

Abstract
Acetate kinase catalyzes the reversible phosphorylation of acetate (CH3COO− + ATP⇄CH3CO2PO32− + ADP). A mechanism which involves a covalent phosphoryl-enzyme intermediate has been proposed, and chemical modification studies of the enzyme from Escherichia coli indicate an unspecified glutamate residue is phosphorylated (J. A. Todhunter and D. L. Purich, Biochem. Biophys. Res. Commun. 60:273–280, 1974). Alignment of the amino acid sequences for the acetate kinases from E. coli (Bacteria domain), Methanosarcina thermophila (Archaea domain), and four other phylogenetically divergent microbes revealed high identity which included five glutamates. These glutamates were replaced in the M. thermophila enzyme to determine if any are essential for catalysis. The histidine-tagged altered enzymes were produced in E. coli and purified to electrophoretic homogeneity by metal affinity chromatography. Replacements of E384 resulted in either undetectable or extremely low kinase activity, suggesting E384 is essential for catalysis which supports the proposed mechanism. Replacement of E385 influenced the Km values for acetate and ATP with only moderate decreases in kcat, which suggests that this residue is involved in substrate binding but not catalysis. The unaltered acetate kinase was not inactivated by N-ethylmaleimide; however, replacement of E385 with cysteine conferred sensitivity to N-ethylmaleimide which was prevented by preincubation with acetate, acetyl phosphate, ATP, or ADP, suggesting that E385 is located near the active site. Replacement of E97 decreased the Km value for acetate but not ATP, suggesting this residue is involved in binding acetate. Replacement of either E32 or E334 had no significant effects on the kinetic constants, which indicates that neither residue is essential for catalysis or significantly influences the binding of acetate or ATP.
Acetate is an end product of most fermentative microbes and is the major growth substrate for the methanoarchaea (22); thus, carbon flow through acetate is of primary importance in anaerobic microbial consortia and the global carbon cycle. Although the metabolisms of fermentatives and acetotrophic methanoarchaea represent the extremes of biochemical divergence in energy-yielding pathways, these microbes have in common the enzymes acetate kinase (reaction 1) and phosphotransacetylase (reaction 2).
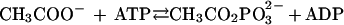 |
1 |
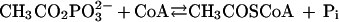 |
2 |
These two enzymes produce acetate from acetyl coenzyme A (acetyl-CoA) in the fermentatives, where a major portion of the energy requirements is obtained through substrate-level phosphorylation catalyzed by acetate kinase. The methanosarcinas utilize acetate kinase and phosphotransacetylase to activate acetate to acetyl-CoA in the first step of the pathway for the fermentation of acetate to methane (5). The acetyl-CoA is cleaved by the CO dehydrogenase–acetyl-CoA synthase enzyme complex, yielding methyl and carbonyl groups (5). The methyl group is reduced to CH4 with electrons derived from oxidation of the carbonyl group to CO2. Transport of electrons through a membrane-bound transport chain generates an electrochemical ion gradient driving ATP synthesis. Thus, acetate kinase and phosphotransacetylase are at the interface of energy-yielding metabolism between fermentatives and the acetotrophic methanoarchaea, which are the principal metabolic groups in anaerobic consortia degrading complex organic matter to methane. In addition to a key intermediate in energy metabolism, acetylphosphate acts as a phosphoryl donor to enzyme I of the phosphoenolpyruvate:glucose phosphotransferase system in Escherichia coli and Salmonella typhimurium via a phosphoenzyme intermediate of acetate kinase (7). Acetylphosphate is also a phosphoryl donor to periplasmic binding proteins (8) as well as to many response regulator proteins of two-component systems (13). It has been proposed that acetylphosphate functions as a global regulatory signal in E. coli (13, 20). Thus, acetate kinase influences the physiology of diverse microbes in a variety of ways.
Comparison of deduced amino acid sequences reveals high identity among acetate kinases (Fig. 1) from the Archaea and Bacteria, which suggests that this enzyme was either highly evolved prior to divergence of the domains or that horizontal gene transfer occurred between domains. The high identity suggests similar mechanisms for these acetate kinases. The acetate kinase from E. coli is phosphorylated with acetylphosphate or ATP (3, 6, 19), and it has been proposed that a phosphoryl-enzyme intermediate is involved in the catalytic mechanism. Acid hydrolysis of the phosphorylated enzyme reduced with [3H]borohydride yields [3H]α-amino-δ-hydroxyvaleric acid, suggesting that an unspecified γ-phosphorylated glutamyl residue is involved in catalysis (19). This hypothesis is novel, as there are no reports of a phosphorylated glutamate in any enzyme reaction mechanism. The proposed mechanism was challenged when it was shown that phosphorylated acetate kinase from E. coli is a phosphoryl donor to enzyme I of the bacterial phosphotransferase system (7), suggesting the possibility of an alternate function for phosphorylated acetate kinase. Furthermore, no evidence has been reported for a glutamate which is essential for kinase activity. Thus, the involvement of glutamate in catalysis of kinase activity by acetate kinases is a matter of controversy.
FIG. 1.

Alignment of the deduced amino acid sequences of acetate kinases. Abbreviations and GenBank accession numbers: M.t., M. thermophila (L23147); E.c., E. coli (M22956); C.a., Clostridium acetobutylicum (U38234); B.s., Bacillus subtilis (L17320); H.i., Haemophilus influenzae (L45839); M.g., Mycoplasma genitalium (L43967). Amino acids are in the single-letter code. Dashes represent gaps introduced for alignment. Closed circles mark the glutamate residues targeted for replacement. Identical residues are shown in bold.
Despite the broad importance of acetate kinase in microbial physiology, the use of biochemical genetics to probe the catalytic site has not been reported. Here we identify an essential glutamate in the Methanosarcina thermophila enzyme which provides support for the previously proposed catalytic mechanism. Two other glutamates are implicated in substrate binding.
MATERIALS AND METHODS
Protein sequence analyses.
Databases were searched at the National Center for Biotechnology Information by using the BLAST network server (2). CLUSTAL W (18) was used for multiple protein sequence alignment at the Human Genome Center of the Baylor School of Medicine.
Site-directed mutagenesis.
Mutagenesis was performed according to the manufacturer’s instructions with the Muta-Gene phagemid mutagenesis kit (Bio-Rad), which employs the oligonucleotide-directed in vitro mutagenesis method (11). The plasmids used are shown in Table 1. The M. thermophila acetate kinase gene was excised from pUC19/ack (12) by KpnI and BamHI digestion and ligated into pTZ18U to generate pTZack. The mutations were verified by double-stranded sequence analysis by using the dideoxy chain termination method (15) and Sequenase version 2.0 (United States Biochemicals).
TABLE 1.
Plasmids used in this study
| Plasmid | Origin | Genotype | Reference or source |
|---|---|---|---|
| pUC19/ack | pUC19 | ack | 12 |
| pTZ18U | Bio-Rad | ||
| pTZack | pTZ18U | ack | This study |
| pET15b | Novagen | ||
| pETack | pET15b | ack | This study |
| pETackE32A | pET15b | ackE32A | This study |
| pETackE97A | pET15b | ackE97A | This study |
| pETackE97D | pET15b | ackE97D | This study |
| pETackE97Q | pET15b | ackE97Q | This study |
| pETackE334A | pET15b | ackE334A | This study |
| pETackE384A | pET15b | ackE384A | This study |
| pETackE384D | pET15b | ackE384D | This study |
| pETackE384Q | pET15b | ackE384Q | This study |
| pETackE385A | pET15b | ackE385A | This study |
| pETackE385C | pET15b | ackE385C | This study |
| pETackE385D | pET15b | ackE385D | This study |
| pETackE385Q | pET15b | ackE385Q | This study |
Heterologous production and purification of acetate kinase.
The unaltered and altered acetate kinase genes were subcloned into the T7-based expression vector pET15b (Novagen) to generate the plasmids listed in Table 1. In these plasmids, a 60-nucleotide leader sequence with six tandem histidine codons was fused in frame to the 5′ end of the unaltered and altered ack genes. E. coli BL21(DE3) was transformed with the expression vectors, inoculated into 50 ml of Luria-Bertani medium containing 100 μg of ampicillin per ml, and grown at 37°C to an A600 of 0.6 to 0.9, at which time IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 1 mM. After 1.5 to 2.0 h of induction, the cells were harvested and stored at −70°C. The unaltered and altered acetate kinases were purified using a Ni-nitrilotriacetic acid silica spin kit (Qiagen) according to the manufacturer’s instructions. The enzymes were eluted in buffer (pH 7.0) containing 50 mM NaH2PO4, 300 mM NaCl, and 250 mM imidazole. Protein concentrations were determined by the Bradford method (4), using protein dye reagent (Bio-Rad) and bovine serum albumin as the standard.
Enzyme activity assays.
Acetate kinase activity was determined by the previously described (1) standard hydroxamate assay, which detects the formation of acetyl phosphate from acetate (200 mM) and ATP (10 mM). The acetate concentrations were 0.7 and 1.5 M for determination of the kinetic constants for ATP for the E385A and E385D enzymes.
The ATP-ADP exchange assays were performed as follows. The reaction mixture (100 μl) contained the following: triethanolamine-HCl, 50 mM, pH 7.0; ATP, 1 mM; ADP, 1 mM; MgCl2, 2 mM; acetate kinase, 10 μg; [α-32P]ATP (3,000 Ci/mmol), 5 μCi. The reactions were initiated by the addition of enzyme, and mixtures were incubated at 20°C. At various times, 5 μl was withdrawn, and the reaction was stopped by the addition of an equal volume of 1 N HCl. A volume of 5 μl was applied to polyethyleneimine cellulose thin-layer chromatography plates (Bakerflex), which were developed ascendingly in 0.52 M potassium phosphate (pH 3.5). The plates were air dried and autoradiographed to visualize the ATP and ADP spots. The ADP spots were excised and counted in 5 ml of Scintiverse (Fisher Scientific) scintillation cocktail. Initial velocities were calculated as described previously (16).
Inhibition by NEM.
N-Ethylmaleimide (NEM) (final concentration, 10 μM) was added to the unaltered acetate kinase (5 μg/ml) and to the E385C enzyme (40 μg/ml) in a final volume of 100 μl at 37°C. Aliquots (10 μl) were removed at the indicated times and assayed for kinase activity. Substrate protection experiments were performed by preincubating the enzyme for 5 min at 37°C with (final concentrations) ADP (10 mM), ATP (10 mM), acetyl phosphate (10 mM), or potassium acetate (200 mM) prior to addition of NEM.
Circular dichroism spectroscopy.
Spectra were acquired at 37°C with an Aviv circular dichroism (CD) spectrometer, model 62DS. Samples (1 to 10 μM) of acetate kinase in 20 mM sodium phosphate (pH 7.5) containing 0.1 M NaCl were placed in a cuvette with a 1-mm path length and data points obtained from 205 to 320 nm in 1.0-nm increments. Five spectra were taken for each sample and averaged. The resulting spectra were normalized for direct comparison.
RESULTS AND DISCUSSION
The sequences of acetate kinases deduced from the genes of widely divergent microbes from the Archaea and Bacteria domains are highly identical, suggesting a similar catalytic mechanism (Fig. 1); for example, the enzymes from E. coli (Bacteria) and M. thermophila (Archaea) have 44% identity. It has been proposed that a phosphorylated glutamate functions in a covalent catalytic mechanism for the E. coli enzyme (19). Five glutamates in the M. thermophila acetate kinase were selected for replacement to determine if any are essential for catalysis. The glutamates selected (Fig. 1) are either highly conserved and within a region of high conservation (E97) or 100% conserved (E32, E334, E384, and E385) with the deduced sequences for five other acetate kinases from extremely diverse microbes. The histidine-tagged altered enzymes were heterologously produced in E. coli and purified by one-step metal affinity chromatography. Each was judged to be homogeneous by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the subunit molecular masses were indistinguishable from that of the unaltered histidine-tagged acetate kinase (not shown). Native gel filtration chromatography (not shown) indicated that the altered enzymes were dimeric, in accord with the authentic acetate kinase purified from M. thermophila (1). The unaltered histidine-tagged acetate kinase displayed a kcat (Table 2) slightly greater than the value previously determined for the authentic enzyme (1) and was most likely a result of the one-step purification procedure yielding more active enzyme. There were no significant differences between the unaltered (Table 2) and authentic acetate kinases with respect to the Km values for acetate or ATP.
TABLE 2.
Kinetic constants of unaltered and altered acetate kinasesa from M. thermophila
| Enzyme | Acetate
|
ATP
|
||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | |
| Authenticb | 22 | 1,050 | 47 | 2.8 | 1,221 | 436 |
| Unaltered | 19 ± 3 | 1,596 ± 140 | 84 | 1.0 ± 0.3 | 1,669 ± 288 | 1,669 |
| E32A | 26 ± 2 | 1,029 ± 97 | 40 | 1.1 ± 0.2 | 1,012 ± 262 | 920 |
| E97A | 2.3 ± 0.5 | 165 ± 3 | 72 | 1.4 ± 0.1 | 302 ± 49 | 216 |
| E97D | 7.2 ± 3.5 | 430 ± 60 | 60 | 1.4 ± 0.6 | 567 ± 175 | 405 |
| E97Q | 4.0 ± 0.4 | 273 ± 56 | 68 | 1.4 ± 0.7 | 276 ± 59 | 197 |
| E334A | 15 ± 6 | 931 ± 35 | 62 | 0.7 ± 0.2 | 1,198 ± 62 | 1,711 |
| E385A | 86 ± 30c | 636 ± 158c | 7 | 6.9 ± 0.5 | 1,308 ± 112 | 190 |
| E385C | 17 ± 4 | 637 ± 139 | 37 | 1.6 ± 0.5 | 813 ± 214 | 508 |
| E385D | 337 ± 81c | 456 ± 132c | 1 | 4.5 ± 0.5 | 671 ± 34 | 149 |
| E385Q | 44 ± 19 | 1,869 ± 479 | 42 | 2.0 ± 0.6 | 1,711 ± 231 | 856 |
Unaltered and altered acetate kinases were produced in E. coli.
Published values for the acetate kinase purified from M. thermophila (1) are given.
Apparent value.
Replacement of E384 and E385.
Among the five glutamates targeted (Fig. 1), all except the enzymes altered at E384 had substantial activity (Table 2). Activity in the standard assay was undetectable (less than 1.0 μmol/min/mg of protein) for the E384D and E384Q enzymes. The E384A enzyme had detectable but extremely low specific activity (4.0 ± 2.0 μmol/min/mg of protein) in the standard assay, which was only 0.5% of the specific activity for the unaltered acetate kinase assayed under the same conditions. The low activity precluded reliable determinations of kinetic constants. These results suggest that E384 is the only essential glutamate. This glutamate is 100% conserved with all other deduced sequences for acetate kinases from widely divergent organisms (Fig. 1), which is consistent with an essential function. The CD spectra of the E384Q enzyme (Fig. 2) and the E384D enzyme (not shown) were similar to that of the unaltered acetate kinase. The ellipticity values at 210 nm for the E384Q and E384D enzymes were 3.6 and 1.2% less negative than that for the unaltered acetate kinase. This result suggests no global conformational change on substitution of E384 with Q or D, which is consistent with the high recovery on purification of the enzymes altered at E384 and suggests that they were dimeric in accord with the authentic enzyme purified from M. thermophila. These results, combined with the loss of kinase activity for the enzymes altered at E384, suggest that E384 is essential for catalysis, which is consistent with the previously proposed covalent mechanism in which a glutamate residue is γ-phosphorylated; however, further experimentation is necessary to prove that E384 is phosphorylated and that the phosphorylated enzyme is kinetically competent to phosphorylate acetate. The complete loss of activity for the E384D enzyme indicates that displacement of the active-site nucleophile by a single methylene carbon is sufficient to completely disrupt catalysis.
FIG. 2.

CD spectra of unaltered (dotted line) and E384Q (solid line) acetate kinase from M. thermophila.
Although the features of the CD spectra were identical, the difference at 210 nm between the unaltered acetate kinase and the E384A enzyme was 14% less negative (not shown), suggesting the possibility that this replacement may have produced secondary structural changes. The thermostability of the E384A kinase was greater than that of the unaltered acetate kinase when the enzymes were preincubated either with or without ATP (Fig. 3A), a result which is consistent with changes in secondary structure. The temperature stability profile for the unaltered acetate kinase is shifted approximately 27°C higher in the presence of ATP (Fig. 3A). A similar shift was observed on preincubation of the E384A enzyme with ATP (Fig. 3B), suggesting that ATP interacts with the altered enzyme in the same way as it does with the unaltered acetate kinase. This result is unlikely if substitution of E384 with alanine produced conformational changes in the active site which abolished activity; however, this possibility cannot be ruled out. Nonetheless, these results do not detract from the results obtained for the E384Q and E384D enzymes which indicate that E384 is essential for catalysis. The small amount of activity recorded for the E384A enzyme is unexplained; however, it is theoretically possible that replacement with alanine may have increased the accessibility of substrate to E385, which functioned as an active-site nucleophile in place of E384.
FIG. 3.

Thermostability profiles of unaltered and E384A acetate kinase from M. thermophila. The enzyme preparations were preincubated without (○) and with (•) 10 mM ATP (final concentration) after which samples were incubated for 15 min at the indicated temperatures, cooled to 4°C, and immediately assayed for kinase activity. (A) Unaltered acetate kinase. One hundred percent activity was 470 μmol/min/mg of protein. (B) E384A acetate kinase. One hundred percent activity was 4.0 μmol/min/mg of protein.
Mechanistically, an exchange of phosphate between ATP and ADP predicts a phosphoryl-enzyme intermediate; thus, linking exchange activity with kinase activity would provide evidence that both activities involve the same intermediate. The authentic and heterologously produced acetate kinase from M. thermophila catalyzed an exchange of phosphate between ATP and ADP (Fig. 4), as reported for other acetate kinases (10, 17). Although the initial rate of exchange (250 ± 40 nmol/min/mg of protein) was nearly 3,000-fold lower than kinase activity (705 ± 70 μmol/min/mg of protein) assayed under the same conditions, the result does not rule out the possibility that a phosphoryl-enzyme intermediate is required for kinase activity. Unknown determinants such as substrate synergism may contribute to relatively low activity of the partial reaction. The previously reported ATP-ADP exchange activity of the E. coli enzyme is consistent with a phosphoryl-enzyme intermediate; however, it was not possible to exclude the possibility that kinase activity occurred by a different mechanism (14). Likewise, it cannot be concluded from the exchange activity alone that kinase activity for the enzyme from M. thermophila involves a phosphoryl-enzyme intermediate; however, the loss of kinase activity on replacement of E384 was paralleled by the inability to detect significant (limit of detection, 10 nmol/min/mg of protein) ATP-ADP exchange activity for any of the enzymes altered at E384 (data not shown), which is consistent with a linkage between the two activities centering on E384.
FIG. 4.

ATP-ADP exchange activity of acetate kinase from M. thermophila. Activity was determined for purified histidine-tagged (○) and untagged (•) M. thermophila acetate kinase produced in E. coli. The ADP fractions contained 35,700 (○) and 92,000 (•) cpm at equilibrium.
Further support for involvement of E384 in the active site of the acetate kinase from M. thermophila was obtained by replacing the neighboring E385 residue. E385 is 100% conserved with the acetate kinase sequences from diverse microbes (Fig. 1); however, other residues with different functionality replaced E385 (Table 2) without the reduction in activity observed for the enzymes altered at E384, suggesting that E385 is not essential for catalysis. Nonetheless, replacement of E385 with alanine or aspartate significantly increased the Km values for acetate and ATP, suggesting that this residue is important for binding these substrates. However, the kinetic constants for the E385Q enzyme were not significantly different from those for the unaltered acetate kinase, suggesting that the negative charge of E385 is inconsequential for substrate binding. The unaltered acetate kinase from M. thermophila was not inactivated by NEM (Fig. 5); however, the E385C enzyme was inactivated in a time-dependent manner, and preincubation with ATP, ADP, acetate, or acetyl phosphate protected against the inactivation (Fig. 6), which suggests that E385 is near the active site.
FIG. 5.

Time course for NEM inactivation of unaltered and E385C acetate kinase from M. thermophila. Kinase activity was determined at the indicated times after addition of NEM to unaltered (▪) or E385C (•) acetate kinase and compared to a control where no inhibitor was added to the E385C enzyme (○).
FIG. 6.

Substrate protection from NEM inactivation of the E385C-altered acetate kinase from M. thermophila. Kinase activity was determined at the indicated times after addition of NEM to the E385C acetate kinase preincubated with ADP (□), ATP (▵), acetate (○), acetyl phosphate (▪), or no substrate (•).
The acetate kinase isolated from E. coli is inactivated by NEM, and the inactivation is protected by preincubation with either ATP or ADP, suggesting that a cysteine residue is located near the active site (21). The inhibition is incomplete, and, therefore, it was concluded that the cysteine is not involved in catalysis. Only one cysteine residue is conserved between the M. thermophila and E. coli acetate kinases (Fig. 1); thus, either the M. thermophila enzyme has no cysteine residue corresponding in location to the modified residue in the E. coli enzyme or the only conserved cysteine residue in the M. thermophila enzyme (C207) is shielded from attack by NEM. Preincubation with acetate or acetyl phosphate does not protect the E. coli acetate kinase from NEM inactivation; however, the pattern by which these substrates prevent protection by AMP, ADP, or ATP suggests that the binding sites for acetate and acetyl phosphate are in close proximity to the phosphoryl binding regions of ADP and ATP (21). The ability of all four substrates to prevent NEM inactivation of the M. thermophila E385C acetate kinase (Fig. 6) is consistent with the results obtained for the E. coli enzyme.
Replacement of E97.
Compared with values obtained with the unaltered acetate kinase, the Km values for acetate of all the E97-altered enzymes decreased with corresponding decreases in kcat, which resulted in relatively smaller differences in the catalytic efficiencies (kcat/Km) (Table 2). These results suggest that E97 is not essential for catalysis but influences the binding of acetate. Replacement of E97 with aspartate was the least effective in lowering the Km for acetate, suggesting that the negative charge of E97 influences binding of the acetate anion by charge repulsion. The results also suggest that displacement of the negative charge by only one methylene carbon significantly influences the binding of acetate. None of the E97 replacements influenced the Km values for ATP, suggesting no involvement of E97 at the ATP binding site.
The Km for acetate of the unaltered acetate kinase is relatively high, which appears to place M. thermophila at a disadvantage when competing for substrate; thus, the ability to decrease the Km nearly 10-fold by simple replacement of a single residue suggests that the enzyme evolved to prefer a high Km, which may be physiologically significant. Indeed, M. thermophila and other Methanosarcina species predominate in environments where the acetate concentrations are in the millimolar range compared with species from the acetotrophic genus Methanothrix, which thrive in habitats with acetate concentrations well below 1 mM (22). The acetotrophic Methanosarcina species outcompete Methanothrix species with higher growth rates and greater conservation of energy. The acetate-activating enzyme acetate thiokinase from Methanothrix soehngenii has a Km for acetate below 1 mM, which allows growth of this species in habitats with low concentrations of acetate where methanosarcinas are unable to compete (9). Apparently, the M. thermophila acetate kinase evolved to favor a high turnover at the expense of a low Km for acetate which supports a faster growth rate in environments with high concentrations of acetate.
Replacement of E32 and E334.
The kinetic constants determined for the E32A and E334A enzymes (Table 2) indicated that neither residue is essential for catalysis or significantly influences the binding of acetate or ATP.
Conclusions.
The first site-specific replacement of amino acids for any acetate kinase has identified a glutamate residue (E384) in the enzyme from M. thermophila which is essential for catalysis and is consistent with the previously proposed covalent mechanism for catalysis of kinase activity. Replacement of two other glutamates (E385 and E97) suggests that they are involved in substrate binding.
ACKNOWLEDGMENTS
This work was supported by grant DE-FG02-95ER20198 to J.G.F. from the Department of Energy-Basic Energy Sciences and by National Institutes of Health Individual National Research Service Award 5F32GM16107 to K.S.-W.
We thank Douglas Berg of Washington University, who graciously accommodated K.S.-W. as a guest in his laboratory, without which this work could not have been completed. We also thank Madeline Rasche for valuable discussions and support.
REFERENCES
- 1.Aceti D J, Ferry J G. Purification and characterization of acetate kinase from Methanosarcina thermophila. Evidence for regulation of synthesis. J Biol Chem. 1988;263:15444–15448. [PubMed] [Google Scholar]
- 2.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Anthony R S, Spector L B. A phosphoenzyme intermediary in acetate kinase action. J Biol Chem. 1970;245:6739–6741. [PubMed] [Google Scholar]
- 4.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Ferry J G. Fermentation of acetate. In: Ferry J G, editor. Methanogenesis. New York, N.Y: Chapman and Hall; 1993. pp. 304–334. [Google Scholar]
- 6.Fox D K, Roseman S. Isolation and characterization of homogeneous acetate kinase from Salmonella typhimurium and Escherichia coli. J Biol Chem. 1986;261:13487–13497. [PubMed] [Google Scholar]
- 7.Fox D K, Meadow N D, Roseman S. Phosphate transfer between acetate kinase and enzyme I of the bacterial phosphotransferase system. J Biol Chem. 1986;261:13498–13503. [PubMed] [Google Scholar]
- 8.Hong J, Hunt A, Masters P, Lieberman M. Requirement for acetyl-phosphate for the binding protein-dependent transport systems in Escherichia coli. Proc Natl Acad Sci USA. 1979;76:1213–1217. doi: 10.1073/pnas.76.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jetten M S M, Stams A J M, Zehnder A J B. Isolation and characterization of acetyl-coenzyme A synthetase from Methanothrix soehngenii. J Bacteriol. 1989;171:5430–5435. doi: 10.1128/jb.171.10.5430-5435.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahane I, Muhlrad A. Purification and properties of acetate kinase from Acholeplasma laidlawii. J Bacteriol. 1979;137:764–772. doi: 10.1128/jb.137.2.764-772.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunkel T A, Roberts J D, Zakour R A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 12.Latimer M T, Ferry J G. Cloning, sequence analysis, and hyperexpression of the genes encoding phosphotransacetylase and acetate kinase from Methanosarcina thermophila. J Bacteriol. 1993;175:6822–6829. doi: 10.1128/jb.175.21.6822-6829.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCleary W R, Stock J B, Ninfa A J. Is acetyl phosphate a global signal in Escherichia coli? J Bacteriol. 1993;175:2793–2798. doi: 10.1128/jb.175.10.2793-2798.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purich D L, Fromm H J. Evaluation of the phosphoryl-enzyme intermediate concept in the acetate kinase and hexokinase reactions from kinetic studies. Arch Biochem Biophys. 1972;149:307–315. doi: 10.1016/0003-9861(72)90326-8. [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segel I H. Enzyme kinetics. New York, N.Y: John Wiley & Sons; 1975. pp. 860–864. [Google Scholar]
- 17.Skarstedt M T, Silverstein E. Escherichia coli acetate kinase mechanism studied by net initial rate, equilibrium, and independent isotopic exchange kinetics. J Biol Chem. 1976;251:6775–6783. [PubMed] [Google Scholar]
- 18.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todhunter J A, Purich D L. Evidence for the formation of a γ-phosphorylated glutamyl residue in the Escherichia coli acetate kinase reaction. Biochem Biophys Res Commun. 1974;60:273–280. doi: 10.1016/0006-291x(74)90201-0. [DOI] [PubMed] [Google Scholar]
- 20.Wanner B L, Wilmes-Riesenberg M R. Involvement of phosphotransacetylase, acetate kinase, and acetyl phosphate synthesis in control of the phosphate regulon in Escherichia coli. J Bacteriol. 1992;174:2124–2130. doi: 10.1128/jb.174.7.2124-2130.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong S S, Wong L-J. Inactivation of Escherichia coli acetate kinase by N-ethylmaleimide. Biochim Biophys Acta. 1980;615:121–131. doi: 10.1016/0005-2744(80)90015-7. [DOI] [PubMed] [Google Scholar]
- 22.Zinder S H. Physiological ecology of methanogens. In: Ferry J G, editor. Methanogenesis. New York, N.Y: Chapman and Hall; 1993. pp. 128–206. [Google Scholar]