

Abstract
Although aging has traditionally been viewed as the most important risk factor for osteoarthritis (OA), an increasing amount of epidemiological evidence has highlighted the association between metabolic abnormalities and OA, particularly in younger individuals. Metabolic abnormalities, such as obesity and type II diabetes, are strongly linked to OA, and they affect both weight-bearing and non-weight-bearing joints, thus suggesting that the pathogenesis of OA is more complicated than the mechanical stress induced by overweight. This review aims to explore the recent advances in research on the relationship between metabolic abnormalities and OA risk, including the impact of abnormal glucose and lipid metabolism, the potential pathogenesis and targeted therapeutic strategies.
Subject terms: Metabolic syndrome, Pathogenesis
Introduction
Osteoarthritis
Osteoarthritis (OA) is a chronic and degenerative joint disease that is prevalent among elderly individuals. It is characterized by cartilage destruction and persistent pain, which can severely impact quality of life. The global incidence of OA has been increasing due to the aging population and other contributing factors, such as metabolic disorders, with an estimated conservative number of 250 million OA patients worldwide1,2. The pathological changes of OA are multifaceted, including synovitis, cartilage degeneration, subchondral bone thickening, osteophyte formation, ligament degeneration, meniscus injury, and structural changes in the surrounding muscles2–5. Unfortunately, current treatment options for OA can only relieve pain and manage symptoms, rather than stopping or reversing disease progression. For most severe cases, joint replacement surgery may be needed, which is expensive, invasive and carries significant risks6,7. Thus, finding new strategies to prevent OA development or improve OA treatment is of utmost importance. Further research is needed to better understand the underlying mechanisms of the disease and to develop new therapies that can effectively treat OA.
The etiology of OA remains elusive, and recent studies have demonstrated that OA is a multifaceted disease influenced by various pathogenic factors8–10. Aging and overweight are well-known risk factors for OA, but metabolic homeostasis imbalance has also been implicated in its pathogenesis11. Inflammation plays a crucial role in the progression of OA, which often originates from the adipose tissue in the joint cavity12. In a cohort study evaluating the incidence of obesity and OA among 1 764 061 subjects, researchers found that the risk of knee osteoarthritis (KOA) in obese individuals was more than three times higher than the risk in healthy individuals13. In patients with obesity, adipose tissue produces adipokines such as leptin, lipocalin, resistin and endolipoproteins, as well as inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin 1 (IL-1) and interleukin 6 (IL-6)14–16. These mediators are released from local or systemic adipose tissue as the result of joint trauma or overuse and can significantly impact the development and progression of OA17. Further research is needed to fully understand the complex mechanisms underlying the relationship between obesity, inflammation and OA, which may lead to the development of novel therapeutic strategies.
Metabolic abnormalities
Metabolic abnormalities encompass a range of conditions, such as obesity, hypertension, dyslipidemia, hyperglycemia, and insulin resistance. These risk factors not only contribute to the onset and progression of OA but also potentially increase the likelihood of developing cardiovascular disease. A multicenter study revealed that individuals with chronic obesity are prone to damage in the medial knee cartilage, leading to the development of OA18. Moreover, there exists a direct association between weight gain and the risk of KOA, as evidenced by a 35% increase in the risk for every five-unit increase in body mass index (BMI)19. It is thus clear that obesity plays a significant role in the manifestation of OA. Nevertheless, subsequent studies have demonstrated that hand osteoarthritis (HOA) can also occur in non-weight-bearing areas of obese individuals, suggesting that OA is not solely influenced by mechanical load but is also associated with metabolic abnormalities20. Chronic hyperglycemia and insulin resistance create an environment that promotes the development of OA, as elevated glucose levels induce the synthesis of proinflammatory cytokines and matrix metalloproteinases in joint tissues, leading to damage to human chondrocytes and the subsequent development of OA21,22. Additionally, dyslipidemia and hypertension have also been identified as causative factors for OA16,23. Consequently, metabolic disorder is one of the key risk factors for OA development and progression.
Association between metabolic disorders and OA risk
A cross-sectional study aimed to examine the association between dietary glycemic index (GI), dietary glycemic load (GL), and knee OA among Korean adults, and the results showed a significant positive association between dietary GI and symptomatic KOA in women24. In addition, a study investigated the relationship between the Mediterranean diet (with lower GI) and prevalence of OA of the knee in a large cohort from North America, where the researchers found that individuals who had higher adherence to the Mediterranean diet usually had a lower risk of KOA25.
Metabolic abnormalities not only augment the susceptibility of OA but also impede the functional recovery of joint replacement surgery. A clinical follow-up study conducted in Canada found that metabolic abnormalities adversely impact patient functionality subsequent to joint replacement surgery, especially in the case of hip surgery26. Furthermore, in another cross-sectional investigation examining the relationship between metabolic syndrome and symptomatic KOA, the results demonstrated a positive correlation between the severity of symptomatic KOA and the metabolic syndrome accumulation factor27.
The adverse effects of diabetes and obesity on OA
OA and diabetes often coexist due to their high prevalence and common risk factors. Nearly 47.3% of individuals with diabetes had some form of arthritis28. The negative impact of diabetes on joints could be explained by the induction of oxidative stress, pro-inflammatory cytokines, chronic high glucose concentration and insulin resistance.
Chronic high glucose concentration
In individuals with type 2 diabetes, elevated levels of blood glucose result in the generation and accumulation of advanced glycation end products (AGEs) due to the maintenance of prolonged hyperglycemia29. This process promotes matrix stiffness. Collagen, which is a key component of various connective tissues, exhibits an exceptionally low turnover rate, rendering it susceptible to modifications by AGEs. Additionally, AGEs bind to the receptor for AGEs (RAGE) on the chondrocyte membrane, initiating intracellular signaling that leads to the overexpression of proinflammatory and prodegradative mediators23,30. In human OA chondrocytes, the specific binding of AGEs to RAGE activates the MAPK signaling pathway, thereby enhancing the expression of IL-6 and IL-831. This, in turn, exacerbates the inflammatory response within chondrocytes31. Activation of RAGE by AGEs in articular chondrocytes prompts an increase in matrix catabolism in articular cartilage, ultimately contributing to the development of OA32. Moreover, elevated levels of AGEs in human articular chondrocytes further impede the turnover of the extracellular matrix in articular cartilage, promote cartilage degradation and diminish proteoglycan synthesis33. Studies have reported significantly higher levels of AGEs in the cartilage of OA patients than in healthy individuals32. Inhibition of the JAK/STAT3 signaling pathway following RAGE activation by AGEs led to a decrease in the expression of matrix metalloproteinase 13 (MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5), resulting in an increase in the synthesis of type II collagen (Col-2) in chondrocytes34. Researchers have also observed that incubation of rabbit chondrocytes with AGEs upregulates reactive oxygen species (ROS) expression, impairs mitochondrial function and induces chondrocyte death35. The accumulation of AGEs renders the collagen network in articular cartilage fragile, thereby increasing the risk of developing OA36.
Proinflammatory cytokines
Proinflammatory cytokines, including IL-1β, TNF-α, and IL-6, are primarily synthesized by activated macrophages and play a crucial role in the inflammatory response associated with OA37,38. Epidemiological studies have identified diabetes and obesity as contributing factors to the development of OA23,39. These conditions induce a local or systemic state of low-grade inflammation in the human body. Hyperglycemic environments and adipose tissue have been shown to increase the in vivo expression of proinflammatory factors, such as IL-1β, IL-6 and TNF-α. This upregulation further activates the nuclear factor-κB (NF-κB) signaling pathway, increases the catabolic activity of articular chondrocytes and promotes degradation of the extracellular matrix (ECM), ultimately leading to OA progression4,16,17,40–42. In a stress-induced mouse model of OA, researchers observed that the group fed a high-fat diet exhibited significantly higher serum levels of TNF-α and more severe cartilage damage than the control group. However, the Toll-like receptor-5-deficient (Tlr5 KO) mouse group, also on a high-fat diet, displayed significantly lower serum levels of IL-6 than the other groups, suggesting that obesity increases the expression of proinflammatory factors, thereby aggravating OA progression43. Another study demonstrated a significant elevation of IL-1β in the serum of mice fed a high-fat diet, with IL-1β inducing an inflammatory response in OA chondrocytes through activation of the NF-κB signaling pathway44,45. Notably, some studies have reported that metformin, a medication used to treat diabetes, not only reduces body mass index (BMI) in obese individuals but also decreases the rate of joint replacement surgery in patients with OA46–48. In murine studies conducted by Li et al., TNF-α and IL-1β markedly increased the mRNA expression levels of matrix metalloproteinase 3 (MMP3), MMP13, metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 in primary articular chondrocytes49. However, the addition of metformin effectively suppressed the expression of MMP13 and MMP3 induced by TNF-α and IL-1β, and it was further revealed that metformin exerted its inhibitory effects on MMP13 and MMP3 expression by attenuating the catabolic responses induced by inflammatory cytokines and promoting the expression of anabolic genes, thereby safeguarding articular chondrocytes49. Consequently, inhibiting the expression of proinflammatory factors through weight reduction or controlling diabetic blood glucose levels can confer positive therapeutic outcomes for the treatment of OA22,50.
Reactive oxygen species
Reactive oxygen species (ROS) are highly reactive molecules that contain oxygen and can cause damage to cells. Oxidative stress and mitochondrial dysfunction are known to be the primary sources of ROS. Extensive research has shown that obesity and diabetes can induce elevated levels of ROS in the body51–54. Adipose tissue and high blood glucose levels create a proinflammatory environment, leading to an increase in M1-type macrophages and proinflammatory cytokines such as IL-1, TNF-α, and IL-6. These factors contribute to tissue damage and further stimulate the secretion of proinflammatory cytokines, exacerbating oxidative stress and mitochondrial dysfunction; consequently, tissues experience heightened levels of ROS55,56. Overproduction of ROS is a contributing factor to the development and progression of OA57. In OA chondrocytes, excessive ROS production activates the MAPK and NF-κB signaling pathways, disrupting the balance between cartilage catabolism and anabolism. This imbalance leads to increased catabolism of articular cartilage, synovial inflammation and subchondral bone thickening56,58. Studies have demonstrated that incubation of chondrocytes with H2O2 results in increased ROS production, chondrocyte death59, degradation of chondrocyte ECM and inhibition of proteoglycan synthesis, which accelerate OA progression60,61. However, the use of ROS inhibitors or scavengers can slow cartilage loss. Jin et al. conducted experiments in a surgically induced mouse model of knee OA and found that intraperitoneal injection of ROS inhibitors significantly reduced the severity of cartilage damage in the knee joints62. Additionally, ROS inhibitors decreased the mRNA levels of MMP13 and ADAMTS5 in OA chondrocytes while increasing the mRNA levels of Col-2 and Aggrecan62. Thus, ROS inhibitors reduce cartilage damage by inhibiting ROS transduction in the MAPK and NF-κB signaling pathways. In the IL-1β-induced human synovial explant of the OA model, the level of the oxidative stress marker 8-OHdG exhibited a fourfold increase, and there was also a significant elevation in MMP13 and ADAMTS5 expression63. Notably, the addition of antioxidants resulted in a significant decrease in the expression of MMP13 and ADAMTS5, indicating that ROS inhibitors may possess the potential to alleviate synovial inflammation in OA63. Lu et al. employed an ACLT surgery-induced rat model of KOA and discovered that the ROS scavenger known as black phosphorus nanosheets (BPNSs) effectively eliminated intracellular ROS while concurrently maintaining cartilage morphology and impeding the reduction of subchondral bone volume in KOA64. Furthermore, it was observed that increased ROS levels in OA chondrocytes could hinder the mitochondrial respiratory chain and give rise to mtDNA mutations. This ROS-induced mtDNA damage subsequently prompted enhanced expression of MMP1 and MMP3 in chondrocytes, thereby further increasing the progression of OA58,65. Consequently, reducing ROS production exerts a significant effect on the retardation of articular chondrocyte senescence or the mitigation of associated damage66.
Insulin resistance of the diabetic synovial membrane
Insulin resistance refers to the diminished physiological response of specific organs or tissues in the body to normal insulin levels, necessitating higher insulin concentrations to maintain normal insulin function67. The presence of insulin resistance underlies the development of type 2 diabetes, and its impact on KOA severity is significantly more pronounced in individuals with type 2 diabetes than in nondiabetic KOA patients68; notably, diabetes induces more severe synovial inflammation or synovial thickening in both diabetic mice and patients with KOA69–71. Extensive expression of insulin receptors (IRs) has been observed in the synovium of both mice and humans69. Moreover, obese KOA patients with type 2 diabetes exhibit elevated levels of TNF in their synovium, whereas this elevation is not observed in obese KOA patients without diabetes69. Fibroblast-like synoviocytes (FLS) respond to increased TNF by upregulating the production of IL-1, TNF-α, IL-6, bone morphogenetic protein 2 (BMP-2), ADAMTS4 and MMP1369,72. Hamada et al. isolated synoviocyte fibroblasts from KOA patients without diabetes and found that insulin inhibited the induction of TNF-mediated cytokines, growth factors and proteases69. However, in diabetic KOA patients, the inhibitory effects of insulin on TNF-induced cytokines, growth factors and protease production are diminished due to impaired signaling by insulin-resistant synovial IRs69. BMP-2, in conjunction with cytokines and proteases, further intensifies the progression of OA, as it promotes the development of osteochondritis dissecans73. Additionally, chronic hyperglycemia triggers oxidative stress, proinflammatory cytokines and excessive production of AGEs within joint tissues. These factors induce the production of vascular endothelial growth factor (VEGF) and aggravate the synovial inflammatory response in human synoviocytes through activation of the RAGE-NF-κB pathway, ultimately leading to joint damage in OA patients30,74,75. In diabetic patients, insulin resistance promotes the progression of OA by impairing the protective and anti-inflammatory effects of insulin within the synovium. Studies have also identified another hepatic metabolic factor, LECT2, which is highly expressed in the liver76. LECT2 mediates glucose metabolism and obesity-related insulin resistance76. In a healthy male population, LECT2 concentrations in the blood increased with the intake of a high-fat diet, suggesting that LECT2 has an important effect on metabolic homeostasis in the body77. The researchers found that LECT2 appeared at high expression levels in OA patients and elderly individuals, suggesting that LECT2 may be involved in OA78. Further studies are needed in the future to determine the pathological mechanisms of LECT2 in OA.
In conclusion, obesity- and diabetes-induced hyperglycemia, oxidative stress, inflammatory response and insulin resistance suggest that disturbance in glucose metabolism may contribute to metabolic OA (Fig. 1).
Fig. 1.

Effect of Glucose Metabolism on Osteoarthritis. a Obesity and diabetes lead to increased pro-inflammatory cytokines IL-1β, IL-6, TNF-α, and ROS, activate the NF-κB pathway and promote the expression of MMP3, MMP13, ADAMTS4 and ADAMTS5. b Normal synovial cells can receive insulin, which acts as an anti-inflammatory. Insulin-resistant synovial cells are insensitive to insulin, resulting in increased levels of the proinflammatory cytokines IL-1β, IL-6, growth factor BMP-2, protease MMP13 and ADAMTS4, aggravating the degree of osteoarthritis. c Hyperglycemia induces the production of AGEs, which promote cartilage matrix stiffness as well as ROS production. AGEs bind to RAGE, activate the MAPK signaling pathway in chondrocytes, and promote the production of IL, MMP and ADAMTS. d Hyperglycemia promotes the expression of ROS and M1 macrophage activation. ROS activate the MAPK and NF-κB pathways, promote MMP13 and ADAMTS5 and inhibit the expression of Col-2 and Aggrecan. ROS inhibitors inhibit the activation of this pathway. IL-1β interleukin-1β, IL-6 interleukin-6, TNF-α tumor necrosis factor-α, ROS reactive oxygen species, NF-κB nuclear factor-κ-gene binding, MMP3 matrix metalloproteinases 3, MMP13 matrix metalloproteinases 13, ADAMTS4 a disintegrin and metalloproteinase protein 4, ADAMTS5 a disintegrin and metalloproteinase protein 5, BMP-2 bone morphogenetic protein-2, AGEs advanced glycation end products, RAGE receptor for advanced glycation end products, MAPK mitogen-activated protein kinase, Col-2 Collagen-2
Lipid metabolism and OA
The process of lipid metabolism includes the synthesis and degradation of lipids in the cell, which is critical for the proper functioning of living organisms. Lipid metabolism involves the digestion, absorption, synthesis, storage and breakdown of fats and the transport of various synthesized substances throughout the body to meet physiological needs, such as the construction of cell membranes. The action of various enzymes and bile salts hydrolyzes fats into glycerol, fatty acids and other substances. Lipids are absorbed through two mechanisms: triglycerides composed of medium-chain and short-chain fatty acids are emulsified and directly absorbed into the blood, while triglycerides containing long-chain fatty acids combine with apolipoproteins and cholesterol to form chylomicrons, which are ultimately absorbed into the blood via the lymphatic system79. After metabolism and absorption, fats are divided into four lipid groups: triglycerides, phospholipids, cholesterol, and plasma lipoproteins. Other substances in the body control the four lipid groups and change them into substances needed for various biochemical processes in the organism. However, when the homeostatic balance of lipid metabolism is disturbed, it will predispose the body to diseases, including OA11. Numerous studies have found that patients with lipid metabolism disorders suffer from a higher risk of OA16,80,81. In a national study in the United States, which focused on the prevalence of OA and metabolic syndrome in subjects with OA and the general population without OA, the results showed that the prevalence of OA was more than twice as high in individuals with metabolic disorders as in the control population82. This review examines the relevance of important lipid metabolites in the development of OA and explores the underlying mechanisms of lipid metabolism disorders in OA pathology, thus providing new insights into the treatment of metabolic OA.
Effects of triglyceride metabolism on OA
A triglyceride is an ester consisting of glycerol and three fatty acids. Elevated levels of serum triglycerides are also a risk factor for the progression of OA83. During moderate- to low-intensity exercise, the breakdown of triglycerides can provide most of the energy needed by the exercising muscles. However, when there is excessive fat intake over a prolonged period, the breakdown and metabolism of fats can exceed the body’s capacity, resulting in the accumulation of fatty acids. Triglycerides stored in adipose tissue are gradually hydrolyzed into glycerol and free fatty acids (FFAs) by lipases and released into the bloodstream. Fatty acids can be classified as saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs) based on the length of their carbon chain and the number of double bonds.
Excessive lipid intake leads to an increased breakdown and metabolism of triglycerides, resulting in elevated levels of SFAs in the blood84. In vitro, studies have shown that treating cartilage explants with SFAs increases the expression of glycosaminoglycans (GAGs), IL-6 and poly (ADP-ribose) polymerase (PARP) and decreases the viability of chondrocytes in the top layer of the explants85. SFAs also induce the upregulation of the autophagy markers microtubule-associated protein and the expression of the p65 protein and activate autophagy and NF-κB signaling pathways in C28/I2 chondrocytes86. In an SFA-induced OA chondrocyte model, IL-1β and MMP13 mRNA expression was increased, while Col-2 and Sox9 mRNA expression was decreased and chondrocyte glucose uptake was reduced87. After feeding Wistar rats an SFA-containing diet for 16 weeks, the results of immunohistochemical (IHC) staining revealed an increase in MMP13 and Col-X expression and a decrease in aggrecan (ACAN) expression in the joint cartilage, and the results of micro-CT showed a decrease in the bone volume fraction of the tibia84. A clinical study of the relationship between diet and OA progression in 2 092 patients with OA showed that as dietary SFA levels in OA patients increased, the width of the joint space decreased by 0.26 mm, 0.27 mm, 0.31 mm and 0.35 mm at 12, 24, 36, and 48 months after feeding with a high SFA diet, respectively, suggesting that high levels of SFA intake may aggravate structural damage in KOA88.
There is relatively limited research on the relationship between MUFAs and the progression of OA. Gas chromatography‒mass spectrometry (GC‒MS) has been used to determine the fatty acid composition of the infrapatellar fat pad in a rabbit model of OA caused by anterior cruciate ligament transection (ACLT). This procedure led to a decrease in the amount of MUFAs in the knee joint, but the link between MUFAs and OA is still not clear89. However, the finding of an in vitro study demonstrated that administration of MUFAs may be able to inhibit cartilage degradation90. This may explain why the proportion of MUFAs is decreased in patients with knee joint OA. In a TNF-α-induced chondrocyte injury model, MUFAs were found to inhibit the mRNA expression of prostaglandin-endoperoxide synthase-2 (PTGS2) and matrix metalloproteinase 1 (MMP1), thus inhibiting cartilage degradation90. In a clinical study, the synovial fluid of 23 OA patients undergoing total knee replacement surgery was analyzed, and it was found that the level of MUFAs in the synovial fluid of the OA group was higher than that in the non-OA group. However, the mechanism underlying the relationship between MUFAs and OA cartilage is still unclear91. The different results obtained from observational studies may be due to differences in the selected study populations. Further research is needed to clarify the relationship between MUFAs and OA.
Polyunsaturated fatty acids (PUFAs) are a unique class of bioactive compounds that play important physiological roles in the human body. PUFAs can be classified into omega-3 (n-3) and omega-6 (n-6) PUFAs based on the position of their double bonds. N-3 PUFAs include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), while n-6 PUFAs include linoleic acid (LA) and arachidonic acid (AA)92,93. N-3 and n-6 PUFAs are precursors for the synthesis of eicosanoids, and the balance of these two molecules in the body plays an important role in stabilizing cell membrane function, regulating gene expression and maintaining cytokine function. In healthy adult chondrocytes cultured with n-6 PUFAs, the secretion of IL-6 was significantly increased94. AA increases PEG production and ADAMTS mRNA expression in canine chondrocytes95. In a male mouse fed a high n-6 PUFA diet, n-6 PUFAs cause the progression of OA, prolong the wound healing time and increase the expression of inflammatory adipokines96. In a prospective cohort study of 5 328 participants, including 42% men, the plasma n-6 PUFA levels in male OA patients were positively correlated with joint effusion and knee structural damage levels97. SFAs and n-6 PUFAs can exacerbate cartilage structure damage by enhancing cell apoptosis and the expression of cartilage degradation-related genes85,97. Studies have shown that increasing n-3 PUFAs and decreasing n-6 PUFAs in the bodies of transgenic fat-1 mice can significantly alleviate cartilage destruction and osteophyte formation in the mouse OA model, reduce the expression of MMP13 and ADAMTS5 in joint cartilage, and stop the loss of chondrocytes and extracellular matrix98. Another study found that n-3 PUFAs significantly reduced the mRNA expression of ADAMTS4, ADAMTS5, MMP3, MMP13, COX-2, IL-1β and TNF-α when bovine chondrocytes were incubated with n-3 PUFAs, while n-6 PUFAs had no effect on the mRNA expression of cartilage degradation-related genes and inflammatory cytokines99. The researchers collected plasma from 167 patients with knee joint OA and found that OA patients with a high n-6:n-3 PUFA ratio had lower pain thresholds and more obvious limitations in joint movement and activities100. In another clinical trial, symptomatic knee OA patients who took different doses of n-3 PUFAs (fish oil supplements) experienced a reduction in clinical pain symptoms and an increase in joint functions and physical activities in the first year of treatment101. The WOMAC score showed that OA patients treated with a low dose of n-3 PUFAs had greater improvement in pain levels and physical functions after 2 years of treatment101 (Fig. 2). We have summarized the fatty acid species and their effects on OA in Table 1.
Fig. 2.
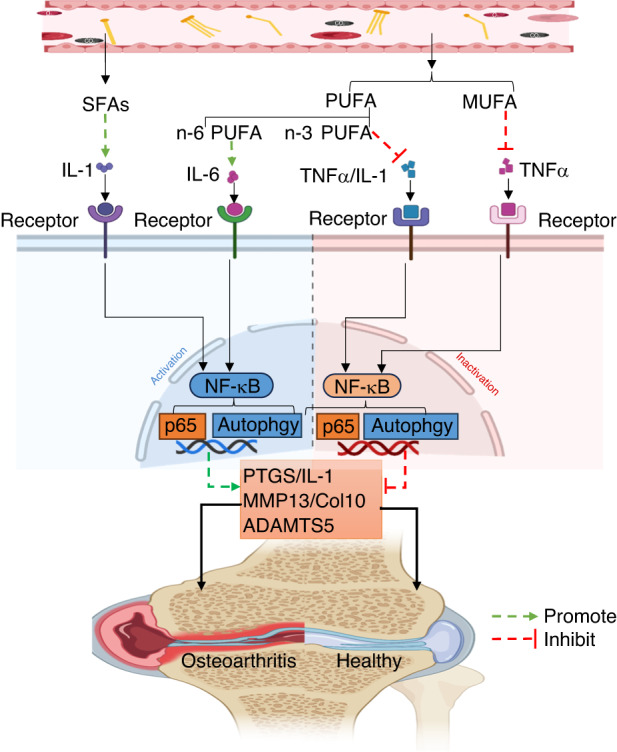
The mechanism of fatty acids in osteoarthritis. Excess SFAs and n-6 PUFAs in the blood directly or indirectly activate the NF-κB pathway by binding to the corresponding receptors, promoting the expression of PTGS, IL-1, MMP13, ADAMTS5, and COL10 and aggravating OA. MUFAs and n-3 PUFAs can inhibit the expression of proinflammatory factors such as TNF-α and IL-1 and block the activation of the MAPK and NF-κB pathways. SFAs saturated fatty acids, n-6 PUFAs omega-6 polyunsaturated fatty acids, NF-κB nuclear factor-k-gene binding, PTGS prostaglandin-endoperoxide synthase, IL-1 interleukin-1, MMP13 matrix metalloproteinases 13, ADAMTS5 a disintegrin and metalloproteinase protein 5, Col-X Collagen-10, MUFA monounsaturated fatty acids, n-3 PUFA omega-3 polyunsaturated fatty acids, TNF-α tumor necrosis factor-α, MAPK mitogen-activated protein kinase
Table 1.
Fatty acids and the development of OA
| Type of fatty acid | Type of study | Role in the pathogenesis of OA | Potential relationship with OA | Reference |
|---|---|---|---|---|
| SFA | In vitro model | Promotes IL-6 release, and ECM degradation, induces chondrocytes death. | Promote OA | 85 |
| In vitro model | Increased activation of autophagy and NF-κB signaling pathways. | 86 | ||
| In vitro model | Increased IL-1β and MMP13 expression, decreased the expression of collagen-II and Sox9. | 87 | ||
| Rat model | Increased MMP13 and Col-X expression, decreased bone volume fraction and ACAN expression. | 84 | ||
| Clinical trial | Reduce the width of the knee gap and aggravate the damage to the cartilage structure of the joint. | 88 | ||
| MUFA | In vitro model | Inhibit expression of PTGS2 and MMP1. | Inhibit OA | 90 |
| Rabbit model | Inhibit cartilage destruction. | 90 | ||
| N-6 PUFA | In vitro model |
1. Increased expression of IL-6. 2. Upregulation of PEG and ADAMTS expression. |
Promote OA | 94,95 |
| Mouse model | Prolongs the time to wound healing, and increases the expression of inflammatory adipokines. | 96 | ||
| Clinical trial | Exacerbates damage to the structure of the knee joint. | 97 | ||
| N-3 PUFA | In vitro model | Reduces the expression of ADAMTS4, ADAMTS5, MMP3, MMP13, COX-2, IL-1β, and TNF-α. | Inhibit OA | 99 |
| Mouse model | Reduces the expression of MMP13 and ADAMTS5, alleviate cartilage destruction and osteophyte hyperplasia. | 98 | ||
| Clinical trial | Reduces clinical pain symptoms and improves motor function. | 101 |
Effects of phospholipid metabolism on OA
Phospholipids are the main components of biological membranes and can be divided into two classes: glycerophospholipids and sphingolipids (SM). Glycerophospholipids are the most abundant type of phospholipids in the body and can participate in cell membrane recognition and signal transduction. Based on LC‒MS plasma lipidomics analysis, Pousinis et al. found 24 lipid spectra differences between the plasma of DMM-induced OA mice and sham-operated mice. The significantly higher SM in the plasma was positively correlated with the degree of DMM-induced joint cartilage injury102. Phosphatidylinositol-4-phosphate 5-kinase type γ (PIP5K1c) is a lipid kinase that catalyzes the synthesis of phosphatidylinositol 4,5-bisphosphate (PIP2) and participates in various cellular processes. It has been reported that mice with Pip5k1c gene deletion exhibit a variety of spontaneous OA pathological phenotypes, including cartilage degeneration, surface fissures, subchondral sclerosis, meniscus deformation, synovial hyperplasia and osteophyte formation103. These findings suggest that Pip5k1c expression in chondrocytes plays a critical role in maintaining joint tissue homeostasis.
In the pathological process of OA, the lubrication of synovial joints could be influenced by mechanical and molecular factors and by changes in synovial fluid. Synovial fluid can reduce joint wear and maintain tissue homeostasis. In healthy individuals, an effective lubricating layer is formed among the surfaces of cartilage and other joint tissues, and changes in the structure and composition of this layer could lead to lubrication abnormalities and dysfunction of joint tissues and OA symptom progression104. Researchers collected synovial samples from 13 OA patients who underwent knee replacement surgery for lipid measurements and found that the spatial distribution of glycerophospholipids was correlated with hypertrophic, inflamed or vascularized synovial regions105. Kosinska et al. used lipidomics and electrospray ionization tandem mass spectrometry methods to analyze synovial fluid (SF) samples from 17 early OA patients, 13 late OA patients, 18 RA patients, and 9 control donors postmortem and identified the following phospholipid categories in SF: phosphatidylcholine (PC), lysophosphatidylcholine (lysoPC), phosphatidylethanolamine, phosphatidylethanolamine-derived aldehyde phospholipid, phosphatidylglycerol, phosphatidylserine, sphingolipids and ceramides. Compared to the median PC concentration in the SF of the control group, the median PC concentration in the SF of early OA is 2.7-fold higher than that of the control, and the median PC concentration in the SF of late OA is 5.4-fold higher than that of the control106. In a clinical cohort study, investigators collected serum from 24 patients with KOA for metabolomics analysis and evaluated the volume of cartilage loss between baseline and 24 months using magnetic resonance imaging (MRI). The results showed that the increased serum ratio of lysoPC to PC was associated with the volume of lateral compartmental cartilage loss in the knee joint and the increase in the joint degradation markers COMP and MMP1107. A recent study has demonstrated the differences in SF phospholipidomics between knee OA patients and non-OA patients, with higher levels of PC, phosphatidylserine and phosphatidylinositol in the SF of OA patients than in non-OA control subjects108. The plasma ratio of lysoPC to PC also significantly increased in KOA patients109. This ratio could be used to predict OA risk, disease progression and treatment response110. This finding suggests that increased conversion of PC to lysoPC, which is catalyzed by phospholipase A2 (PLA2), is associated with OA progression111. Pruzanski et al. found that the concentration of PLA2 in cartilage is higher than that in synovium, suggesting that cartilage may be the main source of PLA2 production112. PLA2 has also been found to play a central role in OA inflammation113.
The impact of cholesterol metabolism on OA
Cholesterol in the human body mainly comes from two sources: endogenous biosynthesis and intestinal absorption. The liver is the major organ for cholesterol synthesis, and other tissues in the body can also synthesize cholesterol114. It has been reported that cholesterol can alter Indian hedgehog (IHH) activity to regulate the development of articular cartilage, suggesting that cholesterol plays an important role in cartilage development115. Numerous studies have shown that promoting cholesterol efflux or increasing cholesterol metabolism may help protect chondrocytes from the influence of inflammation116–118. Nuclear receptors are one of the most abundant transcriptional regulators in animals, as they play important roles in metabolism, differentiation, reproductive development and homeostasis maintenance. Reports have shown that nuclear receptors, such as liver X receptor (LXR), peroxisome proliferator-activated receptor (PPAR) and retinoic acid-related orphan receptor α (RORα), play a key role in the transcriptional regulation of lipid metabolism119–121. They are closely related to the occurrence and development of metabolic OA122.
LXR regulates cholesterol efflux-related genes and plays a key role in the transcriptional regulation of lipid metabolism-related genes as a member of the nuclear hormone receptor superfamily. By activating reverse cholesterol transport (RCT), LXR promotes the conversion of cholesterol into bile acids in the liver, thus protecting the body from hypercholesterolemia79,123,124. Reports have shown that the expression of cholesterol efflux genes in OA patients is significantly reduced and that the expression level of LXR is positively correlated with the expression of cholesterol efflux genes116,125. In a chondrocyte OA model, treatment with the LXR agonist TO-901317 significantly increased the mRNA and protein levels of cholesterol efflux genes ApoA1 and ABCA1 and reduced lipid deposition in OA chondrocytes, suggesting that changes in LXR levels in chondrocytes may be a contributing factor in the regulation of dynamic OA development116. It has also been found that LXR activation regulates the expression of lipid homeostasis-related genes in chondrocytes and the free cholesterol content in chondrocytes through the LXR–Srebp1–Scd1 axis120. After treatment with IL-1β and TNF-α, articular chondrocytes from OA patients showed significantly reduced LXR expression, leading to polysaccharide protein degradation through negative feedback regulation of the activated NF-κB signaling pathway, suggesting that decreased LXR expression levels could promote OA development126. In addition, LXR activation can significantly reduce the expression of proinflammatory cytokines such as TNF-α, COX-2, IL-1β, MMP9 and iNOS127 and inhibit Toll-like receptor-mediated inflammatory responses by promoting cholesterol efflux in macrophages128. Vaspin can inhibit miR-155 expression in rat chondrocytes and promote cholesterol efflux. When Vaspin expression is reduced, LXRα and other cholesterol efflux-related genes are inhibited in chondrocytes, leading to cholesterol accumulation in chondrocytes and worsening OA progression129. Reports have shown that metformin can activate the AMPK/SIRT1 signaling pathway to reverse IL-1β-induced extracellular matrix degradation in chondrocytes130,131. Activation of the AMPK/SIRT1 pathway upregulates LXRα expression, thereby promoting cholesterol efflux in chondrocytes130,131. AMPK signaling could interact with many signaling pathways that may be involved in OA occurrence and progression. For example, it has been shown that metformin inhibits β-cateninS552 phosphorylation and nuclear translocation132. The AMPK activators metformin and berberine inhibit OA progression49,133,134. In an IL-1-treated chondrocyte model, researchers observed that IL-1β significantly downregulated the mRNA and protein expression of cholesterol efflux-related factors ABCA1, ApoA1 and LXR in chondrocytes. Resveratrol (RES) can activate the SIRT1/FoxO1 signaling pathway to promote LXRα expression, reduce cholesterol accumulation in chondrocytes and delay OA progression135.
Due to the inability of peripheral cells to degrade cholesterol, excessive cholesterol efflux is the only way to eliminate cholesterol from these cells. When the LXR transcription factor is activated, it binds to the promoter sequence of the ABCA1 gene. ABCA1 acts as a lipid pump to efflux cholesterol and phospholipids from osteoarthritic chondrocytes to ApoA1 (Fig. 3), generating new high-density lipoprotein (nHDL) particles116,136,137. Lecithin-cholesterol acyltransferase (LCAT) catalyzes the conversion of free cholesterol to cholesterol esters to form mature HDL, which enters the liver through bile secretion and fecal excretion, reducing cholesterol levels in chondrocytes138. Currently, cholesterol efflux agonists have an important impact on inhibiting metabolic OA progression. LCAT deficiency directly affects the normal physiological function of HDL. Researchers found that LCAT-/- mice fed a high-fat diet for 24 weeks developed OA pathology, indicating that the abnormal physiological function of HDL led to the occurrence of an OA phenotype in mice139. There are also reports showing that high levels of HDL have a certain preventive effect on OA progression140. LCAT is mainly produced and secreted by the liver, and upregulation of LCAT expression has been shown to enhance the reverse cholesterol transport (RCT) process in mice with hepatic osteodystrophy, thereby alleviating bone loss141.
Fig. 3.

The role of liver nuclear receptor LXR in osteoarthritis. The liver nuclear receptor LXR binds to ABCA1, promotes the efflux of cholesterol in chondrocytes, inhibits the activation of the NF-κB pathway and reduces the expression of IL-1β, TNF-α, MMP13 and ADAMTS5. LXR agonists can promote LXR expression and strengthen the above two pathways. LXR liver X receptor, ABCA1 ATP-binding cassette transporter A1, NF-κB nuclear factor-k-gene binding, IL-1β interleukin-1β, TNF-α tumor necrosis factor-α, MMP13 matrix metalloproteinases 13, ADAMTS5 a disintegrin and metalloproteinase protein 5
PPAR plays an important role in lipid metabolism, the inflammatory response, and cell apoptosis142,143. PPAR regulates many metabolic processes in cells, including the three subtypes PPARα, PPARγ and PPARδ. Increasing evidence suggests that PPAR is involved in the occurrence and development of OA and is closely related to the regulation of lipid metabolism disorders and OA.
PPARα is present in chondrocytes, endothelial cells and hepatocytes and exhibits anti-inflammatory effects. In aging and surgically induced OA mouse models, it has been shown that the numbers of PPARα-positive chondrocytes decreased gradually in the cartilage with aging and OA progression144. Further IHC analysis showed that PPARα expression in the cartilage of patients with KOA was significantly lower than that in the non-OA individuals, suggesting that PPARα plays an important role in the homeostatic regulation of chondrocytes144. Researchers have also divided the cartilage of OA patients into relatively healthy (non-OA) and severely damaged (OA) groups and found that the lipid deposition area in the OA group was significantly increased compared to that in the non-OA group. Additionally, the expression of PPARα in chondrocytes and cartilage of OA patients was significantly reduced. However, in OA chondrocytes treated with PPARα agonists, lipid deposition was significantly reduced, suggesting that PPARα may be involved in the development of OA by regulating lipid metabolism. The study also found that PPARα regulates the balance of joint cartilage homeostasis through the PPARα–ACOT12 pathway145. More recent studies have demonstrated that PPARα protects against articular cartilage damage in a mouse OA model by inhibiting the inflammatory response146. The mechanism of lipid deposition in articular cartilage and the pathogenesis of OA are not fully understood and require further in-depth investigation in the future.
PPARγ is highly expressed in cartilage tissue, and PPARγ agonists reduce inflammation and prevent cartilage degradation in OA animal models147,148. A study showed that PPARγ expression was downregulated during the progression of OA in STR/Ort mice, which was aggravated under inflammatory conditions in joint cartilage, leading to knee joint cartilage damage and osteophyte formation149. It has been reported that promoting cholesterol efflux through PPARγ-mediated pathways can promote extracellular matrix synthesis in OA chondrocytes in rabbits118. Studies have found that SUMO-modified PPARγ can improve lipid metabolism disorders in chondrocytes. In summary, PPARγ activation has a potential therapeutic effect on OA, but the specific mechanism of PPARγ-regulated lipid metabolism in decelerating OA progression has yet to be fully elucidated150.
Unlike PPARα and PPARγ, which have positive effects on cartilage degradation in OA, current research suggests that PPARδ activation could exacerbate OA progression151. When a PPARδ agonist (GW501516) was used to treat mouse chondrocytes, it was found that PPARδ activation significantly increased the mRNA expression of MMP2, MMP3, ADAMTS2 and ADAMTS5 in chondrocytes compared to that of nontreated cells, and the degree of fatty acid oxidation in chondrocytes was significantly increased152. It has also been shown that deletion of the PPARδ gene in chondrocytes helps alleviate OA symptoms induced by DMM surgery in mice, indicating that PPARδ deficiency has an inhibitory effect on OA development152. The potential mechanism of PPARδ-exacerbated OA could be that PPARδ promotes fatty acid oxidation in chondrocytes, which can induce the production of ROS and accelerate OA progression.
During cholesterol overload, cholesterol signaling activation promotes chondrocyte hypertrophy by upregulating the expression of the nuclear receptor RORα. RORα is a downstream target of the cholesterol metabolism pathway, the cholesterol-25-hydroxylase (CH25H)-oxysterol-7α-hydroxylase (CYP7B1) axis. Overexpression of RORα upregulates cartilage degradation-related genes and downregulates the expression of anabolic metabolic factors153,154. Cui et al. induced effective overexpression of CH25H in the joint by intra-articular injection of adenovirus-CH25H (Ad-CH25H) in mice and found that RORα upregulated the downstream mediators of cholesterol metabolism, leading to severe cartilage damage, osteophyte formation and thickening of the subchondral bone plate in mice, indicating that the CH25H-CYP7B1-RORα axis is involved in cholesterol metabolism and plays a role in the pathological process of OA153. In an ACLT-induced OA mouse model, RORα siRNA delivered by adenovirus was administered into the knee joint two weeks after surgery, the expression of Aggrecan and Col2a1 in joint cartilage was increased, and cartilage damage was partially reversed by RORα siRNA. Researchers found that RORα may regulate the progression of OA through the IL-6/STAT3 signaling pathway155. miR-10a-3p is an upstream target of CH25H. miR-10a-3p can reduce the production of cartilage degradation enzymes in chondrocytes under inflammatory conditions through regulation of the CH25H-CYP7B1-RORα axis and protect cartilage degeneration in a rat OA model156. RORα plays an important role in regulating the cholesterol metabolism pathway and can be a potential target for the treatment of metabolic OA.
Low-density lipoprotein receptor-related protein 3 (LRP3) not only regulates the steady state of blood lipids and fibrinolysis but also participates in the regulation of cholesterol metabolism. It has been shown that LRP3 can positively regulate the metabolism of extracellular matrix in chondrocytes, and the downregulation of LRP3 can activate the Ras signaling pathway and upregulate syndecan-4 protein levels, aggravating mouse knee joint cartilage degeneration157.
High cholesterol levels may play a key role in the pathogenesis of OA. Hypercholesterolemia can lead to atherosclerosis, causing ischemia and hypoxia in the corresponding blood supply area, resulting in inadequate energy supply in the joint tissue. Therefore, when cholesterol accumulates in the joint, the blood supply to the subchondral bone is insufficient, enhancing the insufficient oxygen and nutrient supply to the subchondral bone and worsening the pathological process of OA158. We summarize the functional components of cholesterol in OA and its associated molecular pathways in Table 2.
Table 2.
The role of nuclear receptors associated with cholesterol metabolism in OA
| Nuclear receptor type | Mechanism of action of nuclear receptors | Signaling pathways in OA | Reference |
|---|---|---|---|
| LXR | Increased expression of cholesterol efflux genes ApoA1 and ABCA1 and reduced lipid deposition in chondrocytes. | NA | 116 |
| Promotes the expression of lipid homeostasis genes and reduces free cholesterol in chondrocytes. | LXR-Srebp1-Scd1 signaling pathway activation | 120 | |
| Promotes cholesterol efflux and inhibits inflammatory response. | NA | 128 | |
| Inhibits proteoglycan degradation. | NF-κB signaling pathway inhibition | 126 | |
| Inhibits expression of LXRα leads to the accumulation of cholesterol in cartilage. | NA | 129 | |
| Promotes the efflux of cholesterol inside chondrocytes. | AMPK/SIRT1 signaling pathway activation | 130,131 | |
| Reduces the accumulation of cholesterol in chondrocytes. | SIRT1/FoxO1 signaling pathway activation | 135 | |
| PPARα | Reduces lipid deposition. | PPARα − ACOT12 signaling pathway activation | 145 |
| PPARγ | Reduces micro environmental inflammation and catabolism in articular cartilage. | NA | 149 |
| Promotes extracellular matrix synthesis. | NA | 118 | |
| Inhibits abnormal lipid metabolism in chondrocytes. | NA | 150 | |
| PPARδ |
Increased expression of MMP2, MMP3, ADAMTS2 and ADAMTS5 in chondrocytes. Loss of PPAR-δ protects OA cartilage damage. |
NA | 152 |
| RORα |
1. Upregulation of cartilage degradation-related genes and downregulation of anabolic factors. 2. Exacerbates cartilage damage and subchondral bone thickening. |
CH25H-CYP7B1-RORα axis | 153,154 |
| Inhibition of RORα can promote the elevation of aggregate glycans and Col2a1 in articular cartilage. | IL-6/STAT3 signaling pathway | 155 |
The impact of plasma lipoprotein metabolism on OA
The structure of plasma lipoproteins is mostly spherical, consisting of a core of triglycerides and cholesterol esters, covered by a complex of lipids, phospholipids and free cholesterol molecules on the surface, ensuring the normal transport of lipids in the plasma. Lipoproteins can be classified into chylomicrons (CM), very-low-density lipoproteins (VLDL), low-density lipoproteins (LDL) and high-density lipoproteins (HDL) based on their density. HDL is mainly produced in the liver and is responsible for removing excess cholesterol from cell membranes. The plasma phospholipid cholesterol acyltransferase transfers fatty acid residues from phospholipids to cholesterol to produce cholesterol lipids, and then HDL transports cholesterol lipids to the liver, where excess cholesterol is converted into bile acids, maintaining the homeostasis of normal lipid metabolism in the body. Studies have found that metabolic syndrome and low HDL are associated with decreased medial tibial plateau cartilage volume, while insulin resistance, high waist circumference and low HDL-C are associated with tibial cartilage defects. Interventions targeting these pathogenic factors may prevent or delay knee joint OA progression159,160. However, in a study of the relationship between lipid and lipoprotein levels and OA in the MOST cohort, 337 symptomatic OA patients and 283 radiographic OA patients, of whom 55% were female, were included. The researchers found that the levels of total cholesterol, LDL and HDL in the serum of OA patients were not significantly correlated with joint cartilage loss, synovial inflammation or knee joint pain161. More basic and clinical investigations are needed to determine the role of lipoproteins in metabolic OA.
Lipoproteins are the protein component of plasma lipoproteins and are mainly divided into five categories: A, B, C, D and E. Lipoproteins are proteins that can bind and transport lipids to various tissues in the body for metabolism and utilization.
ApoA-1, the main component of HDL, plays an important role in lipoprotein clearance and cholesterol efflux in chondrocytes154. In a study in which 184 OA patients undergoing knee joint surgery and 180 healthy volunteers were recruited, researchers found that the expression levels of ApoA-1 in the synovial fluid (SF) of OA patients were negatively correlated with the severity of knee OA cartilage damage, radiographic severity, and severity of OA symptoms162. However, in a surgically induced rabbit KOA model, researchers detected a downregulation of ApoA-1 protein levels in the SF after ultrasound treatment163. Further exploration is needed to determine whether ApoA-1 can serve as a reliable marker for OA diagnosis.
ApoB is a basic structural component of CM, VLDL, IDL and LDL. Studies have found significant differences in ApoB levels between OA patients and healthy individuals, with higher levels in OA patients164. However, a bidirectional Mendelian randomization study found that elevated ApoB levels were negatively correlated with the risk of knee and hip OA165. Therefore, further research is needed to determine whether ApoB can serve as a molecular target for OA treatment.
ApoD is a secreted glycosylated protein and is an atypical lipoprotein that can bind to several small molecules, including arachidonic acid, steroids and cholesterol, and has important functions, such as antioxidation, anti-inflammation and anti-stress functions166–168. Qin et al. identified potential biomarkers for OA using weighted gene coexpression network analysis (WGCNA) and confirmed that ApoD was the only gene that was downregulated as a hub gene in multiple tissues169. The researchers collected serum samples from 113 KOA patients and 97 healthy controls for ELISA test and found that ApoD levels were significantly lower in KOA patients than in the control individuals, suggesting that serum ApoD levels may be associated with the severity of OA in OA patients170.
ApoE is an important component of plasma lipoproteins that primarily transports triglycerides and cholesterol to peripheral tissues. Farnaghi et al. generated an OA model in ApoE-deficient mice by feeding the mice a high-cholesterol diet for 4 weeks and found that the mice had increased osteophyte formation, aggravated cartilage degradation and more severe OA pathological symptoms171. In another study, researchers fed APOE-deficient mice with a high-cholesterol diet and found that the synovial membrane thickness increased in these mice172. These studies suggest that ApoE may play an important role in the occurrence and progression of metabolic OA.
ApoC is the main lipoprotein carrier of VLDL and an important regulator of lipoprotein metabolism, but there are currently almost no reports on the relationship between ApoC and OA.
The impact of other lipid metabolism-related proteins on OA
Leptin is a hormone secreted by adipose tissue, and its serum levels are positively correlated with the size of animal fat tissue. Leptin binds to its receptor and induces cellular responses through the JAK-STAT, PI3K, AMPK and MAPK signaling pathways173. Studies have found that leptin has a catabolic effect on cartilage metabolism. In a study using conditioned medium from a patellar fat pad (containing leptin) derived from OA patients to treat chondrocytes, leptin significantly induced collagen release and MMP expression in chondrocytes and activated signaling pathways such as JAK-STAT174. Bao et al. injected recombinant rat leptin (100 μg) into rat knee joints and found that leptin significantly increased the expression levels of MMP2, MMP9, tissue protease D, and Col-2 mRNAs and proteins and observed a decrease in proteoglycans in joint cartilage175. When leptin was used alone or in combination with IL-1β, it upregulated MMP production in human OA chondrocytes through signaling pathways, such as NF-κB and MAPK, leading to protein degradation of cartilage ECM176. In a study of 163 elderly individuals, researchers found that serum leptin levels were negatively correlated with the thickness of joint cartilage, suggesting that leptin may play an important role in changes in cartilage thickness177. These results suggest that leptin plays a catabolic role in cartilage metabolism and may be a detrimental factor in the pathological development of OA. However, in another study, researchers observed a significant increase in proteoglycan synthesis in all cartilage regions of the rat tibial plateau after injection of exogenous leptin (30 μg), indicating a protective effect of leptin on cartilage degradation, and these discrepancies may be dose-related178. Currently, the role of leptin in OA remains unclear, and further investigations are still needed.
Adiponectin, the most abundant adipokine in human plasma, is primarily secreted by white adipose tissue and plays a crucial role in regulating appetite and metabolism. Adiponectin exerts its biological effects through AdipoR1 and AdipoR2 receptors, which are expressed in various tissues, including the liver, articular cartilage, bone and synovium179–181. However, these two receptors have distinct functions in the body, with AdipoR1 mainly associated with AMPK signaling pathway activation, while AdipoR2 is linked to PPAR-α signaling pathway activation182.
In a study containing 12 patients undergoing knee replacement surgery for OA, researchers found that AdipoR1 and AdipoR2 expression levels were significantly higher in the OA cartilage lesion areas than in the non-lesion areas180. Moreover, the growth rate of AdipoR1-positive chondrocytes was significantly higher than that of AdipoR2-positive chondrocytes, suggesting that changes in AdipoR1 expression may better reflect the catabolic metabolism status of cartilage than AdipoR2. Adiponectin may accelerate the degradation of OA cartilage ECM through the activation of the JNK signaling pathway180.
Plasma adiponectin levels and adiponectin release from cartilage were found to be higher in patients with severe OA (Ahlbäck grades 4 and 5) than in those with mild OA (Ahlbäck grades 1 to 3). Adiponectin may activate the MAPK signaling pathway, leading to increased release of inflammatory cytokines and MMP expression in chondrocytes, thereby promoting the destruction of articular cartilage and heightening OA symptoms183. Similarly, in another study of OA patients undergoing total knee replacement surgery, adiponectin in knee synovial fluid significantly inhibited the aggregation of glycosaminoglycans in cartilage, suggesting that synovial adiponectin plays a positive role in cartilage damage184. Based on the current understanding of the relationship between adiponectin and OA, the development of pathway inhibitors related to adiponectin may be a promising avenue for the treatment of metabolic OA (Fig. 4).
Fig. 4.

The mechanisms of leptin and adiponectin in osteoarthritis. The adiponectin and leptin secreted by white adipose tissue bind to the corresponding receptors, activate MAPK and NF-κB pathways, and promote the expression of IL-1β, TNF-α, MMP2 and MMP9. Proinflammatory cytokines and matrix metalloproteinases act on articular cartilage, subchondral bone, synovial membrane and other areas, aggravating the phenotype of osteoarthritis. MAPK mitogen-activated protein kinase, NF-κB nuclear factor-k-gene binding, IL-1β interleukin-1β, TNF-α tumor necrosis factor-α, MMP2 matrix metalloproteinases 2, MMP9 matrix metalloproteinases 9
Lipid metabolism and OA treatment
Treatment of OA remains a challenging issue, with most therapeutic approaches aiming to alleviate pain, improve or restore joint function, enhance patient quality of life, delay disease progression and correct deformities. The treatment of OA requires a combination of pharmacological and nonpharmacological interventions, with surgery being necessary for severe cases in the advanced stages of the disease.
Nonpharmacological interventions include exercise and dietary management. Studies have shown that OA can be treated through nonpharmacological approaches such as aerobic and strength exercises. Regular moderate exercise can alleviate pain, improve physical function and significantly slow disease progression, making it an important component for early intervention of OA185–189. Overweight or obese patients with OA can achieve their ideal body weight by combining dietary adjustments with exercise, thereby reducing the burden on their joints and improving their clinical symptoms190,191. Overweight or obese patients with OA often have lipid metabolic disorders, and weight loss can help slow the progression of metabolic OA.
Pharmacological interventions include the use of nonsteroidal anti-inflammatory drugs (NSAIDs) either locally or systemically, which are commonly used to alleviate mild to moderate pain in OA patients186,192. Intra-articular injection of corticosteroids can provide short-term pain relief in OA patients. A clinical trial showed that physical therapy or corticosteroid injection had similar efficacy in the short term, but physical therapy had better long-term effects, and long-term intra-articular injection may cause some joint damage185,193,194. For late-stage OA patients whose pain cannot be relieved by other treatments, joint replacement surgery is recommended, which can effectively alleviate pain and improve the patient’s quality of life195. For the treatment of metabolic OA, targeting the pathogenesis and correcting lipid metabolism disorders may be an ideal approach.
Lipid-lowering drugs are a class of medications that can reduce plasma triglycerides or lower plasma cholesterol. They include statins, cholesterol absorption inhibitors, fibrates, PCSK9 inhibitors, niacin, bile acid sequestrants and n-3 PUFA. By targeting different types of lipid metabolic disorders, the use of different lipid-lowering drugs is a new treatment approach for metabolic OA.
Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which can effectively lower serum cholesterol levels and are widely used to treat hypercholesterolemia196,197. Members of the statin class include atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin and simvastatin197,198. Studies have shown that statins can delay or inhibit the progression of OA in in vitro cultured OA chondrocytes199–203 and experimental OA animal models in vivo204–207. In a clinical study comparing the progression of OA over 6.5 years between statin users and nonusers, researchers found that use of statin significantly slowed the overall progression of knee OA208. Another clinical follow-up study found that use of statins reduced the risk of joint space narrowing in patients with KOA compared with that of non-statin users209. This may be related to the anti-inflammatory and antioxidant functions of statins.
Cholesterol absorption inhibitors mainly reduce the absorption of cholesterol in the intestine and are represented by ezetimibe, which can lower low-density lipoprotein cholesterol and, when used in combination with statins, can further lower low-density lipoprotein cholesterol levels210,211. Although ezetimibe can significantly lower cholesterol and have therapeutic effects in other diseases, it can only lower serum cholesterol in OA and has no therapeutic effect on OA210,212. Niacin is converted to nicotinamide in the human body, which is a component of coenzyme I and coenzyme II, involved in lipid metabolism in the body. Currently, niacin has not been applied in the treatment of OA. Bile acid sequestration promotes the excretion of cholesterol by inhibiting the reabsorption of cholesterol-rich bile acids. However, due to their many adverse effects, they are now rarely used clinically and have not been used to treat OA.
Fibrates include fenofibrate, bezafibrate and gemfibrozil. Currently, fenofibrate is the only fibrate that is associated with OA. Fenofibrate is a peroxisome proliferator-activated receptor alpha (PPARα) agonist that can lower triglyceride levels in the body. PPARα can regulate the uptake and metabolism of fatty acids, as well as exert anti-inflammatory effects213,214. Studies have found that PPARα is downregulated in the blood and cartilage of surgically induced OA mouse models and KOA patients, indicating that PPARα deficiency may be an intrinsic factor leading to the development of OA, and PPARα agonists can prevent cartilage degradation144. Researchers have found that the addition of PPARα agonists to the infrapatellar fat pad (IPFP) of OA patients can effectively inhibit the production of cytokines, such as IL-6, IL-8, MCP1 and IL-4, induced by IL-1β in IPFP215–217. Patients with HOA showed improvement in pain, hand function, systemic inflammation and lipid status after 4 weeks of treatment with fenofibrate218. However, other studies have found that fenofibrate has no inhibitory effect on cartilage injury in the STR/Ort spontaneous OA mouse model219. Currently, the use of fibrates in the treatment of OA is still unclear and requires more basic and clinical investigations for further clarification.
PCSK9 inhibitors have potent cholesterol-lowering effects by preventing LDL receptor degradation and reducing LDL-C by 50%–70%. However, PCSK9 inhibitors are rarely used alone for lipid-lowering therapy but rather in combination with other lipid-lowering drugs for efficient lipid-lowering therapy. In a study inducing KOA in APOE∗3Leiden. CETP mice fed a high-fat diet, researchers found that cholesterol-lowering therapy with a combination of atorvastatin and PCSK9 inhibitors did not inhibit the progression of cartilage degradation in mice220. This suggests that while PCSK9 inhibitors can be used to treat lipid metabolic disorders, their use in the treatment of metabolic OA is still debatable.
N-3 PUFA is a type of fatty acid that cannot be synthesized by the human body. The main types of n-3 PUFA are alpha-linolenic acid, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which can promote the reduction of triglycerides. In IL-1β-induced chondrocyte and synovial cell inflammation models, the addition of n-3 PUFAs can reduce the expression of inflammatory and degradation markers in chondrocytes and synovial cells93,221. Adler et al. found that n-3 PUFA intervention in IL-induced canine chondrocytes significantly decreased iNOS expression and NO production compared to the control chondrocytes95. Additionally, in human chondrocytes treated with n-3 PUFAs, the expression levels of MMP13 and PGE2 were reduced222,223. Therefore, the current findings suggest that n-3 PUFAs have a positive effect on chondrocytes in treating inflammation. Researchers have injected sustained-release EPA into surgically induced KOA mice and observed that it can effectively prevent the progression of KOA224. In a previous RCT, consuming krill oil improved pain, stiffness and functional movement in patients with mild to moderate KOA225. However, in another RCT, supplementing n-3 PUFAs did not alleviate knee pain, stiffness, or functional movement226. Although a large body of evidence suggests that n-3 PUFAs may have a role in reducing low-grade inflammation related to OA and slowing cartilage degradation, more clinical studies are needed to further clarify the function of n-3 PUFAs. Metabolic OA has been found to be associated with metabolic syndrome because they share common pathogenic factors23. In the future, lipid-lowering drugs combined with nonsteroidal anti-inflammatory drugs have great potential for the treatment of metabolic OA. The studies related to lipid-lowering drugs in the treatment of OA and their efficacy in OA treatment are summarized in Table 3.
Table 3.
Lipid-lowering drugs in the treatment of OA
| Lipid Lowering Drug | Drug function | Potential role in OA | Basic research | Clinical trial |
|---|---|---|---|---|
| Statins |
TC↓ LDL↓ HDL↑ |
Protective effect |
1. Atorvastatin may prevent the damage of the cartilage200. 2. Cindine promotes the repair of damaged chondrocytes201. 3. Pravastatin reduces the expression of MMP, promoting OA chondrocytes cholesterol efflux and protecting the chondrocytes matrix202. 4. Simvastatin reduces IL-1β, MMP-3, and leptin expression203. 5. Simvastatin delays OA progression204. 6. Fluvastatin attenuates the degradation of cartilage in OA206. 7. Lovastatin inhibits apoptosis of rabbit chondrocytes in inflammatory environments207. |
1. Statins can significantly delay disease progression in patients with KOA208. 2. Statins may reduce the risk of narrowing of the joint space in people with KOA209. |
| Ezetimibe | LDL↓ | No effect | Ezetimibe has no effect on inhibiting the development of OA210,212. | NA |
| Fibrates |
TG↓ HDL↑ |
Protective effect | PPARα agonists can prevent cartilage degradation144. |
1. PPARα agonists prevent cartilage degradation144. 2. PPARα agonists downregulates the production of inflammatory factors in the IPFP in patients with OA215–217. 3. Fenofibrate reduces pain, systemic inflammation and lipids in patients with HOA218. |
| No effect | Fenofibrate has no inhibitory effect on the development of cartilage injury in mouse models of OA219. | NA | ||
| PCSK9 Inhibitors | LDL↓ | No effect | PCSK9 inhibitors did not attenuate cartilage degradation in OA mice220. | NA |
| Omega-3 fatty acids | TG ↓ | Protective effect |
1. N-3 PUFA can reduce the expression of inflammatory factors and markers of cartilage degradation93,221. 2. N-3 PUFA reduces the production of iNOS and NO95. 3. N-3 PUFA can reduce the expression of MMP13 and PGE2222,223. 4. N-3 PUFA can mitigate the progression of KOA224. |
N-3 PUFA improves pain, stiffness, and motor function in patients225. |
| No effect | NA | N-3 PUFA cannot alleviate knee pain, stiffness and functional movement226. |
Perspective
Current research on lipid metabolic disorders in OA provides a basis for further exploring the pathogenesis and prognosis of this disease. A large number of studies have confirmed that while age and body weight are related to OA, lipid metabolism disorders are also involved in the progression of OA. Related in vitro and in vivo experiments have been conducted, and molecules important for metabolism, such as LXR120,130,131, PPARα145 and RORα153,154, have been found to play important roles in OA development. Additionally, certain lipid-lowering medications in clinical settings have been found to alleviate OA progression in recent clinical studies144,208,209. A combination of lipid-lowering drugs and anti-inflammatory drugs may be more beneficial for OA patients than anti-inflammatory drugs alone. However, existing data are still limited, a common mechanism for lipid metabolism disorders in OA has not been definitively revealed, and some inconsistent results have been reported. Therefore, more evidence is needed to clearly define the pathological mechanisms of lipid metabolism disorders and their contribution to OA development. Controlling body weight and a balanced diet to maintain the homeostasis of lipid metabolism can play a positive role in maintaining joint health. Finding effective drugs to target metabolic disorders will require further in-depth research in the future.
Acknowledgments
Funding
This work was supported by the National Key Research and Development Program of China (2021YFB3800800) to L.T. and D.C. This project was also supported by the National Natural Science Foundation of China (NSFC) grants (82030067, 82161160342 and 82250710174) to D.C., grant 82360429 to Y.C and grant 82172397 to L.T. This project was also supported by National Science Foundation of Guangxi (2022JJA141126), Advanced Innovation Teams and Xinghu Scholars Program of Guangxi Medical University, China Postdoctoral Science Foundation (2019M650235), Key R&D Project of Qingxiu District, Nanning, Guangxi (2021003) to Y.C., and the Hong Kong RGC grant HKU-17101821 to W.W.L. and D.C., and SIAT Innovation Program for Excellent Young Researchers to K.L.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Guizheng Wei and Ke Lu.
Contributor Information
Yan Chen, Email: cy003@connect.hku.hk.
Liping Tong, Email: lp.tong@siat.ac.cn.
References
- 1.Hu Y, et al. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res. 2021;9:20. doi: 10.1038/s41413-021-00147-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–1759. doi: 10.1016/S0140-6736(19)30417-9. [DOI] [PubMed] [Google Scholar]
- 3.Loeser RF, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen D, et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044. doi: 10.1038/boneres.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong L, et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022;10:60. doi: 10.1038/s41413-022-00226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scuderi GR. Complications after total knee arthroplasty: how to manage patients with osteolysis. J. Bone Jt. Surg. Am. 2011;93:2127–2135. doi: 10.2106/JBJS.9322icl. [DOI] [PubMed] [Google Scholar]
- 7.Grayson CW, Decker RC. Total joint arthroplasty for persons with osteoarthritis. Phys. Med. Rehabil. 2012;4:S97–S103. doi: 10.1016/j.pmrj.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Deveza LA, Loeser RF. Is osteoarthritis one disease or a collection of many? Rheumatology. 2018;57:iv34–iv42. doi: 10.1093/rheumatology/kex417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruyère O, et al. Can we identify patients with high risk of osteoarthritis progression who will respond to treatment? A focus on epidemiology and phenotype of osteoarthritis. Drugs Aging. 2015;32:179–187. doi: 10.1007/s40266-015-0243-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altman R, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29:1039–1049. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 11.Courties A, Sellam J, Berenbaum F. Metabolic syndrome-associated osteoarthritis. Curr. Rheumatol. Rep. 2017;29:214–222. doi: 10.1097/BOR.0000000000000373. [DOI] [PubMed] [Google Scholar]
- 12.Issa R. I. & Griffin T. M. Pathobiology of obesity and osteoarthritis: integrating biomechanics and inflammation. Pathobiol. Aging Age Relat. Dis. 2, (2012). [DOI] [PMC free article] [PubMed]
- 13.Reyes C, et al. Association between overweight and obesity and risk of clinically diagnosed knee, hip, and hand osteoarthritis: a population-based cohort study. Arthritis Rheumatol. 2016;68:1869–1875. doi: 10.1002/art.39707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang J, et al. Systemic and local adipose tissue in knee osteoarthritis. Osteoarthr. Cartil. 2018;26:864–871. doi: 10.1016/j.joca.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Urban H, Little CB. The role of fat and inflammation in the pathogenesis and management of osteoarthritis. Rheumatology. 2018;57:iv10–iv21. doi: 10.1093/rheumatology/kex399. [DOI] [PubMed] [Google Scholar]
- 16.Thijssen E, van Caam A, van der Kraan PM. Obesity and osteoarthritis, more than just wear and tear: pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis. Rheumatology. 2015;54:588–600. doi: 10.1093/rheumatology/keu464. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018;44:38–50. doi: 10.1016/j.cytogfr.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Voinier D, et al. Using cumulative load to explain how body mass index and daily walking relate to worsening knee cartilage damage over two years: the MOST study. Arthritis Rheumatol. 2020;72:957–965. doi: 10.1002/art.41181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lohmander LS, et al. Incidence of severe knee and hip osteoarthritis in relation to different measures of body mass: a population-based prospective cohort study. Ann. Rheum. Dis. 2009;68:490–496. doi: 10.1136/ard.2008.089748. [DOI] [PubMed] [Google Scholar]
- 20.Plotz B, et al. Current epidemiology and risk factors for the development of hand osteoarthritis. Curr. Rheumatol. Rep. 2021;23:61. doi: 10.1007/s11926-021-01025-7. [DOI] [PubMed] [Google Scholar]
- 21.Giri B, et al. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: an update on glucose toxicity. Biomed. Pharmacother. 2018;107:306–328. doi: 10.1016/j.biopha.2018.07.157. [DOI] [PubMed] [Google Scholar]
- 22.Veronese N, et al. Type 2 diabetes mellitus and osteoarthritis. Semin. Arthritis Rheu. 2019;49:9–19. doi: 10.1016/j.semarthrit.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuo Q, et al. Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 2012;8:729–737. doi: 10.1038/nrrheum.2012.135. [DOI] [PubMed] [Google Scholar]
- 24.So MW, Lee S, Kim SH. Association between dietary glycemic index and knee osteoarthritis: the Korean national health and nutrition examination survey 2010-2012. J. Acad. Nutr. Diet. 2018;118:1673–1686.e1672. doi: 10.1016/j.jand.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Veronese N, et al. Adherence to a Mediterranean diet is associated with lower prevalence of osteoarthritis: data from the osteoarthritis initiative. Clin. Nutr. 2017;36:1609–1614. doi: 10.1016/j.clnu.2016.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gandhi R, et al. Metabolic syndrome and the functional outcomes of hip and knee arthroplasty. J. Rheumatol. 2010;37:1917–1922. doi: 10.3899/jrheum.091242. [DOI] [PubMed] [Google Scholar]
- 27.Yasuda E, et al. Association between the severity of symptomatic knee osteoarthritis and cumulative metabolic factors. Aging Clin. Exp. Res. 2018;30:481–488. doi: 10.1007/s40520-017-0808-6. [DOI] [PubMed] [Google Scholar]
- 28.Centers for Disease Control and Prevention (CDC). Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation-United States, 2010-2012. MMWR Morb Mortal Wkly Rep. 62, 869–873 (2013). [PMC free article] [PubMed]
- 29.Jorgensen AE, Kjaer M, Heinemeier KM. The Effect of aging and mechanical loading on the metabolism of articular cartilage. J. Rheumatol. 2017;44:410–417. doi: 10.3899/jrheum.160226. [DOI] [PubMed] [Google Scholar]
- 30.Mobasheri A, et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017;13:302–311. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 31.Rasheed Z, Akhtar N, Haqqi TM. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology. 2011;50:838–851. doi: 10.1093/rheumatology/keq380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steenvoorden MM, et al. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006;54:253–263. doi: 10.1002/art.21523. [DOI] [PubMed] [Google Scholar]
- 33.DeGroot J, et al. Accumulation of advanced glycation endproducts reduces chondrocyte-mediated extracellular matrix turnover in human articular cartilage. Osteoarthr. Cartil. 2001;9:720–726. doi: 10.1053/joca.2001.0469. [DOI] [PubMed] [Google Scholar]
- 34.Huang CY, et al. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology. 2011;50:1379–1389. doi: 10.1093/rheumatology/ker134. [DOI] [PubMed] [Google Scholar]
- 35.Yang Q, et al. Advanced glycation end products-induced chondrocyte apoptosis through mitochondrial dysfunction in cultured rabbit chondrocyte. Fundam. Clin. Pharm. 2015;29:54–61. doi: 10.1111/fcp.12094. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki A, Yabu A, Nakamura H. Advanced glycation end products in musculoskeletal system and disorders. Methods. 2022;203:179–186. doi: 10.1016/j.ymeth.2020.09.012. [DOI] [PubMed] [Google Scholar]
- 37.Gallo J, et al. Inflammation and its resolution and the musculoskeletal system. J. Orthop. Transl. 2017;10:52–67. doi: 10.1016/j.jot.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jrad AIS, et al. Role of pro-inflammatory interleukins in osteoarthritis: a narrative review. Connect. Tissue Res. 2023;64:238–247. doi: 10.1080/03008207.2022.2157270. [DOI] [PubMed] [Google Scholar]
- 39.Anderson JJ, Felson DT. Factors associated with osteoarthritis of the knee in the first national health and nutrition examination survey (HANES I). Evidence for an association with overweight, race, and physical demands of work. Am. J. Epidemiol. 1988;128:179–189. doi: 10.1093/oxfordjournals.aje.a114939. [DOI] [PubMed] [Google Scholar]
- 40.Rogero MM, Calder PC. Obesity, inflammation, toll-Like receptor 4 and fatty acids. Nutrients. 2018;10:432. doi: 10.3390/nu10040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conde J, et al. Adipokines and osteoarthritis: novel molecules involved in the pathogenesis and progression of disease. Arthritis. 2011;2011:203901. doi: 10.1155/2011/203901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fain JN. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediat. Inflamm. 2010;2010:513948. doi: 10.1155/2010/513948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guss JD, et al. The effects of metabolic syndrome, obesity, and the gut microbiome on load-induced osteoarthritis. Osteoarthr. Cartil. 2019;27:129–139. doi: 10.1016/j.joca.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang M, et al. Oral Administration of resveratrol alleviates osteoarthritis pathology in C57BL/6J mice model induced by a high-fat diet. Mediat. Inflamm. 2017;2017:7659023. doi: 10.1155/2017/7659023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, et al. Protective effect of resveratrol against IL-1β-induced inflammatory response on human osteoarthritic chondrocytes partly via the TLR4/MyD88/NF-κB signaling pathway: an “in vitro study”. Int. J. Mol. Sci. 2014;15:6925–6940. doi: 10.3390/ijms15046925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Silva VA, et al. Metformin in prevention and treatment of antipsychotic induced weight gain: a systematic review and meta-analysis. BMC Psychiatry. 2016;16:341. doi: 10.1186/s12888-016-1049-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yerevanian A, Soukas AA. Metformin: mechanisms in human obesity and weight loss. Curr. Obes. Rep. 2019;8:156–164. doi: 10.1007/s13679-019-00335-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu CH, et al. Combination COX-2 inhibitor and metformin attenuate rate of joint replacement in osteoarthritis with diabetes: A nationwide, retrospective, matched-cohort study in Taiwan. PLoS One. 2018;13:e0191242. doi: 10.1371/journal.pone.0191242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann. Rheum. Dis. 2020;79:635–645. doi: 10.1136/annrheumdis-2019-216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conrozier T. How to treat osteoarthritis in obese patients? Curr. Rheumatol. Rev. 2020;16:99–104. doi: 10.2174/1573397115666190625105759. [DOI] [PubMed] [Google Scholar]
- 51.Matsuda M, Shimomura I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pr. 2013;7:E330–E341. doi: 10.1016/j.orcp.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 52.Niemann B, et al. Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution part 3 of a 3-part series. J. Am. Coll. Cardiol. 2017;70:230–251. doi: 10.1016/j.jacc.2017.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed B, Sultana R, Greene MW. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021;137:111315. doi: 10.1016/j.biopha.2021.111315. [DOI] [PubMed] [Google Scholar]
- 54.Paneni F, et al. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur. Heart J. 2013;34:2436–U2434. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Mello AH, et al. Mitochondrial dysfunction in obesity. Life Sci. 2018;192:26–32. doi: 10.1016/j.lfs.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 56.Rendra E, et al. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. 2019;224:242–253. doi: 10.1016/j.imbio.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020;129:110452. doi: 10.1016/j.biopha.2020.110452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lepetsos P, Papavassiliou KA, Papavassiliou AG. Redox and NF-κB signaling in osteoarthritis. Free Radic. Bio. Med. 2019;132:90–100. doi: 10.1016/j.freeradbiomed.2018.09.025. [DOI] [PubMed] [Google Scholar]
- 59.Zhou F, et al. Isorhamnetin attenuates osteoarthritis by inhibiting osteoclastogenesis and protecting chondrocytes through modulating reactive oxygen species homeostasis. J. Cell Mol. Med. 2019;23:4395–4407. doi: 10.1111/jcmm.14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019;132:73–82. doi: 10.1016/j.freeradbiomed.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Dong S. The signaling pathways involved in chondrocyte differentiation and hypertrophic differentiation. Stem Cells Int. 2016;2016:2470351. doi: 10.1155/2016/2470351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han J, et al. Inhibition of NADPH oxidases prevents the development of osteoarthritis. Antioxidants. 2022;11:2346. doi: 10.3390/antiox11122346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gui T, et al. Superoxide dismutase-loaded porous polymersomes as highly efficient antioxidant nanoparticles targeting synovium for osteoarthritis therapy. Biomaterials. 2022;283:121437. doi: 10.1016/j.biomaterials.2022.121437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu HL, et al. Radical-scavenging and subchondral bone-regenerating nanomedicine for osteoarthritis treatment. ACS Nano. 2023;17:6131–6146. doi: 10.1021/acsnano.3c01789. [DOI] [PubMed] [Google Scholar]
- 65.Reed KN, et al. The role of mitochondrial reactive oxygen species in cartilage matrix destruction. Mol. Cell Biochem. 2014;397:195–201. doi: 10.1007/s11010-014-2187-z. [DOI] [PubMed] [Google Scholar]
- 66.Kang D, et al. Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis. Nat. Commun. 2022;13:779. doi: 10.1038/s41467-022-28385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SH, Park SY, Choi CS. Insulin resistance: from mechanisms to therapeutic strategies. Diabetes Metab. J. 2022;46:15–37. doi: 10.4093/dmj.2021.0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eymard F, et al. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthr. Cartil. 2015;23:851–859. doi: 10.1016/j.joca.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 69.Hamada D, et al. Suppressive effects of insulin on tumor necrosis factor-dependent early osteoarthritic changes associated with obesity and type 2 diabetes mellitus. Arthritis Rheumatol. 2016;68:1392–1402. doi: 10.1002/art.39561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ribeiro M, et al. Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthr. Cartil. 2016;24:2116–2125. doi: 10.1016/j.joca.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 71.Schett G, et al. Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes Care. 2013;36:403–409. doi: 10.2337/dc12-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qiao L, Li Y, Sun S. Insulin exacerbates inflammation in fibroblast-like synoviocytes. Inflammation. 2020;43:916–936. doi: 10.1007/s10753-020-01178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blaney Davidson EN, et al. Inducible chondrocyte-specific overexpression of BMP2 in young mice results in severe aggravation of osteophyte formation in experimental OA without altering cartilage damage. Ann. Rheum. Dis. 2015;74:1257–1264. doi: 10.1136/annrheumdis-2013-204528. [DOI] [PubMed] [Google Scholar]
- 74.Courties A, Sellam J. Osteoarthritis and type 2 diabetes mellitus: what are the links? Diabetes Res. Clin. Pr. 2016;122:198–206. doi: 10.1016/j.diabres.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 75.Chen YJ, et al. Advanced glycation end-products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J. Orthop. Res. 2016;34:791–800. doi: 10.1002/jor.23083. [DOI] [PubMed] [Google Scholar]
- 76.Zhu S, et al. The molecular structure and role of LECT2 or CHM-II in arthritis, cancer, and other diseases. J. Cell Physiol. 2022;237:480–488. doi: 10.1002/jcp.30593. [DOI] [PubMed] [Google Scholar]
- 77.Willis SA, et al. Acute hyperenergetic, high-fat feeding increases circulating FGF21, LECT2, and Fetuin-A in healthy men. J. Nutr. 2020;150:1076–1085. doi: 10.1093/jn/nxz333. [DOI] [PubMed] [Google Scholar]
- 78.Ikeda D, et al. iTRAQ-based proteomics reveals novel biomarkers of osteoarthritis. Biomarkers. 2013;18:565–572. doi: 10.3109/1354750X.2013.810667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Y, et al. Lipid metabolism in cartilage and its diseases: a concise review of the research progress. Acta Biochim. Biophys. Sin. 2021;53:517–527. doi: 10.1093/abbs/gmab021. [DOI] [PubMed] [Google Scholar]
- 80.Dahaghin S, et al. Do metabolic factors add to the effect of overweight on hand osteoarthritis? The rotterdam study. Ann. Rheum. Dis. 2007;66:916–920. doi: 10.1136/ard.2005.045724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gkretsi V, Simopoulou T, Tsezou A. Lipid metabolism and osteoarthritis: lessons from atherosclerosis. Prog. Lipid Res. 2011;50:133–140. doi: 10.1016/j.plipres.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 82.Puenpatom RA, Victor TW. Increased prevalence of metabolic syndrome in individuals with osteoarthritis: an analysis of NHANES III data. Postgrad. Med. 2009;121:9–20. doi: 10.3810/pgm.2009.11.2073. [DOI] [PubMed] [Google Scholar]
- 83.Chang HW, et al. Blue mussel (Mytilus edulis) water extract ameliorates inflammatory responses and oxidative stress on osteoarthritis in obese rats. J. Pain. Res. 2020;13:1109–1119. doi: 10.2147/JPR.S244372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sekar S, et al. Saturated fatty acids induce development of both metabolic syndrome and osteoarthritis in rats. Sci. Rep. 2017;7:46457. doi: 10.1038/srep46457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alvarez-Garcia O, et al. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014;66:1779–1788. doi: 10.1002/art.38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sekar S, et al. Saturated fatty acids promote chondrocyte matrix remodeling through reprogramming of autophagy pathways. Nutrition. 2018;54:144–152. doi: 10.1016/j.nut.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 87.Ma H, et al. Myriocin alleviates Oleic/Palmitate induced chondrocyte degeneration via the suppression of ceramide. Eur. Rev. Med. Pharm. Sci. 2020;24:12938–12947. doi: 10.26355/eurrev_202012_24197. [DOI] [PubMed] [Google Scholar]
- 88.Lu B, et al. Dietary fat intake and radiographic progression of knee osteoarthritis: data from the osteoarthritis initiative. Arthritis Care Res. 2017;69:368–375. doi: 10.1002/acr.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mustonen AM, et al. Anterior cruciate ligament transection alters the n-3/n-6 fatty acid balance in the lapine infrapatellar fat pad. Lipids Health Dis. 2019;18:67. doi: 10.1186/s12944-019-1008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bastiaansen-Jenniskens YM, et al. Monounsaturated and saturated, but not n-6 polyunsaturated fatty acids decrease cartilage destruction under inflammatory conditions: a preliminary study. Cartilage. 2013;4:321–328. doi: 10.1177/1947603513494401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van de Vyver A, et al. Synovial fluid fatty acid profiles differ between osteoarthritis and healthy patients. Cartilage. 2020;11:473–478. doi: 10.1177/1947603518798891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang H, et al. Adiponectin, may be a potential protective factor for obesity-related osteoarthritis. Diabetes Metab. Syndr. Obes. 2022;15:1305–1319. doi: 10.2147/DMSO.S359330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Loef M, et al. Fatty acids and osteoarthritis: different types, different effects. Jt. Bone Spine. 2019;86:451–458. doi: 10.1016/j.jbspin.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Frommer KW, et al. Free fatty acids: potential proinflammatory mediators in rheumatic diseases. Ann. Rheum. Dis. 2015;74:303–310. doi: 10.1136/annrheumdis-2013-203755. [DOI] [PubMed] [Google Scholar]
- 95.Adler N, Schoeniger A, Fuhrmann H. Polyunsaturated fatty acids influence inflammatory markers in a cellular model for canine osteoarthritis. J. Anim. Physiol. N. 2018;102:E623–E632. doi: 10.1111/jpn.12804. [DOI] [PubMed] [Google Scholar]
- 96.Wu CL, et al. Serum and synovial fluid lipidomic profiles predict obesity-associated osteoarthritis, synovitis, and wound repair. Sci. Rep. 2017;7:44315. doi: 10.1038/srep44315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Loef M, et al. The association of plasma fatty acids with hand and knee osteoarthritis: the NEO study. Osteoarthr. Cartil. 2020;28:223–230. doi: 10.1016/j.joca.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 98.Huang M-J, et al. Enhancement of the synthesis of n-3 PUFAs in fat-1 transgenic mice inhibits mTORC1 signalling and delays surgically induced osteoarthritis in comparison with wild-type mice. Ann. Rheum. Dis. 2014;73:1719. doi: 10.1136/annrheumdis-2013-203231. [DOI] [PubMed] [Google Scholar]
- 99.Zainal Z, et al. Relative efficacies of omega-3 polyunsaturated fatty acids in reducing expression of key proteins in a model system for studying osteoarthritis. Osteoarthr. Cartil. 2009;17:896–905. doi: 10.1016/j.joca.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 100.Sibille KT, et al. Omega-6: Omega-3 PUFA ratio, pain, functioning, and distress in adults with knee pain. Clin. J. Pain. 2018;34:182–189. doi: 10.1097/AJP.0000000000000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hill CL, et al. Fish oil in knee osteoarthritis: a randomised clinical trial of low dose versus high dose. Ann. Rheum. Dis. 2016;75:23–29. doi: 10.1136/annrheumdis-2014-207169. [DOI] [PubMed] [Google Scholar]
- 102.Pousinis P, et al. Lipidomic identification of plasma lipids associated with pain behaviour and pathology in a mouse model of osteoarthritis. Metabolomics. 2020;16:32. doi: 10.1007/s11306-020-01652-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao W, et al. Label-free and continuous-flow ferrohydrodynamic separation of HeLa cells and blood cells in biocompatible ferrofluids. Adv. Funct. Mater. 2016;26:3990–3998. doi: 10.1002/adfm.201503838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siodmiak J, et al. Molecular dynamic analysis of hyaluronic acid and phospholipid interaction in tribological surgical adjuvant design for osteoarthritis. Molecules. 2017;22:1436. doi: 10.3390/molecules22091436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rocha B, et al. Identification of a distinct lipidomic profile in the osteoarthritic synovial membrane by mass spectrometry imaging. Osteoarthr. Cartil. 2021;29:750–761. doi: 10.1016/j.joca.2020.12.025. [DOI] [PubMed] [Google Scholar]
- 106.Kosinska MK, et al. A lipidomic study of phospholipid classes and species in human synovial fluid. Arthritis Rheum. 2013;65:2323–2333. doi: 10.1002/art.38053. [DOI] [PubMed] [Google Scholar]
- 107.Zhai G, et al. Activation of the phosphatidylcholine to lysophosphatidylcholine pathway is associated with osteoarthritis knee cartilage volume loss over time. Sci. Rep. 2019;9:9648. doi: 10.1038/s41598-019-46185-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rocha B, et al. Targeted phospholipidomic analysis of synovial fluid as a tool for osteoarthritis deep phenotyping. Osteoarthr. Cartil. Open. 2021;3:100219. doi: 10.1016/j.ocarto.2021.100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang W, et al. Lysophosphatidylcholines to phosphatidylcholines ratio predicts advanced knee osteoarthritis. Rheumatology. 2016;55:1566–1574. doi: 10.1093/rheumatology/kew207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhai G. Chapter Three - Clinical relevance of biochemical and metabolic changes in osteoarthritis. In: Makowski G. S. (ed). Advances in Clinical Chemistry, vol. 101. Elsevier, (2021), pp 95-120. [DOI] [PubMed]
- 111.Zhai GJ, Randell EW, Rahman P. Metabolomics of osteoarthritis: emerging novel markers and their potential clinical utility. Rheumatology. 2018;57:2087–2095. doi: 10.1093/rheumatology/kex497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pruzanski W, et al. Enzymatic activity and distribution of phospholipase A2 in human cartilage. Life Sci. 1991;48:2457–2462. doi: 10.1016/0024-3205(91)90381-k. [DOI] [PubMed] [Google Scholar]
- 113.Bomalaski JS, Clark MA. Phospholipase A2 and arthritis. Arthritis Rheum. 1993;36:190–198. doi: 10.1002/art.1780360208. [DOI] [PubMed] [Google Scholar]
- 114.Soccio RE, Breslow JL. Intracellular cholesterol transport. Arterioscler Thromb. Vasc. Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- 115.Wu S, De Luca F. Role of cholesterol in the regulation of growth plate chondrogenesis and longitudinal bone growth. J. Biol. Chem. 2004;279:4642–4647. doi: 10.1074/jbc.M305518200. [DOI] [PubMed] [Google Scholar]
- 116.Tsezou A, et al. Impaired expression of genes regulating cholesterol efflux in human osteoarthritic chondrocytes. J. Orthop. Res. 2010;28:1033–1039. doi: 10.1002/jor.21084. [DOI] [PubMed] [Google Scholar]
- 117.Wang Q, et al. Fermentation supernatant of Staphylococcus aureus drives catabolism in chondrocytes via NF-κB signaling mediated increase of cholesterol metabolism. Exp. Cell Res. 2022;410:112952. doi: 10.1016/j.yexcr.2021.112952. [DOI] [PubMed] [Google Scholar]
- 118.Wang X, et al. Ultrasound-targeted simvastatin-loaded microbubble destruction promotes OA cartilage repair by modulating the cholesterol efflux pathway mediated by PPARγ in rabbits. Bone Jt. Res. 2021;10:693–703. doi: 10.1302/2046-3758.1010.BJR-2021-0162.R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim K, et al. RORα controls hepatic lipid homeostasis via negative regulation of PPARγ transcriptional network. Nat. Commun. 2017;8:162. doi: 10.1038/s41467-017-00215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun MM, Beier F. Liver X receptor activation regulates genes involved in lipid homeostasis in developing chondrocytes. Osteoarthr. Cartil. Open. 2020;2:100030. doi: 10.1016/j.ocarto.2020.100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bougarne N, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018;39:760–802. doi: 10.1210/er.2018-00064. [DOI] [PubMed] [Google Scholar]
- 122.Ratneswaran A, et al. Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J. Mol. Med. 2017;95:431–444. doi: 10.1007/s00109-016-1501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xiao J, et al. Activation of liver X receptors promotes inflammatory cytokine mRNA degradation by upregulation of tristetraprolin. Acta Biochim. Biophys. Sin. 2017;49:277–283. doi: 10.1093/abbs/gmw136. [DOI] [PubMed] [Google Scholar]
- 124.Joseph SB, et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002;277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 125.Kostopoulou F, et al. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015;17:42. doi: 10.1186/s13075-015-0556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Collins-Racie LA, et al. Global analysis of nuclear receptor expression and dysregulation in human osteoarthritic articular cartilage: reduced LXR signaling contributes to catabolic metabolism typical of osteoarthritis. Osteoarthr. Cartil. 2009;17:832–842. doi: 10.1016/j.joca.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 127.Joseph SB, et al. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 128.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015;15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He H, et al. Vaspin regulated cartilage cholesterol metabolism through miR155/LXRα and participated in the occurrence of osteoarthritis in rats. Life Sci. 2021;269:119096. doi: 10.1016/j.lfs.2021.119096. [DOI] [PubMed] [Google Scholar]
- 130.Xing H, et al. Metformin mitigates cholesterol accumulation via the AMPK/SIRT1 pathway to protect osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun. 2022;632:113–121. doi: 10.1016/j.bbrc.2022.09.074. [DOI] [PubMed] [Google Scholar]
- 131.Lin XL, et al. Curcumin enhanced cholesterol efflux by upregulating ABCA1 expression through AMPK-SIRT1-LXRα signaling in THP-1 macrophage-derived foam cells. DNA Cell Biol. 2015;34:561–572. doi: 10.1089/dna.2015.2866. [DOI] [PubMed] [Google Scholar]
- 132.Zhu Z, et al. AMPK activator decelerates osteoarthritis development by inhibition of β-catenin signaling in chondrocytes. J. Orthop. Transl. 2023;38:158–166. doi: 10.1016/j.jot.2022.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li J, et al. Oral administration of berberine limits post-traumatic osteoarthritis development and associated pain via AMP-activated protein kinase (AMPK) in mice. Osteoarthr. Cartil. 2022;30:160–171. doi: 10.1016/j.joca.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yi D, et al. AMPK signaling in energy control, cartilage biology, and osteoarthritis. Front. Cell Dev. Biol. 2021;9:696602. doi: 10.3389/fcell.2021.696602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liang C, et al. Resveratrol improves the progression of osteoarthritis by regulating the SIRT1-FoxO1 pathway-mediated cholesterol metabolism. Mediat. Inflamm. 2023;2023:2936236. doi: 10.1155/2023/2936236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Su Z, et al. Lipid metabolism in cartilage development, degeneration, and regeneration. Nutrients. 2022;14:3984. doi: 10.3390/nu14193984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chambers KF, et al. Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: a review. Nutrients. 2019;11:2588. doi: 10.3390/nu11112588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen L, et al. Molecular mechanisms for ABCA1-mediated cholesterol efflux. Cell Cycle. 2022;21:1121–1139. doi: 10.1080/15384101.2022.2042777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Triantaphyllidou IE, et al. Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthr. Cartil. 2013;21:322–330. doi: 10.1016/j.joca.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 140.Garcia-Gil M, et al. Serum lipid levels and risk of hand osteoarthritis: the chingford prospective cohort study. Sci. Rep. 2017;7:3147. doi: 10.1038/s41598-017-03317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lu K, et al. Defects in a liver-bone axis contribute to hepatic osteodystrophy disease progression. Cell Metab. 2022;34:441–457.e447. doi: 10.1016/j.cmet.2022.02.006. [DOI] [PubMed] [Google Scholar]
- 142.Liu Y, et al. The role of PPAR-δ in metabolism, inflammation, and cancer: many characters of a critical transcription factor. Int. J. Mol. Sci. 2018;19:3339. doi: 10.3390/ijms19113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang H, et al. Glucagon-like peptide-1 attenuated carboxymethyl lysine induced neuronal apoptosis via peroxisome proliferation activated receptor-γ. Aging. 2021;13:19013–19027. doi: 10.18632/aging.203351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nogueira-Recalde U, et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. Ebiomedicine. 2019;45:588–605. doi: 10.1016/j.ebiom.2019.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Park S, et al. PPARα-ACOT12 axis is responsible for maintaining cartilage homeostasis through modulating de novo lipogenesis. Nat. Commun. 2022;13:3. doi: 10.1038/s41467-021-27738-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou Y, et al. Chondroprotection of PPARα activation by WY14643 via autophagy involving Akt and ERK in LPS-treated mouse chondrocytes and osteoarthritis model. J. Cell Mol. Med. 2019;23:2782–2793. doi: 10.1111/jcmm.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sabatini M, et al. Effects of agonists of peroxisome proliferator-activated receptor gamma on proteoglycan degradation and matrix metalloproteinase production in rat cartilage in vitro. Osteoarthr. Cartil. 2002;10:673–679. doi: 10.1053/joca.2002.0827. [DOI] [PubMed] [Google Scholar]
- 148.Vasheghani F, et al. PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 2015;74:569–578. doi: 10.1136/annrheumdis-2014-205743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Watters JW, et al. Inverse relationship between matrix remodeling and lipid metabolism during osteoarthritis progression in the STR/Ort mouse. Arthritis Rheum. 2007;56:2999–3009. doi: 10.1002/art.22836. [DOI] [PubMed] [Google Scholar]
- 150.Wang H, et al. GDF11 inhibits abnormal adipogenesis of condylar chondrocytes in temporomandibular joint osteoarthritis. Bone Jt. Res. 2022;11:453–464. doi: 10.1302/2046-3758.117.BJR-2022-0019.R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang G, et al. Role of peroxisome proliferator-activated receptors in osteoarthritis (Review) Mol. Med. Rep. 2021;23:159. doi: 10.3892/mmr.2020.11798. [DOI] [PubMed] [Google Scholar]
- 152.Ratneswaran A, et al. Peroxisome proliferator-activated receptor δ promotes the progression of posttraumatic osteoarthritis in a mouse model. Arthritis Rheumatol. 2015;67:454–464. doi: 10.1002/art.38915. [DOI] [PubMed] [Google Scholar]
- 153.Choi WS, et al. The CH25H-CYP7B1-RORα axis of cholesterol metabolism regulates osteoarthritis. Nature. 2019;566:254–258. doi: 10.1038/s41586-019-0920-1. [DOI] [PubMed] [Google Scholar]
- 154.Gentili C, et al. Cholesterol secretion and homeostasis in chondrocytes: a liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein A1 expression. Matrix Biol. 2005;24:35–44. doi: 10.1016/j.matbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 155.Liang T, et al. Inhibition of nuclear receptor RORα attenuates cartilage damage in osteoarthritis by modulating IL-6/STAT3 pathway. Cell Death Dis. 2021;12:886. doi: 10.1038/s41419-021-04170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li XC, et al. MicroRNA-10a-3p improves cartilage degeneration by regulating CH25H-CYP7B1-ROR alpha mediated cholesterol metabolism in knee osteoarthritis rats. Front. Pharm. 2021;12:690181. doi: 10.3389/fphar.2021.690181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cao C, et al. Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4. Nat. Commun. 2022;13:7139. doi: 10.1038/s41467-022-34830-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Farnaghi S, et al. Cholesterol metabolism in pathogenesis of osteoarthritis disease. Int. J. Rheum. Dis. 2017;20:131–140. doi: 10.1111/1756-185X.13061. [DOI] [PubMed] [Google Scholar]
- 159.Pan F, et al. Association between metabolic syndrome and knee structural change on MRI. Rheumatology. 2020;59:185–193. doi: 10.1093/rheumatology/kez266. [DOI] [PubMed] [Google Scholar]
- 160.Meng T, et al. Association of glucose homeostasis and metabolic syndrome with knee cartilage defects and cartilage volume in young adults. Semin. Arthritis Rheum. 2020;50:192–197. doi: 10.1016/j.semarthrit.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 161.Schwager JL, et al. Association of serum low-density lipoprotein, high-density lipoprotein, and total cholesterol with development of knee osteoarthritis. Arthrit Care Res. 2022;74:274–280. doi: 10.1002/acr.24455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang KB, et al. High-density lipoprotein cholesterol and apolipoprotein A1 in synovial fluid: potential predictors of disease severity of primary knee osteoarthritis. Cartilage. 2021;13:1465S–1473S. doi: 10.1177/19476035211007919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Luo QL, et al. Effects of ultrasound therapy on the synovial fluid proteome in a rabbit surgery-induced model of knee osteoarthritis. Biomed. Eng. Online. 2019;18:18. doi: 10.1186/s12938-019-0637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sánchez-Enríquez S, et al. Increase levels of apo-A1 and apo B are associated in knee osteoarthritis: lack of association with VEGF-460 T/C and +405 C/G polymorphisms. Rheumatol. Int. 2008;29:63–68. doi: 10.1007/s00296-008-0633-5. [DOI] [PubMed] [Google Scholar]
- 165.Meng H, et al. Causal associations of circulating lipids with osteoarthritis: a bidirectional mendelian randomization study. Nutrients. 2022;14:1327. doi: 10.3390/nu14071327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kurano M, et al. Apolipoprotein D modulates lipid mediators and osteopontin in an anti-inflammatory direction. Inflamm. Res. 2023;72:263–280. doi: 10.1007/s00011-022-01679-8. [DOI] [PubMed] [Google Scholar]
- 167.Bhatia S, et al. Selective reduction of hydroperoxyeicosatetraenoic acids to their hydroxy derivatives by apolipoprotein D: implications for lipid antioxidant activity and Alzheimer’s disease. Biochem. J. 2012;442:713–721. doi: 10.1042/BJ20111166. [DOI] [PubMed] [Google Scholar]
- 168.Zhang Y, et al. Antioxidant activities of recombinant amphioxus (Branchiostoma belcheri) apolipoprotein D. Mol. Biol. Rep. 2011;38:1847–1851. doi: 10.1007/s11033-010-0301-1. [DOI] [PubMed] [Google Scholar]
- 169.Qin Y, et al. Apolipoprotein D as a potential biomarker and construction of a transcriptional regulatory-immune network associated with osteoarthritis by weighted gene coexpression network analysis. Cartilage. 2021;13:1702s–1717s. doi: 10.1177/19476035211053824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li B, et al. A novel serological biomarker are associated with disease severity in patients with osteoarthritis. J. Bone Min. Metab. 2022;40:1007–1013. doi: 10.1007/s00774-022-01364-0. [DOI] [PubMed] [Google Scholar]
- 171.Farnaghi S, et al. Protective effects of mitochondria-targeted antioxidants and statins on cholesterol-induced osteoarthritis. Faseb J. 2017;31:356–367. doi: 10.1096/fj.201600600R. [DOI] [PubMed] [Google Scholar]
- 172.de Munter W, et al. High LDL levels lead to increased synovial inflammation and accelerated ectopic bone formation during experimental osteoarthritis. Osteoarthr. Cartil. 2016;24:844–855. doi: 10.1016/j.joca.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 173.Friedman JM. Leptin and the endocrine control of energy balance. Nat. Metab. 2019;1:754–764. doi: 10.1038/s42255-019-0095-y. [DOI] [PubMed] [Google Scholar]
- 174.Hui W, et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Ann. Rheum. Dis. 2012;71:455–462. doi: 10.1136/annrheumdis-2011-200372. [DOI] [PubMed] [Google Scholar]
- 175.Bao JP, et al. Leptin plays a catabolic role on articular cartilage. Mol. Biol. Rep. 2010;37:3265–3272. doi: 10.1007/s11033-009-9911-x. [DOI] [PubMed] [Google Scholar]
- 176.Abella V, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017;13:100–109. doi: 10.1038/nrrheum.2016.209. [DOI] [PubMed] [Google Scholar]
- 177.Stannus OP, et al. Cross-sectional and longitudinal associations between circulating leptin and knee cartilage thickness in older adults. Ann. Rheum. Dis. 2015;74:82–88. doi: 10.1136/annrheumdis-2013-203308. [DOI] [PubMed] [Google Scholar]
- 178.Dumond H, et al. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003;48:3118–3129. doi: 10.1002/art.11303. [DOI] [PubMed] [Google Scholar]
- 179.Chen TH, et al. Evidence for a protective role for adiponectin in osteoarthritis. Biochim. Biophys. Acta. 2006;1762:711–718. doi: 10.1016/j.bbadis.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 180.Kang EH, et al. Adiponectin is a potential catabolic mediator in osteoarthritis cartilage. Arthritis Res. Ther. 2010;12:R231. doi: 10.1186/ar3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yamauchi T, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 182.Yamauchi T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 183.Koskinen A, et al. Adiponectin associates with markers of cartilage degradation in osteoarthritis and induces production of proinflammatory and catabolic factors through mitogen-activated protein kinase pathways. Arthritis Res. Ther. 2011;13:R184. doi: 10.1186/ar3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hao D, et al. Synovial fluid level of adiponectin correlated with levels of aggrecan degradation markers in osteoarthritis. Rheumatol. Int. 2011;31:1433–1437. doi: 10.1007/s00296-010-1516-0. [DOI] [PubMed] [Google Scholar]
- 185.Sharma L. Osteoarthritis of the knee. N. Engl. J. Med. 2021;384:51–59. doi: 10.1056/NEJMcp1903768. [DOI] [PubMed] [Google Scholar]
- 186.Bannuru RR, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019;27:1578–1589. doi: 10.1016/j.joca.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 187.Kolasinski SL, et al. 2019 American college of rheumatology/arthritis foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Rheumatol. 2020;72:220–233. doi: 10.1002/art.41142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fransen M, et al. Exercise for osteoarthritis of the knee. Cochrane Database Syst. Rev. 2015;1:Cd004376. doi: 10.1002/14651858.CD004376.pub2. [DOI] [PubMed] [Google Scholar]
- 189.Juhl C, et al. Impact of exercise type and dose on pain and disability in knee osteoarthritis: a systematic review and meta-regression analysis of randomized controlled trials. Arthritis Rheumatol. 2014;66:622–636. doi: 10.1002/art.38290. [DOI] [PubMed] [Google Scholar]
- 190.Messier SP, et al. Intentional weight loss in overweight and obese patients with knee osteoarthritis: is more better? Arthritis Care Res. 2018;70:1569–1575. doi: 10.1002/acr.23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hall M, et al. Diet-induced weight loss alone or combined with exercise in overweight or obese people with knee osteoarthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2019;48:765–777. doi: 10.1016/j.semarthrit.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 192.Heidari B. Knee osteoarthritis diagnosis, treatment and associated factors of progression: part II. Casp. J. Intern. Med. 2011;2:249–255. [PMC free article] [PubMed] [Google Scholar]
- 193.Deyle GD, et al. Physical therapy versus glucocorticoid injection for osteoarthritis of the knee. N. Engl. J. Med. 2020;382:1420–1429. doi: 10.1056/NEJMoa1905877. [DOI] [PubMed] [Google Scholar]
- 194.Rutjes AW, et al. Viscosupplementation for osteoarthritis of the knee: a systematic review and meta-analysis. Ann. Intern. Med. 2012;157:180–191. doi: 10.7326/0003-4819-157-3-201208070-00473. [DOI] [PubMed] [Google Scholar]
- 195.Shan L, et al. Intermediate and long-term quality of life after total knee replacement: a systematic review and meta-analysis. J. Bone Jt. Surg. Am. 2015;97:156–168. doi: 10.2106/JBJS.M.00372. [DOI] [PubMed] [Google Scholar]
- 196.Gai Z, et al. Lipid accumulation and chronic kidney disease. Nutrients. 2019;11:722. doi: 10.3390/nu11040722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 198.Saberianpour S, et al. Therapeutic effects of statins on osteoarthritis: a review. J. Cell Biochem. 2022;123:1285–1297. doi: 10.1002/jcb.30309. [DOI] [PubMed] [Google Scholar]
- 199.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017;120:229–243. doi: 10.1161/CIRCRESAHA.116.308537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Juybari KB, Hosseinzadeh A, Sharifi AM. Protective effects of atorvastatin against high glucose-induced nuclear factor-κB activation in cultured C28I2 chondrocytes. J. Recept Signal Transduct. Res. 2019;39:1–8. doi: 10.1080/10799893.2018.1557206. [DOI] [PubMed] [Google Scholar]
- 201.Terabe K, et al. Simvastatin promotes restoration of chondrocyte morphology and phenotype. Arch. Biochem. Biophys. 2019;665:1–11. doi: 10.1016/j.abb.2019.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wu YP, et al. Pravastatin reduces matrix metalloproteinases expression and promotes cholesterol efflux in osteoarthritis chondrocytes. Evid.-Based Compl. Alt. 2022;2022:9666963. doi: 10.1155/2022/9666963. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 203.Du J, et al. Effect of high fat diet and excessive compressive mechanical force on pathologic changes of temporomandibular joint. Sci. Rep.-Uk. 2020;10:17457. doi: 10.1038/s41598-020-74326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tanaka T, et al. Attenuation of osteoarthritis progression in mice following intra-articular administration of simvastatin-conjugated gelatin hydrogel. J. Tissue Eng. Regen. Med. 2019;13:423–432. doi: 10.1002/term.2804. [DOI] [PubMed] [Google Scholar]
- 205.Katole NT, Kale JS, Salankar HV. Evaluation of the antinociceptive action of simvastatin in mice. Cureus. 2022;14:e26910–e26910. doi: 10.7759/cureus.26910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Goto N, et al. Single intra-articular injection of fluvastatin-PLGA microspheres reduces cartilage degradation in rabbits with experimental osteoarthritis. J. Orthop. Res. 2017;35:2465–2475. doi: 10.1002/jor.23562. [DOI] [PubMed] [Google Scholar]
- 207.Zhou B, et al. Proliferation of rabbit chondrocyte and inhibition of IL-1β-induced apoptosis through MEK/ERK signaling by statins. Vitr. Cell Dev.- 2017;53:124–131. doi: 10.1007/s11626-016-0086-1. [DOI] [PubMed] [Google Scholar]
- 208.Clockaerts S, et al. Statin use is associated with reduced incidence and progression of knee osteoarthritis in the Rotterdam study. Ann. Rheum. Dis. 2012;71:642–647. doi: 10.1136/annrheumdis-2011-200092. [DOI] [PubMed] [Google Scholar]
- 209.Haj-Mirzaian A, et al. Statin use and knee osteoarthritis outcome measures according to the presence of heberden nodes: results from the osteoarthritis initiative. Radiology. 2019;293:396–404. doi: 10.1148/radiol.2019190557. [DOI] [PubMed] [Google Scholar]
- 210.Loomba R, et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (mozart trial) Hepatology. 2015;61:1239–1250. doi: 10.1002/hep.27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Cannon CP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 212.Gierman LM, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice-a translational model for atherosclerosis. Ann. Rheum. Dis. 2014;73:921–927. doi: 10.1136/annrheumdis-2013-203248. [DOI] [PubMed] [Google Scholar]
- 213.Szychlinska MA, Ravalli S, Musumeci G. Pleiotropic effect of fibrates on senescence and autophagy in osteoarthritis. Ebiomedicine. 2019;45:11–12. doi: 10.1016/j.ebiom.2019.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb. Vasc. Biol. 2010;30:894–899. doi: 10.1161/ATVBAHA.108.179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Clockaerts S, et al. Cytokine production by infrapatellar fat pad can be stimulated by interleukin 1 beta and inhibited by peroxisome proliferator activated receptor alpha agonist. Ann. Rheum. Dis. 2012;71:1012–1018. doi: 10.1136/annrheumdis-2011-200688. [DOI] [PubMed] [Google Scholar]
- 216.Clockaerts S, et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthr. Cartil. 2010;18:876–882. doi: 10.1016/j.joca.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 217.Distel E, et al. The infrapatellar fat pad in knee osteoarthritis: an important source of interleukin-6 and its soluble receptor. Arthritis Rheum. 2009;60:3374–3377. doi: 10.1002/art.24881. [DOI] [PubMed] [Google Scholar]
- 218.Shirinsky IV, Shirinsky VS. Treatment of erosive osteoarthritis with peroxisome proliferator-activated receptor alpha agonist fenofibrate: a pilot study. Rheumatol. Int. 2014;34:613–616. doi: 10.1007/s00296-013-2766-4. [DOI] [PubMed] [Google Scholar]
- 219.Wei W, et al. Statins and fibrates do not affect development of spontaneous cartilage damage in STR/Ort mice. Osteoarthr. Cartil. 2014;22:293–301. doi: 10.1016/j.joca.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 220.van Gemert Y, et al. Novel high-intensive cholesterol-lowering therapies do not ameliorate knee OA development in humanized dyslipidemic mice. Osteoarthr. Cartil. 2021;29:1314–1323. doi: 10.1016/j.joca.2021.02.570. [DOI] [PubMed] [Google Scholar]
- 221.Wu CL, et al. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann. Rheum. Dis. 2015;74:2076–2083. doi: 10.1136/annrheumdis-2014-205601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yu H, et al. A low ratio of n-6/n-3 polyunsaturated fatty acids suppresses matrix metalloproteinase 13 expression and reduces adjuvant-induced arthritis in rats. Nutr. Res. 2015;35:1113–1121. doi: 10.1016/j.nutres.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 223.Shen CL, et al. Decreased production of inflammatory mediators in human osteoarthritic chondrocytes by conjugated linoleic acids. Lipids. 2004;39:161–166. doi: 10.1007/s11745-004-1214-6. [DOI] [PubMed] [Google Scholar]
- 224.Tsubosaka M, et al. Gelatin hydrogels with eicosapentaenoic acid can prevent osteoarthritis progression in vivo in a mouse model. J. Orthop. Res. 2020;38:2157–2169. doi: 10.1002/jor.24688. [DOI] [PubMed] [Google Scholar]
- 225.Stonehouse W, et al. Krill oil improved osteoarthritic knee pain in adults with mild to moderate knee osteoarthritis: a 6-month multicenter, randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2022;116:672–685. doi: 10.1093/ajcn/nqac125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.MacFarlane LA, et al. The effects of vitamin d and marine omega-3 fatty acid supplementation on chronic knee pain in older us adults: results from a randomized trial. Arthritis Rheumatol. 2020;72:1836–1844. doi: 10.1002/art.41416. [DOI] [PMC free article] [PubMed] [Google Scholar]