

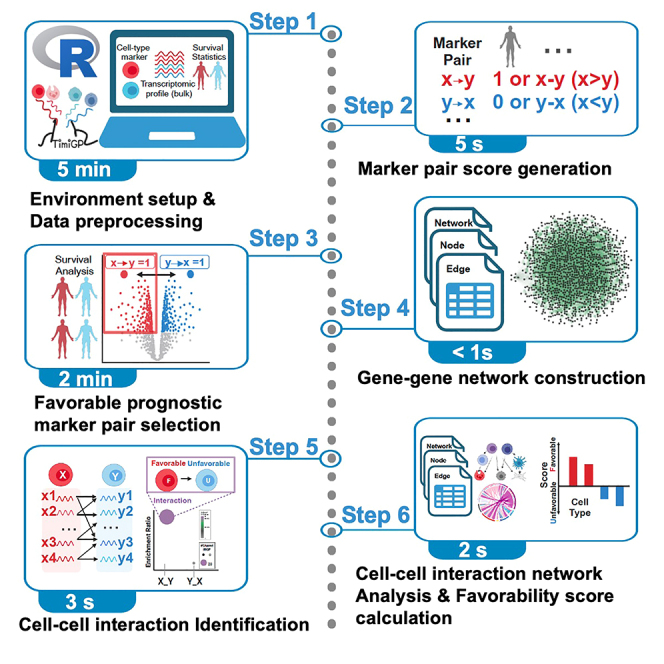
Summary
Exploring the clinical relevance of diverse immune cell types within the tumor microenvironment is pivotal for unraveling cancer intricacies and developing treatments. Here, we present a protocol for using tumor immune microenvironment illustration based on gene pairs, an R package to deduce cell-cell interactions, unveiling the association between immune cell relative abundance and patient prognoses from bulk gene expression and survival data. We describe steps for harnessing cell-type markers derived from single-cell RNA sequencing data to map the tumor immune microenvironment across a spectrum of cancer types.
For complete details on the use and execution of this protocol, please refer to Li et al. (2023).1
Subject areas: Bioinformatics, Single Cell, Cancer, Genomics, RNA-seq, Immunology, Gene Expression, Systems biology
Graphical abstract
Highlights
-
•
A user-friendly R package for TimiGP protocol to infer a cell-cell interaction network
-
•
Integrate bulk transcriptome and survival statistics to gain deeper insights into TIME
-
•
A protocol to introduce data preprocessing, analysis, and visualization
-
•
Customizable parameters to enable pan-cancer analysis at different resolutions
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Exploring the clinical relevance of diverse immune cell types within the tumor microenvironment is pivotal for unraveling cancer intricacies and developing treatments. Here, we present a protocol for using tumor immune microenvironment illustration based on gene pairs, an R package to deduce cell-cell interactions, unveiling the association between immune cell relative abundance and patient prognoses from bulk gene expression and survival data. We describe steps for harnessing cell-type markers derived from single-cell RNA sequencing data to map the tumor immune microenvironment across a spectrum of cancer types.
Before you begin
The tumor microenvironment comprises a complex interplay between cancerous and non-cancerous cells, prominently immune cells. These immune cells encompass both those combatting cancer and those promoting cancer, collectively shaping the tumor immune microenvironment (TIME).2,3 Recent investigations underscore the remarkable impact of TIME on patient prognoses.4,5 However, dissecting these interactions proves intricate due to their interwoven nature, often resulting in conflicting interpretations.
TimiGP (Tumor Immune Microenvironment Illustration based on Gene Pairs) is a computational method that aims at bridging this void.1 This method constructs a network delineating interactions between immune cells from survival statistics and bulk transcriptomics. These interactions spotlight specific associations correlated with favorable patient outcomes, suggesting pivotal immune cell types steering anti-tumor responses.
The adaptability of TimiGP extends to analyses of diverse interaction scales beyond immune cells, using the cell typing results from single-cell RNA sequencing (scRNA-seq). Employing TimiGP across an assortment of solid tumor types unveils distinctive prognostic roles assumed by immune cells across different cancers. This advancement elevates our comprehension of TIME, potentially charting courses for personalized therapeutic interventions.
In this protocol, we provide step-by-step instructions to use the TimiGP R package (v1.2.0) to dissect the association between TIME and prognosis.
Computational requirements for TimiGP
-
1.Computational Requirements:
-
a.RAM: Minimum recommended RAM 8 GB.
-
b.R Version: Minimum required R version 4.0.0.
-
c.Dependencies: In addition to the base R installation, TimiGP relies on several external R packages to ensure its functionality and enhance its capabilities. These dependencies will be automatically installed during the setup process to streamline the installation and configuration of the TimiGP environment. The required external R packages include:
-
i.‘survival’: survival analysis.
-
ii.‘dplyr’: data manipulation.
-
iii.‘doParallel’ and ‘foreach’: parallel computing.
-
iv.‘ggplot2’, ‘circlize’, and ‘RColorBrewer’: data visualization.
-
i.
-
a.
Installation of TimiGP R package
Timing: 5 min
-
2.
Install the TimiGP R package. Troubleshooting 1.
Note: TimiGP R package is not available on the standard R repositories (CRAN). It is public on GitHub under GNU General Public License v3.0. ‘devtools’ is a package that provides tools for package development, including installing packages from GitHub. If you don't have the ‘devtools’ package installed, you need to install it first, and then use the `install_github()` function from the ‘devtools’ package to install the TimiGP package directly from its GitHub repository "CSkylarL/TimiGP".
# Install devtools from CRAN if not already installed.
if(!require(devtools)){
install.packages("devtools")
}
# Install TimiGP R package from GitHub:
# https://github.com/CSkylarL/TimiGP
devtools::install_github("CSkylarL/TimiGP")
Note: In the above code block, the '#' symbol is used to indicate comments, which are not executable as code.
Note: When you install this package, rest assured that the installation process includes the automatic resolution of all required dependencies. The installation of the TimiGP R package, along with its dependencies, takes a total of 5 minutes. However, if the dependencies are already installed, the installation time for the TimiGP R package alone is reduced to just 30 seconds.
-
3.
Load the package.
Note: If the TimiGP R package has been successfully installed, it should be able to load the package into the R session using the `library()` function. Troubleshooting 2
library(TimiGP)
-
4.
Check the package version.
Note: This protocol is for the package version v1.2.0.
packageVersion("TimiGP")
# return
# [1] ‘1.2.0’
Overview of internal data
-
5.The TimiGP R package has several internal data objects that contain preprocessed and curated information necessary for various analyses and functions within the package. These data objects include:
-
a.Example of input data: the transcriptomic profile, and survival statistics of TCGA (The Cancer Genome Atlas) metastatic melanoma samples (TCGA_SKCM06),6 which is preprocessed to the input format for TimiGP analysis.
-
i.`SKCM06rna`: This dataset contains the transcriptome profile for 368 TCGA_SKCM06 samples, which is bulk RNA-seq data for 20,501 genes with RSEM-normalization.
-
ii.`SKCM06info`: This dataset provides survival statistics for 368 TCGA_SKCM06 samples, including the overall survival event and time.
-
i.
-
b.Cell-type markers (Table 1).Note: The cell-type markers provided by TimiGP are HUGO (Human Genome Organization) nomenclature for gene naming.
-
i.`CellType_Bindea2013_cancer`: This dataset contains markers of immune cell types along with markers for one cancer cell type, generated by Bindea et al.7 The special cytotoxic cell markers (e.g., GZMH, GZMA, KLRB1, KLRK1, GNLY, etc.), typically associated with anti-tumor immune responses, serve as a positive control, while the tumor cell markers (e.g., KRT13, RBP1, CCND1, S100A2, KLK6, etc.) are included as a control for pro-tumor cell types.
-
ii.`CellType_Charoentong2017_Bindea2013_Xu2018_Immune`: This dataset compiles immune cell types and their associated markers published by Charoentong et al.,8 Bindea et al.,7 and Xu et al.9 Compared to `CellType_Bindea2013_cancer`, `Bindea2013` solely comprises immune markers while omitting cancer-related markers. In contrast, `Charoentong2017` is characterized by a broader diversity of mutually exclusive cell-subtype markers, while `Xu2018`, the modified cell-type markers developed by Xu et al., are more concise, utilizing only 47 genes to represent a total of 17 distinct cell types. These markers are supplemented with additional well-known immune stimulators, inhibitors, and checkpoints. The comprehensive marker set is saved as `Immune_Marker_n1293`.
-
iii.`CellType_Newman2015_LM22`: This dataset provides markers specific to immune cell types, utilizing the LM22 signature generated by CIBERSORT.10 These markers are valuable for characterizing various immune cells with activated and resting status.
-
iv.`CellType_Zheng2021_Tcell`: This dataset encompasses markers for 40 distinct T cell subtypes generated from scRNA-seq performed by Zheng et al.,11 enabling the high-resolution analysis of diverse T cell populations.
-
v.`CellType_Tirosh2016_melanoma_TME`: This dataset offers insights into the cell types present in the tumor microenvironment of metastatic melanoma, along with their associated markers. It includes melanoma-specific cancer cells, stroma cells, and major immune cell types derived from the scRNA-seq study conducted by Tirosh et al.12
-
i.
-
c.Example of intermediate results.Note: "Intermediate results" refer to pivotal interim outcomes generated during the TimiGP analysis. These intermediate results act as checkpoints or reference points within the analysis, aiding users in both understanding the process and efficiently testing individual components of the TimiGP code.
-
i.`Bindea2013c_COX_MP_SKCM06`: This dataset contains `TimiCOX()` function results using `CellType_Bindea2013_cancer` for the analysis of TCGA_SKCM06 data, revealing the association between each marker pair and favorable prognosis.
-
ii.`Bindea2013c_enrich`: This dataset provides an example result of `TimiEnrich()` and `TimiPermFDR()` functions, allowing for enrichment analysis and defining immune cell interactions.
-
i.
-
a.
Table 1.
Features of internal cell-type markers
| Data | Cell-type marker | No. Cell types | No. Markers | Source | Mutually exclusive | Resolution | Generating codes | Reference |
|---|---|---|---|---|---|---|---|---|
| CellType_Bindea2013_cancer | Bindea2013_cancer | 22 | 539 | Bulk transcriptomic profile | No | Immune cells include cytotoxic cells, plus tumor cells | https://github.com/CSkylarL/TimiGP/blob/master/inst/extdata/build_CellType_Bindea2013_cancer.R"build_CellType_Bindea2013_cancer.R | Bindea et al. (2013)7 |
| CellType_Charoentong2017_Bindea2013_Xu2018_Immune | Bindea2013 | 21 | 502 | No | Immune cell types | https://github.com/CSkylarL/TimiGP/blob/master/inst/extdata/build_CellType_Charoentong2017_Bindea2013_Xu2018_Immune.R"build_CellType_Charoentong2017_Bindea2013_Xu2018_Immune.R | Bindea et al. (2013)7 | |
| Charoentong2017 | 28 | 782 | Yes | Charoentong et al. (2017)8 | ||||
| Xu2018 | 17 | 47 | No | Xu et al. (2018)9 | ||||
| CellType_Newman2015_LM22 | Newman2015 | 22 | 547 | No | Immune cells with activated and resting status | https://github.com/CSkylarL/TimiGP/blob/master/inst/extdata/build_CellType_Newman2015_LM22.R"build_CellType_Newman2015_LM22.R | Newman et al. (2015)10 | |
| CellType_Zheng2021_Tcell | Zheng2021 | 40 | 389 | scRNA-seq | No | T cell subtypes | https://github.com/CSkylarL/TimiGP/blob/master/inst/extdata/build_CellType_Zheng2021_Tcell.R"build_CellType_Zheng2021_Tcell.R | Zheng et al. (2021)11 |
| CellType_Tirosh2016_melanoma_TME | Tirosh2016 | 6 | 391 | Yes | Melanoma, Stroma, and Immune cells | https://github.com/CSkylarL/TimiGP/blob/master/inst/extdata/build_CellType_Tirosh2016_melanoma_TME.R"build_CellType_Tirosh2016_melanoma_TME.R | Tirosh et al. (2016)12 |
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| TimiGP | Li et al.1 | https://github.com/CSkylarL/TimiGP |
| R (v4.3.1) | R CRAN | https://cran.r-project.org/ |
| dplyr R package (v1.1.2) | Wickham et al.13 | https://cran.r-project.org/web/packages/dplyr/index.html |
| devtools R package (v2.4.5) | Wickham et al.14 | https://cran.r-project.org/web/packages/devtools/index.html |
| doParallel R package (v1.0.17) | Analytics and Weston15 | https://cran.r-project.org/web/packages/doParallel/index.html |
| foreach R package (v1.5.2) | Wallig and Weston16 | https://cran.r-project.org/web/packages/foreach/index.html |
| survival R package (v3.5–5) | Therneau and Grambsch17 | https://cran.r-project.org/web/packages/survival/index.html |
| ggplot2 R package (v3.4.3) | Wickham18 | https://cran.r-project.org/web/packages/ggplot2/index.html |
| RColorBrewer R package (v1.1–3) | Neuwirth and Neuwirth19 | https://cran.r-project.org/web/packages/RColorBrewer/index.html |
| circlize R package (v0.4.13) | Gu et al.20 | https://cran.r-project.org/web/packages/circlize/index.html |
| Deposited data | ||
| Bulk RNA-seq of metastatic melanoma cohorts of The Cancer Genome Atlas (TCGA_SKCM06) | Akbani et al.6 | https://gdac.broadinstitute.org/ |
| Other | ||
| Cell-type markers: Bindea2013 | Bindea et al.7 | https://doi.org/10.1016/j.immuni.2013.10.003 |
Step-by-step method details
Here we describe step-by-step details to apply the TimiGP1 R package to TCGA metastatic melanoma data (TCGA_SKCM06) using the cell type markers developed by Bindea et al.7 The code file is available in the example folder (example01). This folder also Includes the step-by-step details to use other cell-type annotations (example02‒05).
Step 1: Data preprocessing
TimiGP requires three inputs: (1) markers representing cell types, (2) transcriptomic profiles, and (3) survival statistics encompassing events (such as death or recurrence) and the corresponding time-to-event within the identical study cohort. We start with a series of systematic steps to prepare input data for TimiGP analysis. The process begins with the loading of input example data of TCGA_SKCM06 samples, including `SKCM06info′ for survival statistics and `SKCM06rna′ for transcriptomic profiles. Next, the `CellType_Bindea2013_cancer` cell type annotation data is loaded as the third input for subsequent analyses.
This data preprocessing step involves preparing the clinical and transcriptomic data for TimiGP analysis. These steps ensure that the data is in a suitable format and quality for subsequent analysis. The preprocessing includes two main functions: `TimiCheckEvent()` and `TimiPrePropress()`, both of which are described below.
-
1.
Clear the workspace and load the TimiGP R package.
rm(list=ls())
library(TimiGP)
Note: Before commencing your analysis, you can use the `setwd()` function to specify a directory path within the parentheses, effectively changing your current working directory in R. As shown below, replacing " Your/Directory/Path/" with the actual path will adjust your working directory to the specified location, facilitating smoother file read and write operations from that directory.
setwd("/Your/Directory/Path/")
-
2.Load the survival statistics dataset and transcriptome profile of TCGA_SKCM06 data.
-
a.The transcriptome profile, labeled as `SKCM06rna′, encompasses a data frame from 20,501 genes (row) collected from a total of 368 metastatic melanoma samples (column). Each row in this dataset corresponds to a specific gene, and each column corresponds to a unique patient identifier (Figure 1A).
-
b.The `SKCM06info′ dataset offers critical clinical information related to survival statistics for a cohort of 368 patients with metastatic melanoma. This dataset is organized in a data frame format, which comprises 368 rows, with each row corresponding to a unique patient identifier. The first column provides the event (indicating patient death) and the second column shows the time (measured in days) to the event (Figure 1B).
-
a.
Note: Events (e.g., deaths, recurrences, etc.) are encoded as "1." These represent the occurrence of the event of interest within the observation time. Censoring, which occurs when there is incomplete data due to subjects not experiencing the event by the end of the study or being lost to follow-up, is encoded as "0." These represent the cases where the event of interest did not occur within the observation time.
Note: Considering the statistic power of survival analysis, the minimum advisable cohort size for this analysis is 30.
Note: Please structure your data in a manner similar to the input example provided, ensuring it aligns with the showcased format, which will facilitate seamless further analysis.
data("SKCM06info")
data("SKCM06rna")
-
3.Load cell type annotations.
-
a.The `CellType_Bindea2013_cancer` data provides immune cell types and their associated marker genes, as annotated by Bindea et al. in 2013.7 This data is organized in a data frame format, comprising 545 rows and 3 columns (Figure 1C). Here are the 3 columns:
-
i."CellType": representing immune and cancer cell types;
-
ii."Gene": denoting the marker gene associated with each cell type;
-
iii."Dataset": indicating the source of the annotation.
-
i.
-
a.
Note: It is flexible to utilize alternative cell type annotations included in the package or create custom cell type annotations in the same standardized format. This allows the user to tailor the analysis to various resolutions or specific research inquiries, adapting the cell type information to suit the unique aspects of your investigation. We recommend 5‒30 markers per cell type, considering the balance between computational efficiency and the robustness of results. Mutually exclusive markers between cell types are preferable but not mandatory.
data("CellType_Bindea2013_cancer")
# The 'geneset' variable will now hold this data,
# making it accessible for further analysis and exploration.
geneset <- CellType_Bindea2013_cancer
# Extract unique marker genes from the 'geneset' data frame
# These unique marker genes will be used for subsequent analysis
marker <- unique(geneset$Gene)
-
4.Preprocess the data using functions `TimiCheckEvent()` and `TimiPrePropress()` functions.
-
a.The `TimiCheckEvent()` function is designed to filter and clean survival statistics for analysis. It takes a data frame containing event and time-to-event information as input. The function performs the following tasks:
-
i.Removes records with missing events or time-to-event data.
-
ii.Filters out records with abnormal time-to-event values less than or equal to 0.
-
iii.Returns the filtered survival information data frame.This function ensures that the clinical data is free from missing values and inappropriate time-to-event values, which are crucial for accurate survival analysis.
-
i.
-
b.The `TimiPrePropress()` function preprocesses transcriptomic profile data for analysis. It takes marker gene list(`marker`), transcriptomic data(`rna`), selected cohort(`cohort`), and two optional normalization parameters as input. The function performs the following tasks:
-
i.Extracts the transcriptomic data for the selected cohort and marker genes. Troubleshooting 3.
-
ii.Removes genes with low expression based on a threshold. The threshold determines the minimum percentage of samples with non-zero expression required for a gene to be considered sufficiently expressed. Genes with expression values above this threshold in at least 50% of the samples are retained, while those falling below this threshold are filtered out due to low expression levels. If no genes meet the threshold, an error message is displayed, and the analysis is halted. Otherwise, it updates the transcriptomic data by keeping only the genes that meet the expression threshold, and a message is printed to inform the user about the number of filtered-out markers with low expressions.
-
iii.Optionally performs log transformation and gene-wise median normalization. If `log = TRUE`, log transformation will be performed; the default value is “TRUE”. If `GMNorm = TRUE`, gene-wise median normalization will be performed; he default value is “TRUE”. The approach of log transformation and gene-wise median normalization is advisable when working with gene expression data that span multiple orders of magnitude, as it can enhance the detection of subtle gene expression differences and mitigate the impact of extreme values. Additionally, enabling gene-wise median normalization (`GMNorm = TRUE`) is recommended when capturing logical relations between marker pairs, while disabling it (`GMNorm = FALSE`) is suitable for more sensitive analyses aimed at capturing continuous relationships between markers. More details regarding marker pair relations can be found in the next step.
-
iv.Returns the preprocessed transcriptomic profile data frame.This function ensures that the transcriptomic data is appropriately filtered, transformed, and normalized, preparing it for subsequent analysis steps.info <- TimiCheckEvent(SKCM06info)rna <- TimiPrePropress(marker = marker, rna = SKCM06rna, cohort = rownames(info), log = TRUE, GMNorm=TRUE)
-
i.
-
a.
Figure 1.

The format to input data
(A) The formats of transcriptomic data; (B) the formats of survival statistics data; (C) the format of cell-type marker data. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Step 2: Define and select marker gene pair matrix
TimiGP is based on the hypothesis that the ratios of expression between Immune Marker Gene Pairs (IMGPs) encapsulate the relative proportions of immune cell infiltration and capture pivotal immune interactions. So first, TimiGP establishes the Marker Pair Score (MPS). This score is formulated by evaluating the expression disparities between IMGPs using the `TimiGenePair()` function.
-
5.Generate Marker Pair Score (MPS) using the `TimiGenePair()` function.
-
a.The `TimiGenePair()` function captures and assesses the logical or continuous relationship between pairs of markers (genes) based on transcriptomic profile data (`rna`) preprocessed by the `TimiPrePropress()` function. This function can be used to generate a matrix of Marker Pair Scores, which indicates whether the expression of one gene is greater than or less than the expression of another gene.
-
b.It offers both logical and continuous analysis options.
-
i.By default (`cont = FALSE`), it captures the logical relationship between marker pairs and produces a matrix of Marker Pair Scores indicating whether the expression of gene A is greater (1 or “TRUE”) or less (0 or FALSE) than that of gene B (Figure 2A).
-
ii.Optionally, it can capture the continuous relationship between marker pairs by setting `cont = TRUE`, producing a matrix of Marker Pair Scores representing the difference between the expression levels of gene A and gene B(Figure 2B).
-
i.
-
a.
Note: If continuous relation analysis is selected (`cont = TRUE`), the function checks whether gene-wise median normalization has been applied and recommends setting `GMNorm = FALSE` in the `TimiPrePropress()` function and then re-running this function.
Optional: In this protocol, we focus on the logical relation-based analysis (default, `cont = FALSE`), which represents a more conservative analysis and results in a list of high-confident cell-cell interactions. Enabling the continuous relation option will preserve more gene pairs and have a higher statistical power in the subsequent survival analysis. Therefore, the optional approach is more sensitive to identifying prognostic gene pairs and cell-cell interactions (Figures 2C‒2F). The step-by-step details for this optional approach are available in the example06, which includes the code to repeat Figures 2B, 2D, and 2F.
mps <- TimiGenePair(rna=rna, cont=FALSE)
Figure 2.

TimiGP results comparison between continuous relation-based analysis (optional), and logical relation-based analysis (default)
(A and B) Output screenshots of marker pair scores using the logical relation-based approach (A) and using the continuous-relation-based approach (B).
(C and D) Chord diagram of cell-cell interactions identified using the logical relation-based approach (C) and the continuous relation-based approach (D).
(E and F) Bar plot of the favorability score to prioritize critical cell type related to favorable (orange) or unfavorable (blue) prognosis using logical relation-based approach (E) and using continues-relation-based approach (F). The default mode represents a more conservative analysis while the optional feature provides a more sensitive choice. In the continuous relation-based analysis, more statistically significant prognostic pairs (8.56% compared to 1.25% in the logical relation-based analysis) are selected by the Cox regression. Accordingly, the identified cell interactions (n = 113) using this approach almost double compared to the default analysis (n = 54). However, the original interactions tend to be the most significant interactions among those identified by the continuous relation-based approach (C, D). The two cell-cell interaction networks resulted in very similar prioritized cell types (cells are sorted according to their favorability scores calculated based on their topological features in the network) (E, F). The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box. Abbreviation of cell types: T, T cell; Th, helper T cell; Th1, type 1 Th; Th2, type 2 Th; Tfh, follicular Th; Tcm, central memory T cell; Tem, effector memory T cell; cytotoxic, cytotoxic cell (common cytotoxic markers of anti-tumor CD8 T cells, Tγδ, and NK cells); NK, natural killer cell; DC, dendritic cell; iDC, immature DC; aDC, activated DC; Mast, mast cell; Tumor, tumor cell.
Step 3: Choose gene pairs associated with a favorable prognosis
The next step of the TimiGP algorithm involves the selection of gene pairs linked to a positive prognosis. TimiGP employs univariate Cox regression to carry out this selection process, that is, the `TimiCOX()` function.
-
6.Perform univariate Cox regression using the `TimiCOX()` function.
-
a.Description: The `TimiCOX()` function is designed to identify marker pairs associated with favorable prognosis using univariate Cox regression analysis. This function takes a matrix of Marker Pair Scores (`mps`), a data frame containing event and days-to-event information (`info`), and an optional parameter for adjusting p-values (`p.adj`). It performs Cox regression analysis for each marker pair as a variable and processes the results to identify pairs associated with favorable prognoses.
-
b.Parameters:
-
i.`mps`: A matrix of Marker Pair Scores, where each row represents a marker pair and each column corresponds to a sample. This is the output of the `TimiGenePair()` function, which is generated in Step 2.
-
ii.`info`: A data frame with two columns, where the first column represents event status (0 for censored, 1 for event occurrence) and the second column represents time to event. This is the output of the `TimiCheckEvent()` function.
-
iii.`p.adj`: A string specifying the method for adjusting p-values. Options include "holm" (Holm’s step-down procedure), "hochberg" (Hochberg’s step-up procedure), "hommel" (Hommel’s method), "bonferroni" (Bonferroni correction), "BH" (Benjamini-Hochberg procedure), "BY" (Benjamini-Yekutieli procedure), "fdr" (False Discovery Rate control), or "none" (No adjustment, p-values are not adjusted). The default is "BH".
-
iv.`parallel`: A logical value indicating whether to enable parallel computing. If set to “TRUE”, the function will utilize parallel processing using the ‘foreach’ and ‘doParallel’ packages to speed up computations. The default value is “FALSE”, meaning that parallel computing is disabled by default. When enabled, the function will distribute tasks across multiple CPU cores to improve efficiency.Note: Depending on the specific conditions of your analysis, particularly the number of markers and the choice of CPU cores, the computational time for this step can vary significantly. If parallel computing is disabled (default, `parallel = FALSE`), this step typically takes around 5 minutes to complete. The computation time may increase if a larger number of cell-type markers are provided. For instance, if there are more than 1000 markers, the process could take more than 30 minutes to finish with this default mode. To expedite the computation, enabling parallel computing by setting the `parallel = TRUE` and specifying the number of CPU cores to use can potentially reduce the execution time. Keep in mind that the effectiveness of parallelization may vary depending on the task and hardware.
-
v.`core`: A numeric value specifying the number of CPU cores to use for parallel execution, when parallel computing is enabled (i.e., when `parallel` is set to “TRUE”). The default value is 1.Note: In this protocol, `core = 20′ is set for the parallelization of the specified task. The choice of the number of cores is highly task-dependent, and it's crucial to consider several factors when making this decision. These factors include hardware limitations, such as the number of physical cores available in the CPU, operating system constraints, the availability of system resources, and the computational load, which can vary based on the number of markers included in the analysis. In many cases, using a more conservative range, such as 2 to 4 cores, proves to be sufficient for efficient and balanced performance.
-
i.
-
c.Return: The function returns a list with two elements (Figure 3):
-
i.“mps”: The first element is the modified marker pair score matrix, where the values of marker pairs associated with poor prognosis have been reversed, and the pair direction and values of the marker pair score have been changed accordingly. For each marker pair, the mps of 1 is associated with a favorable prognosis in the updated matrix (Figure 3A).
-
ii.“cox_res”: The second element is a data frame containing the results of the Cox regression analysis, including three columns: Hazard Ratio (HR), P-Value (PV), and Adjusted P-Value (QV) for each marker pair (row name) (Figure 3B).# Run the TimiCOX functionres <- TimiCOX( mps = mps,info = info,p.adj = "BH",parallel = T,core = 20)# Extract the 'mps' and 'cox_res' objects from the 'res' resultmps <- res$mpscox_res <- res$ cox_res# Now you can use the 'cox_res' variable for further analysisOptional: The above intermediate results `cox_res` has been renamed as `Bindea2013c_COX_MP_SKCM06′ and saved as an example data. The data can be loaded by the below codes:data(Bindea2013c_COX_MP_SKCM06)cox_res <- Bindea2013c_COX_MP_SKCM06
-
i.
-
a.
-
7.
Determine the condition and cutoff to select the prognostic pairs.
Note: By default, the condition is adjusted P value and the cutoff is 0.05. It is influenced by the quality of the survival information. High-quality survival information, including a large cohort size (e.g., > 200) and a reasonable number of events, is preferred to ensure the statistical power of survival analysis. We recommend the percentage of prognostic gene pair within the range of 1%–5%. If you aim for more conservative results, you can choose a stricter cutoff, e.g., adjusted P value < 0.01 by setting `GPcondition <- "QV”` and`GPcutoff <- 0.01`. Otherwise, if you desire more sensitive outcomes and conservative results, you can choose a lenient cutoff, e.g., P value < 0.05 by setting `GPcondition <- "PV"` and `GPcutoff <- 0.05`. Troubleshooting 4
# Count the number of marker pairs with Adjusted P-value (QV) less than 0.05
sum(cox_res$QV < 0.05) # Returns 2773
# Calculate the percentage of significant marker pairs relative to the total number of marker pairs
round(sum(cox_res$QV < 0.05)/ nrow(cox_res) ∗ 100, 2) # Returns 2.52
# Set up a variable to define the condition for selecting marker pairs based on the Adjusted P-value.
GPcondition <- "QV"
# Set up a variable to define the cutoff value for the Adjusted P-value.
GPcutoff <- 0.05
# GP represents Gene Pair
Figure 3.

Output screenshots of the `TimiCOX()` function
`TimiCOX()` returns a list of two data frames: (A) updated “mps” where gene pair and marker pair score are reversed if the original one (Figure 2A) is associated with poor prognosis.
(B) “cox_res”: the result of survival analysis. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Step 4: Construct the directed gene-gene network
Utilizing the prognostic significance of IMGPs, TimiGP establishes pairwise associations between each favorable and unfavorable gene (nodes), creating a directed gene-gene network. The direction of the edges captures the connection between gene expression levels and their influence on patient prognosis. To illustrate, an edge from gene x to gene y (x→y) signifies that a higher relative expression of gene x (favorable gene) compared to gene y (unfavorable gene) correlates with a better prognosis.
-
8.Construct the directed gene-gene Network and export the files for Cytoscape21 visualization using the `TimiGeneNetwork()` function.
-
a.The input data `resdata` of the `TimiGeneNetwork()` function is the cox regression results `cox_res` generated from `TimiCOX()` function.
-
b.The `TimiGeneNetwork()` function uses three parameters to determine the marker pairs associated with a good prognosis for the gene-gene network.
-
i.`condition`: A value in one of "PV" (P.Value) and "QV" (Adjusted P.Value), which is the column name of resdata, which contains information related to statistical significance. The default value is "QV".
-
ii.`cutoff`: This parameter refers to a threshold value associated with the 'condition'. It is used as a filter for gene-gene interactions. Gene pairs with a value less than this cutoff will be considered significant and included in the gene-gene network. The default value is 0.05.
-
iii.`select`: This parameter is a character vector that can contain the names of selected gene pairs. These gene pairs are based on the row names in the `resdata` data structure. By default, this parameter is set to "NULL", indicating that no specific gene pairs are selected. However, if you manually choose gene pairs using this parameter, the automatic filtering based on the 'cutoff' and 'condition' will be overridden for those selected pairs. Troubleshooting 4.Note: Default selection includes all gene-gene interactions with an adjusted p-value < 0.05.
-
i.
-
c.The `TimiGeneNetwork()` function returns a list of network-required data that can be further modified within R. There are three elements in the list: 1) “network”, 2) “node”, and 3) “edge” (Figure 4). Each element is a data frame. This function can also export these data by setting `export = TRUE` (default) and generates below three network files that can be imported to Cytoscape for network analysis and visualization. Those files are saved to the specific location provided through the `path` parameter.
-
i.Network file (gene_network.sif): A simple interaction file exported from “network”, a data frame containing information about the network structure. It has 3 columns: 1) “Source”: Source gene in an interaction (favorable gene); 2) “Interaction”: the type of interaction; 3) “Target”: Target gene in an interaction(unfavorable gene).
-
ii.Node attributes file (gene_node.txt): A file exported from “node”, a data frame containing information about nodes (genes) in the gene network. It is the same as the input cell-type annotation `geneset` (Figure 1C), which has three columns: 1) “CellType”: Type of cell associated with the marker gene; 2) “Gene”: The gene itself; 3) “Dataset”: The dataset or source from which the gene information is derived.
-
iii.Edge attributes file (gene_edge.txt): A file exported from “edge”, a data frame containing information about edges (interactions) in the gene network. It has six columns: 1) “Key”: A key that represents a unique identifier for the interaction between genes; 2) “HR” (Hazard Ratio), 3) “PV” (P-value), 4) “QV” (Adjusted P-value): cox regression results of the interaction obtained from the input `resdata` (Figure 3B); 5) “Favorable.Gene”: One of the interacting genes (source); 6) “Unfavorable.Gene”.0: The other interacting gene (target).Note: If `export = TRUE` but `path = NULL`, those files will be exported to the current working directory.
-
i.
-
d.The `TimiGeneNetwork()` function generates node information based on two parameters.
-
i.`dataset`: This parameter specifies the dataset to be used for generating the gene network files. It expects a value from a predefined set of options: "Bindea2013_Cancer," "Immune3," "Newman2015," "Tirosh2016," "Zheng2021," or "Other." The first five options are accompanied by preset configurations linked to internal cell-type annotations utilized for the analysis. Specifically, these options correspond to: `CellType_Bindea2013_cancer`, `CellType_Charoentong2017_Bindea2013_Xu2018_Immune`, `CellType_Newman2015_LM22, CellType_Zheng2021_Tcell`, and `CellType_Tirosh2016_melanoma_TME`, respectively. If you want to use a different dataset or customize node attributes, please choose "Other" and then provide `geneset`.
-
ii.`geneset`: This parameter is a data frame that contains information about cell markers. It should have at least three columns: the first column represents cell types, the second column represents marker genes, and the optional third column represents the name of the dataset. The `geneset` parameter is used in conjunction with the dataset parameter. If you choose "Other" for the dataset, you need to provide a `geneset` data frame to define the cell markers for node attributes.GeneNET <- TimiGeneNetwork( resdata = cox_res,select = NULL,dataset = "Bindea2013_Cancer",geneset = NULL,condition = GPcondition,cutoff = GPcutoff,export = TRUE,path = "./")# Display the first few rows of the 'network' component in the NET objecthead(GeneNET$network)# Display the first few rows of the 'node' component in the NET objecthead(GeneNET$node)# Display the first few rows of the 'edge' component in the NET objecthead(GeneNET$edge)
-
i.
-
a.
Figure 4.

Output screenshots of the `TimiGeneNetwork()` function
TimiGeneNetwork returns a list of three data frames: (A) “network”, (B) “node”, (C) “edge”. The type of interaction is “TimiGP” representing gene-gene interaction exported by the TimiGP method. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Step 5: Identify cell-cell interactions by enrichment analysis
TimiGP performs enrichment analysis to statistically determine cell-cell interactions that are more prevalent in the gene-gene network compared to random occurrences. The resulting cell-cell interactions offer an understanding of how the varying abundance of cells in the TIME relates to prognosis. This step operates under the immunological premise that the equilibrium between pro- and anti-tumor cells might dictate the TIME’s efficacy or suppressive nature, influencing prognosis as a result.
The `TimiEnrich()` function is designed to perform the enrichment analysis by assessing the significance of inter-interactions within a given gene pair set and cell pair dataset. This section includes step-by-step details to prepare input of the `TimiEnrich()` function and explain its outputs.
-
9.
Select marker pairs significantly associated with good prognosis. Troubleshooting 4.
Note: It selects gene-gene interaction of interest (e.g., x→y) that a higher x-to-y ratio is associated with a positive prognosis, which is thought associated with specific cell-cell interactions. The `GPcondition` and `GPcutoff` are determined previously.
GP <- rownames(cox_res)[which(cox_res[,GPcondition] < GPcutoff)]
-
10.
Prepare marker pairs background for enrichment analysis using the `TimiBG()` function.
Note: The `background` would be the entire set of gene pairs available for analysis, which provides context for evaluating whether the gene-gene interaction is more prevalent than what would be expected by chance. Therefore, the background includes both directions of available gene pairs, such as x→y and y→x. Considering previous intermediate results only have one direction (x→y), TimiGP provides an additional function, `TimiBG()`, to prepare the background. The `TimiBG()` function takes a vector of marker pairs connected by underscores as input (e.g., x_y) and produces background gene pairs by generating complementary pairs (x_y and y_x). The resulting background pairs provide a foundation for subsequent analysis.
background <- TimiBG(marker.pair = row.names(cox_res))
-
11.Generate marker pair annotations of all pairwise cell combinations using the `TimiCellPair()` function.
-
a.Description: The function `TimiCellPair()` generates a predefined annotation set used by enrichment analysis that includes available pairwise cell combination, potentially as cell-cell interactions, which is denoted by marker pairs. In the annotation, a cell interaction X →Y is defined by a list of marker pairs that point from the markers of cell X (e.g., x1, x2) to the markers of cell Y (e.g., y1, y2) as below:Cell X → Cell Y = {x1 → y1, x1 → y2, x2 → y1, x2 → y2,...};Cell Y → Cell X = {y1 → x1, y1 → x2, y2 → x1, y2 → x2,...}
-
b.Parameters:
-
i.`geneset`: A data frame of cell-type annotation, which is the same one used to generate markers for the `TimiPrePropress()` function. As mentioned previously, in this protocol, its internal cell-type markers `CellType_Bindea2013_cancer` are loaded by the `data()` function.
-
ii.`dataset`: If the `geneset` has a third column representing dataset names, the function handles the case where there are multiple datasets. It allows you to specify a dataset for analysis using this optional parameter. This may be needed when using `CellType_Charoentong2017_Bindea2013_Xu2018_Immune` which contains three different immune cell-type annotations. More details and relevant codes were provided in the example02.
-
iii.`core`: The function utilizes parallel processing for certain operations using the ‘foreach’ and ‘doParallel’ packages. This parameter is an integer specifying the number of CPU cores used for parallel execution, which can speed up the analysis when dealing with large amounts of cell types. The default value is 1.
-
i.
-
c.Return: The function returns a data frame where each row contains a cell pair and its associated marker pairs. It has below two columns: (Figure 5A).
-
i.“Cell.Pair”: This column represents the combination of two different cell types that are possibly interacting or being considered together. The cell types are separated by an underscore ("_").
-
ii.“Marker.Pair”: This column represents the combination of two marker genes associated with the corresponding cell pair. The marker genes are separated by an underscore ("_").Note: An underscore separator "_" is used to replace the arrow "→" in the package considering programming readability and compatibility.cell_pair <- TimiCellPair(geneset = geneset, core = 20)
-
i.
-
a.
-
12.Perform enrichment analysis using the `TimiEnrich()` function.
-
a.Description: The `TimiEnrich()` function is designed to perform enrichment analysis. It is used to compare the frequency of gene-gene-directed interaction that belongs to a particular cell-cell interaction with what would be expected by chance. If a statistically significantly higher number of gene pairs from the network are found within a cell-cell interaction `geneset` compared to random chance, it suggests that the cell-cell interaction is enriched in the gene-gene network, implying potential prognostic and immunological relevance.
-
b.Parameters:
-
i.`gene`: A vector of gene symbols or gene pair for which enrichment analysis will be performed. This parameter specifies the set of gene pairs that you want to analyze for enrichment within the provided gene interactions. It’s selected in the previous step in the section.Note: The 'gene' parameter should be in a consistent gene identifier format with the transcriptome data and cell type annotation.
-
ii.`background`: A vector of background genes or pairs. This parameter defines the background against which enrichment is assessed. It represents the universe of gene pairs from which the gene-gene interaction is derived. It’s prepared in the previous step in the section.
-
iii.`geneset`: A data frame with two columns: the first column represents cell pairs, and the second column represents gene pairs. This parameter contains the potential interactions or relationships between cell types and genes that you want to analyze for enrichment. It’s generated in the previous step in the section.
-
iv.`p.adj`: A string specifying the method for adjusting p-values. Enrichment analysis often involves multiple hypothesis testing, so p-values need to be adjusted to control the false discovery rate. Options include "holm" (Holm’s step-down procedure), "hochberg" (Hochberg’s step-up procedure), "hommel" (Hommel’s method), "bonferroni" (Bonferroni correction), "BH" (Benjamini-Hochberg procedure), "BY" (Benjamini-Yekutieli procedure), "fdr" (False Discovery Rate control), or "none" (No adjustment, p-values are not adjusted). The default value is "BH".
-
v.`core`: An integer that indicates the number of cores for parallel execution. This function enables parallel execution via the ‘foreach’ and ‘doParallel’ packages, which can speed up the analysis when dealing with a large number of cell types. The default value is 1.
-
vi.`pair`: A logical value that controls the type of enrichment analysis conducted by the function. It allows the function to cater to two types of enrichment analyses: one focusing on interactions between cell pairs and the other focusing on enrichment within individual cell types. The default value is “TRUE”, that is, the enrichment analysis will be conducted on cell pairs, which is the TimiGP analysis described in this protocol. The results will provide insights into cell-cell interactions. When the parameter is set to “FALSE”, the function performs traditional enrichment analysis. The results will not distinguish between different cell types and will provide insights into enriched gene sets within individual cell types. For this traditional enrichment analysis, other parameters should be changed accordingly. The `gene` and `background` are gene symbols instead of gene pairs. `geneset` is a data frame representing general cell types(1st column) and their associated marker gene(2nd column), e.g., internal cell-type markers `CellType_Bindea2013_cancer` loaded by `data()` function.
-
i.
-
c.Return: The function returns a data frame of enrichment results including below 12 columns (Figure 5B).
-
i.“Index”: The index serves as a unique identifier for each cell-cell interaction in the enrichment analysis results. Unlike the “Rank” (2nd column), which might be shared among interactions with the same p-value, the “Index” is a distinctive identifier that ensures each interaction is uniquely labeled. This unique index can be used as a reference point to specifically identify and distinguish each cell-cell interaction in subsequent analyses, visualizations, or discussions.
-
ii.“Rank”: The rank of the cell-cell interactions is determined based on their p-values. When multiple interactions have the same p-value, the ties are resolved using the "min" method, meaning that the lowest rank among the tied interactions is assigned to all of them. A smaller p-value corresponds to a smaller rank, indicating higher statistical significance. In essence, the rank helps prioritize and order the interactions based on their level of significance.
-
iii."Cell.Interaction", "Favorable.Cell.Type", and "Unfavorable.Cell.Type": This "Cell.Interaction" column represents the interaction or relationship between two cell types (cell pair) that are the subject of the analysis. Each row corresponds to the analysis of a specific cell-cell interaction (e.g., cell X_cell Y), providing statistical insight into the relationship between these two cell types. If the "cell X_cell Y" is statistically significantly enriched, it suggests an interaction from cell X to cell Y, indicating that a high X/Y ratio is associated with a favorable prognosis. In this context, the "Favorable.Cell.Type" column spotlights Cell X, which is favorably poised to embody an anti-tumor cell character linked with optimistic prognostic outcomes. Conversely, the "Unfavorable.Cell.Type" column spotlights Cell Y, indicative of a pro-tumor cell type more inclined toward less favorable clinical outcomes.
-
iv."No.Total.IMGP": The number of immune marker gene pairs (IMGP) associated with the provided cell pair. It reflects the total count of gene interactions that are relevant to the specific cell-cell interaction being analyzed.
-
v."No.Shared.IMGP": The count of gene-gene interactions that are shared within the provided cell pair. It indicates how many of the gene-gene interactions in the total count are specific to the interaction between the two cell types.
-
vi."Enrichment.Ratio": This ratio represents the degree of enrichment for the shared gene interactions within the given cell pair. It indicates how much more frequently these shared interactions occur than would be expected by chance.
-
vii."P.Value": The p-value associated with the enrichment analysis. It quantifies the statistical significance of the observed enrichment. A lower p-value suggests stronger evidence against the null hypothesis of no enrichment.
-
viii."Adjust.P.Value": This column provides the adjusted p-value, which takes into consideration the multiple testing issues when analyzing multiple interactions. It’s often used to control the false discovery rate and provides a more stringent assessment of significance.
-
ix."Shared.IMGP" and "Total.IMGP": These columns contain concatenated lists of gene interaction pairs that are shared and total, respectively, within the specific cell pair. They provide insight into the representative gene-gene interactions contributing to the cell-cell interaction.res <- TimiEnrich( gene = GP,background = background,geneset = cell_pair,p.adj = "BH",core = 20,pair=TRUE)
-
i.
-
a.
-
13.
Determine the condition and cutoff to define the cell-cell interaction.
Note: By default, the cell-cell interaction is defined as a pairwise cell-cell combination whose adjusted P value (condition) is less than 0.05 (cutoff). The percentage of cell-cell interaction is usually around 10%. Troubleshooting 5.# Count the number of marker pairs with an Adjusted P-value less than 0.05sum(res$Adjust.P.Value < 0.05) # Returns 54# Calculate the percentage of cell-cell interaction defined by the above condition and the cutoffround(sum(res$Adjust.P.Value < 0.05)/nrow(res) ∗ 100, 2) # Returns 11.69# Set up a variable to define the condition for cell-cell interaction based on the Adjusted P-value.CIcondition <- "Adjust.P.Value"# Set up a variable to define the cutoff value for the Adjusted P-value.CIcutoff <- 0.05# CI represents Cell-cell InteractionOptional: Enhancing stringency of conditions through permutation for false discovery rate (FDR) estimation using the `TimiPermFDR()` function. (Timing: 4 min)The `TimiPermFDR()` function aims to make more cautious selections of cell interactions or when there’s a need to evaluate the robustness of newly identified cell-type annotations. By permuting sets of marker genes associated with different cell types, the function enables the assessment of cell-type marker quality while effectively controlling the False Discovery Rate (FDR). This permutation-driven approach disrupts the intrinsic link between genes and cell types, facilitating an unbiased evaluation of the interactions. The calculated FDR values for each interaction offer a reliable measure of statistical significance, aiding researchers in identifying interactions that exhibit robustness beyond random chance. Whether refining interaction selections or testing the reliability of novel discoveries, the` TimiPermFDR()` function provides an essential avenue for enhancing the accuracy and credibility of cell interaction analyses.
`TimiPermFDR()` function takes as input the results of `TimiEnrich()` function (`resdata = res`), marker gene information (`geneset = geneset`), gene-gene interactions (`gene = GP`), background information (`background = background`) and other relevant parameters. Through a certain number of gene set permutations (`niter = 100′), the function generates shuffled marker gene sets while maintaining their sizes. It then employs these shuffled sets to analyze cell interactions and compute the FDR for each interaction. As a result, A new column named "Permutation.FDR" is added to the data frame of cell-cell interaction statistics generated from the `TimiEnrich()` function (Figure 5C). The function also utilizes parallel computing by setting the parameter `core`.res <- TimiPermFDR( resdata = res,geneset = geneset,gene = GP,background = background,niter = 100,core = 20)Note: The intermediate results `res` after this step has been renamed as `Bindea2013c_enrich` and saved as example data, which can be loaded by the below codes:data(Bindea2013c_enrich)res <- Bindea2013c_enrich.-
14.Visualize top interactions and enrichment statistics using the `TimiDotplot()` function.
-
a.Description: The `TimiDotplot()` function is designed to visualize the enrichment results of selected cell-cell interaction. It creates a dot plot (Figure 5D) where the x-axis displays chosen cell interactions (The arrow “→” is replaced by an underscore "_"), the y-axis represents the Enrichment Ratio, and the color of the dots corresponds to the P-value or FDR determined by the chosen condition column values, ranging from low (green) to high (purple). Additionally, the size of the dots indicates the number of marker pairs shared by query pairs and annotation pairs.
-
b.Parameters:
-
i.`resdata` (required): TimiGP enrichment result generated from the` TimiEnrich()` function or modified with TimiPermFDR() function.
-
ii.`select`: A numeric vector representing the selected cell interactions based on the "Index" column in `resdata`. The default selection includes the top 5 enrichments.
-
iii.`condition`: A character value specifying the column name in `resdata` for the color representation of the dot plot. It can be one of "P.Value", "Adjust.P.Value", or "Permutation.FDR". The default is "Adjust.P.Value".
-
iv.`cutoff`: The maximum value for the color bar. The default is 0.05 for "Adjust.P.Value", but different values (0.01 for "P.Value" and 0.2 for "Permutation.FDR") are recommended for other conditions.
-
i.
-
c.Return: An enrichment dot plot visualizing the selected cell interactions' enrichment results. It is a ‘ggplot2’ plot since the function requires the ‘ggplot2’ package for plotting. The "Favorable Outcome" label is added to the top left corner of the plot, as an indicator that the cell-cell interaction is associated with favorable clinical outcomes.
-
a.
TimiDotplot ( resdata = res,select = c(1:10),condition = CIcondition,cutoff = CIcutoff) -
14.
Figure 5.

Output example of `TimiCellPair()`, `TimiEnrich()`, `TimiPermFDR()`, and `TimiDotplot()` functions
(A) Output screenshots of `TimiCellPair()` function; (B) Output screenshots of `TimiEnrich()` function; (C) Appended new column "Permutation.FDR" by `TimiPermFDR()` function.
(D) Dot plot generated by the `TimiDotplot()` function. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Step 6: Build and analyze the cell-cell interaction network
Connecting all selected cell-cell interactions, TimiGP constructs a directed network linked to a positive prognosis. This network offers a comprehensive depiction of the association between Tumor Immune Microenvironment (TIME) and clinical outcomes. In this network, each node signifies a specific cell type, while an edge signifies a higher relative abundance between two cell types in the TIME associated with a favorable prognosis. This network demonstrates a hierarchical structure in terms of degrees, referring to the number edges that are linked to each node.
Based on the degree information, TimiGP computes a favorability score, which consists of two components, a favorable score and an unfavorable score. A higher favorable score indicates that the corresponding cell type is generally associated with a positive prognosis and primarily functions as anti-tumor cells in the TIME network. Conversely, a higher unfavorable score suggests an adverse prognostic association and pro-tumor functions.
-
15.Construct the cell-cell network and export the files for ‘Cytoscape’ visualization using the `TimiCellNetwork()` function.Note: The parameters of the `TimiCellNetwork()` function closely resemble those of the `TimiGeneNetwork()` function, simplifying their usage and making it straightforward to work with both functions.
-
a.The input data `resdata` of the `TimiCellNetwork()` function is the enrichment results `res` generated from `TimiEnrich()` or modified with `TimiPermFDR()` functions.
-
b.The `TimiCellNetwork()` function uses three parameters to define the cell-cell interaction from enrichment statistics.
-
i.`condition`: A value in one of "P.Value", "Adjust.P.Value" and "Permutation.FDR", which is the column name of `resdata`, which contains information related to statistical significance. The default value is "Adjust.P.Value".
-
ii.`cutoff`: This parameter refers to a threshold value associated with the 'condition'. It assists in the definition of cell-cell interactions. Cell pairs with a value less than this cutoff will be cell-cell interaction and included in the network. The default value is 0.05.
-
iii.`select`: This parameter is a numeric vector denoting selected cell interactions based on the "Index" column in `resdata`. Default selection encompasses all cell-cell interactions (adjusted p-value < 0.05). By default, this parameter is set to "NULL", indicating that no specific cell-cell interactions are selected. However, if you manually choose cell-cell interactions as research of interest for visualization using this parameter, the automatic filtering based on the `cutoff` and `condition` will be overridden for those selected cell-cell interactions. Troubleshooting 5.Note: The default definition of cell-cell interactions is adjusted p-value (condition) < 0.05(cutoff).
-
i.
-
c.The `TimiCellNetwork()` function returns a list of network-required data that can be further modified within R. There are three elements in the list: 1) "network", 2) "node", and 3) "edge" (Figure 6). Each element is a data frame. This function can also export these data by setting `export = TRUE` (default) and generating the below three network files that can be imported to ‘Cytoscape’ for network analysis and visualization. Those files are saved to the specific location provided through the `path` parameter.
-
i.Network file (cell_network.sif): A simple interaction file exported from “network”, a data frame containing information about the network structure. It has 3 columns: 1) “Source”: Source cell in an interaction (favorable cell); 2) “Interaction”: the type of interaction; 3) “Target”: Target cell in an interaction(unfavorable cell).
-
ii.Node attributes file (cell_node.txt): A file exported from “node”, a data frame containing information about nodes (cells) in the network. It has three columns: 1) “Key”: This column represents different cell types (nodes); 2) “No.Markers”: This column contains the number of markers associated with each cell type. 3) “Group”: This column seems to specify the group or category to which the cell type belongs.
-
iii.Edge attributes file (cell_edge.txt): A file exported from “edge”, a data frame containing information about edges (interactions) in the cell-cell network. It adds a “Key” column that represents a unique identifier for the interaction between cells to the input `resdata` (Figures 5A and 5B).Note: If `export = TRUE` but `path = NULL`, those files will be exported to the current working directory.
-
i.
-
d.The `TimiCellNetwork()` function generates node information based on three parameters.
-
i.`dataset`: This parameter specifies the dataset to be used for generating the cell network files. It expects a value from a predefined set of options: " “Bindea2013_Cancer", "Charoentong2017", Bindea2013", "Xu2018", "Newman2015", "Zheng2021", "Tirosh2016", or "Other". The first seven options are accompanied by preset configurations linked to internal cell-type annotations utilized for the analysis. Specifically, these options correspond to: `CellType_Bindea2013_cancer`, `CellType_Charoentong2017_Bindea2013_Xu2018_Immune` (this includes three cell-type annotations), `CellType_Newman2015_LM22`, `CellType_Zheng2021_Tcell, and CellType_Tirosh2016_melanoma_TME`, respectively. If you want to use a different dataset or customize node attributes, please choose "Other" and then provide `geneset`
-
ii.`geneset`: This parameter is a data frame that contains information about cell-type markers. It should have at least three columns: the first column represents cell types, the second column represents marker genes, and the optional third column represents the name of the dataset. The `geneset` parameter is used in conjunction with the dataset parameter. If you choose "Other" for the dataset, you need to provide a `geneset` data frame to define the cell type and its number of markers for node attributes.
-
iii.`group`: This parameter allows the specification of custom cell-type groups when using the "Other" `dataset` option, which enables the organization of cell types based on specific research needs. It is a vector of self-defined cell groups, whose names are all favorable and unfavorable cell types in selection.CellNET <- TimiCellNetwork( resdata = res,select = NULL,dataset = "Bindea2013_Cancer",group = NULL,geneset = NULL,condition = CIcondition,cutoff = CIcutoff,export = TRUE,path = "./")# Display the first few rows of the 'network' component in the CellNET listhead(CellNET$network)# Display the first few rows of the 'node' component in the CellNET objecthead(CellNET$node)# Display the first few rows of the 'edge' component in the NET objecthead(CellNET$edge)
-
i.
-
a.
-
16.Visualize the cell-cell interaction network with chord using the `TimiCellChord()` function.Note: Besides exporting network files using the `TimiCellNetwork()` function and visualizing through external software, for example, ‘Cytoscape’, TimiGP provides another visualization approach, the `TimiCellChord()` function. This function generates a chord diagram to visualize the cell-cell interaction network.
-
a.The input data `resdata` of the `TimiCellChord()` function is the enrichment results `res` generated from `TimiEnrich()` or modified with `TimiPermFDR()` functions.
-
b.The `TimiCellChord()` function uses three parameters to define the cell-cell interaction from enrichment statistics.Note: The criteria to define the cell-cell interactions of the `TimiCellChord()` function is consistent with the `TimiCellNetwork()` function. So the usage of relevant parameters is the same.
-
i.`condition`: A value in one of "P.Value", "Adjust.P.Value" and "Permutation.FDR", which is the column name of `resdata`, which contains information related to statistical significance. The default value is "Adjust.P.Value".
-
ii.`cutoff`: This parameter refers to a threshold value associated with the 'condition'. It assists in the definition of cell-cell interactions. Cell pairs with a value less than this cutoff will be cell-cell interaction and included in the network. The default value for the 'cutoff' parameter is 0.05.
-
iii.`select`: This parameter is a numeric vector denoting selected cell interactions based on the "Index" column in `resdata`. Default selection encompasses all cell-cell interactions (adjusted p-value < 0.05). By default, this parameter is set to “NULL”, indicating that no specific cell-cell interactions are selected. However, if you manually choose cell-cell interactions as research of interest for visualization using this parameter, the automatic filtering based on the `cutoff` and `condition` will be overridden for those selected cell-cell interactions. Troubleshooting 5.Note: The default definition of cell-cell interactions is adjusted p-value (condition) < 0.05(cutoff).
-
i.
-
c.The `TimiCellChord()` function generates a circular plot of cell-cell interactions through the ‘circlize’ package (Figure 2B).
-
i.Sectors (Nodes): Cell types are represented as sectors along the circumference of the circle. Each sector corresponds to a specific cell type, and the arrangement of these sectors around the circle provides a visual representation of the relationships between different cell types. The color of the outer ring of each sector is consistent with the color used to represent that particular cell type. This color coding makes it easy to identify and associate the color of the outer ring with a specific cell type, allowing viewers to quickly recognize and interpret the plot. The inner ring, which is closer to the center of the circle, is colored to match the unfavorable cell type to which the link points. When there is an interaction between two cell types (e.g., a directional arrow from one favorable cell to an unfavorable cell), the color of the inner ring corresponds to the unfavorable cell type, providing additional visual information about the interaction or relationship between these cell types. In addition, there is a larger gap that creates more space between different groups of cells.
-
i.
-
d.The `TimiCellChord()` function generates node information and enables customized plots based on four parameters.Note: The customization of the `TimiCellChord()` function is similar to the `TimiCellNetwork()` function except for the `color` parameter for visualization.
-
i.`dataset`: This parameter specifies the dataset to be used for generating the cell network files. It expects a value from a predefined set of options: " “Bindea2013_Cancer", "Charoentong2017", Bindea2013", "Xu2018", "Newman2015", "Zheng2021", "Tirosh2016", or "Other". The first seven options are accompanied by preset configurations linked to internal cell-type annotations utilized for the analysis. Specifically, these options correspond to: `CellType_Bindea2013_cancer`, `CellType_Charoentong2017_Bindea2013_Xu2018_Immune` (this includes three cell-type annotations), `CellType_Newman2015_LM22`, `CellType_Zheng2021_Tcell, and CellType_Tirosh2016_melanoma_TME`, respectively. TimiGP package provides internal visualization (group and color) settings for this dataset. If you want to use a different dataset or customize the group and color for each cell type, please choose "Other" and then provide `group` and `color`.
-
ii.`group` and `color`: These parameters allow to specify custom cell-type groups and colors when using the "Other" “dataset” option, which enables organization and visualization of cell types based on specific research needs. They are vectors of cell groups (e.g., “immune cell”) or colors (e.g., “blue” or “#deebf7”), whose names are all favorable and unfavorable cell types in selection.TimiCellChord( resdata = res,select = NULL,dataset = "Bindea2013_Cancer",group = NULL,color = NULL,condition = CIcondition,cutoff = CIcutoff)Optional: Visualize representative marker gene-gene interactions of selected cell-cell interaction using the `TimiGeneChord()` function.This function is designed to create informative chord diagrams that reveal the relationships between genes enriched in favorable and unfavorable cell types in the specific cell-cell interactions (Figure 7A). The diagram elegantly displays directional arrows, pointing from enriched marker genes of one cell type (favorable) to those of another (unfavorable), helping to elucidate gene-gene interaction behind the cell-cell interaction. From the same input `resdata` as the `TimiCellChord()` function, users can specify the cell-cell interaction of interest by setting the `select` parameter according to the "Index" column, and visualize the gene-gene interaction with default or customized colors (`color` parameter) to uniquely identify enriched marker genes. The resulting chord diagram provides a clear and intuitive representation of gene-gene interactions within the chosen cell interaction, aiding researchers in understanding the molecular underpinnings of cell-type-specific behaviors and their impact on biological processes.# `select` the cell-cell interaction #3 according to the index# Default `color`TimiGeneChord(resdata = res,select = 3, color = NULL)
-
i.
-
a.
-
17.Prioritize critical prognostic cell types with favorability scores calculated by the `TimiFS()` function.
-
a.Description: According to the degree of the cell-cell interaction network, the `TimiFS()` function calculates favorability scores to prioritize the association of cell types with clinical outcomes. For each cell type, the score estimates its association with both favorable clinical outcome (favorable score) and unfavorable prognosis, (unfavorable score). A higher favorable score indicates a positive prognosis and anti-tumor cell function, while a higher unfavorable score indicates an adverse prognosis and pro-tumor function in the TIME network.
-
b.Parameters: The parameters of the `TimiFS()` function are similar to those of `TimiCellChord()` and `TimiNetwork()` functions.
-
i.The input data `resdata` is the enrichment results `res` generated from `TimiEnrich()` or modified with `TimiPermFDR()` functions.
-
ii.The criteria to define the cell-cell interactions of `TimiFS()` function is consistent with the `TimiCellChord()` and `TimiCellNetwork()` functions. `condition` is a value in one of "P.Value", "Adjust.P.Value" and "Permutation.FDR", and `cutoff` refers to a threshold value associated with the 'condition'. The default definition of cell-cell interactions is adjusted p-value (`condition`) < 0.05(`cutoff`).
-
i.
-
c.Return: A data frame with information about the favorability scores of various cell types (Figure 7B). Each row in the data frame represents a different cell type, and the columns provide the following information:
-
i.“Index”: An index or identifier for each cell type, used for visualization with the `TimiFSBar()` function.
-
ii.“Rank”: The rank of the cell type based on the difference between its Favorable Score and Unfavorable Score. Cell types with the highest positive differences (indicating Favorable) are ranked higher.
-
iii.“Cell.Type”: The name of the cell type.
-
iv.“OutDegree”: The out-degrees of the cell type, which is the number of outgoing edges of the cell type in the cell-cell interaction network.
-
v.“Favorable.Score”: The favorable score of the cell type calculated from out-degrees, indicating a positive prognosis and anti-tumor cell function.
-
vi.“Indegree”: The in-degree of the cell type, which is the number of ingoing edges of the cell type in the cell-cell interaction network.
-
vii.“Unfavorable.Score”: The unfavorable score of the cell type calculated from the in-degree, indicating a negative prognosis and pro-tumor function.
-
viii.“Difference”: The difference between the Favorable Score and the Unfavorable Score. This value indicates whether the cell type is more favorable (positive value) or more unfavorable (negative value) in terms of prognosis.
-
ix.“Group”: The classification of the cell type based on the Difference value. If the Difference is greater than 0, the cell type is classified as "F" (Favorable). If the difference is negative, the cell type is classified as "U" (Unfavorable).
-
i.
-
a.
score <- TimiFS( resdata = res,
condition = CIcondition,
cutoff = CIcutoff)
-
18.Visualize favorability scores using the `TimiFSBar()` function.
-
a.Description: The `TimiFSBar()` function generates a bar plot of the favorability score of cell types, allowing for the evaluation of their favorable and unfavorable roles in prognosis and anti-tumor response.
-
b.Parameters:
-
i.`score`: Favorability score calculated using the `TimiFS()` function.
-
ii.`select`: A numeric vector of selected cell types according to the "Index" column in `resdata`. The default selection encompasses all cell types.
-
i.
-
c.Return: A bar plot illustrating the favorability scores of different cell types, with orange bars indicating favorable scores (positive) and blue bars indicating unfavorable scores (negative). It is a ‘ggplot2’ plot since the function requires the ‘ggplot2’ package for plotting (Figure 2C).
-
a.
# Visualize favorability scores for selected cell types,
# including the top 5 and bottom 3 cell types.
TimiFSBar(score, select = c(1:5, (nrow(score) - 2):nrow(score)))
Figure 6.

Output screenshots of the `TimiCellNetwork()` function
TimiCellNetwork returns a list of three data frames: (A) “network”, (B) “node”, (C) “edge”. The type of interaction is “TimiGP” representing cell-cell interaction exported by the TimiGP method. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Figure 7.

Output example of TimiGeneChord()` and `TimiFS()` functions
(A) Chord diagram of gene-gene interactions representing Cytotoxic → Tumor cell interaction; (B) Output screenshots of `TimiFS()` function. The screenshot displays white text on a black background within the terminal, with additional manual annotations in red text, arrow, or box.
Expected outcomes
The TimiGP illustrates the tumor microenvironment from the transcriptomic profile by inferring cell-cell interactions and prioritizing the cell type important to the patient’s clinical outcomes. Therefore, the TimiGP R package is expected to generate network files at the gene-gene (Figure 4) or cell-cell interaction (Figure 6) level. The package also provides functions to visualize and analyze the results: 1) Dot plot showing enrichment statistics of cell-cell interactions of interest; 2) Chord diagram of cell-cell interactions (Figure 2C); 3) Chord diagram of gene-gene interactions representing cell-cell interactions of interest (Figure 7A); 4) Bar plot of the favorability score to prioritize critical cell type related to the clinical outcome (Figure 2E).
Limitations
The TimiGP method exhibits two specific limitations. Firstly, for the effective identification of prognostic gene pairs, TimiGP necessitates a substantial gene expression dataset that includes matched survival information. The power of survival analysis relies on factors such as sample size, event percentage, and the accuracy of survival data. In cases where survival data quality is relatively subpar, we recommend employing more lenient statistical thresholds to pinpoint prognostic gene pairs, or alternatively, focusing on the highest-ranking gene pairs. Additionally, the TimiGP package infers the cell-cell interaction network primarily from the prognostic value of marker pairs and does not provide built-in tools for conducting additional analyses to account for potential confounding factors in the survival analysis.
Secondly, the identification of cell-cell interactions in TimiGP is contingent on enrichment analysis, and its sensitivity to detect particular interactions is influenced by the number of associated marker genes. Cell types with a limited number of marker genes may have a diminished likelihood of being recognized as having significant interactions. Fortunately, the widespread utilization of scRNA-seq analysis in cancer immunology has resulted in the availability of high-quality marker gene sets for the majority of immune cell types.
Troubleshooting
Problem 1
You are using a cloud computing platform or environment where you cannot install packages from GitHub directly using ‘devtools’.
Potential solution
-
•
Install through the 'remotes’ package.
Compared to ‘devtools’, the ‘remotes’ package is more streamlined and efficient for package installation, including proper dependency handling.
# Install remotes from CRAN if not already installed.
if(!require(remotes)){
install.packages("remotes")
}
# Install TimiGP R package from GitHub:
-
•
Manually download and install.
You can manually download the package source code from the GitHub repository as a zip file or a tarball. Then, upload the downloaded file to your cloud environment and install it using the `install.packages()` function.
install.packages("path/to/package.tar.gz",
repos = NULL,
type = "source")
Replace "path/to/package.tar.gz" with the actual path to the downloaded package file.
-
•
Git clone and install.
If the cloud environment has Git installed, you can clone the repository directly and install the package from the cloned directory.
git clonehttps://github.com/CSkylarL/TimiGP.git.
R CMD INSTALL TimiGP
-
•
Docker Container.
If you are using a Docker container or another containerization platform, you can create a Docker image with the package installed. Write a Dockerfile that installs the package using one of the above methods, and then build and run the Docker image. For example, you can add the below codes to your Dockerfile to install the TimiGP R package.
RUN R -e "install.packages('remotes')"
RUN R -e remotes::install_github("CSkylarL/TimiGP")
Problem 2
It fails to `library()` the package and return this error message “Error in library(TimiGP) : there is no package called ‘TimiGP’”.
Potential solution
-
•
The error message indicates that R cannot find the package "TimiGP". If you haven’t already installed the "TimiGP" package, please follow the protocol to install the package. If you failed to install the package, please check the computational requirements listed in the protocol, e.g., update the R version greater than 4.0.0.
-
•
If you have successfully installed the package, verify that you have not made any typos in the package name when using `library()` function. R package names are case-sensitive, so "TimiGP" and "timiGP" are considered different packages.
-
•
In addition, ensure that the library path where R is looking for packages is correct. You can use the `.libPaths()` function to check the library paths. Make sure the "TimiGP" package is installed in one of those paths.
Problem 3
The `TimiPrePropress()` fails to extracts the transcriptomic data for the selected cohort and marker genes and return this error message “No markers was found in the transcriptome. Please check marker ID”.
Potential solution
-
•
If you utilize the internal cell-type marker annotation provided by TimiGP, please ensure that each row in the transcriptome data represents genes and that the row names adhere to the HUGO gene nomenclature.
-
•
In the case of external cell-type marker annotation, it is vital to confirm that the "Gene" column in your cell-type annotation follows the same format as the row names in your transcriptome data. For example, if both utilize Ensembl IDs, ensure consistency between them.
Problem 4
It fails to select the prognostic marker pairs with statistical significance for subsequent analysis.
Potential solution
-
•
The survival information may have poor quality (e.g., low percentage of events, small cohort size), so we suggested a P-value ranking-based selection of gene pairs. You can choose the top 1%–5% gene pairs ranked by P value for subsequent analysis, though they might not be able to reach statistical significance after multiple testing corrections.
Problem 5
It fails to obtain more than 5 cell-cell interactions for subsequent analysis.
Potential solution
-
•
Check if the cell type annotation is consistent throughout this analysis.
-
•
If custom cell type annotation is used but fails, the analysis can be repeated with the internal cell type markers with controls (`CellType_Bindea2013_cancer`) to check if the result is caused by the poor quality of custom cell type annotation.
-
•
Check the cohort size and the percentage of events, if any of them are small, the data quality may be poor. You may want to apply the more sensitive approach to capture the continuous relationship between marker pairs by setting the `cont = TRUE` in the `TimiGenePair()` function at the beginning.
-
•
Try a relaxed criterion and threshold (e.g., P value < 0.05) to define the cell-cell interaction.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chao Cheng (chao.cheng@bcm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The datasets and code generated during this study are available at GitHub: https://github.com/CSkylarL/TimiGP. The specific version v1.2.0 in this protocol for the TimiGP R package is also archived in Zenodo: https://doi.org/10.5281/zenodo.8327354.
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health Research Project grants (R01CA234629–01 to J.Z. and R01CA269764 to C.C.), an AACR-Johnson & Johnson Lung Cancer Innovation Science grant (18-90-52-ZHAN to J.Z.), the MD Anderson Physician Scientist Program (J.Z.), the MD Anderson Lung Cancer Moon Shot Program (J.Z.), and the Cancer Prevention and Research Institute of Texas (CPRIT) (RR180061 to C.C.). C.C. is a CPRIT Scholar in Cancer Research. The authors would like to acknowledge the support of the High-Performance Computing for research facility at the University of Texas MD Anderson Cancer Center for providing computational resources that have contributed to the research results reported in this paper. The graphical abstract was created with BioRender.com.
Author contributions
Software, C.L.; formal analysis, C.L.; investigation, C.L.; writing – original draft, C.L.; writing – review and editing, J.Z. and C.C.; visualization, C.L.; supervision, J.Z. and C.C.; funding acquisition, J.Z. and C.C.
Declaration of interests
J.Z. reports grants from Merck, grants and personal fees from Johnson & Johnson and Novartis, and personal fees from Bristol Myers Squibb, AstraZeneca, GenePlus, Innovent, and Hengrui outside the submitted work.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the authors used GPT-3.5 in order to enhance the overall quality of writing, such as identifying and correcting typos and grammar errors, and refining sentence structures. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Contributor Information
Chenyang Li, Email: skylar.li.963@gmail.com.
Jianjun Zhang, Email: jzhang20@mdanderson.org.
Chao Cheng, Email: chao.cheng@bcm.edu.
References
- 1.Li C., Zhang B., Schaafsma E., Reuben A., Wang L., Turk M.J., Zhang J., Cheng C. TimiGP: Inferring cell-cell interactions and prognostic associations in the tumor immune microenvironment through gene pairs. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galon J., Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55–81. doi: 10.1016/j.immuni.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Dunn G.P., Bruce A.T., Ikeda H., Old L.J., Schreiber R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 4.Fridman W.H., Zitvogel L., Sautès-Fridman C., Kroemer G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017;14:717–734. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 5.Bruni D., Angell H.K., Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer. 2020;20:662–680. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 6.Akbani R., Akdemir K., Aksoy B., Albert M., Ally A., Amin S., Arachchi H., Arora A., Auman J., Ayala B., et al. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bindea G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., Angell H., Fredriksen T., Lafontaine L., Berger A., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 9.Xu L., Deng C., Pang B., Zhang X., Liu W., Liao G., Yuan H., Cheng P., Li F., Long Z., et al. TIP: A Web Server for Resolving Tumor Immunophenotype ProfilingTIP: Tracking Tumor Immunophenotype. Cancer Res. 2018;78:6575–6580. doi: 10.1158/0008-5472.CAN-18-0689. [DOI] [PubMed] [Google Scholar]
- 10.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374:abe6474. doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 12.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wickham H., Romain F., Lionel H. 2021. Kirill Müller (2020) Dplyr: A Grammar of Data Manipulation. R Package Version 0.8. 4. [Google Scholar]
- 14.Wickham H., Hester J., Chang W., Hester M.J. 2021. Package ‘devtools’. [Google Scholar]
- 15.Analytics R., Weston S. doParallel: Foreach parallel adaptor for the parallel package. R package version. 2014;1:2014. [Google Scholar]
- 16.Wallig M., Weston S. 2020. Foreach: Provides Foreach Looping Construct. [Google Scholar]
- 17.Therneau T.M., Grambsch P.M. Modeling Survival Data: Extending the Cox Model. Springer; 2000. The cox model; pp. 39–77. [Google Scholar]
- 18.Wickham H. Springer-Verlag New York; 2016. Package ‘ggplot2’: Elegant Graphics for Data Analysis. 10, 978-970. [Google Scholar]
- 19.Neuwirth E., Neuwirth M.E. ColorBrewer Palettes; 2014. Package ‘RColorBrewer’. [Google Scholar]
- 20.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 21.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and code generated during this study are available at GitHub: https://github.com/CSkylarL/TimiGP. The specific version v1.2.0 in this protocol for the TimiGP R package is also archived in Zenodo: https://doi.org/10.5281/zenodo.8327354.