

Abstract
INTRODUCTION:
Tissue plasminogen activator (tPA) is a serine protease that initiates fibrinolysis to remove excessive blood clots and restore blood flow. Intravenous infusion of recombinant tPA is approved as a thrombolytic therapy in thrombotic cardiovascular diseases, including ischemic stroke and myocardial infarction. Low plasma tPA activity is associated with a higher risk of atherosclerotic cardiovascular disease and atherogenic apolipoprotein B (apoB)–lipoprotein cholesterol levels in humans. However, whether low tPA elevates apoB-lipoprotein cholesterol is unknown.
RATIONALE:
Given the central role of hepatocytes in apoB-lipoprotein production and recent studies showing that hepatocytes synthesize tPA, we sought to identify possible links between tPA and apoB-lipoprotein assembly and secretion from hepatocytes.
RESULTS:
To investigate the role of hepatocyte tPA in apoB-lipoprotein metabolism, tPA expression was silenced or deleted in the hepatocytes of mice. These manipulations resulted in higher plasma apoB and cholesterol levels, independent of any changes in hepatic low-density lipoprotein receptor (LDLR) or apolipoprotein E (apoE) expression or Apob mRNA level. The higher plasma cholesterol in these mice was distributed in the very-low-density lipoprotein (VLDL) and low-density lipoprotein (LDL) fractions. In human primary hepatocytes, silencing tPA increased the secretion of newly synthesized [3H]-labeled apoB despite no change in Apob mRNA. Thus, hepatocyte-tPA deficiency increases the secretion of apoB-lipoproteins.
Lipidation is a key factor determining the fate of intrahepatic apoB. Poorly lipidated apoB undergoes intracellular degradation, whereas fully lipidated apoB is efficiently secreted as VLDL particles with larger size and lower density. Silencing hepatocyte tPA in Ldlr−/− mice led to larger VLDL particles in the plasma, with more triglyceride per VLDL particle, indicating increased intrahepatic apoB lipidation. apoB is lipidated in the endoplasmic reticulum (ER) by microsomal triglyceride transfer protein (MTP), which incorporates neutral lipids onto nascent apoB. Transfecting human primary hepatocytes with a plasmid encoding tPA led to lower secretion of newly synthesized [3H]-labeled apoB. Proximity ligation, confocal imaging, and immunoprecipitation assays revealed that tPA interacts with apoB in the hepatocyte ER. In addition, recombinant tPA interacts with solid-phase immobilized LDL, inhibits MTP-apoB interaction, and reduces neutral lipid transfer to apoB. Moreover, the serine protease inactive tPA (S513A) also interacts with solid-phase immobilized LDL, reducing both apoB secretion by human primary hepatocytes and MTP-mediated lipid transfer activity to the same degree as wild-type tPA, which indicates that this action of tPA is independent of its protease activity. The tPA-LDL interaction is inhibited by antibodies against the Kringle 2 (K2) domain of tPA, the MTP-interacting regions at the N terminus of apoB, and the lysine analog tranexamic acid. Further, deleting the K2 domain or mutating the lysine-binding site in the K2 domain of tPA abrogates the effects of tPA on limiting apoB secretion. These data indicate that tPA, partially through the lysine-binding site on its K2 domain, binds to the N terminus of apoB, blocking the interaction between apoB and MTP in hepatocytes. This process reduces VLDL assembly and plasma apoB-lipoprotein cholesterol levels.
Plasminogen activator inhibitor 1 (PAI-1) is a rapidly acting serine protease inhibitor (serpin) of tPA. Upon lipid loading of hepatocytes, PAI-1 forms a complex with tPA and sequesters tPA away from apoB, which allows apoB to be lipidated and facilitates VLDL assembly and secretion. Consistent with these findings, humans with PAI-1 deficiency have smaller VLDL particles and lower plasma levels of apoB-lipoprotein cholesterol.
CONCLUSION:
The findings in this study suggest a mechanism that fine-tunes VLDL assembly through intracellular interactions among tPA, PAI-1, and apoB in hepatocytes, thereby affecting the plasma levels of atherogenic apoB-lipoproteins. Knowledge of this mechanism of hepatic lipoprotein regulation may suggest therapeutic strategies for lowering atherogenic apoB-lipoproteins and cardiovascular risk.
Graphical Abstract
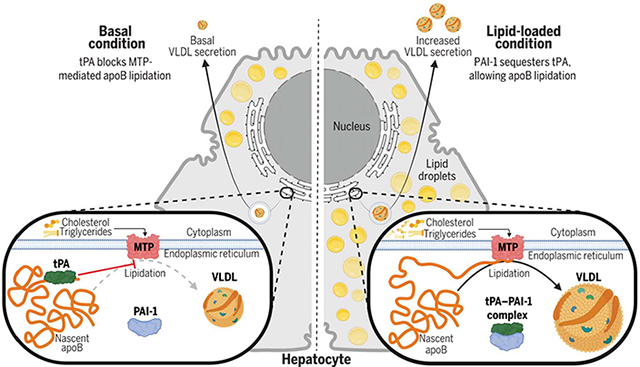
Intracellular tPA–PAI-1 interaction in the ER of hepatocytes determines apoB lipidation, VLDL assembly, and secretion. In the basal state, tPA interacts with apoB and inhibits MTP-apoB interaction in the ER of hepatocytes, limiting MTP-mediated apoB lipidation, VLDL assembly, and secretion. When hepatocytes are loaded with lipid, PAI-1 sequesters tPA away from apoB by forming a complex with tPA, allowing the lipidation of apoB and the assembly and secretion of VLDL. [Figure created with Biorender]
Apolipoprotein B (apoB)–lipoproteins initiate and promote atherosclerotic cardiovascular disease. Plasma tissue plasminogen activator (tPA) activity is negatively associated with atherogenic apoB-lipoprotein cholesterol levels in humans, but the mechanisms are unknown. We found that tPA, partially through the lysine-binding site on its Kringle 2 domain, binds to the N terminus of apoB, blocking the interaction between apoB and microsomal triglyceride transfer protein (MTP) in hepatocytes, thereby reducing very-low-density lipoprotein (VLDL) assembly and plasma apoB-lipoprotein cholesterol levels. Plasminogen activator inhibitor 1 (PAI-1) sequesters tPA away from apoB and increases VLDL assembly. Humans with PAI-1 deficiency have smaller VLDL particles and lower plasma levels of apoB-lipoprotein cholesterol. These results suggest a mechanism that fine-tunes VLDL assembly by intracellular interactions among tPA, PAI-1, and apoB in hepatocytes.
Editor’s summary
Apolipoprotein B (apoB) is a protein that carries lipids and cholesterol. Lipoproteins composed of apoB and its lipid cargo play a key role in the pathogenesis of atherosclerosis. Tissue plasminogen activator (tPA), an anticoagulant protein that can be used to break down a blood clot causing a heart attack or stroke, inversely correlates with the amount of cholesterol-loaded apoB lipoproteins, but it was not clear why this inverse correlation exists. Dai et al. discovered that in both mouse and human cells, tPA produced by the liver binds directly to apoB, preventing it from being loaded with lipids and assembled into lipoproteins. Conversely, an inhibitor of tPA has the opposite effect, binding up free tPA and preventing its interaction with apoB. —Yevgeniya Nusinovich
Apolipoprotein B (apoB)–lipoproteins initiate and promote atherosclerotic cardiovascular disease (CVD) (1, 2). The major source of blood apoB-lipoproteins is hepatocytes, where very-low-density lipoproteins (VLDLs) are assembled and secreted. Circulating VLDL is then progressively hydrolyzed in the blood to form intermediate-density lipoprotein (IDL) and low-density lipoprotein (LDL) (2). Currently recommended lipid-intervention therapies to prevent CVD primarily use statins or protein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, both of which lower LDL by enhancing hepatic LDL receptor (LDLR)–mediated LDL clearance (3). However, these treatments have only a modest effect on other atherogenic apoB-containing lipoproteins, such as VLDL and IDL (4, 5), which contribute to the residual CVD risk in populations with well-controlled LDL cholesterol (6, 7). Thus, therapies that inhibit hepatic VLDL production might be useful in decreasing CVD risk because they would lower all atherogenic apoB-lipoproteins. apoB, a large complex protein, is the structural scaffold for forming VLDL (8). VLDL is assembled in hepatocytes by incorporating triglyceride, cholesteryl ester, and phospholipid onto apoB to form spherical particles (9). This process, known as apoB lipidation, depends on both lipid availability and a neutral lipid transporter, microsomal triglyceride transfer protein (MTP) (10, 11). MTP binds to apoB in the hepatocyte endoplasmic reticulum (ER) and transfers lipids to apoB (10). When MTP and/or lipids are unavailable, VLDL cannot be assembled, and newly translated apoB is then targeted for degradation (12). Although the essential role of MTP in apoB lipidation has been well established, much less is known about the regulation of apoB-MTP interaction and MTP-mediated lipid transfer to apoB.
Previous studies have shown that low plasma tissue plasminogen activator (tPA) activity is associated with a higher risk of atherosclerotic CVD (13-15), but whether decreased fibrinolysis contributes to CVD in this setting remains unknown. Another plausible mechanism linking low tPA to CVD is elevated plasma apoB-lipoprotein cholesterol, which is seen in humans with decreased tPA activity (16-18). However, whether apoB-lipoprotein metabolism and tPA are causally linked and, if so, the mechanisms involved remain unknown. Given the central role of hepatocytes in apoB-lipoprotein production and our recent studies in mice showing that hepatocytes synthesize tPA and contribute ~40 to 50% to both the plasma basal tPA concentration and to fibrinolysis (19, 20), we sought to identify possible links between hepatocyte tPA and apoB-lipoprotein assembly and secretion. Our investigation revealed that endogenous hepatocyte tPA limits VLDL assembly by directly interacting with apoB, interrupting the interaction between apoB and MTP and thereby impairing MTP-dependent neutral lipid transfer and apoB lipidation. We also show that plasminogen activator inhibitor 1 (PAI-1) binds to tPA within hepatocytes and abolishes the effect of tPA on limiting VLDL assembly. Our in vivo data suggest that the tPA–PAI-1 interaction enables physiologic postprandial VLDL assembly. Thus, our findings that tPA can alter the production of atherogenic apoB-lipoproteins have potential therapeutic implications for lowering atherosclerotic CVD risk.
Lowering hepatocyte tPA raises plasma apoB
To investigate the role of hepatocyte tPA in determining plasma apoB-lipoprotein levels in a setting where hepatic clearance by the LDL receptor is not a factor, tPA expression was silenced in the hepatocytes of Ldlr−/− mice fed a high-fat, high-cholesterol Western diet (WD). This silencing was accomplished by administering an adenovirus-associated virus 8 (AAV8) expressing a hairpin RNA against Plat mRNA, which encodes tPA protein, driven by the H1 promoter, AAV8-H1-shPlat (sh-tPA) (19, 20). The hepatocyte tPA–silenced mice showed a 47% higher plasma total cholesterol level (P < 0.01) and 28% higher plasma apoB-100 (P < 0.05) compared with mice receiving AAV8-H1-scrambled RNA (scr) (Fig. 1A and fig. S1A). Plasma lipoprotein fraction profiling by fast protein liquid chromatography (FPLC) revealed that the hepatocyte tPA–silenced mice had more cholesterol and apoB-100 in the VLDL and LDL fractions and more triglyceride in VLDL (Fig. 1A). Using a different hypercholesterolemic model, we showed that silencing hepatocyte tPA in WD-fed Apoe knockout (Apoe−/−) mice led to 56% higher plasma total cholesterol (P < 0.05) and 28% higher apoB-100 (P < 0.05) accompanied by increases in VLDL and LDL cholesterol, triglyceride, and apoB-100 (Fig. 1B and fig. S1B). Similar results were observed in WD-fed wild-type (WT) mice (fig. S1C and fig. S2). To further validate the role of hepatocyte tPA in determining plasma apoB-lipoprotein levels, the hepatocyte tPA–knockout mouse model was generated by administering Platfl/fl mice with an AAV8 expressing a Cre recombinase driven by the thyroxine-binding globulin (TBG) promoter, AAV8-TBG-cre (Cre). The WD-fed hepatocyte tPA–knockout mice showed a 30% higher plasma total cholesterol level (P < 0.01) and 33% higher plasma apoB-100 level (P < 0.05), accompanied by higher cholesterol and apoB-100 in the VLDL and LDL fractions and higher triglyceride in VLDL, compared with mice receiving AAV8-TBG-GFP [green fluorescent protein (GFP)] (Fig. 1C and fig. S1D).
Fig. 1. Silencing hepatocyte tPA increases apoB-lipoprotein cholesterol and apoB independently of LDLR or apoE.

(A) Ldlr−/− mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 8 weeks. The livers were assayed for tPA protein, and plasma samples were assayed for total cholesterol and apoB-100 concentrations and for FPLC profiles of cholesterol, triglyceride, and apoB-100 (n = 9 to 10 mice per group). (B) Apoe−/− mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 8 weeks. The livers were assayed for tPA protein, and plasma samples were assayed for total cholesterol and apoB-100 concentrations and for FPLC profiles of cholesterol, triglyceride, and apoB-100 (n = 5 mice per group). (C) Platfl/fl mice were treated with AAV8-TBG-cre (Cre) or AAV8-TBG-GFP (GFP) and then fed the WD for 8 weeks. The livers were assayed for tPA protein, and plasma samples were assayed for total cholesterol and apoB-100 concentrations and for FPLC profiles of cholesterol, triglyceride, and apoB-100. The cholesterol in the VLDL fractions is shown in a zoomed-in smaller graph (n = 6 mice per group). (D) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. Cell culture medium apoB was quantified by immunoblot. VLDL fractions were isolated by ultracentrifugation, and cholesterol and triglyceride concentrations in VLDL fractions were assayed. (E) McA-RH7777 cells were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. VLDL was isolated from the medium by ultracentrifugation, and cholesterol and triglyceride concentrations in VLDL were assayed. Cell culture medium apoB was assayed by immunoblot. Data are shown as means ± SEMs; *P < 0.05 by two-tailed Student’s t test.
Silencing tPA did not change Apob mRNA in the livers of Ldlr−/−, Apoe−/−, or WT mice (fig. S3, A to C), which suggests that increased plasma apoB was not because of increased apoB synthesis. Moreover, the plasma apoB data were not a result of changes in apoE- or LDLR-mediated pathways because plasma apoE levels and liver LDLR expression remained unchanged (fig. S4, A and B). Collectively, these data show that silencing hepatocyte tPA raises plasma apoB lipoprotein-cholesterol through a mechanism independent of the LDL receptor or apoE.
To examine hepatocyte-intrinsic effects and to link this finding to human relevance, tPA expression was silenced in primary human hepatocytes using small interfering RNA (siRNA) against PLAT mRNA (si-tPA). The si-tPA treated cells had higher levels of apoB-100 in their media compared with control (scr) hepatocytes (fig. S5A and Fig. 1D), whereas hepatocyte APOB mRNA levels were similar in the two groups of cells (fig. S5B). Further, the contents of cholesterol and triglyceride were higher in the VLDL fractions isolated from the culture medium of tPA-silenced human hepatocytes (Fig. 1D). Similar findings were observed in cultured McA-RH7777 cells (Fig. 1E and fig. S5, C and D), a rat hepatoma cell line. The McA-RH7777 cell is an established model system to study VLDL production because it synthesizes VLDL-sized apoB particles similar to human hepatocytes (21). These data show that the ability of tPA to regulate apoB-lipoprotein secretion is a cell-intrinsic property of hepatocytes and is applicable to humans.
tPA limits apoB lipidation in the ER
The data in Ldlr−/− and Apoe−/− mice suggest that tPA limits the production—not the clearance—of apoB-lipoproteins. To examine this point further, we investigated the effect of hepatocyte tPA silencing on VLDL secretion in mice injected with the nonionic surfactant detergent poloxamer 407 (P407). The P407 injection blocks VLDL clearance by inhibiting lipoprotein lipase activity and VLDL lipolysis (22). Hepatocyte tPA–silenced mice had a faster rise in triglycerides (Fig. 2A), which suggests increased VLDL production. Moreover, plasma apoB-100, which is derived only from hepatocytes, increased to a greater degree than plasma apoB-48, which is produced by both hepatocytes and intestinal epithelial cells in mice (23) (Fig. 2B). These data, when combined with those presented above, suggest that silencing hepatocyte tPA increases plasma apoB-containing lipoprotein levels by promoting VLDL production.
Fig. 2. tPA limits apoB lipidation in the ER.

(A) WT mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 14 weeks. Mice were injected with P407 intraperitoneally (i.p.) to assess VLDL secretion. Plasma triglyceride concentration was measured (n = 4 to 5 mice per group). (B) WT mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 14 weeks. Mice were injected with P407 i.p. to assess VLDL secretion. Plasma apoB concentration was measured by ELISA (n = 4 to 5 mice per group). (C) Ldlr−/− mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 8 weeks. VLDL was isolated by ultracentrifugation and visualized by transmission electron microscopy. VLDL (n = 100 for each group) diameter was measured and analyzed using Image-Pro Plus 10.0. Scale bars, 100 nm. (D) Ldlr−/− mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 8 weeks (n = 9 to 10 mice per group). VLDL was isolated by ultracentrifugation, and VLDL diameter was measured by dynamic light scattering. (E) Ldlr−/− mice were treated with AAV8-H1-shPlat (sh-tPA) or AAV8-H1-scrambled control (scr) and then fed the WD for 8 weeks. VLDL was isolated by ultracentrifugation and assayed for the ratio of triglyceride to apoB-100 (n = 9 to 10 mice per group). (F) Whole-body tPA knockout mice (holo-tPA-KO) were treated with AAV8-TBG-Plat (tPA) or AAV8-TBG-lacZ (LacZ) and then fed a normal chow diet for 8 weeks. Plasma samples were assayed for VLDL cholesterol, LDL cholesterol, and apoB-100 concentrations (n = 6 mice per group). (G) Human primary hepatocytes were transduced with a plasmid encoding tPA with C-terminal HA tag (tPA-HA) or GFP. After 48 hours, apoB secretion was measured using a [3H]-labeling method as follows: hepatocytes were incubated for 20 min in [3H]-leucine–containing medium and chased for 3 hours in [3H]-leucine–free medium, and then radioactivity associated with apoB in the cell medium was quantified by scintillation counting. (H) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. apoB secretion was measured using [3H]-labeling, as in (G). Radioactivity associated with apoB in cell medium was quantified by scintillation counting. (I) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. VLDL was isolated by ultracentrifugation, and VLDL diameter was measured by dynamic light scattering. (J) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. VLDL was isolated by ultracentrifugation and assayed for the ratio of triglyceride to apoB-100. (K) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. The ER fraction was isolated, and proteins from the ER were extracted. apoB-lipoproteins extracted from the ER were further separated by density gradient ultracentrifugation and divided into six fractions of increasing density from fraction 1 to 6. apoB from each fraction was measured by immunoblot. Data are shown as means ± SEMs; *P < 0.05 by two-tailed Student’s t test.
Lipidation is a key factor determining the fate of intrahepatic apoB. Poorly lipidated apoB undergoes intracellular degradation, whereas fully lipidated apoB is efficiently secreted as VLDL particles with larger size and lower density (24). Electron microscopic scanning of plasma VLDL particles isolated by density ultracentrifugation showed a shifted distribution of the particles to larger diameters in the hepatocyte tPA–silenced Ldlr−/− mice (Fig. 2C). Moreover, dynamic light-scattering analyses (25-27) revealed that VLDL from sh-tPA mice had a larger hydrodynamic diameter than VLDL from control mice (Fig. 2D). These combined data suggest that the VLDL of the tPA-silenced mice have a higher lipid content, which is further supported by the finding that the VLDL of the tPA-silenced mice had a higher triglyceride/apoB ratio (Fig. 2E).
To explore the impact of increasing hepatocyte tPA expression on plasma apoB-lipoproteins in vivo, whole-body tPA knockout mice (holo-tPA-KO) were injected with AAV8-TBG-Plat (tPA), to restore tPA expression specifically in hepatocytes, or with AAV8-TBG-LacZ control (LacZ) (19, 20, 28-30). As predicted, plasma apoB-100 and VLDL- and LDL-cholesterol were lower in the mice treated AAV8-TBG-Plat versus AAV8-TBG-LacZ (Fig. 2F).
To show relevance to lipoprotein production by human hepatocytes, apoB secretion was measured using a 3[H]-leucine pulse-chase method (31) in human primary hepatocytes. Transfecting these cells with a plasmid encoding tPA led to lower apoB-associated radioactivity in the cell medium, indicating lower secretion of newly synthesized apoB (Fig. 2G). By contrast, silencing tPA increased the secretion of newly synthesized apoB (Fig. 2H). Similar results were observed in tPA-silenced McA-RH7777 cells (fig. S6). However, incubating human primary hepatocytes with recombinant human tPA did not change culture medium apoB-100 levels and cholesterol and triglyceride contents in VLDL (fig. S7), which suggests that the hepatocyte tPA limits apoB lipidation and production intracellularly.
Consistent with the mouse plasma data above, silencing tPA increased the diameter and triglyceride/apoB ratio of VLDL isolated from the cell medium (Fig. 2, I and J). These data suggest that silencing tPA increases apoB lipidation. ER is the major site of apoB lipidation and VLDL assembly (32). The degree of apoB lipidation can be ascertained by the density of apoB lipoprotein particles—namely, greater lipidation of apoB results in lower density of the particle (33, 34). In this context, tPA-silenced human primary hepatocytes had overall higher ER-associated apoB and a higher proportion of apoB in lower-density fractions (fractions 1 and 2, Fig. 2K) than control hepatocytes. Because density and lipidation are inversely related, these findings further support the hypothesis that tPA limits apoB lipidation in the ER.
tPA blocks MTP-mediated VLDL assembly by interacting with apoB
MTP is a chaperone that promotes intrahepatic apoB lipidation by transferring and incorporating neutral lipids, notably triglyceride and cholesteryl esters, to apoB to assemble VLDL in the ER (10, 11). MTP-mediated lipid transfer to apoB involves its direct binding to apoB (10, 35) because inhibiting the apoB-MTP interaction decreases apoB lipidation and secretion (11). We found that tPA silencing in human primary hepatocytes did not alter the level of MTP protein (Fig. 3A, input). However, MTP immunoprecipitated from tPA-silenced cells showed a higher content of apoB compared with immunoprecipitates of MTP from control hepatocytes (Fig. 3A). These data suggest that silencing tPA increases apoB-MTP interaction, which would be expected to increase lipid transfer to apoB (11). Consistent with this idea, microsomal fractions isolated from tPA-silenced hepatocytes had a twofold higher neutral lipid transfer activity (36) compared with microsomes from control cells (Fig. 3B, groups 1 and 2). This lipid transfer activity was due to MTP, as the MTP inhibitor CP-346086 (37) completely abolished the elevated neutral lipid transfer activity in microsomes from tPA-silenced hepatocytes (Fig. 3B, group 3). Similar findings were observed in McA-RH7777 cells (fig. S8). Conversely, when tPA was expressed in human primary hepatocytes, apoB-MTP interaction and neutral lipid transfer activity were decreased (Fig. 3, C and D). Moreover, purified recombinant tPA protein reduced MTP-mediated neutral lipid transfer to LDL in a dose-dependent manner (Fig. 3E). Thus, tPA inhibits apoB-MTP interaction and reduces MTP-mediated lipid transfer activity.
Fig. 3. tPA blocks apoB-VLDL assembly.

(A) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA for 24 hours. Cell lysates (input) and anti-MTP immunoprecipitates (IP: MTP) were assayed for apoB and MTP by immunoblot. (B) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or scrambled RNA (group 1) or tPA mRNA (si-tPA) (groups 2 and 3) for 24 hours. The microsomal fraction was isolated and assayed for neutral lipid transfer activity with DMSO (groups 1 and 2) or without with CP-346086 (10 nM), an MTP inhibitor (group 3). (C) Human primary hepatocytes were transduced with a plasmid encoding tPA with C terminal HA tag (tPA-HA) or GFP for 48 hours. Cell lysates (input) and anti-MTP precipitates (IP: MTP) were assayed for apoB and MTP by immunoblot. (D) Human primary hepatocytes were transduced with a plasmid encoding tPA-HA or GFP for 48 hours. The microsomal fraction was isolated and assayed for neutral lipid transfer activity. (E) The effect of recombinant human tPA on lipid transfer from donor vesicles to human LDL was assayed. (F) Human primary hepatocytes were transduced with a plasmid encoding tPA-HA or GFP for 48 hours. Cell lysates (input) and anti-HA immunoprecipitates (IP: HA) were assayed for apoB and tPA. (G) A proximity ligation assay was used to measure apoB-tPA interaction in human primary hepatocytes. Scale bars, 10 μm. (H) Confocal microscopy immunofluorescence imaging was used to measure the subcellular localization of tPA and apoB in human primary hepatocytes. Scale bars, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (I) Human primary hepatocytes were transduced with a plasmid encoding WT tPA (tPA-WT), an enzymatically inactive mutant of tPA (tPA-S513A), tPA with an ER retention signal sequence (tPA-KDEL), or GFP for 48 hours. apoB secretion was measured by [3H]-labeling, as in Fig. 2. (J) The effect of recombinant WT tPA (tPA-WT) or enzymatically inactive mutant of tPA (tPA-S513A) on lipid transfer from donor vesicles to human LDL was assayed. Data are shown as means ± SEMs; *P < 0.05 by two-tailed Student’s t test (D) or by one-way ANOVA followed by Dunnett’s test [(B), (E), (I), and (J)].
To examine tPA interaction with apoB, we analyzed primary human hepatocytes transfected with tPA with a C-terminal HA-tag and found that apoB eluted from the anti-HA precipitates (Fig. 3F). This finding was further supported by the results of a tPA-apoB proximity-ligation assay in hepatocytes using labeled anti-tPA and anti-apoB. The data showed intracellular punctate fluorescence (Fig. 3G), which indicates that endogenous tPA and apoB interact in these cells. We also found, using confocal immunofluorescence microscopy, that tPA and apoB colocalize in the ER (Fig. 3H). Moreover, expression of a form of tPA that is retained in the ER because of a KDEL sequence (38, 39) at the C terminus of tPA (tPA-KDEL) reduced secretion of newly synthesized apoB (Fig. 3I). Collectively, these data suggest that tPA interacts with apoB in the ER, which, as described below, is relevant to the lowering of apoB-VLDL assembly and secretion by tPA.
The protease activity of tPA depends on the serine residue at position 513, and substituting serine to alanine (S513A) completely abolishes the serine protease activity of tPA (40). We found that the serine protease mutant tPA (S513A) reduced both apoB secretion by human primary hepatocytes and MTP-mediated lipid transfer activity to a similar degree as WT tPA (Fig. 3, I and J), indicating that this action of tPA is independent of its protease activity. We also showed that purified recombinant WT and S513A-tPA directly interacted in a dose-dependent manner with either LDL or apoB-100 immobilized to a microtiter plate surface (Fig. 4A and figs. S9 and S10). By contrast, purified tPA did not interact with solid-phase immobilized MTP (Fig. 4B). Most importantly, preincubating LDL with tPA inhibited the binding of LDL to MTP (Fig. 4C). Finally, surface plasmon resonance (SPR) studies provided further evidence of noncovalent interaction between tPA and LDL (Fig. 4D). These combined data suggest that tPA binds apoB in a tPA-protease–independent manner and that this process blocks the interaction of apoB with MTP, thereby decreasing apoB lipidation and VLDL assembly and secretion.
Fig. 4. The K2 domain of tPA interacts with the N terminus of apoB.

(A) A solid-phase binding assay was used to measure the interaction between immobilized LDL and recombinant human WT tPA (tPA-WT) or enzymatically inactive tPA-S513A. The binding of tPA-WT or tPA-S513A to wells without LDL is also measured under the same conditions as the control. OD, optical density. (B) A solid-phase binding assay was used to measure the interaction between recombinant human tPA and immobilized purified human MTP complex. (C) A solid-phase binding assay was used to measure the ability of tPA to inhibit the binding of MTP to immobilized LDL. (D) SPR was used to measure the interaction between human recombinant tPA and LDL. (E) A solid-phase binding assay was used to test whether anti-apoB N-terminal antibody (1D1) versus control IgG (raised against the β3 domain of apoB) blocks the binding between human recombinant tPA and immobilized LDL. (F) Human primary hepatocytes were transduced with the plasmid encoding WT tPA (tPA-WT), a tPA mutant without the K2 domain (tPA-Δ-K2), or tPA mutated in the K2 domain lysine binding site (tPA-D236, 238N). apoB secretion was measured by [3H]-labeling, as in Fig. 2. (G) A solid-phase binding assay was used to test whether an antibody against tPA-K2 domain interferes with the interaction between human recombinant tPA and immobilized purified LDL. (H) A solid-phase binding assay was used to measure whether tranexamic acid (TXA) interferes with the interaction between recombinant human tPA and immobilized purified LDL. (I) A schematic diagram depicting the interaction between the N terminus of apoB and the K2 domain of tPA. The diagram was generated using biorender.com. Data are shown as means ± SEMs; *P < 0.05 by one-way ANOVA followed by Dunnett’s test. n.s., not significant (P ≥ 0.05).
The Kringle 2 (K2) domain of tPA interacts with the N terminus of apoB
The K2 domain of tPA has a lysine binding site, which is required for its interaction with fibrin (41). Within the K2 domain of human tPA, negatively charged aspartic acid residues at 236 and 238 are responsible for the binding of tPA to positively charged lysine residues (42). The surface-exposed N terminus of apoB (43) has a positively charged lysine-rich region that is responsible for its binding to MTP (44). The MTP-interacting region (amino acids 430 to 570) (45) of the apoB N terminus can be partially blocked using a monoclonal antibody 1D1, developed by immunizing mice with a human apoB fragment comprising amino acids 474 to 539 (46). In this context, we show that the 1D1, but not an epitope control antibody raised against amino acids 4031 to 4080 (α3 domain) of human apoB, inhibited the interaction between tPA and solid surface-immobilized LDL (Fig. 4E). These data suggest that tPA interacts with the N terminus of apoB.
Expressing tPA mutants lacking either the K2 domain (tPA-Δ-K2) or in which aspartic acid residues at positions 236 and 238 were substituted with asparagine (tPA-D236, 238N) blocked the ability of tPA to lower apoB secretion as measured by pulse-chase assay (Fig. 4F). Similarly, in the solid-phase protein binding assay, an antibody against the K2 domain of tPA inhibited the binding of tPA to solid surface-immobilized LDL (Fig. 4G). Furthermore, tranexamic acid (TXA), a lysine analog that inhibits tPA binding to fibrin, partially reduced the interaction between tPA and solid surface-immobilized LDL (Fig. 4H and fig. S11). Taken together, these data support the idea that the K2 domain of tPA, which contains lysine-binding sites at aspartate residues 236 and 238, binds to the lysine-rich region of the N terminus of apoB (Fig. 4I).
PAI-1 sequesters tPA from apoB in hepatocytes
Increased circulating intestinal apoB-48–lipoproteins with oral feeding enhances hepatic production of VLDL in humans (47), yet the regulatory mechanisms are not clearly understood. Obesity, often associated with dyslipidemia, increases tPA synthesis in hepatocytes (19). However, the increased tPA is overcompensated by a larger increase in the serpin inhibitor of tPA, PAI-1, resulting in decreased net functional free tPA in the livers and plasma (19). PAI-1 covalently binds to tPA, forming a stable complex (48). Therefore, we hypothesized that postprandial lipid loading of hepatocytes would increase the intracellular interaction of tPA and PAI-1, leading to reduced free tPA. Reduced free tPA would then promote apoB-MTP interaction, leading to increased VLDL assembly and secretion.
We first immunoblotted tPA in a PAI-1–immunoprecipitate from primary human hepatocytes and found a band at ~120 Kd, representing a covalently bound, sodium dodecyl sulfate (SDS)–stable complex of tPA (~70 kD) and PAI-1 (~50 kD) (Fig. 5A). This interaction was further validated by a proximity-ligation assay using antibodies against tPA and PAI-1 (Fig. 5B). Oleate is a known stimulus for apoB lipidation and VLDL production (49, 50). We observed increased tPA–PAI-1 complex and reduced free tPA as early as 1 hour after oleate treatment of human primary hepatocytes, which is the time needed to observe a stimulatory effect of oleate on VLDL production (50), the tPA–PAI-1 complex increased, and free tPA was reduced (Fig. 5, C and D). Therefore, the tPA–PAI-1 complex forms rapidly in the hepatocytes after oleate treatment. With prolonged oleate treatment for 6 and 24 hours, free tPA was further decreased, accompanied by increased tPA–PAI-1 complex formation (Fig. 5D). These results suggest a type of regulation of apoB lipidation and VLDL production, involving PAI-1 interacting with tPA, when hepatocytes are loaded with lipids.
Fig. 5. PAI-1 sequesters tPA away from apoB, leading to increased VLDL assembly in hepatocytes.

(A) Human primary hepatocytes lysates (input) and anti–PAI-1 immunoprecipitates (IP: PAI-1) were assayed for tPA and PAI-1 by immunoblot. (B) A proximity ligation assay was used to measure tPA–PAI-1 interaction in human primary hepatocytes. Scale bars, 10 μm. (C) Human primary hepatocytes were treated for 6 hours with 0.4 mM oleate complexed with fatty acid–free BSA (oleate group) or fatty acid–free BSA alone (vehicle control). Cell lysates were assayed for tPA by immunoblot using the Jess Simple Western system. (D) Human primary hepatocytes were treated with 0.4 mM oleate complexed with fatty acid–free BSA (oleate group) or fatty acid–free BSA alone (vehicle control). Cell lysates were assayed for tPA–PA-1 complex and PAI-1–free tPA concentrations by ELISA. (E) Human primary hepatocytes were treated with siRNA against PAI-1 mRNA (si-PAI1) or scrambled RNA and then incubated in medium containing either 0.4 mM oleate complexed with fatty acid–free BSA (oleate group) or fatty acid–free BSA alone (vehicle control). Cell lysates were assayed for PAI-1–free tPA concentration by ELISA. (F) Human primary hepatocytes were treated with siRNA against PAI-1 mRNA (si-PAI1) or scrambled RNA and then incubated in medium containing either 0.4 mM oleate complexed with fatty acid–free BSA (oleate group) or fatty acid–free BSA alone (vehicle control). apoB secretion was measured by [3H]-labeling, as in Fig. 2. (G) Human primary hepatocytes were treated with siRNA against tPA mRNA (si-tPA) or against PAI-1 mRNA (si-PAI1) or scrambled RNA. apoB secretion was measured by [3H]-labeling, as in Fig. 2. (H) A solid-phase binding assay was used to measure the interaction between immobilized LDL and recombinant human tPA or tPA–PAI-1 complex. (I) The effect of recombinant human tPA and tPA–PAI-1 complex on lipid transfer from donor vesicles to human LDL was assayed. (J) Normal chow diet–fed WT mice had their food withdrawn for 5 hours and were then euthanized at 0, 1, 2, and 6 hours after oral gavage with olive oil. Liver lysates were assayed for PAI-1–free tPA concentration by ELISA. Data are shown as means ± SEMs; *P < 0.05 by one-way ANOVA followed by Dunnett’s test [(D) to (G), (I), and (J)]. n.s., not significant (P ≥ 0.05).
Silencing PAI-1 in human primary hepatocytes led to higher free tPA and lower apoB secretion, as measured by pulse-chase analysis (Fig. 5, E and F). The inhibitory impact of silencing PAI-1 on apoB secretion was more prominent under oleate treatment conditions, with an ~400% increase in free tPA and an ~60% decrease in apoB secretion. By contrast, without oleate treatment, there was only an ~67% increase in free tPA and an ~25% decrease in apoB secretion. Similar results were observed in the McA-RH7777 hepatocytes (fig. S12). These observations are consistent with the hypothesis that, under basal conditions, most tPA is free and not bound by PAI-1, and thus PAI-1 silencing leads to only a moderate increase in free tPA and a modest reduction in apoB secretion. However, with oleate loading, more tPA is PAI-1–bound, and silencing PAI-1 leads to a robust increase in free tPA and a reduction in apoB secretion. Furthermore, compared with silencing tPA alone, silencing both tPA and PAI-1 at the same time did not lower apoB secretion (Fig. 5G). These data support the idea that PAI-1 facilitates apoB lipidation by sequestering tPA from apoB.
The formation of a complex between tPA and PAI-1 changes tPA’s conformation (51, 52), causing tPA to lose its ability to bind fibrin, which is mediated by the lysine binding site in its K2 domain (53). We therefore reasoned that the binding of PAI-1 to tPA blocks the lysine binding site in K2 and thereby blocks tPA-LDL interaction. Consistent with this idea, the PAI-1–tPA complex was unable to bind to LDL and did not inhibit MTP-mediated neutral lipid transfer activity (Fig. 5, H and I).
To test this finding in vivo, we delivered olive oil to WT mice by oral gavage. Two and 6 hours after oral gavage, liver free tPA was decreased compared with baseline (Fig. 5J) without alteration of liver total tPA and PAI-1 levels (fig. S13). These data suggest that the physiological function of hepatocyte tPA–PAI-1 interaction in fine-tuning apoB lipidation after lipid loading to hepatocytes. As expected, hepatocyte PAI-1 knockout mice (19, 54) had higher liver free tPA and lower plasma apoB, total cholesterol, and cholesterol in VLDL and LDL fractions compared with their littermate controls (Fig. 6, A to D). In contrast to the higher VLDL production observed in hepatocyte tPA–silenced mice (Fig. 2A), hepatocyte PAI-1 knockout mice had a slower rise in triglycerides after detergent P407 injection, suggesting lower VLDL production (Fig. 6E). Moreover, 2 hours after an oral gavage of olive oil, plasma apoB-100 was increased by 46% in control mice but by only 13% in hepatocyte–PAI-1–KO mice (Fig. 6F). These combined data suggest that endogenous hepatocyte PAI-1, by interacting with tPA and sequestering tPA away from apoB, facilitates the physiologic increase in hepatic VLDL production that occurs in response to fat ingestion.
Fig. 6. PAI-1 deficiency leads to lower concentrations of plasma apoB and apoB-cholesterol in mice and humans.

(A) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-LacZ (Ctrl) and then fed a high-fat diet for 8 weeks. Plasma total cholesterol concentration was measured (n = 6 per group). (B) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-LacZ (Ctrl) and then fed a high-fat diet for 8 weeks. Plasma apoB was measured by immunoblot (n = 6 per group). (C) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-LacZ (Ctrl) and then fed a high-fat diet for 8 weeks. Plasma samples were subjected to FPLC fractionation and assayed for cholesterol concentration. The cholesterol in VLDL fractions is shown in the smaller graph in the inset (n = 6 per group). (D) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-LacZ (Ctrl) and then fed a high-fat diet for 8 weeks. Liver lysates were assayed for PAI-1–free tPA concentration by ELISA (n = 6 per group). (E) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-GFP (Ctrl) and then fed a normal chow diet for 4 weeks. Mice were injected with P407 i.p. to assess VLDL secretion (n = 10 per group). (F) Serpine1fl/fl mice were treated with AAV8-TBG-Cre (Cre) or control AAV8-TBG-GFP (Ctrl) and then fed a normal chow diet for 6 weeks. The mice had their food withdrawn for 5 hours and then were euthanized at 0, 2, and 4 hours after oral gavage with olive oil. Plasma apoB-100 concentration was measured by ELISA. (G) Plasma samples from SERPINE1-deficient humans and unaffected age-, gender-, and BMI-matched individuals from the same community were assayed for VLDL cholesterol, LDL cholesterol, and apoB-100 concentrations (n = 10 per group). (H) tPA concentration was measured in VLDL isolated by ultracentrifugation from the plasma of the subjects in (G) (n = 10 per group). (I) VLDL from the plasma of the subjects in (G) was analyzed by dynamic light scattering (n = 10 per group). The correlation between VLDL-associated tPA and VLDL diameter was calculated (n = 10 per group). (J) A schematic diagram depicting how tPA–PAI-1 interaction in hepatocytes determines VLDL assembly. Without lipid stimulation, tPA interacts with apoB and inhibits MTP-apoB interaction in the ER, thereby limiting MTP-mediated apoB lipidation and VLDL assembly. When hepatocytes are loaded with lipid, PAI-1 sequesters free tPA away from apoB and increases VLDL assembly. The diagram was generated using biorender.com. Data are shown as means ± SEMs; P values were calculated by two-tailed Student’s t test [(A), (D), (E), and (F)], paired Student’s t test [(G) and (H)], or Pearson’s correlation analysis (I). *P < 0.05.
PAI-1–deficient humans have lower apoB-cholesterol
We collected plasma samples from humans with a homozygous loss-of-function mutation in SERPINE1, the gene encoding for PAI-1, and compared them with plasma from age-, gender-, and BMI-matched control individuals from the same community (55). None of the individuals were taking lipid-lowering agents or had a known history of CVDs. Plasma from the PAI-1–deficient subjects had 22% lower LDL-cholesterol (P < 0.05), 17% lower apoB-100 (P < 0.05), and 18% lower VLDL-cholesterol (P = 0.06) (Fig. 6G). Compared with control individuals, the PAI-1–deficient individuals had higher tPA in VLDL (Fig. 6H), and VLDL-associated tPA levels were inversely associated with VLDL diameter (r = −0.59, P < 0.01) (Fig. 6I). These data, together with those from hepatocyte PAI-1–deficient mice and PAI-1–silenced human primary hepatocytes (Fig. 5F and Fig. 6D), are consistent with the hypothesis that the PAI-1 deficiency leads to higher free tPA in hepatocytes, which enables more tPA to interact with apoB and thereby limits apoB lipidation and VLDL production.
Taken together, our findings show that tPA directly interacts with apoB within the ER of hepatocytes and that this interaction reduces MTP-mediated VLDL assembly. Lipid loading to hepatocytes induces tPA–PAI-1 complex formation, sequestering tPA from apoB, and thereby facilitating apoB lipidation and VLDL assembly (Fig. 6J).
Discussion
Decades of research have established the role for circulating tPA in initiating the lysis of blood clots (56, 57). Intravenous infusion of recombinant tPA is approved as a thrombolytic therapy to restore blood flow in atherothrombotic diseases, including ischemic stroke and myocardial infraction (58, 59). Conversely, plasma levels of the tPA inhibitor PAI-1 are positively associated with atherothrombotic disease (60). High plasma levels of apoB-lipoproteins promote the arterial retention of these lipoproteins, forming atherosclerotic lesions (61, 62). The atherosclerotic lesions can then progress to a state in which plaque rupture or erosion occurs, leading to acute thrombotic vascular events, such as myocardial infarction and stroke (63). This study establishes a link between tPA, PAI-1, and plasma levels of atherogenic apoB lipoproteins, beyond the well-known roles of tPA and PAI-1 in the occlusive thrombus that is the ultimate manifestation of atherosclerotic vascular disease. The mechanism involves tPA–PAI-1 interaction–mediated regulation of VLDL assembly in the liver through protein-protein interactions rather than through regulation of thrombolysis per se. These findings provide a possible mechanistic explanation behind the correlation between high apoB cholesterol and low tPA in CVD patients (13-15). Moreover, the work has the potential for suggesting LDLR-independent therapeutic strategies for lowering cardiovascular risk.
tPA is a multidomain protein consisting of finger, growth factor, Kringle 1 (K1), K2, and serine protease domains (64). The extracellular function of tPA in fibrinolysis is linked to its serine protease activity, which mediates the enzymatic conversion of plasminogen to plasmin, which carries out fibrinolysis (64). Our data suggest that the K2 domain, independent of its proteolytic activity, confers tPA’s regulatory function in the assembly of VLDL by direct binding to the apoB N terminus, there by inhibiting MTP-apoB interaction. The inhibition of MTP binding to apoB by tPA is likely to be competitive because both tPA and MTP bind to the lysine-rich domains of the apoB N terminus. However, whether apoB interacts with other lysine-binding proteins, such as plasminogen, in the hepatocyte ER, and whether apoB and tPA interact in the circulation represent topics for future studies. If these interactions were found to occur, additional studies would then focus on the possible impact of these interactions on fibrinolysis and the nonfibrinolytic functions of these lysine-binding proteins (65-67).
PAI-1 is a rapidly acting serine protease inhibitor (serpin) of tPA (68). Upon binding to tPA, PAI-1 inactivates the serine protease activity of tPA and inhibits tPA’s other protease-independent functions, such as tPA’s ability to bind fibrin (53) and tPA’s receptor-mediated activities (65). Previous studies have mostly focused on the tPA–PAI-1 interaction in the blood (69) and on the vascular endothelial surface (70), which are essential to maintain a balanced fibrinolytic potential. Our findings show that tPA and PAI-1 also interact within hepatocytes, fine-tuning apoB-VLDL assembly. The intracellular interaction between PAI-1 and tPA has also been observed in other cell types, such as colonic epithelial cells (71). Gerard et al. observed that PAI-1–deficient mice have an increase in free tPA in the colon (71). These data are consistent with our findings that lowering hepatocyte PAI-1 expression leads to an increase in free tPA levels in both human primary hepatocytes and mouse livers. The impact of the intracellular interaction of tPA and PAI-1 in other cell types is worthy of investigating in future studies.
Our findings provide possible mechanistic explanations for the associations of PLAT and SERPINE1 polymorphisms with circulating lipid levels and atherosclerosis (72-75). Congenital tPA deficiency in humans has not been reported, which suggests that null mutations in the tPA gene may be lethal in utero. An insertion/deletion (I/D) polymorphism, rs4646972, is located in the eighth intron of the tPA gene (76). Homozygotes for the deletion (DD) showed lower plasma tPA levels compared with those carrying an insertion locus (DI and II) (77). Moreover, homozygotes for the deletion (DD) had a threefold higher risk of intracranial artery atherosclerosis (72) and higher plasma total cholesterol and LDL cholesterol (73) compared with the homozygotes for the insertion (II). Subjects with the DD genotype have a lower forearm vascular release of tPA compared with the individuals with the II genotype (78). Two SERPINE1 single-nucleotide polymorphisms (SNPs) have also been shown to be related to plasma lipid levels. The rs6950982 is associated with elevated plasma PAI-1 levels (79) and higher total cholesterol, LDL cholesterol, and triglyceride (74). Similarly, the rs2227674 is associated with higher plasma PAI-1 levels (79) and increased triglyceride (75). Genome-wide association studies showed that genetic variants harboring SERPINE1 loci, leading to elevated PAI-1 levels, are associated with a higher risk of coronary artery disease (80, 81). Heterozygous PAI-1 deficiency is associated with a lower composite coronary artery disease risk score (55). Although further investigation is needed, these studies raise the possibility that targeting hepatocyte PAI-1 may have promise in treating atherosclerotic CVDs.
Moreover, because our data suggest that hepatic PAI-1–free tPA limits VLDL assembly and reduces blood apoB cholesterol, future studies should consider the genetic interaction of tPA and PAI-1 SNPs as well as SNPs that affect tPA and PAI-1 expression in hepatocytes. Two SNPs, rs9399599 and rs7301826, are associated with liver expression of STXBP5 and STX2, both of which are exocytosis mediators (82). These SNPs are also associated with circulating tPA levels in humans (82, 83). Future population genetic studies are required to investigate the correlation of these SNPs—their associated expression quantitative trait loci (eQTLs) in hepatocytes—with atherogenic lipoprotein and lipid levels.
We found that the formation of the tPA–PAI-1 complex in the hepatocyte is increased in response to fatty acid loading. Postprandial lipemia results in an influx of fatty acids into hepatocytes and stimulates the lipidation of apoB and subsequent VLDL secretion, which serves as a physiological response to an increased lipid burden within the hepatocyte (50). Fatty acid ingestion increases the degree and duration of the association between MTP and apoB in hepatocytes (35), which could contribute to fatty acid–induced VLDL secretion. However, the mechanisms by which fatty acids promote the association of apoB and MTP have remained unclear. Our in vivo and in vitro studies indicate an acute increase in the formation of the tPA–PAI-1 complexes within the hepatocyte after fatty acid stimulation without an alteration of either total tPA or PAI-1 protein levels in the cells. Moreover, hepatocyte PAI-1 deficiency leads to increased free tPA and reduced fatty acid–induced VLDL secretion. These data offer a mechanistic explanation behind the observed increase in VLDL secretion after postprandial lipid loading of hepatocytes. Additionally, apoB and MTP have similar functions in both hepatocytes and intestinal epithelial cells, where they are involved in chylomicron assembly (84), but whether tPA in the intestine affects chylomicron assembly requires further study.
Obesity increases the risk of atherothrombotic events, such as myocardial infarction and stroke. Elevated PAI-1 in obesity could be a mechanistic link between obesity and increased CVD risk by both reducing fibrinolytic potential and promoting dyslipidemia. Both plasma and liver PAI-1 levels are increased in obesity (19, 85), and hepatocytes are an important resource of plasma PAI-1 (19, 86). Approximately 60 to 70% of obese people have dyslipidemia with increased VLDL production (87). This study provides possible links among obesity, increased VLDL production, and CVD through hepatic PAI-1.
Previous studies have shown that inhibiting VLDL production by using MTP inhibitors increases the risk of steatosis in humans, limiting the wide use of the agents. However, there are no reports to date that PAI-1 deficiency in humans is associated with liver steatosis. Additionally, global PAI-1 inhibition or PAI-1 deficiency in mice attenuates hepatic steatosis (88-90). Notably, our study used a mouse model with hepatocyte-specific PAI-1 deficiency (Fig. 6, A to F), whereas the studies mentioned above used mice with global PAI-1 deficiency (88-90). This distinction could be significant because PAI-1 is expressed in other cell types, including adipocytes (91). Moreover, mice with global PAI-1 deficiency exhibit resistance to diet-induced obesity and display decreased adipocyte size compared with WT control mice (90, 92). Thus, the attenuated hepatic steatosis seen in global PAI-1 deficiency mice might be the result of overall metabolic changes, including body weight loss, which is not seen in mice with hepatocyte PAI-1 deficiency. Therefore, targeting tPA–PAI-1 balance in the hepatocytes may be an ideal therapeutic strategy for dyslipidemia and atherosclerotic CVDs, but this possibility requires more extensive human-based studies.
This study reveals that the tPA–PAI-1 interaction determines VLDL assembly in hepatocytes. These findings provide mechanistic insight into the physiologic response to lipid ingestion and to the atherosclerosis-relevant process of excessive apoB-lipoprotein production by the liver. The latter insight may suggest ideas to therapeutically lower atherogenic lipoproteins to prevent the development of atherothrombotic vascular disease.
Materials and methods summary
Please refer to the supplementary materials for the complete materials and methods.
Plasma from humans with PAI-1 deficiency
Plasma samples were collected from members of the Berne Amish community, who harbor a frameshift mutation in SERPINE1 (SERPINE1−/−; n = 10) (55) and their age-, gender-, and BMI-matched control individuals from the same community (n = 10). The institutional review boards (IRBs) at the Indiana Center for Hemophilia and Thrombosis and Medical College of Wisconsin (MCW) approved the study protocols. The study participants provided written informed consent.
Mice
Platfl/fl mice were generated using WT C57BL6/J mice through the homologous recombination in embryonic stem cell–based approach (Biocytogen, Wakefield, MA). Briefly, a targeting construct is designed to insert loxP sites into the introns 3 and 6, to flox the exons 4 to 6 of Plat gene. This design is to conditionally knock out the exons 4 to 6 of Plat by Cre-loxP system. Hepatocyte tPA knockout mice were generated by administering Platfl/fl mice with an AAV8 expressing a Cre recombinase driven by the thyroxine-binding globulin (TBG) promoter, AAV8-TBG-cre (Cre), and Platfl/fl mice receiving AAV8-TBG-GFP (GFP) were used as controls. To silence tPA expression in heptocytes, mice were intravenously injected with AAV8 virus containing shPlat (AAV8-H1-shPlat) (19). Age-matched control mice were injected with AAV8-H1-scramble silencing control. Ldlr−/−, Apoe−/−, and WT C57BL/6J mice, used for silencing hepatocyte tPA, were purchased from Jackson Laboratory (JAX) (cat. nos. 002207, 002052, and 000664, respectively). Ldlr−/− and Apoe−/− mice were fed with WD (Teklad, cat. no. TD 88137). WT mice were on a standard chow (Lab diet, cat. no. 5053), DIO diet (Research Diets, cat. no. 12492), or WD (Teklad, cat. no. TD 88137). To express tPA in hepatocytes, WT C57BL/6J mice and holo-tPA-KO mice (Jax, cat. no. 002508), on a standard chow diet, received intravenous injection of AAV8-TBG–Plat. For all experiments, mice were maintained on a 12-hour light–12-hour dark cycle with free access to normal chow, WD, or DIO diet and water. Mice of the same age and similar weight were randomly assigned to experimental and control groups. Plasma lipids were assayed in blood collected after a 5-hour withdrawal of food. We use power calculations to determine the number of mice for each experiment. We include both male and female mice. Mice of the same age and weight are randomly assigned to groups; exclusion criteria are death, injury requiring euthanasia, or weight loss >10%, assuming these are rare events that are not statistically different between groups. All endpoint assays and analyses were conducted by researchers who were blinded to the identity of the cohort. All mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of Biomedical Resource Center at MCW and the Institutional Animal Care and Use Committee of Columbia University Irving Medical Center.
Vector constructs
AAV8-TBG-cre and AAV8-TBG-GFP were purchased from Addgene. As previously described (19, 20), AAV8-H1–short hairpin RNA (shRNA) construct targeting murine Plat was made by annealing complementary oligonucleotides and then ligating them into the pAAV-RSV-GFPH1 vector. AAV8-TBG-Plat was purchased from Vector Biolabs. Plasmid expressing WT human tPA, pCMV3-tPA-HA, was purchased from Sino Biologic (Beijing, China). The plasmid constructs to express human tPA mutants were generated by Versiti BRI Core based on the pCMV3-tPA-HA. Constructed tPA mutants include tPA-S513A, tPA-KDEL, tPA-Δ-K2-HA, and tPA-D236, 238N. tPA-Δ-K2 refers to the tPA mutant with the replacement of K2 with K1, leading to the presence of two copies of K1 but no K2. As previously described (93), the purpose of this design is to create a mutant tPA that lacks K2 but mimics normal tPA structure.
Human primary hepatocyte experiments
Human primary hepatocytes were obtained from the Liver Tissue Cell Distribution System at the University of Pittsburgh (Pittsburgh, Pennsylvania, USA). All cells were cultured in Williams’ Medium E supplemented with Hepatocyte Maintenance Supplement Pack (Thermo Fisher Scientific, cat. no. CM4000). Experiments were conducted as described in the figure legends. Cells were harvested, and culture media were collected, snap-frozen in liquid nitrogen, and stored at −80°C until processing. The age and gender information of human donors are listed in table S3.
Pulse-chase assay for apoB secretion
Hepatocyte apoB-100 secretion was assayed using the 3[H] labeling method as previously described (31). Human primary hepatocytes or McA-RH7777 cells were washed with leucine-free media and pulsed with 3[H] leucine (80 uCi/ml; 160 Ci/mmol, Perkin Elmer, cat. no. NET1166005MC) for 20 min. The 3[H] leucine–containing medium was removed, and cells were incubated with fresh Dulbecco’s minimum essential medium (DMEM) for an additional 0.5, 1, or 3 hours. apoB was immunoprecipitated from cell homogenates and media using anti-apoB antibodies (Sigma-Aldrich, cat. no. AB742). Nonlabeled apoB-100 standards were added to the precipitates, and the samples were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel. Gels were silver stained, the bands corresponding to apoB-100 were excised, and radioactivity associated with apoB-100 was quantified by a scintillation counter.
Neutral lipid transfer activity assay
Neutral lipid transfer activity in cultured hepatocyte microsomal fractions was measured using a commercially available kit (Sigma-Aldrich, MAK110) as previously (36). Specifically, hepatocytes were homogenized in low hypotonic buffer (10 mM Tris-HCl, 1 mM EGTA, and 1 mM MgCl2; pH 7.4) using a Polytron homogenizer. Microsomes were isolated by ultracentrifugation (SW55 Ti rotor, 50,000 rpm, 1 hour). Neutral lipid transfer activity was assayed using the kit according to the manufacturer’s manual. Isolated microsomes were incubated with donor vesicles containing fluorescent lipids and the acceptor vesicles–LDL. The fluorescence signal was self-quenched when labeled lipids existed within the donor vesicles but would be detected after lipids transferred to acceptor vesicles.
Solid-phase protein binding assay
Solid-phase binding was performed in polystyrene microtiter plates using an enzyme-linked immunosorbent assay (ELISA). Microtiter plate wells were coated with LDL at 5 μg/ml in coating buffer [tris-buffered saline (TBS)] overnight at 4°C. Unbound sites were blocked with 3% nonfat milk in TBS for 1 hour at 37°C. After washing with TBS containing 0.05% Tween 20 (TBS-Tween), tPA was added to the wells at concentrations from 0 to 20 mg/ml in TBS-Tween. After a 1-hour incubation at 37°C, the wells were washed with TBS-Tween. Bound proteins were reacted with anti-tPA [1 mg/ml immunoglobulin G (IgG) in TBS-Tween in the presence of 3% nonfat milk] followed by goat anti-rabbit IgG conjugated to horseradish peroxidase. Trimethylboron (TMB) substrates were added. After stopping the reaction, the absorbance at 450 nm was measured.
SPR
Studies of the binding of recombinant tPA to purified LDL were performed with a Biacore S200 SPR instrument (Biacore) using a CM5 sensor chip (Cytiva, cat. no. 29149603) (94). LDL was attached to the chip using amine coupling chemistry, according to the manufacturer’s instructions. In brief, the chip surface was prepared by exposing the carboxylated dextran matrix to an aqueous solution containing 0.4 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and 0.1 M N-hydroxysuccinimide (10 μl/min for 7 min). Then LDL (500 μg/ml in 10 mM sodium acetate buffer, pH 5.5) flowed across the chip surface at the same rate for 7 min, followed by 1M ethanolamine-HCl (pH 8.5) at 19 μl/min for 7 min to deactivate excessive reactive groups and remove any noncovalently bound LDL. This procedure led to ~6000 response units (RUs) of LDL immobilized. To monitor LDL association with tPA, tPA solutions in HBS-E buffer (Biacore) (0.01 M Hepes, 0.15 M NaCl, 3 mM EDTA, pH 7.4) in the concentration range of 10 to 500 μg/ml were flowed across the chip at 20 μl/min for 6 min at room temperature. The dissociation of tPA was then monitored by washing the surface for 6 min with HES buffer alone. tPA flowed across an activated, but uncoated CM5 chip under the same conditions as the control for nonspecific binding.
VLDL particle diameters analysis
VLDL particles (the particle density is < 1.006 g/ml) were isolated by KBr density ultracentrifugation (95) followed by two methods to measure their diameters. First, isolated VLDL particles were negatively stained with 20 g/L phosphotungstic acid (pH 7.0) for 2 min and then viewed under a Philips CM10 electron microscope. The mean diameters of VLDL particles were determined using Image-Pro Plus 5.0 image analysis software. Second, the hydrodynamic diameters of isolated VLDL particles were measured using a Zetasizer μV dynamic laser light scattering instrument (Malvern Instruments) at 633 nm. VLDL samples were transferred to a quartz cuvette, and light scatter readings were performed at 20°C (25-27).
Statistics analysis
The data described in the study were generated from biological replicates. The number for human participants research and mouse experiments are described in the figure legends, where applicable. The in vitro cell experiments were repeated at least three times. All results are presented as means ± SEMs. P values were calculated using two-tailed Student’s t test for data that passed the normality test or the Mann-Whitney rank-sum U test for data that were not normally distributed. Oneway analysis of variance (ANOVA) with post hoc Tukey’s test was used to evaluate differences among groups when three or more groups were analyzed.
Study approval
All mouse experiments were conducted with the approval of the IACUC and Institutional Biosafety Committee of MCW and the IACUC of Columbia University Irving Medical Center. The use of human cells and plasma samples in this study was approved by the IRB at the MCW and Indiana Hemophilia and Thrombosis Center. All participants provided written informed consent.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Indiana Hemophilia and Thrombosis Center Research staff. We thank M. Kipp at the Versiti Blood Research Institute (BRI) for assistance with the mouse colonies; E. Michalski and B. Best at Versiti BRI Vector Core for the vector construction; C. Wells at MCW Electron Microscopy Core Facility for the electronic scanning of VLDL particles; R. Bohnsack at the MCW Department of Biochemistry for instructing the SPR; W. Huang at the Versiti BRI for instructing proximity ligation assay; and Y. Chen and G. Stuttgen at MCW, C. Kastrup at Versiti BRI, M. Flick at University of Carolina-Chapel Hill, and G. Reyes-Soffer at Columbia University for helpful discussions. Human primary hepatocytes were obtained from the Liver Tissue Cell Distribution System (University of Minnesota and the University of Pittsburgh) and through the Human Hepatocyte Isolation Distribution (University of Pittsburgh), part of the CBRPC, Pittsburgh, Pennsylvania.
Funding:
This study was supported by National Institutes of Health grant R01HL163516 (Z.Zhe.), American Heart Association Career Development Award 19CDA34660043 (Z.Zhe.), an American Heart Association Collaborative Sciences Award (Z.Zhe.), an American Society of Hematology Fellow Scholar Award (Z.Zhe.), an American Society of Hematology Supplement Award (Z.Zhe.), a Medical College of Wisconsin and Versiti Blood Research Institute Startup Fund (Z.Zhe.), National Institutes of Health grant T32HL007343 (Z.Zhe.; H.N.G., primary investigator), a Medical College of Wisconsin Cardiovascular Center Research Pilot Award (W.D.), National Institutes of Health grant P01HL087123 (I.T.), National Institutes of Health grant R01HL138907 (M.G.S.-T.), American Heart Association grant 19TPA34890023 (M.G.S.-T.), National Institutes of Health grant R35HL135833 (H.N.G.), National Institutes of Health grant HHSN276201200017C (Liver Tissue Cell Distribution System, University of Minnesota, and the University of Pittsburgh), and Pittsburgh Liver Research Center grant P30 DK120531 (Human Hepatocyte Isolation Distribution, University of Pittsburgh).
Footnotes
Competing interests: Z.Zhe., W.D., and H.L. have filed a provisional patent application on recombinant tPA fragments.
All other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Brunner FJ et al. , Application of non-HDL cholesterol for population-based cardiovascular risk stratification: Results from the Multinational Cardiovascular Risk Consortium. Lancet 394, 2173–2183 (2019). doi: 10.1016/S0140-6736(19)32519-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marston NA et al. , Association of Apolipoprotein B–Containing Lipoproteins and Risk of Myocardial Infarction in Individuals With and Without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol. 7, 250–256 (2022). doi: 10.1001/jamacardio.2021.5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stone NJ et al. , 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol 63, 2889–2934 (2014). doi: 10.1016/j.jacc.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 4.Mabuchi H et al. , Effect of an inhibitor of 3-hydroxy-3-methyglutaryl coenzyme A reductase on serum lipoproteins and ubiquinone-10 levels in patients with familial hypercholesterolemia. N. Engl. J. Med 305, 478–482 (1981). doi: 10.1056/NEJM198108273050902 [DOI] [PubMed] [Google Scholar]
- 5.Horton JD, Cohen JC, Hobbs HH, PCSK9: A convertase that coordinates LDL catabolism. J. Lipid Res. 50, S172–S177 (2009). doi: 10.1194/jlr.R800091-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reith C, Armitage J, Management of residual risk after statin therapy. Atherosclerosis 245, 161–170 (2016). doi: 10.1016/j.atherosclerosis.2015.12.018 [DOI] [PubMed] [Google Scholar]
- 7.Wong ND, Residual Risk After Treatment of Patients With Atherosclerotic Cardiovascular Disease With Proprotein Convertase Subtilisin-Kexin Type 9 Monoclonal Antibody Therapy (from the Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk Trial). Am. J. Cardiol 120, 1220–1222 (2017). doi: 10.1016/j.amjcard.2017.06.063 [DOI] [PubMed] [Google Scholar]
- 8.Sniderman AD et al. , Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 4, 1287–1295 (2019). doi: 10.1001/jamacardio.2019.3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sirwi A, Hussain MM, Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J. Lipid Res 59, 1094–1102 (2018). doi: 10.1194/jlr.R083451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidson NO, Shelness GS, Apolipoprotein B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu. Rev. Nutr 20, 169–193 (2000). doi: 10.1146/annurev.nutr.20.1.169 [DOI] [PubMed] [Google Scholar]
- 11.Hussain MM, Shi J, Dreizen P, Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res 44, 22–32 (2003). doi: 10.1194/jlr.R200014-JLR200 [DOI] [PubMed] [Google Scholar]
- 12.Fisher EA, The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta 1821, 778–781 (2012). doi: 10.1016/j.bbalip.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munkvad S, Gram J, Jespersen J, A depression of active tissue plasminogen activator in plasma characterizes patients with unstable angina pectoris who develop myocardial infarction. Eur. Heart J 11, 525–528 (1990). doi: 10.1093/oxfordjournals.eurheartj.a059745 [DOI] [PubMed] [Google Scholar]
- 14.Gram J, Jespersen J, A selective depression of tissue plasminogen activator (t-PA) activity in euglobulins characterises a risk group among survivors of acute myocardial infarction. Thromb. Haemost 57, 137–139 (1987). doi: 10.1055/s-0038-1651081 [DOI] [PubMed] [Google Scholar]
- 15.Gram J, Jespersen J, Kluft C, Rijken DC, On the usefulness of fibrinolysis variables in the characterization of a risk group for myocardial reinfarction. Acta Med. Scand 221, 149–153 (1987). doi: 10.1111/j.0954-6820.1987.tb01259.x [DOI] [PubMed] [Google Scholar]
- 16.Jansson JH, Johansson B, Boman K, Nilsson TK, Hypo-fibrinolysis in patients with hypertension and elevated cholesterol. J. Intern. Med 229, 309–316 (1991). doi: 10.1111/j.1365-2796.1991.tb00352.x [DOI] [PubMed] [Google Scholar]
- 17.Yamada R, Yamada S, Ishii A, Sasamata M, Watanabe S, Association between tissue plasminogen activator and serum lipids in healthy volunteers. Ann. Med 22, 313–318 (1990). doi: 10.3109/07853899009147913 [DOI] [PubMed] [Google Scholar]
- 18.Glueck CJ et al. , Endogenous testosterone, fibrinolysis, and coronary heart disease risk in hyperlipidemic men. J. Lab. Clin. Med 122, 412–420 (1993). [PubMed] [Google Scholar]
- 19.Zheng Z et al. , Interacting hepatic PAI-1/tPA gene regulatory pathways influence impaired fibrinolysis severity in obesity. J. Clin. Invest 130, 4348–4359 (2020). doi: 10.1172/JCI135919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Z et al. , An ATF6-tPA pathway in hepatocytes contributes to systemic fibrinolysis and is repressed by DACH1. Blood 133, 743–753 (2019). doi: 10.1182/blood-2018-07-864843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi J, Gamble MV, Conlon D, Liang JS, Ginsberg HN, The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoproteins in response to oleic acid occurs in the endoplasmic reticulum and not in the Golgi in McA RH7777 cells. J. Biol. Chem 278, 42643–42651 (2003). doi: 10.1074/jbc.M306920200 [DOI] [PubMed] [Google Scholar]
- 22.Millar JS, Cromley DA, McCoy MG, Rader DJ, Billheimer JT, Determining hepatic triglyceride production in mice: Comparison of poloxamer 407 with Triton WR-1339. J. Lipid Res 46, 2023–2028 (2005). doi: 10.1194/jlr.D500019-JLR200 [DOI] [PubMed] [Google Scholar]
- 23.Hinsdale ME, Sullivan PM, Mezdour H, Maeda N, ApoB-48 and apoB-100 differentially influence the expression of type-III hyperlipoproteinemia in APOE*2 mice. J. Lipid Res 43, 1520–1528 (2002). doi: 10.1194/jlr.M200103-JLR200 [DOI] [PubMed] [Google Scholar]
- 24.Tiwari S, Siddiqi SA, Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol 32, 1079–1086 (2012). doi: 10.1161/ATVBAHA.111.241471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng D et al. , Very Low Density Lipoprotein Assembly Is Required for cAMP-responsive Element-binding Protein H Processing and Hepatic Apolipoprotein A-IV Expression. J. Biol. Chem 291, 23793–23803 (2016). doi: 10.1074/jbc.M116.749283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinberg RB, Gallagher JW, Fabritius MA, Shelness GS, ApoA-IV modulates the secretory trafficking of apoB and the size of triglyceride-rich lipoproteins. J. Lipid Res 53, 736–743 (2012). doi: 10.1194/jlr.M019992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakurai T et al. , Measurement of lipoprotein particle sizes using dynamic light scattering. Ann. Clin. Biochem 47, 476–481 (2010). doi: 10.1258/acb.2010.010100 [DOI] [PubMed] [Google Scholar]
- 28.Ozcan L et al. , Hepatocyte DACH1 Is Increased in Obesity via Nuclear Exclusion of HDAC4 and Promotes Hepatic Insulin Resistance. Cell Rep. 15, 2214–2225 (2016). doi: 10.1016/j.celrep.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghorpade DS et al. , Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677 (2018). doi: 10.1038/nature26138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X et al. , Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 31, 969–986.e7 (2020). doi: 10.1016/j.cmet.2020.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahoo D et al. , ABCA1-dependent lipid efflux to apolipoprotein A-I mediates HDL particle formation and decreases VLDL secretion from murine hepatocytes. J. Lipid Res 45, 1122–1131 (2004). doi: 10.1194/jlr.M300529-JLR200 [DOI] [PubMed] [Google Scholar]
- 32.Rustaeus S et al. , Assembly of very low density lipoprotein: A two-step process of apolipoprotein B core lipidation. J. Nutr 129, 463S–466S (1999). doi: 10.1093/jn/129.2.463S [DOI] [PubMed] [Google Scholar]
- 33.Lewis B, Classification of lipoproteins and lipoprotein disorders. J. Clin. Pathol 5, 26–31 (1973). doi: 10.1136/jcp.s1-5.1.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan M, Liang J-S, Fisher EA, Ginsberg HN, The late addition of core lipids to nascent apolipoprotein B100, resulting in the assembly and secretion of triglyceride-rich lipoproteins, is independent of both microsomal triglyceride transfer protein activity and new triglyceride synthesis. J. Biol. Chem 277, 4413–4421 (2002). doi: 10.1074/jbc.M107460200 [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Zhou M, Huang LS, Wetterau J, Ginsberg HN, Demonstration of a physical interaction between microsomal triglyceride transfer protein and apolipoprotein B during the assembly of ApoB-containing lipoproteins. J. Biol. Chem 271, 10277–10281 (1996). doi: 10.1074/jbc.271.17.10277 [DOI] [PubMed] [Google Scholar]
- 36.Athar H, Iqbal J, Jiang XC, Hussain MM, A simple, rapid, and sensitive fluorescence assay for microsomal triglyceride transfer protein. J. Lipid Res 45, 764–772 (2004). doi: 10.1194/jlr.D300026-JLR200 [DOI] [PubMed] [Google Scholar]
- 37.Chandler CE et al. , CP-346086: An MTP inhibitor that lowers plasma cholesterol and triglycerides in experimental animals and in humans. J. Lipid Res 44, 1887–1901 (2003). doi: 10.1194/jlr.M300094-JLR200 [DOI] [PubMed] [Google Scholar]
- 38.Andres DA, Dickerson IM, Dixon JE, Variants of the carboxyl-terminal KDEL sequence direct intracellular retention. J. Biol. Chem 265, 5952–5955 (1990). doi: 10.1016/S0021-9258(19)39273-7 [DOI] [PubMed] [Google Scholar]
- 39.Gerondopoulos A et al. , A signal capture and proofreading mechanism for the KDEL-receptor explains selectivity and dynamic range in ER retrieval. eLife 10, e68380 (2021). doi: 10.7554/eLife.68380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pu H et al. , Protease-independent action of tissue plasminogen activator in brain plasticity and neurological recovery after ischemic stroke. Proc. Natl. Acad. Sci. U.S.A 116, 9115–9124 (2019). doi: 10.1073/pnas.1821979116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Munk GAW et al. , Binding of tissue-type plasminogen activator to lysine, lysine analogues, and fibrin fragments. Biochemistry 28, 7318–7325 (1989). doi: 10.1021/bi00444a026 [DOI] [PubMed] [Google Scholar]
- 42.Weening-Verhoeff EJ et al. , Involvement of aspartic and glutamic residues in kringle-2 of tissue-type plasminogen activator in lysine binding, fibrin binding and stimulation of activity as revealed by chemical modification and oligonucleotide-directed mutagenesis. Protein Eng. Des. Sel 4, 191–198 (1990). doi: 10.1093/protein/4.2.191 [DOI] [PubMed] [Google Scholar]
- 43.Segrest JP, Jones MK, De Loof H, Dashti N, Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res 42, 1346–1367 (2001). doi: 10.1016/S0022-2275(20)30267-4 [DOI] [PubMed] [Google Scholar]
- 44.Hussain MM, Bakillah A, Jamil H, Apolipoprotein B binding to microsomal triglyceride transfer protein decreases with increases in length and lipidation: Implications in lipoprotein biosynthesis. Biochemistry 36, 13060–13067 (1997). doi: 10.1021/bi971395a [DOI] [PubMed] [Google Scholar]
- 45.Hussain MM, Bakillah A, Nayak N, Shelness GS, Amino acids 430-570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J. Biol. Chem 273, 25612–25615 (1998). doi: 10.1074/jbc.273.40.25612 [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Pease R, Bertinato J, Milne RW, Well-defined regions of apolipoprotein B-100 undergo conformational change during its intravascular metabolism. Arterioscler. Thromb. Vasc. Biol 20, 1301–1308 (2000). doi: 10.1161/01.ATV.20.5.1301 [DOI] [PubMed] [Google Scholar]
- 47.Cohn JS, Wagner DA, Cohn SD, Millar JS, Schaefer EJ, Measurement of very low density and low density lipoprotein apolipoprotein (Apo) B-100 and high density lipoprotein Apo A-I production in human subjects using deuterated leucine. Effect of fasting and feeding. J. Clin. Invest 85, 804–811 (1990). doi: 10.1172/JCI114507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawrence DA et al. , Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J. Biol. Chem 270, 25309–25312 (1995). doi: 10.1074/jbc.270.43.25309 [DOI] [PubMed] [Google Scholar]
- 49.White AL, Graham DL, LeGros J, Pease RJ, Scott J, Oleate-mediated stimulation of apolipoprotein B secretion from rat hepatoma cells: A function of the ability of apolipoprotein B to direct lipoprotein assembly and escape presecretory degradation. J. Biol. Chem 267, 15657–15664 (1992). doi: 10.1016/S0021-9258(19)49586-0 [DOI] [PubMed] [Google Scholar]
- 50.Dixon JL, Furukawa S, Ginsberg HN, Oleate stimulates secretion of apolipoprotein B-containing lipoproteins from Hep G2 cells by inhibiting early intracellular degradation of apolipoprotein B. J. Biol. Chem 266, 5080–5086 (1991). doi: 10.1016/S0021-9258(19)67758-6 [DOI] [PubMed] [Google Scholar]
- 51.Perron MJ, Blouse GE, Shore JD, Distortion of the catalytic domain of tissue-type plasminogen activator by plasminogen activator inhibitor-1 coincides with the formation of stable serpin-proteinase complexes. J. Biol. Chem 278, 48197–48203 (2003). doi: 10.1074/jbc.M306184200 [DOI] [PubMed] [Google Scholar]
- 52.Huntington JA, Read RJ, Carrell RW, Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 (2000). doi: 10.1038/35038119 [DOI] [PubMed] [Google Scholar]
- 53.Kaneko M, Sakata Y, Matsuda M, Mimuro J, Interactions between the finger and kringle-2 domains of tissue-type plasminogen activator and plasminogen activator inhibitor-1. J. Biochem 111, 244–248 (1992). doi: 10.1093/oxfordjournals.jbchem.a123744 [DOI] [PubMed] [Google Scholar]
- 54.Jiang C et al. , Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 16, 1114–1124 (2017). doi: 10.1111/acel.12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan SS et al. , A null mutation in SERPINE1 protects against biological aging in humans. Sci. Adv 3, eaao1617 (2017). doi: 10.1126/sciadv.aao1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Astrup T, Stage A, Isolation of a soluble fibrinolytic activator from animal tissue. Nature 170, 929 (1952). doi: 10.1038/170929a0 [DOI] [PubMed] [Google Scholar]
- 57.Collen D, Lijnen HR, The tissue-type plasminogen activator story. Arterioscler. Thromb. Vasc. Biol 29, 1151–1155 (2009). doi: 10.1161/ATVBAHA.108.179655 [DOI] [PubMed] [Google Scholar]
- 58.GUSTO Angiographic Investigators, The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N. Engl. J. Med 329, 1615–1622 (1993). doi: 10.1056/NEJM199311253292204 [DOI] [PubMed] [Google Scholar]
- 59.Wang DZ, Rose JA, Honings DS, Garwacki DJ, Milbrandt JC, Treating acute stroke patients with intravenous tPA. The OSF stroke network experience. Stroke 31, 77–81 (2000). doi: 10.1161/01.STR.31.1.77 [DOI] [PubMed] [Google Scholar]
- 60.Sobel BE, Increased plasminogen activator inhibitor-1 and vasculopathy. A reconcilable paradox. Circulation 99, 2496–2498 (1999). doi: 10.1161/01.CIR.99.19.2496 [DOI] [PubMed] [Google Scholar]
- 61.Tabas I, Williams KJ, Borén J, Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 116, 1832–1844 (2007). doi: 10.1161/CIRCULATIONAHA.106.676890 [DOI] [PubMed] [Google Scholar]
- 62.Borén J, Williams KJ, The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol 27, 473–483 (2016). doi: 10.1097/MOL.0000000000000330 [DOI] [PubMed] [Google Scholar]
- 63.Libby P, The molecular mechanisms of the thrombotic complications of atherosclerosis. J. Intern. Med 263, 517–527 (2008). doi: 10.1111/j.1365-2796.2008.01965.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pennica D et al. , Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature 301, 214–221 (1983). doi: 10.1038/301214a0 [DOI] [PubMed] [Google Scholar]
- 65.Gonias SL et al. , PAI1 blocks NMDA receptor-mediated effects of tissue-type plasminogen activator on cell signaling and physiology. J. Cell Sci 131, jcs217083 (2018). doi: 10.1242/jcs.217083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mantuano E, Lam MS, Shibayama M, Campana WM, Gonias SL, The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J. Cell Sci 128, 3478–3488 (2015). doi: 10.1242/jcs.173765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Samson AL et al. , Tissue-type plasminogen activator requires a co-receptor to enhance NMDA receptor function. J. Neurochem 107, 1091–1101 (2008). doi: 10.1111/j.1471-4159.2008.05687.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thorsen S, Philips M, Selmer J, Lecander I, Astedt B, Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. Eur. J. Biochem 175, 33–39 (1988). doi: 10.1111/j.1432-1033.1988.tb14162.x [DOI] [PubMed] [Google Scholar]
- 69.Chandler WL et al. , Clearance of tissue plasminogen activator (TPA) and TPA/plasminogen activator inhibitor type 1 (PAI-1) complex: Relationship to elevated TPA antigen in patients with high PAI-1 activity levels. Circulation 96, 761–768 (1997). doi: 10.1161/01.CIR.96.3.761 [DOI] [PubMed] [Google Scholar]
- 70.Sakata Y, Okada M, Noro A, Matsuda M, Interaction of tissue-type plasminogen activator and plasminogen activator inhibitor 1 on the surface of endothelial cells. J. Biol. Chem 263, 1960–1969 (1988). doi: 10.1016/S0021-9258(19)77972-1 [DOI] [PubMed] [Google Scholar]
- 71.Kaiko GE et al. , PAI-1 augments mucosal damage in colitis. Sci. Transl. Med 11, eaat0852 (2019). doi: 10.1126/scitranslmed.aat0852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Babu MS et al. , Association of genetic variants of fibrinolytic system with stroke and stroke subtypes. Gene 495, 76–80 (2012). doi: 10.1016/j.gene.2011.12.046 [DOI] [PubMed] [Google Scholar]
- 73.Karadeniz M, Erdogan M, Berdeli A, Saygili F, Yilmaz C, 4G/5G polymorphism of PAI-1 gene and Alu-repeat I/D polymorphism of TPA gene in Turkish patients with polycystic ovary syndrome. J. Assist. Reprod. Genet 24, 412–418 (2007). doi: 10.1007/s10815-007-9160-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sotos-Prieto M et al. , Association between the rs6950982 polymorphism near the SERPINE1 gene and blood pressure and lipid parameters in a high-cardiovascular-risk population: Interaction with Mediterranean diet. Genes Nutr. 8, 401–409 (2013). doi: 10.1007/s12263-012-0327-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bentley AR et al. , Gene-based sequencing identifies lipid-influencing variants with ethnicity-specific effects in African Americans. PLOS Genet. 10, e1004190 (2014). doi: 10.1371/journal.pgen.1004190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ludwig M, Wohn KD, Schleuning WD, Olek K, Allelic dimorphism in the human tissue-type plasminogen activator (TPA) gene as a result of an Alu insertion/deletion event. Hum. Genet 88, 388–392 (1992). doi: 10.1007/BF00215671 [DOI] [PubMed] [Google Scholar]
- 77.Sciacca FL et al. , Genetic and plasma markers of venous thromboembolism in patients with high grade glioma. Clin. Cancer Res 10, 1312–1317 (2004). doi: 10.1158/1078-0432.CCR-03-0198 [DOI] [PubMed] [Google Scholar]
- 78.Jern C, Ladenvall P, Wall U, Jern S, Gene polymorphism of t-PA is associated with forearm vascular release rate of t-PA. Arterioscler. Thromb. Vasc. Biol 19, 454–459 (1999). doi: 10.1161/01.ATV.19.2.454 [DOI] [PubMed] [Google Scholar]
- 79.Kathiresan S et al. , Comprehensive survey of common genetic variation at the plasminogen activator inhibitor-1 locus and relations to circulating plasminogen activator inhibitor-1 levels. Circulation 112, 1728–1735 (2005). doi: 10.1161/CIRCULATIONAHA.105.547836 [DOI] [PubMed] [Google Scholar]
- 80.CARDIoGRAMplusC4D Consortium, A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet 47, 1121–1130 (2015). doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song C, Burgess S, Eicher JD, O’Donnell CJ, Johnson AD, CHARGE Consortium Hemostatic Factor Working Group, ICBP Consortium, CHARGE Consortium Subclinical Working Group, Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J. Am. Heart Assoc 6, e004918 (2017). doi: 10.1161/JAHA.116.004918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang J et al. , Genome-wide association study for circulating tissue plasminogen activator levels and functional follow-up implicates endothelial STXBP5 and STX2. Arterioscler. Thromb. Vasc. Biol 34, 1093–1101 (2014). doi: 10.1161/ATVBAHA.113.302088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Temprano-Sagrera G et al. , Multi-phenotype analyses of hemostatic traits with cardiovascular events reveal novel genetic associations. J. Thromb. Haemost 20, 1331–1349 (2022). doi: 10.1111/jth.15698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iqbal J, Boutjdir M, Rudel LL, Hussain MM, Intestine-specific MTP and global ACAT2 deficiency lowers acute cholesterol absorption with chylomicrons and HDLs. J. Lipid Res 55, 2261–2275 (2014). doi: 10.1194/jlr.M047951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meigs JB et al. , Hyperinsulinemia, hyperglycemia, and impaired hemostasis: The Framingham Offspring Study. JAMA 283, 221–228 (2000). doi: 10.1001/jama.283.2.221 [DOI] [PubMed] [Google Scholar]
- 86.Kietzmann T, Samoylenko A, Roth U, Jungermann K, Hypoxia-inducible factor-1 and hypoxia response elements mediate the induction of plasminogen activator inhibitor-1 gene expression by insulin in primary rat hepatocytes. Blood 101, 907–914 (2003). doi: 10.1182/blood-2002-06-1693 [DOI] [PubMed] [Google Scholar]
- 87.Bays HE et al. , Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. J. Clin. Lipidol 7, 304–383 (2013). doi: 10.1016/j.jacl.2013.04.001 [DOI] [PubMed] [Google Scholar]
- 88.Henkel AS, Khan SS, Olivares S, Miyata T, Vaughan DE, Inhibition of Plasminogen Activator Inhibitor 1 Attenuates Hepatic Steatosis but Does Not Prevent Progressive Nonalcoholic Steatohepatitis in Mice. Hepatol. Commun 2, 1479–1492 (2018). doi: 10.1002/hep4.1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SM et al. , TM5441, a plasminogen activator inhibitor-1 inhibitor, protects against high fat diet-induced non-alcoholic fatty liver disease. Oncotarget 8, 89746–89760 (2017). doi: 10.18632/oncotarget.21120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma LJ et al. , Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 53, 336–346 (2004). doi: 10.2337/diabetes.53.2.336 [DOI] [PubMed] [Google Scholar]
- 91.Lundgren CH, Brown SL, Nordt TK, Sobel BE, Fujii S, Elaboration of type-1 plasminogen activator inhibitor from adipocytes. A potential pathogenetic link between obesity and cardiovascular disease. Circulation 93, 106–110 (1996). doi: 10.1161/01.CIR.93.1.106 [DOI] [PubMed] [Google Scholar]
- 92.Schäfer K, Fujisawa K, Konstantinides S, Loskutoff DJ, Disruption of the plasminogen activator inhibitor 1 gene reduces the adiposity and improves the metabolic profile of genetically obese and diabetic ob/ob mice. FASEB J. 15, 1840–1842 (2001). doi: 10.1096/fj.00-0750fje [DOI] [PubMed] [Google Scholar]
- 93.Longstaff C et al. , The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: Kinetic and microscopic studies. Blood 117, 661–668 (2011). doi: 10.1182/blood-2010-06-290338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Migliorini M et al. , High-affinity binding of plasminogen-activator inhibitor 1 complexes to LDL receptor–related protein 1 requires lysines 80, 88, and 207. J. Biol. Chem 295, 212–222 (2020). doi: 10.1074/jbc.RA119.010449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Basu D et al. , Novel Reversible Model of Atherosclerosis and Regression Using Oligonucleotide Regulation of the LDL Receptor. Circ. Res 122, 560–567 (2018). doi: 10.1161/CIRCRESAHA.117.311361 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.