

Summary
Objective
To investigate the epidemiology of sickle cell disease (SCD) and determinants of knowledge, attitudes and practices (KAP) towards SCD in western Kordofan State, Sudan. methods A community-based, descriptive, cross-sectional study was conducted in three towns. Three hundred and seventy-two households were polled, and blood samples for haemoglobin phenotyping were collected from 1116 individuals. Sociodemographic, socio-economic and KAP data were collected using investigator-administered questionnaires. Descriptive, frequency distribution and multiple regression analyses were performed.
Results
About 50.9% of the study population were Misseriya tribes. Consanguineous marriages were reported by 67.5% of the households. The highest percentage of homozygous SCD was 2.8% among children under 5 years of age. About 24.9% were carriers of HbS allele (HbAS). HbS allele frequency was highest in children aged 5–11 years (18.3%, CI: 13.7–22.9%) and lowest in males >15 years old (12.0%, CI: 6.1–17.9%). The average HbS frequency across all age groups was 14.5% (95% CI: 12.2–16.8%). The most frequent β-globin gene cluster haplotype was the Cameroon (30.8%), followed by the Benin (21.8%), the Senegal (12.8%) and the Bantu (2.2%) haplotypes. About 17.0% of all-cause child deaths were due to SCD. The estimated change in log odds of having the SS genotype per year increase in age was (−) 0.0058 (95% CI −0.0359, 0.0242). This represents a non-statistically significant 2.9% increase in 5-year mortality for individuals with the SS genotype relative to those with AS and AA genotypes. About 46.9% of the households had poor knowledge, 26.1% had satisfactory knowledge, and 26.9% had good knowledge about sickle cell disease. Mothers’ and fathers’ educational levels were significant predictors of good knowledge about SCD (P < 0.05). About 48.0% had a satisfactory attitude towards sickle cell disease while 30.7% had poor attitude and only 21.3 showed good attitudes. Poor knowledge about SCD and low socio-economic status were the strongest positive predictors of poor attitude and practices towards SCD (P < 0.01).
Conclusions
Sickle cell disease is a major health problem in West Kordofan, Sudan. Knowledge, attitude and practices towards the disease are not satisfactory. The development of public health programs is highly recommended to control and manage SCD in western parts of Sudan.
Keywords: sickle cell disease, genetic epidemiology, western Sudan, haplotypes, knowledge attitude and practices
Introduction
While substantial efforts have been made to control communicable diseases in Africa, the burden of non-communicable diseases has generally been neglected. Among these non-communicable diseases, the inherited blood disorder such as sickle cell disease constitutes a major health problem especially in western, central and eastern Africa [1, 2]. Sickle cell disease (SCD) is a group of autosomal recessive genetic blood disorders due to a mutation in the β-goblin gene. This mutation is a structural variant of normal adult haemoglobin (HbA). The resultant haemoglobin S (HbS), which polymerises under low oxygen tension, causes rigid, sickled red blood cells [3]. The main clinical manifestations are vaso-occlusive painful crises and haemolytic anaemia, which lead to life-threatening complications including chronic leg ulcer, priapism, pulmonary hypertension, renal failure, gallstones, autosplenectomy and increased susceptibility to infection, deafness, blindness and stroke [3].
Recent global estimates suggest that more than 300 000 affected children are born annually with SCD, about two-thirds of them in Africa [4]. Accurate survival data for children with SCD are not available [5, 6]. However, studies from Benin and Nigeria suggest mortality rates of up to 50% and 90%, respectively [6, 7]. Major factors thought to contribute to high mortality rate among children with SCD in Africa are the cultural background, lack of medical education and limited healthcare facilities [6]. Nevertheless, simple interventions through comprehensive dedicated SCD programmes in sub-Saharan African countries have reduced SCD morbidity and mortality [5, 6]. Epidemiological information and reliable estimates of the population affected by the disease are key elements for public health priority setting [4] and successful strategies for prevention and management of SCD [8]. Therefore, studies are needed to shed light on the knowledge, attitudes and practices towards SCD in Sudan.
The few pilot studies conducted in Sudan concluded that sickle cell disease is a major health problem in certain parts of the country, particularly the western region where HbS allele frequency among the Misseriya tribe is estimated to range from 18.2% in Kordofan [9] to 30.4% in Darfur [10]. However, huge demographic, socio-economic and cultural changes in Sudan during the last five decades [11] necessitate reliable information about the current SCD prevalence, risk factors and health impact in areas of expected high prevalence of SCD. Thus, we investigated the epidemiology of SCD and determinants of knowledge, attitudes and practices (KAP) towards it in western Kordofan State, Sudan.
Methods
Study design
A community-based, descriptive, cross-sectional survey was conducted in El Fula, Babanusa and El Muglad towns in western Kordofan State, Sudan. The field component of the study was accomplished between 5 and 25th June, 2014. Screening teams were randomly distributed to various districts of three towns, and households were enrolled in accordance with a strict non-skip door to door survey protocol. A total of 372 households were included; 160 from El Fula, 156 from El Muglad and 56 from Babanusa. Blood samples were collected from all household members at the time of interview for haemoglobin phenotyping and complete blood hemogram analysis.
Data collection
Data were collected on demographic, socio-economic status (SES), knowledge, attitudes and practices towards SCD (KAP) using a closed-ended, investigator-administered questionnaire. The questionnaire was developed with items that consider the sociocultural background and that are sensitive to people’s ability to understand it. The study was preceded by a large-scale advocacy effort about its importance and potential benefits especially among community leaders in the study area. A pilot study was conducted in the 2 days preceding the survey to test the questionnaire in three randomly selected residential areas of El Fula. A paper ballot was used for the selection process. The surveyors were trained in data collection before the study start. Four teams, each composed of medical doctor, senior nurses, social workers and a phlebotomist, collected the household data and the blood samples.
Ethics statement
Ethical approval was obtained from the Ethics Committee of the Federal Ministry of Health, Sudan, and Ethical exemption was granted by the Institutional Review Board, Harvard T.H. Chan School of Public Health, and Boston, USA. Self-read or investigator read and explained written consent were obtained from the participants or their guardians.
Demographic and socio-economic characteristics
The ethnic groups and populations inhabiting West Kordofan State were representative of geographic regions from Africa’s Sahel into Chad and West Sudan and encompass the main linguistic families of Africa (Afro-Asiatic, Niger Kordofanian and Nilo-Saharan) [12]. The towns selected for the study are the major settlements sites for the nomadic Bagara, settlers and immigrants tribes from Darfur and West-Central Africa. The residents were chiefly agriculturalists and pastoralists.
All members of a household present at the time of the survey were interviewed collectively; the main informants were the mother (70.4%), the father (23.7%), adult son (2.1%), adult daughter (2.4%) or not determined in (1.4%) of the studied households. A household was defined ‘as a small group of persons who share the same living accommodation, who pool some, or all, of their income and wealth and who consume certain types of goods and share services collectively, mainly housing and food’. The details of the household members, tribe, sub-tribe, parental consanguinity, parents’ level of education, history of SCD, history of child death in general and child death due to SCD, history of having a child with stroke and household assets were recorded.
Wealth index quintiles were derived using principal component analysis (PCA) [13]. The wealth index was assumed to capture the underlying long-term wealth through information on the household assets and was intended to produce a ranking of households by wealth, from poorest to richest. The index was based on the ownership of weighted consumer goods and other characteristics related to the household’s wealth. Each household was assigned a wealth score. The household study population was then ranked according to the wealth score of the household they are living in. Accordingly, the population under study was divided into five equal parts (quintiles) from lowest (poorest) to highest (richest). The assets used in these calculations were as follows: type of fuel regularly in use, possession of electricity, radio, television, non-mobile phone, refrigerator, watch, mobile phone, bicycle, motorcycle, animal drawn cart, car/truck for private use, car/truck to rent, ownership of house, ownership of land and ownership of livestock, herds, other farm animals, or poultry.
Knowledge, Attitude and Practice (KAP) study
Knowledge of sickle cell disease referred to understanding the causes of SCD, its mode of transmission, factors that raise or lower the risk of being a carrier or having the disease. This was composed of six main questions, which included 21 statements. The attitudes and practices towards SCD refer to the degree of positive or negative agreements with statements concerning attitudes and beliefs in the interaction with SCD patients, dietary and breast feeding restrictions, screening test as well as the impact of knowing the risk of having a child with the disease. Attitudes and practices towards SCD were assessed by four main questions which encompassed twelve statements. KAP statements were given a score of 2, 1 or 0 for correct, not sure, or incorrect answers, respectively. Scores were summed for each household and levels of knowledge were categorised as poor [<25th percentile], satisfactory [25 to <75th percentile] and good [>75th percentile]. For the univariate and multivariate binary logistic regression analysis of predictors of SCD KAP, the binary outcome for knowledge was poor knowledge [<25th percentile] and average and above defined as knowledge [>50th percentile]. Similarly, the outcome for attitude and practices was poor attitude and practices [<25th percentile] and average and above average attitude and practices [>50th].
Blood collection
Five ml of whole blood was taken in EDTA tubes from 1116 individuals in the 372 households that participated in the study. The collected blood samples were transferred to a field laboratory in 4 °C cold boxes. Whole blood was fractionated into RBC and plasma by centrifugation at at 450 g for 15 min. The top plasma layer was carefully siphoned off and transferred into another tube. The lower red cell layer was washed twice with physiological saline (0.85% NaCl) and centrifuged to remove traces of plasma and buffy coat. The resulting plasma and RBC pellet were immediately stored at −20 °C. The collected red blood samples were shipped to the research laboratory at the Faculty of Medicine, University of Khartoum, in dry ice boxes and stored at −20 °C until analysis.
Complete blood count
Haemoglobin concentration (Hb), haematocrit (Ht), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH), mean corpuscular haemoglobin concentration (MCHC), total white blood cell (WBC), and red blood cell (RBC) and platelet (PLTs) counts were determined for 904 participants. A total of 212 samples were excluded due to haemolysis, clotting or erroneous labelling. Complete blood count was determined using an automated haematology analyzer (Sysmex KX-21N; Sysmex Corporation, Kobe, Japan). The haemoglobin level below which anaemia (all causes) was considered to be present was defined in accordance to modified WHO recommendations [14]. For children aged 6–59 months, the anaemia cut-off point was 11.0 g/dl; for children aged 5–11 years, 11.5 g/dl; for children aged 12–14 years, 12.0 g/dl; for women >15 years, 12.0 g/dl; and for men >15 years, 13.0 g/dl.
Haemoglobin phenotyping and β-globin gene cluster haplotype
Haemoglobin phenotype HbAA, HbAS, HbSS, HbAC or HbSC was determined with the use of cellulose acetate electrophoresis at pH 8.5. To assess the genetic markers of SCD severity in western Sudan, 5 ml of whole blood was taken in EDTA tubes from 69 HbSS patients who were originally from western Sudan and under regular follow-up at Ibn-Aoaf Paediatric Hospital, Khartoum, Sudan. DNA was extracted by the ReliaPrep™ Blood gDNA Miniprep System (a) [15]. Blood specimens were processed using a binding column in a microcentrifuge tube; 200 μl of blood sample was used per purification. Seven regions around and within the β-gene cluster were amplified by PCR. Seven β-gene restriction fragment length polymorphism (RFLP) regions (ε-HindII, Gγ-XmnI, Gγ-HindIII, Aγ-HindIII, 3ψb-HindII, 5ψb-AvaII and β-HinfI) were used to identify β-globin gene cluster haplotype (n = 2*69 chromosomes), as described by Elderdery et al. [16]. The haplotype findings for each patient were categorised as typical or atypical based on whether the segregation of the two haplotypes could be unequivocally determined as Cameron (CA), Benin (BE), Senegal (SE), Bantu (BA) and Arab-Indian (AI).
Statistical analysis
Data were managed using the epidata software (version 2.0.2.28 r1115), incorporating appropriate skips and range checks. Data were expressed as mean and standard deviation (mean ± SD) or percentages. Mean haemoglobin levels of different haemoglobin phenotypes and age groups were compared by one-way analysis of variance (ANOVA). When statistical differences were indicated, Bon-ferroni’s pairwise multiple comparison post hoc tests were performed. HbS allele frequency was calculated using equation described in Grosse et al. [17]. Departure from Hardy–Weinberg expectations (DHWE) for each age group was calculated using chi-squared goodness-of-fit test.
Excess mortality rate was calculated by subtracting the frequency of HbSS among age group >5 years from HbSS among age group 5—11 years, and the resultant difference was divided by HbSS frequency among age group <5 years [17]. To estimate the excess risk of mortality associated with the SS genotype, we regressed genotype (SS vs. AS and AA combined) on age using a generalised estimating equation with a logit link. Exchangeable correlation structure within the household was used to account for clustering in genotypes among relatives. Individuals with missing either age or genotype were excluded. The age coefficient from the generalised estimating equation is the change in the log odds of having the SS genotype per year increase of age. The additive increase in hazard of mortality associated with the SS genotype was obtained by multiplying the age coefficient by negative one.
For KAP predictors, we analysed the variables by univariate comparisons using chi-squared test. To identify those variables that were independently associated with KAP, all variables were analysed using the logistic regression models. The variables of colinearity were (history of having a child with SCD and child death due to SCD) introduced into the regression model separately. Hosmer–Lemeshow test for goodness of fit was not significant for the two models. Results were expressed as odd ratios (OR) and 95% confidence interval. ‘P’ values of <0.05 were deemed statistically significant. Analyses were conducted with spss for Windows, version 19 (SPSS Ltd., Woking, Surrey, UK), Stata statistical software version 12 and r statistical package.
Results
Demographic and socio-economic characteristics
Households who described themselves as from the Misseriya tribe constituted 50.9% of the study population. About 67.5% of the parents participated in this study were either first cousins (fourth degree consanguinity) or relatives (fifth degree consanguinity and above) (Table 1). Socio-economic analysis revealed that households in Babanusa town had higher socio-economic status than those in El Muglad and El Fula (Figure 1).
Table 1.
Demographic characteristics of the households participated in the study
| Demographic characteristics | Frequency (n = 372) | Percentage |
|---|---|---|
| Area of residence (families) | ||
| El Fula | 160 | 42.7 |
| El Muglad | 156 | 41.6 |
| Babanusa | 56 | 14.9 |
| Educational level (Mothers) | ||
| Illiterate | 72 | 19.2 |
| Basic religious education (Khalwa) | 58 | 15.1 |
| Basic schooling (primary level) | 135 | 36.0 |
| Secondary schooling | 70 | 18.7 |
| Higher education | 39 | 10.4 |
| Educational level (Fathers) | ||
| Illiterate | 62 | 16.5 |
| Basic religious education (Khalwa) | 59 | 15.7 |
| Basic schooling (primary level) | 119 | 38.0 |
| Secondary schooling | 91 | 29.1 |
| Higher education | 42 | 13.4 |
| Employment (Mothers) | ||
| Employed | 89 | 23.7 |
| Unemployed | 279 | 74.4 |
| Employment (Fathers) | ||
| Employed | 315 | 84.0 |
| Unemployed | 49 | 13.1 |
| Number of children | ||
| Zero | 15 | 4.0 |
| 1–2 | 88 | 23.4 |
| 3–4 | 100 | 26.7 |
| 5–6 | 86 | 22.9 |
| 7–8 | 51 | 13.6 |
| ≥9 | 35 | 9.3 |
| Consanguinity | ||
| First-degree cousin | 159 | 42.4 |
| Relative | 94 | 25.1 |
| Not related | 117 | 31.2 |
| Self-described Ethnic group | ||
| Misseriya | 191 | 50.9 |
| Baderiah | 30 | 8.0 |
| Hamer | 22 | 5.9 |
| Borgu | 20 | 5.3 |
| Falata | 13 | 3.5 |
| Berti | 11 | 2.9 |
| Others | 87 | 23.2 |
Figure 1.
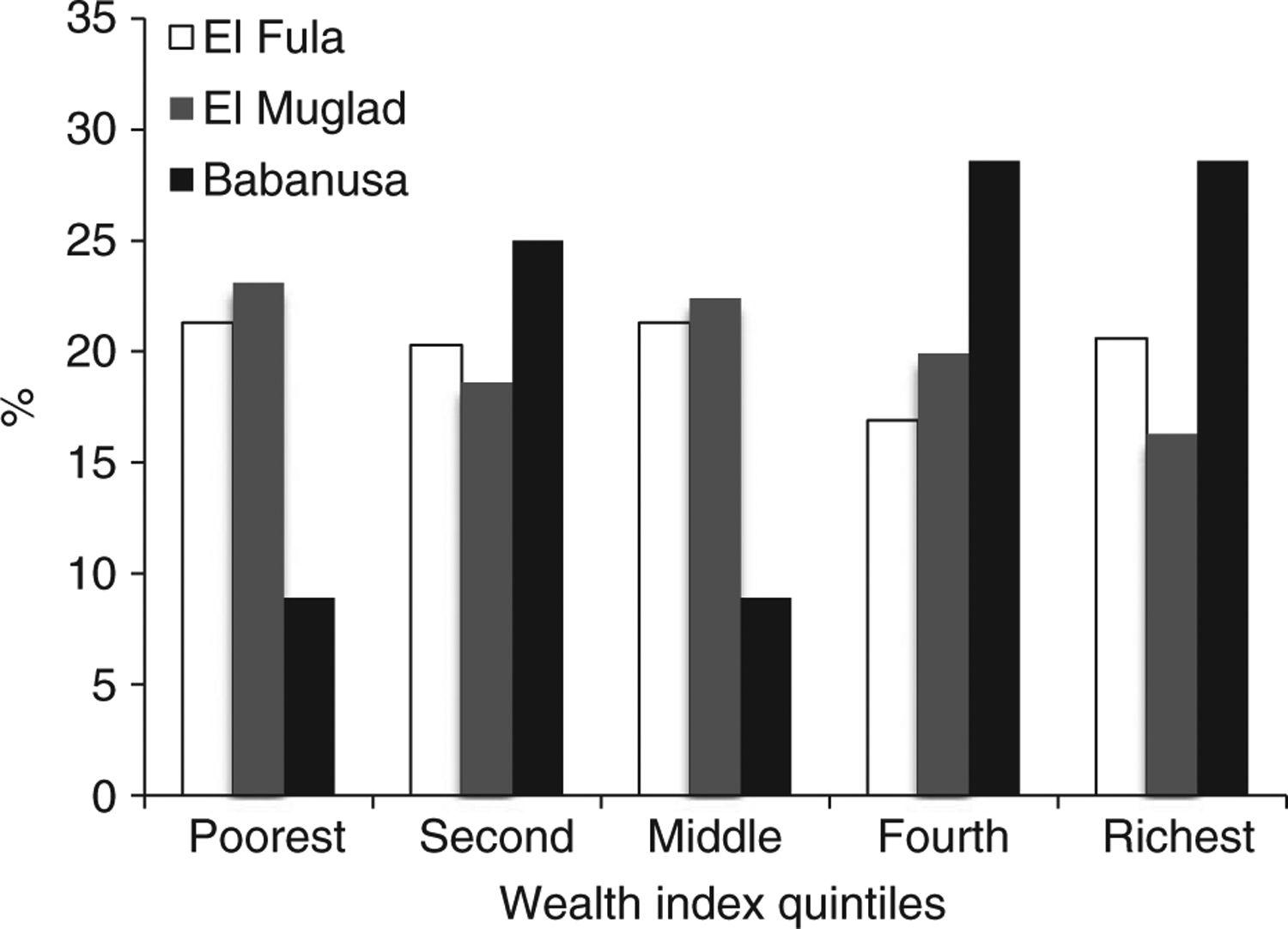
Comparison of households’ socio-economic status according to the area of permanent residence.
Prevalence of SCD in West Kordofan State
About 21.6% of households belonging to Misseriya tribe reported having at least one member with SCD followed by Baderiah (17.2%), Borgu (16.7%) and Hamer (12.5%) tribes. Based on the interview data, SCD prevalence among mothers was 5.1% and among fathers 2.7%. Only 6.1% of the parents reported that they underwent a sickle cell test.
Haemoglobin phenotyping analysis revealed that the percentage of homozygous sickle cell disease was highest in children under 5 years old (2.8%) and lowest among males >15 years old (0.8%). The percentage of sickle cell trait (HbAS) ranged between 21.2% and 30.1% according to the age–gender group (Table 2). The highest HbS allele frequency was detected among children aged 5–11 years (18.3%, CI: 13.7–22.9%) and the lowest among men >15 years old (12.0%, CI: 6.1–17.9%). The average HbS frequency across all age groups was 14.5% (95% CI: 12.2–16.8%). The test for deviation of HbSS proportion in each age group was not statistically significant (Table 3).
Table 2.
Prevalence of homozygous sickle cell diseases, sickle cell trait, HbAC and all-causes anaemia among different age groups in West Kordofan State, Sudan
| Hb phenotype | Blood indices | |||||||
|---|---|---|---|---|---|---|---|---|
| Age group (Years) | NO | HbAA% | HbAS% | HbSS% | HbAC% | Hb (Mean ± SD (mg/l) | Ht (g/dl) | Anaemia (All causes) |
| 0–4 | 104 | 52/72 (72.2%) | 18/72 (25%) | 2.0/72 (2.8%) | (0.0/72) (0.0%) | 11.1 ± 3.5 | 36.7 ± 4.9 | 50.0% |
| 5–11 | 268 | 124/183 (67.8%) | 55/183 (30.1%) | 3.0/183 (1.6%) | 1.0/183 (0.5%) | 11.4 ± 1.7 | 38.1 ± 6.5 | 51.1% |
| 12–14 | 64 | 32/47 (68.1%) | 14/47 (29.8%) | 1.0/47 (2.1%) | 0.0/47 (0.0%) | 11.6 ± 1.5 | 39.1 ± 4.5 | 62.9% |
| Women > 15 | 302 | 161/232 (69.4%) | 65/232 (28.0%) | 6.0/232 (2.6%) | 0.0/232 (0.0%) | 12.9 (2.4) | 39.9 ± 5.7 | 52.2* |
| Men > 15 | 125 | 92/118 (78.0%) | 25/118 (21.2%) | 1.0/118 (0.8%) | 0.0/118 (0.0%) | 12.0 (2.5) | 43.1 ± 7.0 | 50.0% |
| Total | 904 | 644/882 (73.0%) | 220/882 (24.9%) | 16/882 (1.8%) | 2/882 (0.2%) | 11.8 ± 2.2 | 39.2 ± 5.8 | 51.2% |
Pregnant women were included with women>15 years old.
Table 3.
HbS allele frequency among different age groups in West Kordofan State, Sudan
| Age group (Years) | HbS allele frequency % (CI) | Expected HbSS prevalence (HWE%) | Deviation from HWE (P value)† |
|---|---|---|---|
| 0–4 | 14.6% (6.4–22.8%) | 2.3% | NS* |
| 5–11 | 18.3% (13.7–22.9%) | 2.8% | NS |
| 12–14 | 18.1% (7.1–29.1%) | 2.9 | NS |
| Women > 15 | 17.7% (12.8–22.6%) | 4% | NS |
| Men > 15 | 12.0% (6.1–17.9%) | 13.0% | NS |
| Total | 14.5% (12.2–16.8%) | 2.0% | NS |
HWE, Hardy–Weinberg equilibrium.
P > 0.05.
Comparing to a chi-squared distribution with 1 d.f.
Children aged 5–11 years with HbSS had significantly lower Hb concentrations (8.3 ± 2.6 g/dl) than those with HbAA and HbAS, whereas the difference was not significant in other age groups (Table 4). Depending on the age–gender group, all-cause anaemia ranged from 50.0% to 62.9% (Table 2). Sickle cell anaemia constituted 4.8% of all causes of anaemia in the study population.
Table 4.
Mean haemoglobin concentration among different ages and haemoglobin phenotypes in West Kordofan State, Sudan
| Age group (Years) | Hb concentration (mean ± SD) g/dl | ||
|---|---|---|---|
| HbAA | HbAS | HbSS | |
| 1–4 | 10.9 ± 1.9 | 10.5 ± 1.6 | 11.05 ± 2.8 |
| 5–14 | 11.5 ± 1.1 | 10.7 ± 1.8* | 8.3 ± 2.6† |
| Women > 15 | 12.0 ± 2.0 | 12.1 ± 2.2 | 10.8 ± 1.7 |
| Men > 15 | 13.1 ± 2.1 | 12.5 ± 2.1‡ | – |
P < 0.001 HbSS vs. HbAS.
P < 0.001 HbSS vs. HbAA.
P < 0.05 HbAS vs. HbAA.
B-globin gene cluster haplotypes
Haplotypes were assigned unequivocally to 133 chromosomes. Four of the five typical β(S)-globin haplotypes were identified. The most frequent was the Cameroon (30.8%), followed by the Benin (21.8%), the Senegal (12.8%) and the Bantu haplotype (2.2%). The Arab-Indian haplotype was not observed, whereas three atypical haplotypes were identified that occurred in 32.2% of the chromosomes (Figure 2). About 24.6% of the patients were homozygous for CA, whereas 8.7% showed compound heterozygous haplotypes CA/BE.
Figure 2.
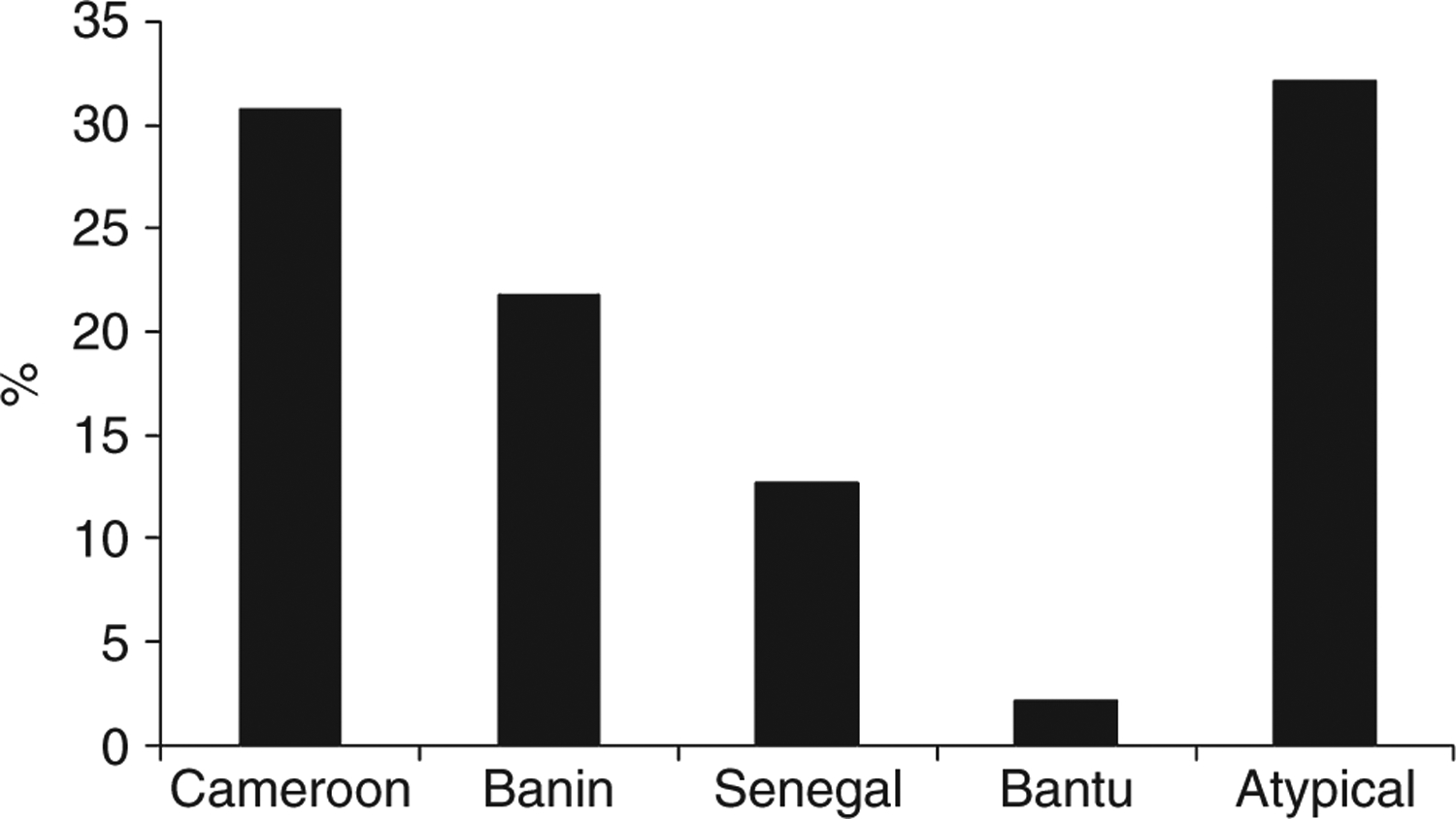
Percentage of β(S)-globin haplotypes among HbSS patients from western Sudan.
Mortality due to sickle cell disease
Among the screened households, 1.9% had alive children with stroke due to SCD. About 11.9% of the families had lost at least one child to SCD. The percentage of child death due to SCD out of all-causes childhood mortality was 17.0%. The frequency of SS decreased from 2.8% among children <5 years of age to 1.6% among children aged 5–11 years, resulting in an apparent excess mortality of 42.9% compared to other Hb genotypes (AA and AS together). The estimated change in log odds of having the SS genotype per year increase in age was (−) 0.0058 (95% CI −0.0359, 0.0242). This represents a non-significant 2.9% increase in 5-year mortality for individuals with the SS genotype relative to those with AS and AA genotypes.
The KAP study
The education level assessment showed that 34.3% of mothers and 32.2% of fathers were illiterate or had obtained Khalwa (basic religious education); 36% of mothers and 38% of fathers had basic education, and 19.1% of mothers and 42.5% of fathers had completed secondary school or higher education (Table 1).
Knowledge about SCD
About 46.9% (175) of the households had poor knowledge, 26.1% had satisfactory knowledge and 26.9% had good knowledge SCD (Figure 3a). About 68.5% of the households indicated that sickle cell disease was an inherited disorder, and 79.9% believed that both mothers and fathers contribute to disease transmission to offspring. About 69.5% agreed that marriage to a close relative might increase the risk of having a child with the disease, whereas only 31.3% agreed that it was useful to know that he or she was a carrier or the partner was carrier of SCD.
Figure 3.

(a) Households’ knowledge about SCD in West Kordofan State, Sudan. (b) Households’ attitude and practices towards SCD in West Kordofan State, Sudan.
Predictors of Knowledge
As shown in Table 5, both univariate and multivariate analyses revealed that mothers’ and fathers’ educational levels were good predictors of knowledge about sickle cell disease (P < 0.05). However, mothers’ educational level had a more pronounced relationship with SCD knowledge than fathers’ educational level. The group in the first quantile of the socio-economic (poorest) status was significantly more likely to have poor knowledge about the disease [OR = 2.0, (CI: 1.1–3.9), P < 0.05] in univariate analysis, but not in multivariate analysis. Parents’ consanguinity, family tribe, history of child with SCD or history of child death due to SCD were not significantly associated with SCD knowledge in either univariate or multivariable analyses (P > 0.05).
Table 5.
Predictors of poor households’ knowledge about sickle cell disease in West Kordofan State, Sudan
| Univariate | Multivariable | |||
|---|---|---|---|---|
| Variable | OR (95% CI) | P value | OR (95% CI) | P value |
| Educational level (Mother) | ||||
| Illiterate | 10.3 (3.8–27.9) | <0.001 | 8.3 (2.7–25.9) | <0.001 |
| Khalwa* | 4.8 (1.7–13.2) | 4.5 (1.4–15.1) | ||
| Primary | 7.1 (2.9–18.0) | 7.3 (2.7–20.4) | ||
| Secondary | 2.2 (0.8–6.2) | 2.3 (0.8–6.8) | ||
| Higher | Ref | Ref | ||
| Educational level (Father) | ||||
| Illiterate | 6.6 (2.8–15.8) | <0.001 | 3.3 (1.2–9.6) | <0.01 |
| Khalwa | 2.1 (0.8–4.9) | 1.4 (0.5–4.1) | ||
| Primary | 2.2 (1.1–4.8) | 1.3 (0.5–3.3) | ||
| Secondary | 1.6 (0.7–3.4) | 1.1 (0.5–2.9) | ||
| Higher | Ref | Ref | ||
| Parents’ degree of relatedness | ||||
| First-degree cousin | 1.0 (0.6–1.6) | 0.9 | 1.0 (0.6–1.7) | 0.4 |
| Relative | 1.6 (0.9–2.8) | 1.6 (0.9–3.2) | ||
| Not relative | Ref | Ref | ||
| Tribe | ||||
| Misseriya | Ref | 0.1 | Ref | 0.3 |
| Baderiah | 1.6 (0.7–3.4) | 1.2 (0.7–2.3) | ||
| Borgu | 1.1 (0.4–2.8) | 0.9 (0.3–3.2) | ||
| Hamer | 1.1 (0.5–2.7) | 3.6 (0.6–19.7) | ||
| Berti | 3.6 (0.9–14.0) | 1.4 (0.5–3.7) | ||
| Falata | 0.85 (0.3–2.6) | 1.6 (0.6–4.4) | ||
| Others | 1.5 (0.9–2.5) | 1.1 (0.4–2.8) | ||
| Having a child with SCD† | ||||
| No | 0.8 (0.4–1.4) | 0.4 | 0.9 (0.4–1.8) | 0.4 |
| Yes | Ref | Ref | ||
| History of child death due to SCD* | ||||
| No | 0.6 (0.3–1.1) | 0.09 | 0.8 (0.4–1.8) | 0.2 |
| Yes | Ref | Ref | ||
| Socio-economic status | ||||
| Poorest | 2.0 (1.1–3.9) | 0.04 | 0.6 (0.2–1.3) | 0.4 |
| Second quantile | 1.1 (0.6–2.1) | 0.5 (0.2–1.1) | ||
| Middle quantile | 1.1 (0.6–2.2) | 0.8 (0.4–1.6) | ||
| Fourth quantile | 0.9 (0.5–1.8) | 0.7 (0.3–1.5) | ||
| Richest | Ref | Ref | ||
OR, odds ratio; 95% CI, 95% confidence interval.
Khalwa is a traditional school in Sudan for basic religious education.
The predictors were included separately in the multivariate analysis model.
Attitudes and Practices towards SCD
In this population, 48.0% had a satisfactory attitude towards sickle cell disease, 30.7% had a poor attitude and 21.3% showed a good attitudes towards the disease (Figure 3b). About 73.1% agreed that conducting sickle cell testing of all family members is important and 52.5% stated that they would not have gotten married to a partner if they knew they were both carriers and may have children with the disease. Only 33.7% of participants believed that a child with SCD can enjoy a relatively normal life, and 15.2% thought that a child with the disease should be restricted from breast feeding or specific types of food.
Predictors of attitudes and practices
Poor knowledge about SCD and low socio-economic status were the strongest positive predictors of poor attitudes and practices towards SCD in univariate and multivariate analyses (P < 0.01). History of no child death due to SCD was associated with good attitudes and practices in univariate and multivariate analysis [OR = 0.2 (CI: 0.1–0.04), P < 0.001]. The educational level of the father was positively associated with poor attitudes and practice in univariate analysis (P < 0.05), whereas mothers’ level of education was not significantly associated with attitudes and practices towards sickle in both univariate and multivariable analyses. Compared with the Misseriya (which had the highest prevalence of SCD), tribes of lower prevalence were more likely to show poor attitudes and practices towards SCD in univariate analysis. No significant association was found between degrees of consanguinity and attitude and practices towards the disease (Table 6).
Table 6.
Predictors of poor households’ attitude and practices towards sickle cell disease in West Kordofan State, Sudan
| Univariate | Multivariable | |||
|---|---|---|---|---|
| Variable | OR (95% CI) | P value | OR (95% CI) | P value |
| Educational level (Mother) | ||||
| Illiterate | 1.8 (0.8–4.4) | 0.09 | 0.5 (0.2–1.6) | 0.4 |
| Khalwa* | 1.1 (0.4–2.8) | 0.6 (0.2–2.3) | ||
| Primary | 1.5 (0.7–3.4) | 0.7 (0.3–2.0) | ||
| Secondary | 0.7 (0.3–1.8) | 0.5 (0.2–1.4) | ||
| Higher | Ref | Ref | ||
| Educational level (Father) | ||||
| Illiterate | 3.3 (1.3–8.2) | 0.02 | 1.9 (0.6–6.6) | 0.8 |
| Khalwa | 1.6 (0.6–4.1) | 1.5 (0.4–5.2) | ||
| Primary | 2.2 (0.9–5.1) | 2.1 (0.7–6.1) | ||
| Secondary | 1.5 (0.6–3.8) | 2.1 (0.7–6.2) | ||
| Higher | Ref | Ref | ||
| Tribe | ||||
| Misseriya | Ref | 0.04 | 0.2 | |
| Baderiah | 1.9 (1.1–3.3) | 1.5 (0.9–3.0) | ||
| Borgu | 0.3 (0.03–2.1) | 0.3 (0.03–2.7) | ||
| Hamer | 3.9 (1.1–12.9) | 1.3 (0.3–6.1) | ||
| Berti | 1.8 (0.7–4.5) | 1.9 (0.7–5.9) | ||
| Falata | 1.7 (0.6–4.5) | 1.9 (0.7–5.9) | ||
| Others | 2.4 (1.1–5.3) | 1.4 (0.5–3.7) | ||
| Parents’ degree of relatedness | ||||
| First-degree cousin | 1.7 (0.9–2.8) | 0.07 | 1.5 (0.8–2.9) | 0.2 |
| Relative | 1.4 (0.7–2.5) | 1.0 (0.5–2.0) | ||
| Not relative | Ref | Ref | ||
| Having a child with SCD† | ||||
| No | 0.2 (0.1–0.4) | <0.001 | 0.2 (0.1–0.4) | 0.9 |
| Yes | Ref | Ref | ||
| History of child death due to SCD* | ||||
| No | 0.2 (0.1–0.4) | <0.001 | 0.2 (0.1–0.4) | <0.001 |
| Yes | Ref | |||
| Socio-economic status | ||||
| Poorest | 2.8 (1.4–5.6) | 0.001 | 2.8 (1.1–7.2) | <0.01 |
| Second quantile | 1.4 (0.7–2.7) | 1.9 (0.8–4.8) | ||
| Middle quantile | 0.7 (0.3–1.5) | 1.0 (0.4–2.6) | ||
| Fourth quantile | 0.8 (0.4–1.7) | 1.1 (0.5–2.5) | ||
| Richest | Ref | Ref | ||
| Knowledge | ||||
| Poor | 4.5 (1.3–8.6) | <0.001 | 3.7 (1.7–7.9) | <0.001 |
| Satisfactory | 3.3 (1.5–6.7) | 2.9 (1.3–6.4) | ||
| Good | Ref | Ref | ||
OR, odds ratio; 95% CI, 95% confidence interval.
Khalwa is a traditional school in Sudan for basic religious education.
The predictor included separately in the multivariate analysis model.
Discussion
Quantitative studies provide essential evidence on which public health decisions can be based [18]. In Sudan, epidemiological studies about sickle cell disease are generally lacking. This study investigated the current status of SCD in western Kordofan and provides evidence that HbS allele frequency in this part of Sudan is one of the highest worldwide [19]. This research work revealed that the prevalence of HbS carriers was greater than the highest sickling rate (percentage of HbAS and HbSS combined) of 24.2% reported from the same areas in western Kordofan four decades ago [10]. The persistence of high carrier percentage in general, and among the older age groups in particular, despite high mortality rate due to SCD in the area, is intriguing. However, it could be a reflection of the vastly prevalent consanguineous marriages, and the heterozygote advantage against fatal malaria [3] in Kordofan area, where the Plasmodium falciparum transmission rate (PfPR2–10 years of age) is > 10–50% (mesoendemic) with pockets of hyperendemicity (PfPR2–10 years of age > 50–75%) [20]. The prevalence of HbSS among adults is less than expected by 22%, which is slightly higher than the 20% reduction observed in other African countries [17, 21]. Moreover, the low percentage of fathers with SCD compared to mothers might suggest a high percentage of male death before marriage age, which is particularly lower among females in this area.
The finding that HbSS associated with non-significant increase in 5-year mortality relative to children with other genotypes is consistent with results reported from Kenya and Ghana [22, 23]. These results could be a reflection of the fact that most of the mortality due to SCD occurs during the first year of life [22]. Two major factors might have resulted in erroneous estimates of increased mortality risk due to SCD in Africa: first, the small sample size of the previous studies, and second, inability to adjust for the potentially huge effect measure modification of age group <1 year due to the absence of the data. Hence, to obtain an accurate estimate of excess mortality risk due SCD in western Kordofan among different age and gender groups, further well-designed cohort or big sample-sized cross-sectional studies are badly needed [17].
In contrast to studies in other African populations, and in accordance with previous studies that investigated β-globin gene cluster haplotype among Sudanese patients with SCD [16, 24], the predominant haplotypes found in this study were Cameroon and Benin. In contrast to Senegal and the Arab-Indian haplotypes, which are absent among Sudanese sickle cell patients [16], the Cameron and Benin haplotypes are generally associated with relatively low HbF percentage and severer clinical course of the disease [25]. This result might partially explain the high excess mortality rate due to SCD in Kordofan State.
Previous reports commonly showed that HbS is more prevalent among Afro-Arab constellation of tribes, Baggara ethnic group [26]. In this study, the prevalence among Misseriya tribe, one component of the Baggara group, is even greater than previously reported [27]. Strikingly, the HbS gene seems to be also widespread among tribes not commonly known to have SCD such as Hamer [26]. This finding indicates the degree of ethnic admixture rapidly taking place in this part of Sudan, the fact that should be taken into account in designing appropriate SCD screening and management programmes.
In addition to SCD, the prevalence of all causes of anaemia is similar to that found among the crisis-affected population of Darfur region [28]. These results highlight the fact that anaemia is exceptionally high in western Sudan, whether compared to other parts of Sudan or Middle Eastern and North African regions [29–31]. Low haemoglobin concentrations and anaemia are important risk factors for the health and development [29]. Therefore, a national health policy to reduce anaemia in Sudan in general and western region in particular is urgently needed.
Apart from anaemia, it has been reported that Sudan is the top country in number of birth defects with prevalence as high as 82 birth defect per 1000 life birth [19]. Indeed, the remarkably high rate of consanguineous marriages in Sudan could be one of the major factors that contribute to causality of birth defect and genetic diseases [27, 32]. In this study, and in agreement with previous studies [33], two-thirds of couples were first cousins or relatives. Hence, an effective approach to minimise the deleterious effects of consanguineous marriage should be considered in any strategy for prevention of birth defect and sickle cell disease.
All households interviewed in this study did know the disease, which points to the fact that SCD is rather common in the area. The KAP analysis applied in this study was designed not to capture the in-depth pathological and clinical knowledge about the disease, but to quantify the elements of knowledge, attitude and practice towards the disease that would reflect on the disease status at public health level. About half of the studied population showed poor knowledge about the disease and only a quarter demonstrated good attitude and practices, which suggests that a significant gap exists in knowledge, attitude and correct practice towards SCD in western Kordofan state.
Interestingly, more than half of the studied population reported that they would not get married to someone if he/she knew beforehand that they were both carriers of SCD and some of their children may have the disease. Similarly, a recent study from Oman showed that 60% of the studied population reported that they would consider results of premarital carrier screening (PMCS) prior to marrying a partner [26]. These findings highlight the degree of acceptance of predominantly Muslim communities to PMCS as an effective tool to prevent genetic diseases [21].
Multivariate analysis demonstrated that fathers’ and mothers’ educational levels were positively correlated with Knowledge, but not the attitude and practices towards SCD. On the other hand, knowledge about SCD was found to be one of the main factors associated with attitude and practices towards SCD. Therefore, and in agreement with KAP studies in communicable diseases, specialised educational and training programmes about SCD are indispensable for appropriate management and control of SCD [34].
High SES was associated with good attitude and practice but showed no significant association with knowledge after multivariate adjustments for covariates. Interestingly, studies on other communicable and non-communicable diseases have shown that family income is a major contributory factor in changing attitudes towards treatment of any disease [34, 35]. The positive effect of SES on attitude and practices towards diseases could be attributed to the fact that better-off households have a better access to health services, which results in a relatively good disease prognosis.
Earlier studies in Sudan concluded that sickle cell disease is highly prevalent in western region, Blue Nile area and Khartoum [10, 36, 37]. To quantify the current magnitude and the impact of SCD in Sudan, a comprehensive national screening programme is warranted. The experience of other countries proved that the institution of newborn screening programme, premarital screening and well-designed management protocol is effective in prevention of the disease and associated complications [38–40]. However, there is ongoing debate about whether SCD management programme should be integrated into existing health service or separate disease-specific programme [40]. WHO recommends that countries where the SCD birth prevalence exceeds 0.5 per 1000 births should develop separate SCD programmes. In Sudan, the prevalence of SCD birth varies considerably between different regions. Consequently, the WHO criteria based on national burden of the disease might help perpetuate the current status of the disease at the bottom of the Sudan’s national health priorities [41]. Therefore, we strongly recommend to WHO to redefine the criteria for SCD national programme development in order to reflect the subnational uneven distribution of the disease in relatively big countries.
In the context of the observed extreme poverty, inadequate education, lack of awareness about SCD, scanty healthcare facilities and high percentage of children with SCD in western Kordofan area, we would suggest urgent introduction of a comprehensive clinical care programme based on two elements: a newborn screening programme and a simple, life-saving and cost-effective home-based intervention. Indeed, studies in Africa have shown that early parental education and counselling on prophylactic penicillin, the detection of enlarged spleen, drinking adequate amounts of water, the dangers of fever and increasing pallor and periodic visits to the physician could help decrease frequency and severity of SCD-related events [6, 42].
Our results indicated that premarital testing for HbS and premarital advice about consanguinity at the primary healthcare level could be a socially acceptable measure to reduce the HbS gene frequency in this part of Sudan [43]. Public awareness about SCD should also be promoted by inclusion of relevant information in the school curricula and well-designed media campaign.
We did not assess the frequency of HbS gene among infants. To obtain a more accurate estimate about the current HbS frequency and mortality to SCD in western Kordofan, further studies based on big sample sizes that include infants are highly warranted.
In conclusion, this study confirms that SCD is a major health problem in West Kordofan and provides basic quantitative measures that are necessary to develop a successful preventive and management programme.
Acknowledgements
Very sincere thanks are due to Dr Abdelhameid Elmugabil, Elshakh Awoda and Mutaz Elhassan for their contribution in data collection and laboratory analysis. We are grateful to Howida Salih, Dr Dawelbeit Alhaj and Gamal Azaz for their role in social mobilisation and community sensitisation, Dr Abdellah Abdelkarim and Professor Anware Kordofani for providing valuable suggestions and support on data collection and laboratory analysis and Dr Yosef Elmaki, Dr Salah Daak and Professor Abdulrahman Maki for logistic support. Special thanks go to Professor Michael Riech and Amy Levin for the invaluable support to AD to write the manuscript.
References
- 1.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008: 86: 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diallo D, Tchernia G. Sickle cell disease in Africa. Curr Opin Hematol 2002: 9: 111–116. [DOI] [PubMed] [Google Scholar]
- 3.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010: 376: 2018–2031. [DOI] [PubMed] [Google Scholar]
- 4.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 2013: 10: e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serjeant GR. Mortality from sickle cell disease in Africa. BMJ 2005: 330: 432–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahimy MC, Gangbo A, Ahouignan G et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anemia in a sub-Saharan African setting. Blood 2003: 102: 834–838. [DOI] [PubMed] [Google Scholar]
- 7.Molineaux L, Fleming AF, Cornille-Brogger R, Kagan I, Storey J. Abnormal haemoglobins in the Sudan savanna of Nigeria. III. Malaria, immunoglobulins and antimalarial antibodies in sickle cell disease. Ann Trop Med Parasitol 1979: 73: 301–310. [DOI] [PubMed] [Google Scholar]
- 8.Yusuf HR, Lloyd-Puryear MA, Grant AM, Parker CS, Creary MS, Atrash HK. Sickle cell disease: the need for a public health agenda. Am J Prev Med 2011: 41(6 Suppl. 4): S376–S383. [DOI] [PubMed] [Google Scholar]
- 9.Vella F. Sickling in the Western Sudan. SMJ 1964: 3: 16–20. [Google Scholar]
- 10.Lauder JRSAI. Sickling in south-West Kordofan. SMJ 1970: 8: 207–214. [Google Scholar]
- 11.UNDP. Sudan national Human development Repot. 2012.
- 12.Bereir RE, Hassan HY, Salih NA et al. Co-introgression of Y-chromosome haplogroups and the sickle cell gene across Africa’s Sahel. Eur J Hum Genet 2007: 1511: 1183–1185. [DOI] [PubMed] [Google Scholar]
- 13.Filmer D, Pritchett LH. Estimating wealth effects without expenditure data–or tears: an application to educational enrollments in states of India. Demography 2001: 38: 115–132. [DOI] [PubMed] [Google Scholar]
- 14.Organization WH, editor. Iron Deficiency Anaemia: Assessment, Prevention and Control, 2001.
- 15.Corporation P. Technical Manual ReliaPrep™ Blood gDNA Miniprep System. Instructions for use of products A5080, A5081 and A5082.: printed in USA.; 2012: p. 1–9.
- 16.Elderdery AY, Mills J, Mohamed BA et al. Molecular analysis of the beta-globin gene cluster haplotypes in a Sudanese population with sickle cell anaemia. Int J Lab Hematol 2012: 34: 262–266. [DOI] [PubMed] [Google Scholar]
- 17.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med 2011: 41(6 Suppl. 4): S398–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dekker LH, Fijnvandraat K, Brabin BJ, van Hensbroek MB. Micronutrients and sickle cell disease, effects on growth, infection and vaso-occlusive crisis: a systematic review. Pediatr Blood Cancer 2012: 59: 211–215. [DOI] [PubMed] [Google Scholar]
- 19.Christianson AHC, Modell B. March of Dimes Global Report on Birth Defects: The Hidden Toll of Dying and Disabled Children. March of Dimes Birth Defects Foundation: New York, 2006. [Google Scholar]
- 20.Noor AM, ElMardi KA, Abdelgader TM et al. Malaria risk mapping for control in the republic of Sudan. Am J Trop Med Hyg 2012: 87: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Hazmi MA. Pre-marital examination as a method of prevention from blood genetic disorders. Community views. SMJ 2006: 27: 1291–1295. [PubMed] [Google Scholar]
- 22.Aidoo M, Terlouw DJ, Kolczak MS et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 2002: 359: 1311–1312. [DOI] [PubMed] [Google Scholar]
- 23.Kreuels B, Kreuzberg C, Kobbe R et al. Differing effects of HbS and HbC traits on uncomplicated falciparum malaria, anemia, and child growth. Blood 2010: 115: 4551–4558. [DOI] [PubMed] [Google Scholar]
- 24.Mohammed AO, Attalla B, Bashir FM et al. Relationship of the sickle cell gene to the ethnic and geographic groups populating the Sudan. Community Genet 2006: 9: 113–120. [DOI] [PubMed] [Google Scholar]
- 25.Akinsheye I, Alsultan A, Solovieff N et al. Fetal hemoglobin in sickle cell anemia. Blood 2011: 118: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Farsi OA, Al-Farsi YM, Gupta I, Ouhtit A, Al-Farsi KS, Al-Adawi S. A study on knowledge, attitude, and practice towards premarital carrier screening among adults attending primary healthcare centers in a region in Oman. BMC Public Health 2014: 14: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.al Husain M, al Bunyan M. Consanguineous marriages in a Saudi population and the effect of inbreeding on prenatal and postnatal mortality. Ann Trop Paediatr. 1997: 17: 155–160. [DOI] [PubMed] [Google Scholar]
- 28.United Nation Children’s Fund (UNICEF). State of the World’s Children 2004. UNICEF: New York, 2004. [Google Scholar]
- 29.Stevens GA, Finucane MM, De-Regil LM et al. Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995–2011: a systematic analysis of population-representative data. Lancet Global Health 2013: 1: e16–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohamed SHM. Prevalence of thinness, stunting and anemia among rural school-aged sudanese children: a cross-sectional study. J Trop Pediatr 2015: 61: 260–265. [DOI] [PubMed] [Google Scholar]
- 31.Munsoor M, Gibreel M, El Karsani M. Nutritional anemia among patients referred to hematology laboratories in Port Sudan city. Clin Med J 2015: 1: 38–42. [Google Scholar]
- 32.Al-Gazali L, Hamamy H, Al-Arrayad S. Genetic disorders in the Arab world. BMJ 2006: 333: 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saha N, Hamad RE, Mohamed S. Inbreeding effects on reproductive outcome in a Sudanese population. Hum Hered 1990: 40: 208–212. [DOI] [PubMed] [Google Scholar]
- 34.Sillah F, Ho HJ, Chao JC. The use of oral rehydration salt in managing children under 5 y old with diarrhea in the Gambia: knowledge, attitude, and practice. Nutrition 2013: 29: 1368–1373. [DOI] [PubMed] [Google Scholar]
- 35.Islam FM, Chakrabarti R, Dirani M et al. Knowledge, attitudes and practice of diabetes in rural Bangladesh: the Bangladesh Population based Diabetes and Eye Study (BPDES). PLoS ONE 2014: 9: e110368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vella F. Sickling in the Western Sudan. SMJ 1964: 3: 6–20. [Google Scholar]
- 37.Ahmed HA, Baker EA. Sickling in the Sudan. Result of surveys in Blue Nile Province. East Afr Med J 1986: 63: 395–399. [PubMed] [Google Scholar]
- 38.Streetly A, Latinovic R, Henthorn J. Positive screening and carrier results for the England-wide universal newborn sickle cell screening programme by ethnicity and area for 2005–07. J Clin Pathol 2010: 63: 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Memish ZA, Saeedi MY. Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and beta-thalassemia in Saudi Arabia. Ann Saudi Med 2011: 31: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makani J, Soka D, Rwezaula S et al. Health policy for sickle cell disease in Africa: experience from Tanzania on interventions to reduce under-five mortality. Trop Med Int Health 2015: 20: 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali LDA, Kararr L, Sorkti S et al. Development of a national package for management of Sickle Cell Disorders. 8 Apr 2013. In: Health FMo, editor. Khartoum, 2013. [Google Scholar]
- 42.Hankins J, Aygun B. Pharmacotherapy in sickle cell disease–state of the art and future prospects. Br J Haematol 2009: 145: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaston MH, Verter JI, Woods G et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986: 314: 1593–1599. [DOI] [PubMed] [Google Scholar]