

Abstract
Background
Variability in datasets is not only the product of biological processes: they are also the product of technical biases. ComBat and ComBat-Seq are among the most widely used tools for correcting those technical biases, called batch effects, in, respectively, microarray and RNA-Seq expression data.
Results
In this technical note, we present a new Python implementation of ComBat and ComBat-Seq. While the mathematical framework is strictly the same, we show here that our implementations: (i) have similar results in terms of batch effects correction; (ii) are as fast or faster than the original implementations in R and; (iii) offer new tools for the bioinformatics community to participate in its development. pyComBat is implemented in the Python language and is distributed under GPL-3.0 (https://www.gnu.org/licenses/gpl-3.0.en.html) license as a module of the inmoose package. Source code is available at https://github.com/epigenelabs/inmoose and Python package at https://pypi.org/project/inmoose.
Conclusions
We present a new Python implementation of state-of-the-art tools ComBat and ComBat-Seq for the correction of batch effects in microarray and RNA-Seq data. This new implementation, based on the same mathematical frameworks as ComBat and ComBat-Seq, offers similar power for batch effect correction, at reduced computational cost.
Keywords: Batch effects, Transcriptomics, Bayesian statistics, Open source
Background
Batch effects are the product of technical biases, such as variations in the experimental design or even atmospheric conditions [1, 2]. They particularly reveal themselves when merging different datasets, which have likely been built under different conditions. If not corrected, these batch effects may lead to incorrect biological insight, since the variability can be wrongly interpreted as the product of a biological process.
Multiple methods exist that address this problem. They include approaches related to frequentist statistics, such as simple normalization [3, 4] or principal component analysis [5]; and machine learning, such as support-vector machines [6]. One of their main flaws is, however, their incapacity to handle low sample sizes or more than two batches at the same time [7].
ComBat, originally implemented in the R library sva [8], is based on the mathematical framework defined in [9]. This tool leverages a parametric and non-parametric empirical Bayes approach for correcting the batch effect in microarray datasets that works for small sample sizes or in the presence of outliers. This approach is based on the fact that microarray expression data are generally distributed according to a log-normal distribution [10]. Note that the parametric method requires strong assumptions but is largely faster than the non-parametric approach.
ComBat-Seq, also implemented in the R library sva, is based on a similar mathematical framework, where normal distributions are replaced by negative binomial distributions, to better reflect the statistical behavior of RNA-Seq raw counts data [11].
We recall the details of the mathematical models of ComBat and ComBat-Seq in Table 1.
Table 1.
Details of the mathematical models of ComBat and ComBat-Seq
| ComBat model | ComBat-Seq model |
|---|---|
denotes the expression value for gene in sample from batch , denotes a gene-wise baseline expression level, is a vector of covariates for sample , is the vector of corresponding regression coefficients for gene , is the additive (impacting the mean) batch effect and is the multiplicative (impacting the variance) batch effect
We introduce in this article pyComBat, a new Python tool implementing ComBat (function pycombat_norm) and ComBat-Seq (function pycombat_seq), following the same mathematical frameworks. Note that the term “pyComBat” implicitly refers to the function pycombat_norm (resp. pycombat_seq) when compared to ComBat (resp. ComBat-Seq). In comparison to both the R implementation and the existing Python implementation of ComBat in the single-cell analysis library Scanpy [12], we show that pyComBat yields similar results for adjusting for batch effects in microarray data, but is generally faster, in particular for the usually slow, but more loose, non-parametric method. Similarly, in comparison to the R implementation, we show that pyComBat yields identical results for adjusting for batch effects in RNA-Seq data, and is generally faster. To our knowledge, it is the sole Python implementation of ComBat-Seq.
Implementation of pyComBat
pyComBat is a Python 3 implementation of ComBat and ComBat-Seq. It mostly uses generic libraries like Pandas [13] or NumPy [14] to mimic ComBat and ComBat-Seq, following the exact same mathematical framework.
Two important features are not directly related to the performance of the software but are of utmost importance. First, pyComBat is available as an open-source software under a GPL-3.0 license, which means anyone can use, modify, distribute and share it. Opening pyComBat to the bioinformatics Python community is the best way for maintaining and improving it, while increasing its robustness. Second, the reliability of pyComBat has been thoroughly checked, using a bench of unit tests (code coverage measured at 88% with Python module “coverage”) serving both as functional tests (to ensure the proper functioning of each submodule) and as non-regression tests (to ease maintenance).
Results: comparing pyComBat with ComBat and ComBat-Seq
Datasets used and preprocessing
For software validation, we created two microarray and two RNA-Seq meta-datasets from public data: one on Ovarian Cancer (6 microarray datasets), one on Multiple Myeloma (4 microarray datasets), one on Breast Cancer (originally used for the validation of ComBat-Seq [11], 2 RNA-Seq datasets) and one on Colon Cancer (2 RNA-Seq datasets) All meta-datasets are described in more detail in Table 2.
Table 2.
Composition of each meta-dataset used for benchmarking pyComBat, Scanpy’s implementation of ComBat, ComBat and ComBat-Seq
| Dataset | Reference(s) |
|---|---|
| Ovarian Cancer (microarray) | |
| GSE18520 | [22] |
| GSE66957 | |
| GSE69428 | [23] |
| GSE9891 | [24] |
| GSE26712 | [25, 26] |
| GSE38666 | [27, 28] |
| Multiple Myeloma (microarray) | |
| GSE5900 | [29–31] |
| GSE66291 | [32, 33] |
| GSE68891 | |
| GSE122231 | [34, 35] |
| Breast cancer (RNA-Seq) | |
| GSE83083 | [36] |
| GSE59765 | [37] |
| Colon cancer (RNA-Seq) | |
| phs000892.v6.p1 | [38] |
| phs000178.v11.p8 | [39] |
Microarray data were normalized with the rma function from the affy R package (v.1.68.0) [15] which applies a log2 transformation and ensures the normal distribution of normalized data, while the raw counts were directly acquired for RNA-Seq data as suggested in the ComBat-seq documentation [11].
We then compared
ComBat, Scanpy’s implementation of ComBat and pyComBat on the microarray datasets on one hand,
ComBat-Seq and pyComBat on the RNA-Seq datasets.
for (i) efficacy for batch effect correction and (ii) computation time.
Batch effect correction
As an implementation of the ComBat and ComBat-Seq algorithms, pyComBat is expected to have similar, if not identical, power in terms of batch effects correction. This is confirmed in Fig. 1A, which shows the distribution of relative differences between the outputs of ComBat and pyComBat, on the Ovarian Cancer dataset (mean = − 1.06 × 10–7, 95% CI = [− 1.28 × 10–3, 1.32 × 10–4]). As expected, the differences are distributed closely around zero with a relative squared error of 1.7 × 10–7, suggesting that the variability relative to the use of ComBat or pyComBat is negligible compared to the intrinsic variability of the data.. The slight variability can be explained by the difference between optimization routines in R and Python (Numpy): while small differences in the fitted distribution parameters have little to no impact for the vast majority of data points, they may be amplified by data adjustment for data points located far away in the distribution tails.
Fig. 1.

Performance of pyComBat vs. Combat vs. Scanpy’s implementation of ComBat. A Distribution of the relative differences between the expression matrices corrected for batch effects, respectively by ComBat and pyComBat (parametric version), on the Ovarian Cancer dataset. The vertical dotted line corresponds to zero. B Computation time in seconds for pyComBat, Scanpy and ComBat for the parametric method, on the Multiple Myeloma dataset. The y-axis is in a log scale. C Computation time in seconds for pyComBat, Scanpy and ComBat for the parametric method, on the Ovarian Cancer dataset. The y-axis is in a log scale. D Computation time in minutes for pyComBat (left) and ComBat (right) for the non-parametric method, on the Ovarian Cancer dataset. The y-axis is in a log scale
Additionally, both ComBat-Seq and pyComBat produce the exact same output on the Breast Cancer and Colon Cancer datasets. Despite the aforementioned differences in optimization routines between R and Python, the slight variations in the fitted distribution parameters are leveled out by rounding to integers during data adjustment as both tools output adjusted integer counts. Given the long-tailed nature of the negative binomial distribution, one would expect that even slight variations in parameter values can impact data points far in the distribution tail, a phenomenon completely absent in our experiments.
To sum up, it is highly unlikely to obtain such similar results unless pyComBat implements the same algorithms as ComBat and ComBat-Seq.
Computation time
Computation time is evaluated by running pyComBat (resp. Scanpy’s implementation of ComBat and ComBat itself) respectively 100 times on both microarray datasets presented in Section “Results: comparing pyComBat with ComBat and ComBat-Seq”, with the parametric approach. As Scanpy doesn’t handle the non-parametric approach, only ComBat and pycombat_norm have been tested with it, on the Ovarian Cancer dataset. As for pycombat_seq and ComBat-Seq, they have been run respectively 50 times on the Breast Cancer dataset and 20 times on the Colon Cancer dataset. The reduced number of runs on RNA-Seq datasets is due to the longer, and less variable, computation times.
Owing to Python (Numpy) efficiency in handling matrix operations and matrix manipulations as well as thorough optimization of our code, pyComBat is as fast or even faster than ComBat. The parametric version of the pyComBat performs 4–5 times as fast as ComBat, and around 1.5 times as fast as the Scanpy implementation of ComBat, in terms of computation time (Fig. 1B, C), on both datasets.
Similar results are observed with the non-parametric version (Fig. 1D), which is inherently more time consuming, but also less dependent on the distribution of the data. In this case, pyComBat is also approximately 4–5 times faster than ComBat, going from more than an hour to around 15 min.
Finally, pyComBat appears to be 4–5 times faster than ComBat-Seq on both RNA-Seq datasets (Fig. 2A, B).
Fig. 2.

Performance of pyComBat vs. ComBat-Seq. A Computation time in seconds for pyComBat and ComBat-Seq, on the Colon Cancer dataset. B Computation time in seconds for pyComBat and ComBat-Seq, on the Breast Cancer dataset
Downstream analysis
The main goal of batch effect correction in transcriptomic is to produce unbiased and more powerful downstream analysis. We show here that the slight differences in corrected gene expression between ComBat and pyComBat have little to no impact on the output of differential gene expression analysis. Such an analysis is not needed on our RNA-Seq datasets, as ComBat-Seq and pyComBat output the exact same adjusted count matrix.
We have applied differential gene expression analysis on the Ovarian Cancer dataset, to compare groups based on sample types: Primary tumors against Normal tissues. Both ComBat and pyComBat corrected data have been analyzed using the Limma R package (v3.56.2) [16], comparing 62 Normal tissue and 615 Primary tumor samples. As shown in Fig. 3, the differences in batch effects corrections have a small impact on the logFoldChange differences (mean = − 1.02·× 10–4, 95% CI = [− 1.15·× 10–3, 7.88·×·10–4]) between both differential analyses. Moreover, with usual thresholds found in the literature (i.e. logFoldChange > 1.5 and fdr < 0.05), the selected genes are the same, which suggests that ComBat and pyCombat can be used interchangeably before downstream analyses.
Fig. 3.
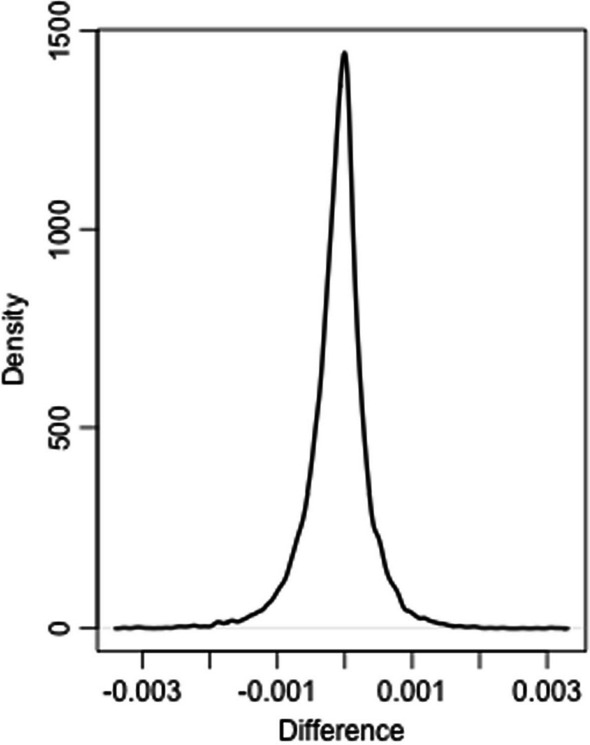
Differences of logFoldChange between ComBat and pyComBat corrected data for the Ovarian microarray dataset, for a differential expression analysis between primary tumor and normal tissue samples using Limma
Discussion and conclusion
We present pyComBat, a new Python implementation of ComBat and ComBat-Seq, the most commonly used software for batch effects correction on high-throughput molecular data. Our implementation offers the same correcting power, with shorter computation time for the parametric method compared to other implementations, and significantly shorter time for the time-consuming non-parametric version compared to the original R implementations. This reduced computing time opens perspectives for a more generic use of the non-parametric approach to a larger range of datasets.
As ComBat and pyComBat (pycombat_norm) assume the normal distribution of the input data, data processing should comply with this assumption. Microarray raw data generally follow a log-normal distribution, and R’s rma function applies a log-transformation after the normalization step, which ensures the normal distribution of the input data. However, other normalization tools, e.g. MAS5, do not log-transform the data. It is up to the user to check the distribution of their data and eventually apply a transformation accordingly. ComBat-seq and pyComBat (pycombat_seq) both work on data that follow a negative binomial distribution, which is the distribution of raw counts in RNA-Seq data. No preprocessing of the raw counts is thus needed.
Despite the historical prevalence of R, Python is gaining momentum in the bioinformatics landscape. Python is a general-purpose programming language, widely used in fields related to bioinformatics: data science, machine learning and AI, orchestration, visualization, etc. The versatility and wide adoption of Python eases interoperability of bioinformatics tools with tools from other domains of expertise. We believe that this interdisciplinary capabilities, along with its general-purpose abilities, gives Python a substantial edge over R, targeted at statistical applications, to grow as the language of choice for bioinformatic tools. By contributing to a single-language unified ecosystem, we hope to eliminate the need to interface several languages (typically R and Python), a common source of technical difficulties and of computational inefficiency. Workflow languages (such as Snakemake [17] or Nextflow [18]) and interoperability libraries (such as rpy2 [19]) still need to translate and copy data from one language to the other. This is a source of inefficiency and of limitations—depending on the supported data formats—especially when the analysis goes back and forth between languages. We thus advocate the necessity to port reference tools from R to Python. Sign of this trend, state-of-the-art tools have been directly developed in Python (e.g. lifelines [20], a library dedicated to survival analysis, scanpy [12] (a library dedicated to the analysis of single-cell omic data) or quickly ported in Python from R (e.g. harmony [21], an R library for integrating single cell data, DESeq2, an R library for differential expression analysis).
Furthermore, porting a tool from one language to another provides an opportunity window to improve both functionality and performance. For instance, pyComBat improves over ComBat-Seq by supporting reference batches.
The recent advent of large language models (LLMs) with coding capabilities, such as Starcoder, GPT4 or GitHub Copilot, represent a great opportunity to accelerate this porting process. Yet, human intervention remains necessary:
to ensure the correctness of the ported code. Beyond hallucinations, LLMs remain statistic-based prediction models, and may thus introduce errors in the implementation, at the risk of changing the mathematical logic;
to take full advantage of the performance and functionality improvement opportunity window. For instance, ComBat-Seq code is a mix of R and C++, and porting it to Python required adapting the C++ code to interface with Python instead of R.
Note however that the work on pyComBat was undertaken before the wide diffusion of LLM-based coding tools.
We have attached importance to making the software open source, coupled with comprehensive documentation. We built a robust set of test cases, in an effort to encourage larger participation from the community. We believe that this will be benefiting the Python bioinformatics community and opening the way towards the translation of other widely used software from R to Python.
Acknowledgements
The authors thank Phuong Pham for his advice about the estimation of the efficiency of the adjustments. The authors also thank all the external contributors on the GitHub repository, for their feedback on the code for pyComBat.
Author contributions
AB implemented pycombat_norm and compared it with ComBat and Scanpy's implementation of ComBat. MC implemented pycombat_seq and compared it with ComBat-Seq. AB and MC wrote this manuscript. JH, AG, GA, CAA and AN reviewed code and provided scientific guidance. All authors read and approved the final manuscript.
Funding
This work was supported by the European Union’s Horizon 2020 research and innovation program under Grant Agreement No. 190185351.
Availability of data and materials
The datasets analyzed during the current study are available in the GEO and dbGAP repositories:—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18520—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66957—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69428—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9891—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26712—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38666—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE5900—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66291—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68891—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122231—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83083—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59765—https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000892.v6.p1—https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v11.p8.
Availability and requirements
Project name: InMoose. Project home page: https://github.com/epigenelabs/inmoose. Operating system: Platform independent. Programming language: Python. Other requirements: Numpy, Scipy License: GNU GPL3. Any restrictions to use by non-academics: None.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abdelkader Behdenna and Maximilien Colange have contributed equally to this work.
Contributor Information
Abdelkader Behdenna, Email: abdelkader@epigenelabs.com.
Akpéli Nordor, Email: akpeli@epigenelabs.com.
References
- 1.Fare TL, Coffey EM, Dai H, He YD, Kessler DA, Kilian KA, et al. Effects of atmospheric ozone on microarray data quality. Anal Chem. 2003;75(17):4672–4675. doi: 10.1021/ac034241b. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES. Array of hope. Nat Genet. 1999;21(1 Suppl):3–4. doi: 10.1038/4427. [DOI] [PubMed] [Google Scholar]
- 3.Tai YC, Speed TP. A multivariate empirical Bayes statistic for replicated microarray time course data. Ann Stat. 2006;34(5):2387–2412. doi: 10.1214/009053606000000759. [DOI] [Google Scholar]
- 4.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen TO, West RB, Linn SC, Alter O, Knowling MA, O’Connell JX, et al. Molecular characterisation of soft tissue tumours: a gene expression study. Lancet Lond Engl. 2002;359(9314):1301–1307. doi: 10.1016/S0140-6736(02)08270-3. [DOI] [PubMed] [Google Scholar]
- 6.Benito M, Parker J, Du Q, Wu J, Xiang D, Perou CM, et al. Adjustment of systematic microarray data biases. Bioinformatics. 2004;20(1):105–114. doi: 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Grennan K, Badner J, Zhang D, Gershon E, Jin L, et al. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. PLoS ONE. 2011;6(2):e17238. doi: 10.1371/journal.pone.0017238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinform Oxf Engl. 2012;28(6):882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 10.Hoyle DC, Rattray M, Jupp R, Brass A. Making sense of microarray data distributions. Bioinformatics. 2002;18(4):576–584. doi: 10.1093/bioinformatics/18.4.576. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genomics Bioinform. 2020;2(3):lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinney W. Data structures for statistical computing in Python. In: Proceedings of 9th Python Sci Conf. 2010;56–61
- 14.van der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng. 2011;13(2):22–30. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- 15.Irizarry RA. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 16.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, et al. Sustainable data analysis with Snakemake. F1000Research. 2021;10:33. doi: 10.12688/f1000research.29032.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 19.rpy2: Python-R bridge [Internet]. [cited 2023 Nov 14]. https://rpy2.github.io/
- 20.Davidson-Pilon C. lifelines, survival analysis in Python [Internet]. Zenodo; 2023 [cited 2023 Nov 14]. https://zenodo.org/record/8341606
- 21.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong K, Kwok, et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell. 2009;16(6):521–532. doi: 10.1016/j.ccr.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto Y, Ning G, Howitt BE, Mehra K, Wu L, Wang X, et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J Pathol. 2016;238(4):519–530. doi: 10.1002/path.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14(16):5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 25.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68(13):5478–5486. doi: 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vathipadiekal V, Wang V, Wei W, Waldron L, Drapkin R, Gillette M, et al. Creation of a human secretome: a novel composite library of human secreted proteins: validation using ovarian cancer gene expression data and a virtual secretome array. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21(21):4960–4969. doi: 10.1158/1078-0432.CCR-14-3173. [DOI] [PubMed] [Google Scholar]
- 27.Huang C, Clayton EA, Matyunina LV, McDonald LD, Benigno BB, Vannberg F, et al. Machine learning predicts individual cancer patient responses to therapeutic drugs with high accuracy. Sci Rep. 2018;8(1):16444. doi: 10.1038/s41598-018-34753-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lili LN, Matyunina LV, Walker LD, Benigno BB, McDonald JF. Molecular profiling predicts the existence of two functionally distinct classes of ovarian cancer stroma. BioMed Res Int. 2013;2013:846387. doi: 10.1155/2013/846387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Driscoll JJ, Pelluru D, Lefkimmiatis K, Fulciniti M, Prabhala RH, Greipp PR, et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115(14):2827–2834. doi: 10.1182/blood-2009-03-211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Wendlandt EB, Darbro B, Xu H, Thomas GS, Tricot G, et al. Genetic analysis of multiple myeloma identifies cytogenetic alterations implicated in disease complexity and progression. Cancers. 2021;13(3):517. doi: 10.3390/cancers13030517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007;109(4):1692–1700. doi: 10.1182/blood-2006-07-037077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lionetti M, Barbieri M, Todoerti K, Agnelli L, Fabris S, Tonon G, et al. A compendium of DIS3 mutations and associated transcriptional signatures in plasma cell dyscrasias. Oncotarget. 2015;6(28):26129–26141. doi: 10.18632/oncotarget.4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lionetti M, Barbieri M, Todoerti K, Agnelli L, Marzorati S, Fabris S, et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget. 2015;6(27):24205–24217. doi: 10.18632/oncotarget.4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan R, Dhodapkar M, Rosenthal A, Heuck C, Papanikolaou X, Qu P, et al. Four genes predict high risk of progression from smoldering to symptomatic multiple myeloma (SWOG S0120) Haematologica. 2015;100(9):1214–1221. doi: 10.3324/haematol.2015.124651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhodapkar MV, Sexton R, Waheed S, Usmani S, Papanikolaou X, Nair B, et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120) Blood. 2014;123(1):78–85. doi: 10.1182/blood-2013-07-515239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahman M, MacNeil SM, Jenkins DF, Shrestha G, Wyatt SR, McQuerry JA, et al. Activity of distinct growth factor receptor network components in breast tumors uncovers two biologically relevant subtypes. Genome Med. 2017;9(1):40. doi: 10.1186/s13073-017-0429-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McQuerry JA, Jenkins DF, Yost SE, Zhang Y, Schmolze D, Johnson WE, et al. Pathway activity profiling of growth factor receptor network and stemness pathways differentiates metaplastic breast cancer histological subtypes. BMC Cancer. 2019;19(1):881. doi: 10.1186/s12885-019-6052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell. 2019;177(4):1035–1049.e19. doi: 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed during the current study are available in the GEO and dbGAP repositories:—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18520—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66957—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69428—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9891—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26712—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38666—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE5900—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66291—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68891—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122231—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83083—https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59765—https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000892.v6.p1—https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v11.p8.
Project name: InMoose. Project home page: https://github.com/epigenelabs/inmoose. Operating system: Platform independent. Programming language: Python. Other requirements: Numpy, Scipy License: GNU GPL3. Any restrictions to use by non-academics: None.