these cells between time points revealed that genes associated with defense response, regulation of cell motility and chemotaxis, antigen processing and presentation, and response to stress were increased in TR-MCs in response to STIA (Figure 7M and Tables S6C and S6D). These included genes such as Stab1 (a scavenger receptor), Apoe, Dusp1 (a phosphatase involved in the MAPK pathway), Psap (a lysosomal protein), and CEBP/b (Figures 7N-7S). These data document that TR-MCs can be identified by conserved features in STIA and that TR-MCs from LFA1−/− mice have an impaired STIA response.
DISCUSSION
Our study identifies a population of Syn tissue Ly6c− cells distinct from circulating NCMs that do not exhibit canonical macrophage or DC markers. First, we described a distinct population of NCMs that are transcriptionally similar to PB NCMs and require NR4A1 but remain attached to the vessel wall even after perfusion. Then, through I.V. injection of antibody we confirmed the e.v. localization of TR-MCs that are negative for surface expression of CD64, F4/80, and CD68 and transcriptionally distinct from circulating monocytes, Syn macrophages, and DCs. These TR-MCs are long-lived, terminal cells that do not require CCR2, NR4A1, or LFA1 for development. In fact, TR-MC development may be NOD2 dependent, as this has previously been linked to restoring circulating NCMs lacking NR4A1.32 Finally, we show that a population of cells corresponding to TR-MCs is present in Syn biopsies from RA patients, and their depletion prevents experimental RA in mice. Taken together, our data suggest that TR-MCs represent a population of Syn mononuclear cells involved in the pathology of RA.
In order to explore the heterogeneity of Syn tissue myeloid cells but exclude macrophages, eosinophils, and neutrophils, we performed scRNA-seq on CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− and CD45+CD11b+Ly6G−SiglecF−CD64−MHCII+ mononuclear phagocytes from the synovium during steady state. Analysis of these data enabled us to distinguish a subpopulation of DCs from NCMs and TR-MCs via MHCII expression. We also compared CD45+CD11b+Ly6G−SiglecF−CD64− subpopulations with six myeloid (CD45+CD11b+Ly6G−) populations harvested from STIA mouse joints on disease day 5.22 Although subpopulations defined by Culemann were annotated as macrophages, their fluorescence-activated cell-sorting (FACS) strategy did not exclude monocytes, thus explaining the high level of overlap between their populations with CD45+CD11b+Ly6G−SiglecF− CD64−MHCII− and MHCII+ cells.33
Further, two recent studies utilized scRNA-seq to characterize human myeloid cells from the joints of RA patients.24,26 By using a module score based on marker genes to annotate cells, we identified MHCII− cells (TR-MCs) among the four monocyte subpopulations defined by Zhang and colleagues.24 The data support a higher presence of a corresponding TR-MC population in human synovium from leukocyte-poor RA and OA tissue, although the study is limited due to the small number of CD14+ cells (750) in the study. The Alivernini group performed scRNA-seq on CD64+CD11b+CD3−CD19−CD20−CD56−CD49−CD117−CD15− Syn cells from healthy controls and RA patients who were treatment-naive/resistant or in clinical remission.26 Based on the transcriptional profile of the cells, we identified the Timd4+ population as TR-MCs. However, these prior studies did not enable us to determine whether TR-MCs could be identified by flow cytometry. Thus, we utilized our CITE-seq data from REASON studies and determined that the TR-MCs corresponded to the TIM4+ cells. Thus, these provide the foundation for future study of our TR-MCs in the context of human disease.
Prior studies on the role of monocytes in RA have presented conflicting results. As has been observed previously and in this study, clo-lips prevent development of RA by ablating all circulating monocytes.8,9 We expand upon these findings by establishing the redundant role of PB monocytes in inflammatory arthritis and identifying the critical TR-MC population susceptible to clo-lip killing. Our previous finding that restoring NCMs following monocyte depletion enables the progression of STIA appears to conflict with the sensitivity of NCM-deficient NR4A1−/− mice to arthritis.13,14 The data presented here resolve this conflict, by confirming the preservation of TR-MCs in NR4A1−/− mice, and by extension of this, PB NCMs may replenish the TR-MC niche when required. This process has been well established in embryonic macrophages, which can be replenished from bone marrow-derived cells following injury.34,35
TR-MCs may represent a terminal monocyte population. Although they lack the canonical macrophage surface markers CD64, F4/80, CD68, and MERTK, TR-MCs express the gene for MafB, and our results indicate that MAFB is highly active as a TF in these cells. MafB has been previously associated with tissue macrophages over monocytes36 and is central for suppressing macrophage proliferation.37 Our experiments determined that TR-MCs maintain their ability to proliferate and do not differentiate into macrophages. The TR-MCs are also negative for the DC master regulator Zbt4638,39 compared with the mono-DCs. Collectively, these data support TR-MCs as a self-renewing terminal monocyte population that is distinct from DCs and macrophages in the tissue.
Increased vascularity and enhanced permeability of the synovium has been associated with RA and experimental models of arthritis.40-42 In steady state, access to the synovium is restricted by the size of the particle,43 consistent with failure of clo-lips to eliminate Syn macrophages. However, since TR-MCs in the synovium are susceptible to depletion by clo-lips, we proposed that these cells have access to the vasculature. By using a second I.V. labeling system, we demonstrated labeling of TR-MCs over time, indicating access to the vasculature by antigen sampling across the endothelium as exhibited by CX3CR1+ macrophages28 or through full reverse transmigration from synovium to i.v. space. One group demonstrated bidirectional transmigration of monocytes across hepatic sinusoidal endothelium,29 and another showed that reverse transmigration contributes to the development of pathogenic foam cells in atherosclerosis.44 Furthermore, access to the vasculature is increased in response to STIA, which may be through activation of TR-MCs or passively through STIA-induced increased vascular permeability. In support of the former, we show that TR-MCs from LFA1-deficient mice do not respond to STIA with proliferation or increased vascular labeling, suggesting a TR-MC-restricted mechanism. These data suggest a role for TR-MCs in surveying the vascular endothelium, maintaining barrier integrity, and responding to potential pathogens. These roles have been established in PB NCMs within the vasculature,5 and therefore TR-MCs may play a complementary role in the e.v. niche.
LFA1 is required for monocyte crawling45 and has been implicated in the pathogenesis of arthritis.30,46 Neutrophils also express LFA1 and are a critical cell for inflammation in STIA, where their influx into joints is LFA1 dependent.31 Therefore, involvement of neutrophils in the TR-MC response to STIA cannot be excluded. Nonetheless, these data support that TR-MCs are involved in inflammatory responses during STIA via LFA1-dependent mechanisms.
Future experiments may be required to further investigate the phenotype of TR-MCs. Given the highly plastic nature of mononuclear phagocytes, culturing cells in vitro results in populations skewed artificially based on culture conditions and cell differentiation. For these reasons, TR-MCs were not cultured in vitro to assess functionality or morphology. However, we showed that the GO processes that are upregulated in TR-MCs at the peak of inflammation in STIA are similar to those that are downregulated in LFA1−/− TR-MCs. These new results suggest that LFA deficiency affects the expression of pathways that are central to the TR-MC response to STIA. These data support the promotion of chemotaxis and cell migration as a critical function of TR-MCs during the development of inflammatory arthritis.
Here we have identified a population of tissue Ly6c− cells distinct from DCs and macrophages in mice, with a corresponding population identified in human RA synovium. These TR-MCs respond rapidly to inflammatory signals, drastically expand in numbers, and traverse the perivascular space via an LFA-dependent mechanism in arthritis. Our data support a role for TR-MCs as instigators of Syn inflammation leading to the pathogenic cascade in inflammatory arthritis.
Limitations of the study
As stated above, a limitation of this study is the lack of in vitro assessment of TR-MC functionality or morphology. Due to the highly plastic nature of myeloid cells, such studies are unlikely to accurately represent the in vivo cell phenotype. Furthermore, the complexities of imaging through bone and hind joints meant that anatomical resolution of TR-MCs was beyond the scope of this study; however, the functional profiling presented here supports the role of TR-MCs as patrolling and lining the vasculature to influence chemotaxis in inflammation.
STAR★TABLE
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Harris Perlman (h-perlman@northwestern.edu).
Materials availability
All mouse lines used in this study are commercially available.
Data and code availability
All sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication using accession code: GSE225803.
This paper analyzes published data that were reported in the original article or obtained directly from the authors.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Breeder pairs were purchased and experimental mice bred in house, and/or acclimated in barrier and specific pathogen-free animal facility at the Center for Comparative Medicine, Northwestern University. Female mice were used for all RA-like studies. All experimental procedures were carried out on mice aged 8-10 weeks (unless stated otherwise in aging studies). All strains were C57Bl/6 background. Littermates of the same sex were randomly assigned to experimental groups. All genotypes are reported in key resource table. All procedures were approved by the Institutional Animal Care and Use committee at Northwestern University.
Highlights.
Single-cell RNA sequencing identifies a tissue-resident monocytic cell in the synovium
TR-MCs are conserved in mice and RA patients
TR-MCs are embryonically derived and do not require CCR2 or NR4A1
TR-MCs require LFA for the induction of inflammatory arthritis
METHOD DETAILS
Induction of serum transfer induced arthritis (STIA)
To induce serum transfer arthritis, 85μL/20g K/BxN serum was given intravenously (I.V) to each mouse.
Flow cytometric analysis
To prepare single-cell suspensions from joint, joints were removed from hind paws following euthanasia, perfused and stored on ice in sterile HBSS. Skin and toes were removed from each paw and bone marrow flushed from exposed tibia with sterile HBSS through a 30G needle. Synovial tissue was then infused with 1.5mL/joint of ankle digestion buffer (2.4mg/mL dispase II, 2mg/mL collagenase D, 0.2mg/mL DNAse I in HBSS pH 7.2-7.6) before incubation at 37°C for 1h with shaking. Cells were then aggravated through a 40-μm mesh filter. Red blood cells were removed with lysis 250μL/sample (1x PharmLyse in sterile water) at room temperature for 1 minute. Dead cells were stained with eFluor 506 viability dye (1:1000 dilution). Cells were incubated with FcBlock (BD Bioscience) and stained with selected antibodies outlined below. Cells were fixed with 10% PFA at 4°C for 20 minutes. To prepare single cell suspensions from blood, 90μL blood collected by cardiac puncture was incubated with FcBlock and selected antibodies. Red blood cells were lysed with FACS lyse at rt for 10 minutes (1x in sterile water) and single-cell suspensions were acquired on BD LSR II or BD Symphony. For all FACSorting studies, cells were acquired on a BD FACSAria. Count eBeads were used in joint preparations to calculate cell numbers. Fluorescence minus one samples were used to set gates. Compensation and analysis of flow-cytometry data was carried out in FlowJo V10.
Bulk RNA sequencing
RNA from FACSorted synovial cells was extracted using PicoPure RNA Isolation kit as per manufacturer’s instructions. Bulk RNA-seq shown in Figure 1 was carried out using QuantSeq 3’ mRNA sequencing kit, while bulk RNA-seq shown in Figure 4 utilized full-length SMART-seq v4 Ultra Low Input Kit for Sequencing. All bulk libraries were sequenced on an Illumina NextSeq 500 to an average depth of 8x106 (QuantSeq) or 2x107 (SMART-seq) reads per sample.
Following the sequencing, libraries in the form of BCL files were obtained from Illumina’s BaseSpace platform and demultiplexed (using bcl2fastq v2.17.1.14) to convert them into FastQ read format for further processing. The QuantSeq reads were then processed further by trimming the adapters, low quality bases and short reads (using BBDuk version 37.22 with the following parameters: k=13 ktrim=r useshortkmers=t mink=5 qtrim=r trimq=10 minlength=20). After trimming, remaining reads were aligned to the mouse genome reference mm10 (Mus Musculus / UCSC assembly GRCm38) using STAR.47 Aligned reads in BAM format were mapped to the reference transcriptome (Mus Musculus GRCm38.87) to obtain exon counts and generate gene expression tables using the tool HTSeq-count.48 The SMART-seq reads were trimmed using Trimmomatic (version 0.36),49 to remove adapter sequences, low quality bases and short reads (minimum length = 20bp). After trimming, remaining reads were aligned to the mm10 genome reference (Mus Musculus / UCSC assembly GRCm38) with Tophat aligner (tophat 2.1.0).50,51 The aligned reads in BAM format were mapped to gene exons by HTSeq-count as above48 using the reference transcriptome GTF file (Mus Musculus GRCm38.87).
All gene expression counts were scaled to read depth using counts per million reads mapped (cpm). To filter out lowly expressed genes, genes with no group mean above 7 cpm in the relevant cell types were excluded from the analysis. Differentially expressed genes (DEG) across multiple cell types were defined as genes with a difference of 2-fold between any two groups. K-means clustering of DEG was carried out in Gene-E. GO enrichment was calculated using Gorilla52,53 on each cluster with all DEGs as background. Expressed genes, K-means clustering, and GO processes for all datasets are provided in supplemental files for each figure. Volcano plots were generated in R (version 3.3.1). Principal Component Analysis (PCA) and Pearson’s correlation were performed on expressed genes and visualized with R (version 3.3.1). In Figure 1, monocyte populations were compared with CD64+ macrophages that were isolated from the same mice and sorted into 4 subpopulations based on the cell surface expression of CX3CR1 and MHCII.
Single-cell RNA sequencing
RNA libraries for single-cell analysis were prepared using 10x Chromium Single Cell 3’ Solution v3. Reads were processed and aligned to mm10 mouse reference genome using mkfastq and count commands of cellranger 3.1.0 pipeline. Subsequent analyses, including quality control, unsupervised clustering, identification of cluster markers, and visualization of gene expression were carried out using Seurat v3.1 package in R. Samples were individually assessed and filtered based on the number of UMI counts and % mitochondrial reads per cell. To account for technical variability, sample-specific thresholds were used as indicated below.
| Genotype | Total Cells Detected |
Median Genes/Cell |
Median UMIs/Cell |
Min UMI/cell |
Max UMI/cell |
Max % MT reads/Cell |
# cells after filtering |
|---|---|---|---|---|---|---|---|
| C57Bl/6 (CD64−) | 9447 | 2681 | 9323 | 4000 | 35000 | 7 | 7160 |
| CCR2−/−(CD64−) | 4867 | 3184 | 12927 | 5500 | 40000 | 7 | 3621 |
| NR4A1−/−(CD64−) | 13375 | 818 | 1494 | 4000 | 38000 | 7 | 2867 |
| LFA1−/−(CD64−) | 7908 | 2622 | 8880 | 3500 | 31000 | 5 | 5444 |
| C57Bl/6 MHCII− CITE-seq | 3529 | 3236 | 11887 | 3500 | 45000 | 5 | 2967 |
| C57Bl/6 MHCII+ CITE-seq | 8753 | 3196 | 11547 | 4500 | 40000 | 5 | 6061 |
| C57Bl/6 MHCII− (Day 7 STIA) CITE-seq | 10131 | 2947 | 10731 | 3500 | 45000 | 5 | 8011 |
| C57BL/6 CD64+ CITE-seq | 9914 | 3001 | 9238 | 5000 | 40000 | 10 | 6051 |
| RA Patient CITE-seq | 2754 | 4270 | 20758 | 8454 | 63404 | 10 | 1614 |
For the initial analysis on the C57BL/6 sample, selection of variable genes was performed using the default vst method with nfeatures set to 2000. UMAP dimensionality reduction and unsupervised graph-based clustering were performed with top 16 principal components (PC) and resolution parameter of 0.2. SingleR package v1.0.5 was used to annotate cells with Immgen reference cell types. Differentially expressed genes for each subpopulation were defined log(fold-change)>∣0.25∣ and adjusted p-value<0.05 by Wilcoxon test with Benjamini-Hochberg procedure for False Discovery Rate. Module scores of cell type signatures based on manually selected genes (Table S2B) were computed using AddModuleScore function with default parameters. Pearson correlations were calculated between the averaged expression profiles of single-cell subpopulations and bulk RNA-seq on monocyte populations (Figure 1). Cell cycle scoring was performed using G2/M and S phase gene sets provided in Seurat, converted to orthologous mouse genes using BioMart R package. Cells in subpopulations 0, 1, and 3 were further classified as either MHCII+ or MHCII− by a threshold of 2 on normalized expression of H2-Eb1. Co-expression of H2-Eb1 with MHCII+/− compartment genes was visualized using DimPlot function with blend=TRUE parameter.
Similarity of our annotation with published clusters in Culemann et al33 was determined by calculating the fraction of their top 20 reported markers that overlapped with the list of differentially expressed genes for each of our subpopulations: Cycling (2), Monocyte (4) and cDC (5) (Table S2A) as well as MHCII+ and MHCII− (Table S2D). Significance of overlap was determined by hypergeometric distribution with the expressed genes as background, defined as those that were present in more than 25% of cells for at least one subpopulation (n = 6344). A FWER cutoff of 0.05 was applied based on Bonferroni correction for 35 comparisons (Table S2E). To assign C57BL/6 myeloid cells to either i.v. or TR-MC, or mono-DC singleR was run with the bulk RNA-seq on these populations as reference.
For the processing of CITE-seq samples, dimensionality reduction was performed with top 10 PCs. Antibody intensities were normalized using CLR method in NormalizeData function. Identities of individual cells were annotated via the FindTransferAnchors and TransferData functions using the top 30 PCs and C57BL/6 sample as reference data. The analysis of scA-TAC-seq experiment was carried out using Signac 1.3.0 package. Quality control was performed with the following cutoffs: fragments in peak region > 4000 and < 40000, percentage of reads in peaks > 40%, ratio reads in ENCODE blacklist regions < 0.02, nucleosome binding signal < 1.2, and transcriptional start site (TSS) enrichment score > 2. Latent semantic indexing (LSI) was performed through RunTFIDF and RunSVD functions, followed by UMAP dimensionality reduction using 2:30 LSI components. Annotation of cell identities was performed by integrating and transferring labels from C57BL/6 CITE-seq data. Per-cell activity scores of JASPAR2020 transcription factor (TF) motifs was inferred through chromVAR wrapper function implemented within Signac package.
For annotation of human myeloid cells, we first merged the MHCII− and the CD64+ CITE-seq data carrying over the annotations from the former. Then, we defined the markers genes for MHCII− cells (TR-MC) using FindAllMarkers and chose the top 10 by fold-change with human orthologs (Table S4F). Next, we calculated the module score in cells from either the AMP CD14+ scRNA-seq dataset24 or our own RA Patient CITE-seq data using AddModuleScore. We also performed de novo clustering (10 PCs and 0.6 resolution) on the latter data to determine which cluster exhibited the highest expression of the MHCII− module and link this with RNA expression and ADT intensity of other genes/surface markers.
Integration of C57BL/6 cells with other samples was executed using Seurat anchoring method with 30 CCA dimensions and visualized by UMAP using top 13 (B6 with CCR2−/− and NR4A1−/−) and 15 (B6 with LFA1−/−) PCs. Subpopulation labels were determined by the majority label of C57BL/6 annotated cells in each cluster. Relative contributions of the integrated samples were calculated by down-sampling to 2000 (B6 with CCR2−/− and NR4A1−/−) and 3000 (B6 with LFA1−/−) cells for each sample after clustering, with significance determined through chi-square test. Enriched GO processes were identified using GOrilla on genes increased or decreased in expression log2(fold-change)>∣0.25∣, adjusted p-value<0.05 by Wilcoxon test with Benjamini-Hochberg) in LFA1−/− MHCII− compartment compared to C57Bl/6 with 14144 expressed genes as background.
For the analysis on the CD45+CD11b+Ly6G−SiglecF−CD64−MHCII− sorted sample from Day 7 STIA, selection of variable genes was performed using the default vst method with nfeatures set to 2000. The Day 7 STIA data was combined with Day 0 steady-state by merging the raw count matrices. UMAP dimensionality reduction and unsupervised graph-based clustering were performed on the merged datasets with top 14 principal components (PC) and resolution parameter of 0.5. Clusters were then annotated based on inclusion of labeled cells from the Day 0 data with the exception of 0, 1, 2, 3, 6, 9 and 13 which were unique to STIA. Annotations were verified by expression of previously observed genes and canonical markers. Differentially expressed genes for each subpopulation were defined log(fold-change)>∣0.25∣ and adjusted p-value<0.05 by Wilcoxon test with Benjamini-Hochberg procedure for False Discovery Rate. Pearson correlations were calculated on the averaged expression profiles of single-cell subpopulations at Day 7 STIA. Enriched GO processes were identified using GOrilla on genes increased in expression log2(fold-change)>0.25, adjusted p-value<0.05 by Wilcoxon test with Benjamini-Hochberg) in MHCII− Day 7 STIA TR-MC compartment compared to MHCII− Day 0 TR-MC compartment with 4533 expressed genes as background.
Intra and extra vascular labeling of immune cells
To label intra-vascular cells, anti-CD45 BUV661 antibody was administered I.V. at 6μg/mouse in 200μl sterile PBS. Mice were then returned to housing environment for 5 minutes before euthanasia or for 1 hour before administration of second I.V. anti-CD43 BUV395 antibody. In studies using STIA, K/BxN serum was administered 5 minutes after anti-CD45 BUV661 antibody, 60 minutes prior to anti-CD43 BUV395 antibody as previously described.
Multiplex immunofluorescence
14plex multiplex staining on FFPE synovium biopsies were performed on COMET™ - automated platform for hyperplex sequential immunofluorescence (seqIF™) (Lunaphore Technologies SA). Brifely, COMET™ workflow includes a PT module required for the preprocessing of FFPE slides, FFeX microfluidic technology with integrated microscope which allows fast and uniform delivery of reagents to a closed staining chamber, where tissues subsequently undergo cycle of staining (Table below with antibody information), imaging and antibody elution in automated manner. Images are delivered as multi-stack ome.tiff files. Lunaphore Viewer was used for Image Visualization and Processing. Autofluorescence subtraction was applied on all channels. The intensity of each channel was normalized independently. Imaging was performed on synovial biopsies obtained by ultrasound guided biopsy from patient fulfilling 2010 RA criteria and a healthy control in the Birmingham Early Arthritis Cohort (BEACON).
Monocyte depletion
For depletion studies, 200μl/mouse clodronate-laden liposomes were given I.V. 24 hours prior to euthanasia. All mice were perfused with 20mL with ice-cold HBSS following euthanasia to remove circulating cells and retain adherent intra-vascular cells.
Bone marrow chimeras
Bone marrow chimeras were generated using C57Bl/6, CX3CR1CreER.zsGFP and/or B6.CD45.1 mice. Recipient mice were maintained on autoclaved water supplemented with antibiotics for 2 weeks prior and 2 weeks post-irradiation. Recipient mice were irradiated with 1000 cGy γ-radiation, 24 hours before I.V. transfer of 10x10^6 donor bone marrow cells. For shielded chimeras, hind joints were shielded with lead during irradiation and mice were treated with Ketamine (100 mg/kg) and Xylazine (16 mg/kg) prior to irradiation to prevent movement. 6 hours after irradiation mice received 30mg/kg busulfan to deplete bone marrow in shielded joints, before transfer of bone marrow. To determine proliferation of monocytes, chimeric mice were treated with 2mg EdU on experimental D0 and 0.5mg EdU for subsequent 14 days. Fate-mapping studies were carried out as described (Yona et al, 2013). Briefly, 50mg/kg tamoxifen and 10mg/mL progesterone were dissolved in corn oil and administered by oral gavage at E15 of gestation to pregnant CX3CR1CreER mice. For Cre induction in adult mice, 50mg/kg tamoxifen in corn oil was administered I.P on D0 and D1. For STIA studies using CX3CR1CreER.zsGFP, 110μL/20g of K/BxN serum was administered to tamoxifen-treated and naive mice to compensate for immunosuppressive effects of tamoxifen. Disease was assessed using clinical scoring 3 times per week for 21 days. Mice were euthanized at 1h and 24 h after serum injection.
QUANTIFICATION AND STATISTICAL ANALYSIS
For in vivo and ex vivo studies, all statistical analysis was carried out in GraphPad Prism V8. P-values less than 0.05 were considered statistically significant using two-tailed unpaired t-test with equal variance. For genomic studies, details on statistical analyses are provided in detail above. All ‘n’ numbers are reported in figure legends and refer to number of mice and/or samples from individual mice.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV421 Flt3 A2F10.1 | BD Bioscience | 562898 RRID:AB_2737876 |
| BV421 Tim4 RMT4-54 | BD Bioscience | 744874 RRID:AB_2742551 |
| BV421 CD64 X54-5/7.1 | Biolegend | 139309 RRID:AB_2562694 |
| BV650 CX3CR1 SA011F11 | Biolegend | 149033 RRID:AB_2565999 |
| BV650 CD105 MJ7/18 | BD Bioscience | 740609 RRID:AB_2740309 |
| BV786 CD64 X54-5/7.1 | BD Bioscience | 741024 RRID:AB_2740644 |
| BV711 Ly6G 1A8 | BD Bioscience | 563979 RRID:AB_2565971 |
| PE Tim4 RMT4-54 | BD Bioscience | 564147 RRID:AB_2738621 |
| PE-CF594 CD105 MJ7/18 | BD Bioscience | 562762 RRID:AB_2737776 |
| PE-CF594 CD4 RM4-5 | BD Bioscience | 56228 RRID:AB_11154410 |
| PE-CF594 CD8 53-6.7 | BD Bioscience | 562315 |
| PE Cy7 MHCII M5/114.15.2 | Biolegend | 107630 |
| PE Cy7 CD62L MEL-14 | Biolegend | 104418 |
| PerCPCy5.5 CD11b M1/70 | BD Bioscience | 550993 RRID:AB_394002 |
| FITC CD43 S7 | BD Bioscience | 553270 RRID:AB_394747 |
| APC MerTK 2B10C42 | Biolegend | 151508 |
| AF647 CX3CR1 SA011F11 | Biolegend | 149004 |
| APC CD43 S7 | BD Bioscience | 560663 RRID:AB_1727479 |
| AF647 CD177 Y127 | BD Bioscience | 566599 RRID:AB_2869790 |
| APC CD19 1D3 | BD Bioscience | 550992 RRID:AB_398483 |
| AF700 CD45 30-F11 | BD Bioscience | 560510 |
| AF700 NK1.1 PK136 | BD Bioscience | 560515 |
| APCCy7 Ly6c AL-21 | BD Bioscience | 560596 |
| BUV395 CD43 S7 | BD Bioscience | 740224 RRID:AB_2739972 |
| BUV661 CD45 30-F11 | BD Bioscience | 612975 |
| BUV737 CD11c HL3 | BD Bioscience | 612796 RRID:AB_2870124 |
| V500 CD45 30-F11 | BD Bioscience | 561487 RRID:AB_10697046 |
| CD68 | BioLegend | 916104 |
| CD11b | Abcam | ab133357 |
| TIMD4 | Atlas | HPA015625 |
| TREM2 | CST | 91068 |
| CCR2 | Proteintech | 16154-1-AP |
| VE-Cad | CST | 93467 |
| MHC II | CST | 68258T |
| MERTK | Abcam | ab176887 |
| CD15 | CST | 4744T |
| CD206 | CST | 91992T |
| Thy1/CD90 | CST | 13801S |
| CD11c | CST | 45581T |
| CD163 | CST | 93498T |
| CD14 | CST | 75181T |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 | Invitrogen | A32727 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 | Invitrogen | A32728 |
| Goat anti-Mouse IgM (Heavy Chain) Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | A21238 |
| Biological Samples | ||
| K/BxN serum was obtained at 8 weeks from the progeny of the KRN X NOD | Mice bred in house | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HBSS | Thermo Fisher | 14190250 |
| Dispase II (neutral protease, grade II) | Millipore Sigma | 4942078001 |
| Collagenase D | Millipore Sigma | 11088858001 |
| DNAse I | Roche | 10104159001 |
| BD PharmLyse | BD Bioscience | 555899 |
| eBioscience™ Fixable Viability Dye eFluor™ 506 | Thermo Fisher | 65-0866-18 |
| Purified Rat Anti-Mouse CD16/CD32 | BD Bioscience | 553141 |
| Paraformaldehyde, 20% solution | VWR | 100496-494 |
| BD FACS lysing solution | BD Bioscience | 349202 |
| PBS | Gibco | 14190250 |
| Clodronate-laden liposomes | Liposoma | C-010 |
| 123count eBeads™ Counting Beads | Thermo Fisher | 01-1234-42 |
| Busulfan | (Sigma #) | B2635-10G |
| ketamine (Vetalar, Ketaset, Ketalar) | Veterinarian supplied | N/A |
| Xylazine (Rompun) | Veterinarian supplied | N/A |
| Trimethoprim/Sulfamethoxazole | Hi-Tech Pharmacal | 50383-823-16 |
| Tamoxifen | Millipore Sigma | T5648-5G |
| Progesterone | Millipore Sigma | P0130 |
| Critical Commercial Assays | ||
| PicoPure RNA Isolation kit | ThermoFisher | KIT0204 |
| QuantSeq 3’ mRNA sequencing kit | Lexagon | N/A |
| SMART-seq v4 Ultra Low Input Kit for Sequencing | Takara | N/A |
| Deposited Data | ||
| Sequencing Data | Gene Expression Omnibus (GEO) | GSE225803 |
| Experimental Models: Organisms/Strains | ||
| C57Bl/6J | Jackson labs | IMSR_JAX:000664 |
| NR4A1−/− (C57BL/6-Rr39em1Ched /J) | Jackson labs | IMSR_JAX:030204 |
| CCR2−/− (B6.129S4-Ccr2tm1lfc /J) | Jackson labs | IMSR_JAX:004999 |
| LFA1−/− (B6.129S7-Itgal tm1.1Bll /J) | Jackson labs | IMSR_JAX:005257 |
| B6.CD45.1 (B6.SJL-PtprcaPepcb/BoyJ) | Jackson labs | IMSR_JAX:002014 |
| CX3CR1f/wtCreER.zsGFP (B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung /J | Jackson labs | IMSR_JAX:020940 |
| Zbt46.zsGFP (B6.129S6(C)-Zbtb46tm1.1Kmm /J) | Jackson labs | IMSR_JAX:027618 |
| MafBCremCherry (B6N(129S4)-Mafb tm1.1(cre)Kmm /J) | Jackson labs | IMSR_JAX:029664 |
| Software and Algorithms | ||
| FlowJo V10 | N/A | N/A |
| BBDuk version 37.22 | N/A | N/A |
| STAR47 | N/A | N/A |
| HTSeq48 | N/A | N/A |
| Trimmomatic (version 0.36),49 | N/A | N/A |
| Tophat aligner (tophat 2.1.0) | N/A | N/A |
| Gorilla50,51 | N/A | N/A |
| cellranger 3.1.0 pipeline | N/A | N/A |
| Seurat v3.1 | N/A | N/A |
| BioMart R package | N/A | N/A |
| singleR R package | N/A | N/A |
| Gene-E | N/A | N/A |
| R version 3.3.1 | N/A | N/A |
| GraphPad Prism V8 | N/A | N/A |
| Other | ||
| BD LSR II | N/A | N/A |
| BD FACSAria | N/A | N/A |
| BD Symphony | N/A | N/A |
| Nordion Cs-137 based Gammacell-40 irradiator | N/A | N/A |
ACKNOWLEDGMENTS
We would like to thank Dr. Steffen Jung for providing critical review of the manuscript. We would also like to thank the Northwestern University Lurie Cancer Center Flow Cytometry Core Facility, which is supported by NCI Cancer Center Support Grant P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center; Dr. Hiam Abdala Valencia, Director of the Next Generation Sequencing Facility for the Divisions of Rheumatology/Pulmonary and Critical Care; the Northwestern NUSeq Core Facility; and Birmingham Tissue Analytics, University of Birmingham, UK. This research was supported in part through the computational resources and staff contributions provided by the Genomics Compute Cluster, which is jointly supported by the Feinberg School of Medicine, the Office of the Provost, the Office for Research, and Northwestern Information Technology. The Genomics Compute Cluster is part of Quest, Northwestern University’s high-performance computing facility, with the purpose to advance research in genomics. S.Y.C. was supported by a predoctoral AHA award (19PRE34380200). C.M.C. was supported by the Lupus Research Alliance (Novel Research Grant), the Rheumatology Research Foundation (Innovative Research Grant), and the Northwestern University Dixon Translational Research Initiative. H.-K.M.M. was supported by KL2 TR001424, HL134375S1, and AR007611. A.V.M. was supported by NIH grants U19AI135964, P01AG049665, PO1HL154998, R56HL135124, R01HL153312, and NUCATS COVID-19 Rapid Response Grant. A.B. was supported by NIH grants HL145478, HL147290, and HL147575. G.R.S.B. was supported by NIH grants U19AI135964, P01AG049665, P01AG04966506S1, and R01HL147575 and Veterans Affairs grant I01CX001777. D.R.W. was supported by funding from the Arthritis National Research Foundation, the American Federation for Aging Research, the American Heart Association (18CDA34110224), and NIH grants AI163742 and AR080513. H.P. was supported by AR074902, AR075423, CA060553, HL134375, AR080513, an RRF Innovative Research Grant, a United States-Israel Binational Science Foundation Investigator Grant, the Precision Medicine Fund, and the Mabel Green Myers Professorship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112513.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJM, Liu YJ, MacPherson G, Randolph GJ, et al. (2010). Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–e80. 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 2.Yona S, and Jung S (2010). Monocytes: subsets, origins, fates and functions. Curr. Opin. Hematol 17, 53–59. 10.1097/MOH.0b013e3283324f80. [DOI] [PubMed] [Google Scholar]
- 3.Guilliams M, Mildner A, and Yona S (2018). Developmental and functional heterogeneity of monocytes. Immunity 49, 595–613. [DOI] [PubMed] [Google Scholar]
- 4.Mildner A, Schönheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, Paul F, Chappell-Maor L, Priller J, Leutz A, et al. (2017). Genomic characterization of murine monocytes reveals C/EBPbeta transcription factor dependence of Ly6C(−) cells. Immunity 46, 849–862.e7. 10.1016/j.immuni.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 5.Narasimhan PB, Marcovecchio P, Hamers AAJ, and Hedrick CC (2019). Nonclassical monocytes in health and disease. Annu. Rev. Immunol. 37, 439–456. 10.1146/annurev-immunol-042617-053119. [DOI] [PubMed] [Google Scholar]
- 6.Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, Farnir F, Pirottin D, Ginhoux F, Boeckxstaens G, et al. (2019). Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun 10, 3964. 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udalova IA, Mantovani A, and Feldmann M (2016). Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol 12, 472–485. 10.1038/nrrheum.2016.91. [DOI] [PubMed] [Google Scholar]
- 8.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, et al. (2014). Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 9, 591–604. 10.1016/j.celrep.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, and Illges H (2005). A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur. J. Immunol 35, 3064–3073. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs JP, Ortiz-Lopez A, Campbell JJ, Gerard CJ, Mathis D, and Benoist C (2010). Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 62, 1921–1932. 10.1002/art.27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puchner A, Saferding V, Bonelli M, Mikami Y, Hofmann M, Brunner JS, Caldera M, Goncalves-Alves E, Binder NB, Fischer A, et al. (2018). Non-classical monocytes as mediators of tissue destruction in arthritis. Ann. Rheum. Dis 77, 1490–1497. 10.1136/annrheumdis-2018-213250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinones MP, Estrada CA, Kalkonde Y, Ahuja SK, Kuziel WA, Mack M, and Ahuja SS (2005). The complex role of the chemokine receptor CCR2 in collagen-induced arthritis: implications for therapeutic targeting of CCR2 in rheumatoid arthritis. J. Mol. Med 83, 672–681. 10.1007/s00109-005-0637-5. [DOI] [PubMed] [Google Scholar]
- 13.Brunet A, LeBel M, Egarnes B, Paquet-Bouchard C, Lessard AJ, Brown JP, and Gosselin J (2016). NR4A1-dependent Ly6C(low) monocytes contribute to reducing joint inflammation in arthritic mice through Treg cells. Eur. J. Immunol 46, 2789–2800. 10.1002/eji.201646406. [DOI] [PubMed] [Google Scholar]
- 14.Liebmann M, Hucke S, Koch K, Eschborn M, Ghelman J, Chasan AI, Glander S, Schädlich M, Kuhlencord M, Daber NM, et al. (2018). Nur77 serves as a molecular brake of the metabolic switch during T cell activation to restrict autoimmunity. Proc. Natl. Acad. Sci. USA 115, E8017–E8026. 10.1073/pnas.1721049115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkling ME, Cytlak U, Lau CM, Lewis KL, Resteu A, Khodadadi-Jamayran A, Siebel CW, Salmon H, Merad M, Tsirigos A, et al. (2018). Notch signaling facilitates in vitro generation of cross-presenting classical dendritic cells. Cell Rep. 23, 3658–3672.e6. 10.1016/j.celrep.2018.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boulet S, Daudelin JF, Odagiu L, Pelletier AN, Yun TJ, Lesage S, Cheong C, and Labrecque N (2019). The orphan nuclear receptor NR4A3 controls the differentiation of monocyte-derived dendritic cells following microbial stimulation. Proc. Natl. Acad. Sci. USA 116, 15150–15159. 10.1073/pnas.1821296116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponichtera HE, Shainheit MG, Liu BC, Raychowdhury R, Larkin BM, Russo JM, Salantes DB, Lai CQ, Parnell LD, Yun TJ, et al. (2014). CD209a expression on dendritic cells is critical for the development of pathogenic Th17 cell responses in murine schistosomiasis. J. Immunol 192, 4655–4665. 10.4049/jimmunol.1400121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, and Gilroy DW (2011). Transcriptomic analyses of murine resolution-phase macrophages. Blood 118, e192–e208. 10.1182/blood-2011-04-345330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung AKL, Ko JMY, Lung HL, Chan KW, Stanbridge EJ, Zabarovsky E, Tokino T, Kashima L, Suzuki T, Kwong DLW, et al. (2011). Cysteine-rich intestinal protein 2 (CRIP2) acts as a repressor of NF-kappaB-mediated proangiogenic cytokine transcription to suppress tumorigenesis and angiogenesis. Proc. Natl. Acad. Sci. USA 108, 8390–8395. 10.1073/pnas.1101747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayan H, Farfel Z, and Karlish SJD (2018). Renal Mg handling, FXYD2 and the central role of the Na,K-ATPase. Physiol. Rep 6, e13843. 10.14814/phy2.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S, Sheng J, Hu JK, Yu W, Kishikawa H, Hu MG, Shima K, Wu D, Xu Z, Xin W, et al. (2013). Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis 16, 387–404. 10.1007/s10456-012-9322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culemann S, Grüneboom A, and Krönke G (2019). Origin and function of synovial macrophage subsets during inflammatory joint disease. Adv. Immunol 143, 75–98. 10.1016/bs.ai.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, et al. (2013). Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39, 599–610. 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, Goodman SM, Tabechian D, Hughes LB, Salomon-Escoto K, et al. (2019). Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol 20, 928–942. 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandelin AM 2nd, Homan PJ, Shaffer AM, Cuda CM, Dominguez ST, Bacalao E, Carns M, Hinchcliff M, Lee J, Aren K, et al. (2018). Transcriptional profiling of synovial macrophages using minimally invasive ultrasound-guided synovial biopsies in rheumatoid arthritis. Arthritis Rheumatol. 70, 841–854. 10.1002/art.40453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, Petricca L, Di Mario C, Bui L, Perniola S, et al. (2020). Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med 26, 1295–1306. 10.1038/s41591-020-0939-8. [DOI] [PubMed] [Google Scholar]
- 27.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, et al. (2005). CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258. 10.1126/science.1102901.307/5707/254. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann HW, Bruns T, Weston CJ, Curbishley SM, Liaskou E, Li KK, Resheq YJ, Badenhorst PW, and Adams DH (2016). Bidirectional transendothelial migration of monocytes across hepatic sinusoidal endothelium shapes monocyte differentiation and regulates the balance between immunity and tolerance in liver. Hepatology 63, 233–246. 10.1002/hep.28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watts GM, Beurskens FJM, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, and Lee DM (2005). Manifestations of inflammatory arthritis are critically dependent on LFA-1. J. Immunol 174, 3668–3675. [DOI] [PubMed] [Google Scholar]
- 31.Monach PA, Nigrovic PA, Chen M, Hock H, Lee DM, Benoist C, and Mathis D (2010). Neutrophils in a mouse model of autoantibody-mediated arthritis: critical producers of Fc receptor gamma, the receptor for C5a, and lymphocyte function-associated antigen 1. Arthritis Rheum. 62, 753–764. 10.1002/art.27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lessard AJ, LeBel M, Egarnes B, Préfontaine P, Thériault P, Droit A, Brunet A, Rivest S, and Gosselin J (2017). Triggering of NOD2 receptor converts inflammatory Ly6C(high) into Ly6C(low) monocytes with patrolling properties. Cell Rep. 20, 1830–1843. 10.1016/j.celrep.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Culemann S, Grüneboom A, Nicolás-Ávila JÁ, Weidner D, Lämmle KF, Rothe T, Quintana JA, Kirchner P, Krljanac B, Eberhardt M, et al. (2019). Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 572, 670–675. 10.1038/s41586-019-1471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, Mack M, Joshi A, Guilliams M, Mowat AMI, et al. (2016). Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat. Commun 7, ncomms11852. 10.1038/ncomms11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrer IR, West HC, Henderson S, Ushakov DS, Santos E Sousa P, Strid J, Chakraverty R, Yates AJ, and Bennett CL (2019). A wave of monocytes is recruited to replenish the long-term Langerhans cell network after immune injury. Sci. Immunol 4, eaax8704. 10.1126/sciimmunol.aax8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly LM, Englmeier U, Lafon I, Sieweke MH, and Graf T (2000). MafB is an inducer of monocytic differentiation. EMBO J. 19, 1987–1997. 10.1093/emboj/19.9.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, and Nussenzweig MC (2012). Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med 209, 1153–1165. 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med 209, 1135–1152. 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atehortúa L, Rojas M, Vásquez GM, and Castaño D (2017). Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Potential Effect of Monocyte Interaction. Mediators Inflamm. 2017, 9680729. 10.1155/2017/9680729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, Weissleder R, Mathis D, and Benoist C (2006). Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat. Immunol 7, 284–292. 10.1038/ni1306. [DOI] [PubMed] [Google Scholar]
- 42.Wipke BT, Wang Z, Kim J, McCarthy TJ, and Allen PM (2002). Dynamic visualization of a joint-specific autoimmune response through positron emission tomography. Nat. Immunol 3, 366–372. [DOI] [PubMed] [Google Scholar]
- 43.Cloutier N, Paré A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, and Boilard E (2012). Platelets can enhance vascular permeability. Blood 120, 1334–1343. 10.1182/blood-2012-02-413047. [DOI] [PubMed] [Google Scholar]
- 44.Angelovich TA, Hearps AC, Maisa A, Kelesidis T, and Jaworowski A (2017). Quantification of monocyte transmigration and foam cell formation from individuals with chronic inflammatory conditions. J. Vis. Exp 10.3791/56293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, and Geissmann F (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670. 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 46.Suchard SJ, Stetsko DK, Davis PM, Skala S, Potin D, Launay M, Dhar TGM, Barrish JC, Susulic V, Shuster DJ, et al. (2010). An LFA-1 (alphaLbeta2) small-molecule antagonist reduces inflammation and joint destruction in murine models of arthritis. J. Immunol 184, 3917–3926. 10.4049/jimmunol.0901095. [DOI] [PubMed] [Google Scholar]
- 47.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eden E, Lipson D, Yogev S, and Yakhini Z (2007). Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol 3, e39. 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 10, 48. 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication using accession code: GSE225803.
This paper analyzes published data that were reported in the original article or obtained directly from the authors.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.