

Abstract
Mitochondrial and peroxisomal anchored protein ligase (MAPL) is a dual ubiquitin and small ubiquitin‐like modifier (SUMO) ligase with roles in mitochondrial quality control, cell death and inflammation in cultured cells. Here, we show that MAPL function in the organismal context converges on metabolic control, as knockout mice are viable, insulin‐sensitive, and protected from diet‐induced obesity. MAPL loss leads to liver‐specific activation of the integrated stress response, inducing secretion of stress hormone FGF21. MAPL knockout mice develop fully penetrant spontaneous hepatocellular carcinoma. Mechanistically, the peroxisomal bile acid transporter ABCD3 is a primary MAPL interacting partner and SUMOylated in a MAPL‐dependent manner. MAPL knockout leads to increased bile acid production coupled with defective regulatory feedback in liver in vivo and in isolated primary hepatocytes, suggesting cell‐autonomous function. Together, our findings establish MAPL function as a regulator of bile acid synthesis whose loss leads to the disruption of bile acid feedback mechanisms. The consequences of MAPL loss in liver, along with evidence of tumor suppression through regulation of cell survival pathways, ultimately lead to hepatocellular carcinogenesis.
Keywords: hepatocellular carcinoma, MUL1, peroxisome, PMP70, SUMO
Subject Categories: Membranes & Trafficking, Metabolism, Post-translational Modifications & Proteolysis
Mitochondrial and peroxisomal anchored protein ligase (MAPL) deficiency in mice leads to loss of SUMOylation of the peroxisomal bile acid transporter ABCD3 and defects in the bile acid metabolism. MAPL knockout mice develop liver stress and spontaneous hepatocellular carcinoma.

Introduction
Mitochondria are an essential signaling platform that contribute to cell fate decisions, from cell cycle transitions to stem cell differentiation, T‐cell activation and pathogen invasion, where both intraorganellar biochemical pathways and signaling complex assembly on the membrane play critical roles (Pfanner et al, 2019; Shen et al, 2022). An underlying reason for the integration of mitochondria within cellular signaling pathways is to signal the transcriptional and post‐transcriptional changes required to alter fuel consumption and/or metabolite generation that drive cellular transitions. Mitochondrial signaling machineries are often shared with peroxisomes, comprising interorganellar functional axes that contribute to the global rewiring of cellular metabolism in response to a variety of extracellular stimuli and intracellular cues (Costello et al, 2018). It is becoming apparent that, akin to other signaling pathways, post‐translational modifications including phosphorylation, ubiquitination and SUMOylation play a central role in the assembly and regulation of mitochondrial signaling complexes (Tait & Green, 2012; Escobar‐Henriques & Langer, 2014; He et al, 2020; Tan & Finkel, 2020). The mechanistic details responsible for these modifications in mitochondrial signaling are however still largely unknown.
MAPL, a mitochondrial and peroxisomal anchored protein ligase (also called MUL1/GIDE/HADES/MULAN) (Li et al, 2008; Neuspiel et al, 2008; Zhang et al, 2008), is part of the shared peroxisome/mitochondrial proteome. MAPL is an E3 ligase that displays preferential SUMOylation and weak ubiquitination activities (Braschi et al, 2009) and has been shown to modulate cellular responses to diverse stresses including mitochondrial division, mitophagy, inflammation and cell death (Neuspiel et al, 2008; Braschi et al, 2009; Ambivero et al, 2014; Yun et al, 2014; Prudent et al, 2015; Rojansky et al, 2016; Doiron et al, 2017; Barry et al, 2018). SUMO and ubiquitin are structurally related and conjugated to their substrates via a similar biochemical cascade, but while ubiquitination often serves as a signal for proteasome‐mediated degradation, SUMOylation acts to modify stability, subcellular localization, or activity of the target (Celen & Sahin, 2020; Vertegaal, 2022). Additionally, mixed SUMO‐ubiquitin chains can be formed with distinct functional consequences (Nie & Boddy, 2016). A common feature of MAPL‐induced SUMOylation is stabilization of substrate complex formation or assembly, making it a prime candidate regulator of signaling platforms on mitochondria and peroxisomes (Prudent et al, 2015; Doiron et al, 2017; Barry et al, 2018). In mitochondria, MAPL is an outer membrane protein carrying two transmembrane domains, a ~40 kDa intermembrane space loop (“beside a membrane” BAM domain) and C‐terminal cytosolic RING finger (Andrade‐Navarro et al, 2009). MAPL is also targeted to peroxisomes in mitochondrial‐derived vesicles (Neuspiel et al, 2008), where it plays a role in regulating peroxisomal fission (Mohanty et al, 2021). However, broader roles of MAPL in peroxisomes are unknown.
To investigate the primary function of MAPL, we explored the proximity interactors using unbiased BioID approaches (Roux et al, 2012; Roux, 2013). In addition to the core mitochondrial and peroxisomal fission machinery, we identified peroxisomal ATP‐binding cassette transporter ABCD3 as an interacting partner and SUMOylated substrate of MAPL. ABCD3 transports the late‐stage steroid bile acid precursors, the C27‐bile acid intermediates 3α,7α‐dihydroxycholestanoic acid (DHCA), 3α,7α,12α‐trihydroxycholestanoic acid (THCA), and dicarboxylic fatty acids from cytosol into peroxisomes (Ferdinandusse et al, 2015; Ranea‐Robles et al, 2021). With the generation of a MAPL knockout mouse model, we uncovered a critical role for this SUMO E3 ligase in restricting the activity of ABCD3, highlighting new links to whole body metabolism. Further analysis of the MAPL‐deficient mice revealed increased hepatocyte proliferation, resistance to programmed cell death, and the development of hepatocellular carcinoma in aging mice. These data provide insight into the post‐translational regulation of bile acid metabolism within the liver and the central role for peroxisomal SUMOylation in metabolic homeostasis.
Results
ABCD3/PMP70 is a MAPL substrate
To identify MAPL‐interacting proteins, we performed an unbiased proximity‐dependent biotin identification (BioID) in HEK 293 cells (Roux et al, 2012; Roux, 2013). As expected, BioID confirmed a robust interaction between MAPL and the fission GTPase DRP1 (Fig 1A) (Braschi et al, 2009; Prudent et al, 2015). Indeed, essentially all of the mitochondrial fission machinery identified to date was identified in this analysis, including the regulator of DRP1 recruitment AKAP1, the cAMP‐dependent protein kinase type II‐alpha regulatory subunit PRKAR2A, the DRP1 receptor MFF, the inverted formin INF2 (Kraus & Ryan, 2017), a regulator of mitophagy and pexophagy USP30 (Bingol et al, 2014; Marcassa et al, 2018) and others. Also present was the antiviral signaling protein MAVS, an interaction we and others have characterized previously (Jenkins et al, 2013; Doiron et al, 2017).
Figure 1. MAPL SUMOylates ABCD3 and modulates complex assembly.

- HEK 293T‐REX (tetracycline‐regulatable expression) cells stably expressing an inducible Tet‐ON fusion construct MAPL‐Flag‐BirA, or Flag‐BirA only, were treated with Dox for 24 h in the presence of biotin, and biotinylated proteins were isolated with streptavidin beads for identification by mass spectrometry. Top hits, by peptide count, are shown.
- Starting material (SM) and MAPL immunoprecipitated (IP) fractions obtained from crude liver extracts were probed for ABCD3, MAPL and Hsp60.
- Cytosol and heavy membrane fractions isolated from MAPLf/f and MAPL−/− livers were solubilized and incubated with SIM beads, and elution fractions probed for ABCD3, DRP1, Hsp70 and MAPL (Cyt = cytosolic fraction, HM = heavy membrane fraction).
- Heavy membrane fractions isolated from livers of MAPLf/f and MAPL−/− animals' tail‐vein injected with adenovirus expressing MAPL‐Flag, MAPL‐ΔRING‐Flag or empty virus (Ad‐rtTA) were solubilized and incubated with SIM beads. Elution fractions were probed for ABCD3, SUMO1 and MAPL (top panel). Quantification from three independent experiments of ABCD3 signals of the heavy membrane SIM‐beads elution fraction from livers isolated from rescued mice (lower panel).
- Two hundred fifty microgram of solubilized protein (from MAPLf/f and MAPL−/− livers) were separated on a 10–50% (w/v) sucrose gradient. Twelve different fractions, as well as the resuspended pellet (P) were analyzed by immunoblot. ABCD3 signals from MAPLf/f and MAPL−/− livers in the different fractions were quantified and plotted as percentage of the total signal, from three biological replicates. *P < 0.05 in a one‐way ANOVA. Data are represented as mean ± SEM.
Source data are available online for this figure.
Potential ubiquitin substrates of MAPL such as MFN1, MFN2, AKT or P53 were absent from the interactome at steady state (Yun et al, 2014; Kim et al, 2015; Lee et al, 2022), possibly due to the degradation of ubiquitinated targets by the 26S proteasome. Therefore, we repeated the BioID experiments in the presence of the proteasome inhibitor MG132 over 24 h to stabilize and accumulate any potential ubiquitinated MAPL‐FlagBirA* substrates (Coyaud et al, 2015) (Table EV1). Notably, the number of peptides identified for the vast majority of MAPL interacting proteins remained unchanged in the presence of MG132 (or were decreased). Only a few additional proteins were detected at very low levels after 24 h of MG132 treatment compared to untreated samples, with peptide counts ranging from 7 to 24 (compared to 612 for AKAP1) including BAX, MIRO1, BNIP3 and MFN2. Collectively, these data suggest that MAPL does not target its primary binding partners for proteasomal degradation, consistent with preferential function of MAPL as a SUMO E3 ligase (Braschi et al, 2009).
Unexpectedly, the top ranked MAPL binding protein detected in this analysis was the peroxisomal transporter ABCD3/PMP70 (Fig 1A). We confirmed this result using western blot analysis of biotinylated ABCD3 captured on streptavidin beads after incubation of MAPL‐BirA expressing cells with biotin (Fig EV1A). ABCD3 is a peroxisomal transporter, suggested to shuttle branched and very long chain fatty acids for oxidation, like ABCD1 and ABCD2 (Ranea‐Robles et al, 2021). However, evidence in mouse models and at least one patient indicates a primary role for ABCD3 in the transport of branched chain acyl‐CoAs and bile acid precursors di‐ and tri‐hydroxycholestanoyl‐CoA (D/THC‐CoA) (Ferdinandusse et al, 2015). Given the specificity of the proximity biotinylation of ABCD3, we chose to validate and interrogate the potential functional consequences of MAPL‐ABCD3 binding in our MAPL knockout (MAPL−/−) mouse line (Doiron et al, 2017; Roque et al, 2023), focusing on liver as a primary organ of bile acid production.
Figure EV1. MAPL SUMOylates ABCD3 and its absence alters ABCD3 complex assembly.

- Representative immunoblot of total lysate and streptavidin‐beads pull‐down fractions of HEK‐293T‐REX cells expressing Flag‐MAPL‐BirA or Flag‐BirA (as control) incubated with biotin and probed for ABCD3, DRP1, MAPL and beta‐actin.
- Quantification from three independent experiments of ABCD3 signals of the heavy membrane SIM‐beads elution fraction (Fig 1C). *P < 0.05 in an unpaired two‐tailed t‐test.
- qRT‐PCR investigating ABCD3 mRNA levels in livers of four different mice (2‐month‐old males).
- Representative immunoblots from whole‐cell liver extracts probed for ABCD3, DRP1 and Mfn2 (Left panel). ABCD3, Drp1 and Mfn2 signals were quantified (n = 3 for each genotype, right panel).
- Representative immunoblots from whole‐cell liver extracts probed for PEX14, ACOX1 and SCP2 (Left panel). PEX14, ACOX1 and SCP2 signals were quantified (n = 3 for each genotype, right panel).
Data are represented as mean ± SEM.
To test for any interaction between native MAPL and ABCD3 proteins, we immunoprecipitated endogenous MAPL from liver of control or MAPL −/− mice. While the anti‐MAPL antibodies did not efficiently precipitate the endogenous protein, we still observed a MAPL‐dependent interaction with ABCD3 (Fig 1B). To monitor the SUMOylation of ABCD3, we isolated cytosol (Cyt) and solubilized the heavy membrane (HM) fraction from MAPLf/f and MAPL−/− livers. These fractions were incubated with agarose beads conjugated to peptides encoding the consensus SUMO interacting motif (SIM) of the nuclear SUMO E3 ligases PIAS1‐4 (Hecker et al, 2006). Notably, while we observed an ABCD3 immunoreactive band in control liver SIM pulldowns, this signal was strikingly decreased in livers from mice lacking MAPL (Fig 1C, quantification Fig EV1B). The molecular weight of ABCD3 on the SIM beads was slightly shifted, consistent with a mono‐SUMOylation event. As a positive control for MAPL‐dependent SUMOylation, we blotted for DRP1 (Fig 1C) (Braschi et al, 2009; Prudent et al, 2015).
To further confirm whether MAPL is responsible for the SUMOylation of ABCD3 in liver we generated adenovirus vectors encoding MAPL‐Flag, or a deletion construct lacking the MAPL C‐terminal RING finger required for SUMO or ubiquitin conjugation (MAPL‐ΔRING‐FLAG). Viruses (including an empty virus [rtTA] as negative control) were injected into the tail vein of wild‐type and MAPL KO mice to rescue the expression of MAPL specifically within liver. ABCD3 SUMOylation was restored upon expression of full length MAPL, but not in MAPL‐ΔRING‐FLAG (Fig 1D). Given the interaction between MAPL and ABCD3 observed in both BioID and immunoprecipitation experiments, and the functional rescue of ABCD3 SUMOylation upon re‐expression of MAPL in the liver of MAPL KO mice, MAPL appears to be an essential regulator of ABCD3 SUMOylation.
We next examined the consequences of the loss of MAPL on the biochemical properties of ABCD3 in liver. Consistent with our evidence that MAPL does not generally regulate protein turnover, the total protein levels of ABCD3 were unchanged in total liver extracts, as were the well‐characterized MAPL targets DRP1 and MFN2 (Fig EV1C and D). We also considered that peroxisomal function, biogenesis or turnover may have been globally altered in MAPL −/− liver but observed no change in the expression levels of the peroxisomal proteins PEX14, ACOX1 and SCP2 (Fig EV1E). This contrasts with the loss of another shared mitochondrial and peroxisomal E3 ligase, March5, whose ubiquitination of ABCD3 led to its degradation and pexophagy (Zheng et al, 2022).
As a half transporter, ABCD3 assembles into both homo‐ and heterodimers (Hillebrand et al, 2007). To examine potential changes in the oligomeric assemblies of ABCD3 in the absence of MAPL, we performed sucrose gradients from solubilized mouse liver extracts. As previously described (van Roermund et al, 2014), the 70 kDa ABCD3 protein migrated at a higher molecular weight, consistent with a higher‐order oligomeric structure (Fig 1E). Consistent with the abundance of ABCD3 in the MAPL BioID, MAPL co‐migrated with ABCD3 in fractions 5, 6 and 7 on the sucrose gradient. Notably, extracts isolated from MAPL −/− mice revealed a change in the migration pattern of ABCD3, with ABCD3 spreading throughout the higher molecular weight fractions, indicating that MAPL SUMOylation activity is required to maintain a stable oligomeric assembly of the ABCD3 transporter.
In sum, these data demonstrate that ABCD3 is SUMOylated by MAPL in liver tissue and that disruption of this process is associated with changes in peroxisomal ABCD3 complex formation. This provides the first evidence of SUMO modifications in the regulation of ABCD3 assembly.
MAPL is required to gate the synthesis of bile acids in liver
Bile acids are cholesterol‐derived steroid acids that play critical roles in the absorption and metabolism of lipids. They are synthesized primarily in the liver, accumulated in the gallbladder and secreted in the small intestine. More than 90% of the pool is reabsorbed by the ileum and recirculated to the liver, and the remaining pool is excreted with the feces (de Boer et al, 2018). To determine functional consequences of the loss of ABCD3 SUMOylation, we quantified circulating bile acids in serum. We observed a 3‐ to 5‐fold increase in the level of total bile acids in MAPL−/− mice (Fig 2A), which was rescued in a RING‐dependent manner upon re‐expression of MAPL in the liver (Fig 2B). This suggests a requirement for MAPL‐mediated SUMOylation in the liver in repressing bile acid metabolism. To investigate whether specific stages of the bile acid metabolic pathway are affected, we employed quantitative mass spectrometry using standards for over 40 selected bile acid conjugate species in serum, liver and feces (see Fig EV2A for schematic of bile acids) (Han et al, 2015). Notably, a significant decrease was observed in the C27 precursors DHCA and THCA with a concomitant increase in C24 mature bile acid species in liver. The increase in C24 mature bile acid species was also observed in circulation, although C27 DHCA and THCA were also slightly elevated in serum. For example, primary C24 beta‐muricholic (b‐MCA) bile acid was increased from ~10,000 to 30,000 fmol/g of liver tissue, and from 600 to 3,500 nM in MAPL −/− serum (Fig 2C, Table EV2). In contrast, both precursors and conjugated bile acids were decreased by more than 50% in feces (Fig 2C, Table EV2), suggestive of bile flow stalling through the liver/gallbladder biliary system (cholestasis, typically resulting in tissue damage) or increased reabsorption from the gut into circulation (Zeng et al, 2023). Histological examination of liver and damage markers showed no evidence of cholestasis, as morphology of bile canaliculi was normal, no signs of necrosis were present and serum alanine transaminase/aspartate aminotransferase (ALT/AST) were not changed (Figs 2C–E and EV2B). Moreover, we measured calories excreted in the feces by bomb calorimetry and found equivalent calories in MAPL−/− feces, suggesting that digestion of food was intact (Fig EV2C). Increased serum and liver levels of gut‐derived secondary bile acids are also indicative of functional enterohepatic circulation of bile acids, and its increased overall flux or reabsorption (Fig 2C). Thus, our data indicate that MAPL loss leads to increased bile acid production in the liver, coupled with increased levels of mature bile acids in circulation and decreased in feces, where the latter likely reflects an increase in gut reabsorption.
Figure 2. MAPL represses bile acid synthesis.

- Total serum bile acids were quantified as described in materials and methods (n = 8 for each genotype, six females (circles) and two males (squares), 2–6 months old). **P < 0.01 in an unpaired two‐tailed t‐test.
- Total serum bile acids in tail vein injected animals were quantified as described in materials and methods (n = 6 with four females and two males, n = 7 with four females and three males, n = 4 with two females and two males and n = 5 with two females and three males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old).
- Bile acid precursors, as well as unconjugated and conjugated primary and secondary bile acids, were quantified from liver (n = 8 for each genotype, 2‐month‐old males). THCA and DHCA: tri‐ and dihydroxycholestanoic acid; CA: cholic acid; a‐MCA, b‐MCA, w‐MCA and d‐MCA: α‐, β‐, γ‐ and δ‐muricholic acid; DCA: deoxycholic acid; MCA: muricholic acid; HDCA: hyodeoxycholic acid; TDCA: taurodeoxycholic acid; td‐MCA: tauro‐δ‐muricholic acid. *P < 0.05 in an unpaired two‐tailed t‐test.
- Representative pictures of livers from MAPL f/f and MAPL −/− mice. Hematoxylin and eosin staining. 40× objective. Scale bar: 100 μm.
- Levels of alanine transaminase (ALT) and aspartate transaminase (AST) (n = 6, 6 month old males).
- Total bile acid production (μM) in cultured primary hepatocytes isolated from MAPLf/f and MAPL−/− mice. Media was collected after 48 h of culture. N = 5 independent experiments. ***P < 0.001 in an unpaired two‐tailed t‐test.
- Total bile acid production (μM) in cultured primary hepatocytes isolated from MAPLf/f and MAPL−/− mice. Hepatocytes were transduced with adenoviruses encoding for rTta, MAPL‐Flag or MAPL‐ΔRing‐Flag. N = 3 independent experiments. **P < 0.01 in a one‐way ANOVA test.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
Figure EV2. Loss of MAPL alters bile acid synthesis.

- A model depicting the generation of the main murine bile acids from THCA and DHCA (tri‐ and dihydroxycholestanoic acids) (G: glyco‐; T: Tauro‐).
- Levels of alanine transaminase (ALT) and aspartate transaminase (AST), markers of liver damage, were quantified and plotted from serum collected from 6‐month‐old females (n = 5 and n = 6 for MAPL f/f and MAPL −/− , respectively for ALT quantification and n = 6 and n = 7 for MAPL f/f and MAPL −/− , respectively for AST quantification). **P < 0.01 in an unpaired two‐tailed t‐test.
- The amount of energy present in 3‐month animal feces has been measured using an oxygen bomb calorimeter, as described in material and methods (n = 7 with four females and three males for each genotype). **P < 0.01 in an unpaired two‐tailed t‐test.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
The tail vein experiments with liver‐specific MAPL re‐expression in MAPL−/− mice showed a return to normal bile acid levels in serum, strongly indicating that systemic bile acid dysregulation is linked to MAPL function in liver and not to altered gut resorption. To further uncouple cell‐autonomous MAPL‐dependent regulation of bile acid production from regulatory feedback in enterohepatic circulation, we isolated primary hepatocytes from control and MAPL−/− mice. Analysis of the conditioned media after 48 h showed that MAPL‐deficient hepatocytes secreted 3‐ to 4‐fold more bile acid into the media than control hepatocytes (Fig 2F). This effect was rescued by re‐expression of MAPL in a RING‐dependent manner (Fig 2G), indicating the role of its ligase activity in regulating hepatocyte bile flux. Together, these data demonstrate that loss of MAPL leads to a cell autonomous increase in bile acid synthesis and secretion from hepatocytes, with an increase in resorption in the gut that contributes to increased circulating bile.
MAPL loss results in a loss of feedback regulation of bile acid biosynthesis
Bile acids are physiological detergents whose levels are controlled by a balance of enzymes involved in biosynthesis and detoxification, where regulation is responsive to both feedback and feed‐forward mechanisms (Zeng et al, 2023). As MAPL−/− mice showed elevated hepatic bile acid production at fed state, but no liver damage or increased bile acid elimination, we explored in detail the multi‐layered regulation of the pathway within the liver. Complete synthesis of primary bile acids from ER‐derived cholesterol requires metabolite flux between the ER, mitochondria, peroxisomes and cytosol (Fig 3A). The cascade is initiated by either ER‐localized cholesterol 7‐alpha‐hydroxylase, CYP7A1 (liver‐specific, “classical” pathway) or mitochondrial 27‐hydroxylase CYP27A1 that oxidizes cholesterol to 27‐hydroxycholesterol (“alternative,” or acidic pathway) (Fig 3A) (Wang et al, 2021). Products of these reactions are consequently converted by ER and cytoplasmic enzymes to late stage C27 precursors DHCA and THCA, activated by SLC27A5 (BACS), transported to peroxisomes and modified into C24 primary bile acids by beta‐oxidation enzymes. Finally, C24 bile acids are conjugated by BAAT with taurine or glycine to be exported via ABCB11 (bile salt export pump, BSEP) on the apical hepatocyte membrane that forms the bile‐draining canaliculi (Fig 3A). Known regulation of the biosynthesis rate occurs via CYP7A1, expression of which is responsive to bile acid levels and regulatory feedback loops through bile acid binding receptors in the ileum and liver (Chiang & Ferrell, 2020). Bile acids can also regulate Cyp27a1 expression transcriptionally (Chen & Chiang, 2003), although less is known about these feedback pathways. Even though bile acids are significantly elevated in MAPL−/− livers, RNAseq analysis showed a minor downregulation of Cyp7a1 and Cyp27a1 transcripts, and no change in CYP7A1 protein expression (Fig 3B and C). In contrast to the stable protein expression of CYP7A1, the level of CYP27A1 protein was 40% lower compared to control mice, consistent with compensation to limit toxicity from elevated bile acids (Fig 3C, Dataset EV1). Levels of Cyp7B1, Hsd3b5, Slc27a5, Baat and Acbc11 were also decreased, presumably as further compensation to reduce bile secretion and uptake (Fig 3C). The near loss of top regulated gene Hsd3b5 was rescued by liver‐specific MAPL re‐expression in a RING‐dependent manner (Fig 3D). The only upregulated gene in the pathway was Cyp39a1, which catalyzes the 7‐alpha hydroxylation of (24S)‐hydroxycholesterol, a neural oxysterol that is metabolized to bile acids (Li‐Hawkins et al, 2000) (Fig 3C). Interestingly, we observed the expected sex‐biased expression of Cyp39a1, Hsd3b5 and Cyp7b1 at basal state (Ahluwalia et al, 2004) (Dataset EV1), yet the mRNA levels of these genes were changed in both sexes of MAPL−/− mice, further pointing to a general upstream signal downregulating gene expression of the bile biosynthesis.
Figure 3. MAPL loss leads to a dysregulation of the bile transcriptional feedback.

- A model depicting the required flux of metabolites between the ER, mitochondria and peroxisomes to facilitate bile acid synthesis in the liver. The initial step of the classical (neutral) pathway is represented with solid arrows, while the alternative (acidic) pathway is represented with dashed arrows. Downstream reactions of the two pathways converge on the same set of enzymes.
- Immunoblots for CYP27A1, CYP7A1, MAPL and ACTB from liver extracts from three animals of each genotypes (two females, one male), as indicated. Bottom panel is the quantification by densitometry. *P < 0.05 in an unpaired two‐tailed t‐test.
- mRNA expression of bile acid biosynthesis enzymes in the liver of MAPL−/− mice.
- qRT‐PCR measuring Hsd3b5 mRNA levels in livers of rescued mice (in triplicate, n = 5 with four females and one male, n = 7 with four females and three males, n = 5 with three females and two males and n = 6 with three females and three males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old).
- Immunoblots for CYP3A11, CYP4A14 and ACTB from liver extracts from three animals of each genotypes (two females, one male), as indicated. Bottom panel is the quantification by densitometry. *P < 0.05 in an unpaired two‐tailed t‐test.
- mRNA expression of bile acid detoxification enzymes in the liver of MAPL KO mice.
- Representative immunoblot of Cyp4A14 protein levels in livers isolated from rescued mice.
Data are represented as mean ± SEM. (C, F) Liver RNAseq, n = 5 per group. log2(FC) of MAPL KO versus Control mice is plotted, genes with P‐adjusted value < 0.05 are marked with pink. See also Fig 5 and Dataset EV1. Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
The toxicity of increased levels of bile acids can be mitigated by hydroxylation, sulfonation, or glucuronidation enzymes that convert these amphipathic species to water soluble, excretable metabolites (Bosma et al, 1994; Wagner et al, 2005). Of these, CYP3A11 and CYP4A14 were increased in MAPL−/− mice (Fig 3E and F) and rescued by MAPL re‐expression in a RING‐dependent manner (Fig 3G).
Bile acid synthesis is tightly coupled to regulatory feedback loops through bile acid binding receptors in the ileum and liver, which target and downregulate gene expression of Cyp7A1 to inhibit production. As CYP7A1 level was unchanged despite elevated bile acid levels, we examined the established components of these regulatory feedback loops. Circulating levels of FGF15, which is secreted from the ileum in response to reabsorbed bile acids and acts as an inhibitor of bile acid biosynthesis in the liver, were unchanged (Fig 4A) (Chiang, 2009). In addition, the liver expression of a key nuclear receptor acting to repress bile acid biosynthesis, transcription factor Farnesoid × receptor (FXR/Nr1h4) was also unchanged (Fig 4B) (Matsubara et al, 2013), consistent with CYP7A1 levels remaining unchanged. This suggested that in addition to increased bile acid biosynthesis, the loss of MAPL also results in a lack of feedback to the high bile acids generated in the liver.
Figure 4. MAPL loss results in a loss of feedback regulation of bile acid biosynthesis.

- Serum FGF15 measured by ELISA (n = 5 and n = 6 for MAPLf/f and MAPL−/− respectively, 2‐month‐old males).
- qRT‐PCR measuring Nr1h4 mRNA levels (FXR) in livers of four different mice (2‐month‐old males).
- Nuclear receptors target gene expression by qRT‐PCR. Livers were harvested from male and female mice fasted (4 h) or fed for 2 h after an overnight fast. N = 4–7 per sex, genotype and timepoint. Representative of two independent cohorts. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 in a 2way ANOVA test followed by a Sidak posthoc test.
- Representative immunoblot of ABCB11 protein levels in livers isolated from three animals of each genotype (one female, two males), as indicated.
- Liver gene expression of muricholic acid‐producing enzyme Cyp2c70. N = 4–7 per sex, genotype and timepoint. Representative of two independent cohorts.
- Liver gene expression of bile transporters Slco1a1, Slco10a1, Slco1b2. N = 4–7 per sex, genotype and timepoint. Representative of two independent cohorts. *P < 0.05, **P < 0.01, ****P < 0.0001 in a 2way ANOVA test followed by a Sidak posthoc test.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
Homeostasis of bile acid biosynthesis, release and absorption is regulated by a concert of transcription factors and transporters responsive to fed states. To investigate the dynamics of bile acid regulatory feedback, we measured the expression of bile responsive nuclear receptor target genes during fasting or feeding. We collected livers from control and MAPL−/− mice after 4 h fasting or 2 h refeeding. Then, we measured FXR, liver × receptor (LXR) (Bideyan et al, 2022), pregnane × receptor (PXR) (Barretto et al, 2019) and peroxisome‐proliferator‐activated receptor alpha (PPARα) gene targets (Rakhshandehroo et al, 2010) in liver by qRT‐PCR. PXR target genes Cyp3a11 and Cyp4a14 are increased by 3‐fold but the target Cyp7a1 (Fig 4C) was not affected, indicating either a differential or non‐activation of PXR transcriptional programs. Importantly, Cyp7A1 expression remained responsive to the fed state (Fig 4C), which indicates that some of the regulatory signaling (Chiang & Ferrell, 2020) was not completely blocked by loss of MAPL. We found an increase in several PPARα gene targets (Acox1, Hmgsc2, Hsb17b4, Acadl, Cd36) suggesting that the increased hepatic bile acid flux is supported by an increased peroxisomal gene transcription program. LXR target genes (Srebf1, Abca1, Abcg5, Abcg8) were unchanged in the liver of MAPL‐deficient mice, either in the fasting or fed state (Fig 4C). We found that the only FXR target gene affected was Baat, down 40% in the knockout mice, again consistent with an adaptation to downregulate bile acid production likely independent of analyzed nuclear receptor activities. An important negative regulator of Cyp7a1 (Goodwin et al, 2000; Lu et al, 2000) and FXR target Small Heterodimer partner (Shp) gene expression was unchanged. The main hepatocyte bile acid exporter (Abcb11/Bsep) and FXR target is not affected by the loss of MAPL at the mRNA and protein levels (Fig 4D). The enzyme responsible for the synthesis of muricholic acid, one of the most abundant bile acid species, CYP2C70 is also the target of Takeda‐G‐protein‐receptor‐5 (TGR5) signaling, a bile‐sensitive G‐Protein Coupled Receptor (Takahashi et al, 2016; Carino et al, 2021). Its expression is unchanged in MAPL‐deficient livers, revealing another dysregulated bile‐sensitive pathway (Fig 4E). We also measured the expression of hepatocyte basolateral bile transporters Slc10a1/Ntcp and Slco1b2/Oatp1b2, that were not changed while Slco1a1/Oatp1a1 was suppressed (Fig 4F).
In summary, while we observe significant increases in bile acids in liver and plasma, the hepatic sensing system for feedback regulation of the classical pathway remained curiously unaltered. Instead, the liver responded with some minor adaptation to limit bile synthesis (Cyp27a1 and Baat) or reabsorption (Slco1a1), and upregulation of detoxification pathways.
Gene expression response to MAPL loss converges on lipid metabolism, ER stress, and cell cycle dysregulation
To better understand the global effects on gene expression in livers lacking MAPL, we performed RNAseq analysis of liver from five mice of each sex and genotype aged 3 months, revealing highly significant changes in both male and female KO mice. Pathway analysis revealed significant changes in bile acid and lipid metabolism pathways of the liver, integrated stress response genes, and genes linked to proliferation (Fig 5A, Dataset EV1). While the overall patterns were similar between male and females in the heat maps, there were several sex‐specific changes, as well (Appendix Fig S1, Dataset EV1). Specifically, 1,063 genes were upregulated and 525 genes were downregulated in both groups, the latter including genetically targeted MAPL (Mul1), and 24 genes were changed in the opposite directions between sexes (Fig 5B). This is consistent with previous work highlighting sexually dimorphic gene expression in liver (Ahluwalia et al, 2004).
Figure 5. Gene expression response to MAPL loss converges on lipid metabolism, ER stress, and cell cycle dysregulation.
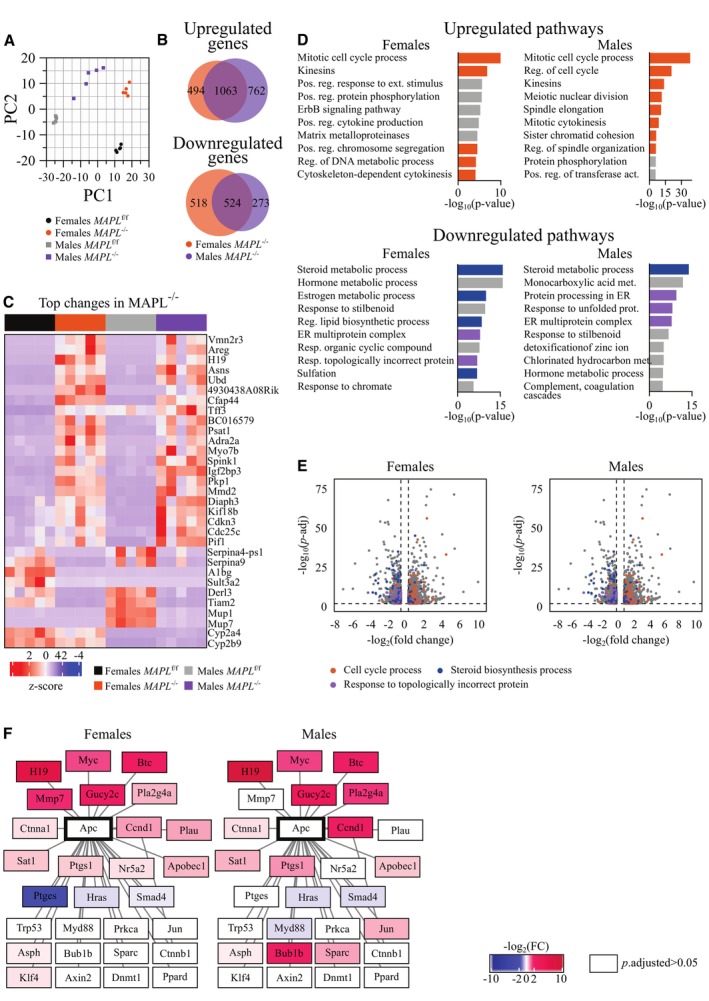
Liver RNAseq of MAPLf/f and MAPL−/− mice, n = 5 per group. See also Dataset EV1.
- PCA analysis of the dataset.
- Venn diagram of genes changed in the dataset (log2(FC) > 0.5, P‐adjusted < 0.05).
- Heatmap of top changes in the dataset (top15 changed (P‐adjusted < 0.05) genes were selected by |log2(FC)| in female and male groups, and non‐overlapping genes from the selection are plotted).
- Pathway analysis of the top500 up‐ or down‐regulated genes. Terms related to cell cycle process (GO:0022402) are marked with orange, to steroid metabolic process (GO:0008202) in dark blue, and to response to topologically incorrect protein (GO:0035966) in purple.
- Volcano plot of genes changed in the dataset (log2(FC) > 0.5, P‐adjusted < 0.05). Genes of the cell cycle process (GO:0022402) are marked with orange, of the steroid metabolic process (GO:0008202) in dark blue, and of the response to topologically incorrect protein (GO:0035966) in purple.
- Expression of APC first‐level interactors (Biogrid gene network database). Genes with P‐adjusted < 0.05 in MAPL−/− compared to MAPLf/f mice are highlighted with color, where intensity corresponds to |log2(FC)|, and blue and red spectrums indicate down‐ and up‐regulated genes, respectively.
Pathway analysis of the 500 most down‐ and up‐regulated genes in female and male MAPL−/− datasets converged on common terms linked to steroid and lipid biosynthesis, as well as sulfonation (Fig 5D and E), further highlighting the signature as a prominent consequence of MAPL loss. ER‐mediated response to unfolded proteins also emerged in the downregulated pathways, possibly indicating engagement of cellular stress responses (Fig 5D and E). Surprisingly, upregulated pathways converged on induced cell proliferation (Fig 5D and E). Exploration of top regulated genes and gene networks revealed a node related to the tumor suppressor gene APC, many members of which were upregulated, including several oncogenes (H19, Myc, Ccnd1, in males – Bub1b) (Fig 5F). Members and ligands of epidermal growth factor receptor signaling pathway also appeared in the list of top upregulated genes and APC node (Areg, Btc), although we did not observe an increase in phosphorylated EGFR by western blot (Appendix Fig S2).
As MAPL is a dual localized mitochondrial and peroxisomal protein, we also examined changes in gene expression profiles of the organelles. Out of 87 peroxisomal genes in our dataset, 18 were changed in females and 9 were changed in males, of which 7 were changed in both sexes (|log2(FC) > 0.5|, P.adj < 0.05). Of 1,070 mitochondrial genes in our dataset, 139 were changed in females and 114 were changed in males, of which 74 were changed in both sexes (|log2(FC) > 0.5|, P.adj < 0.05). Mitochondria harbor a genome that encodes 13 subunits of oxidative phosphorylation complexes, the majority of which also remained unchanged (Fig EV3). Our analysis of protein expression of core mitochondrial or peroxisomal proteins did not indicate any change in overall mass of each organelle in liver (Fig EV1), so the alterations in mRNA encoding ~10–20% of the mitochondrial or peroxisomal proteins would suggest some level of coordinated metabolic or stress responsiveness. The increase in mitochondrial chaperone and protease LONP1 would be consistent with that. However, these proteins did not conform to any specific functional cascade that we could identify (Figs EV1 and EV3).
Figure EV3. Organellar gene expression in MAPL‐deficient mice.
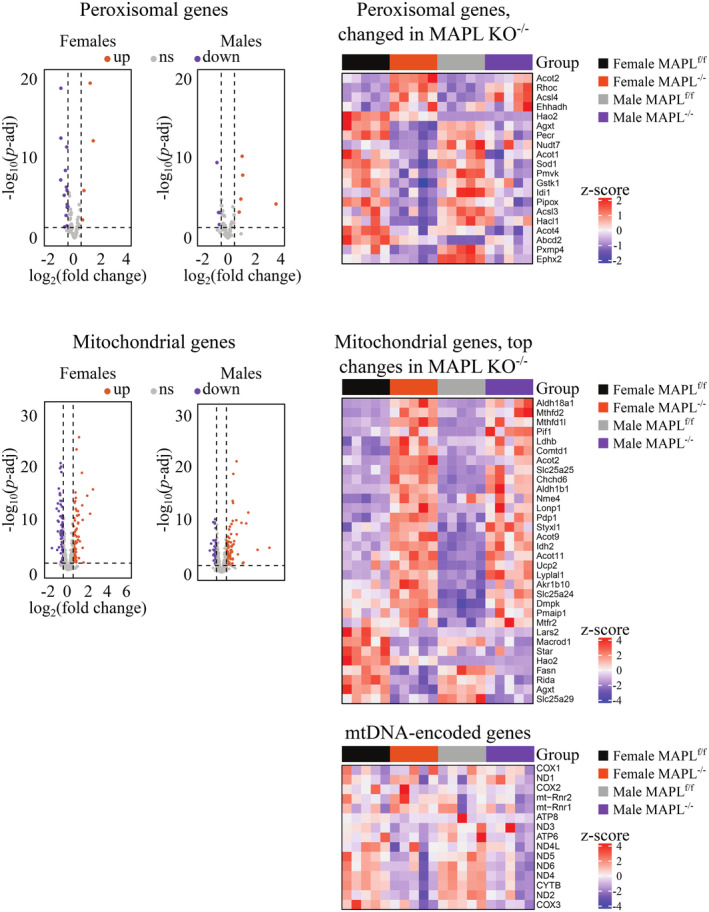
From liver RNAseq (N = 5 per sex and genotype). Volcano plots (left panels) and heat maps (right panels) of peroxisomal (top panels) and mitochondrial (bottom panels) gene expression in the liver of MAPL−/− mice.
Increased bile acid synthesis parallels increased FGF21 secretion that can be uncoupled from ER stress
Almost 90% of the cholesterol in the mouse liver is used to make bile, making this one of the most dominant biochemical processes in liver (Wanders, 2013), serving as the major excretory pathway for excess cholesterol. An accumulation of intracellular bile acids results in ER stress (Bochkis et al, 2008); however, later studies suggested that some of the C27 precursors of bile acids can be toxic, while C24 species protective against ER stress (Perez & Briz, 2009; Fuchs & Trauner, 2022). ER stress is communicated by phosphorylation‐activated ER‐localized PERK kinase onto its target translation initiation factor 2α (EIF2α) to activate the integrated stress response (ISR)—an evolutionarily conserved adaptive pathway responsive to major cell stresses (Costa‐Mattioli & Walter, 2020). The phosphorylation of EIF2α inhibits global cap‐dependent translation, while facilitating the translation of activating transcription factor 4 (ATF4) and other ISR effectors (CHOP, ATF3, ATF5), and consequent transcription of their targets. P‐PERK, p‐IEF2a, and CHOP protein levels were robustly upregulated in MAPL −/− , signifying an ER‐ISR stress response induction (Fig 6A and B, quantification Fig EV4A). RNAseq analysis of the mRNA for ATF3 and several ISR target genes showed significant upregulation, but ATF4 and ATF5 were less affected. Interestingly, the mRNA of the ATF4 target gene DDIT3 encoding CHOP was only mildly increased, relative to the robust upregulation seen at the protein level (Fig 6C). This is consistent with recent work demonstrating that chronic ISR models can lead to a C/EBP‐mediated downregulation of ATF4 (and targets) gene expression (Kaspar et al, 2021).
Figure 6. Loss of MAPL leads to hepatic ER stress, eIF2α activation and Fgf21 expression.

- Representative immunoblots from liver extracts from three animals of each genotype: PERK auto‐phosphorylation along with the total protein and BIP and CHOP expressions are shown (*: unspecific signal).
- Representative immunoblots of eIF2α phosphorylation in liver extracts (n = 3).
- mRNA expression by RNAseq of integrated stress response (ISR) genes in the liver of MAPL−/− mice.
- Fgf21 mRNA expression measured by qRT‐PCR performed on 3 different mouse tissues isolated from MAPLf/f and MAPL−/− males. N = 3 different mice per genotype. *P < 0.05, **P < 0.01, ***P < 0.001 in an unpaired two‐tailed t‐test.
- Representative immunoblots of increased FGF21 protein levels in liver from three pairs of mice from each strain.
- Serum FGF21 was quantified by ELISA (n = 5 for each genotype, 2‐month‐old males). **P < 0.01 in an unpaired two‐tailed t‐test.
- Representative immunoblots from different tissues whole‐cell extracts from one male of each genotype: PERK auto‐phosphorylation, as well as BIP and CHOP expression are shown (Sto: stomach, *: remaining beta‐actin signal).
- Serum FGF21 was quantified by ELISA from tail vein injected adenoviral rescued mice (n = 4 with two females and two males, n = 4 with two females and two males, n = 3 with two females and one male and n = 3 with one female and two males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old). *P < 0.05 in a one‐way ANOVA test.
- Liver Fgf21 gene expression by qRT‐PCR on livers of rescued mice (in triplicate, n = 7 with four females and three males, n = 7 with four females and three males, n = 5 with three females and two males and n = 6 with three females and three males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old). **P < 0.01 in a Kruskal‐Wallis test.
- Representative immunoblot from rescued liver extracts probed for Fgf21, CHOP, P‐eIF2α and eIF2α as indicated (*: unspecific signal).
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
Figure EV4. Loss of MAPL leads to hepatic ER stress, eIF2α activation and Fgf21 expression.

- Quantification of phospho‐PERK, CHOP and phospho‐eIF2α signals presented in Fig 6A and B immunoblots (n = 3 different mouse for each genotype). *P < 0.05, **P < 0.01 in an unpaired two‐tailed t‐test.
- Quantification of Fgf21 signals presented in Fig 6E (n = 3 for each genotype).
- Circulating and liver corticosterone levels were quantified as described in materials and methods (n = 8 for each genotpye, 2‐month‐old males). ***P < 0.001 in an unpaired two‐tailed t‐test.
- Quantification of FGF21, CHOP, phospho‐eIF2α and eIF2α signals presented in Fig 6J (n = 5, 5, 3 and 4 for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old). *P < 0.05, **P < 0.01 in a Kruskal‐Wallis test.
- Representative immunoblot of CHOP protein expression of livers isolated from MAPLf/f and MAPL−/− animals injected or not with the empty virus rtTA.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
However, other ATF4 targets including TRIB3 and FGF21 were robustly increased by RNAseq. Fgf21 gene expression was validated by qRT‐PCR showing a robust ~30‐fold increase (Fig 6D), with a corresponding increase at the protein level in MAPL −/− liver (Fig 6E, quantification Fig EV4B). Fgf21 mRNA was increased in other tissues 2–6‐fold (Fig 6D). Importantly, circulating levels of FGF21, quantified by ELISA showed a 12‐fold increase in MAPL −/− mice (Fig 6F). Circulating FGF21 binds to heterotrimeric surface receptors comprised of FGFR1c, FGFR2c or FGFR3c, in complex with the β‐Klotho receptors (Itoh, 2014; Owen et al, 2014). Initially thought to act primarily to signal the “browning” of white adipocytes, FGF21 binds receptors within the suprachiasmatic nucleus (SCN) of the hypothalamus and the dorsal vagal complex of the hindbrain (Bookout et al, 2013; Owen et al, 2014; Patel et al, 2015). Indeed, the lean phenotype resulting from FGF21 expression was shown to be independent of the uncoupler UCP1 that is central to the browning of white adipocytes (Veniant et al, 2015). FGF21 binding within the suprachiasmatic nucleus leads to dramatic alterations in circulating glucocorticoids, altering circadian rhythm, thirst, blood pressure, and whole‐body metabolism (Bookout et al, 2013; BonDurant & Potthoff, 2018; Pan et al, 2018; Song et al, 2018). Consistent with these findings, we also observe a ~4‐fold increase in circulating corticosterone levels and 10‐fold increase in liver of MAPL−/− mice (Fig EV4C). In addition, evidence in rodents has shown FGF21 as a negative regulator of bile acid synthesis (Chen et al, 2018), again consistent with FGF21 upregulation within MAPL−/− mice playing a potentially compensatory role to reduce bile acid synthesis.
Mitochondrial and peroxisomal anchored protein ligase has been implicated in the regulation of ER/mitochondrial contact sites, mitochondrial quality control and cell survival pathways (Prudent et al, 2015; Rojansky et al, 2016; Roque et al, 2023); therefore, it was possible that the ISR may be activated in other, non‐bile acid‐producing tissues as well. In addition to liver, we observed an increase in the phosphorylation of PERK in the stomach, but this was not accompanied by an increase in CHOP (Fig 6G). Moreover, there was no change in CHOP protein levels in muscle, brain, spleen or embryonic fibroblasts, consistent with a unique, liver‐specific role for MAPL in the regulation of bile acids that can underlie the generation of ER stress through the activation of ATF3 (Sharma et al, 2017).
To further examine the liver specificity of FGF21 secretion, we monitored FGF21 levels following tail vein rescue of MAPL expression in liver. The data showed a complete RING‐dependent restoration of both circulating and hepatic bile acids, and Fgf21 mRNA and protein levels. In this experiment the hepatic ER stress remained (Figs 6H–J and EV4E) since the tail‐vein injection of empty adenovirus (rtTA) induced ER stress, masking any rescue that may have resulted from MAPL expression (Fig EV4E). Instead, the experiment demonstrates an uncoupling of FGF21 expression from the stress response, that instead appears to parallel the levels of circulating bile acids.
MAPL −/− mice are lean and insulin‐sensitive
Elevated levels of circulating FGF21 result in a lean phenotype, involving both autocrine and paracrine signaling pathways between the liver, brain and adipocytes (Zhang et al, 2012; Flippo & Potthoff, 2021). Consistent with elevated FGF21 levels in circulation, mice lacking MAPL are of equal weight upon weaning, but rapidly reveal a lean phenotype, with an inability to gain weight on a high‐fat diet (males in Fig 7A and B, females in Fig EV5A and B). Examining tissues revealed a loss in white adipocyte mass due to decreased lipid content as the primary cause of leanness, where body length and other tissue mass were unaltered (Figs 7C–E and EV5C and D). We observed browning of white fat in ~30% of MAPL−/−mice, defined by expression of UCP1 in white fat by both qRT‐PCR and western blot analysis (Figs 7F and EV5E); however, this did not correlate with all of the mice that were lean. This is consistent with primary targets of FGF21 in the brain and other organs. To further interrogate the metabolic phenotype of MAPL−/− mice, we performed insulin and glucose tolerance tests to monitor glucose metabolism. Insulin administration led to an equivalent reduction in glycemia; however, MAPL−/− mice were slower to restore their glucose levels over the 120‐min time course (males in Fig 7G, females in Fig EV5F). Glucose tolerance tests revealed a more rapid glucose clearance in MAPL−/− compared to control mice, suggesting increased insulin sensitivity in MAPL‐deficient animals (males in Fig 7H, females in Fig EV5G). Indeed, an analysis of circulating insulin revealed a ~3‐fold reduction in insulinemia in MAPL−/− compared to control mice (Fig 7I). In addition, glycogen storage in MAPL‐deficient livers was reduced by ~50% as compared to control livers in ad libitum fed animals (Fig EV5H). Importantly, reconstitution of MAPL expression in liver increased insulinemia, suggesting that the changes in insulin levels stem from the liver rather than effects in muscle, pancreas or the periphery (Fig 7J). Overall, these data show that MAPL−/− mice are profoundly insulin‐sensitive.
Figure 7. MAPL −/− mice are lean and have increased glucose tolerance.

- Body weight (g) of male mice fed with normal chow; MAPLf/f (n = 6), and MAPL−/− (n = 9).
- Body weight (g) of MAPLf/f (n = 8) and MAPL−/− (n = 7) male mice. They were fed normal chow for 5 months (dotted lines), or for 3 months followed by 2 months of a 60% fat diet (solid lines, diet change indicated by HFD arrow).
- Body weight (g, left panel) or length (cm, right panel) of 7‐month‐old MAPLf/f (n = 4 for both parameter) or MAPL−/− (n = 7 for body weight and n = 4 for length) male mice. ****P < 0.0001 in an unpaired two‐tailed t‐test.
- Wet weight of organs including liver, kidney, gastrocnemius muscle, epididymal white fat (WAT) and interscapular brown fat (BAT) isolated from 7‐month‐old male MAPLf/f (n = 4) or MAPL−/− (n = 7) mice. **P < 0.01 in an unpaired two‐tailed t‐test.
- Representative pictures of white adipocytes from MAPLf/f and MAPL−/− mice. Hematoxylin and eosin staining, 40× objective. Scale bar: 100 μm.
- Representative immunoblots of Ucp1 from WAT and BAT whole‐cell extracts from three different pairs of mice (n = 3).
- Insulin tolerance test in male MAPLf/f (n = 7) and MAPL−/− (n = 7) mice. Insulin (0.5 U/kg) was injected intraperitoneally following a 4 h fast and blood glucose was measured at indicated times. Repeated Measures 2way ANOVA analysis showed a statistically significant difference between genotypes (P = 0.005).
- Glucose tolerance test in male MAPLf/f (n = 8) and MAPL−/− (n = 8) mice. Glucose (2 g/kg) was injected intraperitoneally following an overnight fast and blood glucose was measured at indicated times. Repeated Measures 2way ANOVA analysis showed a statistically significant difference between genotypes (P = 0.0043).
- Insulinemia (ng/ml) measured by ELISA in MAPLf/f (n = 4 females and n = 4 males) and MAPL−/− (n = 4 females and n = 4 males) mice. **P < 0.01 in an unpaired two‐tailed t‐test.
- Insulinemia (ng/ml) measured by ELISA from tail vein injected adenoviral rescued mice (n = 6 with three females and three males, n = 7 with four females and three males, n = 5 with three females and two males and n = 6 with three females and three males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old).
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
Figure EV5. MAPL −/− female mice are lean, resistant to weight gain and develop spontaneous hepatocellular carcinoma.

- Female mice fed with normal chow were weighed weekly; MAPLf/f (n = 7) and MAPL−/− (n = 10).
- MAPLf/f and MAPL−/− (n = 8 for each genotype) females were fed with normal chow for 5 months (dotted lines), or for 3 months followed by 2 months of 60% fat chow (solid lines, diet change indicated by HFD arrow).
- Body weight (g, left panel) and length (cm, right panel) of 7‐month‐old MAPLf/f (n = 4 for body weight and n = 3 for length) and MAPL−/− (n = 2) female mice. **P < 0.01 in an unpaired two‐tailed t‐test.
- Wet weight of organs including liver, kidney, gastrocnemius muscle, epididymal white fat (WAT) and interscapular brown fat (BAT) isolated from 7‐month‐old MAPLf/f (n = 4) or MAPL−/− (n = 2) female mice.
- White adipose tissue gene expression performed by qRT‐PCR. N = 3, in triplicate, 2‐month‐old MAPLfl/fl and MAPL−/− male mice.
- Insulin tolerance test in MAPLf/f (n = 8), MAPL−/− (n = 8) female mice. Insulin (0.5 U/kg) was injected intraperitoneally after a 2 h fast and blood glucose was measured at indicated times. Repeated Measures 2way ANOVA analysis for a difference between genotypes P = 0.0135.
- Glucose tolerance test in MAPLf/f (n = 8), MAPL−/− (n = 8) female mice. Glucose (2 g/kg) was injected intraperitoneally after an overnight fast and blood glucose was measured at indicated times. Repeated Measures 2way ANOVA for a difference between genotypes P = 0.0903.
- Liver glycogen measured enzymatically in male MAPL f/f (n = 3), MAPL −/− (n = 3) mice. **P < 0.01 in an unpaired two‐tailed t‐test.
- Survival curve of MAPLf/f (n = 11) and MAPL−/− (n = 10) female mice.
- Cancer‐free survival curve of MAPLf/f (n = 11) and MAPL−/− (n = 10) female mice.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
MAPL −/− mice have increased cell proliferation
The loss of MAPL led to the chronic elevation of bile acids and FGF21, accompanied by a lean and highly insulin‐sensitive phenotype (Figs 2, 6 and 7). High levels of bile acids have been shown to promote cell proliferation and stem cell activation through multiple mechanisms (Sorrentino et al, 2020; Fuchs & Trauner, 2022), and the RNAseq revealed clear signatures of proliferation (Fig 5D–F). Indeed, histology revealed a ~5‐ and ~15‐fold increase in hepatocyte proliferation in 2‐ and 7‐month‐old MAPL−/− relative to control mice, respectively, as evidenced by Ki67 staining (Fig 8A). There was no obvious sign of steatosis or immune infiltration within MAPL−/− livers, commonly linked to liver dysfunction. One of the most upregulated genes was the long noncoding RNA H19 (Fig 5C), which is strongly linked to liver dysfunction and hepatocellular carcinoma (Fuchs & Trauner, 2022; Rojas et al, 2022). This was highly expressed as early as 2 months, and rescue of MAPL expression completely reversed H19 expression levels in a RING‐dependent manner (Fig 8B).
Figure 8. MAPL −/− mice present increased cell proliferation in liver and gut.

- Representative pictures of Ki67 liver staining (top panel) and its quantification (lower panel) in MAPLf/f and MAPL−/− mice (n = 6 males, 2‐ to 6‐month‐old animals). 20× objective. Scale bar: 200 μm. ***P < 0.001 in an unpaired two‐tailed t‐test.
- qRT‐PCR investigating H19 mRNA levels in livers of rescued mice (in triplicate, n = 7 with four females and three males, n = 7 with four females and three males, n = 6 with four females and two males and n = 6 with three females and three males, for MAPLf/f + rtTA, MAPL−/− + rtTA, MAPL−/− + MAPL‐Flag and MAPL−/− + MAPL‐ΔRING‐Flag, respectively, 2–3 months old). *P < 0.05 in a Kruskal‐Wallis test.
- Representative pictures of Ki67 duodenum staining (top panel) and its quantification of crypt length from duodenum, jejunum and ileum (lower panel) in MAPLf/f and MAPL−/− mice (n = 2 females n = 1–2 males, 2‐ to 6‐month‐old animals). 20× objective. Scale bar: 200 μm. *P < 0.05, ***P < 0.001 in an unpaired two‐tailed t‐test.
- Representative results (left panel) of [18F]FDG micro‐PET analysis performed on 6‐month‐old males fed with normal chow as described in material and methods (n = 4 for each genotype). Standard uptake value is the ratio of the tissue radioactivity concentration and the injected radioactivity, divided by the body weight. LV: left ventricle, SM: skeletal muscle (right panel). *P < 0.05 in an unpaired two‐tailed t‐test.
Data are represented as mean ± SEM. Squares represent males whereas circles represent females.
Source data are available online for this figure.
Considering the potential effects of bile acids on cell proliferation (Fuchs & Trauner, 2022), the next highest concentration of bile may then be within the duodenum, where bile empties from the gallbladder. This site has previously been shown to activate proliferation with high levels of bile acids (Sorrentino et al, 2020). Ki67 staining revealed that MAPL−/− mice have increased crypt length and proliferation (Fig 8C). This phenotype lessens as we examined sections from the jejunum and ileum, further consistent with proliferation in duodenum linking to bile concentrations. To extend our analysis in a more global manner to monitor potential changes in proliferation throughout the body of MAPL−/− mice, we turned to whole animal positron emission tomography to quantify FDG‐glucose uptake as a proxy for increased proliferation (Weber et al, 2000). The imaging revealed a striking increase in glucose uptake within the intestines, consistent with the proliferation observed by increased Ki67 staining (Fig 8C). Quantification of the radioactive signals within different organs confirmed a ~3‐fold increase in glucose uptake in the gut, and a 2–3‐fold increase in glucose uptake in the liver, whereas the left ventricle, brain and skeletal muscle were unchanged (Fig 8D). The tissue specificity of these changes suggests the loss of MAPL alone does not drive proliferation, rather it is context‐specific, likely linked to specific metabolic changes including bile metabolism.
MAPL −/− mice develop spontaneous hepatocellular carcinoma
To determine the longer‐term consequences of increased proliferation observed at 2–5 months, we aged these animals up to 30 months. While survival was only slightly reduced in aged MAPL−/− mice relative to wild‐type littermates (males shown Fig 9A, females in Fig EV5I), 87 and 89% (n = 8 and 9) of the MAPL−/− males and females, respectively, presented with liver cancer between 14 and 28 months (Figs 9B and EV5J). Histological analysis from 17 tumors in different mice (both males and females) stained with H&E and reticulin confirmed all had hepatocellular carcinoma; however, some of the tumors had mixed pathology (Fig 9C). We did not observe any tumor formation within the gut, suggesting the metabolic and proliferative changes there were not sufficient to transform enterocytes. In contrast, no littermate controls within this cohort developed any malignancies. The generation of HCC may have been predicted from the RNAseq analysis, which showed high expression of established drivers of HCC including the long noncoding RNA H19, and others (Fig 5F).
Figure 9. MAPL has tumor‐suppressive capability.

- Survival curve of MAPLf/f (n = 8) and MAPL−/− (n = 9) males.
- Cancer‐free survival curve of MAPLf/f (n = 8) and MAPL−/− N (n = 9) males.
- Pathological analysis of liver tumors in MAPLf/f and MAPL−/− mice.
- Survival of Huh7 cells infected with adenoviruses expressing rTta, MAPL‐flag, or MAPL‐ΔRING‐flag constructs. N = 2 independent experiments with three biological replicates each.
- Immunoblots of cleaved caspase 3 and 7 from Huh7 cells infected with adenoviruses expressing rTta, MAPL‐flag, or MAPL‐ΔRING‐flag constructs.
- Ras foci in Rat2 fibroblast cells transfected with empty pcDNA3 or overexpressing MAPL plasmids (left panel). N = 2 independent experiments with three biological replicates each. Immunoblots of PARP, MAPL and beta‐actin in Rat2 fibroblasts transfected with empty pcDNA3 or overexpressing MAPL plasmids (left panel). Immunoblots of PARP, MAPL and beta‐actin in Rat2 fibroblasts treated with DMSO or Staurosporine (STS, 1 μM, 3 h).
- Survival of MAPLf/f and MAPL−/− primary hepatocytes infected with adenoviruses expressing rTta or truncated BID (tBID) (left panel). Survival of MAPLf/f and MAPL−/− primary hepatocytes treated with camptothecin (1 μM, 15 h) or vehicle.
Data are represented as mean ± SEM.
Source data are available online for this figure.
These findings implicate MAPL within a pathway where it exerts a tumor‐suppressive role in the liver. Tumor suppressors are also defined by their ability to drive or promote cell death upon overexpression. Indeed, we previously described critical roles for MAPL and the SUMOylation of DRP1 in the process of apoptosis (Prudent et al, 2015), suggesting that some of the cancer phenotype may arise due to an inhibition of cell death. More recent studies utilized this MAPL KO mouse strain and demonstrated that loss of MAPL within parvalbumin interneurons protected against cell death and memory loss linked to postnatal anesthesia exposure (Roque et al, 2023). To test whether ectopic expression of MAPL may promote cell death directly, we infected a human liver cell line Huh7 with adenovirus coding for MAPL‐Flag, MAPL‐ΔRING‐Flag SUMOylation‐deficient mutant or the empty vector (Ad‐rtTA). Expression of wild‐type but not SUMOylation‐deficient mutant MAPL led to cell death showing ~4% of cells remaining after 24 h (Fig 9D). This was accompanied by the cleavage of caspases 3 and 7, indicating that forced expression of MAPL induced cell death (Fig 9E). Moreover, expression of SUMO competent MAPL in Rat2 fibroblasts stably expressing oncogenic H‐RasV12 also led to an arrest in anchorage‐independent cell growth and cell death initiation, as seen with PARP cleavage (Fig 9F). Therefore, ectopic expression of MAPL promotes cell death and reduces neoplastic growth in vitro.
Lastly, we examined the capacity of MAPL−/− primary hepatocytes to resist cell death induced in two distinct ways. Infection of primary hepatocytes with truncated Bid (tBID) revealed that cells lacking MAPL were more sensitive to this direct induction of death. However, induction of DNA damage upon incubation with the DNA intercalating agent camptothecin showed complete resistance to cell death compared to littermate floxed hepatocytes (Fig 9G). Therefore, while MAPL is not essential for the steps driving apoptosis downstream of activated BAX, loss of MAPL is highly protective against genotoxic stress. These data show that expression of MAPL is a driver of cell death, and loss of MAPL offers significant resistant to cell death, consistent with its liver tumor suppressor activity in vivo.
Discussion
This study provides the first evidence for a core function of MAPL as a key regulator of bile acid metabolism. Mice lacking MAPL present a series of consequential phenotypes from FGF21 production and insulin sensitivity to the generation of hepatocellular carcinoma. While the direct substrates of MAPL as a SUMO and Ubiquitin ligase remain incompletely characterized, we highlight one target, the peroxisomal bile acid transporter ABCD3 as a SUMO substrate where the data suggest that its conjugation acts to gate and repress the transporter. Loss of MAPL led to an uncoupling of bile acid sensing pathways where FGF15 secretion from ileum, and FXR pathways in liver, do not respond to the chronically elevated levels of bile acid. These canonical feedback pathways would normally sense the increase in bile acids and downregulate the key biosynthesis enzyme CYP7A1 in liver. There were instead compensatory changes to mitigate the high levels of bile acids, including the downregulation of Cyp27A1 and Baat to the upregulation of detoxification pathways in the expression of Cyp4A14. The regulation of CYP27A1 expression is tissue‐specific and broad, with links to bile acid feedback, PPAR agonists, insulin signaling, growth hormones and glucocorticoids (Lorbek et al, 2012), most of which are systemically altered in our MAPL−/− model. Ultimately, it remains unclear how MAPL loss could uncouple the canonical CYP7A1 feedback pathways. Possibly, the loss of MAPL in enterocyte peroxisome might also affect these pathways. A challenge of chronic knockout models is the capacity for compensation, which may explain such desensitization, making it complicated to define the direct mechanisms, substrates and signaling cascades responsible. For now, this new mouse model offers an opportunity to study these pathways in future inducible, and tissue‐specific approaches to map the role of MAPL in bile acid circuitry.
Within our current model, tail vein injection of adenoviral‐expressed MAPL in liver allowed us to validate liver autonomous consequences of MAPL loss within a whole‐body knockout and confirm the dependence of these phenotypes on the ligase activity of MAPL. These experiments demonstrated that the elevated bile levels in serum were fully restored, indicating that any changes in reabsorption in the gut were secondary to the global bile acid levels seen in MAPL−/− mice. Moreover, cultured primary hepatocytes lacking MAPL generated ~3‐fold more bile, which returned to control levels upon rescue in a RING‐dependent manner. The restoration of FGF21 and circulating insulin levels upon tail vein rescue strongly suggest that primary changes in liver were the drivers of systemic metabolic changes in insulin sensitivity and the ultimate presentation with a lean phenotype. In addition to liver, considerable changes were observed within the gut with an increased absorption of bile, indicated by a 50% reduction in feces, and the remarkable increase in proliferation observed by FGD‐glucose imaging and histology. We hypothesize that this proliferative increase is linked to the elevated bile exiting the gallbladder and activating bile‐sensitive TGR5 receptors or other bile targets within the duodenum (de Boer et al, 2018). Had this been a cell autonomous response to the loss of MAPL within enterocytes we would predict similar proliferation throughout the gut, not limited to the duodenum.
In addition to the changes in bile acid synthesis, the loss of MAPL resulted in a liver‐specific activation of the ISR. Given the tissue specificity of increased CHOP expression, phosphorylation of PERK and eIF2a, this is not due to core requirements of MAPL to maintain homeostasis in all cell types. Transcriptionally, RNAseq data suggest milder changes in expression of ATF genes and targets, consistent with an activation of CHOP binding to C/EBPβ to repress the ISR in this chronic model, as highlighted recently (de Boer et al, 2018). Unfortunately, our attempts to rescue ISR upon tail vein injection of adenovirus expressing MAPL were confounded by the fact that injection of the empty virus induced CHOP expression and ISR. This makes it unclear exactly how MAPL loss may result in activation of the ISR, although PERK activation suggests an induction due to ER stress. On the other hand, this experiment did demonstrate the full reduction of an ATF4 target gene, FGF21. Our data do not yet distinguish whether FGF21 expression resulted directly from the increase in bile acids through FXR (Cyphert et al, 2012), PPARalpha (Inagaki et al, 2007; Badman et al, 2009) or TGR5 (Donepudi et al, 2017) signaling, but our gene expression analysis points toward an increased PPARalpha activation. It also may relate to an unknown functional target of MAPL in the liver.
Lastly, MAPL−/− mice develop spontaneous hepatocellular carcinoma after 15–20 months. This is consistent with a chronic, long‐term proliferative stress on liver as seen as early as 2 months with Ki67 staining and reflected in the gene expression dataset we generated. The cancers are specific to liver, even though we see significant increases in proliferation in the gut at early ages. The generation of tumors requires proliferation, but additionally inhibition of cell death pathways. Our previous studies investigating substrates and functions of MAPL SUMO E3 ligase activity have shown that SUMOylation of the fission GTPase Drp1 stabilizes the oligomeric state and mitochondrial constriction at ER contact sites, structures that promote calcium flux, cytochrome c release and cell death (Donepudi et al, 2017). Our BioID analysis in this study identified a series of mitochondrial fission proteins as partners of MAPL, including DRP1, MFF, INF2 and AKAP1. Importantly, MAPL is not required for steady‐state mitochondrial division; rather, it is activated upon cell death triggers to SUMOylate DRP1. This explains why the global loss of MAPL did not result in profound mitochondrial dysfunction or morphological aberrations. We demonstrated here that overexpression of MAPL led to very efficient cell death, and loss of MAPL was fully protective against genotoxic stress‐induced cell death. The MUL1 gene lies on chromosome 1p36, which is a very commonly deleted region in human cancer, although a region carrying many genes. The NCI cancer genome atlas reports the most common cancer with loss of MUL1 is cholangiocarcinoma, but patient databases do not identify MAPL as a bona fide tumor suppressor. There are many other substrates and functions of MAPL to be mapped, particularly during stress responses linked to inflammation and survival. Our future work will continue to investigate how the tumor‐suppressive activity of MAPL functions in the regulation of global metabolism in liver, and the potential relevance of MAPL function within human cancers. For now, the MAPL−/− mice provide a new model to better understand the complex signaling pathways in different tissues and under a variety of stimuli.
Materials and Methods
Ethics approval
Animal experimentation was conducted in accordance with the guidelines of the Canadian Council for Animal Care, with protocols approved by the Animal Care Committees of the University of Ottawa and of McGill University.
Generation of floxed MAPL KO mice
The targeting vector and the MAPL WT/flox mice were generated by Ozgene (Australia). The construct contained two loxP sequences inserted in intron 1 and intron 2, excising exon 2, of the MAPL gene, and two FRT sites flanking the neomycin resistance selection cassette. The construct was electroporated into a C57BL/6 ES cell line. Homologous recombinant ES cell clones were identified by Southern hybridization and injected into BALB/cJ blastocysts. Male chimeric mice were obtained and crossed to C57BL/6J females to establish heterozygous germline offspring on pure C57BL/6 background. To remove the Neo‐cassette (neo), the MAPL WT/flox mice were bred with homozygous FlpE‐“deletor” C57BL/6 mice (Ozgene).
To generate MAPL −/− mice, MAPL f/f mice were first bred with CMV‐Cre carrying out mice (The Jackson Laboratory). The resulting MAPL +/−:Cre were then bred with MAPL f/f animals. One‐quarter of the offspring were MAPL f/− . These heterozygous mice, devoid of the CMV‐Cre gene, were used as breeders: their offsprings were composed of 25% MAPL −/− animals, 25% of MAPL f/f animals used as littermate wild‐type controls and 50% of MAPL f/− animals used as littermate heterozygous controls.
Genotyping was performed by PCR of tail DNA (extracted using the DNA Blood & Tissue kit, Qiagen, according to the manufacturer's instruction) using two different primer pairs (Primer1: Fwd: 5′‐GGGAAGTGTGTGCCTTATG Rev: 5′‐AATCCCAAGTCCACAGTGC and Primer2: Fwd: 5′‐CCTCAGAGTTCATTTATCC Rev: 5′‐CCAACACCATCAAAAGGC).
RNAseq analysis confirmed the loss of exon2 within remaining MAPL mRNA.
Mice were fed ad liditum either normal chow or a high‐fat diet (60% fat, 20% proteins, 20% carbohydrate, research diets, for 10 weeks, starting at 12 weeks old). The food intake and body weight of each mouse were recorded weekly. For fasting and refeeding cohorts, mice were starved overnight, refed for 2 h, then fasted again for 4 h. Livers were collected after the 2 h feeding or the 4 h fast.
Metabolic tests
Glucose tolerance tests (GTT) were performed after an overnight (16 h fast). Blood glucose and plasma insulin levels were measured after intra‐peritoneal injection of glucose (2 g/kg of body weight). Insulin tolerance tests (ITT) were performed after intra‐peritoneal injection of human insulin (0.5 U/kg) in 2‐h‐fasted mice.
Primary hepatocytes isolation, culture and bile production
Primary hepatocytes were isolated from 12‐ to 16‐week‐old mice by 2‐step liberase perfusion (Liberase TL; MilliporeSigma #05401020001) and 50% Percoll gradient purification (MilliporeSigma #P1644). 400,000 cells were plated on collagen‐coated plates and cultured in Dulbecco's modified Eagle's medium supplemented with 0.2% bovine serum albumin (fatty acid free; Fisher Scientific), 25 mM glucose, 2 mM sodium pyruvate, 0.1 mM dexamethasone, 1% penicillin/streptomycin and 1 nM insulin for up to 48 h. To measure bile production, conditioned media was harvested after 48 h. Total bile acid concentration released was measured by enzymatic reaction (#MAK309 MilliporeSigma) and normalized to protein content per well.
Adenovirus tail vein injection
2 × 109 PFU/mouse (MAPL‐Flag) or 0.67 × 109 PFU/mouse (MAPL‐ΔRING‐Flag and rtTA) of adenoviruses was injected through the tail vein in a 100 μl final volume of sterile saline solution to 2‐ to 3‐month‐old animals. At day 7 post‐injection, mice were starved overnight and fed back for 3 h (from 8 to 11 AM). Blood was then collected by cardiac puncture and livers were collected for further investigations.
Cesium chloride‐purified adenoviruses expressing MAPL‐ΔRING‐Flag and rtTA were purchased from Vector Biosystems.
Electrophoresis and immunoblot analysis
Tissues were homogenized in ice‐cold lysis buffer (40 mM NaCl, 2 mM EDTA, 1 mM orthovanadate, 50 mM NaF, 10 mM pyrophosphate, 10 mM glycerolphosphate, 20 mM NEM, 1% Triton X‐100, 50 mM Hepes, pH 7.4) supplemented with Complete protease inhibitor cocktail (Roche Molecular Biochemicals) in a borosilicate glass Dounce tissue grinder with tight pestle. After 20 min at 4°C, homogenates were centrifuged at 20,000 g for 20 min at 4°C, and the supernatants were collected. Protein extracts (20 μg) were separated on a Tris‐Glycine 4–20% gradient precast polyacrylamide gel (Invitrogen) and transferred to 0.22 μm pore nitrocellulose membrane (Bio‐Rad). Bands were visualized with Western‐Lightning PLUS‐ECL (Perkin‐Elmer) with an INTAS ChemoCam (INTAS Science Imaging GmbH) and quantified with ImageJ software.
Mitochondrial and peroxisomal anchored protein ligase was detected by rabbit polyclonal antibodies (HPA017681, 1:1,000, Sigma), PERK by rabbit polyclonal antibodies (100‐401‐962, 1:1,000, Rockland antibodies & assays), phospho‐PERK by rabbit monoclonal antibodies (3179, 1:1,000, Cell Signaling), eIF2α by mouse monoclonal antibodies (2103, 1:500, Cell Signaling), phospho‐eIF2α by rabbit polyclonal antibodies (SAB4504388, 1:500, Sigma), Fgf21 by goat polyclonal antibodies (AF3057, 1:500, R&D systems), CHOP by rabbit polyclonal antibodies (5554, 1:500, Cell Signaling), BiP by rabbit polyclonal antibodies (ADI‐SPA‐826‐D, 1:1,000, Enzo), CYP3A11 by rabbit polyclonal antibodies (13384, 1:500, Cell Signaling), CYP4A14 by goat polyclonal antibodies (sc‐46087, 1:500, Santa Cruz), CYP7A1 by rabbit polyclonal antibodies (ab65596, 1:500, Abcam), CYP27A1 by rabbit polyclonal antibodies (NBP2‐16061, 1:500, Novus Biologicals), SUMO1 by mouse monoclonal antibodies (332400, 1:1,000, Invitrogen), Hsp60 by mouse monoclonal antibodies (sc‐136291, 1:1,000, Santa Cruz), Hsp70 by rabbit polyclonal antibodies (ab137680, 1:1,000, Abcam), ABCD3 by mouse monoclonal antibodies (sab4200181, 1:1,000, Sigma), DRP1 by mouse monoclonal antibodies (611113, 1:1,000, BD Transduction Labs), Mfn2 by rabbit polyclonal antibodies (M6319, 1:1,000, Sigma), UCP1 by a polyclonal antibody (U6382, 1:500, Sigma), ACOX1 by a polyclonal antibody (10957‐1‐AP, 1:1,000, Proteintech), SCP2 by a polyclonal antibody (14377‐1‐AP, 1:1,000, Proteintech), PEX14 by a polyclonal antibody (ABC142, 1:1,000, Millipore), vinculin by a monoclonal antibody (V4505, 1:1,000, Sigma), β‐actin by rabbit polyclonal antibodies (SAB4502543, 1:1,000, Sigma) AIF by mouse monoclonal antibody (sc13116 1:1,000, SantaCruz), ABCB11 by polyclonal antibodies (PA527742 1:1,000, Thermo Fisher), EGFR by rabbit polyclonal antibodies (A00023 1:1,000, Boster), phospho‐EGFR by rabbit polyclonal antibodies (369700 1:1,000, Invitrogen) and β‐actin by mouse monoclonal antibodies (A2228, 1:1,000, Sigma).
Cellular fractionation
Liver was collected into ice‐cold PBS and rinsed free of blood. It was minced into small pieces and homogenized using a Dounce homogenizer (3–4 times, 1,600 rpm) into IB isolation buffer (mannitol 200 mM, sucrose 68 mM, Hepes 20 mM pH 7.4, KCl 80 mM, EGTA 0.5 mM, Mg(Acetate)2 2 mM, 2‐chloroacetamide 20 mM and protease inhibitors 1×). Homogenate was centrifuged at 800 g for 10 min to separate nuclear pellet from post‐nuclear supernatant. The nuclear pellet was resuspended into 2 ml of IB buffer and centrifuged once again at 800 g for 10 min. Pellet was resuspended into 200 μl IB and kept as nuclear fraction. The post‐nuclear supernatant was centrifuged at 1,000 g for 10 min. The supernatant was kept and centrifuged at 10,000 g for 20 min to separate mitochondrial heavy membrane pellet and post‐mitochondrial supernatant. Mitochondrial pellet was washed in 1 ml IB and centrifuged at 10,000 g for 10 min. The final mitochondrial pellet was resuspended into 50 μl IB and kept as mitochondrial fraction. The post‐mitochondrial supernatant was centrifuged at 200,000 g in TLA‐110 rotor (Beckman‐Coulter) for 40 min. Supernatant was kept as cytosolic fraction.
Immunoprecipitation
For the MAPL immunoprecipitation, livers from 4‐month‐old males were washed in ice‐cold PBS and homogenized in 5 ml of lysis buffer (50 mM Tris, 150 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 1% triton X‐100, 20 mM NEM, pH 7.5) supplemented with Complete protease inhibitor cocktail (Roche Molecular Biochemicals) in a borosilicate glass Dounce tissue grinder with tight pestle. After 20 min at 4°C rocking, homogenates were centrifuged at 20,000 g for 20 min at 4°C and supernatants were collected. One milligram of proteins (diluted at 2 mg/ml in lysis buffer) was pre‐cleared overnight at 4°C, rocking with 100 μl of Dynabeads protein A beads (Life Technologies). One hundred microliter of Dynabeads protein A beads (resuspended in 200 μl of 0.1 M NaP, 0.08% Tween 20) per condition were incubated overnight with 5 μg of antibodies. The next day, antibodies were covalently bound to the beads using DMP crosslinker (Pierce) 20 mM, for 30 min at RT, rocking in the dark and crosslink reaction was stopped by 50 mM Tris pH 7.5 for 15 min at RT, rocking. Beads were washed two times with 100 μl of 0.1 M glycine pH 2.5. Precleared homogenates (1:20 was saved as starting material, SM) were applied on the antibodies‐bound beads overnight at 4°C, rocking. The next day, homogenates were removed from the beads and beads were washed two times with lysis buffer, two times with high salt buffer (50 mM Tris, 450 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 0.05% triton X‐100, 20 mM NEM, pH 7.5) and two times with low salt buffer (50 mM Tris, 150 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 0.05% triton X‐100, 20 mM NEM, pH 7.5). Proteins were eluted with 50 μl of 0.1 M glycine, pH 2.5, 0.5% Triton X‐100 (two times). Twenty microliter of Tris 1 M pH 7.5 were added to the elution.
SIM‐beads extracts from livers
Mouse livers were washed in ice‐cold PBS and resuspended in cell fractionation buffer. Cells were then broken with a cell cracker (EMBL‐Heidelberg) using ball size 8.002. The samples were centrifuged at 800 g at 4°C for 10 min. Supernatants were re‐cleared at 800 g for 5 min at 4°C. Post‐nuclear supernatants were then centrifuged at 9,000 g for 20 min at 4°C, in order to pellet heavy membrane fraction (supernatant is the cytosolic fraction). The pellets were washed and re‐centrifuged for a further 10 min at 9,000 g. To heavy membrane and cytosolic fractions, Triton X‐100 was added up to 1% concentration, followed by incubation for 20 min at 4°C, rocking. The fraction lysates were then centrifuged for 45 min at 200,000 g at 4°C. Supernatants were then collected and protein concentration determined (80 μg total lysate fraction of each type were separated to run in gel as starting material). Then, 40 μl of SIM beads (AM‐200, Boston Biochemicals) was added to each type of total lysate fractions (containing 0.5–1 mg of proteins) and incubated for at least 1.5 h at 4°C, rocking, followed by centrifugation at 14,000 g for 2 min at 4°C. Beads were then washed five times with cell fractionation buffer containing 1% Triton X‐100. After the last wash was discarded, 1× Laemmli sample buffer was added to the solid beads and run in acrylamide gel, together with the starting material.
BirA and Flag‐MAPL‐BirA stable cells
HEK 293T‐REX cells stably expressing either BirA or Flag‐MAPL‐BirA were maintained in DMEM (Wisent) containing 10% FBS (Wisent), 2 mM L‐glutamine, non‐essential amino acids and 1 mM sodium pyruvate (Life Technologies). The expression of BirA and Flag‐MAPL BirA was induced with 1 μg/ml tetracycline (Sigma) for 24 h in culture at 37°C, accompanied by 50 μM biotin (Sigma) final concentration. After the incubation, the cells on plates were washed three times in PBS, then they were scrapped on ice and lysed for 20 min in buffer: 10 mM Hepes pH 7.4, 200 mM NaCL, 0.5 mM EDTA, 2 mM MgCl2, 1% Triton‐X100. Lysates were centrifuged (20,000 g at 4°C for 15 min), normalized for protein concentration and incubated with streptavidin‐agarose beads (Life Technologies) for 1.5 h rocking at 4°C. Beads were centrifuged, washed three times with lysis buffer, mixed with 1 × Laemmli buffer, and loaded onto a gel, along with starting material. Then electrophoresis was performed, and proteins were transferred to nitrocellulose membranes. Expression of Flag‐BirA‐MAPL, as well other candidate proteins for interacting with MAPL, was determined by western blot with a series of antibodies.
Sucrose gradient
Livers from 2‐month‐old males were washed in ice‐cold PBS and homogenized in 3 ml of homogenization buffer (50 mM Tris, 150 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 20 mM NEM) supplemented with Complete protease inhibitor cocktail (Roche Molecular Biochemicals). A fraction of the homogenates was sonicated in order to quantify protein content. Homogenates were diluted in homogenization buffer to 4.7 mg/ml. Five hundred microliter of homogenate at 4.7 mg/ml were added to 500 μl of homogenization buffer containing 1% DDM. After 20 min at 4°C rocking, homogenates were centrifuged 20 min at 4°C at 21,000 g. Supernatants were collected, and protein content determined. Two hundred fifty microgram of proteins were loaded on the top of the 10–50% sucrose linear gradient (1,800 μl, 200 μl of each) and centrifuged for 4 h at 4°C, at 180,000 g. Twelve fractions were collected and 75 μl of the different fractions were loaded on a 10% acrylamide gel.
Blood analysis
Blood samples from MAPL f/f and MAPL −/− mice were collected by cardiac puncture. Serum was isolated with a serum separator tube (Sarstedt) and analyzed for alanine transaminase (ALT) and aspartate transaminase (AST) (Diagnostic Research Support Service, Animal Resources Centre, McGill University).
Circulating FGF21 was measured from mice starved overnight and re‐fed for 3 h prior cardiac puncture. FGF21 was quantified by ELISA using a mouse/rat Fgf21 kit (R&D systems, MF2100), following manufacturer instructions.
Circulating FGF15 was measured from mice starved overnight and re‐fed for 3 h prior to cardiac puncture. FGF15 was quantified by ELISA using a Fgf15 kit (MyBioSource, MBS052232), following manufacturer instructions.
Glycogen was quantified from MAPL f/f and MAPL −/− livers using the Glycogen assay kit (Abcam, ab65620), following manufacturer instructions.
Quantitation of bile acids and corticosterone from liver and serum
Blood and livers have been collected from MAPL f/f and MAPL −/− mice starved overnight for 16 h and fed back in the morning for 3 h prior to collection (n = 8 for each genotype, 2‐month‐old males). Serum was isolated with a serum separator tube (Sarstedt).
Fifty microliter of each serum was added with 75 μl of water, 50 μl of an internal standard (IS) solution that contained predefined amounts of 14 D‐labeled bile acids (BAs) and D4‐corticosterone. After vortex mixing and 5‐s spin‐down, 350 μl of acetonitrile was added. The samples were vortex‐mixed for 15 s and ultra‐sonicated for 1 min in a water batch. The samples were then centrifuged at 21,000 g and 10°C for 5 min. The supernatants were loaded into wells of a 96‐well phospholipid depletion solid phase extraction (PD‐SPE) plate (50 mg/1 ml each well) for removal of phospholipids using the same protocol as described in a publication (Han et al, 2015). The flow‐through fractions were collected and dried down in a nitrogen evaporator under a gentle nitrogen gas flow. The residues were reconstituted in 200 μl of 50% methanol.
Each liver was ground in liquid nitrogen. Fifty microgram of each ground sample was precisely weighed into a 2‐ml homogenization tube and added with 125 μl of 20% acetonitrile. The samples were then homogenized with the aid of two 5‐mm stainless steel metal balls in a MM400 mixer mill for 45 s × 2 and at a shaking frequency of 30 Hz. After vortex mixing and 5‐s spin‐down, each tube was added with 250‐μl acetonitrile and the samples were homogenized again, followed by centrifugation at 15,000 rpm and 10°C for 5 min. Two hundred microliter of the supernatants were mixed with 50 μl of the same IS solution, followed by PD‐SPE using the same procedure as did for mouse serum. The residues were reconstituted in 200 μl of 50% methanol.
Twenty microliter of each of the above samples was injected onto a 15‐cm long C18 UPLC column for quantitation of bile acids by UPLC‐(−)ESI/MRM/MS with negative‐ion mode detection and with water‐acetonitrile‐formic acid as the mobile phase for binary gradient elution, using the same method as described in the publication. UPLC‐MRM/MS runs were performed on a Dionex Ultimate 3000 UPLC system coupled to a 4000 QTRAP triple‐quad mass spectrometer.
Concentrations of the detected bile acids were calculated with internal standard calibration from calibration curves prepared for individual compounds. For concentration calculation, the 14 D‐labeled bile acids were used as IS for their corresponding non‐D‐labeled forms. For the bile acids, THCA and DHCA, for which there were no D‐labeled analogues as IS, chenodeoxycholic‐D4 acid was used as the common IS for quantitation of the unconjugated bile acids, THCA and DHCA; tauro‐CDCA‐D4 was used as the common IS for quantitation of the taurine‐conjugated species; glyco‐deoxycholic‐D4 acid was used as the common IS for quantitation of the glycine‐conjugated species.
Concentrations of the following bile acids were also estimated in this analysis: glyco‐ω‐MCA, glyco‐α‐MCA, glyco‐β‐MCA, glyco‐λ‐MCA (also as glyco‐γ‐MCA or glycohyocholic acid) or glyco‐allocholic acid. Since there were no standard substances for these compounds, their concentrations were calculated from the calibration curve of glycocholic acid.
Another 20‐μl of each of the same samples was injected again onto the same C18 UPLC column for UPLC‐MRM/MS quantitation of corticosterone, but with positive‐ion (+) mode detection.
Quantitation of bile acids from feces
The authentic compounds of C24 bile acids and 14 D4‐ or D6‐coded bile acids were purchased from Steraloids Inc. (Newport, RI, USA) or Toronto Research Chemicals (Toronto, ON, Canada). Two C27 bile acids, i.e., 3ß,7α‐dihydroxy‐5‐cholestenoic acid (DHCA) and 3α,7α,12α‐trihydroxycholestanoic acid (THCA), were purchased from Avantis Polar Lipids Inc. (Alabaster, AL, USA). Feces were collected daily from seven animals per genotype (three males and four females, 6 month old) and dried for 2 days at 60°C. Dried mouse fecal samples were ground with a mortar and a pestle. A 50‐mg aliquot of each sample was precisely weighed into a 5‐ml borosilicate glass test tube, and 4 ml of 70% aqueous acetonitrile was added. Bile acids were extracted by vortex mixing and sonication of the samples in an ultrasonic ice water bath for 1 min. After clarification by centrifugation at 4,000 g. and 4°C for 15 min in a Beckman Coulter R‐22 centrifuge (Indianapolis, IN, USA), 800 μl of the supernatant was spiked with 200 μl of an internal standard (IS) solution which was composed of a predefined amount of each of the 14 D4‐ or D6‐coded bile acids. The samples were then subjected to phospholipid depletion solid‐phase extraction using a custom‐developed protocol (Han et al, manuscript in preparation). The flow‐through fractions were collected and then dried in a Thermo Savant SPD1010 speed‐vacuum concentrator. The residues were resuspended in 200 μl of 50% methanol. A 20‐μl aliquot was injected for quantitation by ultrahigh‐performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS).
UPLC‐MS/MS using scheduled multiple‐reaction monitoring (MRM) was performed on a Dionex Ultimate 3000 UPLC system (Thermo Scientific Inc. Waltham, MA, USA) coupled to a 4000 QTRAP triple‐quadrupole mass spectrometer (AB Sciex, Concord, ON, Canada). The mass spectrometer was equipped with an electrospray ionization (ESI) source and was operated in the negative‐ion mode. MRM transitions for each analyte were optimized by direction infusion and the scheduled MRM was operated with a retention time window of 1 min. Bile aids were separated on a Waters BEH C18 (2.1 × 150 mm, 1.7 μm) UPLC column with 0.01% formic acid in water (solvent A) and 0.01% formic acid in acetonitrile (solvent B) as the mobile phase using the following binary elution gradient: 12% B for 1 min and 12–75% B in 25 min. The column was then washed with 100% B for 2 min before 25% B column equilibrium for 4 min. The column flow rate was 0.35 ml/min, and the column was maintained at 45°C. Linearly regressed calibration curves of individual bile acids were prepared with the authentic compounds of the bile acids with IS calibration. For those bile acids which had their deuterium‐coded analogues in the IS solution, the D4‐ or D6‐coded analogues were used as the ISs for calculation of their concentrations in the mouse fecal samples. For others, lithocholic acid‐D4 was used as a common IS.
RNA sequencing
MAPL−/− and MAPLwt/wt mice were fasted overnight and refed for 2 h. Pieces of liver were snap‐frozen in liquid nitrogen, and RNA was extracted using TRIzol‐chloroform, followed by purification on RNA‐binding column. Briefly, frozen piece of tissue was transferred to 1 ml of TRIzol (Invitrogen 15596018) and homogenized. Homogenate was spinned at 11,000 g for 10′ at +4°C, mixed with 200 μl of chloroform and incubated at RT for 3′. Aqueous phase was mixed with 75% EtOH and added on RNAeasy column (Qiagen) and proceeded as per manufacturer instructions. RNA integrity was checked using 2100 Bioanalyzer system, and RIN was > 8.5 for all samples. Library was prepared using PolyA selection (Dynabeads Thermo Fisher Scientific) followed by KAPA RNA HyperPrep protocol, using xGen Dual Index UMI Adapters (IDT). Sequencing was done using Nextseq500–1 Flowcell High Output, 75 cycles single end reads (maximum 1 × 85 nt). Library preparation and sequencing was done in Institute for Research in Immunology and Cancer (IRIC), Montreal, Canada.
For analysis, RStudio version 2021.09.1 was used. Reads were filtered to remove adapter sequences, contamination, and low‐quality reads using Trimmomatic (parameters ILLUMINACLIP 2:30:10 (AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC) LEADING:15 TRAILING:15 MINLEN:20) and FastaQC (Bolger et al, 2014). Reads were mapped using HISAT2 (Kim et al, 2019) to reference mouse genome build mm10, and counted using featureCounts (Liao et al, 2014). Differential expression analysis was done using DeSeq2 (Love et al, 2014). Heatmaps were generated using ComplexHeatmaps (Gu et al, 2016), volcano plots using ggplot2 (Wickham, 2016), bar graphs using Prism 9, Venn diagrams using BioVenn (Hulsen et al, 2008) and PCA plot values were computed in R. Pathway enrichment analysis was done using Metascape (Zhou et al, 2019) (genes with P‐adjusted value < 0.05 and |log2(FC)| > 0.5 were selected, sorted by fold change and top500 up‐ or down‐regulated genes were analyzed). Summary terms from the output were plotted. For gene network exploration, Biogrid Mus musculus database and Cytoscape software were used (Shannon et al, 2003; Stark et al, 2006).
BioID
BioID (Stark et al, 2006) was carried out essentially as described previously (Comartin et al, 2013). In brief, the full‐length human MAPL (BC014010) coding sequence was amplified by PCR and cloned into a pcDNA5 FRT/TO BirA*FLAG expression vector (MAPL‐AscI_Fwd: tataGGCGCGCCaATGGAGAGCGGAGGGCGGCCCTCG; MAPL‐NotI_Rev: ttaaGCGGCCGCGCTGTTGTACAGGGGTATCACCCG). Using the Flp‐In system (Invitrogen), 293T‐REx Flp‐In cells stably expressing MAPL‐BirA*Flag were generated. After selection (DMEM + 10% FBS + 200 μg/ml hygromycin B), 10 × 150 cm2 plates of subconfluent (60%) cells were incubated for 24 h in complete media supplemented with 1 μg/ml tetracycline and 50 μM biotin. Five plates were treated with 5 μM MG132. Cells were collected and pelleted (1,000 g, 3 min), the pellet was washed twice with PBS, and dried pellets were snap‐frozen. Pellets were lysed in 10 ml of modified RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 0.1% SDS, 1:500 protease inhibitor cocktail, 250 U Turbonuclease, pH 7.5) at 4°C for 1 h, then sonicated to completely disrupt visible aggregates. The lysates were centrifuged at 35,000 g for 30 min. Clarified supernatants were incubated with 30 μl packed, pre‐equilibrated Streptavidin‐sepharose beads at 4°C for 3 h. Beads were collected by centrifugation, washed six times with 50 mM ammonium bicarbonate pH 8.2, and treated with TPCK‐trypsin (16 h at 37°C). The supernatant containing the tryptic peptides was collected and lyophilized. Peptides were resuspended in 0.1% formic acid and 1/6th of the sample was analyzed per MS run.
Liquid chromatography (LC) analytical columns (75 μm inner diameter) and pre‐columns (100 μm ID) were made in‐house from fused silica capillary tubing from InnovaQuartz and packed with 100 Å C18‐coated silica particles. LC–MS/MS was conducted using a 120 min reversed‐phase buffer gradient running at 250 nl/min (column heated at 35°C) on a Proxeon EASY‐nLC pump in line with a hybrid LTQ‐Orbitrap Velos mass spectrometer. A parent ion scan was performed in the Orbitrap, using a resolving power of 60,000. Simultaneously, up to the 20 most intense peaks were selected for MS/MS (minimum ion count of 1,000 for activation) using standard CID fragmentation. Fragment ions were detected in the LTQ. Dynamic exclusion was activated such that MS/MS of the same m/z (within a 10 ppm window, exclusion list size 500) detected three times within 45 s were excluded from analysis for 30 s. For protein identification, RAW files were converted to the mzXML format using ProteoWizard, then searched using X!Tandem against human RefSeq Version 45 (containing 36,113 entries). Search parameters specified a parent MS tolerance of 15 ppm and an MS/MS fragment ion tolerance of 0.4 Da, with up to 2 missed cleavages allowed for trypsin. Oxidation of the methionine was allowed as a variable modification. Data were analyzed using the trans‐proteomic pipeline via the ProHits 2.0.0 software suite. Proteins identified with a ProteinProphet cut‐off of 0.85 (corresponding to ≤ 1% FDR) were analyzed with SAINT Express v.3.3. Sixteen control runs were used for comparative purposes, comprising eight runs of BioID conducted on untransfected 293T‐REx cells. In each case, four runs were conducted on untreated cells, and four runs were conducted in cells treated with MG132, as above. The 16 controls were collapsed to the highest four spectral counts for each hit.
Histology
Formaldehyde‐fixed, paraffin‐embedded tissues were cut into 4 μm sections and stained with hematoxylin and eosin (H&E).
For Ki67 staining, sections slides were deparaffinized in EZ prep buffer for 8 min at 75°C. Antigen retrieval was performed by incubating slides in cell conditioning buffer 1 (CC1) at 95°C for 64 min. Slides were then blocked with the included Inhibitor CM at 37°C for 8 min. Incubation with primary antibody was conducted at 37°C for 60 min. Incubation with secondary antibody was performed by applying one drop of Omnimap anti‐Rb HRP on slides for 16 min. Staining was revealed by adding one drop of DAB CM for 8 min. Slides were then incubated for 5 min with one drop of Copper CM followed by counterstain with hematoxylin for 8 min. Post‐counterstaining was performed for 8 min with Bluing Reagent. Slides were then dehydrated using increasing concentrations of ethanol, cleaned in xylene and mounted using Acrytol mounting media.
RNA isolation and qRT‐PCR
Total RNAs from various tissues were prepared using TRIzol (Invitrogen). They were treated with DNAse (New England Biolabs) and then reverse transcribed with random primers using the High Capacity sDNA Reverse Transcription Kit (Life Technologies) as described by the manufacturer. Before use, RT samples were diluted 1:5. Gene expression was determined using assays designed with the Universal Probe Library (UPL) from Roche (www.universalprobelibrary.com). For each qPCR assay, a standard curve was performed to ensure the efficacity of the assay is between 90 and 110%. qPCR reactions were performed using 5–25 ng of cDNA samples, the TaqMan Advanced Fast Universal PCR Master Mix (Life Technologies), 2 μM of each primer and 1 μM of the corresponding UPL probe. The Viia7 qPCR instrument (Life Technologies) was used to detect the amplification level and was programmed with an initial step of 3 min at 95°C, followed by 40 cycles of 5 s at 95°C and 30 s at 60°C. All reactions were run in triplicate and the average values of Cts were used for quantification. The relative quantification of target genes was determined using the ΔΔCT method. Briefly, the Ct (threshold cycle) values of target genes were normalized to an endogenous control gene (ΔCT = Cttarget − CtCTRL) and compared with a calibrator: ΔΔCT = ΔCtSample − ΔCtCalibrator. Relative expression (RQ) was calculated using the Sequence Detection System (SDS) 2.2.2 software (Applied Biosystems) and the formula is RQ = 2−ΔΔCT.
qRT‐PCR primers used (5′ to 3′)
| Gene symbol | UPL probe | Forward | Reverse |
|---|---|---|---|
| Abca1 | 26 | atggagcagggaagaccac | gtaggccgtgccagaagtt |
| Abcd3 | 91 | tgttcaggactggatggatg | tgataaaacagtcttgccatcg |
| Abcg5 | 31 | tcctgcatgtgtcctacagc | cccacttctgctggcatgat |
| Abcg8 | 10 | ggacttgaccagcatcgaca | gaacagggctgcaagagact |
| Acadl | 75 | gcttatgaatgtgtgcaactcc | ccgagcatccacgtaagc |
| Acox1 | 45 | cgccagtctgaaatcaagaga | gctgcgtctgaaaatccaa |
| Baat | 2 | atgacctgccttctcgactg | cagaaaggatgccaacacct |
| Cd36 | 75 | ccaagctattgcgacatgatt | tctcaatgtccgagacttttca |
| Cyp2c70 | 33 | acattccaagacttcatggagaa | ggagatagtataaaatagggtaggca |
| Cyp27a1 | 27 | tctaccaccttgccttggaa | gcagtgtcctcaggaatgg |
| Cyp3a11 | 53 | gggactcgtaaacatgaacttttt | ccatgtcgaatttccataaacc |
| Cyp4a14 | 7 | catggcggactctgtcaata | gatctccagagggtggtcct |
| Cyp7a1 | 92 | gatcctctgggcatctcaag | agaggctgctttcattgctt |
| Fgf21 | 67 | agatggagctctctatggatcg | gggcttcagactggtacacat |
| Hmgcs2 | 26 | tgcaggaaacttcgctcaca | atctgttttggccaggggag |
| Hsd17b4 | 7 | aaagcggaatcagcccatga | ttcttgaatggtctgcggct |
| Hsd3b5 | 17 | cgcttccagacagaccatc | gatgaatgttggcacactgg |
| Nr1h4 | 100 | caaaatgactcaggaggagtacg | tccttgatgtattgtctgtctgg |
| Shp‐Nr0b2 | 51 | gcacgatcctcttcaaccca | gacttcacacagtgcccagt |
| Slc10a1 | 5 | cctcaaggcaggcatgatca | cgaacatgatgctgttgccc |
| Slco1a1 | 84 | gagccaaaaaggaaaaccttgga | acactcgtaagactgaacagca |
| Slco1b2 | 5 | caggacaccaagactgctgg | taccctatgccttccaccga |
| Srebf1 | 77 | acaagattgtggagctcaaagac | gcgcaagacagcagatttatt |
Cell death induction in human liver cell line
Huh‐7 cells were plated in 6‐well dish at 5 × 105 cells per well. Cells were infected with adenovirus expressing MAPL‐Flag or MAPL‐ΔRING‐Flag or empty vector (Ad‐rtTA) at 100 pfu per cell. Cells were collected 1 day post‐transfection, and total cell lysate was prepared. Cell survival was determined using Promega CellTiter‐Glow Luminescent Cell Viability assay kit as described by manufacture. Survival was expressed as percentage of rtTA values. For caspase 3 or caspase 7 activation, total cell lysate (30 μg) was analyzed on SDS–PAGE and immunoblotted with antibodies to cleaved caspase 3 (rabbit polyclonal antibody, 9661S, 1:1,000, Cell Signaling) or cleaved‐Caspase 7 (rabbit polyclonal antibody, 9491, 1:1,000, Cell Signaling). Cell was regularly checked for mycoplasma infection.
Foci formation assay
2 × 106 Rat2 cells grown in DMEM/10% FBS were plated in a 10 cm dish. The following day, cells were transfected with 20 μg of cDNA using calcium phosphate (Invitrogen) according to the manufacturer's instructions. Transfection media was removed the following day and media changed every 3 days hereafter. Three weeks after transfection, cells were fixed in 10% formaldehyde/PBS for 30 min at room temperature, stained with 1:25 diluted Giemsa (Sigma) for 5 min and then rinsed with water and air dried. Cell was regularly checked for mycoplasma infection.
Apoptosis induction by MAPL expression
5 × 105 Rat2 cells grown in DMEM/10% FBS were plated in a 60 mm dish. The following day, cells were transfected using 8 μg of MAPL‐Flag or pcDNA3 DNA and 20 μl Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. The following day, cells were washed once in PBS and lysed in 500 μl of RIPA supplemented with cOmplete protease inhibitor (Roche). Protein concentration was measured by Bradford.
Oxygen bomb calorimeter
Feces were collected while mice were single‐housed and dried for 48 h at 60°C. Once fine‐grounded, 0.9–1 g of feces was used to determine the caloric content by means of an oxygen bomb calorimeter (6772 Calorimetric Thermometer, Parr Instrument Company). Analysis was carried out in duplicate, with an intrinsic variation of 2–3%.
Statistical analysis
Normal distribution and homoscedasticity of data were tested by Shapiro‐Wilks and Bartlett tests, respectively. Parametric tests were used if distributions normal and variances equal. Student's t‐test was used to compare 2 groups on 1 variable. One‐way analysis of variance (ANOVA) for univariate multiple comparisons or two‐way ANOVA (for bivariate comparisons) were followed by Tukey's honest significant difference post hoc test. Statistical analyses were performed using GraphPad Prism software (San Diego, CA). Threshold for statistical significance was P < 0.05.
All values are expressed as mean ± SEM.
Author contributions
Vanessa Goyon: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Aurèle Besse‐Patin: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Rodolfo Zunino: Formal analysis; investigation; methodology; writing – review and editing. Olesia Ignatenko: Formal analysis; visualization; writing – review and editing. Mai Nguyen: Conceptualization; investigation; methodology; writing – review and editing. Étienne Coyaud: Investigation; methodology. Jonathan M Lee: Investigation; methodology. Bich N Nguyen: Investigation; methodology. Brian Raught: Conceptualization; investigation; methodology; writing – review and editing. Heidi M McBride: Conceptualization; formal analysis; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Appendix Figure S1–S2
Acknowledgements
The authors would like to thank the IRIC for qRT‐PCR services and Ozgene (Australia) for services in the design and generation of the floxed MAPL strain. We also thank Dr. Atilla Omeroglu (McGill University) for help with histological analysis, Nancy Braverman (McGill University) for advice on bile acid metabolism, and Tharan Srikumar for mass spectrometry technical assistance and the MNI animal care services. This study was funded by Canadian Institute of Health Research and the Canadian Cancer Society Research Institute (to HMM; CIHR#68833, CCSRI#702139). Work in the BR lab was funded by CIHR #130340. HMM holds the Canada Research Chair in Mitochondrial Cell Biology. BR holds the Canada Research Chair in Proteomics and Molecular Medicine. ABP was supported by a postdoctoral fellowship from Fonds de Recherche Québec – Santé, OI was supported by Sigrid Jusélius postdoctoral fellowship.
EMBO reports (2023) 24: e57972
Data availability
The RNA sequencing analysis and dataset is deposited on the NCBI GEO (GSE242501) (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242501). Reagents are available upon requests to the corresponding author.
References
- Ahluwalia A, Clodfelter KH, Waxman DJ (2004) Sexual dimorphism of rat liver gene expression: regulatory role of growth hormone revealed by deoxyribonucleic acid microarray analysis. Mol Endocrinol 18: 747–760 [DOI] [PubMed] [Google Scholar]
- Ambivero CT, Cilenti L, Main S, Zervos AS (2014) Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal 26: 2921–2929 [DOI] [PubMed] [Google Scholar]
- Andrade‐Navarro MA, Sanchez‐Pulido L, McBride HM (2009) Mitochondrial vesicles: an ancient process providing new links to peroxisomes. Curr Opin Cell Biol 21: 560–567 [DOI] [PubMed] [Google Scholar]
- Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos‐Flier E (2009) Fibroblast growth factor 21‐deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 150: 4931–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto SA, Lasserre F, Fougerat A, Smith L, Fougeray T, Lukowicz C, Polizzi A, Smati S, Régnier M, Naylies C et al (2019) Gene expression profiling reveals that PXR activation inhibits hepatic PPARα activity and decreases FGF21 secretion in male C57Bl6/J mice. Int J Mol Sci 20: 3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, Choi J, Kasperkiewicz P, Fernandes‐Alnemri T, Alnemri E et al (2018) SUMO‐mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun 9: 3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bideyan L, Fan W, Kaczor‐Urbanowicz KE, Priest C, Casero D, Tontonoz P (2022) Integrative analysis reveals multiple modes of LXR transcriptional regulation in liver. Proc Natl Acad Sci USA 119: e2122683119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature 510: 370–375 [DOI] [PubMed] [Google Scholar]
- Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH (2008) Hepatocyte‐specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med 14: 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonDurant LD, Potthoff MJ (2018) Fibroblast growth factor 21: a versatile regulator of metabolic homeostasis. Annu Rev Nutr 38: 173–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, Ding X, Elmquist JK, Takahashi JS, Mangelsdorf DJ et al (2013) FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med 19: 1147–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma PJ, Seppen J, Goldhoorn B, Bakker C, Oude Elferink RP, Chowdhury JR, Chowdhury NR, Jansen PL (1994) Bilirubin UDP‐glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem 269: 17960–17964 [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10: 748–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carino A, Marchianò S, Biagioli M, Scarpelli P, Bordoni M, Di Giorgio C, Roselli R, Fiorucci C, Monti MC, Distrutti E et al (2021) The bile acid activated receptors GPBAR1 and FXR exert antagonistic effects on autophagy. FASEB J 35: e21271 [DOI] [PubMed] [Google Scholar]
- Celen AB, Sahin U (2020) Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J 287: 3110–3140 [DOI] [PubMed] [Google Scholar]
- Chen W, Chiang JYL (2003) Regulation of human sterol 27‐hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha). Gene 313: 71–82 [DOI] [PubMed] [Google Scholar]
- Chen MM, Hale C, Stanislaus S, Xu J, Veniant MM (2018) FGF21 acts as a negative regulator of bile acid synthesis. J Endocrinol 237: 139–152 [DOI] [PubMed] [Google Scholar]
- Chiang JYL (2009) Bile acids: regulation of synthesis. J Lipid Res 50: 1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JYL, Ferrell JM (2020) Up to date on cholesterol 7 alpha‐hydroxylase (CYP7A1) in bile acid synthesis. Liver Res 4: 47–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comartin D, Gupta GD, Fussner E, Coyaud E, Hasegan M, Archinti M, Cheung SW, Pinchev D, Lawo S, Raught B et al (2013) CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr Biol 23: 1360–1366 [DOI] [PubMed] [Google Scholar]
- Costa‐Mattioli M, Walter P (2020) The integrated stress response: from mechanism to disease. Science 368: eaat5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JL, Passmore JB, Islinger M, Schrader M (2018) Multi‐localized proteins: the peroxisome‐mitochondria connection. Subcell Biochem 89: 383–415 [DOI] [PubMed] [Google Scholar]
- Coyaud E, Mis M, Laurent EMN, Dunham WH, Couzens AL, Robitaille M, Gingras A‐C, Angers S, Raught B (2015) BioID‐based identification of Skp cullin F‐box (SCF)β‐TrCP1/2 E3 ligase substrates. Mol Cell Proteomics 14: 1781–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert HA, Ge X, Kohan AB, Salati LM, Zhang Y, Hillgartner FB (2012) Activation of the farnesoid × receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem 287: 25123–25138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer JF, Bloks VW, Verkade E, Heiner‐Fokkema MR, Kuipers F (2018) New insights in the multiple roles of bile acids and their signaling pathways in metabolic control. Curr Opin Lipidol 29: 194–202 [DOI] [PubMed] [Google Scholar]
- Doiron K, Goyon V, Coyaud E, Rajapakse S, Raught B, McBride HM (2017) The dynamic interacting landscape of MAPL reveals essential functions for SUMOylation in innate immunity. Sci Rep 7: 107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donepudi AC, Boehme S, Li F, Chiang JYL (2017) G‐protein‐coupled bile acid receptor plays a key role in bile acid metabolism and fasting‐induced hepatic steatosis in mice. Hepatology 65: 813–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar‐Henriques M, Langer T (2014) Dynamic survey of mitochondria by ubiquitin. EMBO Rep 15: 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandusse S, Jimenez‐Sanchez G, Koster J, Denis S, Van Roermund CW, Silva‐Zolezzi I, Moser AB, Visser WF, Gulluoglu M, Durmaz O et al (2015) A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum Mol Genet 24: 361–370 [DOI] [PubMed] [Google Scholar]
- Flippo KH, Potthoff MJ (2021) Metabolic messengers: FGF21. Nat Metab 3: 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs CD, Trauner M (2022) Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat Rev Gastroenterol Hepatol 19: 432–450 [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME et al (2000) A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 6: 517–526 [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849 [DOI] [PubMed] [Google Scholar]
- Han J, Liu Y, Wang R, Yang J, Ling V, Borchers CH (2015) Metabolic profiling of bile acids in human and mouse blood by LC‐MS/MS in combination with phospholipid‐depletion solid‐phase extraction. Anal Chem 87: 1127–1136 [DOI] [PubMed] [Google Scholar]
- He J, Cheng J, Wang T (2020) SUMOylation‐mediated response to mitochondrial stress. Int J Mol Sci 21: 5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I (2006) Specification of SUMO1‐ and SUMO2‐interacting motifs. J Biol Chem 281: 16117–16127 [DOI] [PubMed] [Google Scholar]
- Hillebrand M, Verrier SE, Ohlenbusch A, Schafer A, Soling HD, Wouters FS, Gartner J (2007) Live cell FRET microscopy: homo‐ and heterodimerization of two human peroxisomal ABC transporters, the adrenoleukodystrophy protein (ALDP, ABCD1) and PMP70 (ABCD3). J Biol Chem 282: 26997–27005 [DOI] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W (2008) BioVenn ‐ a web application for the comparison and visualization of biological lists using area‐proportional Venn diagrams. BMC Genomics 9: 488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V et al (2007) Endocrine regulation of the fasting response by PPARalpha‐mediated induction of fibroblast growth factor 21. Cell Metab 5: 415–425 [DOI] [PubMed] [Google Scholar]
- Itoh N (2014) FGF21 as a hepatokine, adipokine, and myokine in metabolism and diseases. Front Endocrinol 5: 107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins K, Khoo JJ, Sadler A, Piganis R, Wang D, Borg NA, Hjerrild K, Gould J, Thomas BJ, Nagley P et al (2013) Mitochondrially localised MUL1 is a novel modulator of antiviral signaling. Immunol Cell Biol 91: 321–330 [DOI] [PubMed] [Google Scholar]
- Kaspar S, Oertlin C, Szczepanowska K, Kukat A, Senft K, Lucas C, Brodesser S, Hatzoglou M, Larsson O, Topisirovic I et al (2021) Adaptation to mitochondrial stress requires CHOP‐directed tuning of ISR. Sci Adv 7: eabf0971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐Y, Kim H‐J, Kang SU, Kim YE, Park JK, Shin YS, Kim YS, Lee K, Kim C‐H (2015) Non‐thermal plasma induces AKT degradation through turn‐on the MUL1 E3 ligase in head and neck cancer. Oncotarget 6: 33382–33396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus F, Ryan MT (2017) The constriction and scission machineries involved in mitochondrial fission. J Cell Sci 130: 2953–2960 [DOI] [PubMed] [Google Scholar]
- Lee M‐S, Lee S‐O, Choi J, Ryu M, Lee M‐K, Kim J‐H, Hwang E, Lee C‐K, Chi S‐W, Ryu K‐S (2022) MUL1‐RING recruits the substrate, p53‐TAD as a complex with UBE2D2‐UB conjugate. FEBS J 289: 3568–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CAP (2008) Genome‐wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One 3: e1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Li‐Hawkins J, Lund EG, Bronson AD, Russell DW (2000) Expression cloning of an oxysterol 7alpha‐hydroxylase selective for 24‐hydroxycholesterol. J Biol Chem 275: 16543–16549 [DOI] [PubMed] [Google Scholar]
- Lorbek G, Lewinska M, Rozman D (2012) Cytochrome P450s in the synthesis of cholesterol and bile acids—from mouse models to human diseases. FEBS J 279: 1516–1533 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6: 507–515 [DOI] [PubMed] [Google Scholar]
- Marcassa E, Kallinos A, Jardine J, Rusilowicz‐Jones EV, Martinez A, Kuehl S, Islinger M, Clague MJ, Urbe S (2018) Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep 19: e45595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T, Li F, Gonzalez FJ (2013) FXR signaling in the enterohepatic system. Mol Cell Endocrinol 368: 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty A, Zunino R, Soubannier V, Dilipkumar S (2021) A new functional role of mitochondria‐anchored protein ligase in peroxisome morphology in mammalian cells. J Cell Biochem 122: 1686–1700 [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade‐Navarro MA, McBride HM (2008) Cargo‐selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18: 102–108 [DOI] [PubMed] [Google Scholar]
- Nie M, Boddy MN (2016) Cooperativity of the SUMO and ubiquitin pathways in genome stability. Biomolecules 6: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, Kliewer SA, Mangelsdorf DJ (2014) FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20: 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, Tian H, Wang B, Wang Y, Zhang Y et al (2018) FGF21 prevents angiotensin II‐induced hypertension and vascular dysfunction by activation of ACE2/angiotensin‐(1‐7) axis in mice. Cell Metab 27: 1323–1337 [DOI] [PubMed] [Google Scholar]
- Patel R, Bookout AL, Magomedova L, Owen BM, Consiglio GP, Shimizu M, Zhang Y, Mangelsdorf DJ, Kliewer SA, Cummins CL (2015) Glucocorticoids regulate the metabolic hormone FGF21 in a feed‐forward loop. Mol Endocrinol 29: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MJ, Briz O (2009) Bile‐acid‐induced cell injury and protection. World J Gastroenterol 15: 1677–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N, Warscheid B, Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20: 267–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM (2015) MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell 59: 941–955 [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Knoch B, Müller M, Kersten S (2010) Peroxisome proliferator‐activated receptor alpha target genes. PPAR Res 2010: 612089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranea‐Robles P, Chen H, Stauffer B, Yu C, Bhattacharya D, Friedman SL, Puchowicz M, Houten SM (2021) The peroxisomal transporter ABCD3 plays a major role in hepatic dicarboxylic fatty acid metabolism and lipid homeostasis. J Inherit Metab Dis 44: 1419–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojansky R, Cha M‐Y, Chan DC (2016) Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 5: e17896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas Á, Gil‐Gómez A, de la Cruz‐Ojeda P, Muñoz‐Hernández R, Sánchez‐Torrijos Y, Gallego‐Durán R, Millán R, Rico MC, Montero‐Vallejo R, Gato‐Zambrano S et al (2022) Long non‐coding RNA H19 as a biomarker for hepatocellular carcinoma. Liver Int 42: 1410–1422 [DOI] [PubMed] [Google Scholar]
- Roque PS, Thorn Perez C, Hooshmandi M, Wong C, Eslamizade MJ, Heshmati S, Brown N, Sharma V, Lister KC, Goyon VM et al (2023) Parvalbumin interneuron loss mediates repeated anesthesia‐induced memory deficits in mice. J Clin Invest 133: e159344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ (2013) Marked by association: techniques for proximity‐dependent labeling of proteins in eukaryotic cells. Cell Mol Life Sci 70: 3657–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Quilty F, Gilmer JF, Long A, Byrne A‐M (2017) Unconjugated secondary bile acids activate the unfolded protein response and induce golgi fragmentation via a src‐kinase‐dependant mechanism. Oncotarget 8: 967–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Pender CL, Bar‐Ziv R, Zhang H, Wickham K, Willey E, Durieux J, Ahmad Q, Dillin A (2022) Mitochondria as cellular and organismal signaling hubs. Annu Rev Cell Dev Biol 38: 179–218 [DOI] [PubMed] [Google Scholar]
- Song P, Zechner C, Hernandez G, Canovas J, Xie Y, Sondhi V, Wagner M, Stadlbauer V, Horvath A, Leber B et al (2018) The hormone FGF21 stimulates water drinking in response to ketogenic diet and alcohol. Cell Metab 27: 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino G, Perino A, Yildiz E, El Alam G, Bou Sleiman M, Gioiello A, Pellicciari R, Schoonjans K (2020) Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology 159: 956–968 [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B‐J, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, Green DR (2012) Mitochondria and cell signalling. J Cell Sci 125: 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ, Gonzalez FJ (2016) Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 57: 2130–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JX, Finkel T (2020) Mitochondria as intracellular signaling platforms in health and disease. J Cell Biol 219: e202002179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roermund CWT, Ijlst L, Wagemans T, Wanders RJA, Waterham HR (2014) A role for the human peroxisomal half‐transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim Biophys Acta 1841: 563–568 [DOI] [PubMed] [Google Scholar]
- Veniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, Moyer C, Lloyd DJ (2015) Pharmacologic effects of FGF21 are independent of the “browning” of white adipose tissue. Cell Metab 21: 731–738 [DOI] [PubMed] [Google Scholar]
- Vertegaal ACO (2022) Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol 23: 715–731 [DOI] [PubMed] [Google Scholar]
- Wagner M, Halilbasic E, Marschall H‐U, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M (2005) CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42: 420–430 [DOI] [PubMed] [Google Scholar]
- Wanders RJ (2013) Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem 69: 23–44 [DOI] [PubMed] [Google Scholar]
- Wang Y, Yutuc E, Griffiths WJ (2021) Cholesterol metabolism pathways ‐ are the intermediates more important than the products? FEBS J 288: 3727–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber WA, Schwaiger M, Avril N (2000) Quantitative assessment of tumor metabolism using FDG‐PET imaging. Nucl Med Biol 27: 683–687 [DOI] [PubMed] [Google Scholar]
- Wickham H (2016) ggplot2: elegant graphics for data analysis. Cham: Springer; [Google Scholar]
- Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, Guo M (2014) MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 3: e01958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Fan J, Zhou H (2023) Bile acid‐mediated signaling in cholestatic liver diseases. Cell Biosci 13: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Huang J, Li H‐L, Liu T, Wang Y‐Y, Waterman P, Mao A‐P, Xu L‐G, Zhai Z, Liu D et al (2008) GIDE is a mitochondrial E3 ubiquitin ligase that induces apoptosis and slows growth. Cell Res 18: 900–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, Katafuchi T, Xiao G, Potthoff MJ, Wei W, Wan Y et al (2012) The starvation hormone, fibroblast growth factor‐21, extends lifespan in mice. Elife 1: e00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Chen X, Liu Q, Zhong G, Zhuang M (2022) Ubiquitin ligase MARCH5 localizes to peroxisomes to regulate pexophagy. J Cell Biol 221: e202103156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 10: 1523 [DOI] [PMC free article] [PubMed] [Google Scholar]