

Abstract

Family 1 glycosyltransferases (GT1s, UGTs) form natural product glycosides with exquisite control over regio- and stereoselectivity, representing attractive biotechnological targets. However, regioselectivity cannot be predicted and large-scale activity assessment efforts of UGTs are commonly performed via mass spectrometry or indirect assays that are blind to regioselectivity. Here, we present a large high performance liquid chromatography screening discriminating between regioisomeric products of 40 diverse UGTs (28.6% average pairwise sequence identity) against 32 polyphenols, identifying enzymes able to reach high glycosylation yields (≥90% in 24 h) in 26/32 cases. In reactions with >50% yield, we observed perfect regioselectivity for 47% (75/158) on polyphenols presenting two hydroxyl groups and for 30% (43/143) on polyphenols presenting ≥3 hydroxyl groups. Moreover, we developed a nuclear magnetic resonance-based procedure to identify the site of glycosylation directly on enzymatic mixtures. We further selected seven regiospecific reactions catalyzed by four enzymes on five dihydroxycoumarins. We characterized the four enzymes, showing that temperature optima are functions of the acceptor substrate, varying by up to 20 °C for the same enzyme. Furthermore, we performed short molecular dynamics simulations of 311 ternary complexes (UGT, UDP-Glc, and glycosyl acceptor) to investigate the molecular basis for regioselectivity. Interestingly, it appeared that most UGTs can accommodate acceptors in configurations favorable to the glycosylation of either hydroxyl. In contrast, evaluation of hydroxyl nucleophilicity appeared to be a strong predictor of the hydroxyl predominantly glycosylated by most enzymes.
Introduction
Pharmaceutically, glycosylation is notably used as a tool to modify the biological properties of small molecules to control uptake and targeted drug delivery systems.1,2 A key effect of glycosylation is the increase in water solubility which is limited in 40% of currently marketed drugs and about 90% of drugs in development.3 A particularly interesting group of potent drugs that is plagued by poor solubility are polyphenols, including coumarin derivatives.4 Some coumarins display anti-inflammatory,5,6 antioxidant,7 anticancer,8 antimicrobial,9 or antiviral properties.10 Furthermore, coumarin glycoside derivatives are used as backbones in fluorescent probes and as inhibitors in α-glucosidase assays.11,12 Befitting their name, polyphenols generally present multiple glycosylation sites. Unfortunately, regiospecific glycosylation is challenging in organic chemistry and often relies on the use of protection groups, resulting in poor atom economy.13 Although significant efforts have been made toward the use of organometallic and transition metal catalysts in stereo and/or regiospecific glycosylation, a generic catalytic system has not been found.14 Conversely, we can emulate glycosylation processes found in nature, where glycosylation reactions are carried out regioselectively and stereoselectively under physiological conditions by glycosyltransferases (GTs).
Glycosylation of natural products is predominantly carried out by enzymes of the UDP-dependent glycosyltransferase family (UGT) that phylogenetically belongs to the glycosyltransferase family 1 (GT1) of the CAZy database.15 These GT1 enzymes use UDP-glucose as the activated sugar donor and are often referred to as Leloir enzymes, as opposed to non-Leloir glycosyltransferases that utilize phosphorylated sugars or other glycosyl donors.16 GT1 enzymes display a GT-B fold, with the catalytic site at the interface of two Rossmann-like domains, the N-terminal domain being more conserved and predominantly involved in the binding of the sugar donor and an aglycone-binding C-terminal domain. Currently (August third, 2023), only 339 of the 39,522 sequences in GT1 have been characterized and new sources of enzymes are continually discovered as genomes are sequenced and annotated.17,18
In order to assess the synthetic capabilities of UGTs for polyphenol glycosylation, 40 UGTs were screened against 32 polyphenols/natural products by reverse phase high performance liquid chromatography (HPLC). For most compounds (26/32), we found GT1s able to glycosylate them in analytic high yields (>90%). We further identified five dihydroxycoumarin derivatives that result in seven distinct products through regiospecific glycosylation by four UGTs (Scheme 1). Additionally, we developed a nuclear magnetic resonance (NMR)-based method to determine the glycoside structures directly in enzymatic mixtures. The Michaelis–Menten kinetic parameters were determined for the seven studied enzyme–acceptor pairs as well as their pH and temperature profiles. Moreover, we analyzed molecular dynamics simulations of 311 possible ternary (UDP-Glc/enzyme/acceptor) Michaelis complexes, showing that almost all of those can adopt configurations that seem potentially reactive in silico—even those for which no product formation was observed in vitro.
Scheme 1. Glycosides Generated in This Study through Regiospecific Glycosylation by RhUGT1 (Blue), OsUGT88C1 (Green), GmUGT88E3 (Purple), and AtUGT78D2 (Red).

Results and Discussion
Polyphenol Glycosylation Screen
The enzyme panel consists of a variety of UGTs with sequence identities ranging from 10 to 90% (Supporting Information identity matrix). The 32 polyphenols are a subset of a natural compound library (TargetMol, USA), selected relative to their molecular weight (200–400 kDa), their reported biological activities, and their ability to present at least 2 glycosylation sites. In vitro reactions were prepared as described in the Method Section and analyzed by RP-HPLC. Most polyphenols (26/32) could be glycosylated with high yields (≥90%) by at least one UGT (Supporting Information data set). Interestingly, the only polyphenol that was not glycosylated with over 50% yield by any of the UGTs was vitexin, the only polyphenol glucoside assayed, with a maximum of 46% conversion by UGT71E5. For the 158 reactions presenting >50% yield, we observed perfect regioselectivity for 47% (75/158) on the 18 polyphenols with two hydroxyl groups, and for 30% (43/143) on the 14 polyphenols with ≥3 hydroxyl groups. Interestingly, no correlation was found between the number of potential glycosylation sites and observed overall glycosylation yields. Moreover, there was also no strong correlation between phylogeny and glycosylation patterns, e.g., the three most related enzymes (having over 80% sequence identity and belonging to the group UGT72B) have numerous differences both in terms of acceptor preference and regioselectivity (Supporting Information data set).19
Dihydroxycoumarin Glucosylation
We further focused on four dihydroxycoumarins (4,7-dihydroxycoumarin) (a), 4-methylesculetin (c), 5,7-dihydroxy-4-methylcoumarin (d), and serratin (e) that resulted in a single product upon reaction with at least one UGT. For each unique reaction, the most efficient enzyme was chosen, resulting in the following panel of enzymes: RhUGT1, OsUGT88C1, GmUGT88E3, and AtUGT78D2.
RhUGT1 has a broad substrate range; the list of acceptors identified by Wang et al. includes flavones, flavonols, flavanones, isoflavones, and chalcones.20 Similarly, GmUGT88E3 is able to glycosylate a broad range of acceptors, including flavones, flavanones, flavonols, an aurone, a coumarin, and a chalcone.21−23AtUGT78D2 is explored less extensively; nevertheless, it is described as a flavonol-3-O-glycosyltransferase for the conversion of kaempferol and quercetin to their corresponding glucosides.24−26OsUGT88C1 is the least-described enzyme used in this study and shown to have activity toward apigenin, resveratrol and scopoletin.27
In addition to the four dihydroxycoumarins that already had been shown to be good substrates giving rise to a single product, we added esculetin (b), which was not part of the initial screen, as a potential acceptor for OsUGT88C1 since b only differs from c at position 4. This resulted in a substrate panel with five dihydroxycoumarin substrates, of which a and b present no additional substitutions, c and d a methyl group at position 4, and e a phenyl group at position 4. In order to identify the optimal reaction conditions for each enzyme–acceptor pair and gain insight into the influence of the acceptor on the reaction conditions, the seven enzyme–acceptor pairs were characterized for pH and temperature dependency, glycosylation site, and kinetic parameters.
Biochemical Characterization
Generally, the enzymes present activity at a broad pH range (Figure 1), with pH optima around 7.5–8. AtUGT78D2 additionally has a second increase in activity at a high pH, observed for both acceptors d and e. Product formation was measured in the temperature range 30 to 54 °C (Figure 1). Interestingly, the acceptor influences the temperature activity profile of the enzyme–acceptor pair. This is most pronounced for OsUGT88C1, where the temperature optimum shifts from <30 °C for c to 40 °C for e. Moreover, the activity of AtUGT78D2 on d is near constant in the range 30–50 °C, whereas the activity on e has a clear optimum at 45 °C. Generally, we observe a decrease in activity above 45 °C, particularly for the later time points, likely due to the thermal instability of UGTs.27−29
Figure 1.

Biochemical characterization. Carried out with 100 μM acceptor and 500 μM UDP-Glc in the presence of UGT as described in the Experimental Section. (A) Initial rate of product formation is plotted against the corresponding pH value. The maximum activity is defined as the highest observed rate of product formation at 293 K. (B) Temperature profiles for each reaction pair corresponding to analytical yields of product formation at optimal pH as determined in (A) and at different time points.
Kinetics
Kinetic analysis was carried out for each enzyme–acceptor pair with an acceptor range 2.9–250 μM (Table 1 and Figure S1). The obtained Km values were quite similar for all enzyme–substrate pairs, in the tens of micromolar range (10–70 μM). However, large variations were observed in terms of kcat, ranging from 321 min–1 for OsUGT88C1 on c to 1.95 min–1 for RhUGT1 on a. The impact of the phenyl group at the 4-position is clearly observed by the decrease in the kcat value between d and e. When we compare the kcat values and temperature profile for OsUGT88C1, we see that the high kcat values with b and c correspond to the lower temperature optimum compared to the reaction with e, which is barely influenced by temperature up to 40 °C but has a lower kcat value. Similarly, AtUGT78D2 has a 7-fold higher kcat with d than with e. However, here the reaction with the highest kcat value is barely influenced by temperature but we see a relatively high optimal temperature for the reaction with e.
Table 1. Michaelis–Menten Kinetic Parameters at 293 K with Optimal pH and Glucoside Products as Determined by 1H NMR.
| enzyme | Km (μM) | kcat (min–1) | product | |
|---|---|---|---|---|
| a | RhUGT1 | 41.2 ± 5.8 | 2.0 ± 0.1 | 7-O-Glc |
| b | OsUGT88C1 | 11.5 ± 2.1 | 183 ± 8.7 | 6-O-Glc |
| c | OsUGT88C1 | 37.8 ± 5.1 | 321 ± 15 | 6-O-Glc |
| d | GmUGT88E3 | 40.9 ± 4.7 | 190 ± 7.7 | 7-O-Glc |
| d | AtUGT78D2 | 34.4 ± 4.3 | 55.9 ± 2.4 | 5-O-Glc |
| e | OsUGT88C1 | 67.1 ± 9.0 | 6.3 ± 0.3 | 5-O-Glc |
| e | AtUGT78D2 | 28.9 ± 4.8 | 7.9 ± 0.4 | 7-O-Glc |
Glucoside Structure Determination
UGTs that catalyzed the formation of a single product with >90% yield in 24 h (Supporting Information data set) were chosen. The glucoside structures were identified directly from the reaction mixture by 1H NMR spectroscopy. Through targeted irradiation of the anomeric α-proton, the neighboring aromatic protons were recognized through the nuclear Overhauser effect (NOE) that resulted in the identification of the glycosylation site on the coumarin backbone S3–S9 (Scheme 1 and Table 1).
Substrate a is glycosylated at position 7 further from the lactone moiety in the coumarin backbone. Moreover, substrates b and c, esculetin and 4-methylesculetin, are glycosylated at position 6, resulting in esculin and 4-methylesculin, respectively. Next to the broad range of flavonoids and other polyphenolic compounds, GmUGT88E3 catalyzes the formation of coumarin-7-O-glycoside, d7. Interestingly, AtUGT78D2 glycosylates the 5-position in d and the 7-position in e. Moreover, OsUGT88C1 glycosylates the 5-position in e, as opposed to the 6-position in b and c.
In order to further investigate what governs UGTs’ regioselectivity, we turned to structural modeling, docking, and molecular dynamics (MD).
In Silico Inspection of Reactive Pairs
MD simulations were carried out on enzyme–acceptor pairs of observed reactions (Figure 1), using AlphaFold2-modeled structures and acceptors docked to the binding sites with UDP-glucose superimposed. We recently showed that such simulation on the ternary complexes could rationalize GmUGT88E3 specificity,30 and visualization of such complexes has proven to be a solid base for UGT engineering.31−34 Based on the reaction mechanism,35,36 we considered a conformation productive when all following criteria are satisfied: (i) the distance between the nucleophilic oxygen (proton donor) and the catalytic histidine (NεHis–Ocoum) is below 3.5 Å; (ii) the angle between mentioned hydrogen, donor, and nitrogen is below 30°; (iii) the nucleophilic attack distance (C1glc–OHcoum) is below 5 Å; and (iv) the angle formed by O1glc–C1glc bond and reactive oxygen of acceptor is above 130°. During simulations, every experimentally observed reactive enzyme/acceptor pair formed productive Michaelis complexes in silico (Figures 2 and S10).
Figure 2.

Selected representative snapshots of MD simulations showing reactive binding poses of every enzyme–acceptor pair. Catalytically relevant distances (Å) are shown.
While one glycosylation site was largely preferred for acceptors a, b, and c across all UGTs, both regioisomers are formed with d and e (Supporting Information data set), hinting that molecular interactions within the active site direct specificity. Coumarin d with GmUGT88E3 presents a strong hydrogen bond between the 5-OH and E329, and electrostatic interaction between H92 and the lactone moiety of d points to the 7-OH to the catalytic dyad. The hydrophobic pocket of AtUGT78D2 enables a tilted fit of d, which, in turn, positions 5-OH in a reactive pose with a hydrogen bond between 7-OH and the carbonyl functionality of F20 in the protein backbone. For e, a hydrophobic pocket of AtUGT78D2 allows the acceptor to fit more tightly in the active site via hydrophobic interactions governed by the F125, W144, A146, F204, and L208. This exposes 7-OH of the substrate for glycosylation. OsUGT88C1, on the other hand, presents a differently oriented and less hydrophobic pocket that allows e to expose 5-OH for glycosylation. The pocket is formed by F121, F122, I147, F201, and Y141, which in AtUGT78D2 is replaced by W144.
In Silico Michaelis Complexes and In Vitro Regioselectivities
Since acceptors d and e can both result in two different glycosidic products with the screened UGT library, we decided to investigate all possible Michaelis complexes for these acceptors for the 34 plant UGTs in our library for all possible regioselectivity—i.e., to also analyze the ones we do not observe experimentally. Initial restraints were applied to form all possible complexes; then relevant geometrical parameters were monitored after constraints were released. Strikingly, 32/34 UGTs appeared able to form a productive catalytic conformation for at least one glycosylation position with d, despite 8 of them being completely inactive on the acceptor in vitro (Table S1). Moreover, >75% of frames displayed productive configurations from 9 and 6 UGTs for forming d5 and d7, respectively. However, no product formation was observed with d as an acceptor for three of these UGTs (AtUGT74F1, LbUGT75L5 and LuUGT85K6) Likewise, 32/34 complexes with e appeared to adopt potentially reactive configurations, including 10 that were found to be inactive in vitro.
To assess the correlation between experimental and simulated regioselectivity, two measures were calculated for each enzyme–acceptor pair: (1) the difference between the experimental yield of 5-O-glucoside and 7-O-glucoside and (2) the difference between fractions of reactive poses along the unrestrained simulations for 5-O-glycosylation and 7-O-glycosylation. Kendall’s correlation coefficients showed that experimental and simulated preferences were not correlated (τ = 0.08; −0.09, for d and e, respectively) (Figure 3).
Figure 3.
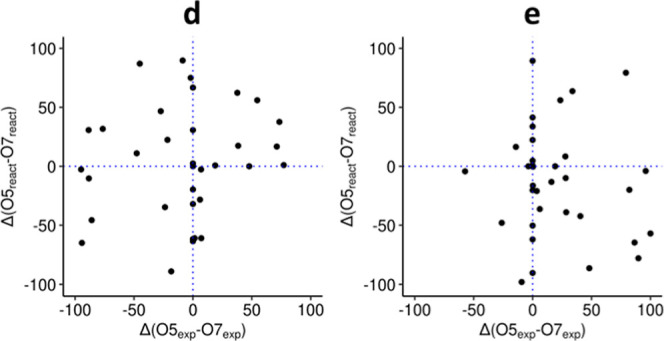
Simulated UGT regioselectivity preferences against experimental preferences with acceptors d and e. Each dot represents the difference between experimentally observed d5 and d7 yields (x-axis), and the difference between the proportion of frames displaying productive configurations for the 7-O- or the 5-O-glucoside (y-axis). Positive values on the x-axis indicate an excess of 5-O-glucoside. Positive values on the y-axis indicate an excess of reactive poses for 5-O-glycosylation. Dotted lines indicate boundaries between preferences.
Additionally, three more sets of simulations were carried out for a subset of 14 enzymes, which showed regioselectivity on dihydroxycoumarins. These simulations included different types of initial restraints, including NεHis–OHcoum hydrogen bond distance (1), C1glc–OHcoum nucleophilic attack distance (2), and the former combined (3). Interestingly, for this set of UGTs, all 56 possible enzyme glycosylation combinations appeared reactive at some point with both acceptors d and e, across all simulations (Tables S1–S3). Moreover, no correlation was observed between experimental yields and reactive pose fractions (Kendall’s τ = 0.21, 0.36, and 0.08 for acceptor d, sets 1, 2, and 3, respectively; τ = 0.04, −0.16, and −0.02 for acceptor e, sets 1, 2, and 3, respectively) (Figure 4).
Figure 4.
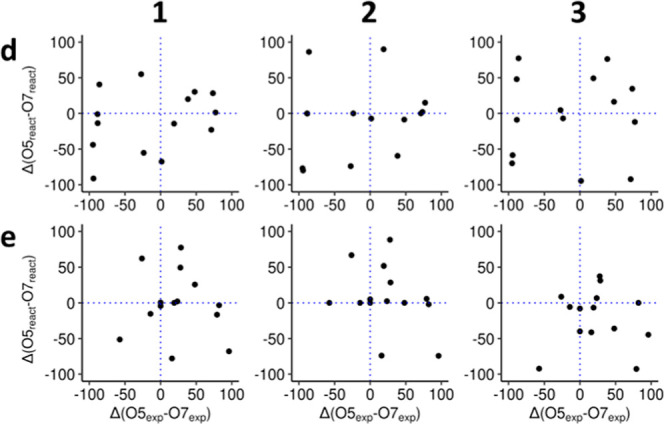
Simulated preferences against experimental preferences for acceptors d and e and the 14 UGTs that show stereospecificity against d or e. Positive values on the x-axis indicate excess of 5-O-glucoside. Positive values on the y-axis indicate excess of reactive poses for 5-O-glycosylation. Dotted lines indicate boundaries between preferences.
This apparent discrepancy between in vitro and in silico is consistent with our previous finding that molecular mechanics alone were not satisfactorily explaining the effect of mutations on reactivity in PtUGT1, which were then only rationalized using first principles (i.e., QM/MM).36 This suggests that reactivity is governed more by the stabilization along the path from Michaelis complex to transition state, rather than the possibility to form productive complexes. Moreover, as products mixtures are experimentally observed for most GT1/acceptor pairs, activation energy differences between reactions yielding different regioisomers are small. Indeed, even a 90/10 ratio indicates a difference in activation energies of only 1.5 kcal mol–1, for reactions which have typically activation energies of the order of 18 kcal mol–1.36 It is also important to point out that the apparent discrepancy could come from statistical noise, as only few short simulations are analyzed—a more exhaustive investigation being out of the scope of this study. Note that obtaining reactive poses was successful for experimentally observed reactions, and that the lack of correlation is to be found with our attempt to evaluate reactions that do not happen. These results stress the limits of docking approaches to evaluate UGTs reactivity and to guide UGTs engineering.
Chemical Reactivity and Regioselectivity
Interestingly, for 21/32 acceptors, the formation of one specific glucoside was overall favored by the enzymes in the data set; e.g., the 6-glucoside of 4-methylesculetin (c) was formed at an average yield of 39% by the 40 enzymes, with a maximum at 97%. Conversely, its 7-O-glucoside was formed with an average yield of 4% and a maximum yield of 22% (Supporting Information data set). Similarly, for 5,7-dihydroxy-4-phenylcoumarin (e), 5-O-glucoside was largely favored (maximum yield of 100%, average yield 36%) over 7-O-glucoside (maximum yield of 59%, average yield 13%). We hypothesize that the keto-enol tautomerization and the consequent low pKa in 4-hydroxycoumarins result in an unfavorable glycosylation site at the 4-OH position.37,38 Similarly, tautomeric forms of b and c have been proposed where the 7-OH tautomerizes to the carbonyl moiety with is structurally not possible to occur at the 6-position.39−42 Hence, chemical properties of the different hydroxyls may appear to be relevant predictors for promiscuous activities of GT1s on polyphenols.
Conclusions
We presented a large data set of UGT activity including regioselectivity data and developed a method for identifying the regioselectivity directly on enzymatic mixtures.
The generated data set resulted in the identification of seven regioselective glycosylation reactions on dihydroxycoumarins, a group of compounds with several applications. Through 311 MD simulations of ternary Michaelis complexes, we observed that most UGTs seem able to accommodate d and e, even though several of the corresponding reactions were not observed experimentally. It is unsurprising that small hydrophobic acceptors would bind the UGTs’ relatively large hydrophobic acceptor subsites ambiguously.43
Across the 1280 observed reactions, we also observed that for most acceptors all UGTs seem to favor the same regioselectivity. It should be stressed that we are investigating and observing here the effects of promiscuous activities and probing their biotechnological interest—not natural activities. Accordingly, glucoside structures predominantly formed regardless of the enzymes could be rationalized by chemical reactivity, e.g., through tautomeric forms of the acceptors.
Experimental Section
Materials
Buffers, chemicals, and reagents were purchased from commercial vendors. The acceptor library originates from a polyphenolic natural product library (L6100, TargetMol, USA).
Expression and Purification
The full-length histidine-tagged DNA sequences were cloned into a pET28a(+) expression vector by GenScript (USA). The plasmids were transformed into E. coli BL21 Star(DE3) (Fisher Scientific), and transformants were stored as glycerol stocks at −70 °C. Overexpression of the gene of interest was induced by the addition of 250 μM IPTG to the E. coli cultures that had reached OD600 = 0.8–1.0 in 2xYT medium at 37 °C (200 rpm). Thereafter, the cultures were incubated for 20 h at 20 °C (200 rpm). The cultures were harvested and stored at −20 °C until further use. For purification, the cell pellet was resuspended in 50 mM Na-phosphate buffer (pH 7.4), and lysis was carried out by 2 rounds of high-pressure homogenization at 10,000 psi (Avestin Emulsiflex C5). After the cell debris was removed by centrifugation (15,000g, 30 min, 4 °C), the cleared and filtered lysate was purified using immobilized metal affinity chromatography on an AKTA Pure with a Histrap FF column (Cytiva). Protein quality was determined by SDS-PAGE and >90% pure protein was stored in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 50 mM NaCl at pH 7.
HPLC Analysis
Samples were analyzed by RP-HPLC on an Ultimate 3000 series apparatus (Dionex) with a Kinetix 2.6 μm C18 100 Å 100 × 4.6 mm analytical column (Phenomenex) maintained at 40 °C. MilliQ water containing 0.1% formic acid and acetonitrile were used as mobile phases A and B, respectively, with the following method in percentages of mobile phase B at 1 mL/min: 0–0.5 min 2%, 0.5–1.5 min 35%, 1.5–3 min 35–80% (gradient), 3–4.2 min 98%, 4.2–5 min 2%. Chromatograms recorded at 300 nm for a and d and at 340 nm for b, c, and e were processed via Chromeleon 7.2.7 (Dionex).
Screening of 40 UGTs against 32 Polyphenols
The following reaction mixture was prepared for each enzyme–acceptor pair; 50 μM acceptor, 60 μM UDP-Glc, and 0.02 mg/mL UGT in 25 mM HEPES with 50 mM NaCl at pH 7. The reaction mixture was incubated for 16 h at 293 K and analyzed by RP-HPLC.
pH Characterization
The reactions were carried out at 293 K in 70 mM Tris–Bis–Tris (TBT) buffer in a pH range from 5 to 10, in the presence of 500 μM sugar donor (UDP-Glc), 100 μM acceptor enzyme. 100 μg/mL enzyme for RhUGT1 + a, OsUGT88C1 + e, AtUGT78D2 + e, 10 μg/mL for GmUGT88E3 + d, AtUGT78D2 + e, 1 μg/mL for OsUGT88C1 + b and c. The reaction was quenched by 25× dilution in 0.1% acetic acid at time points 0, 2, 4, 6, 8, and 10 min.
Temperature Characterization
The reactions were carried out in thermocyclers in a temperature range from 30 to 54 °C for 5, 15, 60, and 180 min, in 70 mM TBT buffer at optimal pH as previously determined and in the presence of 500 μM sugar donor (UDP-Glc) 100 μM acceptor and UGT. 100 μg/mL enzyme for RhUGT1 + a, OsUGT88C1 + e, AtUGT78D2 + e, 10 μg/mL for GmUGT88E3 + d, AtUGT78D2 + e, 1 μg/mL for OsUGT88C1 + b and c. The reactions were quenched by denaturation at 95 °C for 20 s.
Michaelis–Menten Kinetics
A range of acceptor concentrations from 0 to 250 μM was used in 50 mM TBT buffer at optimal pH as previously determined in the presence of 500 μM UDP-Glc. The reactions were carried out at 293 K in a thermocycler for 10 min followed by thermal denaturation at 95 °C for 20 s. The calculated Km and kcat values were based on the ratio between product peak and acceptor peak on the HPLC chromatograms with the assumption the absorbance at given wavelengths is equal. Michaelis–Menten plots were generated and analyzed in R using the drc package.44,45
Structure Determination by NMR
NMR sample preparation a1, b1, and c1: The following mixture was prepared—2.5 μL of 100 mM acceptor in DMSO-d6, 10 μL of 500 mM phosphate buffer at pH 8, 3 μL of 100 mM UDP-Glc, and 0.2 mg/mL of UGT in 1 mL of D2O. The mixture was incubated at 293 K and conversion was tracked by HPLC. The UGT was removed with a centrifugal filter (10 kDa Amicon Ultra 0.5 mL) when >70% conversion was observed. The sample was transferred to an NMR tube and measured accordingly. NMR sample preparation d7, d5, e5, and e7: The following mixture was prepared—40 μL of 100 mM acceptor in DMSO-d6, 40 μL of 500 mM phosphate buffer at pH 8, 50 μL of 100 mM UDP-Glc, and 0.2 mg/mL of UGT in 4 mL of MilliQ water. The mixture was incubated at 293 K, and conversion was tracked by HPLC; when >50% conversion was reached, the sample was stored at −20 °C until completely frozen. The samples were lyophilized by freeze-drying. The dried sample was dissolved in 600 μL of DMSO-d6 and measured accordingly.
NMR Data Acquisition
The NMR data were acquired on a Bruker Avance III (799.75 MHz) equipped with a 5 mm TCI 1H/(13C, 15N) CryoProbe. The 1H NMR spectra were acquired by using the standard Bruker pulse sequence (zg30). Targeted 1D NOESY was carried out using a standard Bruker pulse sequence targeting the anomeric alpha proton as determined by 1H NMR (selnogp). The data were processed using Bruker Topspin 4.1.4.
Computational Analysis
Preparation of the ternary complexes: Protein structural models were generated by using AlphaFold v2.0, using all available structural homologues, and the database search preset was set to “reduced_dbs”.46 After the predictions, built-in model relaxation was performed. Only the highest ranking (in pLDDT score) models were used downstream. Binary complexes of protein and sugar donor were obtained by structurally aligning protein model structures on the crystal structure of PtUGT1 from Polygonum tinctorium, which has a bound UDP-glucose molecule in its active site (6SU6.pdb).36 The acceptor molecules were added by docking into the acceptor binding site of the binary complexes, using gnina v1.0.1 software,47 a fork of smina,48 itself a fork of AutoDock Vina.49 PyMOL (v2.4.0) was used for superimposition and visualization of the resulting structures.
Molecular Dynamics
Simulations were performed on GROMACS (2021.3) software.50 Proteins were parametrized with Amber14SB force field,51 acceptors with gaff2 forcefield,52 and GLYCAM06 was used for glucose moiety. Substrates were prepared with an antechamber module and converted to GROMACS format by using acpype package.53,54 The complex systems were solvated in TIP3P water molecules in a cubic box with minimum 10 Å edge distance.55 Random water molecules were replaced with Na+ and Cl– ions to neutralize the system. Long-range electrostatics were treated with the particle-mesh Ewald method with a cutoff distance of 12 Å.56 Van der Waals interactions were treated in a Verlet scheme with a cutoff distance of 12 Å and a switching function for the forces starting at 10 Å.57 Hydrogen bonds were restrained using the LINCS algorithm.58 Protein with substrates and water with ions were coupled to individual heat baths with a Bussi–Donadio–Parrinello thermostat.59 Pressure coupling was done in a Parrinello–Rahman barostat. Energy minimization was performed with steepest-descent algorithm for 50,000 steps. NVT equilibration was performed for 100 ps with a reference temperature of 300 K, with restraints placed on the protein and substrates. Afterward, NPT equilibration with identical restraints was performed for 100 ps with a reference pressure of 1 bar. Next, the production run was started with flat-bottomed distance restraints of 5000 kJ/mol–1 nm–1 on one or both of nucleophilic attack (4 Å) and/or deprotonation/hydrogen bond (2.8 Å) distances to simulate the process of substrate binding and therefore reduce the dependency on initial simulation conditions. After 0.5 ns, restraints were removed and simulations continued until 2 ns. For every enzyme/acceptor/restraint-type combination, two parallel simulations were executed—one for each glycosylation site and one on another. Trajectories were analyzed with built-in GROMACS command-line tools and visualized with VMD and PyMOL.60
Acknowledgments
The authors thank Lars Boje Petersen for technical assistance in analytical chemistry and Folmer Fredslund for technical support in computational analysis.
Glossary
Abbreviations
- DMSO-d6
deuterated dimethyl sulfoxide
- GT1
family 1 glycosyltransferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- RP-HPLC
reverse-phase high-performance liquid chromatography
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- UDP
uridine diphosphate
- UDP-Glc
uridine diphosphate glucose
- UGT
uridine 5′-diphospho-glucuronosyl-transferase
- NOE
nuclear Overhauser effect
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c08255.
Author Present Address
§ Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen O, Denmark
Author Contributions
R.M.B. and D.V. are contributed equally. The manuscript was written with the contributions of all authors. All authors have approved the final version of the manuscript.
This work was supported by the Novo Nordisk Foundation (grants NNF18OC0034744 to D.H.W., and NNF10CC1016517 and NNF20CC0035580 to the NNF Center for Biosustainability), and the Villum Foundation (DTU NMR Center).
The authors declare no competing financial interest.
Supplementary Material
References
- Chen F.; Huang G. Application of Glycosylation in Targeted Drug Delivery. Eur. J. Med. Chem. 2019, 182, 111612. 10.1016/j.ejmech.2019.111612. [DOI] [PubMed] [Google Scholar]
- Xu L.; Qi T.; Xu L.; Lu L.; Xiao M. Recent Progress in the Enzymatic Glycosylation of Phenolic Compounds. J. Carbohydr. Chem. 2016, 35 (1), 1–23. 10.1080/07328303.2015.1137580. [DOI] [Google Scholar]
- Loftsson T.; Brewster M. E. Pharmaceutical Applications of Cyclodextrins: Basic Science and Product Development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. 10.1111/j.2042-7158.2010.01030.x. [DOI] [PubMed] [Google Scholar]
- Borges F.; Roleira F.; Milhazes N.; Santana L.; Uriarte E. Simple Coumarins and Analogues in Medicinal Chemistry: Occurrence, Synthesis and Biological Activity. Curr. Med. Chem. 2005, 12, 887–916. 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]
- Kontogiorgis C. A.; Hadjipavlou-Litina D. J. Synthesis and Antiinflammatory Activity of Coumarin Derivatives. J. Med. Chem. 2005, 48, 6400–6408. 10.1021/jm0580149. [DOI] [PubMed] [Google Scholar]
- Witaicenis A.; Seito L. N.; di Stasi L. C. Intestinal Anti-Inflammatory Activity of Esculetin and 4-Methylesculetin in the Trinitrobenzenesulphonic Acid Model of Rat Colitis. Chem.-Biol. Interact. 2010, 186 (2), 211–218. 10.1016/j.cbi.2010.03.045. [DOI] [PubMed] [Google Scholar]
- Kostova I.; Bhatia S.; Grigorov P.; Balkansky S.; Parmar V. S.; Prasad A. K.; Saso L. Coumarins as Antioxidants. Curr. Med. Chem. 2011, 18 (25), 3929–3951. 10.2174/092986711803414395. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Singla R.; Dandriyal J.; Jaitak V. Coumarin Derivatives as Anticancer Agents for Lung Cancer Therapy: A Review. Anticancer Agents Med. Chem. 2018, 18 (7), 964–984. 10.2174/1871520618666171229185926. [DOI] [PubMed] [Google Scholar]
- Veselinović J. B.; Veselinović A. M.; Nikolić G. M.; Pešić S. Z.; Stojanović D. B.; Matejić J. S.; Mihajilov-Krstev T. M. Antibacterial Potential of Selected 4-Phenyl Hydroxycoumarins: Integrated in Vitro and Molecular Docking Studies. Med. Chem. Res. 2015, 24 (4), 1626–1634. 10.1007/s00044-014-1245-0. [DOI] [Google Scholar]
- Li Z.; Kong D.; Liu Y.; Li M. Pharmacological Perspectives and Molecular Mechanisms of Coumarin Derivatives against Virus Disease. Genes Dis. 2022, 9, 80–94. 10.1016/j.gendis.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. Y.; Liu T.; Sun J.; Wang X. J. Synthesis and Application of Coumarin Fluorescence Probes. RSC Adv. 2020, 10, 10826–10847. 10.1039/c9ra10290f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. T.; Deng X. Y.; Chen J.; Liang Q. M.; Zhang K.; Li D. L.; Wu P. P.; Zheng X.; Zhou R. P.; Jiang Z. Y.; Ma A. J.; Chen W. H.; Wang S. H. Synthesis and biological evaluation of coumarin derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 2020, 189, 112013. 10.1016/j.ejmech.2019.112013. [DOI] [PubMed] [Google Scholar]
- Bauer E. B. Transition Metal Catalyzed Glycosylation Reactions-an Overview. Org. Biomol. Chem. 2020, 18 (45), 9160–9180. 10.1039/D0OB01782E. [DOI] [PubMed] [Google Scholar]
- Báti G.; He J. X.; Pal K. B.; Liu X. W. Stereo- and Regioselective Glycosylation with Protection-Less Sugar Derivatives: An Alluring Strategy to Access Glycans and Natural Products. Chem. Soc. Rev. 2019, 48, 4006–4018. 10.1039/c8cs00905h. [DOI] [PubMed] [Google Scholar]
- Louveau T.; Osbourn A. The Sweet Side of Plant-Specialized Metabolism. Cold Spring Harbor Perspect. Biol. 2019, 11 (12), a034744. 10.1101/cshperspect.a034744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestrom L.; Przypis M.; Kowalczykiewicz D.; Pollender A.; Kumpf A.; Marsden S. R.; Bento I.; Jarzębski A. B.; Szymańska K.; Chruściel A.; Tischler D.; Schoevaart R.; Hanefeld U.; Hagedoorn P. L. Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 5263. 10.3390/ijms20215263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P. M.; Deleury E.; Davies G. J.; Henrissat B. An Evolving Hierarchical Family Classification for Glycosyltransferases. J. Mol. Biol. 2003, 328 (2), 307–317. 10.1016/S0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- Palcic M. M. Glycosyltransferases as Biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 226–233. 10.1016/j.cbpa.2010.11.022. [DOI] [PubMed] [Google Scholar]
- Mackenzie P. I.; Walter Bock K.; Burchell B.; Guillemette C.; Ikushiro S.-I.; Iyanagi T.; Miners J. O.; Owens I. S.; Nebert D. W. Nomenclature Update for the Mammalian UDP Glycosyltransferase (UGT) Gene Superfamily. Pharmacogenet. Genomics 2005, 15, 677–685. 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- Wang L.; Han W.; Xie C.; Hou J.; Fang Q.; Gu J.; Wang P. G.; Cheng J. Comparing the Acceptor Promiscuity of a Rosa Hybrida Glucosyltransferase RhGT1 and an Engineered Microbial Glucosyltransferase OleDPSA toward a Small Flavonoid Library. Carbohydr. Res. 2013, 368, 73–77. 10.1016/j.carres.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Funaki A.; Waki T.; Noguchi A.; Kawai Y.; Yamashita S.; Takahashi S.; Nakayama T. Identification of a Highly Specific Isoflavone 7-O-Glucosyltransferase in the Soybean (Glycine Max (L.) Merr.). Plant Cell Physiol. 2015, 56 (8), 1512–1520. 10.1093/pcp/pcv072. [DOI] [PubMed] [Google Scholar]
- Noguchi A.; Saito A.; Homma Y.; Nakao M.; Sasaki N.; Nishino T.; Takahashi S.; Nakayama T. A UDP-Glucose:Isoflavone 7-O-Glucosyltransferase from the Roots of Soybean (Glycine max) Seedlings. J. Biol. Chem. 2007, 282 (32), 23581–23590. 10.1074/jbc.M702651200. [DOI] [PubMed] [Google Scholar]
- Farmakalidis E.; Murphy P. A. Oestrogenic Response of the CD-1 Mouse to the Soya-Bean Isoflavones Genistein, Genistin and Daidzin. Food Chem. Toxicol. 1984, 22, 237–239. 10.1016/0278-6915(84)90134-0. [DOI] [PubMed] [Google Scholar]
- An D. G.; Yang S. M.; Kim B. G.; Ahn J. H. Biosynthesis of Two Quercetin O-Diglycosides in Escherichia Coli. J. Ind. Microbiol. Biotechnol. 2016, 43 (6), 841–849. 10.1007/s10295-016-1750-x. [DOI] [PubMed] [Google Scholar]
- Pei J.; Chen A.; Dong P.; Shi X.; Zhao L.; Cao F.; Tang F. Modulating Heterologous Pathways and Optimizing Fermentation Conditions for Biosynthesis of Kaempferol and Astragalin from Naringenin in Escherichia Coli. J. Ind. Microbiol. Biotechnol. 2019, 46 (2), 171–186. 10.1007/s10295-018-02134-6. [DOI] [PubMed] [Google Scholar]
- Kim B. G.; Sung S. H.; Ahn J. H. Biological Synthesis of Quercetin 3-O-N-Acetylglucosamine Conjugate Using Engineered Escherichia Coli Expressing UGT78D2. Appl. Microbiol. Biotechnol. 2012, 93 (6), 2447–2453. 10.1007/s00253-011-3747-8. [DOI] [PubMed] [Google Scholar]
- Teze D.; Bidart G. N.; Welner D. H. Family 1 Glycosyltransferases (GT1, UGTs) Are Subject to Dilution-Induced Inactivation and Low Chemo Stability towards Their Own Acceptor Substrates. Front. Mol. Biosci. 2022, 9, 909659. 10.3389/fmolb.2022.909659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermeier P.; Fortuna C.; Hübschmann K. M.; Bidart G. N.; Tørring T.; Teze D.; Welner D. H.; Kara S. Exploring the in Vitro Operating Window of Glycosyltransferase PtUGT1 from Polygonum Tinctorium for a Biocatalytic Route to Indigo Dye. ACS Sustain. Chem. Eng. 2021, 9 (25), 8497–8506. 10.1021/acssuschemeng.1c01536. [DOI] [Google Scholar]
- Fujiwara R.; Nakajima M.; Yamamoto T.; Nagao H.; Yokoi T. In Silico and in Vitro Approaches to Elucidate the Thermal Stability of Human UDP-Glucuronosyltransferase (UGT) 1A9. Drug Metab. Pharmacokinet. 2009, 24 (3), 235–244. 10.2133/dmpk.24.235. [DOI] [PubMed] [Google Scholar]
- De Boer R. M.; Hvid D. E. H.; Davail E.; Vaitkus D.; Duus J. Ø.; Welner D. H.; Teze D. Promiscuous yet Specific: A Methionine-Aromatics Interaction Drives the Reaction Scope of the Family 1 Glycosyltransferase GmUGT88E3 from Soybean. ChemRxiv 2023, 10.26434/chemrxiv-2023-1sq4z. [DOI] [PubMed] [Google Scholar]
- Chen D.; Chen R.; Wang R.; Li J.; Xie K.; Bian C.; Sun L.; Zhang X.; Liu J.; Yang L.; Ye F.; Yu X.; Dai J. Probing the Catalytic Promiscuity of a Regio- and Stereospecific C-Glycosyltransferase from Mangifera Indica. Angew. Chem. 2015, 127 (43), 12869–12873. 10.1002/ange.201506505. [DOI] [PubMed] [Google Scholar]
- Härle J.; Günther S.; Lauinger B.; Weber M.; Kammerer B.; Zechel D. L.; Luzhetskyy A.; Bechthold A. Rational Design of an Aryl-C-Glycoside Catalyst from a Natural Product O-Glycosyltransferase. Chem. Biol. 2011, 18 (4), 520–530. 10.1016/j.chembiol.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Tam H. K.; Härle J.; Gerhardt S.; Rohr J.; Wang G.; Thorson J. S.; Bigot A.; Lutterbeck M.; Seiche W.; Breit B.; Bechthold A.; Einsle O. Structural Characterization of O- and C-Glycosylating Variants of the Landomycin Glycosyltransferase LanGT2. Angew. Chem., Int. Ed. 2015, 54 (9), 2811–2815. 10.1002/anie.201409792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann A.; Nidetzky B. Switching between O- and C-Glycosyltransferase through Exchange of Active-Site Motifs. Angew. Chem., Int. Ed. 2012, 51 (51), 12879–12883. 10.1002/anie.201206141. [DOI] [PubMed] [Google Scholar]
- Lairson L. L.; Henrissat B.; Davies G. J.; Withers S. G. Glycosyl Transferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Teze D.; Coines J.; Fredslund F.; Dubey K. D.; Bidart G. N.; Adams P. D.; Dueber J. E.; Svensson B.; Rovira C.; Welner D. H. O-/ N-/ S-Specificity in Glycosyltransferase Catalysis: From Mechanistic Understanding to Engineering. ACS Catal. 2021, 11 (3), 1810–1815. 10.1021/acscatal.0c04171. [DOI] [Google Scholar]
- Nowak P. M.; Woźniakiewicz M.; Piwowarska M.; Kościelniak P. Determination of Acid Dissociation Constant of 20 Coumarin Derivatives by Capillary Electrophoresis Using the Amine Capillary and Two Different Methodologies. J. Chromatogr. A 2016, 1446, 149–157. 10.1016/j.chroma.2016.03.084. [DOI] [PubMed] [Google Scholar]
- Traven V. F.; S.Krasavina L.; Dmitrieva E. Yu.; Carberry E. A. Selective O-Glucosylation of 4,7-Dihydroxycoumarin. Heterocycl. Commun. 1996, 2 (4), 309. 10.1515/HC.1996.2.4.309. [DOI] [Google Scholar]
- Ueno K.; Shiraki M.; Sato M.; Saito N. The Crystal and Molecular Structures of Esculetin 6-Glucoside and 7-Glucoside. Bull. Chem. Soc. Jpn. 1985, 58 (1), 230–235. 10.1246/bcsj.58.230. [DOI] [Google Scholar]
- Perel’Son M. E.; Sheinker Y. N. Spectra and Structure of Hydroxycoumarin and Hydroxyfurocoumarin Salts. J. Appl. Spectrosc. 1966, 5, 78–82. 10.1007/BF00604656. [DOI] [Google Scholar]
- Abu-Eittah R. H.; Ali B.; El-Tawil H. The Electronic Absorption Spectra of Some Coumarins. A Molecular Orbital Treatment. Can. J. Chem. 1985, 63, 1173–1179. 10.1139/v85-200. [DOI] [Google Scholar]
- Pina J.; de Castro C. S.; Delgado-Pinar E.; Sérgio Seixas de Melo J. Characterization of 4-Methylesculetin and of Its Mono- and Di-Methoxylated Derivatives in Water and Organic Solvents in Its Ground, Singlet and Triplet Excited States. J. Mol. Liq. 2019, 278, 616–626. 10.1016/j.molliq.2019.01.083. [DOI] [Google Scholar]
- Mobley D. L.; Dill K. A. Binding of Small-Molecule Ligands to Proteins: “What You See” Is Not Always “What You Get.”. Structure 2009, 17 (4), 489–498. 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022.
- Ritz C.; Baty F.; Streibig J. C.; Gerhard D. Dose-Response Analysis Using R. PLoS One 2015, 10 (12), e0146021 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNutt A. T.; Francoeur P.; Aggarwal R.; Masuda T.; Meli R.; Ragoza M.; Sunseri J.; Koes D. R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminf. 2021, 13 (1), 43. 10.1186/s13321-021-00522-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D. R.; Baumgartner M. P.; Camacho C. J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53 (8), 1893–1904. 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Sousa da Silva A. W.; Vranken W. F. ACPYPE-AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi A.; Faller R.; Reith D.; Kirschner K. N. ACPYPE Update for Nonuniform 1–4 Scale Factors: Conversion of the GLYCAM06 Force Field from AMBER to GROMACS. SoftwareX 2019, 10, 100241. 10.1016/j.softx.2019.100241. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Verlet L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159 (1), 98–103. 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. . [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126 (1), 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K.. VMD: Visual Molecular Dynamics, 1996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.