

Abstract
Salts affect the solvation thermodynamics of molecules of all sizes; the Hofmeister series is a prime example in which different ions lead to salting-in or salting-out of aqueous proteins. Early work of Tanford led to the discovery that the solvation of molecular surface motifs is proportional to the solvent accessible surface area (SASA), and later studies have shown that the proportionality constant varies with the salt concentration and type. Using multiscale computer simulations combined with vapor-pressure osmometry on caffeine-salt solutions, we reveal that this SASA description captures a rich set of molecular driving forces in tertiary solutions at changing solute and osmolyte concentrations. Central to the theoretical work is a new potential energy function that depends on the instantaneous surface area, salt type, and concentration. Used in, e.g., Monte Carlo simulations, this allows for a highly efficient exploration of many-body interactions and the resulting thermodynamics at elevated solute and salt concentrations.
Introduction
In biology and chemistry, ion specificity is a key factor for the selective perturbation of molecular matter. The Hofmeister series1−3 has been a governing paradigm to explain the capacity of salts to affect protein solubility. However, Hofmeister’s discovery has a much more universal scope and applies to protein structural stability and numerous equilibria in molecular soft matter such as polymer phase transition and solubility of small molecules.3−5 While the ranking of salts according to their solubilizing (salting-in) or precipitating (salting-out) effect is well established, the molecular mechanisms are still under much investigation.6−11 The predominant hypothesis is related to preferential binding of ions to the solute surface.6 However, free-energy calculations12 suggest that water contributes to ion-specific solvation in ways that cannot be attributed to solute-ion accumulation and/or exclusion.
Furthermore, with the discovery of the reversed Hofmeister series,4,13−15 a mechanistic understanding of ion specificity has become even more complex. Studies of model interfaces show that ion-specific effects are related to surface net charge, polarity, and charge density of the ion.16,17 It is therefore increasingly important to establish predictive models that accurately capture salt-specific effects in complex (bio)molecular solutions.11,18,19
The partitioning concept(20−24) has been particularly important for predicting how salts and osmolytes affect small and large solutes. The main idea is that the solute interacts with the surrounding environment via an inhomogeneous solvent accessible surface area (SASA). Upon adding the cosolute, the solvation free energy is perturbed and assumed linearly dependent on:
-
1.
SASA
-
2.
the concentration of the cosolute
-
3.
a transfer-free energy (TFE), specific for the exposed area and cosolute (see Figure 1A,C)
Figure 1.

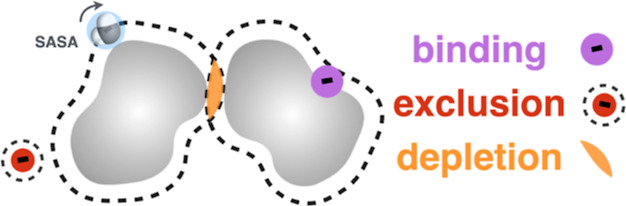
SASA-based description of regimes of a complex solute in neat water (A,B) and in aqueous salt solutions (C,D). For a fully hydrated solute at high dilution, (A) dominantly hydrophobic (gray) solute possesses a fraction of polar (blue) functional groups. SASA is depicted within the dashed line. At elevated solute concentrations of (B), hydrophobic attraction results in a decrease of SASA (pale shaded area). Increased solute association drives an osmolality decrease (right, black curve) compared to ideal behavior (right, green curve). Adding salt to a dilute solute (C), leads to (1) salting-out (TFE > 0) due to a net-ion depletion from the solute vicinity (shown by a small and strongly hydrated cation); and/or (2) salting-in (TFE < 0), due to an excess of ions in the solute vicinity (shown by a large, weakly hydrated anion binding to a hydrophobic region). At higher concentrations of (D), salt further affects solute–solute association. In the salting-out regime (Dout), the solute is more tightly packed due to the excluded volume of depleted salt. Compared to neat water, SASA is further decreased, resulting in decrease of solute osmolality (right, blue curve). In the salting-in regime (Din), solute association allows many-body interactions with the anion, being destabilized, i.e., net solubilized. Consequently, the solute osmolality increases (right gray curve). In this case, decrease in SASA is similar to that in neat water (B), but the dissolved state is favorable due to a larger SASA region available for free-energy beneficial interactions with the anion.
When the cosolute is a salt, it has been shown that cations and anions partition independently to, e.g., air–water or hydrocarbon–water interfaces. Excluded ions exhibit a similar rank order and extent of partitioning at these nonpolar macroscopic and microscopic surfaces.25 The thermodynamic effect of the individual salt ions is additive to a very good approximation.25,26
Chemical denaturation of proteins can, e.g., be assigned to the difference in SASA between the smaller folded state and the larger denatured state. Moreover, a systematic study of proteins of various sizes confirmed that the magnitude of the chemical denaturation (e.g., by urea or guanidinium chloride) correlates with protein size.27 This SASA difference also drives the salt-specific effect on the solute self-association and, consequently, on the precipitation and solubilization processes. The TFE reflects the net cosolute interaction with SASA and have opposite signs for denaturating (salting-in) and stabilizing (salting-out) cosolutes. Figure 1 shows an overview of these mechanisms.
Table 1 shows several models with differing number of TFE values used to describe the cosolute effect on a chemically complex solute molecule. For example, Tanford’s original work28 focused on proteins and has been expanded to provide a quantitative description by assigning individual TFE values to the backbone and 20 amino acid side chains.29−31 Using experimental osmometry and solubility data for a large set of molecules, it has be shown that only a few functional groups (hydrophobic, aromatic, and polar) is sufficient to obtain excellent agreement with stability, equilibria, and kinetics measurements on biomolecules.20−24,32
Table 1. Various Partitioning Models and Their TFE Parameterizationsa.
| reference | number of TFEs | parameterization |
|---|---|---|
| Tanford28 | 21 | amino acid solubilities |
| Bolen29−31 | 21 | amino acid solubilities |
| Record20,21 | 1–4 | VPO; solubilities |
| this work | 1–4 | VPO; MD |
In the present work, we used vapor-pressure osmometry (VPO) and molecular dynamics (MD).
The originally empirical TFE-based models can be rationalized in terms of preferential binding(33−35) which has a solid thermodynamic foundation through Kirkwood-Buff (KB) theory. Recently, the underlying assumptions of additivity and transferability have been probed and verified in silico by molecular dynamics (MD) simulations.36−39 This evidence allowed a rigorous thermodynamic interpretation of salt-specific effects from experimental data as well as direct connection to MD simulation data.9,11,40 Recently, SASA-based arguments were critical in the development of a microscopic origin of coil-to-globule transition in complex mixed solvents, such as under cononsolvency conditions or in the presence of cosolvent surfactants.41,42
Motivated by the above partitioning models,20,28,31 we here devise and incorporate a SASA Hamiltonian into coarse grained (CG) Metropolis-Hastings Monte Carlo (MC) simulations, aiming to accurately capture how solute–solute interactions are affected by salts and osmolytes. The SASA-dependent potential relies on a TFE equivalent to the interaction potential parameter within the solute partitioning model. Ultimately this determines the exclusion or binding of salt species with specific functional groups, thereby affecting thermodynamic properties that are fully accessible in the new simulation scheme.
To test the model, we use caffeine, whose solubility and self-association are strongly influenced by the addition of salts.43−48 For example, weakly hydrated anions (ClO4–, SCN–, and I–) salted caffeine in the aqueous phase, while strongly hydrated anions (CO32–, and SO42–) salted caffeine out. Furthermore, caffeine self-association is promoted by strongly hydrated anions and suppressed by weakly hydrated anions.46,49
Caffeine has high biological significance due to it being one of the key components of coffee, tea, energy drinks, etc., making it probably the most consumed psychoactive drug worldwide. Caffeine is also one of the few naturally occurring chemicals, that has been found to have a plausible positive relationship between intake and reduced risk of Parkinson’s and Alzheimer’s disease.50−52 For these reasons, the chemical and physical properties of caffeine have been studied using a large variety of experimental and computational methods. The existence of oligomers limits the thermodynamic insight, obtainable from experiments performed at caffeine saturation, i.e., from solubility studies.46,53
In this work, we broaden the experimental window and study caffeine–salt interactions from a high dilution to intermediate concentration. To that end, we apply vapor-pressure osmometry (VPO) to aqueous solutions of caffeine at various concentrations in the presence of salting-in (NaSCN) and salting-out (Na2SO4 and NaCl) salts.
Using a new simulation model, we provide insights into the thermodynamics of salting-in and salting-out of caffeine. The model is supported by atomistic MD simulations and VPO data, which allows not only parametrization of the CG model but also gives new insights into salt-specific caffeine-salt and caffeine–caffeine interactions.
Finally, we show that under modest assumptions and known literature data on binary solutions, the results of the CG model can be fine grained to fully recover the thermodynamics of the ternary solution.
Experimental Methodology
Chemicals
Water used as the solvent (labeled 1 from now on) was double-distilled and treated by a Milli-Q ultrapure water purification system from Millipore. The solute (labeled 2) caffeine (Sigma-Aldrich, ≥99.0%, analysis by supplier gives purity 99.6%) was used as received without further purification. The salts (labeled 3) used were sodium chloride (NaCl, Penta p.a.), sodium sulfate (Na2SO4, Penta, >99.0%), and sodium thiocyanate (NaSCN, Fluka, ≥98.0%). NaCl was dried at 398 K for at least 12 h and stored in a desiccator.
Vapor-Pressure Osmometry
All experiments were performed at 310 K and atmospheric pressure using a vapor pressure osmometer, an Osmomat 070 (Gonotec, Germany). VPO measures solution osmolality, which is directly related to water activity via
| 1 |
The osmometer was calibrated before each measurement
with double-distilled water and standard NaCl(aq) solution of known
osmolality. The final osmolality was determined as an average of 10
readings. This protocol was found operational in our earlier study.54 Due to strong influence of the caffeine concentration
on the caffeine–salt interaction, the experiments were designed
so that the caffeine concentration was kept fixed (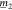 = 10, 25, 50, and
75 mmol/kg), and the
salt concentration was varied. Throughout this work, solely analytical
or total concentrations are used, meaning that we do not distinguish
between different states (monomer, dimer, etc.) of the solutes. Before
each VPO experiment, ternary solutions were heated to 318 K for 1
h under constant stirring to guarantee truly homogeneous solution.
= 10, 25, 50, and
75 mmol/kg), and the
salt concentration was varied. Throughout this work, solely analytical
or total concentrations are used, meaning that we do not distinguish
between different states (monomer, dimer, etc.) of the solutes. Before
each VPO experiment, ternary solutions were heated to 318 K for 1
h under constant stirring to guarantee truly homogeneous solution.
The salt–caffeine interaction was calculated according to
Record et al.20,22 using residual osmolality 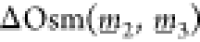 defined as
defined as
| 2 |
where 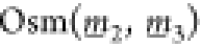 is osmolality
of ternary solution at
is osmolality
of ternary solution at 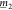 and
and 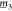 , and
, and 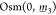 and
and 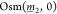 are osmolalities of respective binary solutions.
At low caffeine and salt concentrations, the chemical potential derivative
of caffeine,
are osmolalities of respective binary solutions.
At low caffeine and salt concentrations, the chemical potential derivative
of caffeine, 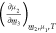 , is proportional
to the residual osmolality20,22 and was described as
, is proportional
to the residual osmolality20,22 and was described as
| 3 |
to be consistent with eqs 1 and 13–15. In eq 3, R is the universal gas
constant, T is the temperature, and C1 and C2 are constants of
the linear fit to the chemical
potential derivative. In case of weakly associating solutes (C2 ≃ 0), eq 3 reduces to the established relation between the residual
osmolality and the salting-out constant 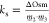 .20,22 In that respect, eq 3 accounts for the effect
of solute concentration on salt–solute interaction, or alternatively, ks becomes a linear function of the solute concentration.
.20,22 In that respect, eq 3 accounts for the effect
of solute concentration on salt–solute interaction, or alternatively, ks becomes a linear function of the solute concentration.
Finally, we remark that alternative approaches to the analysis of osmolality data are possible (raw data provided in Table S2 in the Supporting Information). This includes the generalized isodesmic model, which allows determination of the salt-specific effect on the monomer to multimer states of the solute46 or the application of mathematically exact KB theory of molecular association and aggregation.55
Computational Methodology
MD Simulations
All-atom MD simulations of single caffeine molecule in water and in 1 M salt solutions were performed under ambient conditions (300 K and 0.1 MPa) using Gromacs v4.5.3.56 Temperature and pressure were controlled by a weak velocity rescaling for the canonical sampling coupling scheme57 and the Parinello–Rahmann barostat,58 respectively. Particle mesh Ewald summation was used to account for the long-range electrostatics59 in combination with standard cutoff for Lennard-Jones and short-range electrostatic interactions (1 nm). All bonds containing hydrogen atoms were constrained by the LINCS algorithm,60 while the SETTLE algorithm61 was employed for water molecules.
We used the Amber 11 simulation package62,63 to perform polarizable all-atom MD simulation of caffeine in 1 M Na2SO4 solution64 (with POL3 water65) due to the known issues of the nonpolarizable sulfate force field.64 The simulation parameters were similar to those in Gromacs simulations. The major differences are in the application of Berendsen thermostat and barostat66 and SHAKE algorithm for dealing with constrains.67 Simulation time, time-step, and cut-offs were identical to those in Gromacs.
The caffeine molecule was described by the GROMOS model,68,69 which reproduces well the caffeine solubility in common water models. Various Hofmeister salts were employed, in particular the effect of Na2SO4, NaF, NaCl, NaBr, NaI, and NaSCN aqueous salt solutions on dissolved caffeine molecules were studied, employing the SPC/E water model.70 Salts were described by the force fields which were recently successfully employed in modeling of ion-specific effects in electrophoretic mobility.71
The leapfrog integrator with 2 fs time step was employed, and configurations were gathered every 1 ps for statistical evaluation. Systems were first minimized (to remove potential atomic overlaps) and equilibrated in terms of density and temperature during 1 ns simulation. Subsequently, the ion-distribution around caffeine was equilibrated for 20 ns, followed by 80 ns long production runs when the statistical ensemble was generated. To avoid finite size effects in the solution structure, we investigated sufficiently large systems with caffeine molecules immersed in 2760 water molecules and 55 ion pairs (1 M salt). 3D periodic boundary conditions with an equilibrium box length of 4.5 nm were used.
MD simulation data were analyzed by spatial and proximal distribution functions of ions and water in the proximity of caffeine. Caffeine-hydration and salt–caffeine interaction were quantified by KB integrals Gij calculated from radial distribution functions (RDFs) gij(r), defined in eq 4
| 4 |
| 5 |
where we are reminding that labels 1, 2, and 3 stay for solvent, solute, and salt (cosolvent), respectively. The net salt–caffeine interaction was quantified by a preferential binding coefficient, Γ23 defined in eq 5, where ρ3 is the number density of salt ions. We assigned the thermodynamic value of KB integrals to the plateau value of running KB integral Gij(R) at R = 1.6–2.0 nm (see the shadowed region in Figure S2).
However, in the case of nonspherical molecules, an alternative evaluation of Γ23 via eq 6 is preferred, which requires only the knowledge of running coordination numbers of salt ions (N23) and water (N21) to caffeine.
| 6 |
N01 and N03 are the total
numbers of water
molecules and salt ions in the system, and the ratio 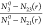 reflects the equilibrium bulk
salt ion
concentration to the local environment of thickness r. This description allows not only to quantify salt interaction with
the whole caffeine molecule but also to evaluate salt-interaction
contributions of individual functional groups (see Supporting Information and Figure S3).
reflects the equilibrium bulk
salt ion
concentration to the local environment of thickness r. This description allows not only to quantify salt interaction with
the whole caffeine molecule but also to evaluate salt-interaction
contributions of individual functional groups (see Supporting Information and Figure S3).
Reminding the
thermodynamic definition in eq 3, an explicit relation between Γ23, kS, and ΔOsm is formulated.20Equation 7 accounts also for the nonideality of the salt solution 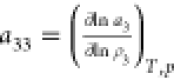 .
.
| 7 |
We note that ρsalt is the concentration of salt, which should not be confused with the concentration of ions ρ3.
SASA Pair Potential
Dividing a molecule
into N fragments or motifs, we assume that the individual
free
energy contributions are additive72 and
proportional to their SASAs, 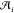
| 8 |
Here γi is a microscopic surface tension that includes the combined effect of solute–solvent and solute–solute interactions in neat water, i.e., free of cosolutes. The term effectively describes short-range attraction stemming from, e.g., hydrophobic interactions. The second term captures the surface tension change as cosolutes are introduced. εi,TFE is a cosolute specific partial TFE and cs is the molar concentration of added cosolute.
In numerical simulations, eq 8 must be evaluated for every microstate, and for large systems containing hundreds of molecules, the many-body SASA calculation becomes prohibitively expensive. We therefore adapt the approximate SASA pair potential: The total surface area of two spheres, i and j of radii R and r ≤ R and with a center-to-center separation, d is
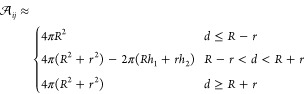 |
9 |
where h1 = (r – R + d)(r + R – d)/2d and h2 = (R – r + d)(R + r – d)/2d are the heights of the two spherical caps comprising
the lens formed
by the overlap. In accordance with eq 8, the pair energy is calculated as 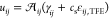 where the radii, R and r, are
the particle radii plus a probe radius, while γij and εij,TFE are
the arithmetic means of the individual values for i and j. Using this approach, γij is treated as a heuristic parameter to effectively capture short-range attraction from, e.g., van
der Waals and hydrophobic interactions.
where the radii, R and r, are
the particle radii plus a probe radius, while γij and εij,TFE are
the arithmetic means of the individual values for i and j. Using this approach, γij is treated as a heuristic parameter to effectively capture short-range attraction from, e.g., van
der Waals and hydrophobic interactions.
Monte Carlo SASA Simulation (MC-SASA)
Metropolis-Hastings MC simulations,73 using Faunus version 2.5,74,75 were conducted on CG caffeine according to the scheme presented in Figure 4, with the potential energy function
| 10 |
Figure 4.
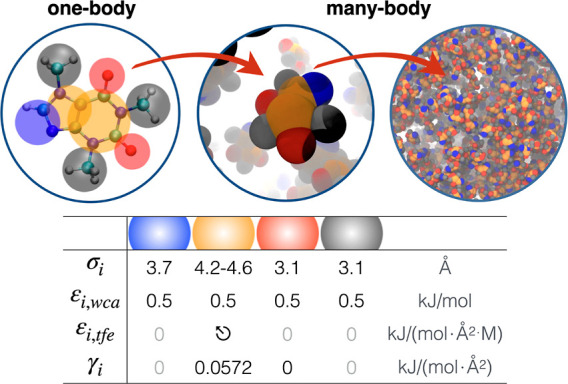
Top: Functional motifs of the atomic structure of caffeine (left) are used to generate a CG representation (middle) for investigating many-body interactions in concentrated caffeine solutions (right). In the CG models, solvent and cosolutes are implicitly accounted for, and beads correspond to the hydrophobic heterocycle (orange); methyl groups (gray); carbonyl oxygen (red); and the —N=CH— moiety (blue). Bottom: Model parameters (cf. eq 10) obtained by fitting the GCMC model to caffeine solubility data in neat water, see Supporting Information. Added osmolytes are subsequently transfer free energy, εi,TFE.
In eq 10, the first term is the Weeks–Chandler–Anderson (WCA) potential,76 a shifted and truncated Lennard-Jones potential, while the second term is the approximate SASA pair potential based on TFEs, εij, and surface tensions, γij. This term is shifted to zero at nonoverlapping separations. While the first two terms are two-body pair potentials, the third and final term is a one-body potential to include the excess chemical potential of transferring a single caffeine molecule from pure water into a salt solution. This term is important only for grand canonical simulations.
The model was calibrated against experimental caffeine solubility data in neat water (cs = 0) using 200 caffeine molecules in a cubic box, with a volume adjusted for the concentration range 0.0096–0.1116 M caffeine. The procedure is detailed in the Supporting Information, and the final parameters are shown in Figure 4.
The MC move set consisted of molecular
rotation and translation
with a maximum translational displacement parameter of 10 Å and
a maximum rotational displacement of 1 rad. The pressure was calculated
though a virtual volume move77 with a volume
perturbation of 5 Å3. The osmotic coefficient, φ,
was calculated from 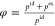 , where pid is
the ideal pressure obtained from the predetermined number density
(pid = NRT/V) and pex is the excess pressure obtained
from virtual volume perturbations.
, where pid is
the ideal pressure obtained from the predetermined number density
(pid = NRT/V) and pex is the excess pressure obtained
from virtual volume perturbations.
The effects of salt in the modulation of caffeine–caffeine interactions were investigated partly in the canonical and grand canonical ensemble. For simulations in the canonical ensemble, caffeine was simulated at different concentrations and values of the product csεTFE. The initial configuration was generated by placing caffeine molecules in a cubic box with a length of 250 Å until the desired concentration was reached. The system energy was equilibrated for 5 × 104 MC iterations, followed by a sampling of the statistical ensemble for a total of 106 MC iterations. The collective variables from the statistical ensemble included the caffeine–caffeine RDF, which was sampled every 10 steps with a resolution of 0.25 Å every 10 MC iterations. The excess pressure is sampled via a virtual volume perturbation77 using a isotropic scaling of the box and center of mass of caffeine, i.e., keeping the conformation of caffeine fixed, with the volume displacement being 50 Å3. Finally, the excess chemical potential is sampled using the Widom method78 with 25 inserts every 10 MC iterations. The MC move set consisted of molecular rotation and translation with a maximum translational displacement parameter of 2.5 Å and maximum rotational displacement parameter of 0.5 rad and a cluster move with molecular rotation and translation with a maximum translational displacement parameter of 7.5 Å and maximum rotational displacement parameter of 1.0 rad and threshold of 6.5 Å. For simulations in the grand canonical ensemble, caffeine was simulated at different activities and values of the product csεTFE. The initial configuration was generated by placing 2.5 × 103 caffeine molecules in a cubic box with a length of 150 Å and equilibrating the system energy and particle density for a maximum of 107 MC iterations. The equilibration was followed by a production run consisting of 5 × 107 MC iterations, in which the statistical ensemble was generated with the sampling of the density every 1000 steps. The MC move set consisted of molecular rotation and translation with a maximum translational displacement parameter of 10 Å and maximum rotational displacement parameter of 1 rad in addition to the insertion and removal of caffeine molecules utilizing the generalized reactive MC move algorithm.79 The difference in excess chemical potential in kT units, Δμex, going from a water to an electrolyte solution, was calculated from Δμex = ln(ρwat/ρsalt) where ρwat and ρsalt are the density of caffeine in water and salt solution, respectively, at constant caffeine activity.
Kirkwood–Buff Analysis of Experimental and/or Simulation Data
The KB analysis was carried out by solving the set of equations eqs 11, 12 for i = 1 and k ≠ 1. These equations relate KB integrals to the chemical potential derivatives and volumetric properties of the solution80
| 11 |
| 12 |
where 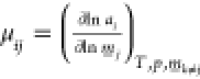 is the activity derivative, δik is the Kronecker δ, n is the number of components, κT is the isothermal compressibility,
is the activity derivative, δik is the Kronecker δ, n is the number of components, κT is the isothermal compressibility, 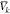 are the partial
molar volumes, and Nij = Gijρj are
the excess coordination numbers related to KB integrals via particle
number density ρj. To obtain necessary
activity derivatives, data from simulations (or experiment) were used
to calculate activity coefficient of caffeine γ2 via
fitting ln γ2 to
are the partial
molar volumes, and Nij = Gijρj are
the excess coordination numbers related to KB integrals via particle
number density ρj. To obtain necessary
activity derivatives, data from simulations (or experiment) were used
to calculate activity coefficient of caffeine γ2 via
fitting ln γ2 to
| 13 |
where A2 and B2 are caffeine binary parameters, C1 and C2 are salt–caffeine
interaction parameters. A2 and B2 were determined from data at 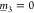 (i.e., independent of the TFE value), while C1 and C2 were specific
for each TFE. Using the Gibbs–Duhem equation, approximate eq 14 for ln a1 consistently determines the form of ln γ2 (eq 13) and of ln
γ±3 (eq 15). With that
the activity derivatives of the remaining components were evaluated.
(i.e., independent of the TFE value), while C1 and C2 were specific
for each TFE. Using the Gibbs–Duhem equation, approximate eq 14 for ln a1 consistently determines the form of ln γ2 (eq 13) and of ln
γ±3 (eq 15). With that
the activity derivatives of the remaining components were evaluated.
| 14 |
| 15 |
where A3 and B3 are salt binary parameters and ν3 is the number of ions in the salt. Partial molar volumes and isothermal compressibility were considered independent of salt concentrations and set to values at infinite dilution.
Results and Discussion
VPO Investigation of Salting Out Constant of Caffeine
Salt-specific effects on sparingly to poorly soluble, small solutes are traditionally investigated by solubility measurements. Using the Setschenow equation,43 the impact of salt on solubility, and thus on the activity coefficient of the dissolved solute in the saturated solution, is directly probed. As the concentration of the dissolved solute at saturation is typically low, it is conveniently measured by UV–vis spectrophotometry. Here, we use VPO on homogeneous solutions to probe the salt–solute interactions as a function of the solute concentration. Optimal VPO experimental conditions are in the range from 0.1 to 1 mol/kg solute and salt concentration, which provide a good signal-to-noise ratio.
Following the protocol
of Record et al.,20,22 we have measured osmolality of
binary solutions for caffeine–water and salt–water,
which were subtracted from the osmolality of the ternary solution
caffeine–water–salt, according to eq 2. Solutions of various caffeine and salt concentrations
were measured (see Tables S2 and S3), and
the calculated ΔOsm is plotted in Figure 2A. Salt specificity leads to different slopes
(see dashed lines), which fit well with the scattered experimental
data obtained at fixed caffeine concentration (squares, 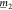 = 50 mmol/kg) via eq 3, and relate to the salting-out
constants,
summarized in Table 2. The VPO data follow the traditional Hofmeister series9 with Na2SO4 being the most
potent salting-out salt, followed by NaCl. Salting-in behavior is
observed for NaSCN.
= 50 mmol/kg) via eq 3, and relate to the salting-out
constants,
summarized in Table 2. The VPO data follow the traditional Hofmeister series9 with Na2SO4 being the most
potent salting-out salt, followed by NaCl. Salting-in behavior is
observed for NaSCN.
Figure 2.
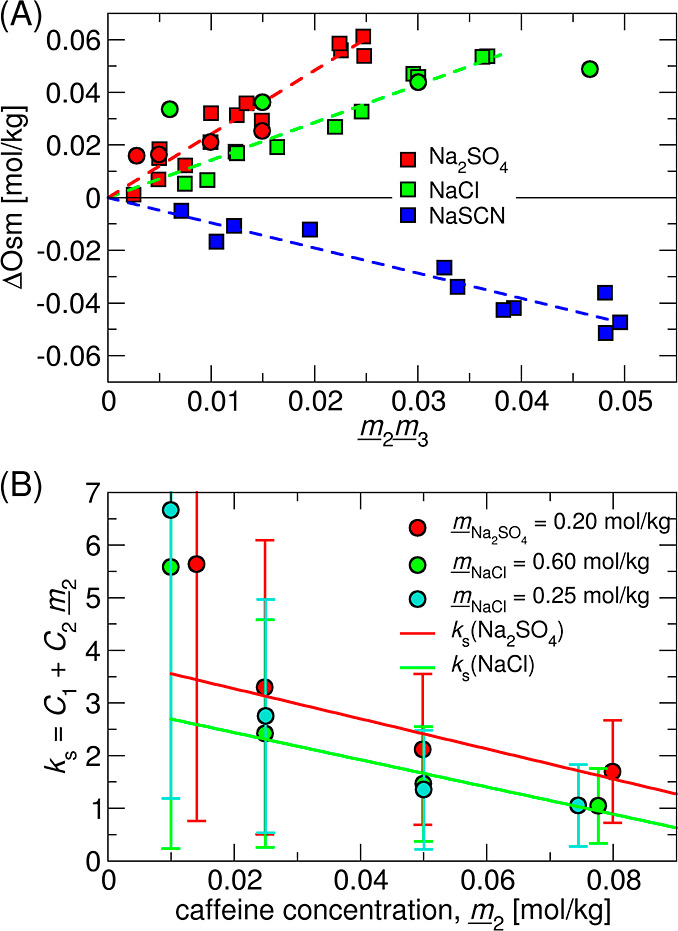
(A) Excess osmolality, ΔOsm, from
VPO measurements of caffeine-salt
solutions: Na2SO4 (red), NaCl (green), and NaSCN
(blue). Data gathered at constant caffeine concentration 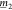 = 50 mmol/kg and
varying salt concentration
are presented in squares. Measurements at constant salt concentration,
NaCl (0.6 mol/kg) and Na2SO4 (0.2 mol/kg) and
varying caffeine concentration are shown with circles. (B) Salting-out
constant, ks, determined
at the fixed salt concentration and varying caffeine concentration
= 50 mmol/kg and
varying salt concentration
are presented in squares. Measurements at constant salt concentration,
NaCl (0.6 mol/kg) and Na2SO4 (0.2 mol/kg) and
varying caffeine concentration are shown with circles. (B) Salting-out
constant, ks, determined
at the fixed salt concentration and varying caffeine concentration 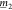 . The linear fit
of the ks dependence on the caffeine concentration
cf. Equation 3 is shown
in red
(Na2SO4) and green (NaCl) lines. Error bars
are determined via error propagation at the 95% confidence level (i.e.,
2 σ). The lowest caffeine concentration was omitted from the
fitting. The potential role of the salt concentration of ks is analyzed in Figure S12 in Supporting Information.
. The linear fit
of the ks dependence on the caffeine concentration
cf. Equation 3 is shown
in red
(Na2SO4) and green (NaCl) lines. Error bars
are determined via error propagation at the 95% confidence level (i.e.,
2 σ). The lowest caffeine concentration was omitted from the
fitting. The potential role of the salt concentration of ks is analyzed in Figure S12 in Supporting Information.
Table 2. Salting Out Constants and Their Caffeine Concentration Dependence (if Available) as Determined from VPO Data in Figure 2A,B, Respectivelya.
| Na2SO4 | NaCl | NaSCN | ||
|---|---|---|---|---|
| C1 | 3.84 | 2.95 | n.a | |
| C2 | –28.59 | –25.83 | n.a | |
| 2.41 | 1.43 | –0.96 |
Ci parameters
are in units of reciprocal molality concentration
of appropriate power, and 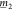 is in mol/kg.
is in mol/kg.
Taking a closer inspection, outlying
points (circles) appear for
Na2SO4 and NaCl (not measured for NaSCN). These
represent measurements at significantly different caffeine concentrations
(10–80 mmol/kg). This can be accounted for by fitting 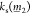 viaeq 3. The results are plotted in Figure 2B where we note that uncertainties
increase with the decreasing caffeine concentration due to increasing
noise and error propagation. For NaSCN, large uncertainties at low
caffeine concentrations prohibited the fitting of
viaeq 3. The results are plotted in Figure 2B where we note that uncertainties
increase with the decreasing caffeine concentration due to increasing
noise and error propagation. For NaSCN, large uncertainties at low
caffeine concentrations prohibited the fitting of 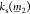 viaeq 3.
viaeq 3.
Salts have the strongest effect at infinite caffeine dilution and weaken approximately linearly with the increasing caffeine concentration. The parameters C1 and C2 of the presumed linear effect in the caffeine concentration are summarized in Table 2. Comparison of salting-out constants at two NaCl concentrations indicates that this effect is due to the variation in the caffeine concentration. The effect of the salt concentration on ks is analyzed in detail in Figure S12 in the Supporting Information and is found to be significantly smaller than that of the caffeine concentration.
Observed weakening of the salting-out effect (C1 > 0) with an increasing caffeine concentration (C2 < 0) may be also interpreted as a strengthening of caffeine–caffeine self-association in the presence of salting-out salts and is consistent with earlier findings based on caffeine solubility and caffeine partitioning study.43
In other words, our data show that the strength of the salting-out effect of the salt in general depends on the solute state (presumably due to differences in SASA, see the next sections). This includes, e.g., native vs denatured state of proteins; coil vs globular state of polymers; or solution structure of solute molecules in the aqueous environment. For caffeine, the latter is manifested as a concentration-dependent self-association.
The correlation between solubility and self-association was reported for hydrophobic molecules (e.g., methane and neopentane) in earlier computational study.81 Moreover, within a statistical-thermodynamic approach, a rigorous relation between the salting-out constant and salt-effect on self-association (virial coefficient) was derived, at least for small hydrophobic solutes.82 In the current work, we use novel MC-SASA simulations to shed light on the experimental data and explore the relation between the salting-out effect and self-association for complex caffeine molecule.
Salting-out constants determined from the slopes in Figure 2A are significantly higher, compared to previously published data.43,46 The difference is, however, only apparent and can be rationalized with the following arguments, exemplified for NaCl: the salting-out constant decreases with the increasing caffeine concentration, thus reaching its lower limit at caffeine saturation. Caffeine solubility steeply increases with temperature, i.e., it is approximately two times higher at 37 °C than that at 25 °C.83 The VPO experiments are thus performed at a rather far from saturation. Assuming that the Setschenow constant is invariant in this temperature window, the estimated caffeine solubility is approximately 0.16 mol/kg at 37 °C in 0.6 mol/kg NaCl.43,83 Extrapolating the results in Figure 2B to saturation, the salting-out constant in NaCl is approximately zero, which recovers the literature value.43,46
All-Atom MD Reveals Anion-Specific Binding Sites of Caffeine
To gain an understanding of the origin of the affinity of various ions to the caffeine molecule, we have conducted all-atom MD simulations of caffeine in aqueous solutions of sodium salts. The anions ranged from salting-in thiocyanate (NaSCN) over weakly salting-out chloride (NaCl) to strongly salting-out sulfate (Na2SO4).
First, detailed insights into the very vicinity of the caffeine molecule are obtained by employing the spatial distribution function (SDF), as shown in Figure 3A. The spatial clouds represent the sites of significantly enhanced probability of water oxygen, anions, and sodium in the proximity of caffeine molecule.
Figure 3.
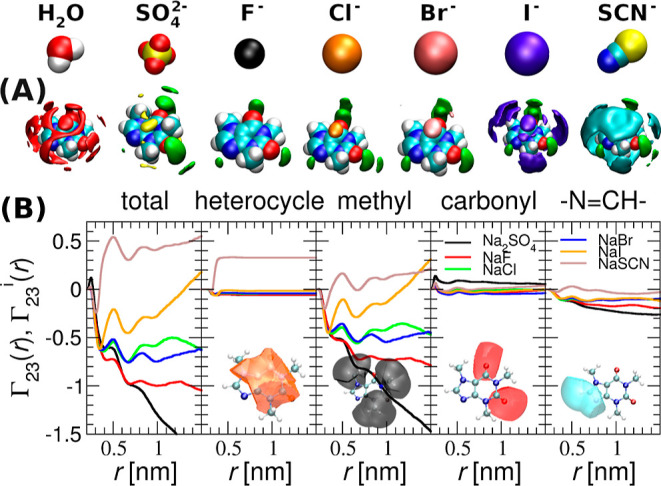
(A) Spatial distribution function (SDF) of water (leftmost panel) and ions around caffeine in salt solutions. Caffeine is depicted in a space filling representation; the distribution of water in red, sodium in green, and anions in indicated colors. The isosurfaces represent twice the bulk density for ions and 1.5 times the bulk density for water. (B) Partial preferential binding coefficients, Γi23, used to quantify salt affinities to functional groups. The total preferential binding coefficients, Γ23 = ∑4i=1 Γi23, are shown in the first column; the following columns show contributions from the hydrophobic heterocycle; methyl groups; carbonyl oxygen; and the —N=CH— moiety. r represents the proximal (closest) distance of salt to heavy atoms of a given functional group of caffeine molecule.
In agreement with the Hofmeister series for anions, weakly hydrated anions (SCN–, I–) are enriched at hydrophobic sites. The latter constitute the three methyl groups and the two heterocyclic aromatic rings. In contrast, the more strongly hydrated SO42– and F– occur in the caffeine vicinity less than water, i.e., they are depleted. Na+ is present near the partially negatively charged amide (C=O) and imine (—N=CH—) moieties. The magnitude of the sodium affinity in the individual salts reflects the excess of its counteranion due to electrostatic correlations. The remainder of the caffeine surface lacks specific interaction sites for salt and water. In summary, anion enrichment or depletion is driven by interactions with the hydrophobic regions of caffeine.
In agreement with previous studies,68,69 the SDF also shows that caffeine is well hydrated by water alone, which is somewhat counterintuitive considering the weak hydrogen-bonding capacity. Tighter hydration is primarily due to the heterocyclic face (owing to effective π–OH interactions) and the amide and imine groups, while methyl groups are poorly hydrated.
The thermodynamic measure of salt excess or depletion near caffeine is captured by the salting-out constant kS. Its origin from the microscopic solution structure stems from KB theory.84 To that end, we have calculated RDFs, evaluated KB integrals, and calculated the preferential binding coefficient Γ23 and salting-out constants. These net-salt effects are summarized in Figures S1 and S2, Table S1, and discussed in detail in the Section S1.
Following our observation in SDFs, we decomposed the caffeine surface into distinct functional groups and evaluated partial proximal distribution functions in Figure S3. From this, we calculate the net preferential binding coefficient, Γ23 (first column of Figure 3B) and introduce partial preferential binding coefficients Γi23 of salts to individual functional groups of caffeine (second to fifth column). The magnitudes of Γi23 in Figure 3B show that the preferential binding to methyl groups (third column) controls the salt specificity. Only SCN– displays some affinity to the heterocyclic aromatic rings (second column). Although some salt specificity is also present in the interactions with amide and imine moieties, quantitatively, these are of minor importance. Strictly speaking Figure 3B presents running preferential binding coefficients Γ23(R), from which plateau region (≈1.5 nm from center of mass, or ≈1.0 nm from the surface of caffeine molecule) the thermodynamic value Γ23 is determined (see also Figure S2).
To summarize, all-atom MD simulations allow us to calculate salting-in/salting-out capability of salts in the limit of infinite dilution of caffeine, where the contributions of caffeine–caffeine interactions are absent. A logical continuation toward the modeling of salt-specific effects at finite caffeine concentrations is the application of an appropriate CG model, which is present in the following sections.
CG Model of Caffeine Using Thermodynamic Data
To investigate the impact of the self-association equilibrium of caffeine on salting-in/salting-out capabilities of salts, a CG model has been constructed. The coarse graining has been conducted using a combined top-down and bottom-up approach. The structural results of anion–caffeine association, obtained from the all-atomic MD simulations, was used to create unified atoms, in which individual methyl groups and heterocycles, constituting the nonpolar groups of caffeine, the imine group, and carbonyl oxygen, constituting the polar groups of caffeine, can be represented by single spheres. The resulting coarse-graining scheme is visualized in Figure 4.
To reproduce the caffeine–caffeine interactions, we calibrated our CG model using experimental data using osmotic coefficients, which represented the average attraction/repulsion between caffeine molecules in salt-free water.85,86 The parametrization of caffeine was conducted by adjusting the atomic surface tension, γi, at a constant value of ϵi = 0.5 kJ/mol for the WCA potential (see eq 10), to reproduce the experimental osmotic coefficients. From atomistic simulations,68 nuclear magnetic resonance spectroscopy,87 and small angle neutron scattering88 experiments, it has been established that the primary mode of association in caffeine oligomerization at room temperature is face-to-face stacking. Consequently, we choose to simplify our model further by limiting the number of attractive sites to the two CG spheres constituting the heterocyclic aromatic groups. The osmotic coefficients obtained from simulations using the best parameter for the atomic surface tension (γ = 0.0572 kJ/mol/Å2) to reproduce the experimental data has been visualized in Figure S4.
At first glance, the osmotic coefficients obtained from the simulation yield good agreement with the experimental data over the available caffeine concentration range. Deviations, however, between experimental data and simulated data do occur at high caffeine concentrations, suggesting that the CG Hamiltonian causes too strong attraction between caffeine molecules. This is likely due to the pair potential being much softer, being nearly linear compared to potentials usually employed for short-ranged attraction. This has the implication that the attraction persists over a longer range compared to, for example, a sixth-inverse-power of Lennard-Jones potentials (see Figure S7). This range is related (i) to the probe radius when calculating the surface area and (2) the magnitude of the WCA potential determined by ϵ. Varying the number of TFE sites to also include the methyl groups by equal and half strength compared to the heterocycles proved to yield highly equivalent results (Figure S8). While the model possessing more TFE sites reproduces the osmotic coefficient better at the increasing caffeine concentration, it will be shown next that the model utilizing only two TFE sites is sufficient to capture the important structural and thermodynamic properties.
In agreement with experimental observations, the most probable mode of caffeine–caffeine association is face–face stacking, with branching being much less probable.88 The stacking to branching ratio can most likely be controlled by the introduction of more interaction sites (CG atoms with nonzero tension), such as the methyl groups. However, the employed model captures the thermodynamics of the caffeine–caffeine interactions. Looking at the RDF between the caffeine molecules (Figure S5), we find the KB integral, G22, being equal to approximately 20 × 103 Å3 in agreement with the experimental data from the literature.85 Consequently, our model possesses qualitative correctness with the ability to capture essential features, laying the foundation for employing the model in investigating how salt alters caffeine’s liquid structure.
Salting-In and Salting-Out from Implicit Salt Hamiltonian
Comparison with Solubility Experiments
With the CG model parametrized to reproduce experimental osmotic coefficients in the 10–100 mM concentration range, we continued to parametrize our model to include salt effects. To do so, we searched for TFE values at constant activity of caffeine that can reproduce experimental differences in the excess chemical potential of caffeine in salt solution at a specific salt concentration and pure water. Within the theory of solute solubility, a convenient experimental measure, pseudo chemical potential (pcp, μ*) was introduced by Ben-Naim.89
The pcp values of caffeine were determined by Shimizu for different cosolutes and cosolute concentrations from caffeine solubility data.46 Since the experiment probes caffeine solutions near saturation, ideally, we are interested in the chemical potential of the liquid phase when the liquid and aggregated phases are in equilibrium at constant temperature and pressure. However, methodologies involving aggregated or solid states of matter in equilibrium with the liquid state are usually not preferred due to either high computational costs (free energy calculations and extended ensembles) or the transition being a rare event, yielding poor statistics (direct sampling). Consequently, TFE parametrization was carried out at a caffeine molar activity of 0.026 in the grand canonical ensemble which is equivalent to a caffeine concentration of 69 mM in pure water (εTFE = 0). This enables us to sufficiently sample converged results without the aggregated phase being sampled. The excess chemical potential, μex, obtained from simulations yielding the best agreement with the experimental data are shown in Figure 5.
Figure 5.

Excess chemical potential of caffeine in salt solutions compared to neat water at a constant molar activity of caffeine (26 mM). Experimental46 and MC-SASA can be mapped onto each other through salt-specific TFE-values (εTFE in units of kJ mol–1 Å–2 M–1): Na2SO4: 0.0178 (blue), NaCl: 0.003 (orange), NaBr: −0.001 (green), NaSCN: −0.008 (red), and NaClO4: −0.0125 (purple). Nonspecific simulation parameters are taken from pure caffeine/water solutions; see Figure S4.
From the simulation, we obtain the excess chemical potential, while the experimental data involve the pseudo chemical potentials. The two are related by μex = μ* – RT ln(q) where q is the internal partition function containing all rotational degrees of freedom. However, for molecules with small conformational ensembles and assuming that the addition of cosolute does not influence that ensemble, the change in pcp can be assumed equal to that in excess chemical potential.84
Different unique TFE values can effectively reproduce the chemical potential of caffeine in salt solutions at different concentrations. Within our TFE model, salting-in and salting-out effects of salts are solely controlled by the sign of the TFE value. A consequence of our model is the linear dependency of excess chemical potential on the additive concentration; however, the analyzed experimental data by Shimizu is fitted to a second-order polynomial, with the fitting parameter in the linear term being identified as the Setschenow constant, while the quadratic term serves to capture nonlinear effects over a wide cosolute concentration range. Conducting linear regression analysis and hypothesis testing on the available experimental data, however, reveals it to be insufficient including the quadratic term (linear r2 > 0.96 for all salts and sucrose), and a linear fit is substantial to describe the experimental data as also suggested by the squared regression coefficients, which adds to the validity of our model.
The excess chemical potential obtained from the grand canonical simulations possesses two contributions: (a) a one-body term representing the TFE of a caffeine monomer from pure solvent to a solvent with an additive solution and (b) a caffeine–caffeine interaction term. These two contributions are shown in Figure 6.
Figure 6.
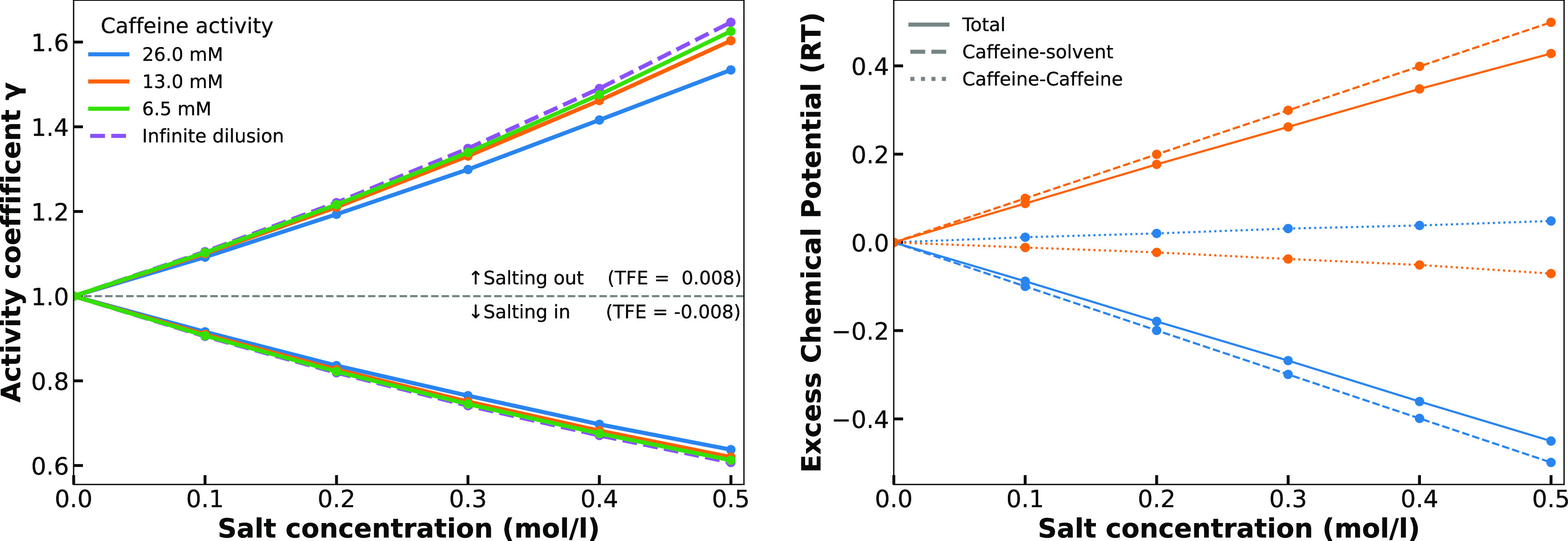
Left: Activity coefficient for caffeine (set to unity in salt-free solution) as a function of salt concentration (εTFE = 0.008 kJ mol–1 Å–2 M–1 for salting-out and εTFE = −0.008 kJ mol–1 Å–2 M–1 for salting-in) for three caffeine activities sampled by MC simulation and infinite dilution calculated analytically using the Hamiltonian in eq 10. Right: Decomposition of the caffeine-solvent and caffeine–caffeine contribution to the difference in excess chemical potential between caffeine in salt solution and caffeine in pure water. The orange curve corresponds to salting out conditions (εTFE = 0.008), while the blue curve is equivalent to salting in (εTFE = −0.008). The molar activity of caffeine is 0.026.
Within our model, the TFE of a caffeine monomer from water to water with an additive opposes the free energy contribution from caffeine–caffeine association. This observation is independent of the sign of the TFE value, while it does, however, determine whether the individual contributions are positive or negative. Conceptually, this is in agreement with the mechanistic understanding of the Hofmeister effect: ions causing salting-in are solute binding species that increase repulsion between solute molecules due to electrostatic repulsion; ions causing salting-out are solute excluding species that increase the attraction between solute molecules due to an enhanced hydrophobic effect by strongly hydrated species.
Within our model, electrostatics are implicitly incorporated into the TFE, assuming that long-range repulsion can be negligible due to short screening lengths at high salt concentrations. The model could however be extended to explicitly include electrostatics by a reactive MC move, in which CG caffeine atoms can change charge upon binding of an (implicit) anion.90
Caffeine Self-Association Modulates the Effect of Salt
The CG model is now tuned to reproduce thermodynamic observables related to the average attraction and repulsion between caffeine molecules in water and the electrolyte solution. We now continue to investigate the effect of elevating the caffeine concentration.
Caffeine possesses a self-association equilibrium, characterized by a specific binding mode between the heterocycle faces of the caffeine molecules. Consequently, by changing the caffeine concentration (or solvent quality, see Figure S6), the relative population of caffeine monomers and caffeine oligomer states is perturbed. Figure 6 shows simulated activity coefficients of caffeine in water with the increasing cosolute (salt) concentration. The effect of cosolute diminishes with increasing activity of caffeine, particularly for positive TFE values which is trivially due to Boltzmann weighting of the TFE values in the Hamiltonian. Experimentally, this has great importance in terms of design and choice of methodology. For determining Setschenow coefficients, common methods include organic-aqueous phase solute partitioning, liquid–solid phase partitioning, and VPO. Each method, however, operates in different concentration regimes which is problematic since our findings suggest that the Setschenow coefficient in general depends on the solute concentration. Consequently, different techniques may yield different Setschenow coefficients, solely because of differences in the solute concentration.
Recovering the Full Solution Thermodynamics
We now
completely recover the thermodynamics of aqueous caffeine-salt solution
using KB theory, applying only modest assumptions on implicitly treated
components. To that end, we have used MC-SASA for a series of caffeine
and salt concentrations, 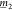 = 0–0.05
mol/kg ×
= 0–0.05
mol/kg × 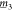 = 0–0.5 mol/kg,
and two types of
salt, i.e., salting out (εTFE = 0.01 kJ mol–1 Å–2 M–1) and salting in
(εTFE = −0.01 kJ mol–1 Å–2 M–1). The direct output of MC-SASA
is the excess chemical potential of caffeine (μex2 = RT ln γ2), as summarized in Table S4. Data were globally fitted to eq 13, yielding A2, B2, C1,
and C2. Known activity data for binary
water-salt solution provided parameters A3 and B3, and standard state partial molar
volumes
= 0–0.5 mol/kg,
and two types of
salt, i.e., salting out (εTFE = 0.01 kJ mol–1 Å–2 M–1) and salting in
(εTFE = −0.01 kJ mol–1 Å–2 M–1). The direct output of MC-SASA
is the excess chemical potential of caffeine (μex2 = RT ln γ2), as summarized in Table S4. Data were globally fitted to eq 13, yielding A2, B2, C1,
and C2. Known activity data for binary
water-salt solution provided parameters A3 and B3, and standard state partial molar
volumes 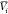 at 37 °C were
employed for dissolved
species and water.91,92 The approximation of constant
at 37 °C were
employed for dissolved
species and water.91,92 The approximation of constant 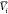 is justified as
the KB integrals are only
weakly dependent on
is justified as
the KB integrals are only
weakly dependent on 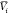 but are dominated
by changes in activity
data.80 A complete set of coefficients
for the thermodynamic description of the salt-caffeine solution in
two representative salts is summarized in Table 3.
but are dominated
by changes in activity
data.80 A complete set of coefficients
for the thermodynamic description of the salt-caffeine solution in
two representative salts is summarized in Table 3.
Table 3. Parameters of the Thermodynamic Model of Caffeine–Salt Solutionsa.
| εTFE = −0.01 | εTFE = 0.01 | description | |
|---|---|---|---|
| A2 | –7.2399 | –7.2399 | caffeine–caffeine |
| B2 | 24.9741 | 24.9741 | caffeine–caffeine |
| A3 | –0.6216 | –0.3506 | salt–salt |
| B3 | 0.4754 | 0.2087 | salt–salt |
| C1 | –1.1145 | 0.8061 | caffeine–salt |
| C2 | 6.1971 | –8.0176 | caffeine–salt–caffeine |
| V̅1 | 18.14 | 18.14 | water partial molar volume |
| V̅2 | 145.9 | 145.9 | caffeine partial molar volume |
| V̅3 | 40.96 | 16.62 | salt partial molar volume |
Ai, Bi, and Ci are in units of reciprocal
molality of appropriate power. Partial molar volumes, 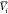 are in in cm3·mol–1. εTFE = −0.01
kJ mol–1 Å–2 M–1 and εTFE = 0.01 kJ mol–1 Å–2 M–1 represent salting-in and salting-out
salts,
respectively. A2, B2, C1, and C2 are obtained by fitting the MC-SASA data (Table S4) to eq 13.
are in in cm3·mol–1. εTFE = −0.01
kJ mol–1 Å–2 M–1 and εTFE = 0.01 kJ mol–1 Å–2 M–1 represent salting-in and salting-out
salts,
respectively. A2, B2, C1, and C2 are obtained by fitting the MC-SASA data (Table S4) to eq 13.
Employing the KB-inversion procedure using eqs 11 and 12, Figures S9 and S10 show all six KB integrals for the two types of salts over the range of caffeine and salt concentrations. Note that KB integrals carry information about the net-solution structure. The most insightful KB integrals are those involving caffeine, i.e., G12, G22, and G23. These are presented for the two types of salts in Figure S11 and discussed in detail in the Supporting Information.
A practically important outcome
of the MC-SASA simulations is the
construction of an in silico VPO experiment, which
allows evaluation the 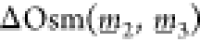 and thus the caffeine–salt
interaction.
Analogous to Figure 2, the simulated Figure 7A shows raw ΔOsm data from individual MC-SASA calculations
(points) along with the slopes, obtained from eq 3 for a series of caffeine concentrations.
Although the two salts differ only in the TFE-sign, the two responses
in ΔOsm are not mirror images.
and thus the caffeine–salt
interaction.
Analogous to Figure 2, the simulated Figure 7A shows raw ΔOsm data from individual MC-SASA calculations
(points) along with the slopes, obtained from eq 3 for a series of caffeine concentrations.
Although the two salts differ only in the TFE-sign, the two responses
in ΔOsm are not mirror images.
Figure 7.
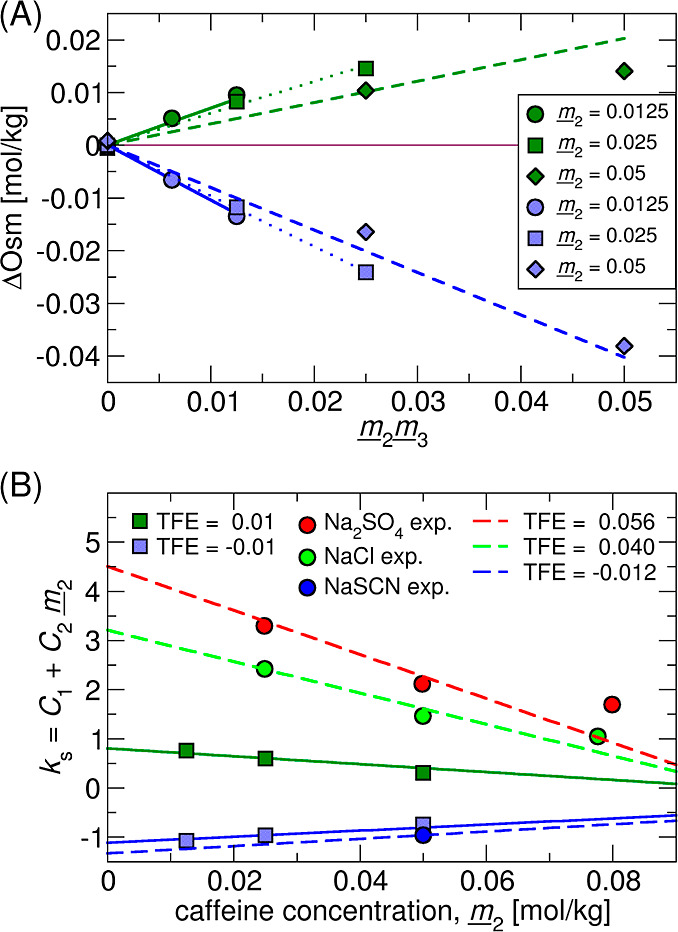
(A) ΔOsm from MC-SASA of caffeine solute using salting-out (εTFE = 0.01 kJ mol–1 Å–2 M–1, in green) and salting-in (εTFE = −0.01 kJ mol–1 Å–2 M–1, in blue). Points present raw MC-SASA data (i.e., μ2ex, Table S4), while lines represent a global fit of MC-SASA data (i.e., with parameters in Table 3). (B) Salting-out constant (cf. eq 3) as a function of the caffeine concentration for εTFE = −0.01 kJ mol–1 Å–2 M–1 (light blue) and εTFE = 0.01 kJ mol–1 Å–2 M–1 (dark green) raw MC-SASA data (squares) along with analytical curves from Table 3 (full lines). Experimental data for Na2SO4 (red circles), NaCl (green), and NaSCN (blue) from Figure 2B fitted by the universal MC-SASA curves (dashed lines) with a single TFE parameter determined (see the legend).
Since only the product εTFEcs enters the MC-SASA calculation, the simulation results are universal and can be used to recalculate ΔOsm curves for the arbitrary TFE value. Figure 7B clearly shows the caffeine concentration dependence of the salting-out constant, kS, as evaluated from raw ΔOsm data together with the curves evaluated via eq 3. Finally, the experimental data for Na2SO4 (red cicrcle), NaCl (green circles), and NaSCN (blue circles) from Figure 2B were fitted by the universal MC-SASA curves (dashed lines), and the caffeine–salt interaction parameter (TFE value) was determined. The relation between C1 and C2 was recently predicted by the statistical-thermodynamic approach for small nonpolar solutes.82 In this work, a simple MC-SASA scheme (see results in Figure 7) captures higher order salt-solute effects, which originate in salt-specific effective interaction with different surface types (functional groups) of complex solutes.
Examplified by caffeine, the present results show that the MC-SASA approach quantitatively captures the changes of the solute chemical potential in the presence of additives. Moreover, the ability of the model to faithfully account for many-body effects, i.e., solute association, points toward its broad applicability from modeling of protein conformational changes up to protein–protein interactions in crowded environments.
Concluding Remarks
Using VPO and molecular scale computer simulations, we have investigated how specific ion effects operate over different solute and salt concentration regimes. Experimental observations were rationalized by introducing an implicit-water, implicit-salt MC model, which uses structural details of solute hydration and solute–salt interactions through the solvent accessible surface area (MC-SASA). Applied to the chemically complex caffeine molecule and salts from the extreme ends of the Hofmeister series, the new method gives quantitative insights into salting-in and salting-out of caffeine over a range of solute and cosolute concentration regimes.
To fine-grain our model, we present a KB theory-based protocol in which complete thermodynamics of the ternary solution is recovered. All KB integrals at any composition are determined from MC-SASA simulation data, employing only modest assumptions on implicitly treated components.
The new MC-SASA algorithm captures the modulation of the magnitude of the salting-in or salting-out effect with the increasing solute concentration in accordance with the new VPO data. The development of our MC-SASA model was motivated by the existing partitioning concept20−24,28 and can be regarded as a generalization where many-body effects are taken into account. While here applied to caffeine salt solutions, the methodology can be expanded to large complex systems in which the implicit treatment of solvent and salt becomes highly efficient. This includes, but is not limited to, polymers, proteins, or protein–ligand complexes at dilute conditions, as well as to concentrated solutions of biomolecules in the presence of crowders.
Acknowledgments
S.H.-H. and M.L. thank the Swedish Research Council (grant 2022-04251); the Swedish Foundation for Strategic Research; the Crafoord foundation; and the European Research Council through the PIPPI consortium. S.H.-H. is grateful to the Postdoctoral Fellowship for Research in Japan (no. 22F21756) from the Japan Society for the Promotion of Science. Computations were enabled by resources provided by LUNARC. J.H. and J.P. thank the Czech Science Foundation (grant 20-24155S) and the financial support from a Specific university research (A1-FCHI-2022-002). This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90140).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.3c01085.
Additional explanations, tables, and plots (PDF)
Author Contributions
# S.H.-H., J.P., J.H., and M.L. contributed equally to this work.
The authors declare no competing financial interest.
Notes
An Electronic notebook for running simulations, analyzing, and plotting the presented data is available at Zenodo, https://doi.org/10.5281/zenodo.10122308.
Supplementary Material
References
- Hofmeister F. Zur Lehre von der Wirkung der Salze. Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. 10.1007/BF01918191. [DOI] [Google Scholar]
- Kunz W.; Henle J.; Ninham B. W. ’Zur Lehre von der Wirkung der Salze’ (about the science of the effect of salts): Franz Hofmeister’s historical papers. Curr. Opin. Colloid Interface Sci. 2004, 9, 19–37. 10.1016/j.cocis.2004.05.005. [DOI] [Google Scholar]
- Kunz W.; Lo Nostro P.; Ninham B. The present state of affairs with Hofmeister effects. Curr. Opin. Colloid Interface Sci. 2004, 9, 1–18. 10.1016/j.cocis.2004.05.004. [DOI] [Google Scholar]
- Traube J. Die Theorie des Haftdrucks (Oberflächendrucks) und ihre Bedeutung für die Physiologie. Pflugers Arch. für Gesamte Physiol. Menschen Tiere 1910, 132–132, 511–538. 10.1007/BF01679763. [DOI] [Google Scholar]
- Kunz W. Specific ion effects in colloidal and biological systems. Curr. Opin. Colloid Interface Sci. 2010, 15, 34–39. 10.1016/j.cocis.2009.11.008. [DOI] [Google Scholar]
- Zhang Y.; Cremer P. S. Chemistry of Hofmeister Anions and Osmolytes. Annu. Rev. Phys. Chem. 2010, 61, 63–83. 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- Deyerle B. A.; Zhang Y. Effects of Hofmeister Anions on the Aggregation Behavior of PEO–PPO–PEO Triblock Copolymers. Langmuir 2011, 27, 9203–9210. 10.1021/la201463g. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Rajkovic I.; Kubicek K.; Quevedo W.; Pietzsch A.; Wernet P.; Föhlisch A.; Techert S. Probing the Hofmeister Effect with Ultrafast Core–Hole Spectroscopy. J. Phys. Chem. B 2014, 118, 9398–9403. 10.1021/jp504577a. [DOI] [PubMed] [Google Scholar]
- Okur H. I.; Hladílková J.; Rembert K. B.; Cho Y.; Heyda J.; Dzubiella J.; Cremer P. S.; Jungwirth P. Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions. J. Phys. Chem. B 2017, 121, 1997–2014. 10.1021/acs.jpcb.6b10797. [DOI] [PubMed] [Google Scholar]
- Kang B.; Tang H.; Zhao Z.; Song S. Hofmeister Series: Insights of Ion Specificity from Amphiphilic Assembly and Interface Property. ACS Omega 2020, 5, 6229–6239. 10.1021/acsomega.0c00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers B. A.; Okur H. I.; Yan C.; Yang T.; Heyda J.; Cremer P. S. Weakly hydrated anions bind to polymers but not monomers in aqueous solutions. Nat. Chem. 2022, 14, 40–45. 10.1038/s41557-021-00805-z. [DOI] [PubMed] [Google Scholar]
- Hervø-Hansen S.; Heyda J.; Lund M.; Matubayasi N. Anion–cation contrast of small molecule solvation in salt solutions. Phys. Chem. Chem. Phys. 2022, 24, 3238–3249. 10.1039/D1CP04129K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries-Kautt M. M.; Ducruix A. F. Relative Effectiveness of Various Ions on the Solubility and Crystal Growth of Lysozyme. J. Biol. Chem. 1989, 264, 745–748. 10.1016/S0021-9258(19)85005-6. [DOI] [PubMed] [Google Scholar]
- Finet S.; Skouri-Panet F.; Casselyn M.; Bonneté F.; Tardieu A. The Hofmeister effect as seen by SAXS in protein solutions. Curr. Opin. Colloid Interface Sci. 2004, 9, 112–116. 10.1016/j.cocis.2004.05.014. [DOI] [Google Scholar]
- Zhang Y.; Cremer P. S. The inverse and direct Hofmeister series for lysozyme. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15249–15253. 10.1073/pnas.0907616106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwierz N.; Horinek D.; Netz R. R. Reversed Anionic Hofmeister Series: The Interplay of Surface Charge and Surface Polarity. Langmuir 2010, 26, 7370–7379. 10.1021/la904397v. [DOI] [PubMed] [Google Scholar]
- Schwierz N.; Horinek D.; Netz R. R. Anionic and Cationic Hofmeister Effects on Hydrophobic and Hydrophilic Surfaces. Langmuir 2013, 29, 2602–2614. 10.1021/la303924e. [DOI] [PubMed] [Google Scholar]
- Xie W. J.; Gao Y. Q. A simple theory for the hofmeister series. J. Phys. Chem. Lett. 2013, 4, 4247–4252. 10.1021/jz402072g. [DOI] [PubMed] [Google Scholar]
- Jungwirth P.; Cremer P. S. Beyond Hofmeister. Nat. Chem. 2014, 6, 261–263. 10.1038/nchem.1899. [DOI] [PubMed] [Google Scholar]
- Record M. T.; Guinn E.; Pegram L.; Capp M. Introductory Lecture: Interpreting and predicting Hofmeister salt ion and solute effects on biopolymer and model processes using the solute partitioning model. Faraday Discuss. 2013, 160, 9–44. 10.1039/C2FD20128C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram L. M.; Wendorff T.; Erdmann R.; Shkel I.; Bellissimo D.; Felitsky D. J.; Record M. T. Why Hofmeister effects of many salts favor protein folding but not DNA helix formation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7716–7721. 10.1073/pnas.0913376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinn E. J.; Pegram L. M.; Capp M. W.; Pollock M. N.; Record M. T. Quantifying why urea is a protein denaturant, whereas glycine betaine is a protein stabilizer. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16932–16937. 10.1073/pnas.1109372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Shkel I. A.; O’Connor K.; Henrich J.; Molzahn C.; Lambert D.; Record M. T. Experimental Atom-by-Atom Dissection of Amide–Amide and Amide–Hydrocarbon Interactions in H2O. J. Am. Chem. Soc. 2017, 139, 9885–9894. 10.1021/jacs.7b03261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Shkel I. A.; O’Connor K.; Record M. T. Experimentally determined strengths of favorable and unfavorable interactions of amide atoms involved in protein self-assembly in water. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 27339–27345. 10.1073/pnas.2012481117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram L. M.; Record M. T. Thermodynamic Origin of Hofmeister Ion Effects. J. Phys. Chem. B 2008, 112, 9428–9436. 10.1021/jp800816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram L. M.; Record M. T. Partitioning of atmospherically relevant ions between bulk water and the water/vapor interface. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14278–14281. 10.1073/pnas.0606256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J. K.; Nick Pace C.; Martin Scholtz J. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford C. Contribution of Hydrophobic Interactions to the Stability of the Globular Conformation of Proteins. J. Am. Chem. Soc. 1962, 84, 4240–4247. 10.1021/ja00881a009. [DOI] [Google Scholar]
- Auton M.; Bolen D. W. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc. Natl. Acad. Sci. 2005, 102, 15065–15068. 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton M.; Bolen D. W.. Application of the Transfer Model to Understand How Naturally Occurring Osmolytes Affect Protein Stability; Elsevier, 2007, pp 397–418. [DOI] [PubMed] [Google Scholar]
- Bolen D. W.; Rose G. D. Structure and Energetics of the Hydrogen-Bonded Backbone in Protein Folding. Annu. Rev. Biochem. 2008, 77, 339–362. 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- Guinn E. J.; Kontur W. S.; Tsodikov O. V.; Shkel I.; Record M. T. Probing the protein-folding mechanism using denaturant and temperature effects on rate constants. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16784–16789. 10.1073/pnas.1311948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellman J. A. Selective binding and solvent denaturation. Biopolymers 1987, 26, 549–559. 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- Timasheff S. N. The Control of Protein Stability and Association by Weak Interactions with Water: How Do Solvents Affect These Processes?. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 67–97. 10.1146/annurev.bb.22.060193.000435. [DOI] [PubMed] [Google Scholar]
- Timasheff S. N.Control of Protein Stability and Reactions by Weakly Interacting Cosolvents: The Simplicity of the Complicated; Elsevier, 1998; pp 355–432. [DOI] [PubMed] [Google Scholar]
- Horinek D.; Netz R. R. Can Simulations Quantitatively Predict Peptide Transfer Free Energies to Urea Solutions?. Thermodynamic Concepts and Force Field Limitations 2011, 115, 6125–6136. 10.1021/jp1110086. [DOI] [PubMed] [Google Scholar]
- Moeser B.; Horinek D. Unified description of urea denaturation: Backbone and side chains contribute equally in the transfer model. J. Phys. Chem. B 2014, 118, 107–114. 10.1021/jp409934q. [DOI] [PubMed] [Google Scholar]
- O’Brien E. P.; Brooks B. R.; Thirumalai D. Molecular origin of constant m-values, denatured state collapse, and residue-dependent transition midpoints in globular proteins. Biochemistry 2009, 48, 3743–3754. 10.1021/bi8021119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Reddy G.; O’Brien E. P.; Thirumalai D. Collapse kinetics and chevron plots from simulations of denaturant-dependent folding of globular proteins. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7787–7792. 10.1073/pnas.1019500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyda J.; Dzubiella J. Thermodynamic Description of Hofmeister Effects on the LCST of Thermosensitive Polymers. J. Phys. Chem. B 2014, 118, 10979–10988. 10.1021/jp5041635. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S.; Nayar D.; Dalgicdir C.; van der Vegt N. F. A. A cosolvent surfactant mechanism affects polymer collapse in miscible good solvents. Commun. Chem. 2020, 3, 165. 10.1038/s42004-020-00405-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S.; Nayar D.; Dalgicdir C.; van der Vegt N. F. A. An interplay of excluded-volume and polymer–(co)solvent attractive interactions regulates polymer collapse in mixed solvents. J. Chem. Phys. 2021, 154, 134903. 10.1063/5.0046746. [DOI] [PubMed] [Google Scholar]
- Al-Maaieh A.; Flanagan D. R. Salt Effects On Caffeine Solubility, Distribution, And Self-Association. J. Pharm. Sci. 2002, 91, 1000–1008. 10.1002/jps.10046. [DOI] [PubMed] [Google Scholar]
- Rogers B. A.; Thompson T. S.; Zhang Y. Hofmeister Anion Effects on Thermodynamics of Caffeine Partitioning between Aqueous and Cyclohexane Phases. J. Phys. Chem. B 2016, 120, 12596–12603. 10.1021/acs.jpcb.6b07760. [DOI] [PubMed] [Google Scholar]
- Rogers B. A.; Zhang Y. Project-Based Experiment in a Physical Chemistry Teaching Laboratory: Ion Effects on Caffeine Partitioning Thermodynamics. J. Chem. Educ. 2020, 97, 4173–4178. 10.1021/acs.jchemed.0c00961. [DOI] [Google Scholar]
- Shimizu S. Caffeine dimerization: effects of sugar, salts, and water structure. Food Funct. 2015, 6, 3228–3235. 10.1039/C5FO00610D. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Paul S. Effects of dilute aqueous NaCl solution on caffeine aggregation. J. Chem. Phys. 2013, 139, 194504. 10.1063/1.4830414. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Paul S. Understanding the Role of Temperature Change and the Presence of NaCl Salts on Caffeine Aggregation in Aqueous Solution: From Structural and Thermodynamics Point of View. J. Phys. Chem. B 2015, 119, 6421–6432. 10.1021/jp512336n. [DOI] [PubMed] [Google Scholar]
- Johnson N. O.; Light T. P.; MacDonald G.; Zhang Y. Anion–Caffeine Interactions Studied by 13C and 1H NMR and ATR–FTIR Spectroscopy. J. Phys. Chem. B 2017, 121, 1649–1659. 10.1021/acs.jpcb.6b12150. [DOI] [PubMed] [Google Scholar]
- Eskelinen M. H.; Kivipelto M. Caffeine as a Protective Factor in Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. 10.3233/jad-2010-1404. [DOI] [PubMed] [Google Scholar]
- Postuma R. B.; Lang A. E.; Munhoz R. P.; Charland K.; Pelletier A.; Moscovich M.; Filla L.; Zanatta D.; Rios Romenets S.; Altman R.; Chuang R.; Shah B. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. 10.1212/wnl.0b013e318263570d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma R. B.; Anang J.; Pelletier A.; Joseph L.; Moscovich M.; Grimes D.; Furtado S.; Munhoz R. P.; Appel-Cresswell S.; Moro A.; Borys A.; Hobson D.; Lang A. E. Caffeine as symptomatic treatment for Parkinson disease (Café-PD). Neurology 2017, 89, 1795–1803. 10.1212/WNL.0000000000004568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavagnacco L.; Brady J. W.; Bruni F.; Callear S.; Ricci M. A.; Saboungi M. L.; Cesàro A. Hydration of Caffeine at High Temperature by Neutron Scattering and Simulation Studies. J. Phys. Chem. B 2015, 119, 13294–13301. 10.1021/acs.jpcb.5b09204. [DOI] [PubMed] [Google Scholar]
- Polák J.; Ondo D.; Heyda J. Thermodynamics of N-Isopropylacrylamide in Water: Insight from Experiments, Simulations, and Kirkwood–Buff Analysis Teamwork. J. Phys. Chem. B 2020, 124, 2495–2504. 10.1021/acs.jpcb.0c00413. [DOI] [PubMed] [Google Scholar]
- Gee M. B.; Smith P. E. Kirkwood–Buff theory of molecular and protein association, aggregation, and cellular crowding. J. Chem. Phys. 2009, 131, 131. 10.1063/1.3253299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Case D.; et al. Amber 11; Amber, 2010.www.ambermd.org.
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernersson E.; Jungwirth P. Effect of water polarizability on the properties of solutions of polyvalent ions: Simulations of aqueous sodium sulfate with different force fields. J. Chem. Theory Comput. 2010, 6, 3233–3240. 10.1021/ct100465g. [DOI] [PubMed] [Google Scholar]
- Caldwell J. W.; Kollman P. A. Structure and Properties of Neat Liquids Using Nonadditive Molecular-Dynamics - Water, Methanol, and N-Methylacetamide. J. Phys. Chem. 1995, 99, 6208–6219. 10.1021/j100016a067. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Sanjeewa R.; Weerasinghe S. Development of a molecular mechanics force field for caffeine to investigate the interactions of caffeine in different solvent media. J. Mol. Struct.: THEOCHEM 2010, 944, 116–123. 10.1016/j.theochem.2009.12.027. [DOI] [Google Scholar]
- Sanjeewa R.; Weerasinghe S. Study of aggregate formation of caffeine in water by molecular dynamics simulation. Comput. Theor. Chem. 2011, 966, 140–148. 10.1016/j.comptc.2011.02.027. [DOI] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Křížek T.; Kubíčková A.; Hladílková J.; Coufal P.; Heyda J.; Jungwirth P. Electrophoretic mobilities of neutral analytes and electroosmotic flow markers in aqueous solutions of Hofmeister salts. Electrophoresis 2014, 35, 617–624. 10.1002/elps.201300544. [DOI] [PubMed] [Google Scholar]
- Hine J.; Mookerjee P. K. Structural effects on rates and equilibriums. XIX. Intrinsic hydrophilic character of organic compounds. Correlations in terms of structural contributions. J. Org. Chem. 1975, 40, 292–298. 10.1021/jo00891a006. [DOI] [Google Scholar]
- Metropolis N.; Rosenbluth A. W.; Rosenbluth M. N.; Teller A. H.; Teller E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. 10.1063/1.1699114. [DOI] [Google Scholar]
- Stenqvist B.; Thuresson A.; Kurut A.; Vácha R.; Lund M. Faunus– a flexible framework for Monte Carlo simulation. Mol. Simul. 2013, 39, 1233–1239. 10.1080/08927022.2013.828207. [DOI] [Google Scholar]
- Lund M.; Trulsson M.; Persson B. Faunus: An object oriented framework for molecular simulation. Source Code Biol. Med. 2008, 3, 1. 10.1186/1751-0473-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks J. D.; Chandler D.; Andersen H. C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. 10.1063/1.1674820. [DOI] [Google Scholar]
- Harismiadis V. I.; Vorholz J.; Panagiotopoulos A. Z. Efficient pressure estimation in molecular simulations without evaluating the virial. J. Chem. Phys. 1996, 105, 8469–8470. 10.1063/1.472721. [DOI] [Google Scholar]
- Widom B. Some Topics in the Theory of Fluids. J. Chem. Phys. 1963, 39, 2808–2812. 10.1063/1.1734110. [DOI] [Google Scholar]
- Johnson J. K.; Panagiotopoulos A. Z.; Gubbins K. E. Reactive canonical Monte Carlo. Mol. Phys. 1994, 81, 717–733. 10.1080/00268979400100481. [DOI] [Google Scholar]
- Smith P. E. On the Kirkwood-Buff inversion procedure. J. Chem. Phys. 2008, 129, 124509. 10.1063/1.2982171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. S.; Elcock A. H. Molecular dynamics simulations of hydrophobic associations in aqueous salt solutions indicate a connection between water hydrogen bonding and the hofmeister effect. J. Am. Chem. Soc. 2007, 129, 14887–14898. 10.1021/ja073097z. [DOI] [PubMed] [Google Scholar]
- Okamoto R.; Koga K. Theory of Gas Solubility and Hydrophobic Interaction in Aqueous Electrolyte Solutions. J. Phys. Chem. B 2021, 125, 12820–12831. 10.1021/acs.jpcb.1c08050. [DOI] [PubMed] [Google Scholar]
- Shalmashi A.; Golmohammad F. Solubility of caffeine in water, ethyl acetate, ethanol, carbon tetrachloride, methanol, chloroform, dichloromethane, and acetone between 298 and 323 K. Lat. Am. Appl. Res. 2010, 40, 283–285. [Google Scholar]
- Smith P. E.; Mazo R. M. On the Theory of Solute Solubility in Mixed Solvents. J. Phys. Chem. B 2008, 112, 7875–7884. 10.1021/jp712179w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žółkiewski M. Kirkwood-Buff integrals and density fluctuations in aqueous solution of caffeine. J. Solution Chem. 1987, 16, 1025–1034. 10.1007/BF00652586. [DOI] [Google Scholar]
- Cesaro A.; Russo E.; Crescenzi V. Thermodynamics of caffeine aqueous solutions. J. Phys. Chem. 1976, 80, 335–339. 10.1021/j100544a026. [DOI] [Google Scholar]
- Falk M.; Chew W.; Walter J. A.; Kwiatkowski W.; Barclay K. D.; Klassen G. A. Molecular modelling and NMR studies of the caffeine dimer. Can. J. Chem. 1998, 76, 48–56. 10.1139/v97-211. [DOI] [Google Scholar]
- Tavagnacco L.; Gerelli Y.; Cesàro A.; Brady J. W. Stacking and Branching in Self-Aggregation of Caffeine in Aqueous Solution: From the Supramolecular to Atomic Scale Clustering. J. Phys. Chem. B 2016, 120, 9987–9996. 10.1021/acs.jpcb.6b06980. [DOI] [PubMed] [Google Scholar]
- Ben-Naim A.Statistical Thermodynamics for Chemists and Biochemists; Plenum: New York, 1992. [Google Scholar]
- Kurut A.; Lund M. Solution electrostatics beyond pH: a coarse grained approach to ion specific interactions between macromolecules. Faraday Discuss. 2013, 160, 271–278. 10.1039/C2FD20073B. [DOI] [PubMed] [Google Scholar]
- Harvey A.; Lemmon E.. NIST/ASME Steam Properties. Version 3.0; NIST, 2013.
- Origlia-Luster M. L.; Patterson B. A.; Woolley E. M. Thermodynamics for self-association of caffeine in water: Apparent molar volumes and apparent molar heat capacities of aqueous caffeine at temperatures from 278.15 to 393.15 K and at the pressure 0.35 MPa. J. Chem. Thermodyn. 2002, 34, 1909–1921. 10.1016/S0021-9614(02)00263-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.