

Abstract
The small size of ciliary structures that underlies photoreceptor function and inherited ciliopathies requires imaging techniques adapted to visualizing them at the highest possible resolution. In addition to powerful super-resolution imaging modalities, emerging approaches to sample preparation, including expansion microscopy (ExM), can provide a robust route to imaging specific molecules at the nanoscale level in the retina. We describe a protocol for applying ExM to whole retinas in order to achieve nanoscale fluorescence imaging of ciliary markers, including tubulin, CEP290, centrin, and CEP164. The results are consistent with those from other super-resolution fluorescence techniques and reveal new insights into their arrangements with respect to the subcompartments of photoreceptor cilia. This technique is complimentary to other imaging modalities used in retinal imaging, and can be carried out in virtually any laboratory, without the need for expensive specialized equipment.
Keywords: Expansion microscopy, Connecting cilium, CEP290, Photoreceptor, Super-resolution
1. Introduction
In recent years the structure of the mouse rod photoreceptor cilium has been defined at an ever-increasing level of detail using electron microscopic and super-resolution fluorescence techniques. The tightly packed submicron structures of rod photoreceptor sensory cilia require optimized staining procedures and super-resolution for accurate localization analyses. 2D stochastic optical reconstruction microscopy (STORM) has been used to localize centrin to the lumen of the CC, surrounded by microtubules and mobile components of the CC, like intraflagellar transport (IFT) and BBSome proteins [1]. Comparative studies using super-resolution fluorescence localization have demonstrated differences in subcellular localization of ciliary proteins in retinal ciliopathy mutants, like Spata7−/− and BBS protein subunit knockout mouse mutants (Bbs2−/−, Bbs4−/−, and Bbs7−/−) [1, 2]. A combination of both structured illumination microscopy (SIM) and STORM was recently used to localize CEP290 along the length of the CC, in comparison with other ciliary proteins like NPHP8 [3].
Expansion microscopy (ExM) [4, 5] is a technique that allows for fluorescence-based imaging, on standard microscopes, at nanoscale resolution. It can be combined with SIM, STORM, and other nanoscopic techniques to potentially improve the spatial resolution achievable beyond the capabilities of each technique alone. ExM is based on the principle of embedding cells and tissues in highly hygroscopic gels polymerized from mixtures of acrylamide and acrylate, which absorb water, allowing the gel to expand [5]. Proteins are cross-linked to the gel, and thus structures containing proteins expand with the gel (Fig. 1). The technique has been adapted and optimized by many different labs over the years, and used for the imaging of a number of ciliary and centriolar structures in cellular cultures [6–13], and very recently for the cilia in retinal tissue [14]. This technique is adaptable to whichever tissue is of interest, and the extent of expansion can be scaled [15]. The materials and instruments used for this technique are relatively accessible as compared to other super-resolution methods.
Fig. 1.
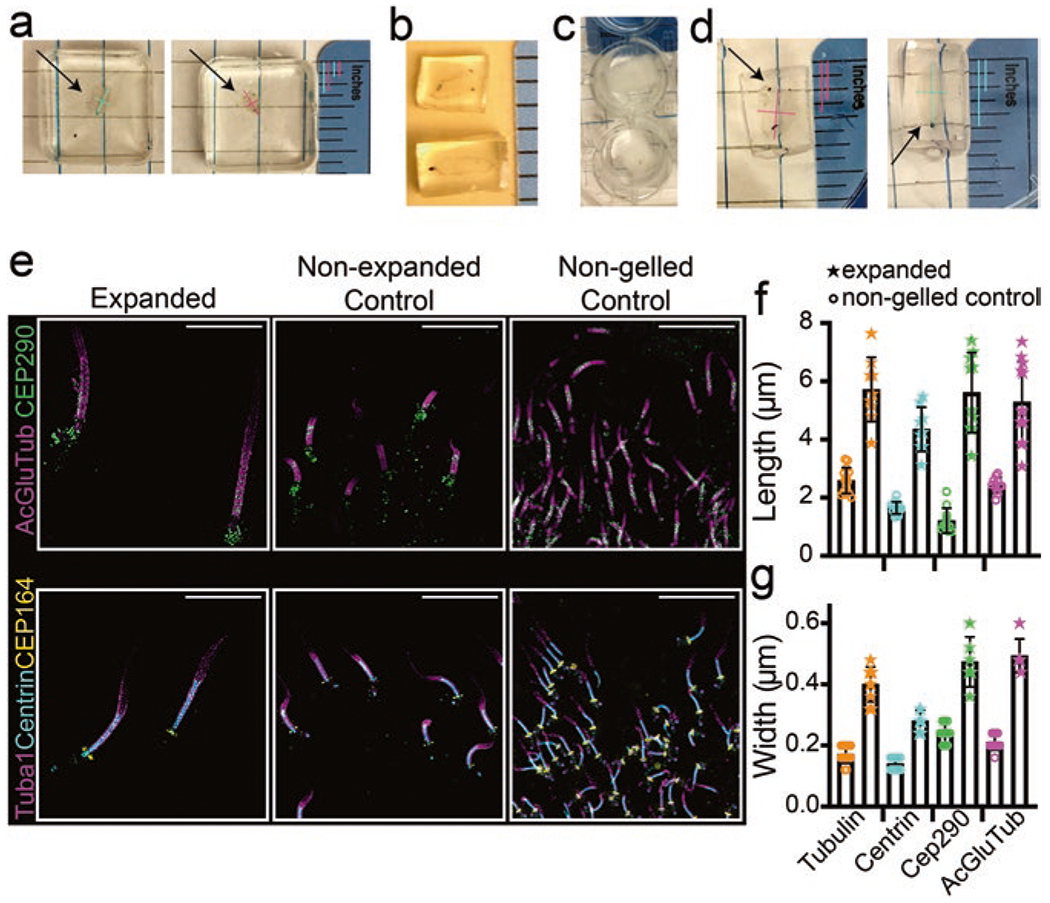
Sample preparation during expansion protocol. (a–d) Display examples of the retina in different steps of the protocol, in order: (a) retina immediately after gelation; (b) retina post-disruption, trimmed and slightly expanded; (c) retina after overnight incubation with primary antibodies (opacity subsides once they warm to RT); and (d) orientation of the retinas for embedding in OCT. Arrows point to retinas within the gels, and colored lines display length and width measurements to show that slight expansion of the retinas during disruption and subsequent steps is, by eye, isotropic. Gelled retinas ~0.08 inches wide, ~0.17 inches long; immediately preceding freezing, retinas ~0.13–0.17 inches wide, and ~0.23–0.27 inches long (~0.08 inch changes in each direction). (e) SIM z-projection images displaying cilia, compared in 512 × 512 pixel images, between an expanded section, a non-expanded control section, and a non-gelled control section stained for the antibodies indicated. Scale bar 5 μm. (f, g) Dot plots with averages and standard deviations of the lengths and widths of staining in connecting cilia pre- and post-expansion
Here we describe an expansion method optimized for visualizing photoreceptor cilia in mouse retina. It can be used with many microscopy modalities. As an example, we have used this modified ExM protocol to improve localization of CEP290 along the microtubules of the connecting cilium (CC) axoneme, and have confirmed and improved localization detail for ciliary proteins CEP164 and centrin (Figs. 1 and 2).
Fig. 2.
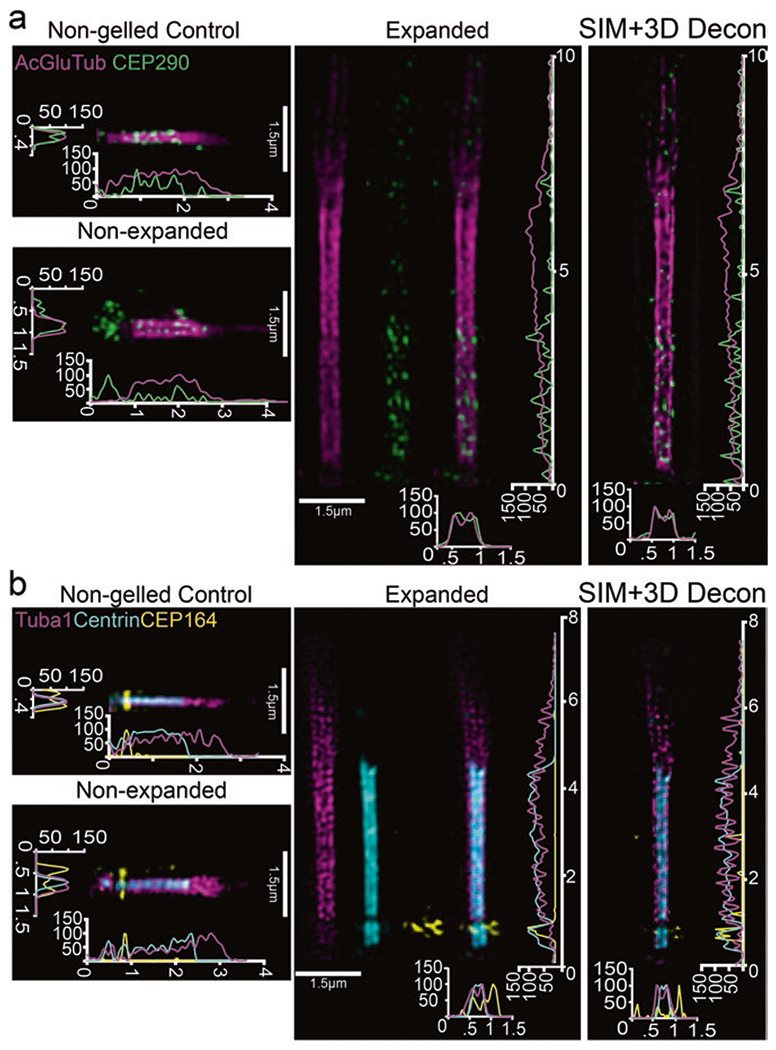
Localization of ciliary markers in connecting cilium following expansion in whole retina. (a, b) SIM z-projection images of single, straightened cilia stained for either (a) acetylated alpha-tubulin, glutamylated-tubulin, and CEP290 or (b) tubulin, centrin, and CEP164 with their corresponding controls. SIM z-projections with 3D deconvolution are shown to the right of each panel. On the axes, numbers less than 10 represent μm
2. Materials and Methods
2.1. Solutions Needed
Ames’ Medium [16] (Sigma A1420)
Ames’ Block Buffer:
15% (v/v) normal goat serum (NGS) (Fitzgerald 88R-NG001) + 5% (w/v) Bovine serum albumin (BSA) (Sigma A9647) + 0.5% (w/v) BSA-c (Aurion 25557) + 5% (w/v) Fish skin gelatin (Sigma G7765) + 1x protease inhibitor cocktail (GenDepot P3100) + 0.2% (w/v) saponin (Calbiochem 558255); remaining volume is 1x Ames’ Medium
Ames’ Wash Buffer:
2% (v/v) NGS in 1x Ames’
Ames’ Secondary Buffer:
2% (v/v) NGS in 1x Ames’ + 1x protease inhibitor cocktail + 0.2% (w/v) saponin
Phosphate buffered saline (PBS), 1x:
144 mg/L KH2PO4, 9000 mg/L NaCl, 421.62 mg/L Na2HPO4, pH = 7.4
PBS Block Buffer:
15% NGS + 5% BSA + 0.5% BSA-c + 5% Fish skin gelatin + 0.2% saponin + 0.01% (w/v) Na-Azide in 1xPBS
PBS Wash Buffer:
2% NGS in 1xPBS
2.2. Gelation Solution
2 M acrylamide (Fisher, BP1402-1), 300 ppm (ug/ml) N,N’-methylene-bis (acrylamide) (Sigma, 146072), 1.1 M Na-Acrylate (Santa Cruz Biotech. sc-236893), 1.5 ppt (mg/ml) ammonium persulfate and tetramethylethylenediamine (TEMED) each, in 1xPBS
Disruption Buffer:
50 mM Tris-Cl pH 7.5, 5% sodium dodecyl sulfate (SDS), 200 mM NaCl
2.3. Method
Unless otherwise noted, all incubation steps at RT and 4 °C were performed with the samples rocking and covered. Note that there are three sequential cycles of incubation with primary antibodies, followed by secondary antibodies, before and after fixation, and before and after expansion, a procedure we have found to enhance sensitivity of immunostaining.
Day 1
We dissected the retinas into ice-cold Ames’ media in 35 mm petri dishes, on ice, and then transferred them via a wide-bore pipette tip into hydrophobic microcentrifuge tubes (GeneMate, C-3302-1) filled with 1 mL of Ames Block, blocked for 2 h. The tubes were positioned so as to allow the retinas to float around in the tube with gentle agitation. We added primary antibodies (5–10 μg each) directly to the block buffer and left the retinas to rock overnight.
Day 2
We washed the retinas, gently, with Ames Wash Buffer six times, 10 min each, on ice. After the last wash, we added Secondary Buffer and 2°ary antibodies and left the retinas to incubate for 2 h. We then washed the retinas with Ames Wash six times, 5 min each, on ice. After rinsing with 1xPBS to remove serum from the wash, we fixed the retinas with 4% (w/v) PFA in 1xPBS, for 20 min, and then quenched with 100 mM glycine HCl in PBS, for 30 min at RT. After one rinse in 1xPBS, we then blocked the retinas for 2 h in PBS Block Buffer at RT. Finally, we added fresh primary antibodies directly to the tubes and stained for 2 full days.
Day 5
We washed the retinas in PBS Wash six times, 10 min each, on ice. We added a fresh 1 mL of PBS Wash Buffer to each tube, with the corresponding 2°ary antibodies, and incubated overnight.
Day 6
We washed the retinas in PBS Wash six times, 5 min each, on ice. We then postfixed the retinas with 3% PFA in PBS for 1 h at RT, followed by quenching with 100 mM glycine HCl in PBS for 30 min at RT. After rinsing in PBS, we carefully cut a small piece of each retina to serve as a non-gelled control. We quickly transferred these pieces to tubes filled with 30% sucrose (made in 1xPBS), added 1:1 30% sucrose-optimal cutting temperature (OCT, Tissue-Tek), and flash froze them in N2(l). We transferred the rest of the retina to a 0.5 dram glass vial containing 1 mL of 0.1 mg/ml acryloyl-X SE (6-((acryloyl)amino) hexanoic acid, succinimidyl ester, Thermo A20770; for cross-linking proteins to hydrogel), diluted in PBS, and left them to incubate on a roller or rocker for 24 h at RT:
If there is no access to a roller, a rocker can be used as before.
Regular eyecup cryosections can also be used for the non-gelled control.
Incubating for less time in Acryloyl-X SE has been used in other protocols expanding cultured cells, but we found a full day of incubation to be best for overall incorporation of acryloyl-X into the entire retinal tissue.
Day 7
Just before use, we prepared gelation solution (1.5 mL per retina) and added 1 mL per retina to a cryomold (Ted Pella, 27147-2), on ice. After rinsing retinas three times in PBS, we placed them in the center of the cryomold to incubate, shaking gently on ice, at 4 °C. We then added the remaining gelation solution to the cryomold before placing a glass coverslip on top (avoiding bubbles) and incubating for 2 h in a humidified 37 °C incubator to induce gelation. After cooling, we gently popped the gels out of the cryomolds using a pipette tip and trimmed excess gel from the retinas (Fig. 1a). We placed each gel into a 15 mL conical tube with 10 mL of disruption buffer (covered with foil), and incubated in an 80 °C chamber for 3 h. We washed the gels in water once and PBS several times to remove SDS (Fig. 1b), then transferred them to wells of a new 24-well plate containing PBS + 0.1% Tween 20 (PBST). We washed the retinas in PBST two times, 30 min each, at RT, then added fresh primary antibodies, and incubated overnight:
Although previous expansion methods call for 3 h of gelation time, we found 2 h to be sufficient.
We found less isotropic expansion and decreased antibody labeling when we used digestion rather than denaturation.
Day 8
We washed the retinas in PBST three times, 20 min each, at RT. We added fresh 1 mL of PBST and 2°ary antibodies to the retinas, and stained them for three and a half hours at RT. DAPI was added at a 1:500 dilution for at least 1 h. After washing the retinas in PBST six times, 15 min each, at RT, we washed the retinas in 30% (w/v) sucrose (prepared in 1xPBS) three times, 15 min each. We then transferred the retinas to 15 ml conical tubes filled with 10 mL of 30% sucrose. Once the retinas sank to the bottom of the tube, we transferred them to a petri dish filled with OCT and swirled them to remove excess sucrose from the gel block, and trimmed the gels if possible. We then immediately transferred the retinas to a cryomold filled with fresh OCT and froze them by placing the cryomolds on a flat surface in a −80 °C freezer:
If the retinas are left in 30% sucrose overnight, the structure of the gel changes slightly. Fluorescence stays intact, but better to not leave it overnight.
During mounting, the gel was pushed to the bottom of the mold for longitudinal sectioning and the mold marked to indicate the gel position.
2.4. Sectioning
Within 3 days after freezing, we cut sections. We placed the cryomolds in a microtome cryochamber set to −27–35 °C and left them to warm for ~30 min. In advance we filled multiple 50 mL beakers with Milli-Q water, prepared coverslips and slides by placing a ~50 ul drop of water on each, and left a thin paintbrush out at RT. We then collected sections onto a Superfrost Plus slide (Fisher, 12-550-15) to check for fluorescence, mounted in ProLong Glass mounting medium (Thermo Fisher P36980), and used those as non-expanded controls. Once the area of interest was reached, we cut 10 μm (or less) sections. We used the RT brush to transfer the section to the glass beaker by dipping the brush in water, lightly touching it to the section, and transferring the section directly into the beaker filled with water. We left the sections to expand for 10 min, covered. We used a loop tool (EMS, 70922-03, or a bigger loop can be made using a wooden stick and silver wire) to transfer the expanded sections from the beaker onto the drop of water on a #1.5 glass coverslip. After wicking away excess water with a Kimwipe (Kimberly-Clark), we mounted the coverslip on the drop of water on a glass slide. We again used a Kimwipe to wick away excess water. We used a fluorescent microscope to choose the best sections for imaging. After carefully sealing the coverslip edges with scotch tape, we marked the positions of the sections with a sharpie. To reduce drying we kept them at 4 °C, protected from light, until and imaged within the day, except for controls which we imaged later that week:
The rest of the block can be stored in −80 °C, but fluorescence signal may decrease over time.
We tested leaving the sections in water longer than 10 min, but the expansion factor did not increase.
Placing the section directly in the water is the best way to get isotropic expansion of the section. Drying it on a coverslip first and then expanding that in water did not produce reliable, consistent expansion.
2.5. Animals
Wild-type mice used for this study were C57BL/6 aged 2–4 months. All procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee and adhered to the Association of Research in Vision and Ophthalmology (ARVO) guidelines for the humane treatment and ethical use of animals for vision research.
2.6. Antibodies
The following commercial antibodies were used: CEP290 (C-terminus), Bethyl Laboratory, A301-659A; acetylated α-tubulin, Santa Cruz, sc-23950; Centrin, pan-centrin antibody from EMD Millipore, 04-1624, raised against Chlamydomonas centrin and reported to recognize all mouse centrin isoforms 1-4 (41, 84); CEP164, Proteintech, 22227-1-AP; CEP164, Santa Cruz, sc-515403; MKS3 (cultured cells) Proteintech, 13975-1-AP; Atto488-Tuba1, Antibodies-online.com, ABIN1169085. Antibodies were used at a concentration of 10 μg/mL, with the exception of Atto488-Tuba1 which was used at a concentration of 7.5 μg/mL. Secondary antibodies used were F(ab’)2-goat anti-rabbit IgG Alexa 488, F(ab’)2-goat anti-mouse IgG Alexa 555, and F(ab’)2-goat anti-mouse IgG Alexa 647 (Thermo Fisher, 8 μg each).
3. Results
We combined our whole retina immunostaining technique developed for STORM [1], with two expansion protocols: whole tissue ExM protocol developed for expanding Drosophila synaptonemal complex during meiosis [17, 18] and further optimized with insights from an ExM method termed TREx (tenfold robust expansion microscopy) that was optimized for both cells and whole tissue [15]. We found that by eye the whole retina expanded isotropically in the gels (Fig. 1). Although cryosections were fragile, the integrity of the retinal tissues was maintained sufficiently to identify correctly localized fluorescently labeled cilia (in reference to DAPI and non-ciliary tubulin staining) for SIM. Though samples were relatively lightly fixed (3% or 4% PFA for a total of 80 min), fixation could affect the sensitivity of some antibodies, which should be taken into account while optimizing this protocol for other antibodies/cells.
With SIM, we collected z-stacks that revealed rod photoreceptor cilia immuno-labeled for post-translationally modified tubulin (acetylated and glutamylated), CEP290, CEP164, centrin, and tubulin that were significantly larger and more expanded than control, unexpanded cilia, as well as control non-gelled retina, imaged with the same settings (Fig. 2a). Reconstructions were performed in Softworx 7 software, and 3D deconvolution of SIM reconstructed z-stacks was performed using Nikon NIS-Elements software. We performed the majority of the analysis thereafter in FIJI/ImageJ. We calculated the expansion factor at the single cilium level by taking ROIs of individual cilia, digitally straightening them, and then taking the row-average intensities, which plots the average intensity across the width of the ROI for each row of pixels along the length of the ROI. Once converted to nm for accurate scaling, these profiles were used to identify the edges of antibody labeling (33% of the maximum intensity value). All measurements were made in a 1.5-μm-wide ROI. We achieved a ~ four-fold expansion factor for length and width (Fig. 1f, g). GraphPad Prism 7 was used to create the graphs. The staining patterns for CEP290, centrin, and CEP164 are all comparable to previously published, non-expanded connecting cilia staining [3, 19, 20], but more detailed analysis is in progress.
4. Discussion
We demonstrate in this report that ExM is a useful super-resolution fluorescence tool for localizing proteins in the rod photoreceptor cilium. In the future, the technique could be applied to localize other critical components of the cilium.
Although ExM is useful for cilia cytoskeleton labeling with reasonably isotropic expansion, there are some potential drawbacks. Some 2°ary antibodies do not work well; as seen in Fig. 2, CEP164 staining in the expanded cilia is less well-maintained than in non-expanded retina, or when compared to the staining under non-gelling protocols [3], possibly due to problems with the Alexa 647-labeled secondary [21]. It remains to be determined which structures expand isotropically. Membranes and protein structures must be disrupted in order to allow for complete gel embedding and subsequent expansion. The gel composition and protein disruption techniques used by different labs vary widely, depending on the structure and/or proteins of interest. Bioactive lipid membranes, such as the inner segment and CC plasma membrane, post-Golgi secretory vesicles, and OS membrane discs, may be adversely affected by the high temperatures used in the protocol. It seems likely there is some anisotropy in the expansion, with some structures expanding more in one direction than in another, but further analysis and possibly further protocol modifications will be required. Improvement in further spatial resolution will require either increasing the expansion factor or by imaging with a more powerful super-resolution microscope like STED or STORM. Such changes will also require additional adaptations to the protocol. Nonetheless, even in its current state, ExM represents a powerful tool for localizing proteins within photoreceptor cilia.
Acknowledgments
This work was supported by NIH grant R01-EY26545 (TGW), F32-EY027171 (MAR), T32-EY007102 (ARM), and F32-EY031574 (ARM) and a grant from the Knights Templar Eye Foundation (MAR). 3D deconvolution analysis was performed in the West Virginia University Microscope Imaging Facility, which has been supported by the WVU Cancer Institute and NIH grants P20RR016440, P30GM103488, and U54GM104942.
Abbreviations
- BBS
Bardet-Biedl syndrome
- BSA
bovine serum albumin
- CC
connecting cilium
- DAPI
4′,6-diamidino-2-phenylindole
- ExM
expansion microscopy
- IFT
intraflagellar transport
- NGS
normal goat serum
- OCT
optimal cutting temperature
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- RT
room temperature
- SDS
sodium dodecyl sulfate
- SIM
structured illumination microscopy
- STORM
stochastic optical reconstruction microscopy
- TEMED
tetramethylethylenediamine
Footnotes
Conflict of Interest The authors have declared that no conflicts of interest exist.
Contributor Information
Abigail R. Moye, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA; Department of Genetics and Ophthalmology, Institute of Molecular and Clinical Ophthalmology Basel, Basel, Switzerland
Michael A. Robichaux, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA; Department of Ophthalmology & Visual Sciences and Department of Biochemistry & Molecular Medicine, West Virginia University, Morgantown, WV USA
Theodore Wensel, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA.
References
- 1.Robichaux MA, Potter VL, Zhang Z, He F, Liu J, Schmid MF, et al. Defining the layers of a sensory cilium with STORM and cryoelectron nanoscopy. Proc Natl Acad Sci U S A. 2019;116(47):23562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dharmat R, Eblimit A, Robichaux MA, Zhang Z, Nguyen TT, Jung SY, et al. SPATA7 maintains a novel photoreceptor-specific zone in the distal connecting cilium. J Cell Biol. 2018;217(8):2851–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potter VL, Moye AR, Robichaux MA, Wensel TG. Super-resolution microscopy reveals photoreceptor-specific subciliary location and function of ciliopathy-associated protein CEP290. JCI Insight. 2021;6(20):e145256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallagher BR, Zhao Y. Expansion microscopy: a powerful nanoscale imaging tool for neuroscientists. Neurobiol Dis. 2021;154:105362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Tillberg PW, Boyden ES. Optical imaging. Expansion microscopy. Science. 2015;347(6221):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambarotto D, Zwettler FU, Le Guennec M, Schmidt-Cernohorska M, Fortun D, Borgers S, et al. Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nat Methods. 2019;16(1):71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Guennec M, Klena N, Gambarotto D, Laporte MH, Tassin AM, van den Hoek H, et al. A helical inner scaffold provides a structural basis for centriole cohesion. Sci Adv. 2020;6(7):eaaz4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katoh Y, Chiba S, Nakayama K. Practical method for superresolution imaging of primary cilia and centrioles by expansion microscopy using an amplibody for fluorescence signal amplification. Mol Biol Cell. 2020;31(20):2195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong D, Loncarek J. Analyzing centrioles and cilia by expansion microscopy. Methods Mol Biol. 2021;2329:249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahabandu N, Kong D, Magidson V, Nanjundappa R, Sullenberger C, Mahjoub MR, et al. Expansion microscopy for the analysis of centrioles and cilia. J Microsc. 2019;276(3):145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gambarotto D, Hamel V, Guichard P. Ultrastructure expansion microscopy (U-ExM). Methods Cell Biol. 2021;161:57–81. [DOI] [PubMed] [Google Scholar]
- 12.Hamel V, Guichard P. Improving the resolution of fluorescence nanoscopy using post-expansion labeling microscopy. Methods Cell Biol. 2021;161:297–315. [DOI] [PubMed] [Google Scholar]
- 13.Zwettler FU, Reinhard S, Gambarotto D, Bell TDM, Hamel V, Guichard P, et al. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat Commun. 2020;11(1):3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercey O, Kostic C, Bertiaux E, Giroud A, Sadian Y, Chang N, et al. The connecting cilium inner scaffold provides a structural foundation to maintain photoreceptor integrity. Plos Bio. 2022;20(6): e3001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damstra HGJ, Mohar B, Eddison M, Akhmanova A, Kapitein LC, Tillberg PW. Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). bioRxiv. 2021:2021.02.03.428837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ames A 3rd, Nesbett FB. In vitro retina as an experimental model of the central nervous system. J Neurochem. 1981;37(4):867–77. [DOI] [PubMed] [Google Scholar]
- 17.Cahoon CK, Yu Z, Wang Y, Guo F, Unruh JR, Slaughter BD, et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proc Natl Acad Sci U S A. 2017;114(33):E6857–E66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Yu Z, Cahoon CK, Parmely T, Thomas N, Unruh JR, et al. Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat Protoc. 2018;13(8):1869–95. [DOI] [PubMed] [Google Scholar]
- 19.Yang TT, Chong WM, Wang WJ, Mazo G, Tanos B, Chen Z, et al. Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat Commun. 2018;9(1):2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi X, Garcia G 3rd, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, et al. Erratum: super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol. 2017;19(11):1379. [DOI] [PubMed] [Google Scholar]
- 21.Tillberg PW, Chen F, Piatkevich KD, Zhao Y, Yu CC, English BP, et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat Biotechnol. 2016;34(9):987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]