

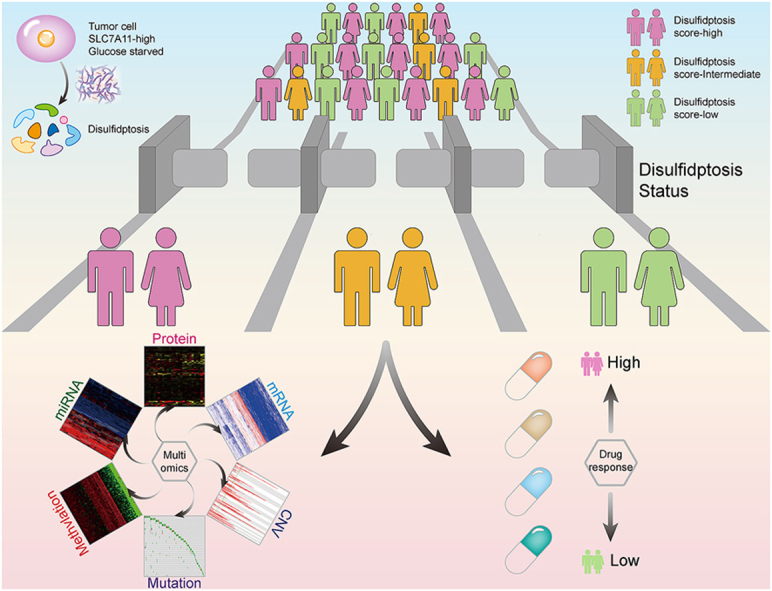
Abstract
The mystery about the mechanistic basis of disulfidptosis has recently been unraveled and shows promise as an effective treatment modality for triggering cancer cell death. However, the limited understanding of the role of disulfidptosis in tumor progression and drug sensitivity has hindered the development of disulfidptosis-targeted therapy and combinations with other therapeutic strategies. Here, we established a disulfidptosis signature model to estimate tumor disulfidptosis status in approximately 10,000 tumor samples across 33 cancer types and revealed its prognostic value. Then, we characterized disulfidptosis-associated molecular features and identified various types of molecular alterations that correlate with both drug-resistant and drug-sensitive responses to anti-tumor drugs. We further showed the vast heterogeneity in disulfidptosis status among 760 cancer cell lines across 25 cancer types. We experimentally validated that disulfidptosis score-high cell lines are more susceptible to glucose starvation-induced disulfidptosis compared to their counterparts with low scores. Finally, we investigated the impact of disulfidptosis status on drug response and revealed that disulfidptosis induction may enhance sensitivity to anti-cancer drugs, but in some cases, it could also lead to drug resistance in cultured cells. Overall, our multi-omics analysis firstly elucidates a comprehensive profile of disulfidptosis-related molecular alterations, prognosis, and potential therapeutic therapies at a pan-cancer level. These findings may uncover opportunities to utilize multiple drug sensitivities induced by disulfidptosis, thereby offering practical implications for clinical cancer therapy.
Keywords: Regulated cell death, Disulfidptosis, Pan-cancer, Drug sensitivity, Multi-omics, Targeted therapy
Graphical abstract
Disulfidptosis was a recently discovered regulated cell death, characterized by NADPH depletion, aberrant disulfide bonding among actin cytoskeleton proteins, and actin network collapse. In this study, we innovatively constructed a disulfidptosis signature to estimate the disulfidptosis status and observed that patients with active disulfidptosis status had significantly better survival. Our findings provide biological insight into the consideration of cancer combination therapeutic strategies based on disulfidptosis.
Highlights
-
•
Disulfidptosis is another new breakthrough in the field of regulated cell death.
-
•
The robustness of disulfidptosis signature was verified in 6 independent glucose-starved datasets.
-
•
Prognostic significance and molecular alterations were characterized at multidimensional levels across 26 cancer types.
-
•
Disulfidptosis score-high cell lines are more susceptible to glucose starvation-induced disulfidptosis.
1. Introduction
Maintenance of the oxidation-reduction (redox) balance is vital for cell survival [1]. Cancer cells often experience elevated levels of oxidative stress compared to non-tumor cells, primarily due to genetic mutations and metabolic reprogramming [2]. To ensure cellular life and proliferation, cancer cells must maintain sufficient glutathione (GSH) levels to counteract excessive intracellular reactive oxygen species (ROS) [1,3,4]. During the GSH synthesis process, cysteine is the key substrate with low content and needs to be supplied through multiple pathways. Most tumor cells mainly rely on solute carrier family 7 member 11 (SLC7A11) to transport extracellular cystine into the cytoplasm and reduce it to cysteine using nicotinamide adenine dinucleotide phosphate (NADPH). Therefore, SLC7A11 is considered a critical oncogene in maintaining cell survival and antioxidant defense of cancer cells [5]. However, recent studies have proposed an unexpected role of SLC7A11 in inducing cell death under glucose deprivation conditions, termed “disulfidptosis” [6,7].
Research on disulfidptosis began in 2017, when Gan and his team found that SLC7A11 overexpression increased glucose dependence in cancer cells and triggered cell death upon glucose starvation [8]. In 2020, they further indicated that protein regulator of cytokinesis 1 (PRC1) inhibition coupled with activating transcription factor 4 (ATF4) induction promotes cell death under glucose starvation conditions [9]. Meanwhile, they demonstrated that key enzymes, including phosphogluconate dehydrogenase (PGD), glucose-6-phosphate dehydrogenase (G6PD), transaldolase 1 (TALDO1), and transketolase (TKT) involved in the glucose-pentose phosphate pathway (PPP), could rescue glucose deprivation-induced cell death in SLC7A11high cancer cells [10]. In 2023, Gan and his team introduced the term disulfidptosis to define this unknown cell death [6], and found glycogen synthase 1 (GYS1), 3-oxoacyl-ACP synthase, mitochondrial (OXSM), nicotinamide adenine dinucleotide hydrogen (NADH): ubiquinone oxidoreductase core subunit S1 (NDUFS1), NADH: ubiquinone oxidoreductase subunit A11 (NDUFA11), nucleotide binding protein-like (NUBPL), leucine rich pentatricopeptide repeat containing (LRPPRC), SLC7A11, solute carrier family 3 member 2 (SLC3A2), ribophorin I (RPN1), and NCK associated protein 1 (NCKAP1) as important participant hits in disulfidptosis through genome-wide CRISPR/Cas9 lose-of-function screening analysis [6]. Notably, glucose starvation is a prerequisite for disulfidptosis in SLC7A11high cancer cells, and as expected, inhibition of glucose uptake by glucose transporter 1/3 (GLUT1/3) inhibitors leads to glucose starvation and disulfidptosis in SLC7A11high cancer cells both in vitro and in vivo [6,10].
Biologically, SLC7A11high cancer cells enhance antioxidant defense capabilities by increasing cystine import and GSH synthesis. This beneficial effect consumes large amounts of NADPH, which is provided by glucose through the PPP [1]. Therefore, when glucose supply is insufficient, tumor cells will experience NADPH depletion and abnormal accumulation of intracellular cystine and other disulfide molecules such as γ-glutamyl-cystine and glutathionyl-cysteine, which results in disulfide stress and aberrant disulfide bonding among actin cytoskeleton proteins, leading to actin network collapse and cell death [6]. This process was verified through direct observation of disulfidptosis-related F-actin contraction and detachment from the plasma membrane by co-staining and unbiased bio-orthogonal chemical proteomic analyses [6]. Cystine is the common amino acid with extremely low solubility, and its large accumulation can lead to the formation of highly toxic crystals in the intracellular lysosomes. However, no clear cystine crystal was observed in cells under disulfide stress through transmission electron microscopy [10]. Additionally, elevated ATP levels, and inability of ROS scavengers to rescue the cell death ruled out the possibility of cell death associated with ATP depletion and ROS accumulation. Thus, this novel form of regulated cell death (RCD) is distinct from other existing forms of cell death, and shed light on new frontiers in RCD and reveal novel mechanisms by which organisms counteract malignant progression of tumors.
RCD plays fundamental roles in cancer therapeutics, including apoptosis, necroptosis, autophagy-dependent cell death, pyroptosis, ferroptosis, and cuproptosis [[11], [12], [13]]. Like other RCDs, the elucidation of disulfidptosis will provide a critical framework for understanding and targeting this unique cell death in cancer therapy [7]. However, current research on disulfidptosis is still limited to the molecular level, and the disulfidptosis status in large populations remains unclear. The hallmark of disulfidptosis status is F-actin contraction and detachment from the plasma membrane, which is difficult to detect in human physiological and pathological conditions. Therefore, it is urgent to develop a robust predictive signature to assess tumor disulfidptosis status in large-scale cancer patients and explore the impact of disulfidptosis on molecular alternations across multiple dimensions. In this study, we innovatively constructed a disulfidptosis signature to estimate the disulfidptosis status and observed that patients with active disulfidptosis status had significantly better survival. We next depicted the molecular characteristics of disulfidptosis across different cancer types from a multi-omics perspective. Moreover, we analyzed the correlation between disulfidptosis status and sensitivities to anti-cancer drugs, and experimentally validated the sensitization to several drugs by disulfidptosis induction in cancer cells. Our findings highlight the role of disulfidptosis in patient prognosis and provide biological insights into the consideration of cancer combination therapeutic strategies based on disulfidptosis.
2. Materials and methods
2.1. Estimating disulfidptosis status across cancer samples by a gene signature
A total of 23 disulfidptosis-related genes were collected from published articles (Table S1) [6,9,10]. Then these genes were divided into two categories according to their regulatory direction, including promoting disulfidptosis genes and inhibiting disulfidptosis genes. The disulfidptosis score model to represent the disulfidptosis status was establish based on the expression data for genes of core promoting disulfidptosis components (pro) including SLC7A11, SLC3A2, RPN1, NCKAP1, cytoplasmic FMR1 interacting protein 1 (CYFIP1), WASP family member 2 (WAVE2), abl interactor 2 (ABI2), haematopoietic stem/progenitor cell protein 300 (HSPC300), Rac family small GTPase 1 (RAC1), ATF4; and negative core components (anti) of NUBPL, NDUFA11, LRPPRC, OXSM, NDUFS1, GYS1, G6PD, PGD, TALDO1, TKT, PRC1, GLUT1, GLUT3. We calculated enrichment score (ES) of pro-disulfidptosis genes and anti-disulfidptosis genes using single sample gene set enrichment analysis (ssGSEA) in the R package ‘GSVA’ [14], the disulfidptosis score to computationally dissect the disulfidptosis status of the tissue samples, and cancer cell line was defined by the differences of ssGSEA score between the ES of pro-disulfidptosis genes minus anti-disulfidptosis genes. The disulfidptosis score model was validated in six independent datasets with known glucose starved status from the Gene Expression Omnibus (GEO): GSE209636, GSE184452, GSE121378, GSE62663, GSE16157, and GSE95097. Wilcoxon rank sum test was used to assess the statistical difference between glucose starved/GLUT1 inhibitor and normal conditions in different cancer cell lines.
2.2. Integration of multi-omics data and clinical data for TCGA samples
Multi-omics data including mRNA expression, miRNA expression, protein expression, somatic mutations, somatic copy number alteration (SCNA) and clinical data across 33 cancer types were downloaded from The Cancer Genome Atlas (TCGA) data portal (https://portal.gdc.cancer.gov/) [15]. The tumor purity of TCGA-tumor samples was obtained from TIMER: Tumor Immune Estimation Resource (http://cistrome.org/TIMER/download.html) and https://doi.org/10.5281/zenodo.253193 [16,17].
2.3. Stratification and multi-omics analysis of tumor samples from TCGA pan cancer cohort
TCGA pan cancer samples were divided into three parts based on the disulfidptosis score distribution of tertiles, defining the top and bottom samples as disulfidptosis-score high and disulfidptosis-score low samples, respectively. We retained a total of 26 cancer types with≥30 samples in both disulfidptosis score-high and disulfidptosis score-low groups for further analysis. METAbolic Flux balance analysis (METAFlux) was used to calculate the glucose metabolic fluxes to investigate the association between the glucose uptake level and disulfidptosis status across cancer types [18]. We further used the matching weights (MW) method of propensity score matching (PSM) algorithm to balance the effects of potential confounders [19,20], including age, gender, tumor purity, race, tumor stage, and examined the balance by comparing standardized difference before and after PSM (standardized difference <0.05). Subsequently, we compared the molecular difference of multi-omics between high disulfidptosis score and low disulfidptosis score in TCGA cohorts. In order to decrease random noise in feature identification, permutation test was repeated 100 times via randomly selecting the high disulfidptosis score or low disulfidptosis score samples. Significant features for four molecular types were identified by the criterion: mRNA expression |fold change| > 2, FDR <0.05; miRNA expression |fold change| > 1.5, FDR < 0.05; somatic mutation and SCNA: FDR <0.05; total protein and DNA methylation: |difference| > 0.1, FDR < 0.05.
2.4. Differential abundance (DA) score
The DA score evaluates the differential regulation of a metabolic pathway between groups with high and low disulfidptosis scores. First, by performing Pearson correlation analysis, we identified 167 metabolites that were significantly positively correlated (pos_cor) with disulfidptosis score (Rs > 0.18, FDR <0.05) and 135 metabolites that were significantly negatively correlated (neg_cor) with disulfidptosis score (Rs < −0.18, FDR <0.05). The DA score for each pathway is calculated as:
Thus, the DA score varies from −1 to 1. A score of −1 indicates that all metabolites in a pathway are negatively correlated with disulfidptosis, while a score of 1 indicates that all metabolites are positively correlated with disulfidptosis. Only pathways with 3 or more significantly altered metabolites were scored.
2.5. Analysis of clinically actionable genes and drug response associated with disulfidptosis status
The area under the dose-response curve (AUC) data and gene expression matrix for cancer cell lines were downloaded from the Genomics of Drug Sensitivity in Cancer (GDSC) (http://www.cancerrxgene.org/downloads) and Cancer Cell Line Encyclopedia (CCLE). Imputed drug response of 138 antitumor drugs in TCGA cancer patients were download from a previous study [21]. The information of clinically actionable genes targeted by Food and Drug Administration (FDA)-approved drugs was downloaded from a previous study [22]. The drug repurposing information with drug-target was downloaded from The Drug Repurposing Hub (https://clue.io/repurposing-app). To assess drug response in cancer cell lines, we calculated the Spearman correlation between the AUC and gene expression of cancer cell lines from GDSC for drug responsiveness (|Rs| > 0.3; FDR <0.05). To assess the effect of disulfidptosis status on drug sensitivity in TCGA tumor samples, Spearman correlation between imputed drug response and disulfidptosis score we calculated (|Rs| > 0.2; FDR <0.05). Among them, drug resistance showed a positive Spearman correlation, while drug sensitivity showed a negative Spearman correlation.
2.6. Pathways enrichment analysis and miRNA-target regulatory networks
In order to study the differences of different disulfidptosis status in cancer hallmarks pathway and Gene Ontology Biological Processes (GOBP). We firstly downloaded the gene set of GOBP and cancer hallmark gene sets from the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/). Then, we used the GSVA method to evaluate these gene sets activity (enrichment scores) of GOBP and cancer hallmark pathways from in each cancer sample. Finally, we calculated the differences of each pathway between the disulfidptosis-high and low groups and screened out the pathways with consistent and significant changes in various cancers. And miRNA-target pairs were downloaded from the miRTarBase [23]. The significantly altered miRNAs and mRNAs (as targets) were used to identify the miRNA-target relationships. Pathway Enrichment analysis of miRNA-target genes was performed by using the clusterProfiler Package [24]. Based on the miRNA-target pairs, we constructed miRNA-target regulatory networks, where the nodes are the miRNAs, or target genes, and the edges are the regulatory pairs.
2.7. Bulk RNA-seq deconvolution analysis
Cell abundance was measured using CIBERSORT with the LM22 matrix from Newman et al. [25] (https://cibersort.stanford.edu/) to quantify the relative abundance of 22 types of immune cells in TCGA pan-cancer. 100 times for permutation test. Expression matrix normalized by FPKM as the input data.
2.8. Cell culture
The human malignant melanoma cell lines A375 and SK-MEL-28, human cervical carcinoma cell line Hela, and human hepatocellular carcinoma cell line Huh-7 were cultured in Dulbecco's modified Eagle medium (DMEM; Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS; Biological Industries) and 1% penicillin-streptomycin solution at 37 °C in an incubator with humid air of 5% CO2. All cell lines were obtained from American Type Culture Collection and free of Mycoplasma contamination (tested by the vendor). None of the cell lines have been found in the International Cell Line Authentication Committee database of commonly misidentified cell lines, based on short tandem repeat profiling performed by the vendor. For the glucose deprivation experiments, cells were incubated with glucose-free DMEM with 10% FBS. The glucose-free DMEM (11966025) was purchased from Thermo Fisher Scientific. The cystine-free and glucose/cystine-double-deprived DMEM were customized from Procell Life Science&Technology.
2.9. Chemicals
Necrostatin-1s (HY-14622A), chloroquine (HY-17589A), Z-VAD-FMK (HY–16658B), liproxstatin-1 (HY-12726), dithiothreitol (HY-15917), etoposide (HY-13629), methotrexate (MTX, HY-14519), and CMK (HY-52101), GW-441756 (HY-18314), ABT-263 (HY-10087), NSC-87877 (HY-18756), staurosporine (HY-15141), and 2-Deoxy-d-glucose (2DG, HY-13966) were purchased from MedChemExpress.
2.10. Cell viability assay
To measure cell viability, 6000 cells per well were seeded in 96-well plates and allowed to adhere. For cell death inhibitor rescue experiments, cells were cultured with glucose-free medium with different cell death inhibitors for about 16 h. For drug sensitivity experiments, cells were cultured with glucose-free or glucose-containing DMEM in indicated concentrations of drugs for about 10–14 h. After treatment, the culture medium in each well of the plate was replaced with 100 μl fresh medium containing 10 μl Cell Counting Kit-8 (CCK-8) (Bimake, B34302). And the culture was returned to the incubator for 2–3 h at 37 °C. Measure the absorbance at 450 nm using a microplate reader and calculate the cell viability according to the manufacturer's instructions.
2.11. Cell proliferation assay
Cell proliferation assay was performed using BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime Biotechnology, C0071L) according to the manufacturer's instructions. Briefly, cells (4 × 104 cells/well) were seeded in 24-well plates and cultured with indicated treatments for about 12 h. Subsequently, cells were incubated with EdU for 2 h, fixed with 4% paraformaldehyde for 15 min, and then permeated with 0.3% Triton X-100 for 15 min. The cells were incubated with the Click Reaction Mixture in the dark for 30 min at room temperature, and then incubated with Hoechst (Beyotime Biotechnology, 33342) for 10 min. Images was detected and captured by Fluorescent Microscopy (Nikon, ECLIPSE Ts2R), and quantification was done using Image J. The results are shown as the ratio of the EdU-positive cells to Hoechst-positive cells.
2.12. Cell cycle
Cell cycle was implemented by cell cycle kit (Beyotime Biotechnology, C1052). After receiving the indicated treatments, cells were collected and fixed overnight in cold 70% ethanol. Then, the cells were stained with propidium iodide and measured by flow cytometry according to the manufacturer's protocols. Cell cycle distribution was assessed by Flow Jo software (version 10.4).
2.13. Caspase-3 activity assay
The activity of caspase-3 was determined using the caspase-3 activity kit (Beyotime Biotechnology, C1116). According to the manufacturer's protocol, cell lysates of SK-MEL-28 cells after indicated treatments were centrifuged at 12, 000×g at 4 °C for 15 min, and protein concentrations were determined by Bradford protein assay. Cellular extracts were incubated in a 96-well plate with Ac-DEVD-pNA (2 mM) for 6 h at 37 °C. Then caspase-3 activity was quantified in the samples with a microplate reader by the absorbance at a wavelength of 405 nm.
2.14. Evaluation of apoptosis by flow cytometry
The apoptosis ratio was analyzed using the Annexin V-FITC Apoptosis Detection Kit (Beyotime Biotechnology, C1062). After indicated treatments, the cells were washed with PBS, digested by trypsin (EDTA depleted), and collected by centrifugation. After being washed with PBS, the cells were resuspended and stained by the binding buffer containing PI and Annexin-V-FITC for 15 min according to the manufacturer's instructions, measured by flow cytometry and finally analyzed by Flow Jo software (version 10.4).
2.15. LDH release assay
Cellular cytotoxicity was monitored by LDH release assay using the LDH Cytotoxicity Assay Kit (Beyotime Biotechnology, C0016) according to the manufacturer's instructions. Briefly, cells were seeded in 96-well plates and then cultured with indicated treatments. Subsequently, the supernatants were collected and transferred to a new plate and incubated with Reaction Mixture in the dark for 30 min at room temperature. Absorbance at 490 nm was measured using a microplate reader to determine the amount of LDH released from cells.
2.16. Statistical analysis
Wilcoxon rank sum test was used to compare the differences. Assessment of statistical significance (p-value) for the association between disulfidptosis-biased mRNA expression and methylation patterns was calculated by Fisher's exact test. Univariate Cox regression model was used to calculate the hazard ratio (HR) of disulfidptosis score. Survminer package was used to determine the cutoff point of survival information for each dataset based on the association between disulfidptosis score and patient overall survival (OS) time, progression free survival (PFS) time, disease free survival (DSS) time and the log-rank test was used to determine the significance of the differences. To find the maximum rank statistic and reduce the calculated batch effect, the “surv-cutpoint” function was used to dichotomy disulfidptosis score and all potential cutting points were repeatedly tested, then the patient samples were divided into the high-disulfidptosis score group and the low-disulfidptosis score group according to the maximum selected log-rank statistics. Kaplan-Meier comparative survival analyses for prognostic analysis were generated, and the log-rank test was used to determine the significance of the differences. Univariate Cox regression model was used to calculate the hazard ratio (HR) of the disulfidptosis status (the high-disulfidptosis score group and the low-disulfidptosis score group). All statistical analysis was two-side. The experimental data was analyzed by GraphPad Prism software and presented as mean ± SD. P values of <0.05 were considered statistically significant and represented as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. All experimental data were obtained from at least three independent biological replicates.
3. Results
3.1. Identification of a gene signature to estimate disulfidptosis status across cancer samples
To estimate the disulfidptosis status in tumor patients, we firstly collected 23 genes coding disulfidptosis regulators and markers from current relevant research [6,9,10], including 10 positive regulators and 13 negative regulators of disulfidptosis (Fig. 1A, Table S1). Next, we collected 6 independent gene expression datasets of cancer cell lines of multiple cancer types under glucose starved/GLUT1 inhibitor-treated and control conditions. To dissect the complexity and heterogeneity of disulfidptosis levels, we then used the ssGSEA algorithm to calculate the disulfidptosis scores based on the enrichment score (ES) of pro-disulfidptosis genes calculated by ssGSEA minus that of the anti-disulfidptosis genes (see Methods) [26]. Notably, the disulfidptosis scores can accurately distinguish the disulfidptosis status in tumor cell lines, characterized by higher disulfidptosis scores in cells under GLUT1 inhibition or glucose starvation, compared with control cells in all 6 independent datasets (Fig. 1B). Consistently, GLUT1 inhibitor BAY-876 or glucose starvation has been reported to induce intracellular disulfidptosis in SLC7A11high cells [6]. To further simulate this state in cancer patient samples, we divided approximately 10,000 tumor samples into two groups: “high-SLC7A11/low-GLUT1” and “low-SLC7A11/high-GLUT1” according to the expression level of SLC7A11 and GLUT1 across 33 cancers from TCGA. We observed that patients in high-SLC7A11/low-GLUT1 group have a significantly higher disulfidptosis scores than those in low-SLC7A11/high-GLUT1 group (Fig. 1C). Furthermore, we calculated glucose metabolic fluxes using METAFlux, a framework for inferring metabolic fluxes from bulk or single-cell transcriptome data [18], to investigate the association between the glucose uptake level and disulfidptosis status across cancer types. As expected, we observed the glucose uptake level was significantly lower in patients with high disulfidptosis score across tumor lineages, comparing differences in glucose uptake levels between the disulfidptosis score-high and disulfidptosis score-low groups (Fig. S1). These results demonstrate the robustness of the 23-gene signature to define disulfidptosis status across different cancer types.
Fig. 1.

Validation of a gene signature for estimating disulfidptosis status among cancer samples. (A) Cell model diagram of positive and negative disulfidptosis regulators. (B) Disulfidptosis scores of cancer cell lines under glucose deprivation/fasting-mimicking/CLUT1 inhibitor-treated conditions (navy blue) and control diets conditions (magenta) in six datasets. Wilcox test was used to assess the difference. (C) Difference of disulfidptosis score between “high-SLC7A11/low-GLUT1” and “low-SLC7A11/high-GLUT1” groups among 33 cancer types. Pie charts show the percentage of cancer types with significant (magenta) and non-significant (grey) alteration. Wilcox test was used to assess the difference. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Clinical relevance of the disulfidptosis signature across cancer types
Furthermore, we compared the differences in disulfidptosis levels between tumor and normal tissues (Fig. 2A). Of note, significant differences were found in most cancers, such as lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), uterine corpus endometrial carcinoma (UCEC), and lower disulfidptosis levels were observed in most tumors compared to normal tissues, except for head and neck squamous cell carcinoma (HNSC). Then, we evaluated the disulfidptosis levels of over 10,000 samples across 33 cancer types from TCGA. The distribution of disulfidptosis level was different in most of cancer types. Low-grade glioma (LGG), glioblastoma (GBM) and prostate adenocarcinoma (PRAD) exhibited higher disulfidptosis levels, whereas lymphoid neoplasm diffuse large B-cell lymphoma (DLBC) and acute myeloid leukemia (LAML) showed lower disulfidptosis levels (Fig. 2B). These results reflect the varying sensitivities to disulfidptosis across tumor types and suggest which tumor types may benefit from disulfidptosis-targeted therapy. In addition, we analyzed the difference of disulfidptosis scores among cancer types based on body location/system. We observed the cancer types located in the neurologic system (including LGG, GBM) had the highest disulfidptosis score, whereas tumors in the hematologic/blood system (such as LAML and DLBC) had the lowest disulfidptosis score (Fig. S2A). The brain relies on glucose as its main energy source and maintains a high level of glucose metabolic state at all times [27]. However, due to the presence of the blood-brain barrier, glucose concentrations in the brain interstitial fluid may only reach 20–30% of circulating levels [28]. Thus, neurological tumors, such as LGG and GBM, may be more susceptible to experiencing glucose starvation, leading to a high disulfidptosis status. In contrast, hematologic/blood system tumors have an abundant supply of glucose and thus do not experience glucose starvation [29], resulting in a low disulfidptosis status. This indicates that the process of disulfidptosis is possible closely related to physiological discrepancies in different parts of the body, which may be due to differences in the expression of transporters and metabolic environment. We further analyzed the relationship between tumor staging and disulfidptosis score. As shown in Figs. S2B–S2C, significant differences in disulfidptosis score were also observed among different tumor stages in cancer types. In BLCA, BRCA, KIRC, and THCA, tumor stage I exhibited the highest disulfidptosis score, while stage IV had the lowest disulfidptosis score (Figs. S2B–S2D). This result highlights that clinical factors such as tumor staging have tumor-specific effects on disulfidptosis score and helps explain why patients with high disulfidptosis scores tend to have better prognosis.
Fig. 2.

The clinical relevance of disulfidptosis across cancer types. (A) The different disulfidptosis score between paired normal and tumor tissues among cancers. (B) Disulfidptosis scores based on the 23-mRNA abundance signature for 33 tumor types, sorted by the median disulfidptosis score for each tumor type. (C) Association of disulfidptosis status with patient overall survival times (OS), progression free survival times (PFS) and disease free survival times (DSS) based on both univariate Cox proportional hazards models in different cancer types. Color means the hazard ratio; size means statistical significance at a given FDR. (D) Kaplan-Meier curves show that patients with higher disulfidptosis score (red) exhibited better overall survival compared to patients with lower disulfidptosis score across cancer types. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To assess the clinical relevance of disulfidptosis, we examined the correlations of disulfidptosis status classification with the OS, PFS and DSS of cancer patients. We consistently observed that disulfidptosis score-high tumors were associated with a better prognosis across various cancer types by using the Cox proportional hazards model (Fig. 2C) and performing survival analysis (Fig. 2D, Fig. S3A–B), such as skin cutaneous melanoma (SKCM, log-rank test, P = 9.8 × 10−4; Fig. 2D), and pancreatic adenocarcinoma (PAAD; log-rank test, P = 3.6 × 10−4; Fig. 2D). These results suggest that disulfidptosis has potential prognostic power and might play critical roles in cancer survival.
3.3. Global landscape of multi-omics alterations associated with disulfidptosis status across tumor types
To explore global landscape of molecular alterations associated with disulfidptosis, samples from TCGA were classified into disulfidptosis score-high, score-intermediate, and score-low groups using the distribution of score tertiles. For the accuracy of statistical analysis, tumor types with less than 30 samples in each group were excluded for subsequent analysis. PSM algorithm was applied to balance the confounders [20], including age at initial pathologic diagnosis, sex, race, pathologic stage, histological type, and tumor purity (Fig. S4A). In general, we identified a large number of molecular alterations related to disulfidptosis in 26 cancer types, including mRNA, miRNA, protein, methylation, mutation, and SCNA (Fig. S4B). The workflow of our study is depicted in Fig. 3A.
Fig. 3.

Overview of six molecular patterns associated with disulfidptosis across cancer types. (A) The overview of integrated analysis of disulfidptosis-related multi-omics and drug response across different cancer types. (B) Number of each altered molecular feature (mRNA, miRNA, methylation, mutation, protein, and SCNA) and total altered molecular features in disulfidptosis score-high (red) and disulfidptosis score-low (green) groups from TCGA tumor samples. Pie plot in the upper panel means the percentage of cancer types with significantly altered disulfidptosis-related features in each molecular data. Pie plot in the left panel means the percentage of molecular layers with significantly altered disulfidptosis-related features in each cancer type. The navy-blue point denotes the percentage of significant features over the total features in each cancer. SCNA, somatic copy number alterations. (C) Pie charts display percentages of genes with both significant disulfidptosis-related mRNA expression and DNA methylation patterns. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The impacts of disulfidptosis status on molecular alterations varied significantly across different cancer types (Fig. 3B). For example, mRNA expression showed the highest number of variations (8912 mRNA variations), ranging from 33 genes in pheochromocytoma and paraganglioma (PCPG) to 5709 genes in thymoma (THYM), whereas SCNAs exhibited minimal variation, with only 79 alterations in 26 cancer types, ranging from 1 SCNAs in kidney chromophobe (KICH) to 29 SCNAs in breast cancer (BRCA). The degree of molecular alterations affected by disulfidptosis status differed across tumor types. For example, several cancer types, including THYM, SKCM, STAD, and LUAD, showed a larger number of variations between the disulfidptosis score-high and score-low groups in all 6 molecular layers, while PCPG, KICH, ovarian serous cystadenocarcinoma (OV) demonstrated minimal variations, limited to partial molecular levels. The total number of molecular alterations across multiple layers also varied greatly. For example, THYM had various molecular alterations across six molecular layers, including 5709 mRNAs, 520 miRNAs, 105 proteins, 4799 methylation probes, 1 gene mutation and 2 SCNAs, whereas PCPG only showed 2 molecular layers changes including 33 mRNAs and 11 miRNAs (Fig. 3B; Fig. S4B). These differences may be attributed to tumor heterogeneity and targeting disulfidptosis could be more suitable for the treatment of certain tumors (e.g., THYM, BRCA and SKCM). Subsequently, we analyzed the specific changes and proportions of methylation in different tumors under the influence of disulfidptosis (Fig. 3C). The results indicated that HNSC is more likely to experience reduced methylation, while PAAD exhibited an increased methylation pattern. Furthermore, the mRNA expression of disulfidptosis-biased genes tended to be contrary to their DNA methylation levels, in line with the concept that hypomethylation generally leads to overexpression of gene, while hypermethylation results in gene silencing [30,31].
In addition, we also investigate the global metabolism alterations associated with disulfidptosis in cancers. To this end, we collected a clear cell renal cell carcinoma (ccRCC) cohort containing patient transcriptome data and 571 metabolite data and calculated the disulfidptosis score for each sample to explore the impact of disulfidptosis on patient metabolism [32]. By performing Pearson correlation analysis, we identified 167 metabolites that were significantly positive correlated with disulfidptosis score (Fig. S4C; Rs > 0.18, FDR <0.05) and 135 metabolites that were significantly negative correlated with disulfidptosis score (Fig. S4C; Rs < −0.18, FDR <0.05). As stated previously, disulfidptosis occurs when the synthesis of NADPH is inhibited under glucose starvation. Overexpression of SLC7A11 at this time will lead to an increase in cystine pumping into cells, causing the accumulation of disulfide compounds, and triggering cell death caused by protein folding errors. Consistent with this process, we observed that a high disulfidptosis score was positively correlated with cystine abundance level while negatively correlated with glucose abundance level, suggesting the robustness of the disulfidptosis score model for defining disulfidptosis status (Fig. S4D). Among the disulfidptosis-related metabolites, we found several interesting metabolites that vary with the disulfidptosis state and deserve further exploration in the future. For example, glucose 6-phosphate is negatively correlated with the disulfidptosis score, and we speculate that this may be due to its role in NADPH generation through shuttling into the PPP of glucose catabolism [10]. Similarly, lactate also exhibits a negative correlation with disulfidptosis score, which may be attributed to its contribution to the production of NADPH by isocitrate dehydrogenase 1 under glucose-deprived conditions [33]. On the contrary, phenylacetylglutamine is positively correlated with the disulfidptosis score, which may be due to the fact that cells in a high disulfidptosis state consume a large amount of glutamate when transporting cystine, thus being highly dependent on glutamine uptake [1]. In addition, 3-methylglutarylcarnitine is a metabolite of branched-chain amino acids (BCAAs), which can provide energy for tricarboxylic acid (TCA) cycle and oxidative phosphorylation to maintain abnormal proliferation of tumor cells. This may be an important method of energy compensation in tumor cells experiencing glucose starvation and is positively correlated with the disulfidptosis score [34] (Fig. S4F).
Additionally, we explored the changes in metabolic pathways associated with disulfidptosis status. Through differential abundance (DA) analysis, we observed that metabolites negatively correlated with disulfidptosis were more enriched in pathways such as glycogen metabolism (DA score = −1), sphingosines metabolism (DA score = −1) and fructose, mannose, galactose, starch, and sucrose metabolism (DA score = −0.6), while metabolites that positively correlated with disulfidptosis were more enriched in pathways such as fatty Acid, dihydroxy (DA score = 1), phosphatidylethanolamine (DA score = 0.67) and glycerolipid metabolism (DA score = 0.5) (Fig. S4G). Overall, our results provide a global landscape of molecular alterations associated with disulfidptosis status across various tumor types.
3.4. Association between disulfidptosis and mRNA expression and signaling pathway
To investigate the manners by which disulfidptosis exert biological processes, we focused on the significantly altered pathways across different cancers. The results showed that the changing trends of various functional pathways remain consistent in 26 cancer types (Fig. 4A-B). As previously reported, actin cytoskeleton organization formed the most prominent interaction cluster in the disulfidptosis process, and increasing NADPH supply could rescue disulfidptosis [6]. Pathways associated with positive regulation of actin nucleation, alpha actinin binding, and actin cytoskeleton reorganization, were strikingly enriched in disulfidptosis score-high samples. In contrast, energy metabolism-related pathways such as NADH dehydrogenase complex assembly, oxidoreduction coenzyme metabolic process and adenosine triphosphate (ADP) metabolic process were commonly enriched in disulfidptosis score-low samples across cancer types (Fig. 4A). To explore the potential mechanisms underlying disulfidptosis in cancer pathogenesis, we also analyzed the relationship between cancer hallmark pathway activity calculated by GSVA algorithm (details see the Methods) and disulfidptosis. Disulfidptosis is characterized by glucose starvation and a lack of redox substrates. Consistent with this status, ROS pathway, glycolysis, and oxidative phosphorylation were all found to be downregulated in the disulfidptosis score-high samples, thus confirming the accuracy of our analysis (Fig. 4B). Additionally, we observed that pathways involving ultraviolet (UV) response signaling, transforming growth factor (TGF) beta signaling were likely to be upregulated in disulfidptosis score-high samples, whereas pathways related to MYC targets v1/2, fatty acid metabolism, PI3K/AKT/MTOR signaling, and DNA repair were prone to be upregulated in disulfidptosis score-low samples (Fig. 4B). These findings suggest that alterations in signaling pathways may be related to disulfidptosis status in cancer patients, and their specific interactions require further investigation.
Fig. 4.

Relationships between disulfidptosis and Signaling Pathways and drug response. (A–B) Heatmap showing normalized difference for Gene sets activity in Gene Ontology Biological Processes (GOBP) (A) and cancer hallmark pathways activity (B) between high- and low-disulfidptosis score tumor tissues. (C) Spearman's rank correlation between disulfidptosis-related mRNA expression levels in different signaling pathways and drug sensitivity (AUC value) across 1074 cancer cell lines. The red dots indicate disulfidptosis-related genes; the bar plot denotes the number of drugs correlated with the genes. The grey circle denotes cancer-related signaling pathway; the orange dots along the grey circle indicate drugs targeting pathways and the point size shows the number of genes correlated with drug sensitivity (|Rs| > 0.3, FDR <0.05); the magenta and green lines indicate positive and negative correlation, respectively. AUC, Area Under Curve; FDR, False discovery rate. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To assess the roles of disulfidptosis-related features in drug response, we focused on 137 genes with differential mRNA levels between disulfidptosis score-high and score-low samples in at least 5 cancer types. We calculated the correlations between the expression of these genes in 1074 cancer cell lines and the AUC values of 252 anti-tumor drugs targeting multiple cancer-related pathways obtained from the CCLE and GDSC databases. The results demonstrated that 137 disulfidptosis-related genes in 19 cancer signaling pathways were significantly associated with the sensitivity of 180 anti-tumor drugs (|Rs| > 0.3, FDR <0.05), and most genes (86.8%; n = 119) were related to sensitization of anti-tumor drugs (Fig. 4C). The related pathways included the cytoskeleton signaling pathway, chromatin signature, cell cycle, DNA replication, EGFR signaling pathway, MAPK signaling pathway, PI3K signaling pathway, and RTK signaling pathway (Fig. 4C and Fig. S5). For instance, the MAPK signaling pathway has been reported to modulate drug efficacy through ERK, and JNK and p38 signaling pathway [35]. We found that 32 genes in the MAPK signaling pathway are associated with the sensitization of 10 anti-tumor drugs (Rs < −0.3, FDR <0.05; Fig. S5A), and 37 genes in the MAPK signaling pathway are associated with the resistance of 12 anti-tumor drugs (Rs > 0.3, FDR <0.05; Fig. S5A). Regulation of the cytoskeleton signaling pathway has been reported to enhance drug resistance in multiple cancer types through sub-families of proteins including microtubules, actin, and intermediate filaments [[36], [37], [38]]. We found that 32 genes in the cytoskeleton signaling pathway are associated with the sensitization of 5 anti-tumor drugs (Rs < −0.3, FDR < 0.05; Fig. S5B), and 39 genes in the cytoskeleton signaling pathway are associated with the resistance of 6 anti-tumor drugs (Rs > 0.3, FDR < 0.05; Fig. S5B). Taken together, these results show extensive interactions between disulfidptosis-related molecular features and drug responses, which broaden a comprehensive perspective for exploring therapeutic strategies via targeting genes in signaling pathways.
3.5. Regulatory network of disulfidptosis-related miRNAs, target genes and proteins
To further understand the potential regulatory network in disulfidptosis, we analyzed differences in miRNA expression among different disulfidptosis status (Fig. S6A) and constructed a miRNA-target regulatory network to investigate the impacts of miRNAs on disulfidptosis. 34 miRNAs significantly differentially expressed in at least four cancers between disulfidptosis score-high and score-low samples were identified (Fig. S6A), and enrichment analysis of the signaling pathways of their target genes was performed (Fig. 5A, Fig. S6B). Intriguingly, the expression of most miRNAs was significantly down-regulated in disulfidptosis score-high samples, while their target genes were significantly up-regulated (Fig. 5A). These target genes are mainly enriched in the terms related to the regulation of actin cytoskeleton, B cell receptor (BCR), T cell receptor (TCR), chemokine signaling pathway, p53 signaling pathway and other immune-regulated signaling pathways (Fig. 5A). Among them, hsa-miR-183-5p and hsa-miR-7-5p are closely associated with the regulation of actin cytoskeleton and may serves as important regulators of actin network collapse in the disulfidptosis process [39,40] (Fig. 5A). Furthermore, the expression of hsa-miR-155-5p and hsa-miR-93-5p was found to be reduced in tumors with high disulfidptosis status and significantly associated with activation of the BCR and TCR pathways (Fig. 5A) [[41], [42], [43], [44], [45], [46]]. The above results provide valuable insights into the mechanism by which miRNA regulates BCR and TCR, thereby affecting immune cell infiltration in different disulfidptosis status. To further explore the relationship between disulfidptosis and immune cell infiltration, we used the CIBERSORT tool to calculate the infiltration degree of 22 immune cell types across pan-cancer samples. As expected, we found a real correlation between disulfidptosis and certain immune cell populations. For example, disulfidptosis was positively correlated with the infiltration of resting memory CD4 T cells, B cells, mast cells, and plasma cells, and negatively correlated with the infiltration of regulatory T cells (Tregs), follicular helper T cells, and M0 macrophages (Fig. S6C). Therefore, our study reveals the possible impact of disulfidptosis on the tumor microenvironment, and the specific mechanism requires further exploration.
Fig. 5.

Disulfidptosis-related miRNA and protein signatures. (A) Significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched by mRNAs in the miRNA-target regulatory network. The magenta line indicates an upregulate of miRNA in the high disulfidptosis score group, and the green line indicates a downregulate of miRNA the high disulfidptosis score group. Yellow dots correspond to miRNA-targeted genes highly expressed in the high disulfidptosis score group, and purple dots correspond to miRNA-targeted genes highly expressed in the disulfidptosis score group. The circle represents a signaling pathway enriched with targeted genes. Statistical analysis was performed using a propensity score algorithm to identify disulfidptosis-related miRNAs and proteins; details are given in the Methods. (B) Heatmap at the bottom panel display alterations of protein or protein phosphorylation in disulfidptosis score-high samples in at least three cancer types. Color represents the degree of difference between the disulfidptosis score-high vs low groups. Statistical analysis was performed using a propensity score algorithm to identify disulfidptosis-associated proteins (details are given in the Methods). The barplot at the upper panel shows the accumulated number of drugs positively (purple) or negatively (dark green) correlated with disulfidptosis-related proteins in different cancer types. The correlation was performed by Spearman's rank correlation. (C) Altered signaling pathways based on functional proteomics data of reverse-phase protein arrays in the disulfidptosis score-high group vs. the disulfidptosis score-low group for multiple cancers. Color indicates the difference in pathway score; point size indicates FDR for pathway score. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To explore the potential impact of disulfidptosis on protein expression, we collected proteins from functional proteomics data on TCGA reverse-phase protein arrays, including key total and phosphorylated proteins associated with cancer. The results showed that 119 proteins were significantly related to the disulfidptosis process in at least three cancer types (Fig. 5B, bottom panel). Among these, programmed cell death 4 (PDCD4), a novel tumor suppressor known to inhibit cell growth, tumor invasion, and metastasis [47], was significantly upregulated in disulfidptosis-score high samples in seven cancer types. Additionally, transferrin receptor (TFRC), a specific ferroptosis marker, was significantly downregulated in disulfidptosis-score high samples in eight cancer types [48], and Cyclin B1 (CCNB1), a protein essential for cell proliferation, was significantly downregulated in disulfidptosis-score high samples in nine cancer types [49] (Fig. 5B). Furthermore, disulfidptosis-related proteins were correlated with the response to anti-tumor drugs (Fig. 5B up panel). Of these, Yes1 associated transcriptional regulator (YAP) was negatively correlated (drug-sensitive) with the response to PHA665752 in 4 cancer types, which is a selective and ATP-competitive c-Met kinase inhibitor targeting the RTK signaling pathway, but positively correlated (drug-resistant) with CCT018159, a HSP90 inhibitor (Fig. S6D). Moreover, we examined the pathway scores based on protein expression in 10 cancer-associated signaling pathways. We found that the RAS/MAPK, hormone ER, hormone AR, RTK, and PI3K/AKT signaling pathways were upregulated, while epithelial mesenchymal transition (EMT), cell cycle, and apoptosis pathways were downregulated in disulfidptosis score-high samples (Fig. 5C). These results elucidate the related proteins that may play important roles in the process of disulfidptosis and provide an important direction for further research into the detailed mechanisms underlying disulfidptosis.
3.6. Disulfidptosis-related somatic copy number alterations across cancers
Given that SCNAs largely mediate aberrant gene expression, we explored the effects of amplification and deletion on disulfidptosis regulator expression across various cancer types (Fig. S7A-C). The results showed that SCNAs varied in different tumor types, which may be an important reason for tumor heterogeneity. For example, RAC1, a key molecule involved in promoting lamellipodia formation and disulfidptosis in SLC7A11high cells [6], is mainly located on chromosome 7p22.1. We found that 7p22.1 copy number amplification leads to RAC1 overexpression mostly in GBM and testicular germ cell tumors (TGCT) (Frequency as 80% and 83%), while 7p22.1 copy number deletion leads to RAC1 low expression mostly occurred in OV (Frequency as 43%) (Fig. S7A-C). Subsequently, we investigated the global pattern of disulfidptosis-related SCNAs in tumors and identified 112 significantly altered SCNAs in 23 cancer types, containing 82 deletions in 22 cancer types with 280 genes located in these regions and 32 amplifications in 17 cancer types with 87 genes located in these areas (Fig. 6A). Furthermore, 14 clinically actionable genes were harbored in these altered SCNAs (Fig. 6B), and they were targeted by 30 anti-tumor drugs of three categories, including targeted therapy, hormone therapy and immunotherapy (Fig. 6C). For example, 2q37.3 deletion is the most frequent SCNA in the disulfidptosis score-high samples of three cancers (SKCM, UCEC and BRCA; Fig. 6A). The programmed cell death protein 1 (PDCD1), as the target of nivolumab and pembrolizumab for cancer immune checkpoint inhibitors, is located in the 2q37.3 region in SKCM, UCEC and BRCA (Fig. 6B). Phosphatase and tensin homolog (PTEN), as a negative regulator of GLUT1 [50], is located in the 10q23.31 deletion region in disulfidptosis score-high samples of PRAD and BRCA (Fig. 6B). Cyclin dependent kinase inhibitor 2A (CDKN2A), located in the 9p23 deletion region in disulfidptosis score-high samples of kidney clear cell carcinoma (KIRC) and LGG (Fig. 6B), was found to regulate the formation of homotypic cell-in-cell structures correlated with F-actin rearrangement [51]. Notably, CDKN2A is targeted by the CDK inhibitor palbociclib (Fig. 6C), and loss-of-function CDKN2A alterations had an effect on the response and survival in patients treated with immunotherapy in multiple studies [52]. Fibroblast growth factor receptor 1 (FGFR1), located in the 8p11.21 amplification region in disulfidptosis score-high samples of rectum adenocarcinoma (READ), was known to induce rapid reorganization of the actin cytoskeleton and stimulate the formation of stress fibers [53]. These results highlight that there exists heterogeneity in the effect of SCNAs on disulfidptosis across tumor types, and disulfidptosis-associated SCNAs are associated with tumor response to anti-tumor drugs including targeted therapy, hormone therapy and immunotherapy.
Fig. 6.

Disulfidptosis-related SCNAs among cancer types. (A) Disulfidptosis-related SCNAs across multiple cancers. The upper bar denotes the number of cancer types with amplifications (magenta) or deletions (green); the right bar denotes the number of amplifications (magenta) or deletions (green) in each cancer. Upper triangles denote the disulfidptosis score-high; Inverted triangles denotes the disulfidptosis score-low. (B) Chromosome plot displays locations of disulfidptosis-related SCNAs with significant alterations or harbored genes in the disulfidptosis score-high group vs. the disulfidptosis score-low group. Magenta indicates amplification in the disulfidptosis score-high group; green indicates amplification in the disulfidptosis score-low group. Pink indicates deletion in the disulfidptosis score-high group; light blue indicates deletion in the disulfidptosis score-low group. The purple label shows clinical actionable genes (CAGs). (C) FDA-approved drugs targeting CAGs in disulfidptosis-related SCNAs. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.7. Disulfidptosis status associated with drug response and their therapeutic liability
To comprehensively characterize the functional effects of disulfidptosis status on the response of current clinical drugs, we calculated the association analysis between the disulfidptosis score and 138 estimated drug susceptibility data in TCGA. The results showed that the number of drugs associated with disulfidptosis status ranged from 1 in OV to 71 in THYM (Fig. S8; FDR <0.05). Furthermore, we evaluated the effects of disulfidptosis on drug response using imputed drug data from patient samples. We found that disulfidptosis score-high cancer samples were more sensitive to various drugs. For example, patients with higher disulfidptosis score demonstrated greater sensitivity to cytarabine in five cancer types (Fig. S8), which is consistent with the previous observation that glucose uptake restriction, such as treatment with GLUT1 inhibitors, makes acute myeloid leukemia cells more sensitive to cytarabine [54]. In addition, a total of 99 clinically actionable genes (CAGs) were targeted by 83 clinically available drugs, including 27 FDA-approved drugs (Fig. S9). These CAGs were altered at typically five different molecular levels across 26 cancer types (DNA methylation, mRNA expression, protein expression, somatic mutation and SCNAs) (Fig. S9). For example, the androgen receptor (AR) was significantly altered at the mRNA and protein levels in 12 cancer types, including PRAD. The AR, targeted by anti-androgen drugs (enzalutamide and flutamide), has been consistently reported to promote the progression of prostate cancer [55]. Moreover, previous studies identified a negative regulatory domain in the AR hinge region that interacts with the actin-binding cytoskeletal protein-filamin A (FLNa) [55], suggesting AR as a potential disulfidptosis-related gene. Overall, we found that disulfidptosis might also contribute to drug resistance, suggesting that disulfidptosis has a complex impact on drug responses, and the combined therapies need to be based on preliminary experiments and in-depth studies of the mechanism (Figs. S8–S9).
To directly validate our multi-omics findings in vitro, we firstly investigated whether glucose starvation-induced disulfidptosis differs between disulfidptosis score-high and score-low cell lines based on the Gan et al.'s findings. We obtained the gene expression matrix for cancer cell lines from GDSC. We then assessed disulfidptosis levels in each cancer cell line and divided the cancer cell lines into high- and low-disulfidptosis score group based on the median disulfidptosis score (Fig. 7A, Details see Table S2). In order to determine whether cell lines with different disulfidptosis scores have different sensitivities to low glucose, we selected 4 cancer cell lines for subsequent experimental verification based on currently available materials. We tested two disulfidptosis score-low cell lines including the human cervical carcinoma cell line Hela and the human hepatocellular carcinoma cell line Huh-7. We found that glucose starvation for 16 h could not trigger cell death in Hela and Huh-7 cells (Fig. 7B). Conversely, glucose depletion for 16 h triggered a striking decrease in cell viability in disulfidptosis score-high melanoma cell lines (A375 and SK-MEL-28) (Fig. 7C). Notably, glucose starvation-induced cytotoxicity could be largely abrogated by the disulfide-reducing agent dithiothreitol (DTT), but not by inhibitors of ferroptosis (liproxstatin-1), apoptosis (Z-VAD-FMK), necroptosis (necrostatin-1s), or autophagy (chloroquine) (Fig. 7C). To further corroborate this finding, we conducted lactate dehydrogenase (LDH) release experiments as a direct indicator of cell death. We found that treating SK-MEL-28 cells with glucose-free medium induced a significant increase in LDH release, and this increased cell death could be completely abolished by DTT (Fig. 7D). 2DG, a glucose analog, has been reported to produce NADPH through the PPP and further inhibit disulfidptosis [6,10]. Consistent with this finding, treatment with 2DG significantly mitigated cytotoxicity induced by glucose deprivation in disulfidptosis score-high melanoma cell lines A375 and SK-MEL-28, but not in disulfidptosis score-low Hela and Huh-7 cells (Fig. S10A). The extensive import of cystine and its subsequent reduction to cysteine contribute to NADPH depletion and disulfidptosis during glucose starvation [6,10]. Therefore, the short-term removal of cystine from the culture medium could rescue disulfidptosis. As expected, we observed that removal of cystine from the culture medium indeed suppressed glucose deprivation-induced cytotoxicity in A375 and SK-MEL-28 cells, whereas Hela and Huh-7 cells showed minimal response (Fig. S10B). Prolonged culturing of A375 and SK-MEL-28 cells in cystine-depleted medium eventually resulted in significant cell death, which could be rescued by the ferroptosis inhibitor liproxstatin-1 (Fig. S10C), further supporting the well-established notion that cystine depletion induces ferroptosis in cancer cells over a long period of time [10]. Furthermore, glucose depletion neither inhibited cell proliferation nor promoted cell cycle arrest in SK-MEL-28 cells (Fig. 7E and F). Likewise, glucose starvation did not induce apoptosis in SK-MEL-28 cells detected by caspase-3 activity and Annexin V/PI apoptosis assays (Fig. 7G and H). Therefore, our data strongly suggest that glucose deprivation-induced cytotoxicity is not associated with inhibition of cell proliferation or increased apoptosis but drives a unique form of cell death known as disulfidptosis in A375 and SK-MEL-28 melanoma cells.
Fig. 7.

Disulfidptosis-related molecular features in clinically actionable genes and drug response associated with disulfidptosis in vitro. (A) Disulfidptosis scores for 25 types of cancer that including 760 cancer cell lines. Cancer types are sorted by the median disulfidptosis score (horizontal black line) for each cancer type. Each dot represents one cancer cell line. Sample sizes for each cancer type are listed near the bottom. The horizontal black dotted line means the median value of disulfidptosis score in all the 760 cancer cell lines that divide all the cancer cell lines into disulfidptosis score-high (red) and disulfidptosis score-low (green) groups. (B–C) Relative cell viability in the HeLa, Huh-7 (B; Green label; Disulfidptosis-score low), A375 and SK-MEL-28 (C; Red label; Disulfidptosis-score high) cells cultured in glucose-containing (+Glucose) or glucose-free (-Glucose) medium with or without indicated concentrations of liproxstatin-1 (Lip-1), Z-VAD-FMK (Z-VAD), necrostatin-1s (Nec-1s), chloroquine (CQ), and dithiothreitol (DTT) for about 16 h. (D) LDH release analysis of SK-MEL-28 cells after indicated treatments for about 14 h. (E) Representative EdU staining images and the ratio of EdU positive SK-MEL-28 cells after indicated treatments for about 12 h. The nuclei were stained with Hoechst (blue). Scale bar, 50 μm (200 × ). (F) Cell cycle distribution of SK-MEL-28 cells after indicated treatments for about 12 h. (G) Apoptosis analysis of SK-MEL-28 cells by flow cytometry at 14 h after receiving indicted treatments and stained by PI and Annexin V. (H) Caspase-3 activity of SK-MEL-28 cells after indicated treatments for about 14 h. STS, staurosporine, the apoptosis inducer. (I) Drug response associated with the disulfidptosis status in SKCM. The green point denotes drug sensitivity; the magenta point denotes drug resistance. (J–K) Dose-response curves for relative cell viability of etoposide, methotrexate, CMK (E; drug sensitivity), GW-441756, ABT-263, and NS-87877 (F; drug resistant) in glucose-containing (+Glucose) and glucose-free (-Glucose) conditions in the SK-MEL-28 cells. Quantification data are presented as mean ± SD and compared with one-way ANOVA in B–H and two-way ANOVA in J-K. NS, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Subsequently, we predicted the effect of disulfidptosis on drug sensitivity in melanoma (Fig. 7I and S10D). For drug compounds that predicted increased sensitivity under disulfidptosis status, we selected three drugs (etoposide, methotrexate, and CMK) for subsequent experiments, excluding OSI.906 due to unavailability. Consistent with our computational prediction, SK-MEL-28 cells became significantly sensitive to etoposide, methotrexate and CMK under the glucose starvation-induced disulfidptosis status compared with normal conditions (Fig. 7J). Moreover, A375 cells also tend to be more sensitive to methotrexate and CMK in the context of glucose deprivation-triggered disulfidptosis (Fig. S10E). Among the drug compounds that predicted decreased sensitivity under disulfidptosis conditions, we chose three of the top seven drugs (GW-441756, ABT-263, and NSC-87877), excluding others due to unavailability. Consistently, glucose starvation-induced disulfidptosis status conferred resistance to GW-441756, ABT-263, and NSC-87877 in SK-MEL-28 cells (Fig. 7K). Taken together, these results suggest that disulfidptosis induction could indeed affect the sensitivity of tumor cells to anti-tumor drugs, and the combination of disulfidptosis inducers and anti-tumor drugs may provide an alternative direction for cancer therapy.
4. Discussion
Disulfidptosis is a groundbreaking discovery in the field of RCD, uncovering a novel mechanism for organisms to combat malignant progression of tumors [7]. Therefore, elucidating the effect of disulfidptosis on molecular alternations is crucial for the development of effective therapeutic strategies. The hallmark of disulfidptosis is F-actin contraction and detachment from the plasma membrane, which is not easily detectable under human physiological and pathological conditions [6]. Hence, it is urgent to establish a robust predictive signature for assessing tumor disulfidptosis status in large-scale cancer patients and explore the impact of disulfidptosis on molecular alternations in multiple dimensions.
In this study, we first proved the stability of a 23 gene signature for defining disulfidptosis status across various cancer types [6,9,10]. Our findings show the extensive heterogeneity exists in disulfidptosis levels within and across cancer types. The location and staging of tumors may be one of the potential causes of the phenomenon, suggesting that tumor location and staging should be considered when developing treatment strategies targeting disulfidptosis. Thus, this may provide a reference for selecting accurate cancer patients to use disulfidptosis targeting drugs in future prospective trials. Based on above signatures, we stratified cancer patients into disulfidptosis score-high, score-intermediate, and score-low groups and PSM algorithm was applied to eliminate the influence of confounding factors on the analysis [19,20,26]. Subsequently, we identified molecular alternations at multidimensional levels, including mRNA, miRNA, protein, methylation, mutation, and SCNA, in patients with different disulfidptosis status across 26 cancer types. Importantly, we found that disulfidptosis is associated with cancer patient prognosis and drug sensitivity. Our comprehensive analysis provides a strong multi-omics foundation for the clinical translation of disulfidptosis, as demonstrated by previous rigorous research [20,26].
Particularly, we observed that 99 out of 121 (81.8%) clinically actionable genes exhibited significant alterations, resulting in altered sensitivity of patients to 83 clinical drugs under the influence of disulfidptosis. These drugs contain 27 FDA-approved anti-tumor drugs, covering a variety of treatment types such as immunotherapy, chemotherapy, hormone therapy and targeted therapy. Possibly due to the tumor heterogeneity, disulfidptosis had differential effects on molecular alterations in different tumor types, but the majority of clinically actionable genes were biased towards patients with active disulfidptosis status, suggesting that disulfidptosis-targeted treatment had synergistic effects on multiple cancer treatments and thus had the potential to combine currently clinically actionable genes to develop new therapeutic strategies. At present, there are limited studies on disulfidptosis [6,9,10,56], so we directly validate our multi-omics findings in vitro. As expected, disulfidptosis score-high cell lines, such as melanoma cells, were more susceptible to glucose starvation-induced cell death, which could be abolished by the disulfide reducing agent DTT. Previous studies indicated that glucose uptake supports cell proliferation, and its depletion could induce apoptosis [57,58]. Here, we found that glucose starvation had no effect on cell proliferation and apoptosis in melanoma cells within a short induction time. This discrepancy may be attributed to differences in the glucose-free induction time and cancer types because glucose starvation could induce rapid cell death as disulfidptosis in disulfidptosis-sensitive cells, while cause apoptosis with much delayed kinetics in disulfidptosis-resistant cells [6]. We further found that glucose depletion-induced disulfidptosis status enhances cell sensitivity to certain anti-tumor drugs (etoposide, methotrexate, and CMK). These findings could facilitate the development of relevant clinical treatments based on disulfidptosis status.
There are still several limitations in our research. Firstly, although disulfidptosis status can be directly evaluated by specific inhibitors or co-staining, this method is challenging in large-scale patient samples. Therefore, here we assessed the disulfidptosis status of patients by a 23 gene signature and verified its robustness in 6 glucose-starved datasets, which still hold significant biological significance. Secondly, although we collected about 10,000 tumor samples from 33 cancer types, only 26 tumor types were finally included in the follow-up study due to the small number of samples in some tumor types. Further studies are needed to explore the effect of disulfidptosis on the tumors not analyzed in this study. Finally, our observations lack further validation in more rigorous clinical settings due to the limited data from most clinical trials. Nonetheless, our study emphasizes the significance of monitoring tumor disulfidptosis status in future clinical studies.
5. Conclusion
In conclusion, we firstly uncover a comprehensive profile of disulfidptosis-related molecular alterations at the pan-cancer levels and provide opportunities to utilize multiple drug sensitivities induced by disulfidptosis through multi-omics analysis. Our work highlights the important clinical application potential of disulfidptosis and lays the foundation for developing personalized treatment strategies for pan-cancer patients.
Ethics statement
Not Applicable.
Authors' contributions
G.D, Y.H, X.C. and F.Z conceived and designed the study; D.Z, Y.H contributed to analysis and interpretation of data and drafting of the manuscript. Y.M conducted the experiments. Y.D, Q.Z, Y.S, J.L, F.Z, Y.M, G.D revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National key research and development program (2022YFC2504700 to X.C.), the National Natural Science Foundation of China (Grant Nos. 82103183 to F.Z., 82102803, 82272849 to G.D.), Natural Science Foundation of Hunan Province (Grant Nos. 2022JJ40767 to F.Z., 2021JJ40976 to G.D.), and National Science Fund for Outstanding Youths in Hunan Province (2023JJ20093 to G.D.).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was supported by the National Key Research and Development Program (2022YFC2504700 to X.C.), the National Natural Science Foundation of China (Grant Nos. 82103183 to F.Z., 82102803, 82272849 to G.D.), Natural Science Foundation of Hunan Province (Grant Nos. 2022JJ40767 to F.Z., 2021JJ40976 to G.D.), and National Science Fund for Outstanding Youths in Hunan Province (2023JJ20093 to G.D.).
We are very grateful to Gene-Expression Omnibus (GEO) and the Cancer Genome Atlas (TCGA) database for providing the transcriptome and clinical information. We also thank BioRender (www.biorender.com) for the assistance for the schematic and regulatory mechanisms illustration.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102966.
Contributor Information
Furong Zeng, Email: zengflorachn@hotmail.com.
Xiang Chen, Email: chenxiangck@126.com.
Yi He, Email: yihe0902@163.com.
Guangtong Deng, Email: dengguagntong@outlook.com.
Abbreviations:
- Redox
oxidation–reduction
- GSH
glutathione
- ROS
Reactive oxygen species
- PPP
pentose phosphate pathway
- ES
enrichment score
- ssGSEA
single sample gene set enrichment analysis
- GEO
Gene Expression Omnibus
- SCNA
somatic copy number alteration
- TCGA
The Cancer Genome Atlas
- PSM
propensity score matching
- DA score
Differential abundance score
- GDSC
Genomics of Drug Sensitivity in Cancer
- CCLE
Cancer Cell Line Encyclopedia
- LAML
Acute Myeloid Leukemia
- ACC
Adrenocortical carcinoma
- BLCA
Bladder Urothelial Carcinoma
- LGG
Brain Lower Grade Glioma
- BRCA
Breast invasive carcinoma
- CESC
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL
Cholangiocarcinoma
- LCML
Chronic Myelogenous Leukemia
- COAD
Colon adenocarcinoma
- CNTL
Controls
- ESCA
Esophageal carcinoma
- FPPP
FFPE Pilot Phase II
- GBM
Glioblastoma multiforme
- HNSC
Head and Neck squamous cell carcinoma
- KICH
Kidney Chromophobe
- KIRC
Kidney renal clear cell carcinoma
- KIRP
Kidney renal papillary cell carcinoma
- LIHC
Liver hepatocellular carcinoma
- LUAD
Lung adenocarcinoma
- LUSC
Lung squamous cell carcinoma
- DLBC
Lymphoid Neoplasm Diffuse Large B-cell Lymphoma
- MESO
Mesothelioma
- MISC
Miscellaneous
- OV
Ovarian serous cystadenocarcinoma
- PAAD
Pancreatic adenocarcinoma
- PCPG
Pheochromocytoma and Paraganglioma
- PRAD
Prostate adenocarcinoma
- READ
Rectum adenocarcinoma
- SARC
Sarcoma
- SKCM
Skin Cutaneous Melanoma
- STAD
Stomach adenocarcinoma
- TGCT
Testicular Germ Cell Tumors
- THYM
Thymoma
- THCA
Thyroid carcinoma
- UCS
Uterine Carcinosarcoma
- UCEC
Uterine Corpus Endometrial Carcinoma
- UVM
Uveal Melanoma
- DTT
dithiothreitol
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- 1.Liu X., Zhuang L., Gan B. Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol. 2023 doi: 10.1016/j.tcb.2023.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Hayes J.D., Dinkova-Kostova A.T., Tew K.D. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung E.C., Vousden K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer. 2022;22(5):280–297. doi: 10.1038/s41568-021-00435-0. [DOI] [PubMed] [Google Scholar]
- 4.Harris I.S., DeNicola G.M. The complex interplay between antioxidants and ROS in cancer. Trends Cell Biol. 2020;30(6):440–451. doi: 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Koppula P., Zhuang L., Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein & cell. 2021;12(8):599–620. doi: 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X., Nie L., Zhang Y., Yan Y., Wang C., Colic M., et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023;25(3):404–414. doi: 10.1038/s41556-023-01091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng T., Liu Q., Xing F., Zeng C., Wang W. Disulfidptosis: a new form of programmed cell death. J. Exp. Clin. Cancer Res. : CR. 2023;42(1):137. doi: 10.1186/s13046-023-02712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koppula P., Zhang Y., Shi J., Li W., Gan B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017;292(34):14240–14249. doi: 10.1074/jbc.M117.798405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y., Shi J., Liu X., Xiao Z., Lei G., Lee H., et al. H2A monoubiquitination links glucose availability to epigenetic regulation of the endoplasmic reticulum stress response and cancer cell death. Cancer Res. 2020;80(11):2243–2256. doi: 10.1158/0008-5472.CAN-19-3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X., Olszewski K., Zhang Y., Lim E.W., Shi J., Zhang X., et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020;22(4):476–486. doi: 10.1038/s41556-020-0496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koren E., Fuchs Y. Modes of regulated cell death in cancer. Cancer Discov. 2021;11(2):245–265. doi: 10.1158/2159-8290.CD-20-0789. [DOI] [PubMed] [Google Scholar]
- 12.Tong X., Tang R., Xiao M., Xu J., Wang W., Zhang B., et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol. 2022;15(1):174. doi: 10.1186/s13045-022-01392-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadian K., Stockwell B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 2023 doi: 10.1038/s41573-023-00749-8. [DOI] [PubMed] [Google Scholar]
- 14.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., et al. The cancer genome Atlas pan-cancer analysis project. Nat. Genet. 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X., Zhang N., Wu H.J., Wu H. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genome Biol. 2017;18(1):17. doi: 10.1186/s13059-016-1143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T., Fu J., Zeng Z., Cohen D., Li J., Chen Q., et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1) doi: 10.1093/nar/gkaa407. W509-w14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y., Mohanty V., Dede M., Tsai K., Daher M., Li L., et al. Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux. Nat. Commun. 2023;14(1):4883. doi: 10.1038/s41467-023-40457-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo M., Ye L., Chang R., Ye Y., Zhang Z., Liu C., et al. Multi-omics characterization of autophagy-related molecular features for therapeutic targeting of autophagy. Nat. Commun. 2022;13(1):6345. doi: 10.1038/s41467-022-33946-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Y., Hu Q., Chen H., Liang K., Yuan Y., Xiang Y., et al. Characterization of hypoxia-associated molecular features to aid hypoxia-targeted therapy. Nat. Metab. 2019;1(4):431–444. doi: 10.1038/s42255-019-0045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geeleher P., Zhang Z., Wang F., Gruener R.F., Nath A., Morrison G., et al. Discovering novel pharmacogenomic biomarkers by imputing drug response in cancer patients from large genomics studies. Genome Res. 2017;27(10):1743–1751. doi: 10.1101/gr.221077.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Allen E.M., Wagle N., Stojanov P., Perrin D.L., Cibulskis K., Marlow S., et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014;20(6):682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H.Y., Lin Y.C., Cui S., Huang Y., Tang Y., Xu J., et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022;50(D1) doi: 10.1093/nar/gkab1079. D222-d30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3) doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B., Khodadoust M.S., Liu C.L., Newman A.M., Alizadeh A.A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 2018;1711:243–259. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y., Dong Y., Chen Y., Zhang G., Zhang H., Lei G., et al. Multi-omics characterization and therapeutic liability of ferroptosis in melanoma. Signal Transduct. Targeted Ther. 2022;7(1):268. doi: 10.1038/s41392-022-01067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mergenthaler P., Lindauer U., Dienel G.A., Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36(10):587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentsen M.A., Mirzadeh Z., Schwartz M.W. Revisiting how the brain senses glucose-and why. Cell Metabol. 2019;29(1):11–17. doi: 10.1016/j.cmet.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wasserman D.H. Four grams of glucose. Am. J. Physiol. Endocrinol. Metabol. 2009;296(1):E11–E21. doi: 10.1152/ajpendo.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiyama A., Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. : TIG (Trends Genet.) 2021;37(11):1012–1027. doi: 10.1016/j.tig.2021.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Mattei A.L., Bailly N., Meissner A. DNA methylation: a historical perspective. Trends Genet. : TIG (Trends Genet.) 2022;38(7):676–707. doi: 10.1016/j.tig.2022.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Tang C., Xie A.X., Liu E.M., Kuo F., Kim M., DiNatale R.G., et al. Immunometabolic coevolution defines unique microenvironmental niches in ccRCC. Cell Metabol. 2023;35(8) doi: 10.1016/j.cmet.2023.06.005. 1424-40.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ying M., You D., Zhu X., Cai L., Zeng S., Hu X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021;46 doi: 10.1016/j.redox.2021.102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivanand S., Vander Heiden M.G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell. 2020;37(2):147–156. doi: 10.1016/j.ccell.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S., Rauch J., Kolch W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020;21(3) doi: 10.3390/ijms21031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong M.S., Deng S., Halim C.E., Cai W., Tan T.Z., Huang R.Y., et al. Cytoskeletal proteins in cancer and intracellular stress: a therapeutic perspective. Cancers. 2020;12(1) doi: 10.3390/cancers12010238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 38.Bambang I.F., Lu D., Li H., Chiu L.L., Lau Q.C., Koay E., et al. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp. Cell Res. 2009;315(11):1964–1974. doi: 10.1016/j.yexcr.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira-Rizzo C., Ottati M.C., Fort R.S., Chavez S., Trinidad J.M., DiPaolo A., et al. Hsa-miR-183-5p modulates cell adhesion by repression of ITGB1 expression in prostate cancer. Non-coding RNA. 2022;8(1) doi: 10.3390/ncrna8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q., Wu X., Guo L., Shi J., Li J. MicroRNA-7-5p induces cell growth inhibition, cell cycle arrest and apoptosis by targeting PAK2 in non-small cell lung cancer. FEBS open bio. 2019;9(11):1983–1993. doi: 10.1002/2211-5463.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo F., Yuan Y., Chen Z., Gao F., Li X., Wang H., et al. Downregulation of the long non-coding RNA MALAT1 in tenofovir-treated pregnant women with hepatitis B virus infection promotes immune recovery of natural killer cells via the has-miR-155-5p/HIF-1α axis. Int. Immunopharm. 2022;107 doi: 10.1016/j.intimp.2022.108701. [DOI] [PubMed] [Google Scholar]
- 42.Li X., Wang S., Mu W., Barry J., Han A., Carpenter R.L., et al. Reactive oxygen species reprogram macrophages to suppress antitumor immune response through the exosomal miR-155-5p/PD-L1 pathway. J. Exp. Clin. Cancer Res. : CR. 2022;41(1):41. doi: 10.1186/s13046-022-02244-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quero L., Tiaden A.N., Hanser E., Roux J., Laski A., Hall J., et al. miR-221-3p drives the shift of M2-macrophages to a pro-inflammatory function by suppressing JAK3/STAT3 activation. Front. Immunol. 2019;10:3087. doi: 10.3389/fimmu.2019.03087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dragomir M.P., Fuentes-Mattei E., Winkle M., Okubo K., Bayraktar R., Knutsen E., et al. Anti-miR-93-5p therapy prolongs sepsis survival by restoring the peripheral immune response. J. Clin. Investig. 2023;133(14) doi: 10.1172/JCI158348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan C., Shi W., Zhang Y., Liu C., Hu T., Chen D., et al. MiR-93-5p inhibits retinal neurons apoptosis by regulating PDCD4 in acute ocular hypertension model. Life Sci. Alliance. 2023;6(9) doi: 10.26508/lsa.202201732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong Z.R., Cai J.B., Shi G.M., Yang Y.F., Huang X.Y., Zhang C., et al. Oncogenic miR-93-5p/Gal-9 axis drives CD8 (+) T-cell inactivation and is a therapeutic target for hepatocellular carcinoma immunotherapy. Cancer Lett. 2023;564 doi: 10.1016/j.canlet.2023.216186. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q., Yang H.S. The role of Pdcd4 in tumour suppression and protein translation. Biol. Cell. 2018 doi: 10.1111/boc.201800014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng H., Schorpp K., Jin J., Yozwiak C.E., Hoffstrom B.G., Decker A.M., et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10) doi: 10.1016/j.celrep.2020.02.049. 3411-23.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Androic I., Krämer A., Yan R., Rödel F., Gätje R., Kaufmann M., et al. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer. 2008;8:391. doi: 10.1186/1471-2407-8-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu R., Yun Q., Zhang J., Wang Z., Zhang X., Bao J. Knockdown of circular RNA tousled-like kinase 1 relieves ischemic stroke in middle cerebral artery occlusion mice and oxygen-glucose deprivation and reoxygenation-induced N2a cell damage. Bioengineered. 2022;13(2):3434–3449. doi: 10.1080/21655979.2021.2024684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang J., Fan J., Wang M., Niu Z., Zhang Z., Yuan L., et al. CDKN2A inhibits formation of homotypic cell-in-cell structures. Oncogenesis. 2018;7(6):50. doi: 10.1038/s41389-018-0056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adib E., Nassar A.H., Akl E.W., Abou Alaiwi S., Nuzzo P.V., Mouhieddine T.H., et al. CDKN2A alterations and response to immunotherapy in solid tumors. Clin. Cancer Res. 2021;27(14):4025–4035. doi: 10.1158/1078-0432.CCR-21-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim N., Kim J.M., Lee M., Kim C.Y., Chang K.Y., Heo W.D. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem. Biol. 2014;21(7):903–912. doi: 10.1016/j.chembiol.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 54.Åbacka H., Hansen J.S., Huang P., Venskutonytė R., Hyrenius-Wittsten A., Poli G., et al. Targeting GLUT1 in acute myeloid leukemia to overcome cytarabine resistance. Haematologica. 2021;106(4):1163–1166. doi: 10.3324/haematol.2020.246843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loy C.J., Sim K.S., Yong E.L. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc. Natl. Acad. Sci. U. S. A. 2003;100(8):4562–4567. doi: 10.1073/pnas.0736237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X., Zhang Y., Zhuang L., Olszewski K., Gan B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis. 2021;8(6):731–745. doi: 10.1016/j.gendis.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu J., Thompson C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019;20(7):436–450. doi: 10.1038/s41580-019-0123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El Mjiyad N., Caro-Maldonado A., Ramírez-Peinado S., Muñoz-Pinedo C. Sugar-free approaches to cancer cell killing. Oncogene. 2011;30(3):253–264. doi: 10.1038/onc.2010.466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.