

Abstract
The inhibitor of apoptosis protein BIRC2 regulates fundamental cell death and survival signaling pathways. Here we show that BIRC2 accumulates in the nucleus via binding of its second and third BIR domains, BIRC2BIR2 and BIRC2BIR3, to the histone H3 tail and report the structure of the BIRC2BIR3–H3 complex. RNA-seq analysis reveals that the genes involved in interferon and defense response signaling and cell-cycle regulation are most affected by depletion of BIRC2. Overexpression of BIRC2 delays DNA damage repair and recovery of the cell-cycle progression. We describe the structural mechanism for targeting of BIRC2BIR3 by a potent but biochemically uncharacterized small molecule inhibitor LCL161 and demonstrate that LCL161 disrupts the association of endogenous BIRC2 with H3 and stimulates cell death in cancer cells. We further show that LCL161 mediates degradation of BIRC2 in human immunodeficiency virus type 1-infected human CD4+ T cells. Our findings provide mechanistic insights into the nuclear accumulation of and blocking BIRC2.
Human inhibitor of apoptosis (IAP) proteins are main regulators of apoptotic pathways and are implicated in inflammatory and innate immune signaling. Aberrant activities particularly of cellular IAPs (cIAPs)—cIAP1, also named BIRC2, and cIAP2 (BIRC3)—have been associated with cancer, inflammation and immunopathogenesis1–3. Acute or latent human immunodeficiency virus type 1 (HIV-1) infection increases the expression of IAPs4 leading to the inhibition of HIV-1 transcription5–7. cIAPs contain three baculovirus IAP repeats (BIRs), a ubiquitin associating (UBA) domain, a CARD domain and a RING finger (Fig. 1a). The second and third BIR repeats of cIAPs form complexes with caspases, whereas the RING finger of cIAPs functions as an E3 ubiquitin ligase, which mediates autoubiquitination and ubiquitination of protein substrates8–10. The anti-apoptotic activity of cIAPs is countered by IAP antagonists, such as second mitochondria-derived activator of caspase (SMAC, also known as DIABLO)11–13. The upregulation of IAPs has been shown to reduce autophagy through the ubiquitination of BECN1, inhibit apoptosis through the antagonization, ubiquitination and neddylation of caspases, and increase the ubiquitination of RIPK1 (refs. 1,2,9,14). The latter promotes prosurvival NF-κB signaling while also inhibiting the assembly of distinct RIPK1 complexes that regulate cell death. Beyond the regulation of apoptosis, cIAPs were found to promote cell proliferation, migration and invasion and to mediate the cell cycle, chromosome segregation in mitosis, DNA damage response and transcription15–20.
Fig. 1 |. BIRC2 recognizes histone tails.
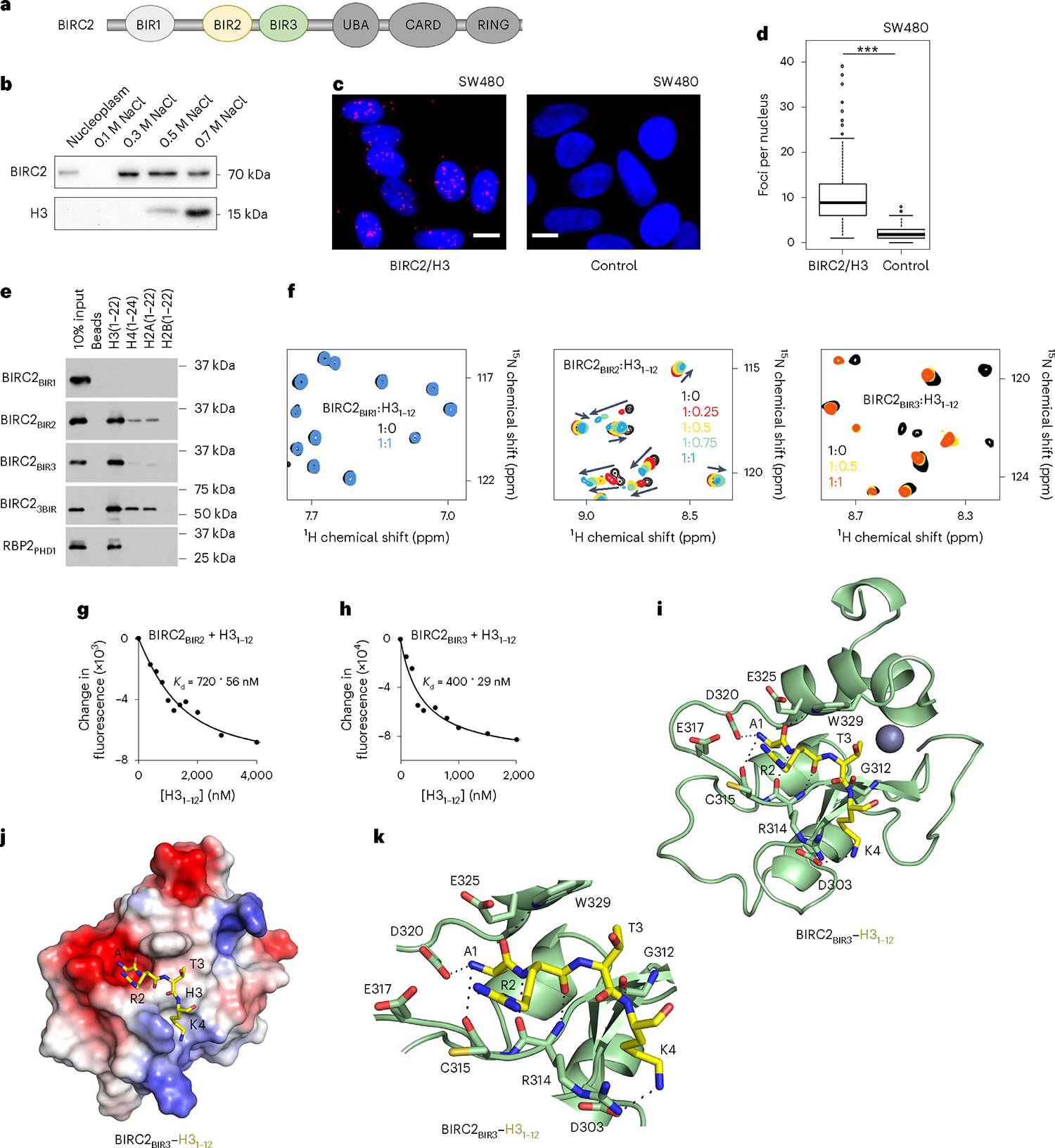
a, BIRC2 domain architecture. b, Western blot analysis of the salt gradient chromatin fractionation of SW480 nuclei. The experiment was performed independently three times. c, Representative images of PLA (red) between BIRC2 and H3 in SW480 cells. Nuclei are stained with DAPI (blue). Scale bars, 10 μm. d, Quantification of the PLA signal with at least 75 cells analyzed per condition. Statistical analysis is described in Statistics and reproducibility. Two-sided Wilcoxon tests. P < 2.2 × 10−16. e, Western blot analysis of pulldowns of the GST-tagged BIR domains of BIRC2 with the indicated histone peptides. RBP2-PHD, control. The experiment was performed independently three times. f, Superimposed 1H,15N HSQC spectra of 15N-labeled BIR domains of BIRC2, collected while the H31–12 peptide was titrated in the NMR samples. Spectra are color-coded according to the protein:peptide molar ratio. g,h, Representative binding curves used to determine values by tryptophan fluorescence. values were averaged over three separate experiments, with error calculated as s.d. between the runs. i, Ribbon diagram of BIRC2BIR3 (green) in complex with the H3 peptide (yellow). Dashed lines represent hydrogen bonds. The zinc ion is shown as a gray sphere. j, Electrostatic surface potential of BIRC2BIR3 is colored blue and red for positive charge and negative charge, respectively. The bound H3 peptide is shown as yellow sticks. k, Ribbon diagram of the BIRC2BIR3 (green) in complex with the H3 peptide (yellow). Dashed lines represent hydrogen bonds.
Unlike other IAP family members that are found primarily in the cytosolic fraction, BIRC2 can localize to the cell nucleus through an unknown mechanism21,22. Furthermore, because BIRC2 is overexpressed in response to HIV-1 infection and in various cancers, it has emerged as an attractive therapeutic target23–28. Among many cIAP targeting small molecule compounds developed in the past few years, LCL161, designed to mimic SMAC, has received considerable attention25. LCL161 is characterized by favorable pharmacological properties and minor toxicity and is currently being investigated in many preclinical and phase 1 and 2 clinical studies25,26. It shows potent anticancer activity in at least two dozen tumor types and sensitizes cancer cells to conventional chemotherapy with cisplatin and vincristine25,26, yet our understanding of the biochemical mechanisms underlying the LCL161 effect is lacking.
Here, we show that BIRC2 engages with chromatin through binding to the H3 tail and determine the mechanistic details and biological consequences of this engagement. We also describe the structural basis for targeting of BIRC2 by LCL161 and highlight the molecular mechanism by which LCL161 triggers cell death in cancerous and HIV-1-infected T cells.
Results and discussion
BIRC2 recognizes histone tails
The association of BIRC2 with chromatin was originally observed in salt gradient fractionation of the isolated nuclei from SW480 cells and proximity ligation assay (PLA). Endogenous BIRC2 was detected in the nuclear fraction containing 0.3–0.5 M NaCl (Fig. 1b), and it was bound to H3 in PLA (Fig. 1c,d). The histone binding function of BIRC2 was substantiated by pulldown experiments using BIR domains of BIRC2 and histone peptides (Fig. 1e). The third BIR domain (BIRC2BIR3) bound to the N terminus of H3 (residues 1–22 of H3) but did not recognize other histones. The second BIR domain (BIRC2BIR2) associated mostly with H3 and to a lesser extent with the N termini of H4 and H2A, whereas the first BIR domain (BIRC2BIR1) did not show histone binding activity. The histone selectivity, that is, the strong binding to H3 and weaker binding to H4 and H2A, was maintained in the construct containing all three BIRs (BIRC23BIR), which represented a unique histone reader capable of recognizing all but H2B tails. To characterize the binding of BIRs in detail, we produced 15N-labeled domains and monitored their interactions with the H31–12 (residues 1–12 of H3) peptide by NMR spectroscopy (Fig. 1f). 1H,15N HSQC (heteronuclear single quantum coherence) spectra of BIRs were recorded while the H3 peptide was added stepwise to the NMR samples. Large chemical shift perturbations (CSPs) in the spectra of BIRC2BIR3 and BIRC2BIR2 were induced by the peptide and indicated formation of the BIR–H3 complexes. CSPs were in the slow exchange regime on the NMR timescale for BIRC2BIR3 and in the intermediate exchange regime for BIRC2BIR2 and suggested tight binding to both domains. In agreement, binding affinity of BIRC2BIR3 for the H31–12 peptide, measured by fluorescence and microscale thermophoresis (MST) assays, was found to be 400 nM and 340 nM, respectively, and binding affinity of BIRC2BIR2 for the H31–12 peptide was measured as 0.72 μM and 1 μM, respectively (Fig. 1g,h and Supplementary Fig. 1). These values were in the range of binding affinities of the BIR3 and BIR2 domains of IAPs to the established ligands, SMAC and caspase-9 (refs. 29–31), and of other histone binding modules32–35, supporting the finding that BIRs are epigenetic readers with selectivity for H3.
Molecular mechanism for the BIRC2BIR3 association with H3
To gain insight into the molecular basis for the recognition of the H3 tail, we cocrystallized BIRC2BIR3 and H31–12 peptide and obtained a 1.8 Å resolution crystal structure of the BIRC2BIR3–H3 complex (Fig. 1i–k and Table 1). In the complex, BIRC2BIR3 folds into a compact globular structure consisting of five short α-helices, a three-stranded antiparallel β-sheet and a zinc-binding cluster (Fig. 1i). The histone peptide is bound in a groove formed by the β3 strand, α3 helix and the β3–α3 loop. Analysis of the structure reveals an extensive network of hydrogen-bonding, hydrophobic and electrostatic contacts that stabilize the complex. Ala1 of the H3 peptide occupies a highly negatively charged pocket lined by the side chains of Glu317, Asp320 and Glu325 of the protein (Fig. 1i,j). The N-terminal amino group of Ala1 of H3 is restrained through hydrogen bonds with the carboxyl group of Asp320 and the backbone carbonyl group of Cys315 (Fig. 1k). The backbone carbonyl of Ala1 of H3 forms a hydrogen bond with the indolyl nitrogen atom of Trp329, whereas the backbone amino and carbonyl groups of Arg2 of H3 are involved in hydrogen-bonding contacts with the backbone carbonyl and amino groups of Arg314. The side chain ε-amino moiety of Lys4 donates a hydrogen bond to the carboxyl group of Asp303, and the backbone amino group of Lys4 is hydrogen-bonded to the carbonyl oxygen atom of Gly312.
Table 1.
Data collection and refinement statistics
| BIRC2BIR3-H3 | BIRC2BIR3-LCL161 | |
|---|---|---|
| Data collection | ||
| Space group | P1211 | P321 |
| Cell dimensions | ||
| a, b, c (Å) | 33.7, 35.0, 35.6 | 104.7, 104.7, 27.1 |
| α, β, γ (°) | 90.0, 103.9, 90.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 1.73 (1.76–1.73)a | 2.4 (2.44–2.4)a |
| Rpim | 2.6 (29.3) | 3.8 (13.0) |
| I/σ(I) | 21.8 (2.0) | 21.0 (5.7) |
| Completeness (%) | 97.0 (77.7) | 100 (100) |
| Redundancy | 4.4 (1.7) | 13.5 (11.1) |
| Refinement | ||
| Resolution (Å) | 1.74 | 2.4 |
| No. reflections | 7,635 | 13,010 |
| Rwork / Rfree | 0.1623 / 0.2060 | 0.1725 / 0.2222 |
| No. atoms | 835 | 807 |
| BIR3 | 744 | 714 |
| H3/LCL161 | 32 | 35 |
| Zn | 1 | 1 |
| EDO | 24 | 3 |
| Water | 34 | 45 |
| B factors (Å2) | 22.7 | 37.6 |
| BIR3 | 22.0 | 37.2 |
| H3/LCL161 | 27.5 | 38.0 |
| Zn | 17.6 | 34.2 |
| EDO | 27.3 | 46.4 |
| Water | 30.9 | 41.3 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.017 | 0.007 |
| Bond angles (°) | 1.39 | 0.922 |
| Ramachandran plot | ||
| Most favored (%) | 95.6 | 97.65 |
| Allowed (%) | 4.4 | 2.35 |
| Outliers (%) | 0 | 0 |
Values in parentheses are for highest-resolution shell.
Overlay of the structures of BIRC2BIR3 in complex with H3, SMAC and caspase-9 reveals that these ligands occupy the same binding site of BIRC2BIR3 (Supplementary Fig. 2). The position of the backbone and even various side chain atoms of the three bound to BIRC2BIR3 sequences—ARTK of H3, AVPI of SMAC and ATPF of caspase-9—is remarkably similar, despite the fact that only the first alanine residue is conserved. The deletion of Ala1 and Arg2 in the H3 peptide (H3–33) notably reduced binding of BIRC2BIR2, BIRC2BIR3 and BIRC23BIR (Fig. 1a). Posttranslational modifications (PTMs), commonly found in H3, including methylation, acetylation and crotonylation of lysine residues, did not alter binding of BIRs, but methylation of Arg2 reduced binding of BIRC2BIR3 to H3 (Supplementary Fig. 3). Yet, the most notable PTM-induced effect was caused by phosphorylation of Thr3, which abrogated binding of BIRC2BIR3 but only slightly reduced binding of BIRC2BIR2, as seen in pulldown assays and NMR titration experiments (Fig. 2b–d). The structure of the BIRC2BIR3–H3 complex provides an explanation to the observed sensitivity of BIRC2BIR3. Thr3 is in close proximity to the negatively charged Ala1-binding pocket, therefore the presence of the negatively charged phosphate group should lead to electrostatic repulsion. Additionally, phosphorylation of Thr3 is unfavorable due to steric clashes with the bulky side chain of Trp329, the position of which is fixed as it forms a hydrogen bond with the carbonyl group of Ala1 (Fig. 1k). By contrast, BIRC2BIR2 contains less bulky His243 in place of Trp329, which may tolerate Thr3 phosphorylation, especially if it is at least partially protonated.
Fig. 2 |. BIRs mediate histone and DNA binding functions of BIRC2.
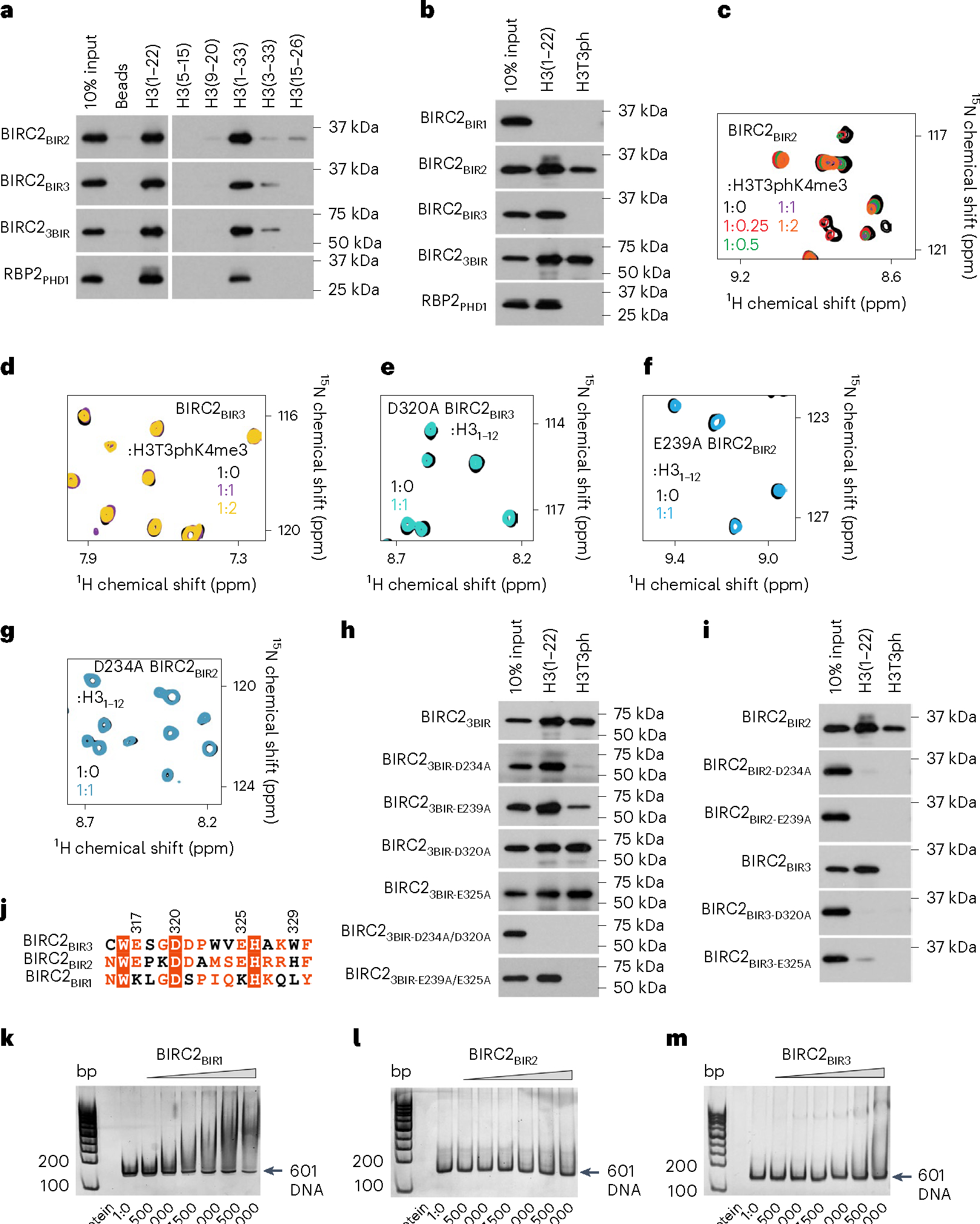
a,b, Western blot analysis of pulldowns of the GST-tagged BIR domains of BIRC2 with the indicated histone peptides. RBP2-PHD, control. The experiments were performed independently three times. c,d, Overlays of 1H,15N HSQC spectra of the BIR domains of BIRC2 collected before (black) and after addition of the indicated peptides. Spectra are color-coded according to the protein:peptide molar ratio. e–g, Overlays of 1H,15N HSQC spectra of the mutated BIRC2 BIR domains collected before (black) and after addition of the indicated peptides. Spectra are color-coded according to the protein:peptide molar ratio. h,i, Western blot analysis of pulldowns of the GST-tagged mutated BIRC2 BIR domains with the indicated histone peptides. The experiments were performed independently twice. j, Alignment of a fragment of the amino acid sequences of the BIRC2 BIR domains. Identical residues are highlighted by red boxes and moderately conserved residues are colored red. BIRC2BIR3 residues are labeled. k–m, EMSAs of 147 bp 601 DNA in the presence of increasing amounts of the indicated BIR domains of BIRC2. DNA:protein ratio is shown below gel images.
BIRs mediate histone and DNA binding functions of BIRC2
The importance of caging Ala1 of H3 for the interaction of BIRC2BIR3 and BIRC2BIR2 with the histone tail was corroborated by mutagenesis. We generated D320A and E325A mutants of BIRC2BIR3 and the corresponding D234A and E239A mutants of BIRC2BIR2 and tested them by NMR (Fig. 2e–g) and pulldown assays (Fig. 2h,i). As shown in Fig. 2e–i, substitution of the negatively charged residues with an alanine abrogated binding of individual BIRC2BIR2 and BIRC2BIR3 to H3 and H3T3ph; however, mutations in both domains (D234A/D320A) concurrently were needed to disrupt binding of the triple BIRC23BIR (Fig. 2i). Notably, the BIRC2BIR1 sequence contains positively charged lysine residues in place of Glu317 and Glu325 in BIRC2BIR3 (Fig. 2j), which likely accounts for the inability of BIRC2BIR1 to recognize H3 (Fig. 1e,f). As genetic mutations within BIRC2BIR2 and BIRC2BIR3 have been identified in various cancers, we also generated a set of cancer-relevant mutants and examined their histone binding activities in pulldown assays (Supplementary Fig. 4a). The cancer-relevant mutations in BIRC2BIR3, including L313F, W316R and D320N, eliminated binding to H3, and the BIRC2BIR2 H243W mutant lost its ability to tolerate Thr3 phosphorylation, pointing to a potential role of the BIR–H3 interactions in oncogenesis.
A number of chromatin-binding proteins have been shown to associate with both histone tails and DNA36. We tested whether BIRC2 has a DNA binding function using electrophoretic mobility shift assays (EMSA). Increasing amounts of BIRC2BIR1, BIRC2BIR2 and BIRC2BIR3 were incubated with 601 DNA, and the reaction mixtures were resolved on a native polyacrylamide gel (Fig. 2k–m). A shift of the free 601 DNA indicated formation of the BIRC2BIR1–DNA complex, whereas no DNA binding activity was detected for BIRC2BIR2 and BIRC2BIR3. These data indicate that BIRC2 can bivalently engage with chromatin through binding of BIRC2BIR2 and BIRC2BIR3 to H3 and binding of BIRC2BIR1 to DNA.
BIRC2 regulates cell defense and cell-cycle signaling
To identify genes regulated by BIRC2, we knocked out endogenous BIRC2 in the MCF7 breast cancer cell line using CRISPR–Cas9 and two sgRNAs targeting BIRC2 (Supplementary Fig. 4b) and extracted total RNA for next-generation sequencing. RNA-seq analysis identified 526 genes that were upregulated and 443 genes that were downregulated in cells expressing BIRC2 sgRNAs (Fig. 3a and Supplementary Table 1). Gene ontology (GO) analysis revealed that the genes involved in the interferon and defense response signaling pathways and cell-cycle regulation were most affected by the depletion of BIRC2 (Fig. 3b and Supplementary Table 2). In support of GO analysis, in the interferon-γ (IFNγ) stimulated ex vivo differentiated monocyte-derived macrophages, inhibition of IAPs with the XIAP inhibitor embelin led to an increase of interleukin-6 (IL-6), as observed in the cytokines antibody array (Fig. 3c). We also investigated the DNA damage response and cell-cycle regulation in BIRC2-overexpressing HeLa cells (Supplementary Fig. 5a–d). Cells were briefly treated with a low concentration of etoposide until the DNA damage marker γH2AX was detected (Supplementary Fig. 5b,c) and then cultured in a drug-free medium to allow for DNA repair and restoration of the normal cell cycle. Overexpression of BIRC2 delayed the DNA damage repair judging by the persistent level of γH2AX in western blot and flow cytometry assays, even 6 h after the drug removal (Supplementary Fig. 5b,c). The delay in the DNA damage repair was further corroborated by a flow cytometry analysis of the cell cycle (Supplementary Fig. 5d). Overexpression of BIRC2 delayed the recovery of cell-cycle progression and extended etoposide-induced G2/M cell-cycle arrest (Fig. 3d and Supplementary Fig. 5d).
Fig. 3 |. BIRC2 mediates cell defense and cell-cycle signaling.
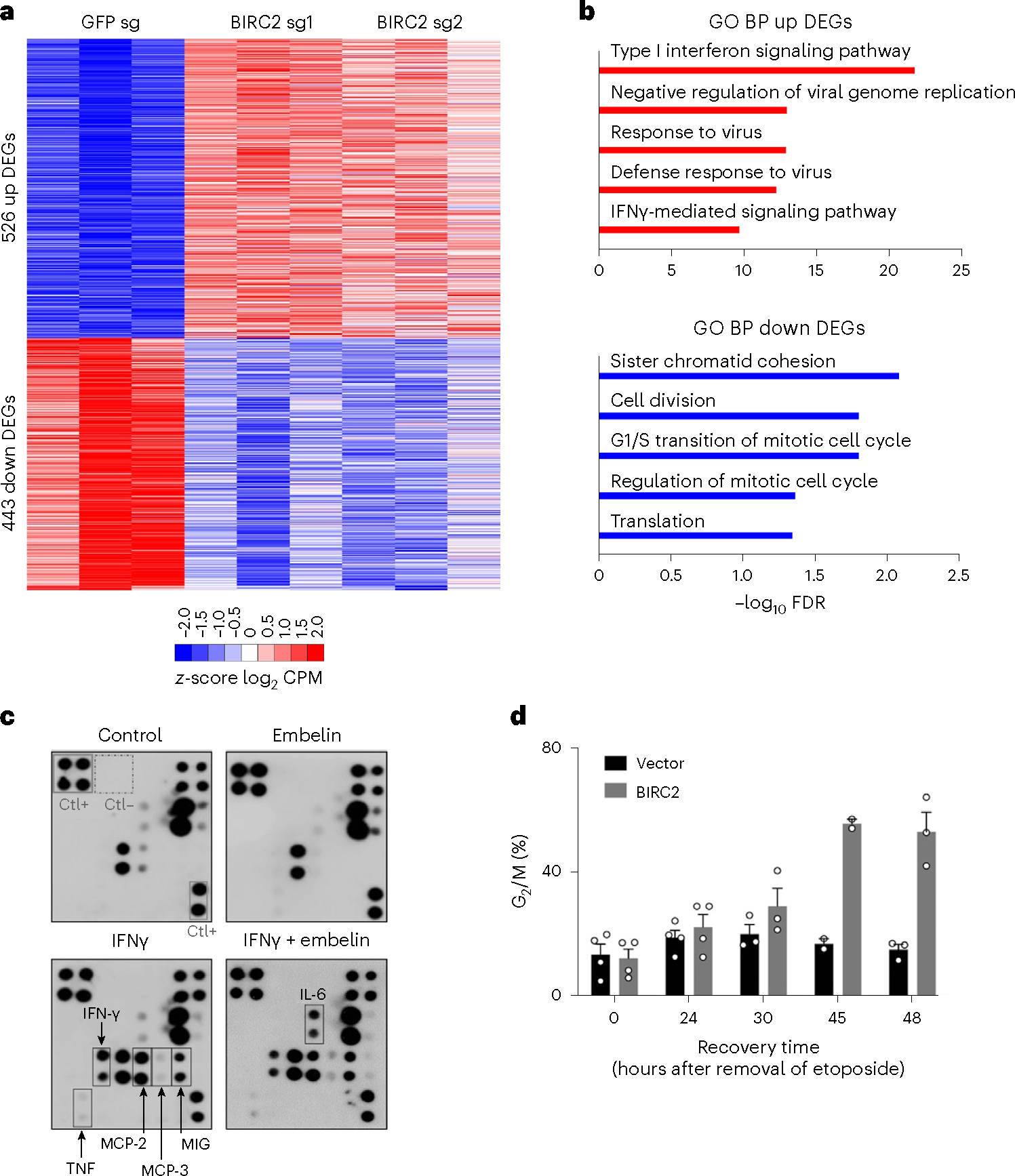
a, Heatmap of common DEGs in MCF7 cells treated with BIRC2 sgRNAs (sg1 and sg2) or the control GFP sgRNA. Genes are shown as z-score log2CPM (counts per million reads mapped). Upregulated genes are shown in red and downregulated genes in blue. Adjusted P values for DEGs were calculated by two-sided Exact test model. b, Top-ranked GO biological process terms of up (red) and down (blue) regulated genes of DEGs as in a in BIRC2-knockout cells. The full lists are shown in Supplementary Tables 1 and 2. c, Cytokine antibody array with supernatants derived from human macrophages obtained by ex vivo differentiation of normal blood monocytes and stimulated with IFNγ for 24 h in the presence of 10 μM embelin or vehicle as control. d, HeLa cells transfected with either BIRC2-encoding or empty vector were untreated or treated with etoposide, then washed and incubated in a drug-free medium. Percentage of cells in G2/M. Data represent mean ± s.e.m. from at least three independent biological replicates (two for 45 h).
LCL161 binds to BIRC2BIR3 and BIRC2BIR2 but not to BIRC2BIR1
Among several SMAC mimetic compounds developed in the past few years, LCL161 (Fig. 4a) displays one of the most promising antiproliferation and bioavailability properties25,26,37. While therapeutic potency of LCL161 has been established in preclinical and clinical studies, little is known about its binding mechanism. To better understand the molecular basis underlying its inhibitory activity, we investigated how LCL161 interacts with each BIR domain of BIRC2. LCL161 was titrated into NMR samples of individual 15N-labeled BIRC2BIR1, BIRC2BIR2 and BIRC2BIR3 while 1H,15N HSQC spectra were recorded, processed and overlaid (Fig. 4b–d). LCL161 caused CSPs in the spectra of BIRC2BIR3 and BIRC2BIR2 but not in the spectrum of BIRC2BIR1, revealing that BIRC2BIR3 and BIRC2BIR2 but not BIRC2BIR1 are direct targets of the compound. values, measured by tryptophan fluorescence, were found to be 490 nM for BIRC2BIR3 and 2.2 μM for BIRC2BIR2 (Fig. 4e,f).
Fig. 4 |. LCL161 binds to BIRC2BIR2 and BIRC2BIR3.
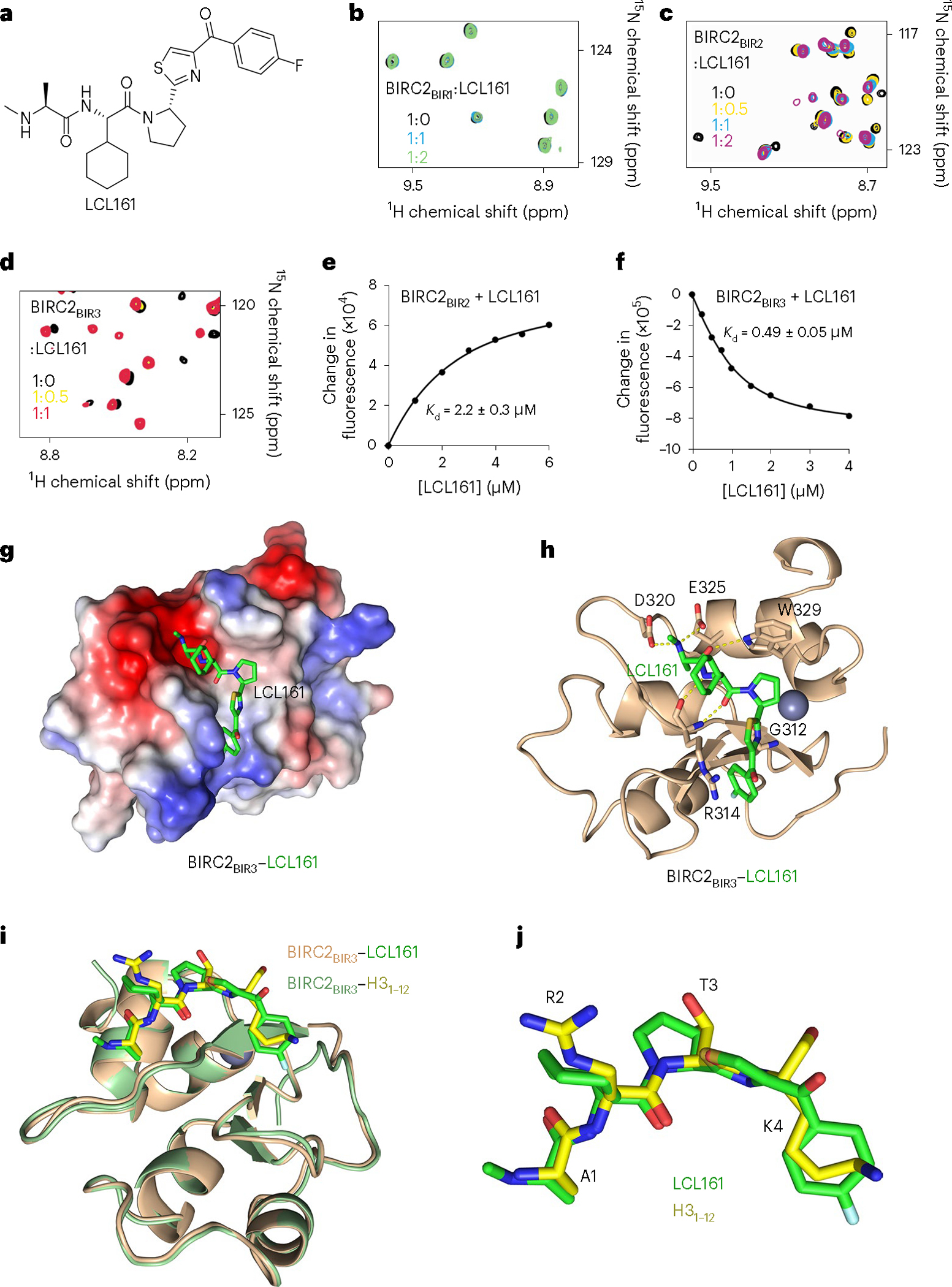
a, Chemical structure of LCL161. b–d, Superimposed 1H,15N HSQC spectra of 15N-labeled BIRC2BIR1, BIRC2BIR2 and BIRC2BIR3, collected while LCL161 was titrated in the NMR samples. Spectra are color-coded according to the protein:inhibitor molar ratio. e,f, Representative binding curves used to determine values by tryptophan fluorescence. The values were averaged over three separate experiments, with error calculated as s.d. between the runs. g, Electrostatic surface potential of BIRC2BIR3 is colored blue and red for the positive charge and the negative charge, respectively. The bound LCL161 compound is shown as green sticks. h, A ribbon diagram of the BIRC2BIR3 (wheat) in complex with LCL161 (green). Dashed lines represent hydrogen bonds. The zinc ion is shown as a gray sphere. i, Structural overlay of BIRC2BIR3 bound to LCL161 (green) and the H3 peptide (yellow). j, Superimposed LCL161 (green) and the H3 peptide (yellow) bound to BIRC2BIR3.
The mechanistic details of the interaction with LCL161 were obtained from the crystal structure of the BIRC2BIR3–LCL161 complex (Fig. 4g,h and Table 1). The complex is stabilized by a large set of hydrogen bonds. These include contacts between the side chain carboxyl groups of Asp320 and Glu325 and the methylamino group of LCL161, and between the carbonyl group of alanine in LCL161 and the indolyl moiety of Trp329. The amino group and the carbonyl group of cyclohexylglycine in LCL161 are hydrogen-bonded with the backbone carbonyl group and the amino group of Arg314, respectively. Additionally, the side chain guanidino group of Arg314 is near the p-fluorobenzoyl group in LCL161, whereas the thiazole ring of LCL161 is in close proximity to Gly312. LCL161 lies in the H3-binding site, ideally tracing the geometry of the H3 peptide (Fig. 4i,j), and overlays with GDC0152—another BIRC2 inhibitor (Supplementary Fig. 6).
LCL161 disrupts binding of BIRC2 to H3 and stimulates cell death
The impact of LCL161 on endogenous BIRC2 was tested in colon adenocarcinoma SW480 cells. As shown in Fig. 5a,b, treatment of the cells with LCL161 disrupted the association of BIRC2 with H3 in PLA. This result can be attributed to the binding of LCL161 to BIRC2BIR3 and BIRC2BIR2, as incubation of the isolated GST-tagged BIRC2BIR3 and BIRC2BIR2 with LCL161 abolished binding of BIRC2BIR3 to the H3 peptide and decreased binding of BIRC2BIR2 in pulldown experiments (Fig. 5c). Western blot analysis of BIRC2 following the treatment of SW480 cells with LCL161 for 1 h showed that LCL161 induces BIRC2 degradation (Fig. 5d). LCL161 treatment of IFNγ stimulated ex vivo differentiated monocyte-derived macrophages led to an increase in IL-6 and RANTES expression, supporting the cytokines antibody array data (Fig. 3c) and BIRC2 degradation (Fig. 5e–g).
Fig. 5 |. LCL161 disrupts binding of BIRC2 to H3 and induces cell death.

a, Representative images of the PLA (red) between BIRC2 and H3 in SW480 cells treated with or without 10 μM LCL161 for 1 h. Nuclei are stained with DAPI (blue). Scale bars, 10 μm. The experiment was performed independently three times. b, Quantification of the PLA signal with at least 75 cells analyzed per condition. Statistical analysis is described in Statistics and reproducibility. Two-sided Wilcoxon tests. P < 2.2 × 10−16. c, Western blot analysis of peptide pulldowns of GST-tagged BIRC2BIR3 with the H3 peptide (amino acids 1–22) in the presence of increasing concentrations of LCL161. The experiment was performed independently three times. d, Western blot analysis of BIRC2 in SW480 cells treated with or without 10 μM LCL161 for 1 h. β-actin was used as loading control. The experiment was performed independently three times. e, Western blot analysis of BIRC2 in human macrophages obtained by ex vivo differentiation of normal blood monocytes and stimulated or nonstimulated with IFNγ for 24 h in the presence or absence of 10 μM LCL161. β-actin was used as loading control. The experiment was performed independently three times. f,g, RT–qPCR analysis of IL6 (f) and RANTES (g) mRNA expression levels in human macrophages stimulated or nonstimulated with IFNγ for 24 h in the presence or absence of 10 μM LCL161. Data represent mean ± s.e.m. of at least three independent experiments. Paired two-sided t-test. h,i, MTT assay showing survival of mouse MEF (h) or human glioma LN18 cells (i) incubated for 24 h in medium containing the indicated concentrations of LCL161 and/or TNF. Data represent mean ± s.e.m. from three independent biological replicates.
To explore the effect of LCL161 on prosurvival function of BIRC2, we monitored cell survival following treatment with LCL161, tumor necrosis factor (TNF, previously known as TNFα), or both in a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay. Consistent with its ability to divert TNF receptor 1 (TNFR1) signaling toward cell death pathways, LCL161 sensitized mouse embryonic fibroblasts (MEFs) and human glioma LN18 cells to killing by TNF (Fig. 5h,i).
LCL161 mediates BIRCs degradation in HIV-1-infected CD4+ T cells
The mechanism by which LCL161 promotes cell death was examined in in vitro HIV-1-infected human CD4+ T cells (HIV-TCM). As identified previously, these HIV-TCM have increased expression of BIRC2 and BIRC3 (cIAP2), the TNF receptor superfamily (TNFRSF) death receptor FAS and both membrane-bound and soluble FAS ligand (FASLG) compared with TCM (Fig. 6a)38,39 and most express the lineage markers CD45RO, CD62L, CCR7 and CD27 while not expressing high levels of activation (HLA-DR and CD25) or cellular proliferation (MKI67) markers after 30 days culture with IL-7 (Supplementary Fig. 7). Under physiological conditions, the interaction of TNFRSF with its cognate ligand leads to receptor trimerization and the recruitment of FADD and caspase-8 to form a prominent death-inducing signaling complex (DISC). IAPs can inhibit this effect through the posttranslational ubiquitination of RIPK1, which activates canonical NF-κB prosurvival signaling pathways40,41. In cells with high expression of FAS and FASLG, loss of IAPs can result in the deubiquitination and autophosphorylation of RIPK1 at Ser166 that activates and increases RIPK1 recruitment to the FAS DISC alongside FADD, caspase-8 and cFLIP isoforms42,43. As the activation of FAS-induced apoptosis is known to be inhibited by IAPs, and FAS, FASLG, BIRC2 and BIRC3 are upregulated in HIV-TCM, we treated HIV-TCM and uninfected TCM with LCL161. LCL161 induced the rapid degradation of BIRC2 and BIRC3 in both HIV-TCM and uninfected cells (Fig. 6b). This correlated with the deubiquitination and Ser166 phosphorylation and thus activation of RIPK1 (Fig. 6b,c). We then examined the FAS DISC using coimmunoprecipitation for FAS. We readily detected the association of FADD, procaspase-8, activated caspase-8, RIPK1 (total, Ser166 phosphorylated and the cleaved form (p39)), cFLIPS (CFLARS) and the p43 fragment of cFLIPL (CFLARL) with FAS in HIV-TCM after LCL161 treatment (Fig. 6b), whereas FADD, cFLIPS and Ser166 phosphorylated RIPK1 did not associate with FAS in LCL161-treated TCM.
Fig. 6 |. LCL161 mediates BIRCs degradation in HIV-1-infected CD4+ T cells.
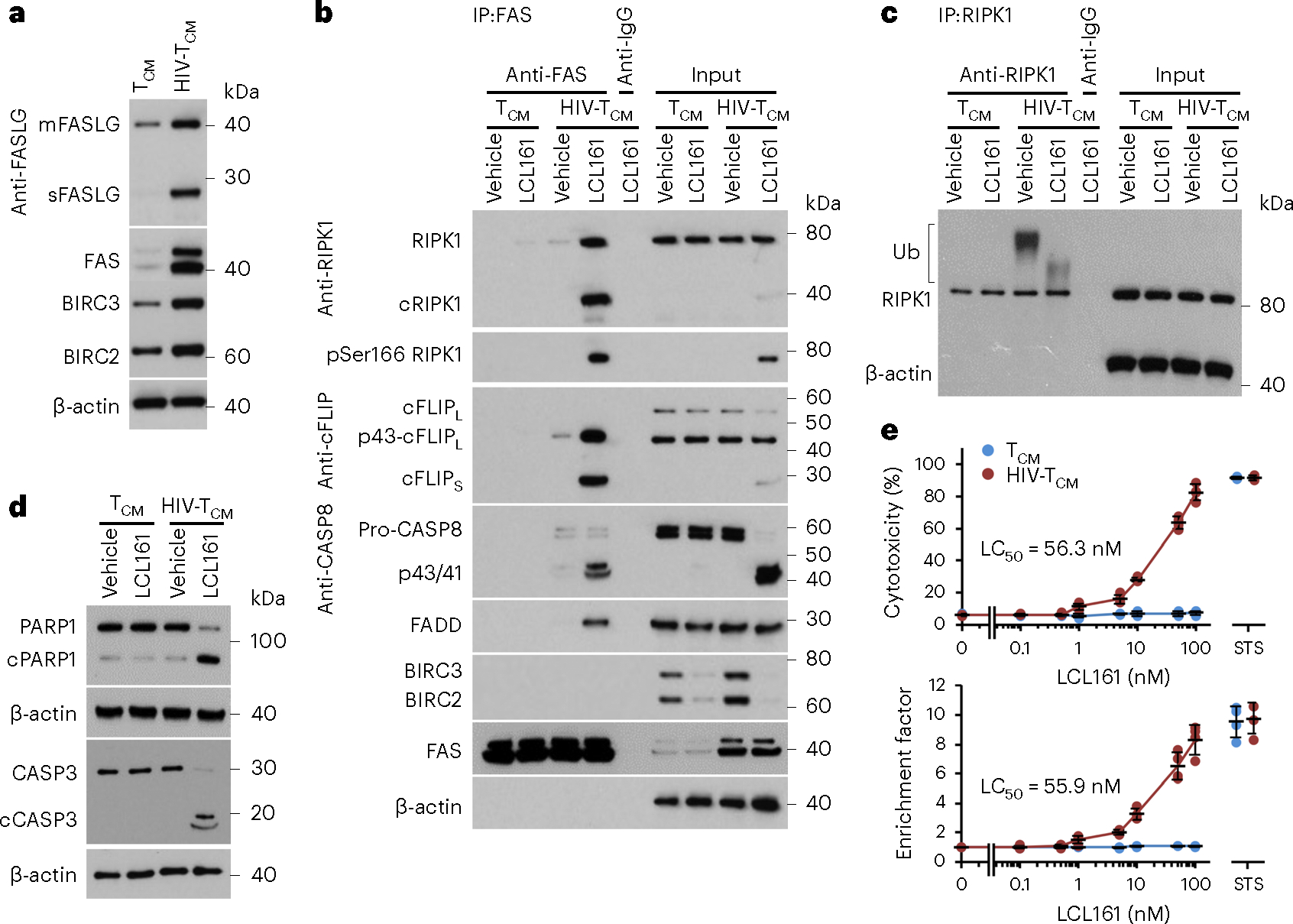
a, Representative western blots of FAS, membrane-bound FASLG (mFASLG), soluble FASLG (sFASLG), BIRC2 and BIRC3 in uninfected CD4+ T cells (TCM) and HIV-1-infected CD4+ T cells (HIV-TCM); n = 4. b, TCM and HIV-TCM were treated with 100 nM LCL161 for 4 h. FAS was immunoprecipitated using anti-FAS with a mouse IgG1 (anti-IgG as a negative control). Immunoprecipitates and cell lysates were analyzed by western blot using the indicated antibodies; n = 4. c, TCM and HIV-TCM were treated with 100 nM LCL161 for 5 min. RIPK1 was immunoprecipitated using anti-RIPK1 with a rabbit IgG (anti-IgG) as a negative control, and endogenous RIPK1 ubiquitination detected by western blotting using an RIPK1 antibody. n = 4. d, TCM and HIV-TCM were treated with 100 nM LCL161 for 4 h. Cell lysates were analyzed by western blot using the indicated antibodies; n = 4. e, TCM and HIV-TCM were treated with LCL161 or 2 μM STS for 4 h. Top, aliquots of supernatants were tested spectrophotometrically for LDH as a measure of cell death. Bottom, cells were harvested and cytoplasmic fractions assayed for cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) by ELISA (see Methods for calculation of enrichment factor); n = 4 biologically independent replicates. Data are presented as means of biologically independent replicates with grand mean values ± s.d.
The formation of the FAS DISC in HIV-TCM correlated with the cleavage of caspase-3, the proteolysis of poly(ADP-ribose) polymerase 1 (PARP1) (Fig. 6d), an increase in cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) (Fig. 6e), the translocation of phosphatidylserine from the inner to the outer leaflet (extracellular side) of the plasma membrane, permeabilization of the plasma membrane (Supplementary Fig. 8) and an increase in lactate dehydrogenase (LDH) release from HIV-TCM (Fig. 6e) but not in the uninfected cells, indicating that the cleavage of procapsase-8 within the FAS DISC resulted in the execution of apoptosis in the HIV-TCM. This suggests that, although RIPK1 was cleaved and cFLIPS was recruited to the FAS DISC, it was insufficient to prevent LCL161-mediated cell death44. In agreement with previous studies, a specific increase in HIV-TCM cell death was not accompanied by an increase in HIV-1 p24 antigen release from infected cells (Supplementary Fig. 7c,d)6,45. To confirm a role for the LCL161-mediated degradation of BIRC2 in the observed cell death, we performed RNA interference for BIRC2 (Supplementary Fig. 9a,b), which resulted in the death of HIV-TCM but not TCM (Supplementary Fig. 9c). Collectively, these data demonstrate that the presence of BIRC2 inhibits FAS-mediated cell death of HIV-TCM, and that the LCL161-mediated degradation of BIRC2 in HIV-TCM leads to the assembly of the FAS DISC and subsequent cell death.
In conclusion, in this study we elucidate a mechanism by which BIRC2 localizes to the cell nucleus. We found that BIRC2 engages with chromatin in a multivalent manner: while its BIRC2BIR2 and BIRC2BIR3 domains robustly recognize H3 tail, BIRC2BIR1 binds to DNA. We show that the nuclear pool of BIRC2 regulates a large set of the genes involved in the interferon and defense response signaling pathways and the cell-cycle regulation. In support, immunoprecipitation and flow cytometry assays demonstrate that BIRC2 negatively regulates DNA damage repair and recovery of cell-cycle progression and extends etoposide-induced G2/M cell-cycle arrest. The crystal structure of BIRC2BIR3 in complex with a small molecule inhibitor LCL161 along with binding affinities of BIRC2BIR2 and BIRC2BIR3 toward LCL161 provide mechanistic insights into the inhibition of BIRC2, which is instrumental in the development of a new generation of SMAC-derived inhibitors of IAPs with the goal of destroying cancer and HIV-infected cells.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-023-01044-1.
Methods
Cell culture
Human HeLa and SW480 cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle medium (DMEM) (Cellgro) supplemented with 10% FBS (Sigma), 2 mM l-glutamine and 100 U ml−1 penicillin/streptomycin. Human MCF7 and LN18 cells (purchased from ATCC) and SV-40 transformed MEF cells (a gift from A. Jabbour and P. Ekert) were cultured in DMEM with high glucose (Invitrogen) supplemented with 10% FBS (Sigma or Scientifix Life) and cells maintained at 37 °C in air supplemented with 5% CO2. HIV-1NL4–3 obtained from M. Martin via the National Institutes of Health (NIH) HIV Reagent Program was prepared and titered, and multiplicity of infection calculated as described39. Whole blood was drawn from HIV-1-seronegative healthy male and female volunteers, aged between 18 and 65 years, at University of California (UC) San Diego Health Sciences using protocols approved by the Human Research Protections Program of UC San Diego (approval no. 180485AW) in accordance with the requirements of the Code of Federal Regulations on the Protection of Human Subjects (45 CFR 46 and 21 CFR 50 and 56). All volunteers gave written informed consent before their participation, all samples were deidentified, and donors remained anonymous. Peripheral blood mononuclear cells (PBMC) were isolated from whole blood by density gradient centrifugation over Ficoll-Paque Plus (GE Healthcare) and HIV-1-infected, resting CD4+ T cells (HIV-TCM) were prepared from these PBMC and characterized as previously described39. Briefly, CD4+ T cells were isolated from PBMC using the CD4+ T cell isolation kit (Miltenyi Biotec, catalog no. 130–096-533) and incubated for 48 h in GM (RPMI 1640 supplemented with 10% (v/v) heat-inactivated FBS, 50 μM 2-sulfanylethan-1-ol (both Sigma), 100 μM nonessential amino acids, 1 mM sodium pyruvate, 0.1 mg ml−1 streptomycin, 100 U ml–1 penicillin (all Gibco)) supplemented with 29 nM CCL19 (R&D Systems) before infection with 0.4 multiplicity of infection HIV-1NL4–3 for 3 h. Cells were washed then plated at 5 × 105 cells ml–1 in GM supplemented with 250 ng ml−1 staphylococcal enterotoxin B (Sigma) and 25 U ml–1 IL-2 (Roche) and cultured for 3 days in 5% CO2 at 37 °C. Cells were then washed and resuspended at 5 × 105 cells ml–1 in GM supplemented with 25 U ml–1 IL-2 and cultured at 5% CO2 at 37 °C. Every 2–3 days, cell concentration was adjusted back to 5 × 105 cells ml–1 with 25 U ml−1 IL-2-containing GM and supernatant HIV-1 p24 release was monitored using the Perkin Elmer Alliance HIV p24 antigen ELISA. After 12 days memory CD4+ T cells were isolated by negative selection using a memory CD4+ T cell isolation kit (Miltenyi Biotec, catalog no. 130–091-893). Memory CD4+ T cells were then cultured in RPMI 1640 supplemented with 10% (v/v) heat-inactivated FBS, 0.1 mg ml−1 streptomycin, 100 U ml–1 penicillin and 1 ng ml IL-7 (R&D Systems) for 30 days at 37 °C, 5% CO2. Phenotyping and phytohemagglutinin-M (PHA; Sigma, catalog no. L8902) inducible virus was assessed by flow cytometry, and blinded quantification of HIV-1 integrated DNA was performed using a previously described nested Alu-LTR quantitative PCR (qPCR) assay39. LCL161 was obtained from Selleck Chemicals. Cell cytotoxicity was calculated using a LDH release assay according to the manufacturer’s protocol (Takara Bio).
Protein expression and purification
The human BIRC2 BIR domains BIR1 (aa 37–123), BIR2 (aa 175–260) and BIR3 (aa 261–346) were cloned into pGEX 6p-1 vectors. All constructs were expressed in Escherichia coli BL21(DE3) RIL cells in 15N-labeled M9 minimal medium or LB medium supplemented with 50 μM ZnCl2. Cultures were grown to an OD600 of 0.8 and induced with 0.5 mM IPTG for 18 h at 16 °C. Bacteria were harvested by centrifugation and lysed by sonication. GST-tagged proteins were purified on Pierce glutathione agarose beads (Thermo Scientific) using first 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 0.5% Triton X-100 and 2 mM DTT buffer, and then 20 mM Tris-HCl (pH 7.5), 150 mM NaCl and 2 mM DTT buffer. The GST-tag was cleaved with PreScission protease overnight at 4 °C. Proteins were further purified by size exclusion chromatography (SEC) and concentrated in Millipore concentrators (Millipore). For crystallographic studies of BIR3 and LCL161, 20 mM Bis-Tris Propane (pH 7.5), 150 mM NaCl and 2 mM DTT was used in SEC.
Chromatin fractionation
SW480 cells were lysed in 10 mM HEPES pH 7.8 buffer, supplemented with 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 0.2% (v/v) NP-40, 10% (v/v) glycerol and protease inhibitors (Complete protease inhibitor cocktail tablet, Merck, catalog no. 11697498001), and lysates were centrifuged. The nuclear fraction (pellet) was suspended in 3 mM EDTA buffer, supplemented with 0.2 EGTA and protease inhibitors, and centrifuged to separate supernatant (nucleoplasm) from the pellet. The pellet was successively subjected to gradient salt extraction with increasing concentrations of NaCl (0.1 M to 0.7 M) in 50 mM Tris buffer supplemented with 0.05% NP-40. Proteins of nucleoplasm and chromatin-associated fractions were then quantified and subjected to western blot analysis.
Proximity ligation assay
SW480 cells grown on No. 1.5 coverslips were fixed using 4% PFA in PBS for 10 min at room temperature. Cells were permeabilized by chilled methanol at −20 °C for 10 min. The PLA was carried out using Duolink protocol and anti-BIRC2 (Atlas Antibodies, catalog no. HPA005513, RRID:AB_1846746), anti-H3 (Cell Signaling, catalog no. 14269, 1B1B2, RRID:AB 2756816, dilution 1:1,000), anti-rabbit plus (Sigma-Aldrich, catalog no. DUO92002) and anti-mouse minus (Sigma-Aldrich, catalog no. DUO92004) PLA probes and PLA detection kit orange (Sigma-Aldrich, catalog no. DUO92007) in the presence or absence of 10 μM LCL161. Microscopy images were acquired using a Zeiss Axio Imager 2 (Zeiss GmBH) equipped with ×20 0.46 numerical aperture (NA), ×40 0.6 NA and ×63 1.45 NA Plan Apochromatic objectives and an mRM4 CCD (charge-coupled device) camera. Illumination was provided by an HXP120 metal halide lamp (Zeiss GmBH). Filter sets used were 49 HE (DAPI; catalog no. 488049–0000), 38 HE (Alexa488, catalog no. 489038–0000) and 43 HE (Alexa568, catalog no. 489043–0000). Image analysis was carried out using the ICY software (http://icy.bioimageanalysis.org) on ten randomly taken fields (based on DAPI). Nuclei were segmented using the Active Contour plugin on DAPI signal; PLA were detected using the Spot Detector plugin. Fluorescence signals were measured in maximal projections of unmodified images. Western blotting of endogenous BIRC2 was performed before and after the treatment of SW480 cells with 10 μM LCL161 for 1 h. SW480 cells were lysed in RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, supplemented with a protease and phosphatase inhibitor cocktail). Anti-BIRC2 and β-actin peroxidase conjugated antibodies were from R&D system (catalog no. AF8181) and Sigma-Aldrich (catalog no. 3854, clone AC-15, dilution 1:1,000).
IFNγ stimulation
Human peripheral blood monocytes were provided by French blood establishment (EFS, agreement no. deco-140037). They were obtained from healthy donors with informed consent, purified and differentiated ex vivo into macrophages as previously described46. Macrophages were then stimulated by 100 UI ml−1 IFNγ for 24 h with or without 10 μM embelin (Sigma-Aldrich) or 10 μM LCL161 in RPMI 1640 medium (Lonza), 10% fetal calf serum (Lonza). The supernatants were analyzed for the cytokines content by an antibody array (RayBiotech) following the manufacturerʼs protocol. The cytokines arrays were performed in duplicate. The arrays include positive control, positive control, negative control, negative control, GCSF, GM-CSF, GRO, GRO-α, IL-1α, IL-2, IL-3, IL-5, IL-6, IL-7, IL-8, IL-10, IL-13, IL-15, IFNγ, MCP-1, MCP-3, MIG, RANTES, TGFβ, TNF, lymphotoxin-α (LT-α, previously known as TNFβ) and positive control. IL6 and RANTES expression were quantified by RT–qPCR as previously described46. Western blotting of endogenous BIRC2 was performed before and after the treatment of macrophages with 10 μM LCL161 for 24 h as above.
DNA damage assay
HeLa cells were transfected with the pCI or pCI-BIRC2 vectors using JetPEI (Plyplus transfection, Ozyme). Cells were then treated with 1 μM etoposide for 3 h, washed and cultured for 0 to 48 h in a drug-free medium. DNA damage was evaluated by western blot and immunofluorescence analysis of γH2AX using anti-phospho-H2AX (Ser139) (Millipore catalog no. DAM1546024, Clone JBW301, Merckmillipore, dilution 1:100). For immunofluorescence, cells were fixed using 4% PFA in PBS for 10 min, washed, permeabilized for 10 min with 100% methanol at −20 °C and washed again. Nonspecific binding was blocked by 3% BSA for 1 h. Samples were then incubated overnight with anti-γH2AX (Cell Signaling, catalog no. 7631, clone D17A3, dilution 1:1,000), washed four times, incubated with Alexa Fluor 647 Donkey anti-rabbit IgG Biolegend (catalog no. 406414, dilution 1:5,000) for 1 h at room temperature, rinsed in water and analyzed on BD LSR II flow cytometer (BD Biosciences) (excitation: 635 nm laser light, detection: bandpass filter 620/20). For the cell-cycle analysis, cells were fixed in cold 70% ethanol for 30 min, washed, treated with ribonuclease and stain with propidium iodide solution (25 μg ml−1) before flow cytometry analysis (excitation: 488 nm laser light, detection: bandpass filter 575/26).
NMR experiments
NMR experiments were performed at 298 K on a Varian INOVA 600 MHz spectrometer equipped with a cryogenic probe. The NMR samples contained 0.1 mM uniformly 15N-labeled wild-type or mutated BIR domains of BIRC2 in 20 mM Tris-HCl (pH 7) buffer supplemented with 150 mM NaCl, 5 mM DTT and 10% D2O. Binding was characterized by monitoring chemical shift changes in 1H,15N HSQC spectra of the proteins induced by the addition of histone peptides (aa 1–12 of H3, synthesized by Synpeptides) or LCL161.
Fluorescence spectroscopy
Spectra were recorded at 25 °C on a Fluoromax-3 spectrofluorometer (HORIBA). The samples containing 1 μM BIR domains of BIRC2 (in 20 mM Tris (pH 6.8), 150 mM NaCl and 1 mM DTT buffer) and progressively increasing concentrations of peptides or LCL161 were excited at 295 nm. Emission spectra were recorded between 330 and 360 nm with a 0.5 nm step size and a 1 s integration time. The values were determined using a nonlinear least-squares analysis and the equation:
where [] is concentration of the peptide or inhibitor, [] is concentration of the protein, is the observed change of signal intensity and is the difference in signal intensity of the free and bound states of the protein. values were averaged over three separate experiments, and error was calculated as the s.d. between runs.
Microscale thermophoresis
MST experiments were performed using a Monolith NT.115 instrument (NanoTemper). The experiments were carried out on BIRC2BIR2 and BIRC2BIR3 in a buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM DTT. The final concentration of the C-terminal fluorescein-labeled H3 peptide (1–12, Synpeptide) was kept at 40 nM for the BIRC2BIR2-H31–12 interaction and 80 nM for the BIRC2BIR3-H31–12 interaction. Dissociation constants were determined using the binding assay where the FAM-labeled H31–12 peptide was added to the serially diluted samples of BIRC2BIR2 and BIRC2BIR3. For all measurements, the samples were loaded into standard capillaries, and the measurements were performed at 60% LED and 40% MST power. All the experiments were performed in triplicates and the values were determined using the MO.Affinity Analysis software (NanoTemper Technologies GmbH). Figures were generated using the GraphPad PRISM software with error bars shown as s.e.m. from average for each protein concentration. values were averaged over three separate experiments, and error was calculated as the s.d. between runs.
X-ray crystallography
The BIR3 domain of human BIRC2 (residues 261–346) was concentrated to 10 mg ml–1 and incubated on ice in a 1:1.2 molar ratio with either the H3 peptide (aa 1–12) or LCL161 for 1 h. Crystals of the BIRC2BIR3–H3 complex were grown using the sitting-drop vapor-diffusion method at 18 °C by mixing equal volumes of protein solution with well solution composed of 0.1 M SPG buffer (2:7:7 molar ratio of succinic acid:sodium dihydrogen phosphate: glycine) pH 7.0, supplemented with 25% PEG 1500. Crystals of the BIRC2BIR3–LCL161 complex were grown at 4 °C using the sitting-drop vapor-diffusion method in 0.1 M Bis-Tris Propane (pH 7.5), 0.2 M sodium citrate and 20% PEG 3350. X-ray diffraction data were collected from single crystals on the CU Anschutz Medical Campus X-ray crystallography core facility Rigaku Micromax 007 high-frequency microfocus X-ray generator equipped with a Pilatus 200K 2D area detector. The phase solutions for the BIRC2BIR3:H3 (Phenix) and BIRC2BIR3–LCL161 (CCP4) complexes were obtained by molecular replacement using the BIRC2BIR3 structure (PDB ID: 3D9T with the peptide, solvent and the zinc ion removed) as a search model. Indexing and scaling was completed using HKL3000. Manual model building was performed using Coot47, and the structure was refined using Phenix48. The final structure was verified on the PDB validation server. The X-ray diffraction and structure refinement statistics are summarized in Table 1.
Peptide pulldown assays
Biotinylated histone peptides (1 μg; synthesized by CPC scientific) with different modifications were incubated with 1 μg GST-fused BIRC2 BIR domains in binding buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.1% NP-40 and 1 mM PMSF) overnight with rotation at 4 °C. Streptavidin magnetic beads (Pierce) were added to the mixture, and the mixture was incubated for 1 h with rotation at 4 °C. The beads were then washed three times using a magnetic stand and the bound proteins were analyzed using SDS–PAGE and Western blotting. Anti GST antibodies were from Santa Cruz (catalog no. sc-459, AB_631586, final dilution 1:1,000).
RNA-seq
Total RNAs were extracted from MCF7 cells by RNeasy Plus Mini Kit (QIAGEN, catalog no. 74136). Illumina NovaSeq 6000 were used for generating polyA-plus 50 bp pair-end reads. Fastq reads were mapped to hg38 human genome by HISAT2 (v.2.1.0)49 with -k 1. Differentially expressed genes (DEGs) were calculated by edgeR (v.3.16.5)50 with Exact test model (false discovery rate (FDR) < 0.05). GO term enrichment was done by DAVID 6.8 (ref. 51).
Electrophoretic mobility shift assay
EMSAs were performed by mixing increasing amounts of BIR domains of BIRC2 with 0.25 pmol of 601 DNA per lane in 25 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5 mM EDTA in a 10 μl reaction volume. Reaction mixtures were incubated at room temperature for 10 min (2 μl of loading dye was added to each sample) and loaded onto a 5% native polyacrylamide gel. Electrophoresis was performed in 0.2× Tris-borate-EDTA (TBE) at 80–100 V on ice. The gels were stained with SYBR Gold (Thermo Fisher Sci) and visualized by Blue LED (UltraThin LED Illuminator-GelCompany).
MTT assays
Two thousand MEF or LN18 cells were seeded into wells of 96-well plates. Equal volumes of media containing LCL161 and/or TNF were added to the cells and incubated for 24 h. Tetrazolium (MTT) reduction by viable cells to produce formazan crystals was quantitated as a measure of survival52.
Flow cytometry
CD4+ T cells (1 × 105) were harvested, washed in PBS supplemented with 1 mg ml–1 bovine serum albumin and 100 μg ml−1 NaN3 (FACS buffer) and stained with the following fluorescently conjugated antibodies in FACS buffer for 30 mins at 4 °C: allophycocyanin (APC)-eFluor 780 conjugated anti-CD3 (catalog no. 47–0036-41, RRID:AB_10718679) or isotype control IgG (catalog no. 47–4714-80, RRID:AB_1271993), eFluor 450 conjugated anti-CD4 (catalog no. 48–0047-41, RRID:AB_1603232) or isotype control IgG (catalog no. 48–4714-80, RRID:AB_1271995), Super Bright 600 conjugated anti-CD25 (catalog no. 63–0259-42, RRID:AB_2637187) or isotype control IgG (catalog no. 63–4714-80, RRID:AB_2637447), peridinin chlorophyll protein (PerCp)-cyanine (Cy) 5.5 conjugated anti-CD25 (catalog no. 45–0251-80, RRID:AB_914323) or isotype control IgG (catalog no. 45–4301-80, RRID: AB_906256), Super Bright 645 conjugated anti-CD27 (catalog no. 64–0279-42, RRID:AB_2688222) or isotype control IgG (catalog no. 64–4714-80, RRID:AB_2665350), phycoerythrin (PE)-Cy7 conjugated anti-CD45RO (catalog no. 25–0457-42, RRID:AB_10718534) or isotype control IgG (catalog no. 25–4724-81, RRID:AB_470203), APC conjugated anti-CD45RO (catalog no. 17–0457-41, RRID:AB_1907398) or isotype control IgG (catalog no. 17–4724-81, RRID:AB_470188), APC conjugated anti-CD45RA (catalog no. 17–0458-42, RRID:AB_1907398) or isotype control IgG (catalog no. 17–4732-81, RRID:AB_763656), fluorescein isothiocyanate (FITC) conjugated anti-CD62L (catalog no. 11–0629-41, RRID:AB_10669578) or isotype control IgG (catalog no. 11–4714-81, RRID:AB_470021), PE-conjugated anti-CCR7 (catalog no. 12–1979-41, RRID:AB_10667886) or isotype control IgG (catalog no. 12–4321-80, RRID:AB_1834380), and PE-Cy5.5 conjugated anti-HLA-DR (catalog no. MHLDR18, RRID:AB_10372966) or isotype control IgG (catalog no. 35–4732-80, RRID:AB_11218699) (all from Invitrogen). Cells were then washed in FACS buffer, then fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) for 30 mins at 4 °C followed by staining with APC conjugated anti-MKI67 (catalog no. 17–5699-41, RRID:AB_2573217) or isotype control IgG (catalog no. 17–4714-81, RRID:AB_763650) (both from Invitrogen) or PE-conjugated anti-HIV-1 core antigen (Beckman Coulter, catalog no. 6604667, RRID:AB_1575989) or isotype control IgG (Invitrogen, catalog no. 12–4714-42, RRID:AB_1944423) for 30 mins at 4 °C. Cells were then washed in FACS buffer before blinded acquisition using a BD FACSAria II SORP flow cytometer with FACSDiva v.8.0.1 acquisition software (BD Biosciences) using 405 nm laser excitation with a 605/40 BP filter to detect Super Bright 600 and a 662/15 BP filter to detect Super Bright 645, a 488 nm laser excitation with a 530/30 BP filter to detect FITC, a 585/15 BP filter to detect PE, a 680/60 BP filter to detect PE-Cy5.5, and a 705/70 BP filter to detect PerCp-Cy5.5, a 561 nm yellow/green laser and a 660/20 BP filter to detect APC and a 780/60 BP filter to detect PE-Cy7, and a 633 nm red laser with a 450/40 BP filter to detect eFluor 450 and a 780/60 BP filter to detect APC-eFluor 780. Analysis was performed using FlowJo v.10 (BD Biosciences) using FMO and INC to determine gating strategy.
For cell death flow cytometry analysis of CD4+ T cells, 1 × 105 CD4+ T cells were harvested and stained with Alexa Fluor 488-conjugated annexin V (catalog no. A13201; Thermo Fisher) and 1 μg ml–1 propidium iodide (catalog no. P3566; Thermo Fisher) in binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) for 15 mins at 4 °C. Samples were then diluted 1:4 in additional binding buffer before blinded acquisition using a BD FACSCalibur flow cytometer with CellQuest Pro v.5.2.1 acquisition software (BD Biosciences) using the 488 nm blue laser for excitation and 530/30 BP filter for detection of Alexa Fluor 488 emission and 670/LP filter for propidium iodide. Analysis was performed using FlowJo v.10.8.1.
Lactate dehydrogenase assay
To assess the extent of cell death, LDH activity of supernatants was measured using a mixture of diaphorase/NAD+ and 3-(4-iodophenyl)-2-(4-nitrophenyl)-5-phenyl-2H-tetrazol-3-ium chloride/sodium 2-hydroxypropanoate according to the manufacturer’s protocol (Takara Bio). Staurosporine (STS) (Sigma-Aldrich catalog no. S6942) was used as a control.
Cell death detection enzyme-linked immunosorbent assay
To assess the qualitative and quantitative determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) after LCL161 or staurosporine (positive control) treatment, the Cell Death Detection ELISA (Roche) was used according to the manufacturer’s protocol. Enrichment factor is calculated using the following equation when mU is absorbance (10−3):
siRNA transfection
CD4+ T cells were transfected with Mission BIRC2 esiRNA (ID no. EHU002711) or control (catalog no. EHUEGFP) siRNA (siNS) using lipofectamine RNAiMAX transfection reagent (all Invitrogen) in Opti-MEM (Gibco) according to the manufacturer’s instructions; 48 h later, cells were analyzed for target gene silencing by western blot. Transfection efficiency was assessed with BLOCK-iT Alexa Fluor Red fluorescent control (catalog no. 14750100, Invitrogen) using a BD FACSCalibur flow cytometer with the 488 nm blue laser for excitation and a 585/42 BP filter.
Western blotting
Cell lysis, coimmunoprecipitation and western blotting of CD4+ T cells were performed as described previously39,53. Briefly, cell lysates were prepared using 20 mM HEPES, 150 mM NaCl and 1 mM EDTA supplemented with 1% Triton X-100 and 1% Thermo Scientific Halt protease and phosphatase inhibitor cocktail. Coimmunoprecipitation was performed using the Pierce Co-Immunoprecipitation Kit with 50 μg cell lysates and either 50 μg anti-FAS IgG1 (catalog no. 8023, RRID:AB_10860778) with a mouse IgG1 (catalog no. 5415, RRID:AB_10829607) as a negative control, or 50 μg anti-RIPK1 IgG XP (catalog no. 3493, RRID:AB_2305314) with a rabbit IgG XP (catalog no. 3900, RRID:AB_1550038) as a negative control (all from Cell Signaling) according to the manufacturer’s directions. Cell lysates and immunoprecipitates were resolved using 2-[bis(2-hydroxyethyl) amino]-2-(hydroxymethyl)propane-1,3-diol buffered polyacrylamide gels, transferred to 0.2 μm PVDF membranes, probed with primary antibodies overnight at 4 °C, followed by detection using alkaline phosphatase tagged secondary antibodies (Invitrogen) and 0.25 mM CDP-Star supplemented with 5% Nitro-Block II (both Applied Biosystems). The primary antibodies raised against the following were used in CD4+ T cell western blots: BIRC2 (catalog no. 7065, RRID:AB_10890862), BIRC3 (catalog no. 3130, RRID:AB_10693298), cFLIP (catalog no. 56343, RRID:AB_2799508), CASP3 (catalog no. 14220, RRID:AB_2798429), CASP8 (catalog no. 9496, RRID:AB_561381), FADD (catalog no. 2782, RRID:AB_2100484), FAS (catalog no. 4233, RRID:AB_2100359), FASLG (Cat# 4273, RRID:AB_2100652), PARP1 (catalog no. 9532, RRID:AB_659884), RIPK1 (catalog no. 3493, RRID:AB_2305314), Phospho-RIPK1-Ser166 (catalog no. 44590, RRID:AB_2799268) from Cell Signaling Technologies and ACTB (Sigma catalog no. A2228, RRID:AB_476697). Relative densities of the target bands were compared with ACTB and were calculated using Fiji v.1.53k (RRID:SCR_002285).
Anti-GAPDH antibodies were from Santa Cruz (catalog no. sc-32233, RRID:AB_627679, final dilution 1: 200), and anti-BIRC2 antibodies were from Abcam (catalog no. ab108361, RRID:AB_10862855, final dilution 1:1,000).
Statistics and reproducibility
Independent biological replicate sample size (n) of CD4+ T cell data was determined using a two-sample two-sided equality test with power (1 – β) = 0.8, α = 0.05 and preliminary data where the minimum difference in outcome was at least 70%. CD4+ T cell samples were assigned to experimental groups through simple random sampling. Statistical significance in Figs. 1d and 5b was assessed by a two-sided Wilcoxon test (n ≥ 75 cells analyzed per condition; P < 2.2 × 10−16). Box-and-whiskers plots are presented with interquartile box bounds (Q1: 25% and Q3: 75%); middle line represents the median. The whiskers extend to the most extreme value in the dataset in the Q3 + 1.5 × (Q3–Q1) range (upper whisker) or Q1–1.5 × (Q3–Q1) range (lower whisker). Adjusted P values for DEGs in Fig. 3a and Supplementary Table 2 were calculated by two-sided Exact test model. Statistical significance in Fig. 5f,g was determined using two-sided paired t-test. P values = 0.0191 for LCL161 versus IFNγ + LCL161 and 0.0092 for IFNγ versus IFNγ + LCL161 (Fig. 5f). P values = 0.0265 for vehicle versus IFNγ, 0.0147 for vehicle versus IFNγ + LCL161, 0.0235 for LCL161 versus IFNγ + LCL161, and 0.0454 for IFNγ versus IFNγ + LCL161 (Fig. 5g). MTT assay data represent mean ± s.e.m. from three independent biological replicates. No statistical methods were used to predetermine sample sizes. Except where indicated, investigators were not blinded to allocation during experiments and outcome assessment and the experiments were not randomized. No data were excluded from the analyses.
Supplementary Material
Acknowledgements
This work was supported in part by grants from the NIH nos. GM125195, GM135671, HL151334, CA252707 and AG067664 to T.G.K., nos. CA204020 and CA268440 to X.S., nos. MH128021 to G.R.C., nos. NS104015 to S.A.S. and no. CA255506 to H.W., by the Ligue Contre le Cancer to L.D., the European Union and the ‘Conseil Régional de Bourgogne,’ a French Government grant managed by the French National Research Agency under the program ‘Investissements d’Avenir’ (ANR-11-LABX-0021) to L.D. and the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (impaactnetwork. org) to S.A.S. Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network is provided by the National Institute of Allergy and Infectious Diseases of the NIH under award numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), with cofunding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Institute of Mental Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-023-01044-1.
Peer review information Nature Structural & Molecular Biology thanks Francisco Blanco, Ji-Joon Song and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Carolina Perdigoto, in collaboration with the Nature Structural & Molecular Biology team.
Reprints and permissions information is available at www.nature.com/reprints.
Code availability
This paper does not report original code.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 7TRL and 7TRM. RNA-seq data are deposited in the NCBI Gene Expression Omnibus (GEO) database with the accession number GSE23028. Source data and Supplementary Data files are provided with this paper. All other relevant data supporting the key findings of this study are available within the article, its Supplementary Information or from the corresponding authors upon reasonable request.
References
- 1.Gyrd-Hansen M & Meier P IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 10, 561–574 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Estornes Y & Bertrand MJ IAPs, regulators of innate immunity and inflammation. Semin. Cell Dev. Biol. 39, 106–114 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Mehrotra S et al. IAP regulation of metastasis. Cancer Cell 17, 53–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swingler S, Mann AM, Zhou J, Swingler C & Stevenson M Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 3, 1281–1290 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Ragupathy V, Zhao J & Hewlett I Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem. Biophys. Res. Commun. 414, 20–24 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Pache L et al. BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by Smac mimetics to promote reversal of viral latency. Cell Host Microbe 18, 345–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarnegar BJ et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dueber EC et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334, 376–380 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Bertrand MJ et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Varfolomeev E et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131, 669–681 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Verhagen AM et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Du C, Fang M, Li Y, Li L & Wang X Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Vince JE et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131, 682–693 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Huang X et al. XIAP facilitates breast and colon carcinoma growth via promotion of p62 depletion through ubiquitination-dependent proteasomal degradation. Oncogene 38, 1448–1460 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Varfolomeev E et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 283, 24295–24299 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uren AG et al. Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc Natl Acad. Sci. USA 96, 10170–10175 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glorian V et al. DNA damage and S phase-dependent E2F1 stabilization requires the cIAP1 E3-ubiquitin ligase and is associated with K63-poly-ubiquitination on lysine 161/164 residues. Cell Death Dis. 8, e2816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauer M et al. Induction of the DNA damage response by IAP inhibition triggers natural immunity via upregulation of NKG2D ligands in Hodgkin lymphoma in vitro. Biol. Chem. 394, 1325–1331 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Dogan T et al. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat. Cell Biol. 10, 1447–1455 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Cartier J et al. Cellular inhibitor of apoptosis protein-1 (cIAP1) can regulate E2F1 transcription factor-mediated control of cyclin transcription. J. Biol. Chem. 286, 26406–26417 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel T et al. cIAP1 localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 65, 210–218 (2005). [PubMed] [Google Scholar]
- 22.Plenchette S et al. Translocation of the inhibitor of apoptosis protein c-IAP1 from the nucleus to the Golgi in hematopoietic cells undergoing differentiation: a nuclear export signal-mediated event. Blood 104, 2035–2043 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Fulda S & Vucic D Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11, 109–124 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Chen SM et al. Targeting inhibitors of apoptosis proteins suppresses medulloblastoma cell proliferation via G2/M phase arrest and attenuated neddylation of p21. Cancer Med 7, 3988–4003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang YC & Cheung CH An updated review of Smac mimetics, LCL161, birinapant, and CDC-0152 in cancer treatment. Appl. Sci. 11, 335 (2021). [Google Scholar]
- 26.Fulda S Smac mimetics to therapeutically target IAP proteins in cancer. Int Rev. Cell Mol. Biol. 330, 157–169 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bai L, Smith DC & Wang S Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 144, 82–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell GR, To RK, Zhang G & Spector SA SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected macrophages. Cell Death Dis. 11, 590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welsh K et al. Characterization of potent SMAC mimetics that sensitize cancer cells to TNF family-induced apoptosis. PLoS ONE 11, e0161952 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu G et al. Structural basis of IAP recognition by Smac/DIABLO. Nature 408, 1008–1012 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Srinivasula SM et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410, 112–116 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Zeng L et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466, 258–262 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eustermann S et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 18, 777–782 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Tencer AH et al. Molecular mechanism of the MORC4 ATPase activation. Nat. Commun. 11, 5466 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musselman CA et al. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem. J. 423, 179–187 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vann KR, Klein BJ & Kutateladze TG Mechanistic similarities in recognition of histone tails and DNA by epigenetic readers. Curr. Opin. Struct. Biol. 71, 1–6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pemmaraju N et al. Final results of a phase 2 clinical trial of LCL161, an oral SMAC mimetic for patients with myelofibrosis. Blood Adv. 5, 3163–3173 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silvestris F et al. Overexpression of Fas antigen on T cells in advanced HIV-1 infection: differential ligation constantly induces apoptosis. AIDS 10, 131–141 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Campbell GR, Bruckman RS, Chu YL, Trout RN & Spector SA SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T cells. Cell Host Microbe 24, 689–702.e687 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McEleny K et al. An antisense oligonucleotide to cIAP-1 sensitizes prostate cancer cells to fas and TNFalpha mediated apoptosis. Prostate 59, 419–425 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Dynek JN et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laurien L et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11, 1747 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feltham R & Silke J The small molecule that packs a punch: ubiquitin-mediated regulation of RIPK1/FADD/caspase-8 complexes. Cell Death Differ. 24, 1196–1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwarzer R, Laurien L & Pasparakis M New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr. Opin. Cell Biol. 63, 186–193 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Campbell GR et al. CD4+ T cell-mimicking nanoparticles encapsulating DIABLO/SMAC mimetics broadly neutralize HIV-1 and selectively kills HIV-1-infected cells. Theranostics 11, 9009–9021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dupoux A et al. cIAP1-dependent TRAF2 degradation regulates the differentiation of monocytes into macrophages and their response to CD40 ligand. Blood 113, 175–185 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Tada H, Shiho O, Kuroshima K, Koyama M & Tsukamoto K An improved colorimetric assay for interleukin 2. J. Immunol. Methods 93, 157–165 (1986). [DOI] [PubMed] [Google Scholar]
- 53.Velikkakath AK, Nishimura T, Oita E, Ishihara N & Mizushima N Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 7TRL and 7TRM. RNA-seq data are deposited in the NCBI Gene Expression Omnibus (GEO) database with the accession number GSE23028. Source data and Supplementary Data files are provided with this paper. All other relevant data supporting the key findings of this study are available within the article, its Supplementary Information or from the corresponding authors upon reasonable request.