

Abstract
E3 SUMO‐protein ligase CBX4 (CBX4), a key component of polycomb‐repressive complexes 1 (PRC1), has been reported to regulate a variety of genes implicated in tumor growth, metastasis, and angiogenesis. However, its role in T‐cell‐mediated antitumor immunity remains elusive. To shed light on this issue, we generated mice with T‐cell‐specific deletion of Cbx4. Tumor growth was increased in the knockout mice. Additionally, their tumor‐infiltrating lymphocytes exhibited impaired tumor necrosis factor‐alpha (TNF‐α) and interferon‐gamma (IFN‐γ) production, with an elevated programmed cell death protein 1 (PD‐1) level. In fact, dysregulated Pdcd1 expression was observed in all major subsets of peripheral T cells from the knockout mice, which was accompanied by a functional defect in response to T‐cell receptor (TCR) stimulation. In support of a direct link between CBX4 and PD‐1, Cbx4 overexpression resulted in the downregulation of Pdcd1 expression. Epigenetic analyses indicated that Cbx4 deficiency leads to diminished accumulation of inhibitory histone modifications at conserved region (CR)‐C and CR‐B sites of the Pdcd1 promoter, namely mono‐ubiquitinated histone H2A at lysine 119 (H2AK119ub1) and trimethylated histone H3 at lysine 27 (H3K27me3). Moreover, inhibition of either the E3 ligase activity of polycomb‐repressive complexes 1 (PRC1) or the methyltransferase activity of polycomb‐repressive complexes 2 (PRC2) restores Pdcd1 expression in Cbx4‐transfected cells. Cumulatively, this study reveals a novel function of CBX4 in the regulation of T‐cell function and expands our understanding of the epigenetic control of Pdcd1 expression.
Keywords: antitumor immunity, CBX4, H2AK119ub1, PD‐1, PRC1, T‐cell response
E3 SUMO‐protein ligase CBX4, a key component of polycomb‐repressive complexes 1, has been reported to regulate a variety of genes implicated in tumor growth, metastasis, and angiogenesis. We found a novel function of CBX4 in the regulation of histone modification, H2AK119ub1, at the Pdcd1 locus in activated T cells, affecting antitumor immunity. This finding helps expand our understanding of the epigenetic control of Pdcd1 expression.
Abbreviations
- AP‐1
activator protein‐1
- BrdU
5‐bromo‐2‐deoxyuridine
- CBX4
E3 SUMO‐protein ligase
- CCNE1
cyclin E1
- ChIP
chromatin IP
- CR‐B
conserved region ‐B
- CR‐C
conserved region ‐C
- CTLA‐4
cytotoxic T lymphocyte‐associated antigen‐4
- EED
embryonic ectoderm development
- EZH2
enhancer of Zeste homolog
- FoxO1
forkhead box O1
- GCN5
lysine Acetyltransferase 2A
- GzmB
granzyme B
- H2AK119ub1
mono‐ubiquitinated histone H2A at lysine 119
- H3K27me3
trimethylated histone H3 at lysine 27
- HDAC2
histone deacetylase 2
- HDAC3
histone deacetylase 3
- IFN‐γ
interferon gamma
- LAG‐3
lymphocyte‐activation gene 3
- NFATc1
nuclear factor of activated T cells 1
- PBS
phosphate‐buffered saline
- PD‐1
programmed cell death protein 1
- PRC1
polycomb‐repressive complexes 1
- PRC2
polycomb‐repressive complexes 2
- SIM
SUMO‐interacting motifs
- SUZ12
suppressor of Zeste 12 Homolog
- TCR
T‐cell receptor
- TIM‐3
T‐cell immunoglobulin domain and mucin domain‐3
- TNF‐ɑ
tumor necrosis factor‐alpha
- ZAP‐70
tyrosine‐protein kinase ZAP‐70
1. Introduction
Polycomb group proteins are crucial transcriptional repressors that mediate epigenetic gene silencing [1, 2]. They are organized into two forms of polycomb‐repressive complexes (PRC), PRC1, and PRC2. While PRC1 acts to monoubiquinate lysine 119 of histone 2A (H2AK119ub1), PRC2 mainly targets histone H3 lysine 27 for mono‐, di‐, and trimethylation. Despite the functional distinction, these two complexes are tightly interconnected and show remarkable synergism in transcriptional repression [3, 4, 5, 6]. The polycomb chromobox (CBX) family, including CBX2, CBX4, CBX6, CBX7, and CBX8, are important components of the PRC1 complex [7]. Much effort has been devoted to dissecting their role in tumorigenesis. In a context‐dependent manner, they act as either oncogenes or tumor suppressors. CBX7, for example, shows an antitumor activity in lung cancer through interacting with histone deacetylase 2 (HDAC2) and inhibiting CCNE1 expression [8]. In gastric cancer and lymphoma, however, it primarily exerts a tumor‐promoting activity, most likely by repressing the transcription of the INK4a/ARF locus [9, 10, 11]. Similarly, CBX4 plays opposite roles in colon cancer versus osteosarcoma. While inhibiting the metastasis of colon cancer by recruiting histone deacetylase 3 (HDAC3) to the Runx2 promoter [12], CBX4 promotes the metastasis of osteosarcoma by recruiting GCN5, which catalyzes H3K27 acetylation of the Runx2 promoter [13]. Of the CBX family members, CBX4 is of particular interest in that, in addition to the canonical polycomb function, it possesses two SUMO‐interacting motifs (SIM) and has the capacity to catalyze the sumoylation of a variety of substrates, such as CtBP [14], Dnmt3a [15], and BMI‐1 [16]. This SUMO E3 ligase activity has been demonstrated to be critical for DNA damage response [16] and epidermal stem cell activation and differentiation [17].
Adaptive immune responses, mainly mediated by T cells, have been shown to play a vital role in the control of tumor development and progression [18, 19, 20, 21]. Tumor antigens arising from somatic mutation or aberrant expression are captured and processed by antigen‐presenting cells. Peptides derived from these antigens are then displayed bound to class I or II MHC molecules for recognition by CD8+ and CD4+ T cells, respectively. As the principal component of immune protection against tumors, activated CD8+ T cells directly lysed tumor cells. CD4+ T cells, on the contrary, secrete an array of cytokines, which promote the generation and effector functions of CD8+ T cells. Notably, immune responses frequently fail to control tumor growth as tumors develop the capacity to evade antitumor immunity [21, 22, 23]. One of the best‐studied mechanisms is the inhibition mediated by programmed cell death protein 1 (PD‐1) on TCR signals. Physiologically, after stimulation, phosphorylation of Lck and ZAP‐70 will initiate the TCR signaling cascade. PD‐1 signals directly targeting on the phosphorylation of Lck and ZAP‐70 are normally involved in the fine‐tuning of T‐cell functions and prevention of their over‐activation [24, 25, 26, 27]. This pathway, however, is hijacked by tumors. High levels of PD‐1 expression are often detected in tumor‐infiltrating T cells and these tumor‐associated PD‐1+ T cells are functionally impaired [28]. Blocking this inhibitory pathway, on the contrary, leads to the development of strong T‐cell responses against a variety of tumors [29, 30, 31, 32, 33].
Previous studies of CBX proteins in tumorigenesis have been focused on their role in the regulation of malignant behaviors of tumor cells. Their impact on antitumor immunity is largely ignored. Li et al. [34] have reported that CBX7 promotes activation‐induced cell death of CD4+ T cells by regulating DNA demethylation of the FasL promoter. In addition, data from our group indicate that CBX4 are absolutely required for thymic organogenesis. Loss of Cbx4 impairs the formation and maintenance of the thymic epithelium, leading to the disruption of T‐lymphopoiesis [35]. In view of the vital role of T‐cell‐mediated immunity in the protection against tumors, here we explored the regulatory role of CBX4 in T‐cell function and antitumor immunity. In a murine model of colorectal carcinoma, T‐cell‐specific deficiency of Cbx4 resulted in accelerated tumor growth. T cells from the knockout mice were defective in the response to TCR stimulation, most likely due to dysregulated Pdcd1 expression. We further showed that, by promoting the accumulation of H2AK119ub1 and H3K27me3 histone modifications around the Pdcd1 promoter, CBX4 served as an epigenetic repressor for Pdcd1 expression.
2. Materials and methods
2.1. Experimental animals
Cbx4 flox/flox mice were gifted by Guoliang Xu at the Shanghai Institutes of Biological Sciences. CD4‐Cre mice were purchased from Jackson Lab. Mice are kept in the specific‐pathogen‐free animal facility of the Peking University Health Science Center. To generate mice with selective Cbx4 deficiency in T cells, Cbx4 flox/flox mice were crossed with CD4‐Cre mice, both on C57BL/6 background. Cbx4 flox/floxCD4‐Cre (called ‘knockout’ here) and the Cbx4 flox/flox (called ‘wild‐type’ here) littermates were used at the age of 10–12 weeks (divided by sex), both sexes were used in the experiments. The experimental procedures on use and care of animals were approved by the Ethics Committee of Peking University Health Science Center.
2.2. Cell lines
The cell lines MC38 (RRID: CVCL_0A68), EL‐4 (RRID: CVCL_0255), and Jurkat (RRID: CVCL_0065) were used in this study and have been authenticated within the past 3 years. The cell lines of EL‐4 (RRID: CVCL_0255) and Jurkat (RRID: CVCL_0065) were purchased from ATCC (Rockefeller, MD, USA; https://www.atcc.org). The cell line of MC38 (RRID: CVCL_0A68) was purchased from BTCC (http://www.btcccell.com/). Authentication was conducted using the widely accepted method of short tandem repeat (STR) profiling to ensure the integrity and reliability of the cell lines. We confirm that all experiments conducted in this study were performed using mycoplasma‐free cells. To verify the absence of mycoplasma contamination, regular screenings were performed using polymerase chain reaction (PCR) tests specifically designed for detecting the presence of mycoplasma DNA. These screenings were conducted by a reputable laboratory with expertise in mycoplasma detection. Cells were cultured in RPMI medium 1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum at 37 °C, 5% CO2.
2.3. Reagents and antibodies
Purified anti‐CD3 and anti‐CD28 antibodies, and fluorochrome‐conjugated antibodies against PD‐1, LAG‐3, CTLA‐4, and TIM‐3 were purchased from BioLegend (San Diego, CA, USA); fluorochrome‐conjugated antibodies against CD3, CD4, CD8, CD19, TCR‐β, CD44, CD62L, CD69, CD25, and Ki‐67, and Annexin V‐7AAD apoptosis staining kit were purchased from BD Corporation (Franklin, NJ, USA); fluorochrome‐conjugated antibodies against Foxp3, IL‐2, IL‐4, IL‐17, GzmB, IFN‐γ, TNF‐α, CD45, and human PD‐1 were purchased from eBioscience (San Diego, CA, USA). The anti‐CBX4 antibody and anti‐β‐actin antibody were purchased from Proteintech (Wuhan, China). Antibodies against EED, SUZ12, and EZH2 were purchased from Cell Signaling Technology (Danvers, MA, USA). IL‐2, IL‐4, IL‐6, IL‐12, and TGF‐β1 were purchased from R&D Systems (Minneapolis, MN, USA). PRT4165 and GSK126 were purchased from Selleck (Houston, TX, USA). Human CBX4 (NM_003655) cDNA plasmid, and mouse CBX4 (NM_007625) cDNA plasmid were purchased from YouBio (Changsha, China).
2.4. Flow cytometry
Single‐cell suspension from the spleen, lymph nodes, or tumor tissues was incubated with the appropriate antibodies for 30 min, washed, and resuspended with PBS. Flow cytometry analysis was performed on a FACS Canto Flow Cytometer (BD Corporation) and analyzed with kaluza software (Beckman Coulter, Brea, CA, USA).
2.5. T‐cell sorting and culture
CD3+ T cells were purified from lymph nodes using CD3ε MicroBead Kit (Miltenyi Biotec, Cologne, Germany) according to the manufacturer's instructions. To isolate naïve CD4+ or CD8+ cell, single‐cell suspension from lymph nodes was stained with fluorochrome‐conjugated antibodies for 30 min at 4 °C, washed twice, and resuspended at a density of 1 × 108 cells·mL−1. CD62L+CD44lowcells (naive T cells), CD62L−CD44high (effector memory T cells), and CD62L+CD44high (central memory T cells) were sorted using BD FACS Aria II. T cells were then placed onto anti‐CD3‐coated 96‐well flat‐bottom plates at a density of 1 × 105 cells/well with the addition of 1 μg·mL−1 anti‐CD28. In polarization cultures, cytokines and cytokine antibodies were added as follows: Th1 (10 ng·mL−1 IL‐12 and 2 ng·mL−1 IL‐2); Th2 (20 ng·mL−1 IL‐4, 2 ng·mL−1 IL‐2 and 10 μg·mL−1 anti‐IFN‐γ); Th17 (10 ng·mL−1 TGF‐β1, 10 μg·mL−1 IL‐6, 10 μg·mL−1 anti‐IFN‐γ, and 10 μg·mL−1 anti‐IL‐4); and Treg (1 ng·mL−1 TGF‐β1 and 10 ng·mL−1 IL‐2).
2.6. CD45RBhi adoptive transfer colitis model
Half a million FACS‐purified CD4+CD45RBhi naïve T cells were intravenously injected into Rag1 −/− mice. The receiving mice were monitored and weighed each week. At Week 7, the mice were sacrificed and measured for colon length. Colon tissues were then fixed in Bouin's fixative solutions. Hematoxylin and eosin staining was performed on paraffin sections of the colon.
2.7. Tumor model
MC38 cells were cultured in the DMEM medium (Gibco, Grand Island, NY, USA) and harvested in the logarithmic phase. One million cells were subcutaneously injected into the flanks of the mice. Tumor growth was measured daily or every other day over a period of 20 days using a caliper, and the tumor volume was calculated using the following formula: Tumor volume = length × width2/2. For the anti‐PD‐1 therapy, tumor‐bearing mice were inoculated intraperitoneally with anti‐PD‐1 (RMP1‐14, 200 μg per mouse, dissolved in PBS) or PBS on Days 8, 11, and 14. CD3+ T cells were purified from draining lymph nodes using CD3ε MicroBead Kit (Miltenyi Biotec) according to the manufacturer's instructions.
2.8. BrdU incorporation
Cells were incubated in medium containing 0.1 mm 5‐bromo‐2‐deoxyuridine (BrdU) for 1 h. BrdU incorporation was detected by flow cytometry following staining with BrdU staining kit (eBioscience) according to manufacturers' instructions.
2.9. Quantitative RT‐PCR
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA) and chloroform, and cDNA was generated using the Rever TraAce™ qPCR RT Master Mix (Toyobo, Osaka, Japan) according to the manufacturer's instructions. Quantification of mRNA was conducted by qPCR using KOD SYBR® qPCR Mix (Toyobo) with primers listed in Table S1. Reactions were performed on the Eco TM Real‐time PCR system. Results were normalized to the expression of the housekeeping gene β‐actin.
2.10. Chromatin immunoprecipitation for histone modification analysis
Chromatin immunoprecipitation assay was conducted with 1 × 107 cells using Chromatin IP (ChIP) Assay Kit (Merck KGaA, Darmstadt, Germany) according to the manufacturer's instructions. In brief, T cells were fixed in 1% formaldehyde. Chromatin was sonicated to produce fragments of 200–1000 bp long. After preclearing by incubation with Protein A Agarose/Salmon Sperm DNA (Merck KGaA), fragmented chromatin was incubated with antibody‐bound beads. Antibodies used were anti‐H3K27me3, anti‐H2AK119ub1, or the isotype control rabbit IgG (Biodragon, Suzhou, China). After washing, DNA was eluted and cross‐linking was reversed before DNA was purified using the PCR purification kit according to the manufacturer's instructions (Tiangen, Beijing, China). Eluted DNA was quantified using qPCR. Primer sequences for ChIP‐qPCR are listed in Table S2. Fold enrichment over background was calculated using values normalized to input.
2.11. Western blot analysis
T cells were lysed in a cell lysis buffer containing a cocktail of protease inhibitors. Proteins were separated on SDS/PAGE gel and transferred onto a PVDF membrane. Subsequently, the PVDF membranes were probed by incubation with primary antibodies, followed by incubation with horseradish peroxidase (HRP)‐conjugated secondary antibodies. The protein bands were visualized with a SuperSignal WestPico Kit (Thermo Fisher) according to the manufacturer's instructions. Antibodies against EED (85322S; clone: E4L6E; antibody ratio 1 : 1000), SUZ12 (3737S; clone: D39F6; antibody ratio 1 : 1000), EZH2 (5246S; D2C9; antibody ratio 1 : 1000), anti‐H3K27me3 (9733S; clone: C36B11; antibody ratio 1 : 5000), and anti‐H2AK119ub1 (8240S; clone: D27C4; antibody ratio 1 : 5000) were purchased from Cell Signaling Technology.
2.12. Statistical analysis
Statistical analyses were performed with graphpad prism 5. Data are presented as the mean ± standard deviation (SEM). Two‐tailed Student's t‐test or paired t‐test were used for single comparison. In all tests, P < 0.05 were considered statistically significant.
2.13. Ethics statement
This study was carried out in accordance with the recommendations of and the approval (protocol No. LA2018106) by the Ethics Committee of Peking University Health Science Center.
3. Results
3.1. T‐cell‐specific deletion of Cbx4 impairs antitumor immunity
To explore the potential role of CBX4 in T‐cell‐mediated antitumor immunity, we generated T‐cell‐specific Cbx4‐deficient mice by crossbreeding mice with loxP‐flanked Cbx4 alleles (Cbx4 fl/fl) and CD4Cre transgenic mice and the efficiency and specificity of the deletion were confirmed (Fig. S1A). CD4‐specific depletion of Cbx4 leads to its depletion not only in CD4+ T cells but also CD8+ T cells as a likely consequence of the co‐expression of CD4 and CD8 markers in DP cells, precursors of the CD4+ or CD8+ (SP) thymocytes. Flow cytometric analysis indicated that T‐cell‐specific deletion had no significant effect on thymocyte development (Fig. S1B) and the peripheral T‐cell compartment (Fig. S1C). The KO mice and the wild‐type (WT) littermates were subcutaneously inoculated with MC38 tumor cells. As measured by volume and weight, tumors in KO mice showed accelerated growth (Fig. 1A–C). At the end of observation, tumor‐infiltrating lymphocytes (TIL) were isolated and subjected to flow cytometric analysis. As shown in Fig. 1D, T cells from tumors in KO mice produced significantly lower levels of effector cytokines, such as TNF‐α and IFN‐γ (Fig. 1D). On the contrary, comparable Ki‐67 staining was observed for both CD4+ and CD8+ T cells from WT and KO mice as well as frequency of Foxp3+CD4+ cells (Fig. 1E,F), excluding the possibility of altered proliferation or Tregs recruitment. We next examined the expression of immune checkpoint inhibitors. In the absence of Cbx4, increased percentages of PD‐1+ cells were found among CD8+ and CD4+ T cells in tumor tissues (Fig. 1G) and from draining lymph nodes (Fig. 1H), whereas no difference was detected for LAG3 (Fig. S2). Collectively, these results indicate that Cbx4 deficiency in T cells impairs antitumor immunity. To further clarify that the effect of CBX4 on T cells is through the regulation of PD‐1 in tumors, we performed anti‐PD‐1 checkpoint blockade assay to treat subcutaneous engraftment in WT and CBX4 KO mice. As indicated, PD‐1 antibody therapy resulted in significant tumor suppression both in WT and KO mice. Notably, anti‐PD‐1 therapy diminished the difference in tumor growth between WT and CBX4 KO mice (Fig. 1I,J), suggesting that the accelerated tumor development due to Cbx4 deficiency is mainly determined by their enhanced PD‐1 expression.
Fig. 1.
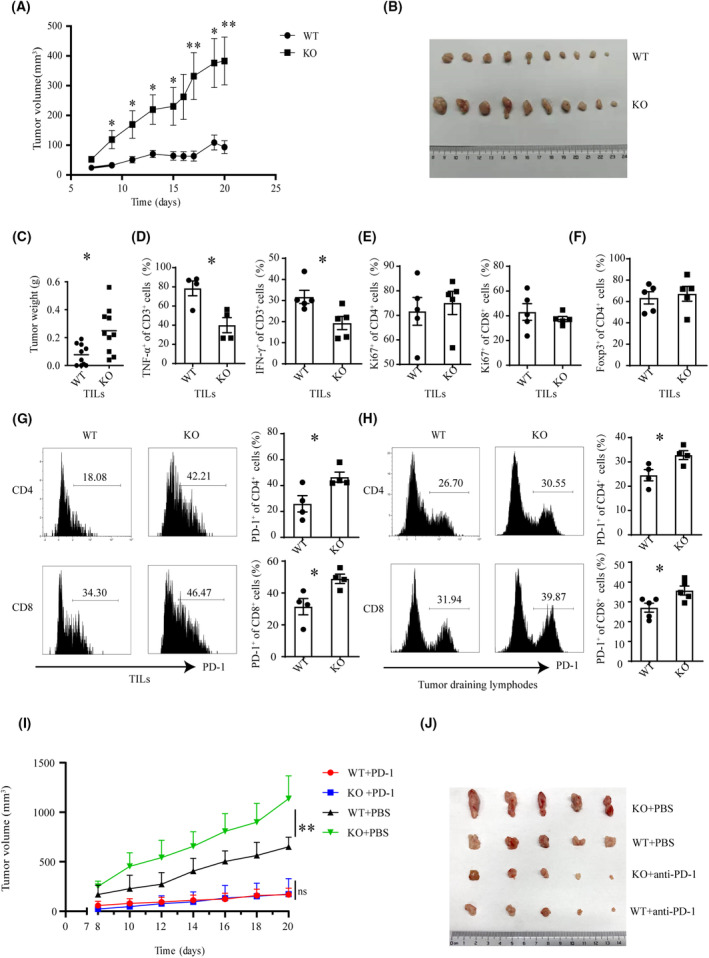
Suppressed antitumor immunity following T‐cell‐specific deletion of Cbx4. WT (wild‐type) and Cbx4 KO (knockout) mice were subcutaneously injected with MC38 cells (1 × 106 cells per mouse). Tumor volume was monitored over a period of 20 days. Tumors were removed at the end of experiment and weighted. Tumor‐infiltrating lymphocytes were isolated and analyzed for the expression of surface markers, intracellular cytokines, Foxp3, and Ki‐67 using flow cytometry. (A) Tumor volume over the course (n = 10). (B) Tumor size (scale bar = 1 cm, n = 10). (C) Tumor weight (n = 10). (D) The percentage of TNF‐α+ and IFN‐γ+ cells in CD3+ T cells (n = 4). (E) The percentage of Ki‐67+ cells among CD4+ or CD8+ T cells (n = 5). (F) The percentage of Foxp3+ cells in CD4+ T cells (n = 5). (G) The percentage of PD‐1+ cells among CD4+ or CD8+ T cells (n = 4). The horizontal axis represents fluorescence intensity. (H) Tumor‐draining lymph nodes were removed. Flow cytometry was performed to examine PD‐1 expression in CD4+ or CD8+ T cells (n = 4). The horizontal axis represents fluorescence intensity. Data are presented as Mean ± SE. Unpaired t‐test was used for comparison. *P < 0.05, **P < 0.01. (I and J) WT and CBX4 KO mice were subcutaneously injected with MC38 cells. Tumor growth was monitored over a period of 20 days. Mice were intraperitoneally injected with PBS or anti–PD‐1 (RMP1‐14, 200 μg per mouse, dissolved in PBS) every 3 days (three times in total) 8 days after MC38 inoculation and tumor sizes were recorded every day afterward (scale bar = 1 cm, n = 5), **P < 0.01, two‐way ANOVA, Statistical data are presented as Mean ± SE.
3.2. Loss of Cbx4 leads to upregulation of PD‐1
The elevated levels of PD‐1 in tumor‐infiltrating T cells prompted us to scrutinize the regulatory effect of CBX4 on the expression of PD‐1 and other known negative regulators in resting and activated T cells. We sorted out the naïve T cells as Fig. S3A and examined whether Cbx4 deficiency had impact on Pdcd1 mRNA transcription. Our results showed that Cbx4 KO naïve T cells had increased basal levels of Pdcd1 mRNA in comparison with the WT counterparts (Fig. 2A), and the difference was maintained upon TCR stimulation (Fig. 2B). On the contrary, Cbx4 deficiency had no significant impact on other negative regulators, including Lag3, Ctla4, Havcr2, Egr2/3, Ikzf1, and Tle4 (Fig. S3B,C). Increased Pdcd1 mRNA was also detected in purified CD44highCD4+ and CD8+ T cells (Fig. 2C). Flow cytometry was next engaged to analyze PD‐1 protein expression. In line with mRNA expression, PD‐1+ cells were present at an increased frequency among Cbx4‐deficient T cells (Fig. 2D), including central and effector memory cells enriched as Fig. S3A (Fig. S4A,B). When stimulated with anti‐CD3 and anti‐CD28 in vitro, naïve T cells quickly upregulated PD‐1 expression and significantly more PD‐1+ cells were generated in the cultures of Cbx4‐deficient T cells (Fig. 2E). Other than PD‐1 TIM‐3, LAG‐3 and CTLA‐4 are also important inhibitory roles in T‐cell function. In the process of rapid T‐cell activation, TIM‐3, LAG‐3, and CTLA‐4 will make sure T cells are not overactivated. Our data demonstrated there was comparable expression of TIM3, LAG3, and CTLA4 in WT and KO peripheral T cells (Fig. S4C,D), suggesting the effect of Cbx4 was relatively specific for PD‐1. So was the frequency of CD25+Foxp3+ Treg cells and the PD‐1 expression in these cells (Fig. S4E).
Fig. 2.
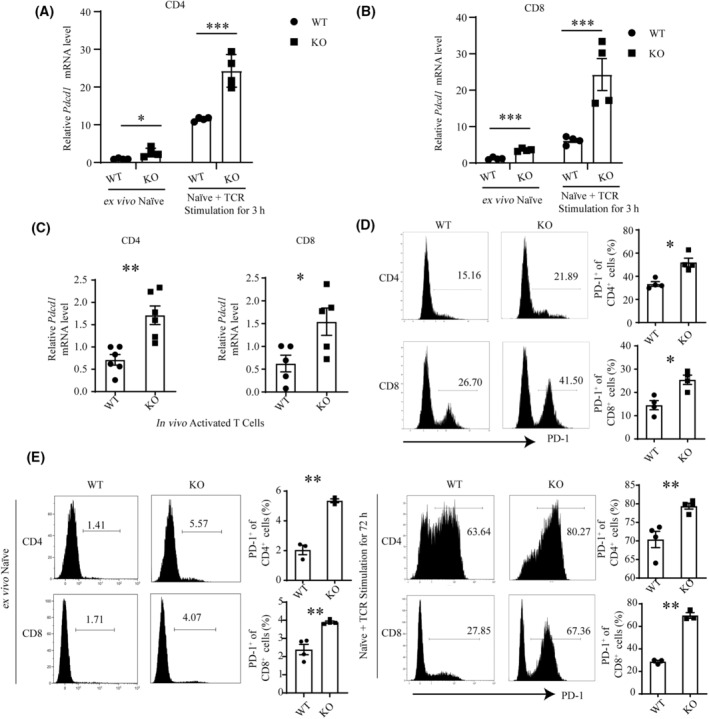
Upregulation of PD‐1 induced by Cbx4 deficiency. (A–C) Relative Pdcd1 mRNA levels in Cbx4‐deficient versus WT (wild‐type). (A) CD4+CD62L+CD44− naïve or TCR‐stimulated (3 h) (n = 4) (B), CD8+CD62L+CD44− naïve or TCR‐stimulated (3 h) (n = 4), (C) and freshly isolated CD44+ effector/memory CD4+ and CD8+ T cells as determined by RT‐qPCR (n = 5). (D, E) PD‐1 protein expression in Cbx4‐deficient versus WT splenic total (D), purified naïve, and TCR‐stimulated (72 h) (n = 4). (E) CD4+ and CD8+ T cells as determined by flow cytometry (n = 3). Representative histograms are shown on the left. Statistical data are presented as Mean ± SE on the right. Each experiment was repeated at least three times. Unpaired t‐test was used for comparison, *P < 0.05, **P < 0.01, ***P < 0.001.
To further elucidate the regulatory role of CBX4, we analyzed its expression over the course of T‐cell activation. Cbx4 mRNA was seen to be briefly upregulated at the early stage, however reduced below basal levels thereafter (Fig. 3A). Accordingly, all effector T‐cell subsets, including Th0, Th1, Th2, Th17, and iTreg displayed low expression of Cbx4 (Fig. 3B). Importantly, after stimulation, PD‐1 expression upregulation was consistent with Cbx4 expression pattern, although higher PD‐1 expression was found in KO T cells all the time points (Fig. 3C). In support of a direct role of CBX4 in PD‐1 expression, transfection of Jurkat cells and EL‐4 cells with a Cbx4‐expression construct were produced and purified respectively (Fig. S5A). High expression of Cbx4 reduced the level of PD‐1 on their surface (Fig. 3D,E).
Fig. 3.
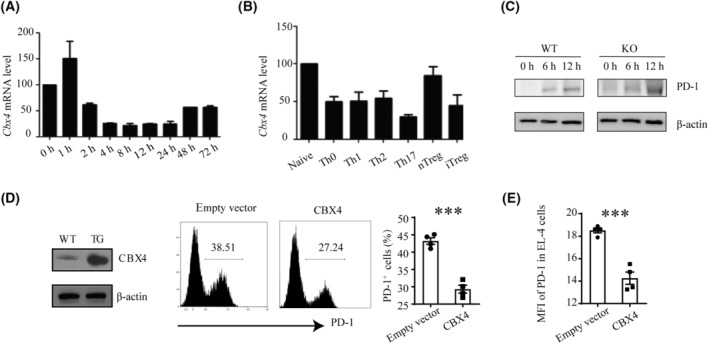
Inhibition of PD‐1 expression by CBX4. (A) Relative levels of Cbx4 mRNA expression in naïve (0 h) versus TCR‐stimulated CD4+ T cells at different time points as measured by quantitative RT‐PCR. Data from three independent experiments are shown. Error bars indicate SEM. (B) Relative levels of Cbx4 mRNA expression in naïve T cells, naturally occurring Treg (nTreg) cells, and effector T cells generated under various polarizing conditions for 72 h. The experiment was repeated three times. (C) Naive CD4+ T cells from WT and KO mice were stimulated with anti‐CD3 (2 μg·mL−1) and anti‐CD28 (1 μg·mL−1) for 12 h. PD‐1 levels were detected 6 h and 12 h after stimulation by western blotting (n = 2). (D) Jurkat cells were transfected with Myc‐CBX4 or empty vectors. CBX4 expression was verified western blotting (left). Following stimulation with anti‐CD3 (2 μg·mL−1) and anti‐CD28 (1 μg·mL−1) for 24 h, PD‐1 expression in the transfectants was detected by flow cytometry (n = 4). (E) EL‐4 cells were transfected with Flag‐CBX4 or empty vectors. Following stimulation with anti‐CD3 (2 μg·mL−1) and anti‐CD28 (1 μg·mL−1) for 24 h, PD‐1 expression in the transfectants was detected by flow cytometry (n = 4). Representative histograms are shown in the middle. Statistical data are presented as Mean ± SE Each experiment was repeated at least three times. Unpaired t‐test was used for comparison, ***P < 0.001.
3.3. Cbx4‐deficient T cells exhibit activation defects
We next investigated the impact of dysregulated PD‐1 expression on T‐cell activation. Naïve T cells were purified and stimulated with anti‐CD3 and anti‐CD28. In comparison with WT cells, both CD4+ and CD8+ T cells from KO mice showed reduced proliferation as measured by BrdU incorporation (Fig. 4A) and slightly increased apoptosis in Annexin V/7‐AAD staining (Fig. 4B). Moreover, Cbx4 KO CD4+ T cells produced less Th1 cytokines, such as IL‐2 and IFN‐γ (Fig. 4C). On the contrary, differentiation into Th2, Treg, or Th17 cells was not affected (Fig. S5B). Impaired function was also observed for Cbx4 KO CD8+ T cells in terms of IFN‐γ, GzmB, and TNF‐α expression (Fig. 4D). To reveal the functional consequence of the activation defect, a murine model of colitis was established by transferring CD4+CD45RBhi naïve T cells into Rag1‐deficient hosts. Since the ratio of CD4+ in CD45+ cells showed no difference in peripheral blood in the course of colitis, we believed Rag1‐deficient mice received similar donor cells from WT and KO mice (Fig. S5C). Adoptive transfer of WT T cells caused rapid weight loss (Fig. 4E), significant colon shortening (Fig. 4F, Fig. S5D), and apparent more severe colonic pathomorphology, including more transmural inflammation, increased proliferation, crypt abscesses, and obvious infiltration (Fig. 4G). Minimal signs of colitis, however, were observed following adoptive transfer of KO T cells (Fig. 4E–G), indicating that they were less pathogenic. Briefly, these results support that CBX4 is required for the proper activation of naïve T cells.
Fig. 4.
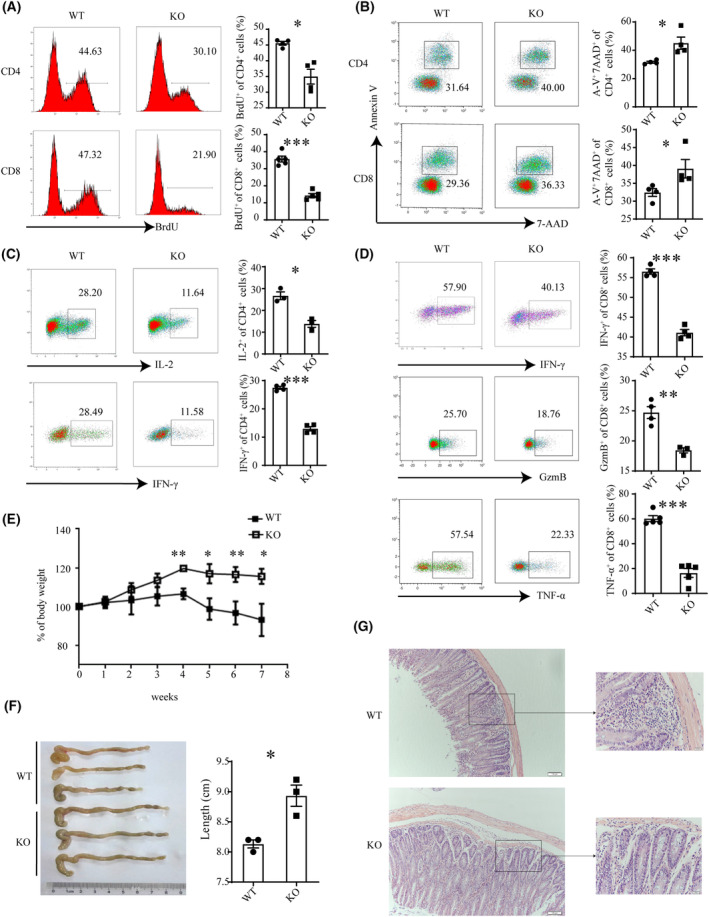
Activation defects of Cbx4‐deficient T cells. Naive CD4+ and CD8+ T cells from WT (wild‐type) and KO (knockout) mice were stimulated with anti‐CD3 (2 μg·mL−1) and anti‐CD28 (1 μg·mL−1) for 72 h. (A–D) Cell proliferation was measured by BrdU incorporation (A). Cell survival was analyzed with Annexin V and 7‐AAD staining (n = 4) (B). IL‐2 and IFN‐γ production by CD4+ T cells (n = 4) (C) and IFN‐γ, GzmB, and TNF‐α expression by CD8+ T cells (n = 4) (D) was examined by flow cytometry. Representative histograms (left) and statistical data (right) from three to four independent experiments are shown. (E–G) Body weight of Rag1 −/−recipient mice given wild‐type or Cbx4 KO CD4+CD45RBhi naïve T cells were adoptively transferred into Rag1 −/−recipient mice (n = 3 for each group). Body weight was monitored weekly (E). At the end of experiment, colon length was recorded (F) and the colonic sections were examined by HE staining (scale bar = 1 cm) (G). Scale bar = 100 μm. Data are presented as Mean ± SE. Unpaired t‐test was used for comparison, *P < 0.05, **P < 0.01, ***P < 0.001.
3.4. CBX4 promotes the formation of inhibitory histone modifications at the Pdcd1 locus
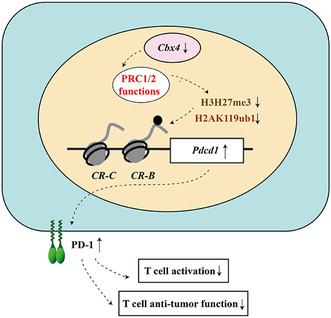
CBX4 primarily regulates gene transcription through epigenetic mechanisms. Therefore, our subsequent studies were focused on altered epigenetic modifications associated with Cbx4 deficiency. It is well established that PRC1 complexes mainly catalyzes the deposition of monoubiquitination of lysine 119 of histone H2A (H2AK119ub1). There was no significant difference in total H2AK119ub1 levels between Cbx4 KO and WT T cells at all the time points we tested, including before or 0.5 h and 6 h after TCR engagement (Fig. 5A). ChIP analysis, however, demonstrated that H2AK119ub1 deposition was significantly reduced at the CR‐C and CR‐B sites in the Pdcd1 promoter region in the absence of Cbx4 (Fig. 5B) and con, a genomic region void of enrichment as a negative control for site‐specific enrichment of H2AK119ub1 showed no difference (Fig. S6G). PRT4165 is an inhibitor of PRC1‐mediated H2A ubiquitylation [36]. When added into cultures of activated Jurkat or EL‐4 cells, either percentage or intensity of PD‐1 expression was seen to be enhanced (Fig. 5C, Fig. S6A). Furthermore, PRT4165 overcame the suppressive effect on PD‐1 expression induced by enforced expression of Cbx4 (Fig. S6B,C). These data support a critical role of the CBX4‐induced H2A119ub1 modification in the control of PD‐1 expression.
Fig. 5.
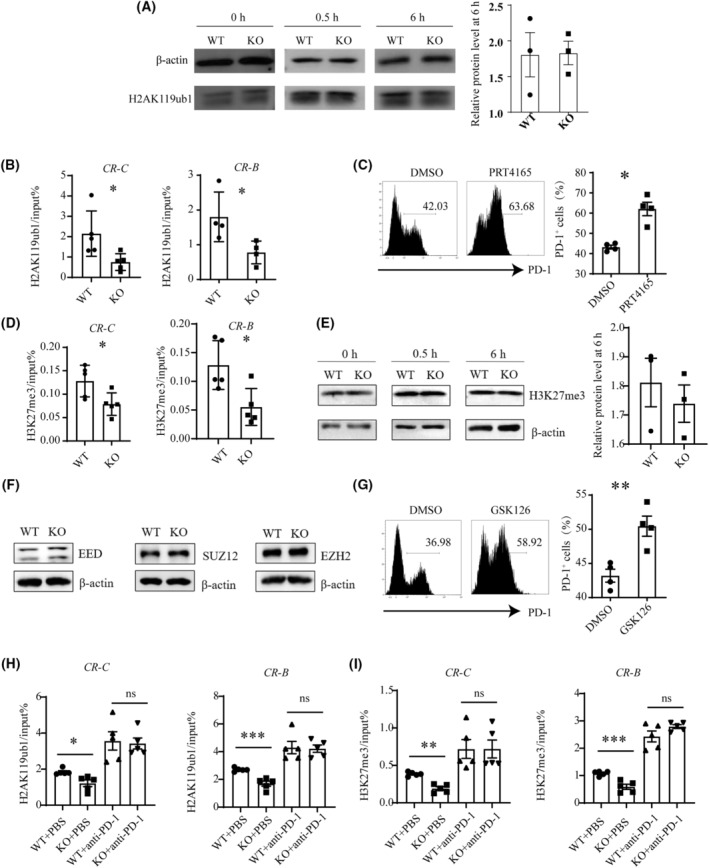
Reduced deposition of inhibitory histone modifications at the Pdcd1 locus in Cbx4‐deficient activated T cells. (A and B): (A) MACS‐purified CD3+ T cells from WT (knockout) and Cbx4 KO (knockout) mice were stimulated with anti‐CD3 and anti‐CD28. Total H2AK119ub1 levels were detected before and 0.5 h and 6 h after stimulation by western blotting. Representative results are shown from three independent experiments, and relative protein level was shown on the right side at the point of 6 h (n = 3). (B) H2AK119ub1 modification at CR‐C and CR‐B of the Pdcd1 locus at 0.5 h were analyzed by ChIP‐qPCR. Six and five pairs of WT and KO mice were used for the analysis of the CR‐C and CR‐B site, respectively (n = 4). (C) PD‐1 expression on Jurkat cells stimulated with anti‐CD3 and CD‐28 for 24 h with the addition of PRT4165 or DMSO (n = 5). (D) Levels of H3K27me3 modification at the CR‐C and CR‐B site (n = 5). (E) Total H3K27me3 levels in activated T cells were shown on the left, and relative protein level at 6 h was shown on the right side. Representative blots of three independent experiments are shown. (F) Expression of PRC2 components in T cells treated with anti‐CD3 and CD28 for 0.5 h. Representative blots of three independent experiments are shown. (G) PD‐1 expression on TCR‐stimulated Jurkat cells in the presence or absence of GSK126 (n = 4). (H) Levels of H2AK119ub1 modification at the CR‐C and CR‐B site in CD3+ T cells of draining lymph nodes in MC38 subcutaneous tumor models (n = 5). (I) Levels of H3K27me3 modification at the CR‐C and CR‐B site in CD3+ T cells of draining lymph nodes in MC38 subcutaneous tumor models (n = 5). Statistical data are presented as Mean ± SE. Each experiment was repeated at least three times. Unpaired t‐test for A–E, G–I. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
H2AK119 ubiquitination has been reported to facilitate the recruitment of PRC2 complex and stimulate its methyltransferase activity, leading to the trimethylation of lysine 27 of histone H3 (H3K27me3) [15, 17]. Indeed, KO T cells showed less accumulation of H3K27me3 at the CR‐C and CR‐B sites around the Pdcd1 promoter (Fig. 5D), while they showed similar accumulation of H3K27me3 at the con site as a control (Fig. S6G). Again, this effect appears to be specific to Pdcd1 as total H3K27me3 levels were comparable in WT and KO T cells, either in the resting state or upon activation (Fig. 5E). Besides, the major components of the PRC2 complex, such as EZH2, EED, and SUZ12, were equally present in WT and KO cells (Fig. 5F). To verify the functional relevance of the altered H3K27me3 modification at the Pdcd1 promoter, GSK126 was used to block the methyltransferase activity of EZH2 [37]. Like PRT4165, GSK126 further increased PD‐1 expression on Jurkat or EL‐4 cells following TCR stimulation and counteracted the inhibitory effect on PD‐1 expression imposed by Cbx4 overexpression (Fig. 5G, Fig. S6D–F).
To further confirm the effect of Cbx4 on Pdcd1 promoter region in tumor, we sorted out CD3+ T cells in draining lymph nodes from MC38 tumor‐bearing mice and checked their H2AK119ub1 and H3K27me3 deposition in the Pdcd1 promoter region. As expected, in line with higher PD‐1 expression, statistically less H2AK119ub1 and H3K27me3 deposition at the CR‐C and CR‐B sites in the Pdcd1 promoter region was found in CBX4 KO CD3+ T cells (Fig. 5H,I). Collectively, these data suggest that Cbx4‐induced downregulation of PD‐1 is largely attributable to the accumulation of inhibitory histone modifications at the promoter.
4. Discussion
CBX4 has been reported to have a cell‐intrinsic role in the regulation of tumor growth and invasion [12, 13, 38, 39]. From a new prospect, the present study investigated the possibility that CBX4 had additional roles in tumorigenesis by modulating T‐cell‐mediated antitumor immunity. Indeed, tumor growth was found to be accelerated in mice with T‐cell‐specific deletion of Cbx4 and the tumor‐infiltrating T cells from these mice were defective in IFN‐γ and TNF‐α production. When stimulated with anti‐CD3 and anti‐CD28 in vitro, Cbx4‐deficient naive T cells showed impaired proliferation and reduced expression of multiple effector factors. Moreover, the knockout mice were more susceptible to lymphopenia‐induced colitis. These results highlight a novel function of CBX4 as a negative regulator of T‐cell responses.
Accumulating evidence suggests the importance of PD‐1 signaling in tumor evasion of immune attacks. Blockade of this pathway elicits durable antitumor responses and long‐term remissions in a subset of patients with a broad spectrum of cancers [40, 41, 42, 43, 44, 45, 46, 47, 48, 49]. Along with the impaired antitumor immunity, increased levels of PD‐1 were detected in tumor‐infiltrating CD4+ and CD8+ T cells from Cbx4 knockout mice. In fact, higher percentages of PD‐1+ cells were observed in all major subsets of peripheral T cells or upon TCR‐mediated T‐cell activation in the absence of Cbx4. In contrast, enforced expression of Cbx4 in Jurkat cells resulted in downregulation of PD‐1, rendering further support for a direct role of CBX4 in the regulation of PD‐1 expression. While the immune suppressive effect of PD‐1 is well established, its contribution to the dampened T‐cell responses in the absence of Cbx4 remains to be verified. TCR signal transduction is an important condition for T‐cell activation, and the phosphorylation of downstream kinases Lck and ZAP‐70 of TCR is the initial signal of T‐cell cascade signaling pathway. Further investigation is needed to determine whether this effect is dependent or independent of the enhanced PD‐1 expression.
PD‐1 expression is dynamically regulated in T cells. Upon acute antigen exposure, its expression is transiently induced, but is quickly downregulated to allow for normal T‐cell responses. Chronic antigen stimulation, however, leads to sustained PD‐1 expression and immune exhaustion. A number of cis‐DNA elements, transcription factors, and epigenetic components are implicated in the transcriptional control of Pdcd1 [50]. Two conserved regions (CR‐B and CR‐C), located 100‐bp and 1.1‐kb upstream of the transcription start site of Pdcd1, are essential for the observed patterns of gene expression [51]. Transcription factors AP‐1 [52], NFATc1 [51], and FoxO1 [53] bind to these sites to activate Pdcd1 expression, whereas T‐bet [54] and Blimp‐1 [55] primarily function as repressors. As much as epigenetic mechanisms are concerned, an inverse correlation has been documented between PD‐1 expression and DNA methylation [56]. The landscape of histone modifications is more complicated. Briefly, H3K9ac and H3K27ac enrichment at the CR‐C site and the promoter region is associated with PD‐1 induction [55, 56], whereas loss of PD‐1 expression is accompanied by the appearance of repressive marks, such as H3K9me3, H3K27me3, and H4K20me3 at CR‐C [55]. Here, we demonstrated the presence of another form of histone modification, H2AK119ub1, at the Pdcd1 locus in activated T cells, which was reduced in the absence of Cbx4. In addition, Cbx4 deficiency was featured by less deposition of H3K27me3 at CR‐C and CR‐B. On the contrary, levels of histone acetylation, including H3ac, H4ac, H3K27ac, and H3K9ac, were not affected by Cbx4 deficiency (data not shown), indicative of the dominant effect of repressive histone modifications. In support of the importance of CBX4‐induced histone modifications, inhibition of H2A ubiquitylation restored Pdcd1 expression under conditions of Cbx4 overexpression. Of note, the reduction in H2AK119ub1 and H3K27me3 appeared to be rather selective for the Pdcd1 locus, as their total levels were comparable in WT and KO T cells. The mechanism underlying the selectivity is unclear. Chromatin immunoprecipitation assay detected no direct interaction of CBX4 with the Pdcd1 promoter (data not shown). It is most likely that CBX4 is targeted to Pdcd1 through other factors.
5. Conclusions
In conclusion, the present study reveals a previously unrecognized function of CBX4 in the regulation of T‐cell responses and antitumor immunity. The finding that CBX4‐mediated H2AK119ub1 modification regulates Pdcd1 expression expands our understanding of the epigenetic control of antitumor immunity. This, and recent studies of the enhanced antitumor immunity with EZH2 inhibitors [57], highlights the potential of targeting epigenetic mechanisms in tumor therapy.
Funding information
This work was supported by grants from Beijing Municipal Natural Science Foundation (7212058), the National Natural Sciences Foundation of China (31970840, 31872735 and 32230037), and Clinical Medicine Plus X‐Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (71006Y2823 and 71006Y3025).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
LR, ZL, YZhou, YZhang, and WW designed the project. LR, ZL, and WW did the experiment. LR, YZhang, and WW wrote the manuscript. YJ, ZW, YZhou, and YZhao helped with mouse breeding and establishment of disease models. ZZ and JZ contributed to data analysis. XP and XS helped to perform the flow cytometric analysis and sorting.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/1878‐0261.13516.
Supporting information
Table S1. Primer sequences for RT‐PCR.
Table S2. Primer sequences for ChIP‐qPCR.
Fig. S1. (related to Fig. 1). Absence of Cbx4 did not affect T‐cell development.
Fig. S2. (related to Fig. 1). LAG‐3 expression in tumor‐infiltrating CD4+ and CD8+ T cells from WT and Cbx4 KO mice.
Fig. S3. (related to Fig. 2). Cbx4 deficiency had no significant impact on other negative regulators.
Fig. S4. (related to Fig. 2). PD‐1+ cells were present at an increased frequency among Cbx4‐deficient T cells.
Fig. S5. (related to Figs 3, 4). Cbx4‐deficient T cells exhibit activation defects.
Fig. S6. (related to Fig. 5). CBX4 promotes the formation of inhibitory histone modifications at the Pdcd1 locus.
Acknowledgments
We thank Prof. Guoliang Xu of the Shanghai Institutes of Biological Sciences for the gift of Cbx4 flox/flox mice; and all members of Yu Zhang's laboratory for insightful discussion and technical assistance. We thank Beijing Municipal Natural Science Foundation and the National Natural Sciences Foundation of China for providing the funding.
Liwei Ren and Ziyin Li contributed equally to the work.
Contributor Information
Wei Wang, Email: wangwei83427@bjmu.edu.cn.
Yu Zhang, Email: zhangyu007@hsc.pku.edu.cn.
Data accessibility
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simon JA, Kingston RE. Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10(10):697–708. [DOI] [PubMed] [Google Scholar]
- 3. Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14(5):637–646. [DOI] [PubMed] [Google Scholar]
- 4. Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17(15):1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl‐lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17(15):1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalb R, Latwiel S, Baymaz HI, Jansen PW, Müller CW, Vermeulen M, et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol. 2014;21(6):569–571. [DOI] [PubMed] [Google Scholar]
- 7. Vandamme J, Völkel P, Rosnoblet C, Le Faou P, Angrand PO. Interaction proteomics analysis of polycomb proteins defines distinct PRC1 complexes in mammalian cells. Mol Cell Proteomics. 2011;10(4):M110.002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forzati F, Federico A, Pallante P, Abbate A, Esposito F, Malapelle U, et al. CBX7 is a tumor suppressor in mice and humans. J Clin Invest. 2012;122(2):612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X‐W, Zhang L, Qin W, Yao X‐H, Zheng L‐Z, Liu X, et al. Oncogenic role of the chromobox protein CBX7 in gastric cancer. J Exp Clin Cancer Res. 2010;29(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scott CL, Gil J, Hernando E, Teruya‐Feldstein J, Narita M, Martinez D, et al. Role of the chromobox protein CBX7 in lymphomagenesis. Proc Natl Acad Sci U S A. 2007;104(13):5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernard D, Martinez‐Leal JF, Rizzo S, Martinez D, Hudson D, Visakorpi T, et al. CBX7 controls the growth of normal and tumor‐derived prostate cells by repressing the Ink4a/Arf locus. Oncogene. 2005;24(36):5543–5551. [DOI] [PubMed] [Google Scholar]
- 12. Wang X, Li L, Wu Y, Zhang R, Zhang M, Liao D, et al. CBX4 suppresses metastasis via recruitment of HDAC3 to the Runx2 promoter in colorectal carcinoma. Cancer Res. 2016;76(24):7277–7289. [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Qin G, Liang X, Wang W, Wang Z, Liao D, et al. Targeting the CK1α/CBX4 axis for metastasis in osteosarcoma. Nat Commun. 2020;11(1):1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kagey MH, Melhuish TA, Wotton D. The Polycomb protein Pc2 is a SUMO E3. Cell. 2003;113(1):127–137. [DOI] [PubMed] [Google Scholar]
- 15. Li B, Zhou J, Liu P, Hu J, Jin H, Shimono Y, et al. Polycomb protein Cbx4 promotes SUMO modification of de novo DNA methyltransferase Dnmt3a. Biochem J. 2007;405(2):369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ismail IH, Gagné JP, Caron MC, McDonald D, Xu Z, Masson JY, et al. CBX4‐mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40(12):5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luis Nuno M, Morey L, Mejetta S, Pascual G, Janich P, Kuebler B, et al. Regulation of human epidermal stem cell proliferation and senescence requires polycomb‐dependent and ‐independent functions of Cbx4. Cell Stem Cell. 2011;9(3):233–246. [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Conejo‐Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. [DOI] [PubMed] [Google Scholar]
- 19. Pagès F, Berger A, Camus M, Sanchez‐Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–2666. [DOI] [PubMed] [Google Scholar]
- 20. Galon J, Costes A, Sanchez‐Cabo F, Kirilovsky A, Mlecnik B, Lagorce‐Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. [DOI] [PubMed] [Google Scholar]
- 21. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. [DOI] [PubMed] [Google Scholar]
- 24. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. [DOI] [PubMed] [Google Scholar]
- 25. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7‐CD28 ligand‐receptor family. Immunity. 2016;44(5):955–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Riley JL. PD‐1 signaling in primary T cells. Immunol Rev. 2009;229(1):114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, et al. Programmed death‐1 blockade enhances expansion and functional capacity of human melanoma antigen‐specific CTLs. Int Immunol. 2007;19(10):1223–1234. [DOI] [PubMed] [Google Scholar]
- 30. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen‐specific CD8 T cells infiltrating the tumor express high levels of PD‐1 and are functionally impaired. Blood. 2009;114(8):1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7‐H1/programmed death‐1 interactions. Cancer Res. 2009;69(20):8067–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuzaki J, Gnjatic S, Mhawech‐Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Li Y, Cao Y, Yuan M, Gao Z, Guo X, et al. Polycomb chromobox (Cbx) 7 modulates activation‐induced CD4+ T cell apoptosis. Arch Biochem Biophys. 2014;564:184–188. [DOI] [PubMed] [Google Scholar]
- 35. Liu B, Liu YF, Du YR, Mardaryev AN, Yang W, Chen H, et al. Cbx4 regulates the proliferation of thymic epithelial cells and thymus function. Development. 2013;140(4):780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, et al. A cellular chemical probe targeting the chromodomains of polycomb repressive complex 1. Nat Chem Biol. 2016;12(3):180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature. 2012;492(7427):108–112. [DOI] [PubMed] [Google Scholar]
- 38. Jiang N, Niu G, Pan Y‐H, Pan W, Zhang M‐F, Zhang CZ, et al. CBX4 transcriptionally suppresses KLF6 via interaction with HDAC1 to exert oncogenic activities in clear cell renal cell carcinoma. EBioMedicine. 2020;53:102692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Z, Fang Z, Chen G, Liu B, Xu J, Li F, et al. Chromobox 4 facilitates tumorigenesis of lung adenocarcinoma through the Wnt/β‐catenin pathway. Neoplasia. 2021;23(2):222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti‐PD‐L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. [DOI] [PubMed] [Google Scholar]
- 41. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, et al. Durable cancer regression off‐treatment and effective reinduction therapy with an anti‐PD‐1 antibody. Clin Cancer Res. 2013;19(2):462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, et al. Disabling immune tolerance by programmed death‐1 blockade with pidilizumab after autologous hematopoietic stem‐cell transplantation for diffuse large B‐cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N Engl J Med. 2013;369(2):134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bally AP, Austin JW, Boss JM. Genetic and epigenetic regulation of PD‐1 expression. J Immunol. 2016;196(6):2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD‐1 expression upon T cell activation. J Immunol. 2008;181(7):4832–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T‐cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A. 2012;109(38):15419–15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD‐1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41(5):802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T‐bet represses expression of the inhibitory receptor PD‐1 and sustains virus‐specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011;12(7):663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, et al. Blimp‐1 represses CD8 T cell expression of PD‐1 using a feed‐forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211(3):515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McPherson RC, Konkel JE, Prendergast CT, Thomson JP, Ottaviano R, Leech MD, et al. Epigenetic modification of the PD‐1 (Pdcd1) promoter in effector CD4(+) T cells tolerized by peptide immunotherapy. eLife. 2014;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents anti‐PD‐1 resistance in head and neck cancer. Clin Cancer Res. 2020;26(1):290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences for RT‐PCR.
Table S2. Primer sequences for ChIP‐qPCR.
Fig. S1. (related to Fig. 1). Absence of Cbx4 did not affect T‐cell development.
Fig. S2. (related to Fig. 1). LAG‐3 expression in tumor‐infiltrating CD4+ and CD8+ T cells from WT and Cbx4 KO mice.
Fig. S3. (related to Fig. 2). Cbx4 deficiency had no significant impact on other negative regulators.
Fig. S4. (related to Fig. 2). PD‐1+ cells were present at an increased frequency among Cbx4‐deficient T cells.
Fig. S5. (related to Figs 3, 4). Cbx4‐deficient T cells exhibit activation defects.
Fig. S6. (related to Fig. 5). CBX4 promotes the formation of inhibitory histone modifications at the Pdcd1 locus.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.