

Abstract
Type 2 diabetes mellitus (T2DM), characterized by hyperglycemia and dyslipidemia, leads to nonproliferative diabetic retinopathy (NPDR). NPDR is associated with blood-retina barrier disruption, plasma exudates, microvascular degeneration, elevated inflammatory cytokine levels, and monocyte (Mo) infiltration. Whether and how the diabetes-associated changes in plasma lipid and carbohydrate levels modify Mo differentiation remains unknown. Here, we show that mononuclear phagocytes (MPs) in areas of vascular leakage in DR donor retinas expressed perilipin 2 (PLIN2), a marker of intracellular lipid load. Strong upregulation of PLIN2 was also observed when healthy donor Mos were treated with plasma from patients with T2DM or with palmitate concentrations typical of those found in T2DM plasma, but not under high-glucose conditions. PLIN2 expression correlated with the expression of other key genes involved in lipid metabolism (ACADVL, PDK4) and the DR biomarkers ANGPTL4 and CXCL8. Mechanistically, we show that lipid-exposed MPs induced capillary degeneration in ex vivo explants that was inhibited by pharmaceutical inhibition of PPARγ signaling. Our study reveals a mechanism linking dyslipidemia-induced MP polarization to the increased inflammatory cytokine levels and microvascular degeneration that characterize NPDR. This study provides comprehensive insights into the glycemia-independent activation of Mos in T2DM and identifies MP PPARγ as a target for inhibition of lipid-activated MPs in DR.
Keywords: Inflammation, Ophthalmology
Keywords: Diabetes, Macrophages, Microcirculation
Introduction
Type 2 diabetes mellitus (T2DM) affects a growing part of the population worldwide (1). Fifteen years after the onset of diabetes, more than 60% of patients will develop some form of diabetic retinopathy (DR) (2). DR stages are clinically characterized by the observation of retinal vascular damage (3). Alterations in the blood-retina barrier and the resulting progressive degeneration of retinal capillaries, ensuing retinal ischemia, extravasation, and bleeding occur in nonproliferative DR (NPDR), the early stage of the disease. Severe NPDR can ultimately progress to sight-threatening proliferative DR (PDR), which is characterized by the formation of neovascular tufts around the ischemic retinal tissue. These areas of vascular remodeling are associated with neurodegenerative processes and innate immune dysregulation (4, 5). High intraocular levels of inflammatory cytokines are found in patients with DR, and cytokines such as TNF-α, IL-1β, IL-6, and IL-8 (CXCL8) have been proposed to be involved in the progression of DR (5, 6). Consistent with such a role for cytokines, treatments targeting inflammation have been shown to be protective against diabetes-induced neurodegeneration and vascular remodeling (7). Activation of retinal mononuclear phagocytes (MPs) has been described in patients with diabetes (8) and in animal models of T2DM (9, 10). The retinal MP population in DR is composed not only of resident microglia but also of infiltrating monocyte-derived (Mo-derived) macrophages. The latter play a prominent role in inflammation and vascular remodeling in DR (11–14).
DR is a multifactorial disease, and hyperglycemia only accounts for a limited proportion of the risk of developing DR (15). Indeed, a number of other glycemic-independent risk factors are associated with the development and progression of DR (16–18).
Dyslipidemia has been a largely neglected field of study but has recently attracted attention and is now considered to be an important area of research in DR (17, 18). Indeed, numerous studies (19–25), as well as a meta-analysis (26), have demonstrated correlations between plasma lipid levels and DR or diabetic edema in the past 10 years. Moreover, the lipid-lowering drug fenofibrate was shown to slow DR progression independently of glycemic control in 2 large randomized, controlled clinical trials (27, 28). Saturated free fatty acids (FFAs) are generally considered to be proinflammatory, whereas unsaturated FFAs are classically considered to be antiinflammatory (29–32). Palmitate (PA) (saturated 16-carbon FA [C16:0]) is the most abundant FFA in the plasma (33), and its concentration is found to be elevated among patients with T2DM (34, 35). MPs can take up and store lipids in intracellular lipid droplets stabilized by the amphiphilic protein perilipin 2 (PLIN2) (36). PLIN2+ lipid-laden MPs (PLIN2+ MPs) are found in atherosclerotic lesions and in fat lesions after trauma (37). In the brain, PLIN2+ MPs were found in grade 2 cerebellar infarcts, whereas they were absent in control and low-grade infarcts (37). More recently, lipid-laden MPs were found to progressively accumulate in the aging mouse and human brain (38). PLIN2+ MPs exhibit alterations in their metabolism, phagocytosis, and autophagy, leading to the production of ROS and inflammatory cytokines (38–42). The presence and role of MPs exposed to plasma lipids in the retina in DR remain to be determined.
In the present study, MPs expressing PLIN2 were found in the retinas of patients with DR in regions with active vascular leakage and vascular remodeling. Next, we aimed to understand the molecular cues that drive Mos to differentiate into PLIN2+ MPs and their role in vascular remodeling. We show that exposure of naive human Mos to heat-inactivated plasma from patients with diabetes or to FFAs was sufficient to induce PLIN2 expression and secretion of DR-associated inflammatory cytokines. Experimentally, we show that lipid-exposed MPs had strong vasodegenerative properties that were inhibited by a pharmacological PPARγ inhibitor. Overall, these results pave the way for potential treatment targeting MP PPARγ signaling in dyslipidemia-mediated inflammation, such as DR, and potentially beyond retinal vasodegeneration.
Results
Human diabetic retina postmortem analysis reveals PLIN2+ MP accumulation in regions with vascular leakage.
PLIN2+ MPs have been shown to progressively accumulate in the brains of aging mice and humans and in regions of cerebellar infarcts. Lipid-laden macrophages have been reported in the retina following blood-retina barrier breakdown (43), but the localization of PLIN2+ MPs in the diabetic retina has not been reported thus far. We identified regions with potential PLIN2+ cells by staining retinas from a collection of postmortem diabetic donor retinas (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI161348DS1) with anti–collagen IV (COL4), anti-albumin (anti-ALB), anti-IBA1, a pan-MP antibody, and anti-PLIN2 antibodies. COL4 and ALB staining revealed regions where ALB was found only in the vessel lumen (Figure 1A) and regions with microaneurysms where extraluminal ALB was found (Figure 1B). IBA1 staining identified numerous MPs in contact with retinal aneurysms, and 2 anti-PLIN2–stained samples showed identical localization and cellular patterns (Figure 1, C–F). All PLIN2+ cells were also stained with IBA1 and ALB, which clearly identified PLIN2+ cells as intraretinal MPs (Figure 1, C–F) that had taken up plasma leakage material. Higher magnification showed the presence of both PLIN2+ and PLIN2– MPs at the sites of the aneurysm (Figure 1E). To determine the proportion of PLIN2+ MPs in retinal tissue obtained from postmortem donors, we used anti-IBA1 and anti-PLIN2 antibodies to stain retinal MPs and Ulex europaeus I agglutinin (UEA1, a marker of human vascular endothelial cells) to costain vessels (Figure 1, F and G). No PLIN2+ cells were observed in regions without microaneurysms in the diabetes mellitus (DM) retina (Figure 1F), nor were these cells found in the control patient’s retina (data not shown). We then counted the number of MPs that fell into each category in large-field retinal flat-mount images from the 4 different postmortem DM donors (Figure 1, G–I). In the diseased retina, we detected between 4 and 21 aneurysms per field. We analyzed a minimum of 4 aneurysms and up to 7 per DM donor for a total of 19 aneurysms analyzed. On average, we observed 10 MPs per aneurysm, 2 of which were PLIN2+. In detail, we found 7 aneurysms that were exclusively composed of PLIN2– MPs (Figure 1H). The remaining 12 aneurysms (63.2%) were made up of 29.8% PLIN2+ MPs (Figure 1I). Similar to other reports on the aging or infarcted brain, PLIN2+ MPs were found in the vicinity of damaged capillaries in human samples of DR retinas (Figure 1J).
Figure 1. PLIN2+ MPs are found in regions with retinal plasma leakage in a patient with DR.

(A–G) Representative immunofluorescence (IF) images of human flat-mounted retinas from 4 postmortem DM donors. (A) IF staining of ALB (plasma protein) and COL4 (vessels) within a healthy vasculature area of human retina. Scale bar: 200 μm. (B) IF staining for ALB and COL4 within an area showing vascular leakage. Scale bar: 200 μm. (C) IF staining for ALB, IBA1, and PLIN2 within an area with a leaking microaneurysm. Scale bar: 40 μm. (D) IF staining for COL4, IBA1 (MPs), and PLIN2 (neutral lipid droplets) within an area with a microaneurysm. Scale bar: 30 μm. (E and F) IF staining for IBA1 and PLIN2 and labeling with UEA1 (vessels). (E) IF staining for IBA1 and PLIN2. The arrows show a PLIN2– (blue) and a PLIN2+ (red) MP within the same region. Scale bar: 10 μm. (F) Representative image outside the area with the microaneurysms. Scale bar: 200 μm. (G) Representative image within the area of the microaneurysms. (H) Total MP count and PLIN2+ MP count around microaneurysms in 4 different DM donors (each color represents a different donor). (I) Percentage of PLIN2+ MPs within microaneurysms containing at least 1 PLIN2+ MP. (J) Schematic depiction of a ruptured retinal microaneurysm leading to plasma leakage in a DR retina.
PLIN2 expression in PA-exposed MPs is associated with changes in lipid metabolism genes and expression of key DR markers.
NPDR is characterized by blood-retina barrier breakdown, Mo infiltration, and plasma extravasation into the retina. Our PLIN2 immunohistochemical studies (Figure 1) suggested that resident MPs and/or infiltrating Mos had taken up extravasated lipids. We investigated the molecular mechanism by which plasma lipids drive the differentiation of Mos into lipid-associated MPs (PLIN2+ MPs) by exposing naive Mos to a blood-mimicking (500 μM) concentration of PA (34). We first compared the cellular FA chain composition in early Mo-derived MPs 18 hours after exposure of naive Mos to PA bound to BSA or BSA alone (Figure 2A). Lipidomic analysis showed that PA was rapidly internalized by differentiating Mos and represented 47.9% of the total FA chains in PA-treated, early-differentiated MPs, whereas it represented only 14.7% of the FA in the BSA-only condition (Figure 2B). PA stimulation did not affect the relative abundance of total saturated (other than PA), monounsaturated, total polyunsaturated, or total omega-3 or total omega-6 polyunsaturated FAs (Figure 2, C and D) or induce a change in the composition of individual non-PA FA chains (Figure 2E and Supplemental Table 2). Concomitant with PA influx, PLIN2 expression significantly increased in MPs, whereas the addition of methyl-PA (PACH3), a PA analog that can be internalized but that is less biologically active and not readily metabolizable, resulted in partial inhibition of this induction (Figure 2F). Overall, our results suggest that naive Mos actively adapted their transcriptomic profile in response to PA exposure. How Mos and early Mo-derived MPs adapt to a lipid-rich microenvironment is unknown. We aimed to obtain an unbiased view of the transcriptomic changes induced by a lipid-rich environment during differentiation. We thus exposed naive human Mos from healthy donors to PA (500 μM) or to BSA (Figure 2A) for 18 hours, a time point representative of early Mo differentiation (44), and compared their transcript expression levels by RNA-Seq. The expression level, in transcripts per million (TPM), of each transcript in each condition was determined (Supplemental Table 3), and the means are presented as scatter plots for the BSA condition (x axis) and the PA condition (y axis) (Figure 2G). DESeq2 analysis showed that PA treatment induced a statistically significant (adjusted P ≤ 0.05) differential expression of 9,794 transcripts (Supplemental Table 3). Among them, 528 (5.4 %) were regulated with a log2 fold change (FC) of greater than 2, of which 37 transcripts (0.4%, red dots) were upregulated more than 16-fold (Figure 2G). The top 10 up- and downregulated transcripts are shown in Figure 2H. We then performed a gene ontology (GO) enrichment analysis of these 528 regulated transcripts to determine the major functions affected by PA treatment. Thirteen GO terms were significantly enriched, 8 of which were related to lipid metabolism (red dots), consistent with the adaptation of Mo metabolism to the lipid-rich environment (Figure 2I). This observation was further confirmed by specific analysis of the broad GO term “fatty acid metabolic process” (414 genes). Among them, 229 of the 414 transcripts (55.3%) were found to be differentially expressed following PA versus BSA treatment. Sixteen (7.8%) were among the 37 highly regulated genes (FC ≥4) (Figure 2J and Supplemental Table 4). Only 2 were downregulated (SMPD3 and CD36; log2 FCs = –2.29 and –2.22, respectively) (Figure 2H and Supplemental Table 4). Our analysis confirmed the upregulation of PLIN2 and identified CXCL8 as the third most highly expressed transcript in PA-stimulated Mos, with a log2 FC of 2.87 (Figure 2, G and J). PA stimulation also induced a strong upregulation of ANGPTL4 (log2 FC = 2.14). ANGPTL4 acts as an endogenous inhibitor of lipoprotein lipase (LPL), an enzyme responsible for breaking down triglycerides (TGs) in circulating lipoproteins, and its levels in aqueous humor have been shown to correlate with areas of nonperfusion in patients with DR (45, 46).
Figure 2. PA stimulation triggers the expression of key DR markers by MPs.

(A) Schematic representation of Mo isolation, treatment, and preparation. (B–D) Chromatographic analysis of FA chain composition after BSA (n = 3) or PA (n = 3) treatment. (B) Mean percentage of PA (C16:0) chains relative to total FAs. P s were calculated using Welch’s t test. (C) Mean percentage of monounsaturated FAs (MUFAs), polyunsaturated FAs (PUFAs), and saturated FAs (SFAs) (minus PA). (D) Mean percentage of ω−3 and ω–6 PUFAs. (C and D) P values were calculated using multiple Welch’s t tests corrected for multiple comparisons using the Holm-Šidák method. (E) Heatmap representation of the percentage of individual FA chains relative to total FA chains (minus PA). (F) RT-qPCR quantification of PLIN2 after treatment with BSA (n = 4), PA (n = 4), or PACH3 (n = 4). P values were determined by 1-way Welch’s ANOVA (P = 0.0021) followed by Dunnett’s T3 multiple-comparison test. (G–I) RNA-Seq transcriptomics analysis after BSA (n = 3) or PA (n = 3) treatment. (G) Scatter plot of the mean TPM value for all transcripts detected after BSA (x axis) or PA (y axis) treatment. The red and blue dots represent transcripts upregulated with a log2 FC of 4 or higher or a log2 FC of 4 or lower. (H) Heatmap representation of the log2 variance stabilizing transformation (vst) of the top 10 upregulated and downregulated transcripts. (I) GO enrichment analysis representing the fold enrichment of the 528 transcripts with a log2 FC of 2 or higher; red dots represent pathways related to lipid metabolism. Numbers in parenthesis represents the number of genes regulated by PA stimulation. Resp., response; Neg., negative; reg., regulator; act. activation; lipoprot., lipoprotein; Cell., cellular; stim., stimulation; Pos., positive; org., organization; diff., differentiation; drvd, derived. (J) Heatmap representation of the log2 vst of transcripts with a log2 FC of 4 or higher and belonging to the GO pathway “fatty acid metabolic process.” (K) Schematic representation of the biological function of the markers selected as a signature of lipid exposure.
Overall, our transcriptomic analysis showed a modification in lipid metabolism and DR-related cytokine expression in Mos in response to PA. We chose 4 markers that are highly regulated by PA exposure for downstream analysis: PLIN2 (for its role in intracellular lipid droplet stabilization, log2 FC = 4.13), PDK4 (an inhibitor of pyruvate conversion into acetyl-CoA, which was the most highly regulated transcript by PA treatment, log2 FC = 7.27), ACADVL (for its role in lipid β-oxidation, log2 FC = 2.7), and ANGPTL4 (for its role in regulating TG uptake and its involvement in DR physiopathology, log2 FC = 2.14) (Figure 2K). To ensure that our results were not biased by the ability of BSA to bind FFAs released by the cells, we also performed RNA-Seq analysis of gene expression in naive Mos stimulated with PA in the presence of BSA, compared with a control medium without BSA. PLIN2, PDK4, ACADVL, ANGPTL4, and CXCL8 were also upregulated when MPs grown in BSA-free conditions were used as controls, and a very similar regulation of the genes involved in the “fatty acid metabolic process” was also observed (Supplemental Figure 1).
We then studied the effect on MP polarization of other FFA species that are also found to be elevated in patients with T2DM, such as stearate (SA) (C18:0), the second most abundant saturated FFA in the blood, and palmitoleate (PoA) (C16:1 n-7), an unsaturated form of PA. SA and PoA also demonstrated the ability to upregulate PLIN2, PDK4, ACADVL, ANGPTL4, and CXCL8 transcripts (Supplemental Figure 2A) (34, 35).
To gain a deeper understanding of the influence of FFA mixtures on MP polarization, we implemented an expanded set of stimulations. We then investigated the potential modulatory role of oleic acid (OA) (C18:1 n-9), one of the most abundant circulating FAs traditionally considered to be antiinflammatory, in PA stimulation. Naive Mos were stimulated with an equimolar mixture of PA and OA (250 μM each, totaling 500 μM FFA), and transcript expression was compared with that induced by 500 μM PA alone. Interestingly, we found that the equimolar mixture significantly increased the expression of PLIN2 and PDK4 compared with expression following stimulation with PA alone, whereas, in sharp contrast, CXCL8 was strongly downregulated. However, despite the presence of OA, we still observed overexpression of our selected markers PLIN2, PDK4, ACADVL, ANGPTL4, and CXCL8 (Supplemental Figure 2B).
Considering the complexity of the FFA milieu in vivo, we developed a custom FFA blend with a target concentration of 500 μM to allow a meaningful comparison with standard PA stimulation. According to Yi et al., the 4 most abundant FAs in the blood are PA (136 μM), SA (46 μM), OA (117 μM), and linoleic acid (LA) (132 μM), collectively constituting 84% (430 μM) of the total FAs (509 μM) (34). The custom blend of these 4 major FAs was therefore formulated as follows to mimic their relative abundance in the bloodstream: PA, 157 μM; SA, 53 μM; OA, 136 μM; and LA, 153 μM (as detailed in Supplemental Table 5). As with the PA-OA equimolar mixture, this plasma representative blend also induced a trend toward higher expression of PLIN2, PDK4, and ACADVL (although these changes were statistically significant only for PDK4) when compared with 500 μM PA alone. Interestingly, ANGPTL4 was found to be significantly downregulated after FFA blend stimulation. Consistent with what we observed with the PA-OA equimolar mixture, the FA mixture also induced an overexpression of all of our selected markers (PLIN2, PDK4, ACADVL, ANGPTL4, and CXCL8) compared with the BSA condition (Supplemental Figure 2B).
Last, we replaced PA with an unreadily metabolizable form of PA, PACH3, in the blend. Under these conditions, we observed a nonstatistically significant trend toward lower expression of lipid-related transcripts, which correlated with a statistically significant reduction in expression of the inflammatory cytokine CXCL8 (Supplemental Figure 2B).
Stimulation of naive Mos with T2DM plasma increases the expression of PLIN2, which is correlated with the expression of lipid metabolism and DR marker genes.
We next analyzed the expression of the key markers defined above in naive Mos exposed to plasma from patients with diabetes. We established a cohort of donors (Table 1 and Figure 3A) consisting of control nondiabetic (ND) donors (n = 10) and diabetic patients with no signs of retinopathy (T2DM no DR, n = 10), early-to-mild NPDR (T2DM NPDR, n = 10), or PDR (T2DM PDR, n = 7) and collected plasma from each (total: n = 10 ND donors and n = 27 patients with T2DM). The mean leukocyte count and plasma creatinine concentration were similar between the control and T2DM groups (Table 1). The patients with TD2M (n = 27) had been diagnosed with T2DM for a median duration of 15 years, and their median age was also higher than that of the ND donors (63 vs. 50 years) (Table 1). The median glycemia and glycated hemoglobin levels for the T2DM donors were 185 mg/dL and 8.4%, respectively, and 85 mg/dL and 4.85%, respectively, for the ND donors. The same purification of naive Mos from a healthy human donor was sampled and exposed for 18 hours to 20% heat-inactivated plasma from each of the 37 donors in the cohort. Heat inactivation for 1 hour at 60°C (hi) was performed to denature and inactivate endogenous plasma cytokines that could have interfered with the Mo and cytokine measurements (47) (Figure 2A). Reverse transcription quantitative PCR (RT-qPCR) analysis showed that expression of the PA-inducible transcripts PLIN2, PDK4, ACADVL, and ANGPTL4 (Figure 2) was significantly higher in Mos exposed to T2DM patients’ hi-plasma than in those exposed to ND donor hi-plasma (Figure 3B). Although our study did not reveal significant differences between the T2DM subgroups (Supplemental Figure 3), we found a highly significant correlation between the expression of PDK4, ACADVL, ANGPTL4, and CXCL8 and of PLIN2 in the entire T2DM group (Figure 3C). This finding links the expression of lipid overload markers, such as PLIN2, in early differentiating Mos with known DR-associated cytokines, such as ANGPTL4 and CXCL8 (37, 45).
Table 1. Cohort structure.
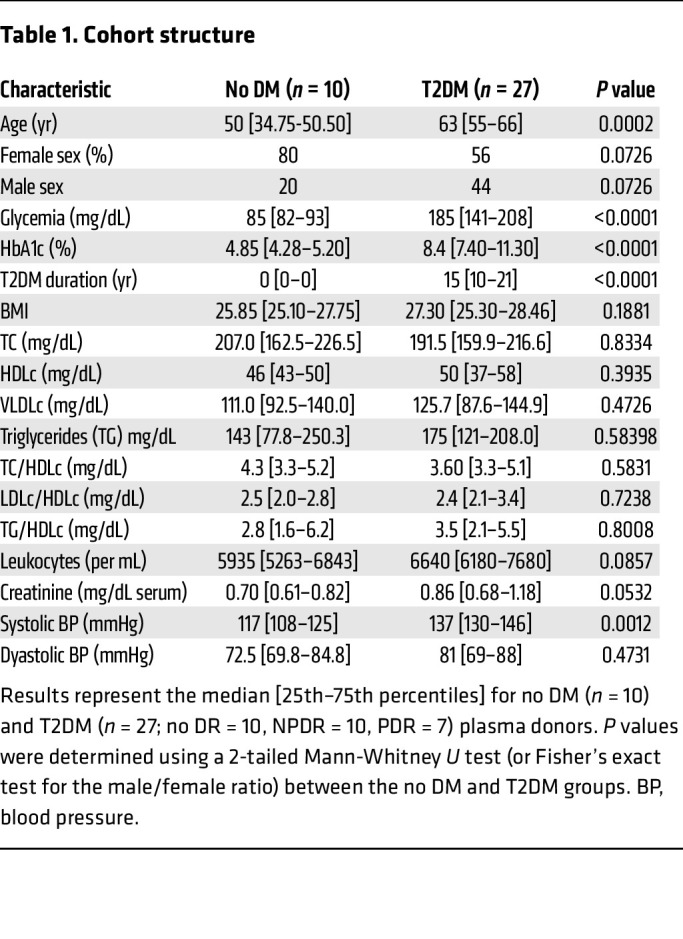
Figure 3. T2DM plasma, but not glucose, induces a lipid-associated phenotype in MPs.

(A) Schematic representation of donor phenotyping and group attribution, plasma preparation, and naive Mo treatment. (B and C) RT-qPCR quantification of healthy donor naive Mos treated for 18 hours with donor plasma from ND individuals (n = 10 [blue dots]) or patients with T2DM (n = 27, no DR [light pink dots], NPDR [pink dots], PDR [dark red dots]). (B) Violin plot representation of the relative expression of the indicated genes in response to individual donor plasma exposure. Dashed lines represent the median and quartiles. P values were determined using a 2-tailed Mann-Whitney U test. (C) Simple linear regression representation of PDK4, ACADVL, ANGPTL4, and CXCL8 (y axis) and PLIN2 expression (x axis). Correlations between expression levels were analyzed using Spearman’s correlation; the linear regression equation, Spearman’s r [95% CI], and 2-tailed P values are given below each correlation graph. (D) Schematic representation of naive Mo treatment with PA and increasing concentrations of glucose. (E) Scatter plot representation of RT-qPCR expression of selected markers in healthy donor naive Mos treated for 18 hours (or 42 hours for ANGPTL4) with either BSA (unbound BSA, blue dots) or PA (BSA-bound PA, red dots) and various concentrations of glucose. Values represent the mean ± SEM of 4 independent cultures. Statistical differences were analyzed by 2-way ANOVA interaction, and P values for the PA and glucose treatments are given below each graph.
As glycemia also differs between T2DM and ND plasma (34, 35), we next tested the effect of 2.5 mM, 5 mM, and 25 mM glucose in the presence or absence of PA on the differentiation of naive Mos (Figure 3D). The expression of PLIN2, PDK4, ACADVL, and CXCL8 was again induced by the presence of PA but was not altered by increasing glucose concentrations, and a high glucose concentration did not potentiate the effect of PA (Figure 3E). ANGPTL4 was only weakly expressed in Mos after 18 hours of PA stimulation but was highly expressed at 42 hours. As for the other transcripts, ANGPTL4 expression was not potentiated by high glucose (Figure 3E).
In summary, our results show that heat-resistant components of T2DM plasma induce an expression pattern in human Mos very similar to that seen with PA. The lack of induction by glucose and the strong induction of lipid overload marker expression by the T2DM plasma strongly suggest that elevated plasma levels of FFA, such as PA, are responsible for the T2DM-induced shift in Mo polarization.
Dose-dependent and long-term effects of PA stimulation on naive Mos.
We next explored naive Mo differentiation by assessing the effects of different concentrations and different durations of PA stimulation. Concentrations above 100 μM PA induced dose-dependent expression of PLIN2, PDK4, ACADVL, ANGPTL4, and CXCL8 transcripts after 42 hours (Figure 4, A and B). We next compared the expression of PLIN2 and CXCL8, which we demonstrated to be markers of lipid overload in early differentiating Mos, after 42 hours by Mos stimulated with PA for only the first 18 hours and compared the expression levels with expression by Mos continuously stimulated for 42 hours (Figure 4C). Medium renewal after 18 hours (PA-to-BSA condition) resulted in a reduction of PLIN2 expression compared with the PA-to-PA condition (PA exposure from t0 to t42 h), but CXCL8 expression remained elevated (Figure 4D). Our results demonstrate that initial PA stimulation may be sufficient to induce a sustained inflammatory response.
Figure 4. Lipid-associated MPs produce inflammatory cytokines.

(A) Schematic representation of naive Mo treatment with increasing concentrations of PA. (B) Nonlinear regression representation of the RT-qPCR relative expression of selected markers of healthy donor naive Mos treated for 42 hours with increasing concentrations of PA (0–500 μM). Values represent the mean ± SEM of 5 independent culture points. Goodness of fit is indicated by R2 (0.9577, 0.9413, 0.7958, 0.9067, and 0.8298, respectively). (C) Schematic representation of 2-phase treatment of naive Mos to test the long-term effect of PA. (D) Scatter plot representation of RT-qPCR expression of PLIN2 and CXCL8 in healthy donor naive Mos subjected to the 2-phase treatment. Values represent the mean ± SEM of a minimum of 4 independent culture points. P values were determined by 1-way Welch’s ANOVA (P = 0.0010 for PLIN2; P = 0.0002 for CXCL8) followed by Dunnett’s T3 multiple-comparison test. (E) Schematic representation of the preparation CM of Mos. (F) Scatter plot of cytokines and growth factor protein concentrations in pg/mL detected by multiplex measurements in CM of naive Mos from a healthy donor differentiated with PA (PA-bound BSA, y axis) or BSA (unbound BSA, x axis) (left graph) or 25 mM glucose plus BSA (y axis) or 5 mM glucose plus BSA (x axis) (right graph).
Lipid-stimulated MPs secrete inflammatory cytokines.
The presence of lipid-laden MPs in the vicinity of vascular leaks could be a source of vasoactive cytokines. We thus collected conditioned medium (CM) 42 hours after an initial 18-hour stimulation of naive Mos with PA or 25 mM glucose (Figure 4E). Using multiplex technology, the cytokine content of PA-stimulated PA-free CM (PAstimCMPAfree) (PA-to-BSA) was compared with CM from MPs previously stimulated with control CM (CtlCM) (BSA-to-BSA) or with the CM from MPs previously stimulated with 25 mM glucose (Figure 4E). Early PA stimulation upregulated the expression of the cytokines CCL2, FGF2, GMCSF, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IFN-γ, and TNF-α, which have been found to be elevated in the vitreous of patients with DR (5). By contrast, these cytokines were only slightly modulated or not expressed after a similar early stimulation with 25 mM glucose (Figure 4F).
These results highlight that key DR inflammatory cytokines were upregulated in the secretome of MPs after an initial stimulation with PA. We have previously showed that PACH3, the unreadily metabolizable analog of PA, reduces lipid polarization of MPs (Figure 2E and Supplemental Figure 2B). We therefore hypothesized that PACH3 could reduce the expression of a number of DR-related cytokines compared with PA. We thus examined the expression of CXCL8, TNF, IL1B, and IL6 and found that all of these inflammatory cytokine transcripts were significantly decreased when naive Mos were exposed to PACH3 instead of PA (Supplemental Figure 4A).
Lipid-associated MPs show vasodegenerative properties.
We tested the activity of the CM on human umbilical vein endothelial cells (HUVECs). PAstimCMPAfree resulted in a 4-fold reduction in cell numbers after 24 hours compared with CtlCM (Figure 5, A–C). We next quantified the capillary degeneration induced by PAstimCMPAfree using our specially designed ex vivo assay (48). Rat aortic rings were allowed to sprout for 6 days and were then treated with PAstimCMPAfree, Ctl-CM, or basal medium. Sprouts from all rings were counted daily from day 4 (2 days before treatment) to day 8 (2 days after treatment) (Figure 5D). CtlCM did not affect the sprouting rate of aortic rings. On the contrary, the addition of PAstimCMPAfree resulted in a decrease in the mean number of branches starting from day 7 (Figure 5E), and a paired analysis of the numbers of branches between day 6 and day 8 showed severe loss of the organized endothelial cell network in each individual aortic ring (Figure 5F). Lectin-stained images of day-8 fixed aortic rings confirmed capillary degeneration after the addition of PAstimCMPAfree (Figure 5G). Stimulation with PACH3 did not correlate with increased expression of inflammatory cytokines (Supplemental Figure 4A). Conversely, PACH3stim-CMPACH3free (where naive Mos were initially stimulated with PACH3 instead of PA) demonstrated significantly lower vasodegenerative activity compared with PAstimCMPAfree (Supplemental Figure 4, B and C). Given the design of our study, we concentrated only stable macromolecules larger than 10 kDa that were present in the CM. Consequently, our results suggest the likely involvement of macromolecules such as inflammatory cytokines secreted by MPs in the observed vasodegenerative properties (Figure 5H).
Figure 5. Lipid-associated MPs show vasodegenerative properties.

(A) Schematic representation of the preparation of CtlCM and PAstimCMPAfree from healthy donor Mos treated with PA (PA-bound BSA) or BSA (unbound BSA). (B and C) HUVECs were stimulated with either CtlCM or PAstimCMPAfree. (B) Epifluorescence images of HUVEC nuclei stained with Hoescht (white). Scale bars: 100 μm. (C) Scatter plot of normalized nuclei counts. Values represent the mean ± SEM of 6 independent culture points. The P value was determined using Welch’s 2-tailed t test. (D–G) Capillary degeneration was quantified in an ex vivo assay (48). (E) Time-course of the mean ± SEM sprout number between day 4 (D4) and day 8 (D8) in the 3 treatment groups: basal medium (n = 10, black), CtlCM (n = 7, blue), and PAstimCMPAfree (n = 7, red). (F) Violin plot of the log2 FC of sprout numbers between paired day-6 and day-8 rings; dots represent individual aortic rings, and dashed lines represent the median and quartiles. P values were determined by 1-way Welch’s ANOVA test (P < 0.0001) followed by Dunnett’s T3 multiple-comparison test. (G) Aortic rings and sprouts treated with CtlCM or PAstimCMPAfree on day 6 and stained with COL4 on day 8. Left: Epifluorescence micrographs of COL4 (white). Scale bars: 500 μm. Right: Higher-magnification confocal micrographs of COL4 (green). Scale bars: 200 μm. Nuclei were stained with Hoechst (blue). (H) Schematic of the biological changes in Mos after lipid exposure and their acquired properties.
Inhibiting PPARγ signaling normalizes the PA-induced phenotype of differentiated MPs.
We found a strong association between PLIN2 and CXCL8 expression in the presence of T2DM plasma or PA. However, we show that lipid removal led to a reduction in PLIN2 expression, whereas CXCL8 overexpression remained persistent, suggesting that the regulation of PLIN2 and CXCL8 may be uncoupled after the initial period of lipid exposure (Figure 4D). We therefore next aimed to determine whether an initial PLIN2 upregulation is necessary for CXCL8 induction. Given the short lifespan of primary human Mos and the challenges associated with their transfection for siRNA assays, we used as a model the more easily transfectable THP-1 cells, a human monocytic cell line derived from a patient with acute monocytic leukemia. The response of THP-1 cells transfected with a control siRNA to PA was similar to that of primary human Mos with a strong upregulation of PLIN2 and CXCL8 (Supplemental Figure 5, A and B). siRNA targeting PLIN2 reduced PLIN2 expression by 77% (Supplemental Figure 5A) and reduced PLIN2 induction in THP-1 cells by 69 % in the presence of PA (Supplemental Figure 5B). This reduced expression of PLIN2 did not reduce inflammatory CXCL8 expression (Figure 6A) or lipid metabolism markers in THP-1 cells (Supplemental Figure 5C).
Figure 6. Inhibition of PPARγ signaling normalizes the PA-induced lipid-associated phenotype.

(A) Scatter plot of RT-qPCR quantification of CXCL8 in THP-1 cells transfected with the indicated siRNA and treated with BSA (unbound BSA) or PA (PA-bound BSA). P values were determined by 1-way Welch’s ANOVA test (P = 0.0136) followed by Dunnett’s T3 multiple-comparison test. (B) Left: Graph of high-confidence predicted PPAR target genes (49) differentially expressed in naive Mos treated with PA. Middle and right: Heatmaps of the expression level (TPM) of curated PPARα target genes with a log2 FC of 2 or higher (middle) and curated PPARγ target genes with a log2 FC of 2 or higher (right). (C) Schematic of the stimulation of naive Mos with PA or BSA and PPAR modulators. (D–G) Scatter plots of RT-qPCR quantification of CXCL8 in healthy donor naive Mos treated with either PA or BSA and with (D) fenofibric acid PPARα agonist (P values for interaction, P = 0.0790; PA effect, P < 0.0001; and drug effect, P = 0.0760); (E) GW6471 PPARα antagonist (P = 0.0570, P < 0.0001, and P = 0.9448); (F) pioglitazone PPARγ agonist (P = 0.0027, P = 0.0002, and P = 0.0021); or (G) T0070907 (T007) PPARγ antagonist (all P < 0.0001). Interactions and P values and were determined by 2-way ANOVA followed by Tukey’s multiple-comparison test. (H) Scatter plots of RT-qPCR quantification of the indicated genes in healthy donor naive Mos treated with PA plus T007. P values were determined using a t test with Welch’s correction. (I) Representative images and scatter plot of TUNEL+ HUVECs after stimulation with CtlCM, PAstimCMPAfree, or PAstim+T007-CMPA&T007-free. P values were determined by 2-way Welch’s ANOVA (P < 0.0046) followed by Dunnett’s T3 multiple-comparison test. (J) Violin plot of the log2 FC of sprout numbers between paired rings before and after stimulation with CtlCM, PAstimCMPAfree, or PAstim+T007-CMPA&T007free. Dashed lines indicate the median and quartiles. P values were determined by 1-way Welch’s ANOVA (P < 0.0001) followed by Dunnett’s T3 multiple-comparison test.
Therefore, we considered a broader approach targeting signaling pathways associated with lipid exposure. Our comprehensive bulk RNA-Seq analysis revealed that naive, healthy MPs swiftly responded to PA exposure by upregulating the expression of PPARG, a gene encoding a member of the PPAR transcription factor family. Conversely, the expression of PPARA, which encodes the therapeutic target of fenofibrate — a clinical diabetes treatment — was marginally downregulated (log2 FC = –0.44). Given the critical role of PPARs in controlling the transcription of lipid metabolism–associated genes, we hypothesized that a potentially large-scale, PPAR-mediated modulation of lipid-associated phenotypic characteristics occurs in MPs. Consequently, we examined the expression of PPARα and PPARγ transcriptional targets in PA-stimulated Mos. We assessed the expression of 448 high-confidence PPAR target genes that were identified by Fang et al. using a combination of transcriptomics data and the presence of the PPAR-responsive element (PPRE) in the regulatory regions of these target genes (49). We found that 252 of them (56.2%) were statistically differentially expressed after PA exposure (Figure 6B and Supplemental Table 6). Next, we analyzed the regulation of the 83 manually curated transcript targets of PPARα and the 104 manually curated transcript targets of PPARγ (49). PA regulated 38 transcripts (45.7%) on the curated PPARα target list, including 7 (8.4%) with a log2 FC of 2 or higher (Figure 6B and Supplemental Table 7). PA also regulated 50 transcripts (49.0%) on the curated PPARγ list, including 6 (5.8%) with a log2 FC of 2 or higher (Figure 6B and Supplemental Table 8). Although ACADVL was not included in the curated PPAR target list, the other 3 previously identified PA-associated transcripts (PLIN2, PDK4, and ANGPTL4) were all represented in the highly regulated target list (Figures 2 and 3). These findings suggest that PPARα and PPARγ are potential targets to blunt lipid-induced proinflammatory MP polarization.
We therefore tested the effects of fenofibric acid and GW6471 (a PPARα agonist and antagonist, respectively) and pioglitazone and T0070907 (a PPARγ agonist and antagonist, respectively) on the expression of the selected prototypical CXCL8 transcript (Figure 6, C–G). Neither the PPARα agonist nor the antagonist affected CXCL8 expression in the BSA condition, and these agents failed to reduce its induction after PA stimulation (Figure 6, D and E). Pioglitazone, a PPARγ agonist, increased CXCL8 expression in the BSA condition and had no effect after PA stimulation (Figure 6F). Finally, the PPARγ antagonist T0070907 strongly reduced CXCL8 expression after PA stimulation and also reduced its basal expression in the BSA condition (Figure 6G). Consistent with these results, T0070907 reduced the expression of the PPAR targets PLIN2, PDK4, and ANGPTL4 but not of the non-PPAR–curated target ACADVL (Figure 6H). CXCL8 downregulation was associated with decreased expression of the DR-associated cytokines TNF, IL1B, and IL6 (Supplemental Figure 6).
We have previously shown that PAstimCMPAfree leads to a reduction in the number of HUVECs relative to CtlCM (Figure 5, A–C). To gain further knowledge about the potential protective effect of T0070907 on endothelial cell viability, we next analyzed HUVEC apoptotic cell death by TUNEL assay 48 hours after addition of CtlCM, PAstimCMPAfree, or (PA+T0070907)stimCM(PA+T0070907)free. PAstimCMPAfree induced a statistically significant 75% increase in TUNEL+ HUVECs compared with CtlCM, whereas (PA+T0070907)stimCM(PA+T0070907)free completely rescued HUVECs undergoing apoptotic cell death (Figure 6I). Finally, we compared the effect of these 3 CMs on the integrity of the capillary network in the rat aortic ring model. Again, we found that (PA+T0070907)stimCM(PA+T0070907)free was protective of the vascular network compared with PAstimCMPAfree (Figure 6J).
Discussion
In this study, we show that PLIN2+ MPs were present in the vicinity of leaky capillaries, identified by extraluminal ALB, in postmortem human T2DM donor retina with histopathologic features of NPDR. Importantly, we demonstrate that hi-plasma from patients with T2DM (with or without DR) strongly upregulated the expression of lipid overload markers in Mos from healthy donors, which strongly correlated with the expression of DR biomarkers such as ANGPTL4 and CXCL8. We also show that PA, an abundant plasma FFA that is elevated in dyslipidemic plasma of patients with T2DM, induced a nearly identical expression pattern in naive human Mos. We characterized the PA-induced transcriptome and secretome changes and show that the PA-stimulated Mo secretome induced endothelial apoptosis and ex vivo capillary degeneration. A PPARγ antagonist blunted PA-induced cytokine expression and reversed the vasodegenerative properties of the PA-stimulated Mo secretome. This study provides comprehensive insights into the glycemia-independent activation of Mos in T2DM and identifies MP PPARγ as a target for dyslipidemia-activated MPs in DR.
Plasma lipids are internalized by Mos.
FFAs are abundant in the blood, and their levels are elevated in T2DM plasma (34, 50). TGs, another lipid component found in the blood, are composed of 3 FAs that can be hydrolyzed by extracellular LPL and further increase the pool of FFAs available to cells. Lipoprotein TGs and ALB-bound FFAs do not extravasate in the absence of vascular leakage. However, years of diabetes can compromise the integrity of the blood-retina barrier, leading to increased permeation of plasma components such as ALB, carbohydrates, and lipids into the retinal environment. These elements not only come into direct physical contact with local cells such as Müller glial cells, leading to their robust activation (51, 52), but also shape the milieu that drives the differentiation of infiltrating Mos. Using chromatographic techniques, we demonstrated that Mos internalized PA. We show that the proportion of PA chains increased markedly, while the proportion of other FA chains did not change. Together with the increased expression of PLIN2, this suggests that PA was stored intracellularly. Interestingly, we show that Mo activation persisted even after PA removal. This long-term effect suggests that the resolution of in vivo inflammation may lag even after extravasated lipids have been taken up intracellularly.
Clinically, elevated plasma TG levels have been associated with an increased risk of DR, with a clear association between TG levels and an increased frequency of hard exudates (53). We performed classical lipid analysis of plasma from our donors in the clinic (total cholesterol [TC], HDL cholesterol [HDLc], LDLc, VLDLc, and TGs) and observed no significant difference in overall plasma lipid load between the controls and T2DM donors in our cohort. However, we found that plasma-derived nonglucose cues could induce lipid-associated MP polarization. Our study thus suggests that there may be undiagnosed risks (i.e., the available PA that we showed had a detrimental effect on Mo polarization) of Mo activation that cannot be detected by performing classical lipid analysis in patients with T2DM. Our study focused on PA, the most abundant FFA in the blood. PA is not the only elevated FFA in diabetes, and numerous FFAs are also found to be elevated in T2DM (34, 35). We also show that SA, the second most abundant saturated FFA in the blood, PoA, the n-7 unsaturated form of PA, and a plasma representative lipid blend also induced PLIN2 overexpression.
Lipid-laden MPs are associated with vascular remodeling during DR.
PLIN2+ MPs are found in atherosclerosis plaques, cholesterol polyps of the gall bladder, and fat necrosis (37). In the present study, in DR, we found PLIN2+ MPs in regions of microvascular disease characterized by ALB extravasation, suggesting that MPs were involved in the uptake of extravasated lipids and lipid storage. A similar observation has been made in the central nervous system, where PLIN2+ cells accumulate with age in the hippocampus and can be stained with lipid dyes (38). Consistent with our observation, MPs have been found in the red oil+ hard exudates of the retina in DR (54). More recently, adaptive optics funduscopy has revealed the presence of round motile particles that resemble MPs in hard exudates, a structure found in the periphery of regions of active vascular leakage (43). Collectively, these findings indicate that lipid-polarized MPs are found in regions of active vascular remodeling in the DR retina, that they may progressively accumulate in regions exposed to plasma leakage, and that they could be a source of vasoactive cytokines.
In an effort to understand how plasma leakage may influence MP activation, in particular the fate of infiltrating Mos, we differentiated naive Mos in the presence of hi-plasma from healthy and T2DM donors. We show that hi-plasma from patients with T2DM rapidly induced a lipid-associated polarization of MPs characterized by overexpression of lipid-related genes. We also show a correlation between PLIN2 expression (used as a proxy for MP lipid load) and other lipid-related genes as well as DR biomarkers such as ANGPTL4 and CXCL8.
Such differentiation of Mos could be mimicked by PA, but not by glucose exposure. PA stimulation was associated with an increase in the production of at least 14 proinflammatory cytokines, 11 of which have been shown to be directly involved in DR. Similarly, Müller glial cells, the major glial cells of the retina, have been shown to have a greater increase in the expression of cytokines associated with DR when stimulated with PA than with glucose (51, 52).
ANGPTL4 is a protein that regulates lipid processing but also has vasoactive properties. ANGPTL4 was notably upregulated when naive Mos were stimulated with PA or T2DM plasma, and ANGPTL4 expression showed a strong correlation with PLIN2 expression. ANGPTL4 plays an important role in the pathogenesis of DR (55, 56). Its expression has been found to correlate with the nonperfused area (45); in line with this finding, we found that the net effect of the PA-stimulated MP secretome was vasodegenerative, which was highlighted by a decrease in vascular sprouting in a 3D model and an increase in endothelial cell apoptosis.
Interestingly, we found that MPs remained activated and secreted inflammatory cytokines for at least 24 hours after the initial lipid stimulation. This effect may lead to a long-term vascular remodeling activity of lipid-stimulated MPs. Such vasodegenerative activity is consistent with the results of previous reports showing an overall antiangiogenic activity of MPs in the context of retinal ischemia. Indeed, MPs are associated with early vascular regression in models of ischemic retinopathy and contribute to the resolution of vascular proliferation (10, 57). In our hands, mouse Mos were less sensitive to PA than were human Mos, and our attempts to establish a mouse model of lipid-associated DR have so far been unsuccessful. This difference is consistent with the overall difference in the severity of DR between mice and humans. Mice do not develop clear early signs of NPDR, such as severe retinal capillary loss or massive blood retina-barrier breakdown and thus are not prone to progress to PDR like humans are. Hence, although our ex vivo aortic ring model clearly identified vasodegenerative properties of lipid-exposed MPs, this could not yet be validated in vivo. In contrast, subretinal MPs in exudative age-related macular degeneration show proangiogenic activity (58, 59). The exact mechanism by which the addition of CM from PA-stimulated MPs leads to endothelial cell loss is yet to be determined. Cytokines found to be increased in CM from PA-stimulated MPs (CCL2, FGF2, IL-1β, IL-2, IL-4, IL-5, IL-6, CXCL8, IL-10, IFN-γ, and TNF-α) have been implicated in the pathogenesis of DR and are probably responsible for this activity.
The PPARγ signaling pathway is involved in vasodegenerative activities of lipid-associated MPs.
We demonstrated a potent correlation between the expression of PLIN2 and CXCL8 during the early stages of MP differentiation in the context of plasma lipids and exposure to various isolated FFAs. This may suggest a regulatory role for PLIN2 in CXCL8 expression. Similar correlations between PLIN2 and inflammatory cytokines have been observed in tumor-associated macrophages as well as in THP-1 cells (39, 60). Notably, PLIN2+ MPs are observed around retinal microaneurysms and in numerous pathological contexts, suggesting that therapeutic inhibition of PLIN2 may serve as a strategy to curb inflammation-associated vascular remodeling. However, our results show that siRNA-based silencing of PLIN2 transcript expression in the THP-1 cell line, a model of Mos, neither inhibited CXCL8 transcription nor altered the regulation of other selected markers of lipid metabolism. Similarly, the production of cytokines by bone marrow–derived macrophages stimulated with various lipids remained unaltered following PLIN2 deletion (61), whereas Chen et al. reported a slight reduction in inflammatory cytokine expression in unstimulated THP-1 cells following PLIN2 silencing (39). Altogether, these results suggest that, while PLIN2 serves as an excellent marker of lipid polarization in vivo in the human retina and in vitro with Mos stimulated by T2DM plasma or PA, its inhibition by siRNAs is insufficient to prevent inflammatory polarization of at least lipid-exposed THP-1. Considering that we observed persistent overexpression of CXCL8, even after PA removal, and given the high stability of PLIN proteins when incorporated into lipid droplets (62, 63), therapeutic strategies targeting the inhibition of PLIN2 protein production may prove ineffective in situations in which MPs harboring lipid droplets have already accumulated.
Our transcriptomics analysis of differentiating Mos in the presence of PA or plasma from patients with T2DM showed upregulated expression of the genes PLIN2, PDK4, and ANGPTL4. These genes are all transcriptional targets of PPAR transcription factors. Consistent with this role, our RNA-Seq analysis identified PPARG as one of the most highly upregulated genes (log2 FC = 4.07), whereas PPARA was slightly downregulated (log2 FC = –0.44). Members of the PPAR family are nuclear transcription factors that bind to the retinoid X receptor α (RXRα) to regulate the expression of numerous genes, most of which are part of the lipid metabolism pathway. PPARs act as repressors when not bound to a ligand. When natural (i.e., FFA agonists) or synthetic ligands bind to the ligand-binding domain (LBD) of PPARs, they promote the binding of coactivator or corepressor factors, resulting in ligand-dependent modulation of gene expression. We investigated the role of the PPARα and PPARγ pathways in the differentiation of Mos in the presence of lipids using PPARα and PPARγ agonists and antagonists. Fenofibrate, which is metabolized to the PPARα agonist fenofibric acid, is a widely used systemic treatment to lower plasma lipid concentrations. Fenofibrate has successfully proven its protective effect in patients with DR (27, 28). It has also been shown to reduce systemic inflammation in patients with NPDR, suggesting a potential role of circulating lipids or lipid metabolism in the chronic inflammatory status of patients with T2DM (64). Fenofibric acid and GW6471, a PPARα antagonist, did not regulate the expression of the DR prototypical cytokine CXCL8 in MPs. However, in animal models of T1DM, systemic fenofibrate treatment has a protective effect on the blood-retina barrier and protects against neovascularization in the oxygen-induced retinopathy model, notably by attenuating the overexpression of VEGF, CCL2, and ICAM-1 (39). Overall, this suggests that the beneficial effects of PPARα agonists in vivo could be due to effects on nonmyeloid cells. For example, PPARα agonists have been shown to directly protect endothelial cells (65, 66).
In contrast to the PPARα agonist T0070907, a PPARγ antagonist, attenuated PA-induced CXCL8 overexpression and the CM of naive Mos treated with PA+T0070907 showed reduced vasodegenerative properties. Activation of the PPARγ pathway in MPs is generally considered to be antiinflammatory (67, 68), and there is consensus for a beneficial effect of systemic PPARγ agonist treatment in a wide range of diseases, including diabetes and its comorbidities. However, PPARγ antagonism has been reported to protect against the progression of nonalcoholic fatty liver disease (NAFLD) (69) and to reduce foam cell formation (70–72). In BV2 microglia cells treated with LPS, and thus in the absence of DR-relevant stress, Pparg knockdown or T0070907 treatment was also shown to be associated with antiinflammatory polarization (73). In a study using the human myeloid cell line Mono Mac 6, the small molecule bindarit was shown to promote, in a PPARγ-dependent manner, overexpression of the lipid-associated protein FABP4 along with the CXCL8 transcript (74). In accordance with this result, we found that pioglitazone, a PPARγ agonist, increased the expression of CXCL8 in the BSA-only condition, suggesting that PPARγ upregulation may result in adverse effects even in the absence of exogenously added PPAR ligand. Locally in the eye, inflammation in retinal pigment epithelial cells treated with N-retinylidene-N-retinylethanolamine, a byproduct of the visual cycle associated with age-related macular degeneration, was also shown to be inhibited by T0070907 (75). A possible explanation for this discrepancy between the beneficial effects of PPARγ agonists and antagonists may be that normalization of circulating lipid levels and restoration of lipid storage in adipocytes by systemic PPARγ agonism is responsible for the observed beneficial effect, whereas in our setting, PPARγ antagonism was applied only to MPs. Overall, growing evidence suggests that the definition of PPARγ pathway activation as an inhibitor of M1 inflammatory polarization and an inducer of M2 antiinflammatory polarization may be an oversimplification. The mechanism by which these plasma lipids induce Mo differentiation into MPs with proinflammatory properties is still unknown. However, and in accordance with our finding that PA was rapidly internalized, PA-induced polarization of MPs to a proinflammatory profile has recently been shown to be independent of direct TLR4 binding (76). Our results further suggest that the differentiation of Mos in the presence of lipids leads to a modification of their metabolism, as (a) the use of PACH3, a PA analog that can be internalized but that is not readily metabolizable, did not trigger CXCL8 secretion, nor did it induce the degeneration of aortic ring sprouts, and (b) the PDK4 gene was found to be highly upregulated in the presence of PA, suggesting a potential switch to aerobic glycolysis (the so-called Warburg effect), a situation found in proinflammatory MPs. Of note, PPARδ antagonism is also beneficial in the ocular context. The lipid-induced Müller glial cell response can be inhibited by GSK0660, a PPARδ antagonist (77). Overall, this suggests that PPAR antagonists could be used locally in addition to systemic PPARα agonist treatment to inhibit the pathological differentiation of local and infiltrating cells in the retina in DR.
Study limitations.
In this study, we focused on identifying the major class of lipids present in the blood of our cohort of patients. However, we must acknowledge that detailed information regarding lipid chain composition and concentrations of specific FFAs, including PA, was not obtained. Future investigations will be necessary to establish possible correlations between individual plasma lipid species, the activation level of naive Mos, and DR and its progression.
Additionally, our findings demonstrated that the secretome of PA-stimulated MP exhibits antiangiogenic activity, suggesting a potential link between lipid extravasation and vascular degeneration in vivo. However, it is essential to recognize that our study lacked relevant in vivo models of vascular permeability. As a result, we were unable to assess the relative importance of MP-induced degeneration compared with other known circulating factors involved in vascular remodeling, such as high glucose, advanced glycation endproducts (AGEs), oxidized lipids, and metabolites. More recently, diabetes-induced microbiote modification has also been recognized as an important player in disease progression. Exploring additional in vivo models and investigating the interactions of various factors contributing to vascular remodeling will provide valuable insights into this complex phenomenon.
In conclusion, DR is a neurodegenerative, microvascular, and inflammatory complication of T2DM, in which breakdown of the blood-retina barrier and vascular degeneration lead to the extravasation of plasma proteins, lipids, and leukocytes. We showed that MPs expressing PLIN2 were found in retinas with active vascular leakage and vascular remodeling. Exposure of naive Mos to hi-plasma from patients with diabetes or PA was sufficient to induce PLIN2 expression and trigger long-term expression of DR-related proinflammatory cytokines. The secretome of lipid-exposed naive Mos showed strong vasodegenerative properties that could be blunted by a PPARγ inhibitor. Our study sheds light on the harmful role of lipid-associated MP accumulation in the retina in DR and the role of pathogenic lipids, rather than glucose, in the activation of these cells.
Methods
Detailed Methods can be found in the Supplemental Methods.
FFA solubilization and medium preparation.
To allow FFA solubilization in culture medium, FFAs were bound to FFA-free BSA. FFAs were dissolved in absolute ethanol (EtOH) and added to a BSA-containing culture medium (0.88% w/v of BSA) to obtain the working concentration (500 μM FFA, 0.5 % v/v EtOH; FFA/BSA molar ratio of 3.8).
Preparation of CM.
To obtain CM free of BSA and PA, Mos were isolated as described in the Supplemental Methods and stimulated for the first 18 hours. The culture medium was then removed, and early differentiated MPs were cultured for another 24 hours in a fresh control culture medium.
Statistics.
GraphPad Prism 8 (GraphPad Software) was used for all graphical representations and for all but the RNA-Seq statistical analysis. A detailed explanation of the specific statistical choices made is provided in the Supplemental Methods. A P value of less than 0.05 was considered significant.
Study approval.
Plasma samples from patients with T2DM and healthy donors were collected from volunteer donors at the Institute of Ophthalmology Foundation Conde de Valenciana (Mexico City, Mexico), in accordance with Declaration of Helsinki principles and with the approval of the Investigation Committee (registry 13 CI 09 015 261, protocol number CI-051-2015); the Research Ethics Committee (registry 13 CEI 09 015 095, protocol number CEI-2015-11-05); and the Biosafety Committee (registry 13 CB 31 050 269, protocol number CB-051-11-2015) (all in Mexico City, Mexico). Individuals provided written informed consent prior to their participation in the study.
Data and materials availability.
Experimental materials are available upon request with no restrictions. The RNA-Seq raw fastq files and count files were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE239512). Values for all data points in graphs can be found in the Supplemental Supporting Data Values file.
Author contributions
GB, RK, FB, and XG conceived the study. GB, HCM, FB, AJC, and XG curated data. GB, HCM, FS, and XG acquired funding. GB, RK, LP, TMS, HCM, PN, SA, FPMR, LV, and XG collected data. GB, RK, and XG designed the study methodology. AC, DRDLP, AJC, YG (patient recruitment and phenotyping), NA (lipidomics), and JAS provided resources. GB, NA, YG, FS, CD, and XG supervised the study. GB and XG wrote the original draft of the manuscript. GB, RK, HCM, RK, FPMR, DRDLP, AJC, CD, YG, FS, and XG reviewed and edited the manuscript.
Supplementary Material
Acknowledgments
Funding for this study was provided by INSERM (to FS and XG); the Agence Nationale pour la Recherche Labex Lifesenses (ANR-10-LABEX-65, to XG); IHU FOReSIGHT (ANR-18-IAHU-01, to XG); UNADEV (INSERM-UNADEV-22UU146-00, to XG); and ED394 – Sorbonne University (to GB and HCM, salary). We wish to thank the cell culturing facility and Stéphane Fouquet of the imaging facility at the Institut de la Vision and Wassila Carpentier and Maureen Hayibor, Pierre Duquesne, Antoine Auvergne, and Mariana Garcia, who provided technical assistance. Certain elements of the schemes were adapted from Servier Medical Art, licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). The figure color codes are based on the Vibrant qualitative color scheme of Paul Tol (https://personal.sron.nl/~pault/).
Version 1. 10/02/2023
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2023, Blot et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2023;133(19):e161348.https://doi.org/10.1172/JCI161348.
Contributor Information
Guillaume Blot, Email: guillaume_blot@outlook.com.
Rémi Karadayi, Email: remi.karadayi@inserm.fr.
Lauriane Przegralek, Email: lauriane.przegralek@inserm.fr.
Thérèse-Marie Sartoris, Email: tmsartoris@yahoo.fr.
Hugo Charles-Messance, Email: hugo.charles.messance@gmail.com.
Pierre Negrier, Email: p.negrier@icloud.com.
Frédéric Blond, Email: frederic.blond@inserm.fr.
Lucile Vignaud, Email: lucile.vignaud@inserm.fr.
Aude Couturier, Email: audecouturier6@gmail.com.
José-Alain Sahel, Email: j-sahel@quinze-vingts.fr.
Niyazi Acar, Email: niyazi.acar@inrae.fr.
Aida Jimenez-Corona, Email: aidaajc@gmail.com.
Cécile Delarasse, Email: cecile.delarasse@inserm.fr.
Yonathan Garfias, Email: yogarfias@institutodeoftalmologia.org.
Florian Sennlaub, Email: florian.sennlaub@inserm.fr.
Xavier Guillonneau, Email: xavier.guillonneau@inserm.fr.
References
- 1.(NCD-RisC) NRFC. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet. 2016;387(10027):1513–1530. doi: 10.1016/S0140-6736(16)00618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahlqvist E, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–369. doi: 10.1016/S2213-8587(18)30051-2. [DOI] [PubMed] [Google Scholar]
- 3.Group ER. Grading diabetic retinopathy from stereoscopic color fundus photographs--an extension of the modified Airlie House classification. ETDRS report number 10. Early treatment diabetic retinopathy study research group. Ophthalmology. 1991;98(5 suppl):786–806. doi: 10.1016/S0161-6420(13)38012-9. [DOI] [PubMed] [Google Scholar]
- 4.Pan WW, et al. The innate immune system in diabetic retinopathy. Prog Retin Eye Res. 2021;84:100940. doi: 10.1016/j.preteyeres.2021.100940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubsam A, et al. Role of inflammation in diabetic retinopathy. Int J Mol Sci. 2018;19(4):942. doi: 10.3390/ijms19040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAuley AK, et al. Vitreous biomarkers in diabetic retinopathy: a systematic review and meta-analysis. J Diabetes Complications. 2014;28(3):419–425. doi: 10.1016/j.jdiacomp.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Semeraro F, et al. Diabetic retinopathy, a vascular and inflammatory disease: therapeutic implications. Diabetes Metab. 2019;45(6):517–527. doi: 10.1016/j.diabet.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Zeng HY, et al. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126(2):227–232. doi: 10.1001/archophthalmol.2007.65. [DOI] [PubMed] [Google Scholar]
- 9.Gaucher D, et al. Microglial changes occur without neural cell death in diabetic retinopathy. Vision Res. 2007;47(5):612–623. doi: 10.1016/j.visres.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 10.Kermorvant-Duchemin E, et al. Neonatal hyperglycemia inhibits angiogenesis and induces inflammation and neuronal degeneration in the retina. PLoS One. 2013;8(11):10. doi: 10.1371/journal.pone.0079545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakravarthy H, et al. Role of acid sphingomyelinase in shifting the balance between proinflammatory and reparative bone marrow cells in diabetic retinopathy. Stem Cells. 2016;34(4):972–983. doi: 10.1002/stem.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rangasamy S, et al. Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One. 2014;9(10):e108508. doi: 10.1371/journal.pone.0108508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G, et al. Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes. 2012;61(12):3294–3303. doi: 10.2337/db11-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hazra S, et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia. 2013;56(3):644–653. doi: 10.1007/s00125-012-2781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lachin JM, et al. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial--revisited. Diabetes. 2008;57(4):995–1001. doi: 10.2337/db07-1618. [DOI] [PubMed] [Google Scholar]
- 16.Do DV, et al. Blood pressure control for diabetic retinopathy. Cochrane Database Syst Rev. 2015;1(3):CD006127. doi: 10.1002/14651858.CD006127.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eid S, et al. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia. 2019;62(9):1539–1549. doi: 10.1007/s00125-019-4959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer SS, Busik JV. The role of dyslipidemia in diabetic retinopathy. Vision Res. 2017;139:228–236. doi: 10.1016/j.visres.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benarous R, et al. Differential association of serum lipids with diabetic retinopathy and diabetic macular edema. Invest Ophthalmol Vis Sci. 2011;52(10):7464–7469. doi: 10.1167/iovs.11-7598. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, et al. Risk factors of diabetic retinopathy and sight-threatening diabetic retinopathy: a cross-sectional study of 13 473 patients with type 2 diabetes mellitus in mainland China. BMJ Open. 2017;7(9):e016280. doi: 10.1136/bmjopen-2017-016280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pugliese G, et al. High prevalence of advanced retinopathy in patients with type 2 diabetes from the renal insufficiency and cardiovascular events (RIACE) Italian multicenter study. Diabetes Res Clin Pract. 2012;98(2):329–337. doi: 10.1016/j.diabres.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Romero-Aroca P, et al. Ten-year incidence of diabetic retinopathy and macular edema. Risk factors in a sample of people with type 1 diabetes. Diabetes Res Clin Pract. 2011;94(1):126–132. doi: 10.1016/j.diabres.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Raman R, et al. Influence of serum lipids on clinically significant versus nonclinically significant macular edema: SN-DREAMS report number 13. Ophthalmology. 2010;117(4):766–772. doi: 10.1016/j.ophtha.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, et al. Prevalence and risk factors of diabetes and diabetic retinopathy in Liaoning province, China: a population-based cross-sectional study. PLoS One. 2015;10(3):e0121477. doi: 10.1371/journal.pone.0121477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agroiya P, et al. Association of serum lipids with diabetic retinopathy in type 2 diabetes. Indian J Endocrinol Metab. 2013;17(suppl 1):S335–S337. doi: 10.4103/2230-8210.119637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yau JW, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–564. doi: 10.2337/dc11-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keech AC, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 28.Group AS. et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010;363(3):233–244. doi: 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon B, et al. Oleate prevents palmitate-induced mitochondrial dysfunction, insulin resistance and inflammatory signaling in neuronal cells. Biochim Biophys Acta. 2014;1843(7):1402–1413. doi: 10.1016/j.bbamcr.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Weigert C, et al. Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J Biol Chem. 2004;279(23):23942–23952. doi: 10.1074/jbc.M312692200. [DOI] [PubMed] [Google Scholar]
- 31.Ishiyama J, et al. Unsaturated FAs prevent palmitate-induced LOX-1 induction via inhibition of ER stress in macrophages. J Lipid Res. 2011;52(2):299–307. doi: 10.1194/jlr.M007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palomer X, et al. Palmitic and oleic acid: the Yin and Yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab. 2018;29(3):178–190. doi: 10.1016/j.tem.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Abdelmagid SA, et al. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS One. 2015;10(2):e0116195. doi: 10.1371/journal.pone.0116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi L, et al. Simultaneously quantitative measurement of comprehensive profiles of esterified and non-esterified fatty acid in plasma of type 2 diabetic patients. Chem Phys Lipids. 2007;150(2):204–216. doi: 10.1016/j.chemphyslip.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Sobczak AIS, et al. Changes in plasma free fatty acids associated with type-2 diabetes. Nutrients. 2019;11(9):2022. doi: 10.3390/nu11092022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlicky DJ, et al. Multiple functions encoded by the N-terminal PAT domain of adipophilin. J Cell Sci. 2008;121(pt 17):2921–2929. doi: 10.1242/jcs.026153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Straub BK, et al. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology. 2013;62(4):617–631. doi: 10.1111/his.12038. [DOI] [PubMed] [Google Scholar]
- 38.Marschallinger J, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23(2):194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen FL, et al. Adipophilin affects the expression of TNF-alpha, MCP-1, and IL-6 in THP-1 macrophages. Mol Cell Biochem. 2010;337(1–2):193–199. doi: 10.1007/s11010-009-0299-7. [DOI] [PubMed] [Google Scholar]
- 40.Qiao L, et al. Deficient chaperone-mediated autophagy promotes lipid accumulation in macrophage. J Cardiovasc Transl Res. 2021;14(4):661–669. doi: 10.1007/s12265-020-09986-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saliba-Gustafsson P, et al. Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed-forward loop between LXR and autophagy. J Intern Med. 2019;286(6):660–675. doi: 10.1111/joim.12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan Y, et al. Adipophilin involved in lipopolysaccharide-induced inflammation in RAW264.7 cell via extracellular signal-regulated kinase 1/2-peroxisome proliferator-activated receptor gamma pathway. DNA Cell Biol. 2017;36(12):1159–1167. doi: 10.1089/dna.2017.3706. [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi M, et al. High-resolution imaging by adaptive optics scanning laser ophthalmoscopy reveals two morphologically distinct types of retinal hard exudates. Sci Rep. 2016;6:33574. doi: 10.1038/srep33574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu SJ, et al. Upregulation of P2RX7 in Cx3cr1-deficient mononuclear phagocytes leads to increased interleukin-1 beta secretion and photoreceptor neurodegeneration. J Neurosci. 2015;35(18):6987–6996. doi: 10.1523/JNEUROSCI.3955-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon SH, et al. Aqueous levels of angiopoietin-like 4 and semaphorin 3E correlate with nonperfusion area and macular volume in diabetic retinopathy. Ophthalmology. 2015;122(5):968–975. doi: 10.1016/j.ophtha.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Dijk W, Kersten S. Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol Metab. 2014;25(3):146–155. doi: 10.1016/j.tem.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 47.Marques-Vidal P, et al. Association between circulating cytokine levels, diabetes and insulin resistance in a population-based sample (CoLaus study) Clin Endocrinol (Oxf) 2013;78(2):232–241. doi: 10.1111/j.1365-2265.2012.04384.x. [DOI] [PubMed] [Google Scholar]
- 48.Blot G, et al. Modifications to the classical rat aortic ring model to allow vascular degeneration studies. STAR Protoc. 2021;2(1):100281. doi: 10.1016/j.xpro.2020.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang L, et al. PPARgene: a database of experimentally verified and computationally predicted PPAR target genes. PPAR Res. 2016;2016:6042162. doi: 10.1155/2016/6042162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, et al. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(28):2817–2825. doi: 10.1016/j.jchromb.2010.08.035. [DOI] [PubMed] [Google Scholar]
- 51.Couturier A, et al. Reproducing diabetic retinopathy features using newly developed human induced-pluripotent stem cell-derived retinal Müller glial cells. Glia. 2021;69(7):1679–1693. doi: 10.1002/glia.23983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Capozzi ME, et al. Palmitic acid induces müller cell inflammation that is potentiated by co-treatment with glucose. Sci Rep. 2018;8(1):5459. doi: 10.1038/s41598-018-23601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang YC, Wu WC. Dyslipidemia and diabetic retinopathy. Rev Diabet Stud. 2013;10(2–3):121–132. doi: 10.1900/RDS.2013.10.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cusick M, et al. Histopathology and regression of retinal hard exudates in diabetic retinopathy after reduction of elevated serum lipid levels. Ophthalmology. 2003;110(11):2126–2133. doi: 10.1016/j.ophtha.2003.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Babapoor-Farrokhran S, et al. Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc Natl Acad Sci U S A. 2015;112(23):E3030–E3039. doi: 10.1073/pnas.1423765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xin X, et al. Hypoxic retinal Muller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc Natl Acad Sci U S A. 2013;110(36):E3425–E3434. doi: 10.1073/pnas.1217091110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies MH, et al. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest Ophthalmol Vis Sci. 2008;49(9):4195–4202. doi: 10.1167/iovs.07-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guillonneau X, et al. On phagocytes and macular degeneration. Prog Retin Eye Res. 2017;61:98–128. doi: 10.1016/j.preteyeres.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Mettu PS, et al. Incomplete response to Anti-VEGF therapy in neovascular AMD: exploring disease mechanisms and therapeutic opportunities. Prog Retin Eye Res. 2021;82:100906. doi: 10.1016/j.preteyeres.2020.100906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He Y, et al. Lipid droplet-related PLIN2 in CD68+ tumor-associated macrophage of oral squamous cell carcinoma: implications for cancer prognosis and immunotherapy. Front Oncol. 2022;12:824235. doi: 10.3389/fonc.2022.824235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Son SH, et al. Perilipin 2 (PLIN2)-deficiency does not increase cholesterol-induced toxicity in macrophages. PLoS One. 2012;7(3):e33063. doi: 10.1371/journal.pone.0033063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen KT, et al. N-terminal acetylation and the N-end rule pathway control degradation of the lipid droplet protein PLIN2. J Biol Chem. 2019;294(1):379–388. doi: 10.1074/jbc.RA118.005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gimenez-Andres M, et al. Exceptional stability of a perilipin on lipid droplets depends on its polar residues, suggesting multimeric assembly. Elife. 2021;10:e61401. doi: 10.7554/eLife.61401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ju HB, et al. Effects of fenofibrate on inflammatory cytokines in diabetic retinopathy patients. Medicine (Baltimore) 2017;96(31):e7671. doi: 10.1097/MD.0000000000007671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim J, et al. Fenofibrate regulates retinal endothelial cell survival through the AMPK signal transduction pathway. Exp Eye Res. 2007;84(5):886–893. doi: 10.1016/j.exer.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Tomizawa A, et al. Fenofibrate suppresses microvascular inflammation and apoptosis through adenosine monophosphate-activated protein kinase activation. Metabolism. 2011;60(4):513–522. doi: 10.1016/j.metabol.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 67.Christofides A, et al. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism. 2021;114:154338. doi: 10.1016/j.metabol.2020.154338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toobian D, et al. Parsing the role of PPARs in macrophage processes. Front Immunol. 2021;12:783780. doi: 10.3389/fimmu.2021.783780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maruyama H, et al. Palmitate-induced regulation of PPARγ via PGC1α: a mechanism for lipid accumulation in the liver in nonalcoholic fatty liver disease. Int J Med Sci. 2016;13(3):169–178. doi: 10.7150/ijms.13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oppi S, et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARγ signature. Eur Heart J. 2020;41(9):995–1005. doi: 10.1093/eurheartj/ehz667. [DOI] [PubMed] [Google Scholar]
- 71.Park KS, et al. The natural compound dansameum reduces foam cell formation by downregulating CD36 and peroxisome proliferator-activated receptor-gamma; expression. Pharmacogn Mag. 2018;13(suppl 4):S868–S874. doi: 10.4103/pm.pm_112_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu J, et al. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep. 2016;6:30053. doi: 10.1038/srep30053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ji J, et al. Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell. 2018;17(4):e12774. doi: 10.1111/acel.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oddi S, et al. The anti-inflammatory agent bindarit acts as a modulator of fatty acid-binding protein 4 in human monocytic cells. Sci Rep. 2019;9(1):15155. doi: 10.1038/s41598-019-51691-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fontaine V, et al. A2E-induced inflammation and angiogenesis in RPE cells in vitro are modulated by PPAR-α, -β/δ, -γ, and RXR antagonists and by norbixin. Aging (Albany NY) 2021;13(18):22040–22058. doi: 10.18632/aging.203558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lancaster GI, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018;27(5):1096–1110. doi: 10.1016/j.cmet.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 77.Capozzi ME, et al. The peroxisome proliferator-activated receptor-β/δ antagonist GSK0660 mitigates retinal cell inflammation and leukostasis. Exp Eye Res. 2020;190:107885. doi: 10.1016/j.exer.2019.107885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.