

Abstract
CASP15 added a new category of ligand prediction to promote the development of protein/RNA-ligand modeling methods, which have become important tools in modern drug discovery. A total of 22 targets were released, including 18 protein-ligand targets and 4 RNA-ligand targets. We applied our recently developed template-guided method to the protein-ligand complex structure predictions. The method combined a physicochemical, molecular docking method and a bioinformatics-based ligand similarity method. The Protein Data Bank was scanned for template structures containing the target protein, homologous proteins, or proteins sharing a similar fold with the target protein. The binding modes of the co-bound ligands in the template structures were used to guide the complex structure prediction for the target. The CASP assessment results show that the overall performance of our method was ranked second when the top predicted model was considered for each target. Here, we analyzed our predictions in detail, and discussed the challenges including protein conformational changes, large and flexible ligands, and multiple diverse ligands in a binding pocket.
Keywords: CASP, protein-ligand interactions, molecular docking, template-guided, ligand similarity, drug design
1. INTRODUCTION
Computational methods for predicting protein-ligand interactions play important roles in understanding many biological processes and developing therapeutic agents [1–4]. Such methods usually include the prediction of the three-dimensional (3D) protein-ligand complex structure (so-called the binding mode) and the interaction energy (referred to as the binding score). They can be broadly categorized into two groups, physicochemical methods (e.g., molecular docking) [5,6] and information-based methods (e.g., empirical fitting, statistical potential-based, machine learning and deep learning) [7–10]. Despite their great successes, the existing methods are far from perfect mainly due to the challenges of protein flexibility and binding free energy calculations.
In the last decade, several community-wide efforts have been devoted to accelerating the development of new strategies and improvement of the existing methods for the prediction of protein-ligand interactions. Two prominent events are Drug Design Data Resource (D3R, https://www.drugdesigndata.org, 2015-2019) [11–14] and Community Structure-Activity Resource (CSAR, http://www.csardock.org, 2010-2014) [15–18]. Both D3R and CSAR released valuable benchmarking datasets containing refined experimentally-determined protein-ligand complex structures and binding affinity data. During the CSAR and D3R challenges, we developed several strategies for predicting protein-ligand interactions [19–24], among which a template-guided strategy yielded good performance on protein-ligand binding mode predictions [21,22]. This template-guided method combined a physicochemical, molecular docking method and a bioinformatics-based ligand-similarity method. The latter benefits from abundant protein-ligand complex structures deposited in the Protein Data Bank (PDB) [25].
Very recently, a new modeling category called “protein-ligand complex modeling” was added to Round 15 of the community-wide experiment CASP (Critical Assessment of Structure Prediction), which was previously focused on protein structure modeling. The CASP15 ligand prediction category released a total of 22 targets, including 18 protein-ligand targets and 4 RNA-ligand targets. Most ligands in the CASP15 targets are endogenous, in contrast to the ligands in D3R and CSAR which are drug-like small molecules. Moreover, the CASP15 targets consist of a wide variety of proteins and ligands, while each round of D3R/CSAR usually focused on a few therapeutic protein targets with a few dozens to hundreds of ligand molecules.
During CASP15 protein-ligand modeling, we participated in predicting 17 out of 18 protein-ligand targets. We did not participate in T1118v1, because it was marked as “server/ligand” which we thought it belonged to a different category. We did not participate in the RNA-ligand targets, either. Our template-guided method performed well on the targets with templates. Out of the 17 submitted protein-ligand targets, the template-guided method was applied to 13 of them. When considering the top predicted model for each query ligand, our group ranked second out of more than 30 participating groups in the CASP evaluation. These results suggest that reliable template structures can significantly benefit most predictions, though templates could mislead the predictions are noticed in several cases. We noticed several problems and challenges in the prediction, including the importance of the conformation of the binding pocket in determining the protein-ligand complex structure. Large conformational changes in binding pockets remain a major challenge for protein-ligand complex structure predictions. Additionally, the presence of highly flexible ligands, multiple co-binding ligands in the binding pocket, and ions in the binding site make accurate prediction a daunting task. The detailed analysis is presented in the following sections.
2. METHODS
2.1. Construction of protein and ligand structures
For each protein-ligand target, CASP released the amino acid sequence(s) for the protein(s) and the SMILES strings for the ligand(s). To model each protein structure, AlphaFold v2.2.0 [26] was employed with the default setting and five models were generated. For the ligand, OMEGA2 (Version 3.0.1.2, OpenEye of Cadence Molecular Sciences, Santa Fe, NM, USA. http://www.eyesopen.com) [27, 28] was used to build its 3D structures. Up to 200 conformers were generated for a query ligand when the template-guided method was used for the protein-ligand complex structure prediction.
2.2. Template searching
The Protein Data Bank was searched for template structures containing the target protein or its homologs (sequence identity ≥ 30%). Specifically, web servers, Protein BLAST [29] or UVA FASTA [30], were employed with the protein sequence as the inputs. The searching database was set as PDB (Protein Data Bank proteins). Experimentally determined structures of the target protein or its homologies were downloaded from the PDB, and the structures containing co-bound ligands were kept.
When no structure with co-bound ligands was found in the above step, the program TM-align [31] was employed to search in the protein-ligand complex structure database for proteins sharing similar structures (even if the sequences differ significantly) with the target protein. This protein-ligand database collected all the protein structures containing at least one non-covalently bound ligand from the Protein Data Bank. For a CASP target, the structural similarity between the target protein and each protein in the protein-ligand database was evaluated by the TM-score, which ranges from 0 to 1. The two proteins are considered to have the same fold when the TM-score value is larger than 0.5, and distinct fold when the TM-score is less than 0.3. In this study, we focused on the protein structures with TM-score > 0.5 (normalized by the length of the target protein) and those protein structures containing co-bound ligands.
In summary, to search templates for a protein-ligand target, we first looked for experimental structures of the target protein containing co-bound ligands (different to the target ligand). If there was no template found in this step, we would use homologous proteins with co-bound ligands as the templates. Furthermore, if no template was found from homologous proteins, we would use the protein complex structures having similar protein structures but low sequence identities with the target protein as the templates.
2.3. Template-guided method
The template-guided method consists of three steps: superimposition, local refinement, and ranking. Details of each step are described as follows.
For a protein-ligand target, each conformer of the query ligand was superimposed onto the co-bound structure of the template ligand using SHAFTS (http://lilab-ecust.cn/chemmapper/download.html) [32]. SHAFTS calculates 3D similarity between two ligands using a hybrid method that combines molecular shape overlay and pharmacophore feature match. Its similarity score consists of a ShapeScore (shape-densities overlap) and a FeatureScore (pharmacophore feature fit values). Both scores are normalized to the range of [0, 1]. Therefore, the SHAFTS score, which is the sum of the ShapeScore and the FeatureScore, is scaled to [0, 2]. 0 means no similarity, and 2 means identical ligand. Next, the superimposed ligand conformers were ranked by their SHAFTS scores. The top ranked superimposed conformers in complex with the target protein were used as the input for the following local refinement step. Here, the target protein structure modeled by AlphaFold2 was matched to that of the template protein when the template was searched from homologous proteins or proteins with similar folds.
Then, the molecular docking program AutoDock Vina [33] was employed to refine the complex structures generated in the superimposition step. AutoDockTools was used to prepare the input files, which converts the PDB files to the PDBQT files. The proteins were treated as rigid bodies, and the ligands were treated to be fully flexible. For the refinement purpose, the option “local_only” was set for Vina, and other parameters were set to default values.
Finally, the refined protein-ligand complex structures were ranked by our hybrid scoring function, Hybrid_score = Vina_score + w*SHAFTS_score. Here, Vina_score is the protein-ligand binding score calculated by AutoDock Vina, and SHAFTS_score is the 3D molecular similarity between the query ligand and the template ligand calculated by SHAFTS. w is the weight to balance the contributions from the binding score and the ligand similarity score. Specifically, w = min(Vina_scores)/2, in which min(Vina_scores) was the minimum value of the Vina scores for the target protein and 2 is the maximum value of the SHAFTS scores. Here, Vina scores were obtained by docking the query ligand to the predefined binding site on the target protein using AutoDock Vina. During the docking process, the protein structure was considered rigid, and the ligand was considered fully flexible. Remaining parameters were set as default.
More details regarding our template-guided method and systemic evaluations of the method based on a protein-ligand dataset were described in References 21 and 22.
2.4. Molecular docking
When there was no template found for a protein-ligand target, the molecular docking program AutoDock Vina [33] was employed for protein-ligand binding mode prediction. First, the binding site information about the query ligand was searched from the homologous proteins in the Protein Data Bank. Local dockings were performed with AutoDock Vina using the default parameters if the binding sites were known. If the binding location was unknown, global docking was performed using AutoDock Vina by increasing the exhaustiveness value (e.g., from the default 8 to 100 independent runs). For both local and global docking, the protein structures were treated as rigid bodies, and the ligand structures were fully flexible.
2.5. Models for submission
The top 10 predicted complex structures either from the template-guided method or molecular docking were further inspected for electrostatic, polar, and nonpolar interactions. Finally, 5 models were selected and further optimized with a simple force-field minimization of the energy in Maestro, Version 12.9.137 (Schrödinger, LLC) [34]. The minimization process was carried out utilizing the OPLS_2005 force field. The minimization was considered complete when either the maximum change in the distance between atoms did not exceed 0.1Å or when a total of 75 iterations were performed. They were then submitted to CASP15.
3. RESULTS
During the CASP15 ligand prediction, we submitted modeled complex structures for 17 protein-ligand targets with a total of 33 ligands. The performance of our methods on these targets is reported in Table 1 based on the CASP evaluation results. Two criteria, lDDT_pli and RMSD, were used to compare a predicted protein-ligand binding mode with the complex structure determined by experimental methods. The Local Distance Difference Test (lDDT) is a superposition-free score that evaluates local distance differences of all atoms in a model [35]. Here, lDDT_pli considers only the contacts between the ligand and the protein (binding site radius: 4 Å; lDDT inclusion radius: 6 Å). lDDT_pli score values range from 0 to 1, with 0 corresponding to the lowest quality model and 1 corresponding to the highest quality model. For the RMSD (root-mean-square deviation), a predicted complex structure was superimposed onto the experimental structure based on the binding site residues (i.e., binding site superposition), and the ligand RMSD was then calculated between the predicted pose and the one in the experimental structure. More details regarding the calculations of the two criteria can be found on the CASP15 website (https://predictioncenter.org/casp15).
Table 1.
Performance of our modeling methods in CASP15 ligand predictions. The results were based on the top submitted model for each target.
| Target | Ligands | lDDT_pli | RMSD (Å) | Template |
|---|---|---|---|---|
| H1114 | 3NI/F3S/FCO/MG/MQ7 | 0.86/0.82/0.95/0.93/0.01 | 0.70/1.06/0.43/0.50/15.07 | 1e3d |
| H1135 | K | 0.99 | 0.06 | 6r15 |
| T1124 | SAH/TYR | 0.62/0.39 | 3.02/5.23 | 4a6e |
| T1127v2 | COA | 0.66 | 4.19 | 3bj7 |
| T1146 | NAG | 0.97 | 0.47 | 4q68 |
| T1152 | NAG | 0.54 | 5.06 | 4b8v |
| T1158v1 | XPG | 0.42 | 7.54 | - |
| T1158v2 | P2E | 0.39 | 6.40 | - |
| T1158v3 | XH0 | 0.00 | 21.11 | 6uy0 |
| T1158v4 | ATP/MG | 0.77/0.88 | 1.55/2.54 | 6uy0 |
| T1170 | ADP/AGS/MG | 0.90/0.86/0.92 | 0.88/1.12/1.27 | 6oax |
| H1171 | ADP/AGS/MG | 0.82/0.73/0.75 | 0.89/1.70/0.74 | 6oax |
| H1172 | ADP/AGS/MG | 0.82/0.74/0.97 | 1.82/1.63/1.19 | 6oax |
| T1181 | CA/OAA/ZN | 0.00/0.18/0.30 | 138.32/23.45/4.55 | - |
| T1186 | LIG | 0.62 | 3.9 | 6cyq |
| T1187 | NAG | 0.00 | 13.65 | - |
| T1188 | CD/CO/DW0 | 0.00/0.00/0.64 | 21.67/29.96/4.58 | 2ybt |
The following subsections provide the details of our modeled structures, including both successful and failed cases, and the discussions about what were learned from these studies.
3.1. Targets with templates
In our submitted models, a total of 24 query ligands in 13 targets were predicted by the template-guided method. PDB IDs of templates used for each target are reported in the last column of Table 1. When the top predicted model is considered for each query ligand, the mean and median values of lDDT_pli are 0.76 and 0.82, respectively. Using the RMSD as the criterion, the corresponding mean and median values are 2.74 and 1.41 Å, respectively. Slightly better performance was yielded when the top 5 models are considered for each target (see Table S1). Specifically, the mean and median lDDT_pli values increase to 0.80 and 0.86, respectively. The mean and median RMSD values decrease to 2.40 and 1.13 Å, respectively.
Figure 1 presents six examples of the predicted models compared to the experimentally determined structures. As shown in Fig. 1A, the target H1135 was to predict the binding modes of the K+ ion in the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex formed by SUN domain-containing protein 1 (SUN1) and a peptide of 25 amino acids. The sequence of SUN1 in the template 6r15 is identical to the one in the target, but the co-bound peptides in the two structures are different. The binding site of the K+ ion is located on SUN1. Our modeling results show that the binding modes of the K+ ion in the two structures (bound with two distant peptides) are very close (lDDT_pli = 0.99).
Figure 1.
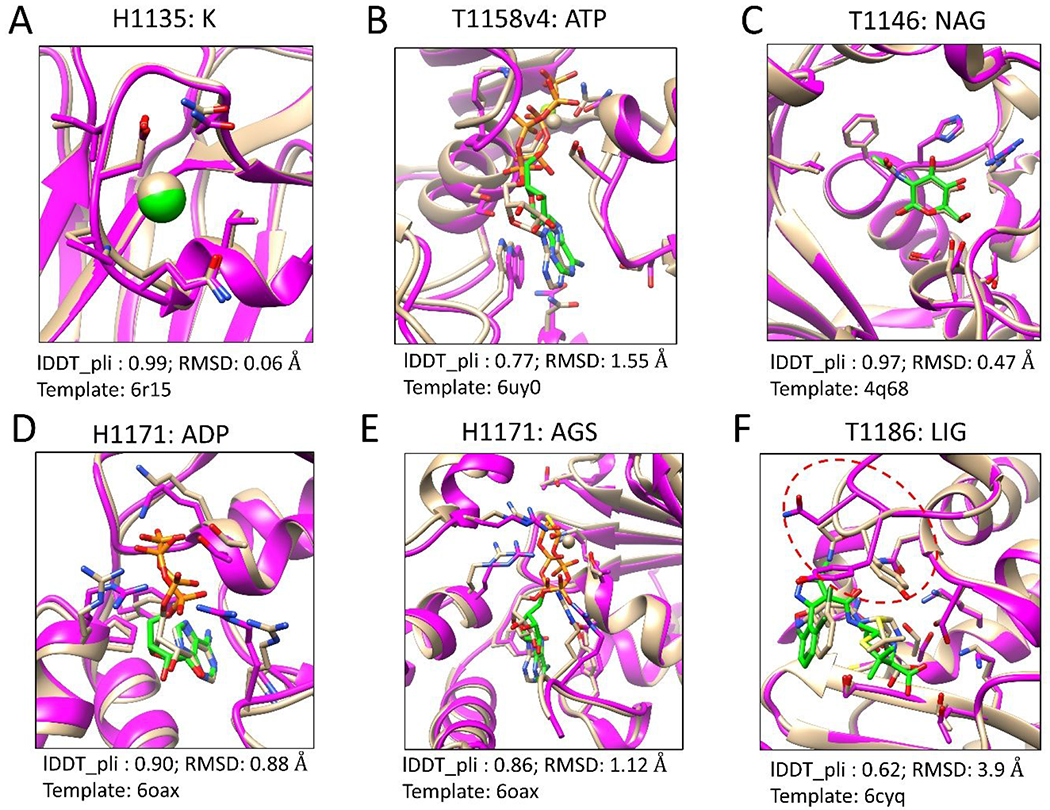
Six examples of the top modeled structures by our template-guided method. The experimentally determined structures are colored tan. The predicted protein structures are colored magenta. The ligands and their contact residues in the proteins are represented by the stick model. The carbons of the ligands in the predicted models are colored green. The figures were prepared using UCSF Chimera [36].
Fig. 1B shows the top predicted model of T1158v4 with ATP on the multidrug resistance-associated protein 4 (MRP4). The template 6uy0 is a complex structure of ATP and MRP1, which shares a sequence identity of 37% with MRP4. The lDDT_pli and RMSD values of the top model compared to experimental structure are 0.77 and 1.55 Å, respectively.
Fig. 1C displays the top model of T1146, which is a complex of NAG (2-acetamido-2-deoxy-beta-D-glucopyranose) and DUF1460 domain-containing protein. The complex structure in the template (PDB 4q68) contains the same ligand NAG and a homogeneous protein sharing a sequence identity of 30.4% with the target protein. The high-quality structure was modeled, whose lDDT_pli and RMSD are 0.97 and 0.47 Å, respectively.
Fig. 1D and E present the modeling results of two ligands, ADP and AGS, in the target H1171, respectively. The protein in the template structure (PDB 6oax) does not show significant sequence similarity with the target protein, but they share similar 3D structures with a TM-score of 0.66. Good performance was achieved for both ligands, ADP and AGS, with lDDT_pli values of 0.90 and 0.86, respectively. Comparable results were obtained for two related targets, T1170 and H1172.
Fig. 1F shows a predicted model of T1186, which is a complex structure of a small molecule and a class A beta-lactamase, CTX-M-14. The template structure (PDB 6cyq) contains the same protein as the target protein but a different co-bound ligand. The 3D similarity between the query ligand and the template ligand is 1.24 (SHAFTS score). The lDDT_pli and RMSD values of the top model compared to experimental structure are 0.62 and 3.9 Å, respectively. The relatively large RMSD value was mainly caused by the conformational changes of a flexible loop (red circle in Fig. 1F) near the binding pocket.
3.2. Misleading templates
The use of reliable templates can greatly improve the accuracy of protein structure predictions. However, there are situations where templates can be misleading, as in the case of target T1158v3, which involves the multidrug resistance protein 4 (MRP4). In this case, a homologous protein MRP1 (PDB 6uy0) was used as the template (PDB 6uy0) with a sequence identity of 37%. The co-bound ligand CLR in the template structure shares a similar 3D structure with the query ligand XH0 (SHAFTS score = 1.3), suggesting that they could have a similar binding site and pose.
However, the solved structure of MRP4 showed that the binding site of the query ligand is located in a different region than the selected template ligand CLR in MRP1 (as shown in Fig. 2). While CLR is located on the surface of transmembrane domains (TMDs) 1/2 in MRP1 (transmembrane region), the binding site of the query ligand XH0 in MRP4 is located between TMD1 and TMD2, which is the same location as the substrate binding site in MRP1. The complex structure of MRP1 and its substrate was revealed in PDB 5uja, which was overlooked due to the high structural similarity between CLR and the query ligand. The query ligand has more hydrophilic groups than CLR, which likely contributes to its location in the substrate binding pocket of MRP4, while CLR remains outside of the substrate binding site in MRP1 due to the hydrophobic environment of the membrane structure. Thus, it is important to consider not only structural similarity between templates and queries, but also protein function, surrounding environment, and ligand properties when predicting the binding sites.
Figure 2.
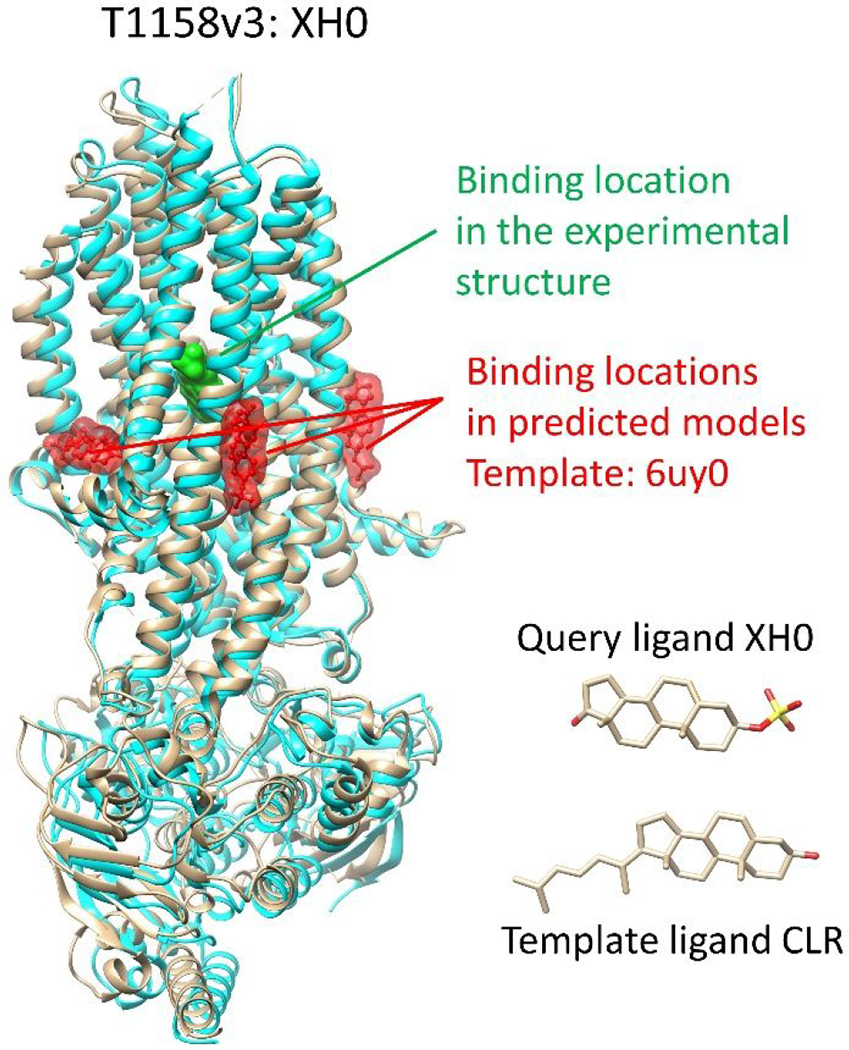
An example of misleading templates. The experimentally determined protein structure is colored tan. The protein structure in the predicted model is colored cyan. The 3D molecular similarity score (SHAFTS score) between the query ligand and the template ligand is 1.3.
3.3. Accuracy in the modeled protein structures
One of the challenges in predicting protein-ligand complex structures is the prediction of the conformation of the protein binding pocket. While AlphaFold shows outstanding performance in predicting protein structures, it remains challenging to accurately predict binding pocket conformations. One example is target T1181, which is a tail protein in bacteriophage G7C. The AlphaFold2 prediction of its structure was generally in agreement with the solved structure. However, the flexible regions of the protein, such as loops, were difficult to predict accurately due to their inherent flexibility and lack of structure. Unfortunately, some of these loops are important components of the binding site for the query ligand OAA, as shown in Fig. 3A. With the binding pocket conformation inaccurately predicted, accurate prediction of the ligand binding pose becomes extremely challenging. Another challenge for target T1181 is that the query ligand is large and highly flexible and that there is no template available for predicting its binding mode. Large and flexible ligands have large degrees of freedom, which means a huge conformational space needs to be sampled and evaluated in molecular docking. Due to the complexity of this target, no group submitted correct models.
Figure 3.
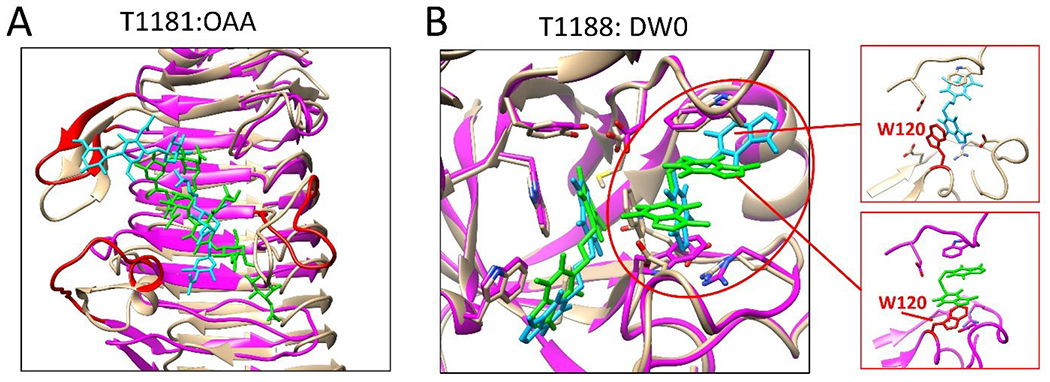
Two examples to demonstrate the importance of accurate protein structures to ligand binding mode prediction. Proteins in the experimental structures and in the predicted models are colored tan and magenta, respectively. Ligands in the experimental structures and in the predicted models are colored cyan and green, respectively. (A) shows the best modeled structure (model 5, lDDT_pli: 0.31, RMSD: 9.47 Å) of the query ligand OAA of T1181. Flexible loops involved in ligand binding are colored red. (B) shows a case in which the side chain conformations of the residues in the binding pocket can significantly affect the ligand binding mode prediction.
Another example that highlights the importance of an accurate binding pocket conformation in ligand binding mode prediction is target T1188, which is the chitinase in Clostridium perfringens. A template (PDB 2ybt) was identified using the TM-align with a TM-score of 0.56. The sequence identity and query coverage between the target and template protein were 27% and 63%, respectively. Additionally, the query ligand DW0 is included in the template’s complex structure. Although the key residues in the binding pocket of the template were conserved in the predicted target structure, the solved structure showed a conformational variation in residue W120 in the target, leading to a change in the binding mode of the query ligand DW0, as shown in Fig. 3B. The rotated side chain of W120 formed parallel stacked π–π interaction with both DW0 ligands in the binding pocket, while the corresponding W99 formed parallel stacked π–π interaction with only one ligand in the template structure. The conformational variation and the presence of an extra cadmium ion in the DW0 binding pocket of the query target compared to the template resulted in the difference in the binding pose between the query and template ligands.
3.4. Multiple diverse ligands in a binding pocket
Figure 4 shows two targets, T1124 and T1127v2, in which two diverse ligands bind in a protein pocket. Fig. 4A displays the binding modes of two ligands, SAH and TYR, on the target protein MfnG in the top submitted model. The binding modes were built based on the template 4a6e, which is the structure of N-acetylserotonin methyltransferase (ASMT) in complex with SAM and N-acetylserotonin. The sequence identity between the target protein and the template protein is 23%, while the TM-score is 0.76 (i.e., similar fold). The RMSD values of SAH and TYR are 3.02 and 5.23 Å, respectively, for which the binding site residues were superimposed when the RMSD between two ligands were calculated by CASP assessment team. The RMSD values of SAH and TYR reduced to 1.7 and 1.9 Å, respectively, when the superimposition was performed on the whole protein rather than on the binding site residues. By comparing the modeled protein structure with the experimental structure, the modeled structure shows a misalignment region (color red in Fig. 4A) near the ligand binding site. The backbone RMSD of the misalignment region in the two structures is 8.3 Å, which may explain why relatively large RMSD values were obtained in the CASP assessment results.
Figure 4.
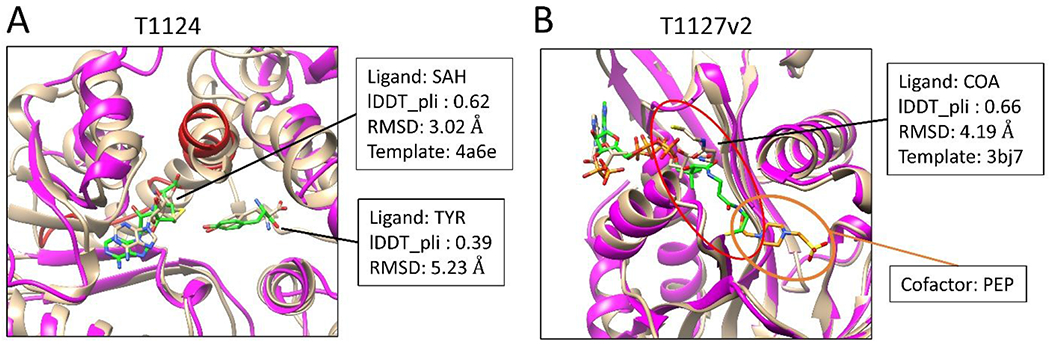
Targets with two diverse ligands binding in a protein pocket. The experimentally determined protein structures are colored tan. The predicted protein structures are colored magenta. The ligands are shown in stick representation. The carbons of the predicted ligands are colored green.
As another example, Fig. 4B presents a modeled structure of T1127v2. The sequence identity and structure similarity (TM-score) between the protein in the template (PDB 3bj7) and the one in the target are 26% and 0.68, respectively. The co-bound ligand, COA, in the template structure is identical to one of the co-bound ligands in the target structure. Differently, a cofactor, PEP, is observed in the binding pocket of COA in the experimental structure of T1127v2 (orange circle in Fig. 4B), while the template structure does not contain any cofactor. The lDDT_pli and RMSD values of the predicted binding mode of COA are 0.66 and 4.19 Å, respectively. The relatively large RMSD value is mainly contributed by the flexible tail of COA, which partially overlaps with the cofactor PEP in our modeled structure as we did not consider PEP in our modeling.
3.5. Other targets
In Table 1, large RMSD values are found for H1114_MQ7, T1158v3, T1181, T1187, and T1188. There are two major reasons for these failed cases, either because of using a low-quality protein structure or modeling with a wrong binding site. For the query ligand MQ7 in H1114, AlphaFold2 failed to model the co-bound protein structure. For the other four targets, the ligands were built on the protein sites that were far from those in the experimental structures.
4. DISCUSSION AND CONCLUSION
During CASP15 ligand prediction, a total of 18 protein-ligand targets were released. We submitted modeled structures for 17 of them and did not submit Target T1118v1 by error. Our template-guided method was applied to 24 query ligands in 13 targets. The assessment results show that template structures are useful for binding mode predictions. In addition to the templates containing homologous proteins of the target proteins, the templates with low sequence identity but similar structural fold can result in good performance in the protein-ligand binding mode prediction (e.g., T1170, H1171, and H1172). This is mainly due to the structure conservation of the target protein, especially for the residues in the binding site. Using complex structures with similar protein folds as templates would significantly expand the applications of our template-guided method.
On the other hand, template structures may also mislead predictions. For example, the query ligand XH0 was modeled on a wrong binding site on the target protein MRP4 in Target T1158v3 based on the template structure (PDB 6uy0). Another example is T1188, in which the query ligand DW0 shows distinct binding modes in the template structure and in the target structure.
In our method, protein structures were generated and treated as rigid bodies during modeling. However, protein conformations may change dramatically upon ligand binding, particularly when flexible loops are near the ligand binding site (e.g., T1181). It is also challenging to model binding modes for highly flexible ligands (e.g., T1158v1, T1158v2, T1181, and T1187). Other challenges include the binding mode prediction for multiple diverse ligands in a protein pocket (e.g., T1124 and T1127v2) and the binding site prediction for ions (e.g., T1181 and T1188).
In summary, our template-guided strategy achieved good performance during the CASP15 protein-ligand complex structure prediction. The method combines a physicochemical, molecular docking method and a bioinformatics-based ligand similarity method. The method benefits from the abundant protein-ligand complex structures deposited in the Protein Data Bank. However, sometimes the template structures may mislead the predictions. The challenges on protein-ligand structure prediction include protein conformational changes, large and flexible ligands, and multiple diverse ligands in a binding pocket. These issues point to the future research directions to improve our modeling methods.
Supplementary Material
Acknowledgement
Support to XZ from OpenEye of Cadence Molecular Sciences (Santa Fe, NM, http://www.eyesopen.com) is gratefully acknowledged. Professor Jianlin Cheng is also thankfully acknowledged for providing the AlphaFold2 modeling structure of T1158. This work was supported by NIH R35GM136409 (PI: XZ) and NIH R01 HL142301 (PI: Jonathan Silva). The computations were performed on the HPC resources supported by the University of Missouri Bioinformatics Consortium (UMBC).
References
- 1.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004; 3: 935–947. [DOI] [PubMed] [Google Scholar]
- 2.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Ann Rev Biophys Biomol Struct 2003; 32(1): 335–373. [DOI] [PubMed] [Google Scholar]
- 3.Grinter SZ, Zou X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules 2014; 19: 10150–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu X, Huang M, Zou X. Docking-based inverse virtual screening: methods, applications, and challenges. Biophys Rep 2018; 4: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang SY, Zou X. Advances and challenges in protein-ligand docking. Int J Mol Sci 2010;11(8): 3016–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys Chem Chem Phys 2014; 12: 12899–12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen DD, Gao K, Wang M, Wei GW. MathDL: mathematical deep learning for D3R Grand Challenge 4. J Comput Aided Mol Des 2020; 34: 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang C, Zhang Y. Delta Machine Learning to Improve Scoring-Ranking-Screening Performances of Protein–Ligand Scoring Functions. J Chem Inf Model 2022; 62: 2696–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isert C, Atz K, Schneider G. Structure-based drug design with geometric deep learning. Curr Opin Struct Biol 2023; 79: 102548. [DOI] [PubMed] [Google Scholar]
- 10.Kotelnikov S, Alekseenko A, Liu C, et al. Sampling and refinement protocols for template-based macrocycle docking: 2018 D3R Grand Challenge 4. J Comput Aided Mol Des 2020; 34: 179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parks CD, Gaieb Z, Chiu M, et al. D3R grand challenge 4: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des 2020; 34, 99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaieb Z, Parks CD, Chiu M, et al. D3R Grand Challenge 3: blind prediction of protein–ligand poses and affinity rankings. J Comput Aided Mol Des 2019; 33: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaieb Z, Liu S, Gathiaka S, et al. D3R Grand Challenge 2: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des 2-18; 32: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gathiaka S, Liu S, Chiu M, et al. D3R grand challenge 2015: evaluation of protein–ligand pose and affinity predictions. J Comput Aided Mol Des 2016; 30: 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlson HA, Smith RD, Damm-Ganamet KL, et al. CSAR 2014: a benchmark exercise using unpublished data from pharma. J Chem Inf Model 2016; 56:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith RD, Damm-Ganamet KL, Dunbar JB Jr, et al. CSAR benchmark exercise 2013: evaluation of results from a combined computational protein design, docking, and scoring/ranking challenge. J Chem Inf Model 2016; 56: 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damm-Ganamet KL, Smith RD, Dunbar JB Jr. et al. CSAR benchmark exercise 2011-2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model 2013; 53:1853–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith RD, Dunbar JB Jr, Ung PM, et al. CSAR benchmark exercise of 2010: combined evaluation across all submitted scoring functions. J Chem Inf Model 2011; 51: 2115–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Yan C, Zou X. Improving binding mode and binding affinity predictions of docking by ligand-based search of protein conformations: evaluation in D3R grand challenge 2015. J Comput Aided Mol Des 2017; 31: 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan R, Xu X, Zou X. (2018) Lessons learned from participating in D3R 2016 grand challenge 2: compounds targeting the farnesoid X receptor. J Comput Aided Mol Des 2018; 32: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Ma Z, Duan R, Zou X. Predicting protein–ligand binding modes for CELPP and GC3: workflows and insight. J Comput Aided Mol Des 2019; 33: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, Zou X. Dissimilar ligands bind in a similar fashion: a guide to ligand binding-mode prediction with application to CELPP studies. Int J Mol Sci 2021; 22: 12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan C, Grinter SZ, Merideth BR, Ma Z, Zou X. Iterative knowledge-based scoring functions derived from rigid and flexible decoy structures: evaluation with the 2013 and 2014 CSAR benchmarks. J Chem Inf Model 2016; 56: 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang SY, Zou X. Scoring and lessons learned with the CSAR benchmark using an improved iterative knowledge-based scoring function. J Chem Inf Model 2011; 51: 2097–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000; 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021; 596: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with omega: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J Chem Inf Model 2010; 50: 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawkins PC, Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model 2012; 52: 2919–2936 [DOI] [PubMed] [Google Scholar]
- 29.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform 2009; 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 1988; 85: 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 2005; 33: 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Jiang H, Li H. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J Chem Inf Model 2011; 51: 2372–2385. [DOI] [PubMed] [Google Scholar]
- 33.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrödinger Release 2021-3: Maestro, Schrödinger, LLC, New York, NY, 2021. [Google Scholar]
- 35.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013; 29: 2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, et al. (2004) UCSF chimera - a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.