

Summary
Intercalated motifs or i-Motifs (iMs) are nucleic acid structures formed by cytosine-rich sequences, which may regulate cellular processes and have broad applications in nanotechnology due to their pH-dependent nature. We have developed an iM-specific nanobody (iMbody) that can recognize iM DNA structures regardless of their sequences, making it a versatile research tool for studying iMs in various contexts. Here, we provide a protocol for the bacterial expression and His-tag purification of iMbody. We then describe procedures for performing ELISA and immunostaining using iMbody.
Subject areas: Genomics, Microscopy, Molecular Biology, Antibody, Molecular/Chemical Probes, Protein Biochemistry, Protein expression and purification, Biotechnology and bioengineering
Graphical abstract
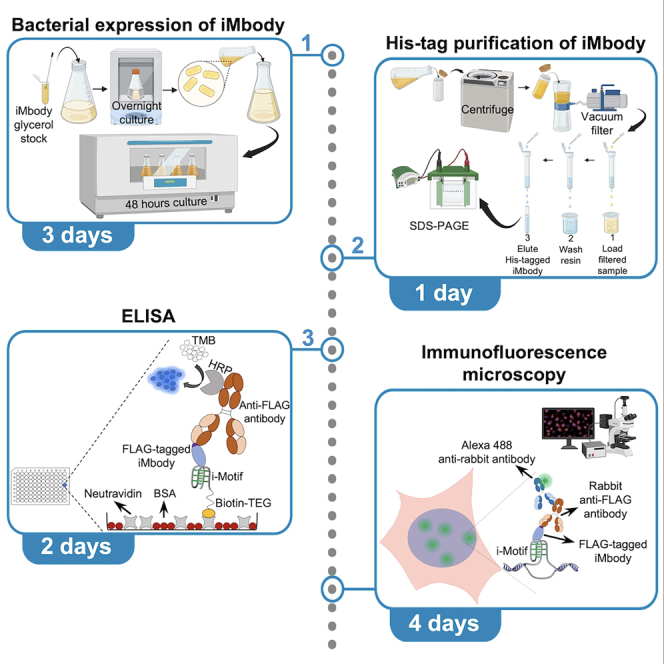
Highlights
-
•
An i-Motif-specific nanobody (iMbody) that can be utilized in i-Motif research
-
•
A detailed protocol outlining the production and purification of iMbody
-
•
A step-by-step guide for performing ELISA and confocal microscopy using iMbody
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Intercalated motifs or i-Motifs (iMs) are nucleic acid structures formed by cytosine-rich sequences, which may regulate cellular processes and have broad applications in nanotechnology due to their pH-dependent nature. We have developed an iM-specific nanobody (iMbody) that can recognize iM DNA structures regardless of their sequences, making it a versatile research tool for studying iMs in various contexts. Here, we provide a protocol for the bacterial expression and His-tag purification of iMbody. We then describe procedures for performing ELISA and immunostaining using iMbody.
Before you begin
Protocol overview
DNA sequences rich in cytosine can adopt a pH-dependent conformation known as the intercalated motif or i-Motif (iM)1 (Figure 1A). The pH dependency of iMs has enabled their utility in DNA nanotechnology.2 Moreover, accumulating evidence demonstrates the formation of iMs in cells and their involvement in regulatory processes.3,4 Therefore, the development of probes that specifically recognize iMs can greatly advance iM research in both biology and nanotechnology. Here, we present the development of an iM-specific nanobody, termed iMbody, based on the hs2dAb nanobody scaffold5 (Figure 1B), following a procedure we described previously.6 iMbody can recognize iM DNA structures regardless of their sequences, making it a versatile research tool for studying iMs in various contexts. We believe that iMbody will prove valuable in other applications beyond those mentioned here. To expand its potential applications, we provide the protein sequence of iMbody (Figure 1C) and its corresponding plasmid (Figure 1D) (Addgene plasmid #184496). This allows for easy modification of its gene by adding various tags, cloning it into a preferred vector, and optimizing codons for specific applications in other model organisms. It is important to note that careful optimization of experimental conditions is necessary to ensure reliable outcomes and interpretations.
Figure 1.

An i-Motif specific nanobody (iMbody)
(A) Schematic of a canonical i-Motif DNA structure. Green bars represent hemi-protonated cytosine base pairs (C·C+).
(B) Schematic of a camelid antibody and a nanobody.
(C) Protein sequence of iMbody.
(D) iMbody-pSANG10-3F vector map available from Addgene (plasmid #184496).
Here, we provide a protocol detailing the bacterial expression and His-tag purification of iMbody. The pelB leader sequence directs iMbody to the periplasm. However, overnight incubation leads to its release from the periplasm into the cell culture media. Purification of His-tagged iMbody from the supernatant using Ni-NTA agarose resins yields a pure product. To enhance the yield, simultaneous purification from periplasm is feasible, but it frequently yields an impure product, necessitating an additional purification step. Ensuring a pure sample is crucial as impurities can impact calculations needed for diluting iMbody in subsequent experiments and can impact their reproducibility. This protocol specifically delineates iMbody purification from the supernatant. In addition to the production of iMbody, we provide detailed procedures for utilizing iMbody in enzyme-linked immunosorbent assay (ELISA) and immunostaining of iMs in the nuclei of HeLa cells as example applications.
Preparation of reagents
Timing: 1 day
Prepare all the required reagents as described in the materials and equipment section.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG M2-peroxidase (HRP) antibody (1:15,000 dilution for ELISA) | Sigma-Aldrich | Cat# A8592; RRID: AB_439702 |
| Rabbit monoclonal anti-FLAG antibody (1:600 dilution for immunostaining) | Cell Signaling Technology | Cat# 14793S; RRID: AB_2572291 |
| Goat anti-rabbit Alexa Fluor 488 conjugated antibody (1:400 dilution for immunostaining) | Cell Signaling Technology | Cat# 4412S; RRID: AB_1904025 |
| Bacterial and virus strains | ||
| BL21(DE3) competent E. coli | New England Biolabs | Cat# C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma-Aldrich | Cat# k1377 |
| BD Difco dehydrated culture media: LB agar, Miller (Luria-Bertani) | Fisher Scientific | Cat# 244520 |
| BD Difco dehydrated culture media: LB broth, Miller (Luria-Bertani) | Fisher Scientific | Cat# 244620 |
| Glycerol | Sigma-Aldrich | Cat# G5516 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | Cat# 16758 |
| Ni-NTA agarose | QIAGEN | Cat# 30210 |
| Dulbecco’s phosphate-buffered saline (DPBS) | Thermo Fisher Scientific | Cat# 14190144 |
| NuPAGE 4%–12%, Bis-Tris, mini protein gels | Thermo Fisher Scientific | Cat# NP0322BOX |
| NuPAGE MES SDS running buffer, 20× | Thermo Fisher Scientific | Cat# NP0002 |
| NuPAGE LDS sample buffer, 4× | Thermo Fisher Scientific | Cat# NP0007 |
| NuPAGE sample reducing agent, 10× | Thermo Fisher Scientific | Cat# NP0009 |
| PageRuler Plus prestained protein ladder, 10–250 kDa | Thermo Fisher Scientific | Cat# 26619 |
| EZBlue gel staining reagent | Sigma-Aldrich | Cat# G1041 |
| NaH2PO4·H2O | Sigma-Aldrich | Cat# S9638 |
| Sodium chloride (NaCl) | Ajax Finechem | Cat# AJA465 |
| Sodium hydroxide (NaOH) | Ajax Finechem | Cat# AJA482 |
| Potassium chloride (KCl) | Ajax Finechem | Cat# AJA383 |
| Imidazole | Sigma-Aldrich | Cat# I2399 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| Nuclease-free duplex buffer | Integrated DNA Technologies | Cat# 11-05-01-03 |
| NeutrAvidin | Thermo Fisher Scientific | Cat# 31000 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | Cat# A9647 |
| Tween 20 | Sigma-Aldrich | Cat# P9416 |
| Tris(hydroxymethyl)aminomethane | Merck Millipore | Cat# 108382 |
| Carbon nanotube, single-walled, carboxylic acid functionalized (SWNT-COOH) | Sigma-Aldrich | Cat# 652490 |
| N,N-Dimethylformamide (DMF) | Sigma-Aldrich | Cat# D4551 |
| UltraPure salmon sperm DNA solution | Thermo Fisher Scientific | Cat# 15632011 |
| Tetramethylbenzidine (TMB) chromogen solution | Thermo Fisher Scientific | Cat# 002023 |
| Hydrochloric acid 37% (HCl) | RCI Labscan | Cat# RP1107 |
| Pure ethanol | Sigma-Aldrich | Cat# E7023 |
| Dulbecco’s modified Eagle’s medium (DMEM), high glucose, pyruvate | Thermo Fisher Scientific | Cat# 11995065 |
| Fetal bovine serum (FBS) | Sigma-Aldrich | Cat# F9423 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat# P4333 |
| Poly-L-lysine solution 0.01%, sterile-filtered | Sigma-Aldrich | Cat# P4707 |
| Trypsin-EDTA solution | Sigma-Aldrich | Cat# T3924 |
| Pierce 16% formaldehyde (w/v), methanol-free | Thermo Fisher Scientific | Cat# 28908 |
| Triton X-100 (10% w/v) | Sigma-Aldrich | Cat# 11332481001 |
| ProLong Gold antifade reagent with DAPI | Cell Signaling Technology | Cat# 8961S |
| Alexa Fluor 555 conjugated phalloidin (1:20 dilution for immunostaining) | Cell Signaling Technology | Cat# 8953S |
| Experimental models: Cell lines | ||
| HeLa cells | ATCC | Cat# CCL-2 |
| Oligonucleotides | ||
| T7 promoter forward primer | Integrated DNA Technologies | 5′-TAATACGACTCACTATAGGG |
| T7 terminal reverse primer | Integrated DNA Technologies | 5′-GCTAGTTATTGCTCAGCGG |
| Recombinant DNA | ||
| pSANG10-3F-iMbody plasmid | This paper | Addgene plasmid #184496, www.addgene.org/184496/ |
| Software and algorithms | ||
| BioRender | BioRender | https://biorender.com/ |
| Prism | Dotmatics | https://www.graphpad.com/scientific-software/prism/ |
| SnapGene | Dotmatics | http://www.snapgene.com |
| Fiji | Schindelin et al.7 | https://imagej.net/software/fiji/downloads |
| Other | ||
| 125 mL Erlenmeyer cell culture flasks with vented cap | Sigma-Aldrich | Cat# CLS431143 |
| 0.2-μm syringe filter | Sartorius | Cat# 16534-K |
| Disposable cuvettes with 220–1600 nm spectral range | Eppendorf | Cat# 0030106300 |
| Nalgene centrifuge bottles, style 3120 | Sigma-Aldrich | Cat# B1283 |
| 0.22-μm vacuum filter units | Sigma-Aldrich | Cat# 431098 |
| Gravity-flow chromatography column | Bio-Rad | Cat# 7372522 |
| Amicon Ultra-15 centrifugal filter units, 10 kDa | Millipore | Cat# UFC901024 |
| Costar Spin-X centrifuge tube filters | Sigma-Aldrich | Cat# CLS8161 |
| 96 well MicroWell MaxiSorp flat-bottom plate | Sigma-Aldrich | Cat# M9410 |
| SealPlate film | Sigma-Aldrich | Cat# Z369659 |
| TC-treated 12-well plate | Sigma-Aldrich | Cat# CLS3513 |
| 75 cm2 cell culture flask | Sigma-Aldrich | Cat# CLS430641U |
| Hemocytometer | Sigma-Aldrich | Cat# Z359629 |
| Miniprep kit | QIAGEN | Cat# 27104 |
| Shaking incubator (CO2 regulator is not required) | Eppendorf | New Brunswick Innova 42R |
| Erlenmeyer baffled cell culture flasks | Sigma-Aldrich | Cat# CLS431256 |
| UV-visible spectrophotometer | Thermo Fisher Scientific | NanoDrop One/Oneᶜ |
| High-speed refrigerated centrifuge with R10A5 rotor | Hitachi | CR22G III |
| Vacuum pump and a connecting system | QIAGEN | QIAvac system |
| Roller | IKA | Roller 6 digital |
| Electrophoresis power supply | Bio-Rad | PowerPac Basic |
| Mini gel tank | Thermo Fisher Scientific | Cat# A25977 |
| Microplate shaker | Neuation | iShak Quattro |
| Microplate reader | PerkinElmer | EnSight |
| Tissue culture incubator with CO2 regulator | Thermo Fisher Scientific | Heracell Vios 160i CR |
| Menzel coverslips, 13 mm, #1.5 | Fisher Scientific | Cat# 11588492 |
| Universal microscope glass slide, frosted one end two sides, thickness: 1.0–1.2 mm | Livingstone | Cat# 7107-U |
| Confocal microscope | Leica Microsystems | TCS SP8 DLS |
Materials and equipment
-
•
NPI-20 buffer.
| Reagent | Final concentration | Amount |
|---|---|---|
| NaH2PO4·H2O | 50 mM | 6.90 g |
| NaCl | 300 mM | 17.54 g |
| Imidazole | 20 mM | 1.36 g |
| ddH2O | N/A | Up to 1 L |
| Total | N/A | 1 L |
Dissolve NaH2PO4, NaCl and imidazole in 600 mL ddH2O. Adjust the pH to 8.0 using 5 M NaOH and 5 M HCl. Bring it to a final volume of 1 L using ddH2O. Filter the solution using a 0.22-μm vacuum filter unit. Can be stored at room temperature (∼22°C) for several months.
CRITICAL: Work in a fume hood and wear suitable personal protective equipment while handling imidazole. Avoid skin contact and inhalation.
-
•
NPI-500 buffer.
| Reagent | Final concentration | Amount |
|---|---|---|
| NaH2PO4·H2O | 50 mM | 6.90 g |
| NaCl | 300 mM | 17.54 g |
| Imidazole | 500 mM | 34.04 g |
| ddH2O | N/A | Up to 1 L |
| Total | N/A | 1 L |
Dissolve NaH2PO4, NaCl and imidazole in 600 mL ddH2O. Adjust the pH to 8.0 using 5 M NaOH and 5 M HCl. Bring it to a final volume of 1 L using ddH2O. Filter the solution using a 0.22-μm vacuum filter unit. Can be stored at room temperature (∼22°C) for several months.
-
•
Blocking buffer for immunostaining.
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 2% | 1 g |
| Skim milk | 1% | 0.5 g |
| Tween 20 | 0.1% | 50 μL |
| Triton X-100 (10% w/v) | 0.01% | 50 μL |
| DPBS | N/A | Up to 50 mL |
| Total | N/A | 50 mL |
Make a fresh blocking buffer for each immunostaining experiment. Can be kept at 4°C for 24 h.
-
•
50 mg/mL Kanamycin stock solution.
Dissolve 0.5 g of kanamycin in up to 10 mL of molecular biology grade water. Filter through a 0.2 μm syringe filter. Dispense in 1 mL aliquots. Can be stored at −20°C for up to 1 year.
-
•
LB-kanamycin agar plates.
Dissolve 12 g of Difco LB agar in 300 mL of ddH2O. Autoclave the solution for 15 min at 121°C. Cool the solution to 50°C and add 300 μL of 50 mg/mL kanamycin stock solution (Final concentration of kanamycin is 50 μg/mL). Pour the solution into plates. The plates can be stored at 4°C for up to 4 weeks.
-
•
LB-kanamycin broth.
Dissolve 25 g of Difco LB broth in 1 L of ddH2O. Autoclave the solution for 15 min at 121°C. Cool down the solution to room temperature (∼22°C) and add 1 mL of 50 mg/mL kanamycin stock solution (Final concentration of kanamycin is 50 μg/mL). The medium can be stored at 4°C for up to 4 weeks.
-
•
1 Molar IPTG stock solution.
Dissolve 2.38 g of IPTG in 8 mL of molecular biology grade water. Bring to a final volume of 10 mL using molecular biology grade water. Filter through a 0.2 μm syringe filter. Dispense in 1 mL aliquots. Can be stored at −20°C for up to 1 year.
-
•
5 Molar hydrochloric acid (HCl).
Add 41.7 mL of 37% HCl to 58.3 mL of ddH2O. This can be stored at room temperature (∼22°C) for several months.
-
•
5 Molar sodium hydroxide (NaOH).
Dissolve 20 g of NaOH pellets to 80 mL of ddH2O. When cooled, bring to a final volume of 100 mL using ddH2O. This can be stored at room temperature (∼22°C) for several months.
-
•
1 Molar Tris-HCl buffer, pH 6.
Dissolve 121.14 g of Tris base in 700 mL of ddH2O. Adjust the pH to 6 using 5 M NaOH and 5 M HCl. Bring to a final volume of 1 L using ddH2O. Filter using a 0.22-μm vacuum filter unit. Can be stored at room temperature (∼22°C) for several months. The buffer is diluted fivefold for the dilution of iM-forming oligonucleotides.
-
•
1 Molar Tris-HCl buffer supplemented with 500 mM KCl, pH 7.2.
Dissolve 121.14 g of Tris base and 37.3 g of KCl in 700 mL of ddH2O. Adjust the pH to 7.2 using NaOH and HCl. Bring to a final volume of 1 L using ddH2O. Filter using a 0.22-μm vacuum filter unit. Can be stored at room temperature (∼22°C) for several months. The buffer is diluted fivefold for the dilution of G-quadruplex-forming oligonucleotides.
-
•
50 μg/mL NeutrAvidin solution.
Dissolve 10 mg NeutrAvidin in 1 mL molecular biology grade water (final concentration is 10 mg/mL). Dispense in 26 μL aliquots. Can be stored at −20°C for up to 2 years. For coating a 96-wells Nunc MaxiSorp plate, thaw one aliquot and dilute in 5.2 mL DPBS (final concentration is 50 μg/mL).
Step-by-step method details
Preparing iMbody plasmid
This section describes the initial steps for propagating and purifying the iMbody plasmid received from Addgene.
-
1.Propagation and purification of the plasmid.
-
a.Upon receiving the bacterial stab from Addgene, streak the bacteria on a LB-kanamycin agar plate and grow overnight (16–24 h) at 37°C while the plates are upside down to avoid condensation on the culture.Note: The Addgene stab culture can be stored at 4°C for up to two weeks.
-
b.Select five colonies to inoculate five separate 5 mL LB-kanamycin cultures and grow overnight (16–24 h) at 37°C while shaking at 230 revolutions per minute (RPM).
-
c.Purify the plasmids using a miniprep kit and store at −20°C.
-
a.
Making a glycerol stock
This section encompasses steps for transforming competent E. coli cells with pSANG10-3F-iMbody plasmids and creating a glycerol stock of iMbody. The glycerol stock serves as a starting point for subsequent steps in the protocol.
Note: The precise timing is contingent upon the sequencing result that verifies the sequence of iMbody.
-
2.Transforming BL21(DE3) competent E. coli cells with pSANG10-3F-iMbody plasmids prepared in Step 1.Note: The iMbody gene is controlled by a T7 promoter within the pSANG10-3F plasmid. BL21(DE3) cells are highly suitable for T7 expression, although other bacterial strains compatible with T7 expression may also be considered. However, optimizing the expression conditions may be necessary for other strains.
-
a.Add 50 ng of each purified plasmid to 25 μL of competent cells separately, and gently mix by flicking the tubes.
-
b.Incubate on ice for 30 min.
-
c.Perform a heat shock by incubating the tubes at 42°C for 30 s.
-
d.Incubate on ice for 5 min.
-
e.Add 250 μL LB broth (without kanamycin) to each tube and incubate at 37°C while shaking at 230 RPM for 1 h.
-
f.Spread each mixture on a prewarmed LB-kanamycin agar plate, let the plates air-dry on the bench and then grow overnight (16–24 h) at 37°C while the plates are upside down to avoid condensation on the culture.Note: Perform b-f for 25 μL untransformed competent cells as a control. If colonies are observed on the control plates, then repeat Step 2 using a fresh LB-kanamycin agar plate and make sure the concentration of kanamycin is correct.
-
a.
-
3.Making an iMbody overnight culture.
-
a.Using sterile pipette tips, pick one colony from each transformed BL21 plate (five plates in total) to inoculate five separate 10 mL LB-kanamycin cultures and grow overnight (16–24 h) at 37°C while shaking at 230 RPM.
-
b.Make a glycerol stock for each transformation by adding 250 μL glycerol to 1 mL overnight culture in a cryovial tube. Mix thoroughly and store at −80°C.
-
c.Purify the plasmids from remaining 9 mL overnight culture for each transformation using a miniprep kit and store at −20°C.
-
d.For each transformation, verify the sequence of iMbody by sequencing utilizing T7 promoter forward and T7 terminal reverse primers.
-
e.Proceed to Step 4 if the iMbody sequence is confirmed by the sequencing results; otherwise, discard unverified minipreps and repeat Steps 2 and 3. Refer to problem 1 in the troubleshooting section if the issue persists.
-
a.
Production of iMbody
This section outlines the process for iMbody expression using a verified glycerol stock, with a focus on a 1-L culture. The protocol can be adapted by adjusting reagent quantities for larger cultures.
-
4.iMbody expression.
-
a.Use a sterile pipette tip to get some bacteria from a verified iMbody glycerol stock and inoculate 50 mL of LB-kanamycin broth and grow overnight (16–24 h) at 37°C while shaking at 230 RPM.
-
b.Inoculate 990 mL of LB-kanamycin broth with 10 mL of the overnight culture in a 2-L Erlenmeyer flask.
-
c.Grow at 37°C while shaking at 230 RPM. Measure the optical density at 600 nm (OD600nm) initially after 1 h, and then every 10 min thereafter.
-
d.When OD600nm reaches to ∼ 0.4–0.5, induce the expression of iMbody by adding 1 mL of 1 M IPTG (final concentration of IPTG in the culture is 1 mM).
-
e.Continue growing at 30°C while shaking at 230 RPM for 24 h.Note: Reducing the temperature from 37°C to 30°C decreases the likelihood of iMbody aggregation in the culture.
-
f.Next day, reinduce the expression of iMbody by adding 0.5 mL of 1 M IPTG.
-
g.Continue growing at 30°C while shaking at 230 RPM for another 24 h.
-
h.Divide the culture over 3 × 500 mL polypropylene bottles and centrifuge at 10,000 × g for 30 min at 4°C.
-
i.Filter the supernatant using a vacuum filter system and a 0.22-μm vacuum filter unit. Discard the pellets.Note: The filtered supernatant can be stored at 4°C for up to one week before purification.
-
a.
Purification of iMbody
This section provides instructions for the His-tag purification of iMbody and the subsequent concentration of the purified sample.
-
5.Capturing His-tagged iMbody using Ni-NTA agarose resins.
-
a.Shake the Ni-NTA agarose resins container to mix it well.
-
b.Transfer 0.5 mL resins to a 15 mL tube.
-
c.Wash the resins twice with 10 mL DPBS.
-
d.Resuspend the resin pellet in 1 mL of DPBS and add it to the filtered supernatant.
-
e.Incubate for 1 h at 4°C on a roller at 40 RPM .
-
a.
-
6.Running the sample through a gravity column.
-
a.Prepare a gravity-flow chromatography column by running two column volume (CV) ddH2O and one CV DPBS through the column.
-
b.Flow the supernatant-Ni-NTA resins mixture through the column.
-
c.Collect the flow-through and store it at 4°C until running an SDS-PAGE gel.
-
d.Close the gravity-flow column and incubate the resins with 30 mL of NPI-20 buffer in the column for 10 min.
-
e.Open the column and collect the NPI-20 and add it to the flow-through.
-
a.
-
7.Elution of iMbody.
-
a.Close the gravity-flow column and incubate the resins with 5 mL of NPI-500 buffer in the column for 5 min.
-
b.Collect the eluted iMbody.
-
c.Measure the absorbance at 280 nm (A280nm).
-
d.Continue with elution in 5 mL fractions until the A280nm value drops below 0.02. Typically, around five fractions are sufficient to elute all the bound iMbody from 1 L of supernatant.
-
e.Combine all fractions in a 50 mL tube, measure the A280nm and keep the tube on ice.
-
a.
-
8.Regenerating the Ni-NTA agarose resins.
-
a.Close the gravity-flow column and incubate the resins with 20 mL of NPI-500 buffer in the column for 20 min.
-
b.Collect the NPI-500 buffer and dispose of it according to toxic material handling guidelines.
-
c.Incubate the resins with 30 mL of 0.5 M NaOH for 30 min in the gravity-column.
-
d.Flow-through the NaOH solution and wash the resins with 1 CV ddH2O.
-
e.Close the gravity-flow column and collect the resins using 10 mL of 20% ethanol.Note: Regenerated Ni-NTA agarose resins appear bluish. Use regenerated resins only for purifying His-tagged iMbody. The resin can be stored at 4°C for several months.
-
f.Rinse the gravity-flow column using ddH2O.
-
a.
-
9.Buffer exchange and concentration of purified iMbody.
-
a.Equilibrate an Amicon ultra-15 centrifugal filter unit (10 kDa cutoff) using 10 mL of DBPS supplemented with 10% glycerol (DPBS-glycerol).
-
b.Add 5 mL DPBS-glycerol to the Amicon tube, top it up with 10 mL of eluted iMbody sample, and centrifuge at 3000 × g and 4°C for 7 min.
-
c.Collect the flow-through, top up with more iMbody sample, and repeat the process until no iMbody sample remains.
-
d.Top up the remaining iMbody sample in the Amicon tube with DPBS-glycerol and centrifuge at 3000 × g and 4°C for 7 min.
-
e.Repeat Step 9-d until 50 mL of flow-through is achieved, ensuring the exchange of NPI-500 buffer with DPBS-glycerol.
-
f.Continue the centrifugation until the upper chamber of the Amicon tube contains between 0.5 mL to 1 mL of concentrated sample.
-
g.Equilibrate two Costar Spin-X centrifuge tubes with DPBS-glycerol.
-
h.Transfer the concentrated iMbody sample to Costar Spin-X centrifuge tubes and centrifuge for 1 min at 10000 × g and 4°C.
- i.
-
a.
-
10.Running an SDS-PAGE to check the purity of purified iMbody.
-
a.Add 2 μg iMbody, 4 μL NuPAGE LDS sample buffer, 2 μL NuPAGE sample reducing agent and up to 16 μL molecular biology grade water into a tube.
-
b.Heat the sample at 95°C for 5 min.
-
c.Load the sample on a NuPAGE 4%–12% Bis-Tris mini protein gel.
-
d.Load 7 μL prestained protein ladder in a separate well.
-
e.Perform electrophoresis at 200 V using MES-SDS running buffer for 30 min or until well-separated ladder bands appear on the gel.
-
f.Rinse the gel using ddH2O.
-
g.Stain the gel using 15 mL EZBlue gel staining reagent for 1 h at room temperature (∼ 22°C) with gentle orbital shaking.
-
h.Destain extensively using ddH2O overnight (16–24 h) or until the blue background is faded at room temperature (∼ 22°C) with gentle orbital shaking.
-
i.A blue band corresponding to 17.3 kDa (between 15 and 25 kDa) appears (Figure 2). See problem 2 in troubleshooting section if sample contains impurities.
-
j.After confirming the purity of iMbody through SDS-PAGE, dispense the purified iMbody sample into 50 μL aliquots and store them at −80°C.
-
a.
Note: Purified samples stored at −80°C remain stable for a minimum of one year, but it's advisable to prevent frequent freeze-thaw cycles. Upon thawing an aliquot, it should be filtered using a Costar Spin-X centrifuge tube, and the concentration should be determined following the instructions in Step 9. Purified samples can be stored in a refrigerator (2°C–8°C) for up to one week. We strongly recommend filtering the sample and assessing the iMbody concentration before each use.
Table 1.
Physical parameters of iMbody
| Extinction coefficient | Molecular weight | |
|---|---|---|
| iMbody | 34045 | 13158.60 |
| iMbody-6×His-3×FLAG | 38515 | 17306.87 |
Figure 2.

SDS-PAGE result for purified iMbody-6×His-3×FLAG (17.3 kDa)
Lane 1: iMbody purified from periplasm contains impurities.
Lane 2: iMbody purified from supernatant.
In each lane, 2 μg of purified sample was loaded.
Performing ELISA using iMbody
This section delineates the ELISA procedure utilizing iMbody for the characterization of a putative iM-forming oligonucleotide. By incorporating the positive and negative controls specified in this protocol, this method can serve as a complement to other techniques, such as circular dichroism spectroscopy, used in the characterization of iM-forming oligonucleotides.
-
11.Preparing DNA oligonucleotides.
-
a.Order 5′ Biotin-TEG conjugated DNA oligonucleotides as lyophilized samples.Note: Oligonucleotides used in this protocol are listed in Table 2.
-
b.Briefly centrifuge the tube before opening it to avoid losing samples that might be attached to the cap.
-
c.Reconstitute the oligonucleotide in nuclease-free water to make a 100 μM stock solution. This solution can then be further diluted using a preferred buffer.
-
d.Vortex thoroughly. If it is difficult to resuspend, heat the tube at 55°C for 10 min followed by vortexing and centrifugation.
-
e.Measure the concentration and purity of the stock solution. A ratio above 1.8 for Abs260nm/Abs280nm and Abs260nm/Abs230nm is generally considered as pure for DNA samples.
-
f.The stock solution of a DNA oligonucleotide can be stored at −80°C for at least 2 years.
-
a.
-
12.Annealing DNA oligonucleotides.
-
a.Dilute an iM-forming oligonucleotide in 200 mM Tris-HCl pH 6 to a final concentration of 70 nM. Prepare a 60 μL DNA solution for each well and ensure at least duplicates for each condition.
-
b.Anneal the oligonucleotide by heating at 95°C for 10 min followed by slowly cooling down to room temperature (∼ 22°C), and then keep it at 4°C overnight (16–24 h).
-
a.
Note: Control oligonucleotides should be diluted and annealed in appropriate buffers. For example, we dilute and anneal G-quadruplexes in 200 mM Tris-HCl pH 7.2 supplemented with 100 mM KCl and double-stranded DNA in IDT nuclease-free duplex buffer.
-
13.Immobilizing DNA oligonucleotides on a plate.
-
a.Coat a 96-well Nunc MaxiSorp plate by adding 50 μL per well of 50 μg/mL NeutrAvidin diluted in DPBS.
-
b.Shake the plate gently, seal it with a SealPlate film, and incubate overnight (16–24 h) at 4°C.
-
c.Aspirate the contents and wash one time with DPBS (250 μL per well). After washing, turn the plate upside down and gently tap on absorbent paper to eliminate any extra liquid.
-
d.Block the plate by adding 200 μL per well of DPBS supplemented with 2% BSA and 0.1% Tween 20 for 2 h at room temperature (∼ 22°C) while shaking at 300 RPM on a plate shaker or overnight (16–24 h) at 4°C.
-
e.Aspirate the blocking buffer and wash one time with 200 mM Tris-HCl pH 6 or appropriate buffers for the control oligonucleotides (250 μL per well). After washing, turn the plate upside down and gently tap on absorbent paper to eliminate any extra liquid.
-
f.Add 60 μL of annealed DNA samples to each well and incubate at room temperature (∼22°C) for 1 h while shaking at 300 RPM on a plate shaker.
-
g.Aspirate the contents and wash one time with DPBS (250 μL per well). After washing, turn the plate upside down and gently tap on absorbent paper to eliminate any extra liquid.
-
a.
-
14.Adding iMbody.
-
a.Thaw the required volume of iMbody, based on the concentration determined after His-tag purification and the number of samples in the planned ELISA experiment.
-
b.Filter using a Costar Spin-X centrifuge tube and determine the concentration again, following the instructions in Step 9.Note: The new concentration might be slightly different due to aggregation and loss of the purified iMbody caused by freeze-thawing.
-
c.Make a serial dilution of iMbody starting from 10 nM to 19.5 pM in DPBS supplemented with 5% BSA, 0.1% Tween 20 and 100 μg/mL salmon sperm DNA. Incubate at room temperature (∼22°C) for 15 min.
-
d.Add 60 μL of serially diluted iMbody per well and incubate at room temperature (∼22°C) for 45 min while shaking at 300 RPM on a plate shaker.
-
e.Aspirate the contents and wash three times with DPBS supplemented with 0.1% Tween 20 and once with DPBS (250 μL per well). After each washing, turn the plate upside down and gently tap on absorbent paper to eliminate any extra liquid.
-
a.
-
15.Adding secondary antibody.
-
a.Dilute the HRP-conjugated anti-FLAG antibody 1:15000 in DPBS supplemented with 2% BSA, 0.1% Tween 20. Incubate at room temperature (∼22°C) for 15 min.
-
b.Add 50 μL diluted HRP-conjugated anti-FLAG antibody per well and incubate at room temperature (∼22°C) for 45 min while shaking at 300 RPM on a plate shaker.
-
c.Aspirate the contents and wash twice with DPBS supplemented with 0.1% Tween 20 and once with DPBS (250 μL per well). After each washing, turn the plate upside down and gently tap on absorbent paper to eliminate any extra liquid.
-
a.
-
16.Developing the enzymatic reaction.
-
a.Add 50 μL of TMB substrate solution to each well and incubate at room temperature (∼22°C) with occasional gentle shaking until a dark blue color develops for the highest concentration of iMbody.
-
b.Stop the reaction by adding 50 μL of 0.5 M HCl per well.
-
c.Read the absorbance at 450 nm using a UV-vis plate reader within 10 min of adding HCl.
-
d.Make the graph in GraphPad Prism (Figure 3A). See problem 3 in troubleshooting section if significant cross-reactivity with negative controls is observed.
-
a.
Note: To explore potential competition between a small molecule and iMbody, the iM-forming oligonucleotide should be annealed in the presence of the small molecule. For instance, we anneal iM-Myc oligonucleotide with final concentrations of 5, 10, and 20 μg/mL SWNT-COOH (dispersed in DMF), which has been demonstrated to recognize iMs.19,20 Additionally, we include a control sample containing only DMF (Figure 3B). The rest of the protocol remains unchanged as described above.
Table 2.
List of oligonucleotides used in this study
| Name | Sequence (5′ end on the left) | Reference |
|---|---|---|
| iM-Myc | CCCCACCTTCCCCACCCTCCCCACCCTCCCC | Sun et al.9 |
| iM-Nano | CCCCCCCCTTTCCCCCCCC | Li et al.10 |
| iM-hTelo | CCCTAACCCTAACCCTAACCCT | Phan et al.11 |
| iM-1A83 | CCTTTCCTTTACCTTTCC | Han et al.12 |
| iM-RET | CCCCGCCCCGCCCCGCCCCTA | Guo et al.13 |
| G4-KIT2 | CGGGCGGGCGCTAGGGAGGGT | Kuryavyi et al.14 |
| G4-TBA | GGTTGGTGTGGTTGG | Schultze et al.15 |
| G4-2JSM | TAGGGTTAGGGTTAGGGTTAGGG | Phan et al.16 |
| G4-BCL2 | GGGCGCGGGAGGAATTGGGCGGG | Dai et al.17 |
| G4-Myc-46 | GGGAGGGGCGCTTATGGG GAGGGTGGGGAGGGTGG GGAAGGTGGGG |
Sun et al.9 |
| G4-1KF1 | AGGGTTAGGGTTAGGGTTAGGG | Parkinson et al.18 |
| DNA hairpin | AGTCAGTCAAAATGACTGACT | This paper |
| dsDNA | Sense: /5′BiotinTEG/GATCGTTAACTAGTTAC Antisense: GTAACTAGTTAACGATC |
This paper |
All oligonucleotides are 5′-Biotin-TEG conjugated.
Figure 3.

Using iMbody in ELISA
(A) ELISA represents the binding of 10 nM iMbody to four different iM-forming oligonucleotides and a range of controls.
(B) ELISA shows the binding of serially diluted iMbody to iM-Myc oligonucleotide, which was annealed in the presence of various concentrations of SWNT-COOH or DMF (used to disperse SWNT-COOH). The plotted values represent the mean ± standard error of two replicates, obtained through a serial dilution of iMbody starting from 10 nM to 19.5 pM. Curve fitting was conducted using the "One site - Specific binding" equation in GraphPad Prism. The oligonucleotide sequences are provided in Table 2.
Performing immunostaining using iMbody
This section provides a detailed procedure for visualizing iMs in HeLa cells using iMbody. This includes preparing the coverslips for the cell culture, seeding HeLa cells, and performing fixation and permeabilization to prepare them for immunostaining. The subsequent steps involve adding iMbody and appropriate secondary and tertiary antibodies, followed by mounting the coverslips on slides. Finally, the section outlines the microscopy and image acquisition process and provides guidance on post-imaging processing to visualize iMs within the cells.
Note: The precise timing varies depending on the cell growth rate. The mentioned timing starts from the seeding day of HeLa cells.
-
17.Preparing coverslips.
-
a.Acid treat glass coverslips (#1.5) by submerging them in 1 M HCl and incubating at room temperature (∼22°C) at least for 3 h.
-
b.Rinse the coverslips three times using ddH2O, followed by three rinses with pure ethanol.
-
c.Air dry the coverslips in a biosafety cabinet.
-
d.Place each coverslip into an individual well of a 12-well plate containing 1 mL of 0.001% poly-L-lysine solution diluted in molecular biology grade water. Allow them to incubate at room temperature (∼22°C) for 20 min.
-
e.Aspirate the poly-L-lysine solution. Rinse with 1 mL molecular biology grade water per well and air dry the coverslip in a biosafety cabinet.
-
f.Sterilize under the UV light in a biosafety cabinet for at least 1 h.
-
a.
-
18.Seeding HeLa cells.Note: Below is the protocol for the immunostaining of HeLa cells. The ideal iMbody concentration for immunostaining other cells might vary and should be established through a preliminary pilot experiment involving a serial dilution on synchronized cells.
-
a.Grow HeLa cells in a T75 cell culture flask using DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (complete media) until they reach 70%–80% confluency.
-
b.Rinse the cells twice with 15 mL of warm (∼37°C) DPBS.
-
c.Add 4 mL warm (∼37°C) trypsin-EDTA to the flask and incubate at 37°C for 5 min or until all cells are detached.
-
d.Add 8 mL warm (∼37°C) complete media to the flask to deactivate trypsin and collect the cells into a 15 mL tube.
-
e.Centrifuge for 1 min at 500 × g at room temperature (∼22°C).
-
f.Discard the supernatant and gently resuspend the cells with up to 1 mL complete media.
-
g.Count the number of cells using a hemocytometer. Keep the tube containing the remaining cells in a 37°C water bath.
-
h.Seed 1 × 104 cells resuspended in 1 mL complete media in each well that contains poly-L-lysine coated coverslip.
-
i.Incubate the cells at 37°C and 5% CO2 until they reach 70%–80% confluency, which may take 1–2 days after seeding 1 × 104 cells per well.
-
a.
-
19.Fixation and permeabilization of the cells.
-
a.Pre-fix the cells by adding 1 mL of 1% formaldehyde diluted in DPBS directly to the well containing media and incubating for 2 min at room temperature (∼22°C).CRITICAL: Formaldehyde is toxic. Work in a fume hood and wear suitable personal protective equipment while diluting 16% formaldehyde to 1% using DPBS. Avoid skin contact and inhalation. Prepare fresh 1% formaldehyde for each immunostaining.
-
b.Aspirate the content and rinse the cells once with 1 mL ice-cold DPBS per well.
-
c.Fix the cells by adding 1 mL 1% formaldehyde in each well and incubating for 15 min at 4°C without shaking.
-
d.Rinse the cells twice with 1 mL ice-cold DPBS per well.
-
e.Permeabilize the cells by adding 1 mL of ice-cold 0.1% Triton X-100 diluted in DPBS in each well and incubating for 15 min at 4°C without shaking.
-
f.Rinse the cells twice with 1 mL ice-cold DPBS per well.
-
g.Block the coverslip and cells with 1 mL DPBS supplemented with 2% BSA, 1% skim milk, 0.1% Tween 20 and 0.01% Triton X (blocking buffer) for 3 h at 4°C.
-
a.
-
20.Adding iMbody.
-
a.Thaw the required volume of iMbody, based on the concentration determined after His-tag purification and the number of samples planned for the immunostaining experiment.
-
b.Filter using a Costar Spin-X centrifuge tube and determine the concentration again, following the instructions in Step 9.Note: The new concentration might be slightly different due to aggregation and loss of the purified iMbody caused by freeze-thawing.
-
c.Prepare a serial dilution of iMbody ranging from 2 μM to 500 nM in the blocking buffer. For each well, 350 μL is required.Note: Always include control wells including no iMbody and no secondary antibody.
-
d.Aspirate the blocking buffer and dispense 350 μL per well of a serially diluted iMbody. Incubate overnight (16–20 h) at 4°C.
-
e.Next day, perform four washes on the cells using 1 mL of ice-cold DPBS supplemented with 0.1% Tween 20 per well. Each wash should last for 5 min and involve gentle shaking at 200 RPM on a plate shaker.
-
f.Rinse the cells with 1 mL per well of ice-cold DPBS.
-
a.
-
21.Adding secondary antibody.
-
a.Dispense 350 μL of rabbit anti-FLAG antibody (1:600 dilution) per well, after diluting in the blocking buffer.
-
b.Incubate at room temperature (∼22°C) for 30 min while shaking at 200 RPM on a plate shaker.
-
c.Perform two washes on the cells using 1 mL of ice-cold DPBS supplemented with 0.1% Tween 20 per well. Each wash should last for 5 min and involve shaking at 200 RPM on a plate shaker.
-
d.Rinse the cells with 1 mL per well of ice-cold DPBS.
-
a.
-
22.Adding tertiary antibody and phalloidin.
-
a.Dispense 350 μL of Alexa Fluor 488-conjugated anti-rabbit (1:400 dilution) and Alexa Fluor 555-conjugated phalloidin (1:20 dilution) per well, after diluting in the blocking buffer.
-
b.Cover the plate with aluminum foil and incubate at room temperature (∼22°C) for 30 min while shaking at 200 RPM on a plate shaker.
-
c.Perform two washes on the cells using 1 mL of ice-cold DPBS supplemented with 0.1% Tween 20 per well. Each wash should last for 5 min and involve shaking at 200 RPM on a plate shaker.
-
d.Rinse the cells twice with 1 mL per well of molecular biology grade water.
-
a.
-
23.Mounting the coverslips.
-
a.Carefully remove the coverslip from the plate and place it onto a paper towel, such as a Kimwipe, allowing it to air dry within a biosafety cabinet. Ensure that the side of the coverslip with attached cells does not come into contact with the paper towel.
-
b.Dispense 15 μL of ProLong Gold antifade reagent with DAPI onto the center of a slide.
-
c.Mount the coverslip onto the slide in a way that the side of the coverslip with attached cells faces downward and makes contact with the mounting reagent.
-
d.Place the slides within a cassette and store them in a dark location at room temperature (∼22°C) overnight (16–24 h).
-
e.Seal the edge of the coverslip with nail polish and transfer the slides to 4°C protected from light for a long-term storage. Staining will last for a few weeks.
-
a.
Note: Sealing is not required if the image acquisition takes place within one week after the staining process.
-
24.Microscopy and image acquisition.
-
a.Acquire images on a Leica TCS SP8 confocal microscope using a 63×/1.40 oil (HC PL APO CS2) objective or a similar confocal microscope with the following settings:
-
i.Use a 405 nm laser (laser power 3%) to excite and image DAPI (nucleus).
-
ii.Use a 488 nm laser (laser power 50%) to excite and image Alexa Fluor 488 (iMs).
-
iii.Use a 561 nm laser (laser power 5%) to excite and image Alexa Fluor 555 (actin filaments).
-
iv.Use HyD detector with counting mode.
-
v.Speed 400 Hz.
-
vi.Pinhole 1 airy unit.
-
vii.Sequential frame scanning.
-
viii.Frame accumulation 3.
-
i.
-
b.Identify the cell boundary using actin filaments and zoom in when investigating iMs within an individual cell.Note: Maintain a consistent frame size when examining the formation of iMs under various treatment conditions.
-
c.Identify the starting and ending points of a nucleus, then employ the “system optimized” to determine the required number of z-stacks and intervals.
-
a.
-
25.Post-imaging processing.Note: The following procedure outlines the generation of Figure 4 in this paper. Post-imaging processing is contingent upon the experimental settings and the research questions being addressed by the experiment.
-
a.Open the microscopy image in Fiji with following setting:
-
i.View stack with: Hyperstack.
-
ii.Dataset: Open files individually.
-
iii.Color options: Colorized, Autoscale.
-
iv.Split into separate windows: Split channels.
-
i.
-
b.Select a captured image.
-
c.Adjust the Brightness/Contrast:
-
i.Navigate to the brightest image in blue (nucleus) and red (F-actin) channels and choose “Auto” in the B&C pop up.
-
ii.Navigate to the brightest image in the green (iMs) channel and choose the “Auto” option in the B&C pop-up. Continue refining the “Minimum” and “Maximum” settings until distinct foci become visible and there is no background interference.Note: Maintain a consistent “Minimum” and “Maximum” setting in the green channel (iMs) for different concentrations of iMbody used in the immunostaining, as well as when examining the impact of various treatments or conditions on the iM formation.
-
i.
-
d.Smooth the channels (optional).
-
e.Merge the channels and create a composite while keeping the source images.
-
a.
Figure 4.

Visualization of i-Motifs in the nuclei of HeLa cells
(A) Confocal images of HeLa cells stained with different concentrations of iMbody. Maximum intensity projection of images is displayed. Nuclei are shown in blue (stained with DAPI), actin filaments (F-actin) are shown in red (stained with Alexa Fluor 555 phalloidin), and i-Motifs are shown in green (stained with FLAG-tagged iMbody, rabbit anti-FLAG secondary antibodies, and Alexa Fluor 488 anti-rabbit tertiary antibodies). The green channel in all images is smoothed using FIJI, and maximum and minimum displayed values are set to 100 and 70, respectively.
(B) Confocal images of a HeLa cell stained with 1 μM of iMbody. Six frames of a z-stack are displayed. i-Motifs are present in N2, N3 and N4 images. The green channel is smoothed using FIJI, and maximum and minimum displayed values are set to 100 and 70, respectively. The scale bars represent 10 μm. Images were captured using a Leica TCS SP8 confocal microscope.
Expected outcomes
This protocol is anticipated to produce approximately 350 μg of pure iMbody-6×His-3×FLAG from a 1-L culture when the pSANG10-3F-iMbody plasmid and BL21 (DE3) cells are utilized.
Limitations
Antibodies could potentially exhibit off-target interactions when utilized outside their optimal concentration range.21 Within the concentration range of iMbody and buffer conditions described in this protocol, we have not observed significant cross-reactivity with other tested biomolecules. Changes in the binding conditions might affect nucleic acid structures and the iMbody folding. Possible deviations could be made with this caveat in mind. Moreover, iMbody could potentially recognize partially folded or non-canonical iMs, as these structures might include the epitope to which iMbody binds.
The fixation, permeabilization and further treatment of cells prior to iMbody staining might affect iM structures. These steps should be done at 4°C to minimize artifacts. Comparisons involving treatments necessitating higher temperatures and distinct buffer conditions from those outlined in this protocol, either before or during iMbody staining, should be conducted alongside control samples subjected to identical conditions.
It is possible to explore the impact of a small molecule or a biomolecule on the in vivo formation of iMs through iMbody staining. However, we advise conducting a competition assay between iMbody and these molecules for iMs binding via ELISA or comparable techniques before immunostaining. In case of observed competition, as demonstrated with SWCN-COOH in this study (Figure 2B), the interpretation of immunostaining outcomes should be adjusted accordingly.
iMbody can be employed in a colocalization assay to examine the presence of endogenous proteins near iMs. In this case, it is crucial to verify the absence of undesired cross-reactivity among the primary, secondary, and tertiary antibodies used in the colocalization assay.
Troubleshooting
Problem 1
The sequence of iMbody is not verified by sequencing.
Potential solution
-
•
Repeat Step 1 and screen five additional colonies.
-
•
If the iMbody sequence remains unverifiable, report the plasmid issue to Addgene.
Problem 2
Purified iMbody sample contains impurities.
Potential solution
-
•
Increase the concentration of imidazole in NPI-20 buffer used in Step 6 from 20 mM to50 mM and incubate for 20 min.
-
•
Prepare NPI buffer with various imidazole concentrations (e.g., 300, 400, 500, 600 mM) and use them in small-scale productions to find an optimized concentration of imidazole for elution in Step 7.
Problem 3
iMbody significantly cross-reacts with negative controls.
Potential solution
-
•
Always include no iMbody and no antigen controls to rule out the potential for unintended cross-reactivity of the secondary antibody with the target or the plate.
-
•
Include additional and more stringent washings after iMbody incubation in Step 14.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marcel Dinger (marcel.dinger@sydney.edu.au).
Materials availability
Plasmids generated in this study have been deposited to Addgene (iMbody_pSANG10-3F, Plasmid #184496).
Data and code availability
This study did not generate/analyze datasets/code.
Acknowledgments
The authors would like to thank Katharina Gaus Light Microscopy Facility and MEDRIOS team at UNSW Sydney. This project was supported by a Cancer Institute NSW Fellowship (2022/ECF1421) awarded to M.Z., a Tour de Cure Early Career Research Grant (RSP-164-2020) awarded to B.A., and an ARC Discovery Grant (DP210103811) awarded to M.E.D. and C.K.
Author contributions
M.Z. and M.E.D. conceptualized the project. M.Z. generated iMbody and performed all the experiments. S.E.R., B.A., and A.G.R. assisted with the experiments and analyses. M.Z. wrote the manuscript. C.K. and M.E.D. supported and supervised the project and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Mahdi Zeraati, Email: mahdi.zeraati@sydney.edu.au.
Marcel E. Dinger, Email: marcel.dinger@sydney.edu.au.
References
- 1.Abou Assi H., Garavís M., González C., Damha M.J. i-Motif DNA: structural features and significance to cell biology. Nucleic Acids Res. 2018;46:8038–8056. doi: 10.1093/nar/gky735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong Y., Yang Z., Liu D. DNA Nanotechnology Based on i-Motif Structures. Acc. Chem. Res. 2014;47:1853–1860. doi: 10.1021/ar500073a. [DOI] [PubMed] [Google Scholar]
- 3.Zeraati M., Langley D.B., Schofield P., Moye A.L., Rouet R., Hughes W.E., Bryan T.M., Dinger M.E., Christ D. I-motif DNA structures are formed in the nuclei of human cells. Nat. Chem. 2018;10:631–637. doi: 10.1038/s41557-018-0046-3. [DOI] [PubMed] [Google Scholar]
- 4.Zanin I., Ruggiero E., Nicoletto G., Lago S., Maurizio I., Gallina I., Richter S.N. Genome-wide mapping of i-motifs reveals their association with transcription regulation in live human cells. Nucleic Acids Res. 2023;51:8309–8321. doi: 10.1093/nar/gkad626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moutel S., Bery N., Bernard V., Keller L., Lemesre E., de Marco A., Ligat L., Rain J.-C., Favre G., Olichon A., Perez F. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. Elife. 2016;5 doi: 10.7554/eLife.16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeraati M., Dinger M.E., Christ D. In: Antibody Engineering: Methods and Protocols Methods in Molecular Biology. Nevoltris D., Chames P., editors. Springer; 2018. Selection of Antibody Fragments Against Structured DNA by Phage Display; pp. 197–209. [DOI] [PubMed] [Google Scholar]
- 7.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Protein Identification and Analysis Tools in the ExPASy Server | SpringerLink. https://link.springer.com/protocol/10.1385/1-59259-584-7:531 [DOI] [PubMed]
- 9.Sun D., Hurley L.H. The Importance of Negative Superhelicity in Inducing the Formation of G-Quadruplex and i-Motif Structures in the c-Myc Promoter: Implications for Drug Targeting and Control of Gene Expression. J. Med. Chem. 2009;52:2863–2874. doi: 10.1021/jm900055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li T., Famulok M. I-Motif-Programmed Functionalization of DNA Nanocircles. J. Am. Chem. Soc. 2013;135:1593–1599. doi: 10.1021/ja3118224. [DOI] [PubMed] [Google Scholar]
- 11.Phan A.T., Guéron M., Leroy J.-L. The solution structure and internal motions of a fragment of the cytidine-rich strand of the human telomere11Edited by I. Tinoco. J. Mol. Biol. 2000;299:123–144. doi: 10.1006/jmbi.2000.3613. [DOI] [PubMed] [Google Scholar]
- 12.Han X., Leroy J.-L., Guéron M. An intramolecular i-motif: the solution structure and base-pair opening kinetics of d(5mCCT3CCT3ACCT3CC)11Edited by I. Tinoco. J. Mol. Biol. 1998;278:949–965. doi: 10.1006/jmbi.1998.1740. [DOI] [PubMed] [Google Scholar]
- 13.Guo K., Pourpak A., Beetz-Rogers K., Gokhale V., Sun D., Hurley L.H. Formation of Pseudosymmetrical G-Quadruplex and i-Motif Structures in the Proximal Promoter Region of the RET Oncogene. J. Am. Chem. Soc. 2007;129:10220–10228. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuryavyi V., Phan A.T., Patel D.J. Solution structures of all parallel-stranded monomeric and dimeric G-quadruplex scaffolds of the human c-kit2 promoter. Nucleic Acids Res. 2010;38:6757–6773. doi: 10.1093/nar/gkq558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schultze P., Macaya R.F., Feigon J. Three-dimensional Solution Structure of the Thrombin-binding DNA Aptamer d(GGTTGGTGTGGTTGG) J. Mol. Biol. 1994;235:1532–1547. doi: 10.1006/jmbi.1994.1105. [DOI] [PubMed] [Google Scholar]
- 16.Phan A.T., Kuryavyi V., Luu K.N., Patel D.J. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K + solution. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai J., Chen D., Jones R.A., Hurley L.H., Yang D. NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkinson G.N., Lee M.P.H., Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 19.Li X., Peng Y., Ren J., Qu X. Carboxyl-modified single-walled carbon nanotubes selectively induce human telomeric i-motif formation. Proc. Natl. Acad. Sci. USA. 2006;103:19658–19663. doi: 10.1073/pnas.0607245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y., Qu K., Zhao C., Wu L., Ren J., Wang J., Qu X. Insights into the biomedical effects of carboxylated single-wall carbon nanotubes on telomerase and telomeres. Nat. Commun. 2012;3:1074. doi: 10.1038/ncomms2091. [DOI] [PubMed] [Google Scholar]
- 21.Ausserwöger H., Schneider M.M., Herling T.W., Arosio P., Invernizzi G., Knowles T.P.J., Lorenzen N. Non-specificity as the sticky problem in therapeutic antibody development. Nat. Rev. Chem. 2022;6:844–861. doi: 10.1038/s41570-022-00438-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.