

Abstract
Faithful DNA replication requires specific proteins that protect replication forks and so prevent the formation of DNA lesions that may damage the genome. Identification of new proteins involved in this process is essential to understand how DNA lesions accumulate in cancer cells and how they tolerate them. Here, we show that human GNL3/nucleostemin, a GTP‐binding protein localized mostly in the nucleolus and highly expressed in cancer cells, prevents nuclease‐dependent resection of nascent DNA in response to replication stress. We demonstrate that inhibiting origin firing reduces resection. This suggests that the heightened replication origin activation observed upon GNL3 depletion largely drives the observed DNA resection probably due to the exhaustion of the available RPA pool. We show that GNL3 and DNA replication initiation factor ORC2 interact in the nucleolus and that the concentration of GNL3 in the nucleolus is required to limit DNA resection. We propose that the control of origin firing by GNL3 through the sequestration of ORC2 in the nucleolus is critical to prevent nascent DNA resection in response to replication stress.
Keywords: DNA replication stress, DNA resection, GNL3, ORC2, origin firing
Subject Categories: DNA Replication, Recombination & Repair
GNL3 limits the number of fired replication origins by sequestering ORC2 within the nucleolus. In the absence of GNL3, increased replication origins firing triggers nuclease‐mediated resection of newly synthesized DNA in response to replication stress.
Introduction
In all cells, DNA replication must occur precisely before their division to ensure faithful transmission of the genome. In humans, accurate DNA replication is particularly important for stem cells and for preventing premature aging and/or cancer (Macheret & Halazonetis, 2015; Schumacher et al, 2021). Replication must occur correctly in space and time to ensure that the whole genome is copied entirely once per cell cycle with no under‐replicated or over‐replicated regions.
DNA replication initiates from specific sites distributed all over the genome, called replication origins (Mechali, 2010; Fragkos et al, 2015). Initiation of replication is a two‐step process. First, the origins are “licensed” for replication by binding of the origin recognition complex (ORC, composed of six subunits, ORC1–6) and the replicative helicase MCM2–7, which forms the pre‐replicative complex. Second, origin firing (the start of DNA synthesis) requires activation of cyclin‐dependent kinases and CDC7/DBF4 kinases. Although the ORC complex is mainly responsible for initiating DNA replication, it also has other functions. For example, one of the ORC subunits, ORC2, plays roles at centromeres and in sister chromatid cohesion independently of the ORC complex (Prasanth et al, 2004; Shimada & Gasser, 2007; MacAlpine et al, 2010; Huang et al, 2016; Bauwens et al, 2021).
After DNA replication starts, the progression of the replisome is perturbed by a variety of impediments that lead to replication fork stalling and therefore creates replication stress that may result in the formation of DNA lesions such as single‐strand gaps or DNA double‐strand breaks (DSBs) (Lambert & Carr, 2013). The main pathway activated to prevent fork collapse and genomic instability, the ATR–Chk1 checkpoint, prevents further progress through S phase, thus providing time for stalled forks to be stabilized to avoid formation of DNA lesions (Zeman & Cimprich, 2014). Many other proteins, for example BRCA1, protect stalled forks by preventing the action of specific nucleases like MRE11 or CtIP (Liao et al, 2018; Rickman & Smogorzewska, 2019; Berti et al, 2020). ATR–Chk1 maintains genomic stability by limiting the firing of replication origins in response to replication stress (Blow et al, 2011; Toledo et al, 2013; Courtot et al, 2018). WEE1, a kinase that limits entry into mitosis by inhibiting CDK1, acts in a similar way (Beck et al, 2012; Toledo et al, 2013; Moiseeva et al, 2019).
We previously used the iPOND (isolation of proteins on nascent DNA) method coupled with mass spectrometry (iPOND‐MS) to identify novel factors associated with replication forks (Lossaint et al, 2013; Ribeyre et al, 2016; Lebdy et al, 2023). Here, we performed a small siRNA screen to identify those novel factors whose depletion increases the number of DNA lesions, such as DSBs or single‐strand gaps, in response to exogenous replication stress using H2A.X phosphorylation (γH2A.X) as a readout. The protein whose depletion had the greatest effect was GNL3 (G protein nucleolar 3, also known as nucleostemin), a GTP‐binding protein localized mainly in the nucleolus which is highly expressed in stem cells and cancer cell lines (Tsai & McKay, 2002). Previous studies found that GNL3 depletion leads to activation of the DNA damage response during S phase (Lin et al, 2013; Meng et al, 2013; Yamashita et al, 2013). GNL3 is recruited to DSBs, and its depletion prevents RAD51, a key protein for DSBs repair by homologous recombination, from being recruited at DSBs and hydroxyurea (HU)‐induced lesions (Lin et al, 2013; Meng et al, 2013). Consistent with this, GNL3‐depleted cells are more sensitive to HU (Lin et al, 2014) and are less able to repair DSBs by homologous recombination (Meng et al, 2013). The current model suggests that GNL3 in the nucleoplasm maintains genome stability in S phase by being recruited to DNA lesions to stabilize RAD51 (Tsai, 2014). The partners of GNL3 and its functions during S phase, DNA replication and genome stability remain poorly understood. In this report, we demonstrate that GNL3 is required to protect stalled replication forks from resection by limiting replication origin firing possibly through regulation of some ORC2 functions.
Results
GNL3 prevents DNA resection of stalled replication forks
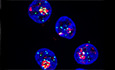
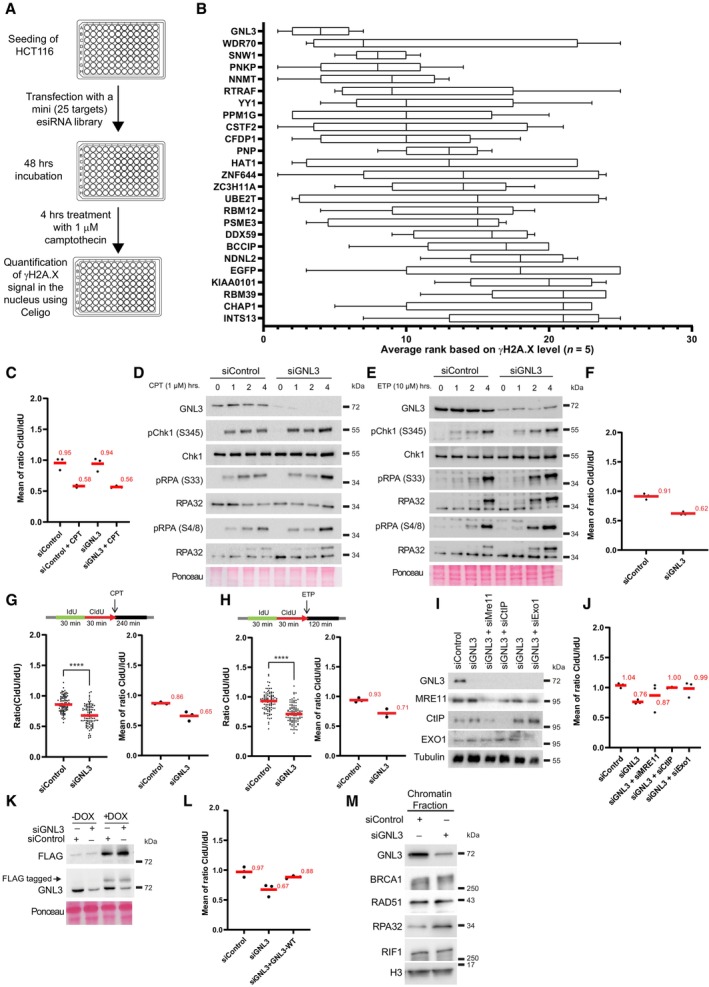
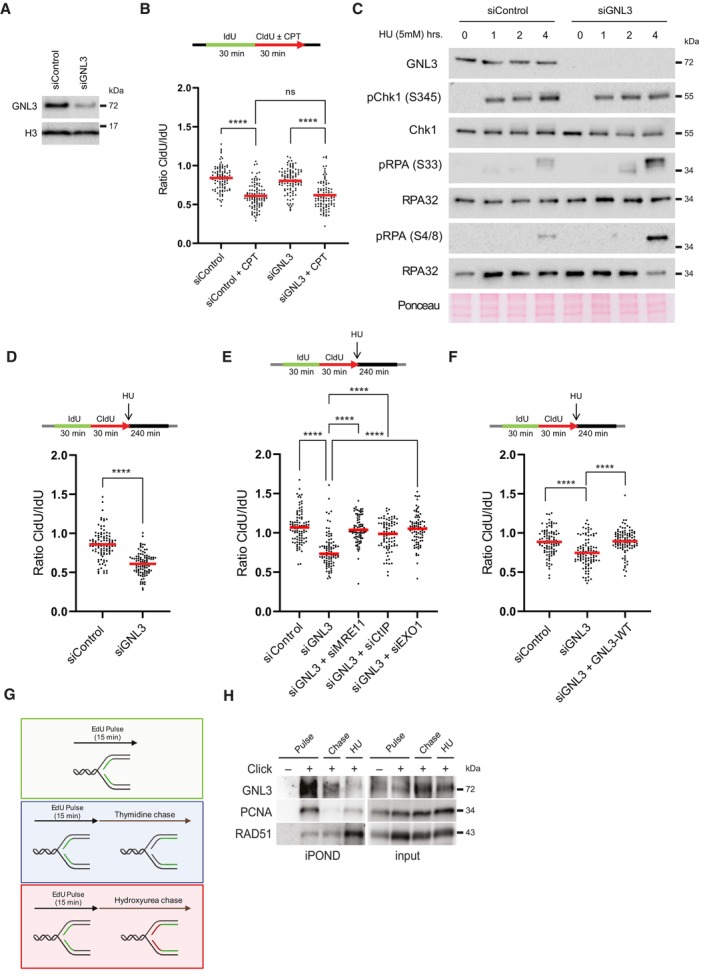
We reported previously our use of the iPOND method to identify novel factors associated with replication forks (Lebdy et al, 2023). Briefly, we pulse‐labeled newly synthesized DNA in Hela S3 cells with 5‐ethynyl‐2′‐deoxyuridine (EdU, a nucleoside analog of thymidine that can be labeled by Click chemistry) or pulsed with EdU then chased for 2 h with thymidine, then we purified the proteins associated with EdU. Those proteins that were significantly enriched in the pulse‐labeled samples when compared to the chase were defined as components of the replisome (Lebdy et al, 2023). These components included many proteins that were not previously known to be associated with nascent DNA. To select candidates for further analysis, we designed an orthogonal approach based on a mini screen using 25 individual endoribonuclease‐prepared siRNAs (esiRNAs; against 24 candidates plus a negative control esiRNA against EGFP). We wished to focus on proteins required to protect DNA integrity, in this case their depletion should increase the number of DNA lesions upon treatment with exogenous molecules that enhance replication stress. We analyzed DNA lesions by quantifying the amount of γH2A.X phosphorylation after 4 h of replication stress due to treatment with 1 μM camptothecin (CPT, an inhibitor of DNA topoisomerase 1). Briefly, HCT116 cells growing in 96 well plates were transfected with each of the 25 esiRNAs. Forty‐eight hours after transfection, the cells were treated for 4 h with 1 μM CPT and the amount of γH2A.X in the nucleus was analyzed by immunofluorescence microscopy using a Celigo high‐throughput microscope (Fig EV1A). We ranked the effects of the 25 esiRNAs based on the amount of γH2A.X and found that GNL3 ranked highest, suggesting that it may be important to tolerate replication stress (Fig EV1B). This is consistent with earlier results showing that GNL3 depletion leads to activation of the DNA damage response during S phase and GNL3‐depleted cells are more sensitive than control cells to hydroxyurea (HU), an inducer of replication stress (Lin et al, 2013, 2014; Meng et al, 2013; Yamashita et al, 2013). Since we found more γH2A.X in the nucleus of CPT‐treated cells depleted of GNL3 than in control cells (Fig EV1B), we investigated further whether GNL3 regulates replication fork progression in the presence of CPT. To do so we depleted GNL3 (Fig 1A) and labeled cells for 30 min with IdU followed by labelling for 30 min with CldU in the presence or absence of 1 μM CPT and measured the length of both tracks to obtain the CldU/IdU ratio (Fig 1B). As expected, addition of CPT strongly reduced the CldU/IdU ratio, however, depletion of GNL3 had no additional impact (Figs 1B and EV1C). This indicates that GNL3 has no great influence on replication fork progression during brief treatments with CPT. When the cells were treated with CPT for 1, 2, and 4 h (Fig EV1D), CPT treatment induced rapid phosphorylation of the DNA damage response kinase Chk1 on Ser 345, as expected; however, the kinetics of its phosphorylation was not markedly affected by GNL3 depletion, further supporting our conclusion that GNL3 does not affect fork progression in response to CPT. By contrast, after 4 h of treatment with CPT, the level of phosphorylation of RPA on both Ser 33 and Ser 4/8 was higher in the absence of GNL3 than in the controls (Fig EV1D). To determine if this effect was specific to CPT, we performed the same experiment but treated the cells with HU or etoposide (ETP), a topoisomerase 2 inhibitor. Treatment with 5 mM HU or 10 μM ETP‐induced phosphorylation of Chk1 on serine 345 in control cells but, as with CPT, no obvious difference was seen when GNL3 was depleted (Figs 1C and EV1E). Also, as with CPT, we observed stronger phosphorylation of RPA on Ser 33 and Ser 4/8 in the absence of GNL3 than in control cells after 4 h treatment with HU (Fig 1C) and after 2 h treatment with ETP (Fig EV1E). Thus, we hypothesized that GNL3 depletion may not impact replication stress signaling through Chk1 but, rather, the stability of stalled replication forks, since RPA phosphorylation is a marker of DNA resection (Soniat et al, 2019). Several proteins, including BRCA1, BRCA2, and FANCD2, have been shown to protect nascent DNA from resection in response to replication stress (Rickman & Smogorzewska, 2019). To test if GNL3 protects nascent strand DNA, we sequentially labeled cells with IdU and CldU for 30 min each and then treated the cells with HU for 4 h (Fig 1D). In the controls, the CldU/IdU ratio was close to 1, indicating that the nascent DNA was protected from extensive degradation, as expected. In cells depleted of GNL3, by contrast, the CldU/IdU ratio was significantly lower (Figs 1D and EV1F), indicating DNA resection at the fork by nuclease(s). Likewise, we saw similar effects in response to CPT (Fig EV1G) and ETP (Fig EV1H), consistent with the increased level of RPA phosphorylation induced by these agents in GNL3‐depleted cells.
Figure EV1. GNL3 prevents DNA resection of stalled replication forks (related to Fig 1).
- Scheme explaining the mini esiRNA screen to determine the best candidates for further characterization. HCT116 cells were transfected in 96‐wells plate with each esiRNA from the library (Dataset EV1). After 48 h cells were treated with 1 μM camptothecin for 4 h and subjected to immunofluorescence using an antibody directed against γH2A.X. The level of γH2A.X within nuclei was analyzed using a Celigo high‐throughput microscope.
- The γH2A.X level upon depletion and camptothecin treatment in five biological replicates was used to rank the candidates, GNL3 was ranked first and EGFP (negative control) was ranked at the end of the list (21st). The bounds of the box are the 25th and 75th percentiles, the line in the center is the median and the bounds of the whiskers are the maxima and the minima.
- Graphic representation (related to Fig 1B) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of HeLa S3 cells treated with 1 μM camptothecin (CPT) during the indicated time.
- Western‐blot analysis of HeLa S3 cells treated with 10 μM etoposide (ETP) during the indicated time.
- Graphic representation (related to Fig 1D) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 1 μM CPT for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. At least 100 individual DNA fibers was counted for each condition, the graphic representation of the medians of CldU/IdU ratios in three biological replicates with the average indicated in red is shown.
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 10 μM ETP for 120 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. At least 100 individual DNA fibers was counted for each condition, the graphic representation of the medians of CldU/IdU ratios in three biological replicates with the average indicated in red is shown.
- Western‐blot analysis of HeLa S3 cells depleted for GNL3, MRE11, CtIP or EXO1.
- Graphic representation (related to Fig 1E) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of Flp‐in T‐Rex HeLa cells expressing GNL3‐WT tagged with FLAG. Cells were first transfected with siControl or siGNL3 for 48 h then expression of GNL3‐WT (resistant to the siRNA against GNL3) was induced using 10 μg/ml of doxycycline (DOX) for 16 h.
- Graphic representation (related to Fig 1F) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of the indicated proteins upon chromatin fractionation of Hela S3 cells treated 4 h with 5 mM HU.
Source data are available online for this figure.
Figure 1. GNL3 prevents DNA resection of stalled replication forks.
- Western‐blot analysis of HeLa S3 cells depleted with a pool of four siRNA‐targeting GNL3 (siGNL3) or not (siControl).
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU with or without 1 μM CPT. Ratios between CldU and IdU are plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV1C.
- Western‐blot analysis of HeLa S3 cells treated with 5 mM HU during the indicated time.
- HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV1F.
- HeLa S3 were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. At least 80 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV1J.
- Flp‐in T‐Rex HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV1L.
- Experimental set‐up of iPOND experiment.
- iPOND experiment analyzed by Western‐blot. Cells were pulsed with 15 min EdU and chased for 2 h with 10 μM thymidine or 5 mM HU. In no click sample, biotin‐TEG azide was replaced by DMSO.
Source data are available online for this figure.
The resection observed in the absence of fork protectors is most probably initiated by the nuclease activities of MRE11, CtIP, and EXO1 (Rickman & Smogorzewska, 2019). To test further the function of GNL3 as a fork protector, we depleted GNL3 and MRE11, GNL3 and CtIP, or GNL3 and EXO1 and found that loss of the nucleases prevented the resection seen upon depletion of GNL3 alone (Figs 1E and EV1I and J), further supporting our conclusion that GNL3 protects nascent strand degradation by nucleases. To show definitively that GNL3 protects against DNA resection at stalled replication forks, we depleted the endogenous GNL3 with a specific siRNA and complemented its function by expressing an siRNA‐resistant, doxycycline (DOX)‐inducible GNL3‐FLAG gene in Flp‐In T‐Rex HeLa cells (Fig EV1K). We treated these cells with HU and analyzed the level of resection by IdU and CldU incorporation, as before. Expression of siRNA‐resistant GNL3‐FLAG suppressed almost completely the increased resection due to GNL3 depletion (Figs 1F and EV1L).
Other proteins known to protect replication forks (BRCA1, RAD51, and FANCD2, for example) accumulate on HU‐stalled forks (Lossaint et al, 2013; Dungrawala et al, 2015; Zellweger et al, 2015), suggesting that they may protect them directly from the action of nucleases. To determine whether GNL3 protects stalled replication forks from nucleases in the same way, we used iPOND to identify the proteins on nascent DNA. Cells were pulse labeled for 15 min with EdU and then chased for 2 h with thymidine or with HU (Fig 1G). As already shown (Sirbu et al, 2011; Dungrawala et al, 2015), treatment with HU increased the recruitment of RAD51 (Fig 1H). By contrast, recruitment of GNL3 was strongly decreased in response to HU, as was PCNA (Fig 1H), indicating that GNL3 does not accumulate at stalled forks. This suggests that the ability of GNL3 to protect from resection may not rely on direct protection from nucleases. Since GNL3 is required for RAD51 recruitment upon 24 h treatment with HU (Meng et al, 2013), it may be possible that GNL3 regulates RAD51 recruitment in response to HU to prevent resection. However, we failed to detect any impact of RAD51 recruitment on chromatin upon GNL3 depletion (Fig EV1M). Also, we could not see any impact of GNL3 depletion on the recruitment of BRCA1 and RIF1 (Fig EV1M) two known fork protectors (Schlacher et al, 2011; Mukherjee et al, 2019). From our experiments we conclude that GNL3 does not protect directly stalled forks nor is required for the recruitment of known forks protectors.
GNL3 depletion increases the firing of replication origins
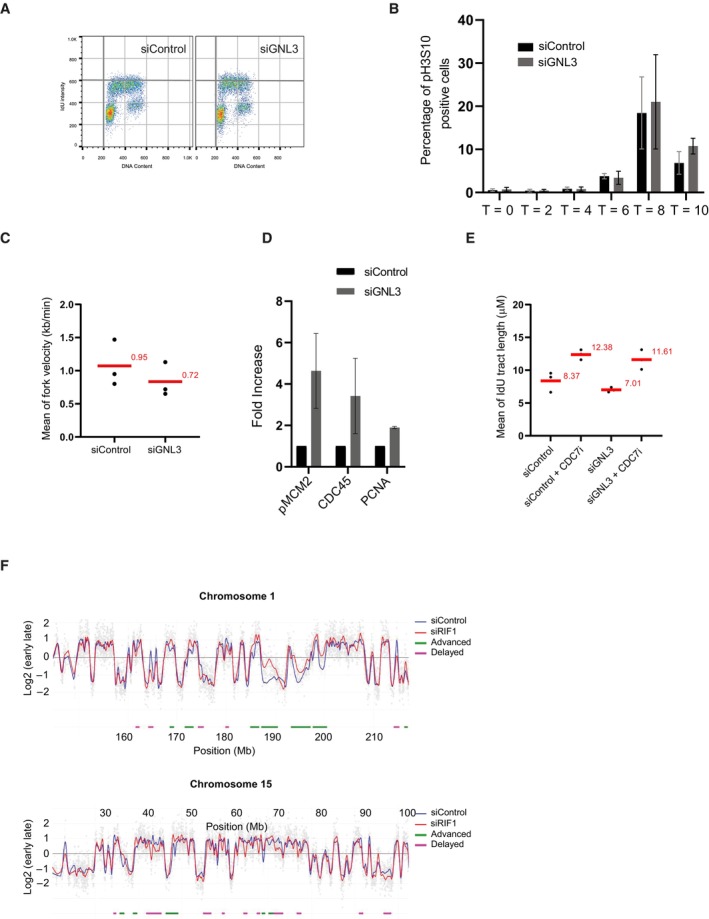
To try to understand how GNL3 might protect stalled replication forks from resection we analyzed the impact of GNL3 depletion on DNA replication in basal conditions. We found no obvious effect of GNL3 depletion, however, either on the distribution of cells in various phases of the cell cycle whether in an unsynchronized population (Fig EV2A) or in a population synchronized with a thymidine block and released into S‐phase (Fig 2A). To confirm this conclusion, we measured the length of S phase by examining the timing of entry into mitosis after a thymidine block, as indicated by phosphorylation of histone H3 on Ser 10 (Prigent & Dimitrov, 2003). Confirming that the length of S phase was unaffected by GNL3 depletion, no sign of early mitotic entry was detected 8 h after release (Fig EV2B). Ten hours after release, however, we noticed a small increase in the percentage of pH3S10‐positive cells in GNL3‐depleted cells when compared to the control, suggesting the cells accumulate in mitosis in the absence of GNL3, a phenomenon observed also in breast cancer cells lacking GNL3 (Lin et al, 2014). In those cells, loss of GNL3 increased the number of foci containing the DNA damage response protein 53BP1 (Yamashita et al, 2013; Lin et al, 2014), potentially an indicator of incomplete replication (Harrigan et al, 2011). To test if GNL3 depletion perturbs DNA replication, we analyzed its dynamic with DNA combing (Fig 2B): we labeled the cells with IdU for 20 min and then with CldU, for 20 min and observed that GNL3 depletion reduced fork velocity by about 25% (Figs 2C and EV2C). Since the length of S phase is not affected by GNL3 depletion, this may reflect a change in the number of active replication origins. To investigate this possibility, we determined the number of forks per megabase of combed DNA by using a highly accurate assay named Global Instant Fork Density (GIFD). This method considers cell cycle distribution and indicates precisely the number of active forks which directly reflects the density of fired replication origins (Bialic et al, 2015). A significant increase in the number of forks per megabase in GNL3‐depleted cells indicated that indeed more origins fire in the absence of GNL3 than in control cells (Fig 2D). To confirm this observation, we isolated the chromatin from cells depleted of GNL3 and from control cells and analyzed the presence of markers of origin firing by western blotting. We found more CDC45, MCM2 phosphorylated at Ser 40/41 (pMCM2 S40/41) and PCNA in the chromatin fraction of cells depleted of GNL3 than in control cells (Figs 2E and EV2D) confirming that more origins are firing in the absence of GNL3. The increased level of origin firing may be a consequence of reduced fork velocity (Conti et al, 2007). To determine if the primary effect of GNL3 impairment is on replication origin firing, we chemically inhibited CDC7 to limit replication origin firing (Montagnoli et al, 2008; Rodriguez‐Acebes et al, 2018). As expected, pre‐treatment with CDC7 inhibitor increased the length of IdU tracts (Figs 2F and EV2E) in control conditions to compensate for reduced origin activation. Strikingly, the impact of CDC7 inhibition was similar in the absence of GNL3 (Figs 2F and EV2E). This results strongly suggests that the decreased fork velocity observed in the absence of GNL3 is a consequence of the increased firing of replication origins. To investigate whether GNL3 affects the firing of replication origins globally or only at specific regions, as does RIF1 (Yamazaki et al, 2012), we analyzed the effect of GNL3 depletion on replication timing. As expected from previous studies (Cornacchia et al, 2012; Yamazaki et al, 2012), depletion of RIF1 had a substantial impact on replication timing; some regions were delayed and others advanced when compared to the control (Fig EV2F). GNL3 depletion, by contrast, had little or no effect on replication timing (Fig 2G). We conclude that GNL3 depletion increases the firing of replication origins globally without affecting the replication timing.
Figure EV2. GNL3 depletion increases the firing of replication origins (related to Fig 2).
- Flow cytometry experiment of HeLa S3 cells. Nascent DNA was labeled with IdU and total DNA stained with propidium iodide.
- Percentage of HeLa S3 cells positive for histone H3 phosphorylated on Histone 10 (pH3S10) upon thymidine block and release. The error bars represent the standard deviation between three biological replicates.
- Graphic representation (related to Fig 2C) of the average fork velocity in three biological replicates, the average is indicated in red.
- Quantification of chromatin fractionation based on three biological replicates (related to Fig 2E), the error bars represent the standard deviation.
- Graphic representation (related to Fig 2F) of the medians of IdU lengths in three biological replicates, the average is indicated in red. Average length of IdU tracts (indicated in red) in three independent experiments. The average value is indicated in red.
- Loss of RIF1 has effect on replication timing in specific genomic loci. Cells were pulse‐labeled with BrdU for 90 min and sorted by flow cytometry in two fractions, S1 and S2, corresponding to early and late S‐phase. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies. Early and late neo‐synthesized DNAs were labeled with Cy3 and Cy5 and hybridized on microarrays. After processing analyzing with the START‐R software, replication‐timing profiles can be obtained from two replicates. Shown are the zoomed microarray profiles of the timing of replication on chromosome 1 and chromosome 15 as example. Blue lines represent replication timing from siControl cells and red lines represent siRIF1 cells and gray spots represent the log ratio intensity for each probe of the microarray. Significantly disturbed regions are detected by START‐R software and advanced regions are indicated with green line and delayed regions by a pink line.
Figure 2. GNL3 depletion increases the firing of replication origins.
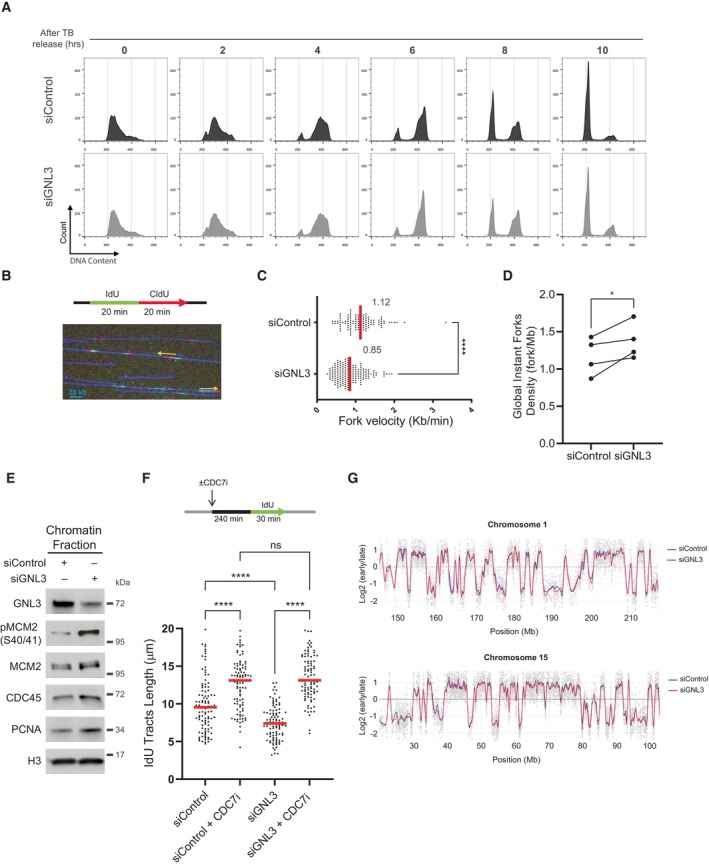
- Analysis of HeLa S3 cells subjected to thymidine block and released at different timepoints by flow cytometry. DNA was stained with propidium iodide.
- DNA combing experiment. HeLa S3 cells were subjected to two consecutive 20 min pulses of IdU and CldU and analyzed by DNA combing. A representative microscopy image of combed DNA molecules containing IdU (red) and CldU (green) tracks in presented with arrows indicating the direction of replication.
- Analysis of replication forks velocity by DNA combing. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. At least 100 individual replication forks were counted for each condition, biological replicates are shown in Fig EV2C.
- Analysis of GIFD (Global Instant Fork Density) by DNA combing in HeLa S3 cells. For statistical analysis paired t‐test was used; *P < 0.05. The error bars indicate the standard deviation.
- Western‐blot analysis of the indicated proteins upon chromatin fractionation.
- HeLa S3 cells were pretreated for 4 h with 10 μM of CDC7 inhibitor PHA‐767491 and labeled for 30 min with IdU. The length of IdU tracts is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. At least 100 individual replication forks were counted for each condition, and biological replicates are shown in Fig EV2E.
- Replication timing experiment. HeLa S3 cells were pulse‐labeled with BrdU for 90 min and sorted by flow cytometry in two fractions, S1 and S2, corresponding to early and late S‐phase. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies. Early and late neo‐synthesized DNAs were labeled with Cy3 and Cy5 and hybridized on microarrays. After analyzing with the START‐R software, replication‐timing profiles can be obtained from two replicates. Shown are the zoomed microarray profiles of the timing of replication on chromosome 1 and chromosome 15 as example. Blue lines represent replication timing from siControl cells, and red lines represent siGNL3 cells and gray spots represent the log ratio intensity for each probes of the microarray. Any significantly disturbed regions are detected by START‐R software.
Source data are available online for this figure.
DNA resection in the absence of GNL3 is a consequence of increased origin firing
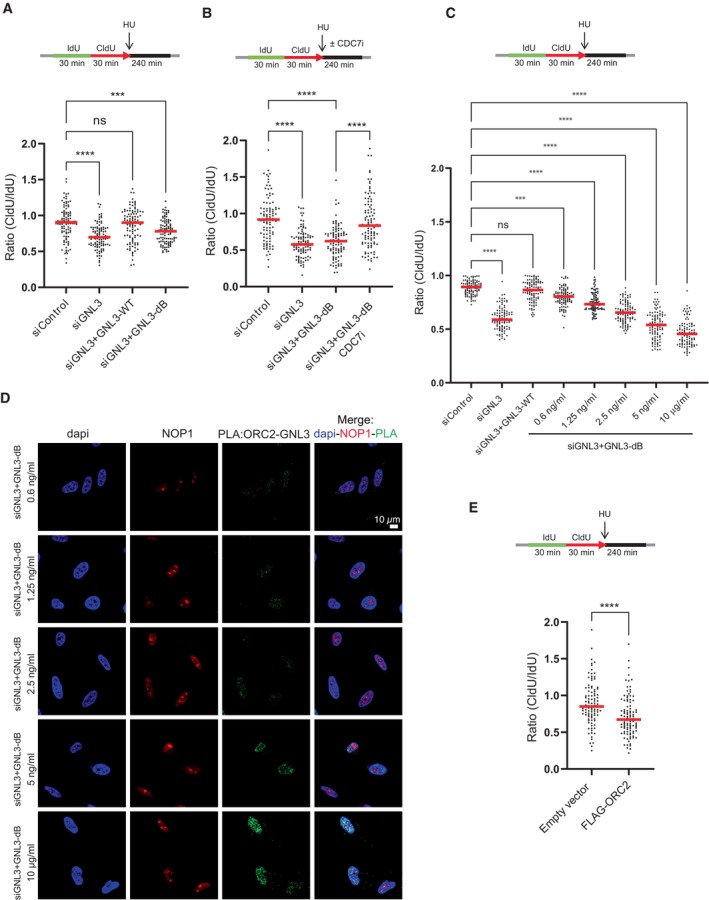
So far, we show that GNL3 depletion increases replication origin firing and increases DNA resection in response to exogenous inducers of replication stress. Interestingly, the inhibition of WEE1 or ATR increases replication origin firing (Beck et al, 2012; Moiseeva et al, 2017, 2019) and induces DNA lesions in response to HU (Toledo et al, 2013). Importantly, this phenotype is partially suppressed by inhibition of origin firing (Toledo et al, 2013), suggesting that increased resection may be a consequence of increased origin firing. We therefore tested the effect of inhibiting ATR or WEE1 on resection in response to HU by sequentially labeling cells with IdU and CldU and then treating them with HU for 4 h, as before, but in the presence of an inhibitor of ATR or an inhibitor of WEE1 (Figs 3A and EV3A). As predicted, inhibition of ATR (Figs 3B and EV3B) or inhibition of WEE1 (Figs 3C and EV3B) increased resection in response to HU (Elbaek et al, 2022; Leung et al, 2023). Moreover, inhibiting the increased origin firing with an inhibitor of CDC7, partially reversed this effect (Figs 3B and C, and EV3B and C). This experiment demonstrates that limiting the number of origins that fire is crucial to preventing resection in response to replication stress. If so, inhibiting origin firing might suppress the HU‐induced resection observed upon GNL3 depletion. To test this, we sequentially labeled cells with IdU and CldU for 30 min each and then treated them with HU for 4 h in the presence of an inhibitor of CDC7 to inhibit replication origin firing. Resection was strongly decreased when CDC7 was inhibited, indicating that in the absence of GNL3 an excess of origin firing in response to HU accounts for the increased resection (Figs 3D and EV3D). Consistent with the decrease in DNA resection, CDC7 inhibition also decreased the phosphorylation of RPA on Ser4/8 (Figs 3E and EV3E).
Figure 3. DNA resection in the absence of GNL3 is a consequence of increased origin firing.
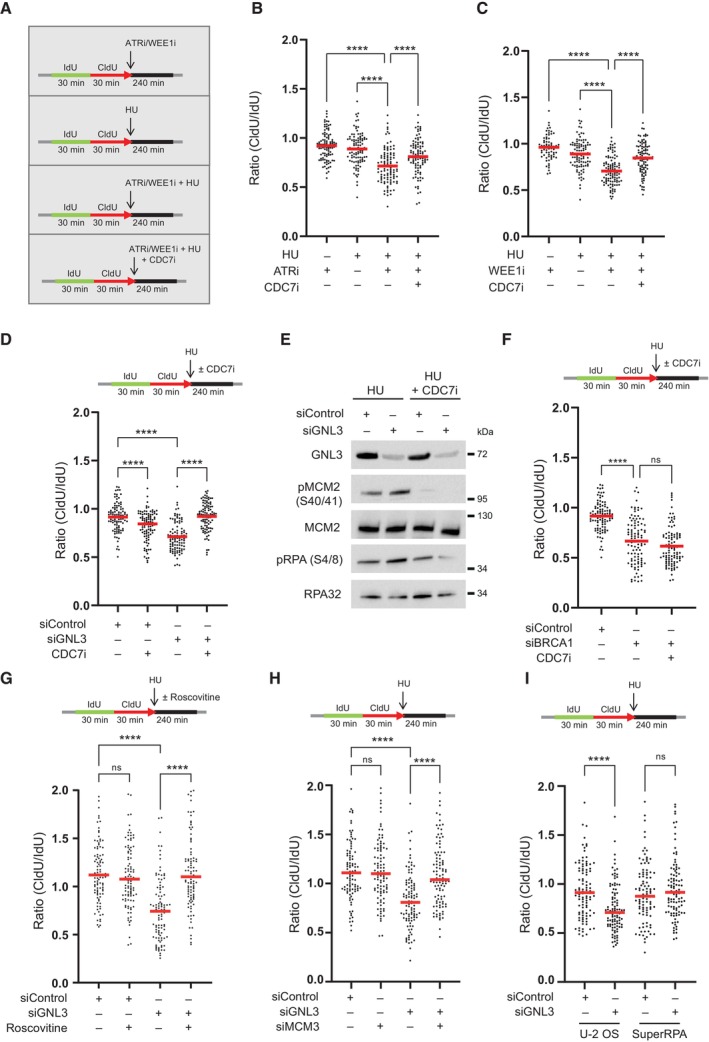
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated or not with 5 mM HU for 240 min with or without 10 μM of ATR VE‐821 inhibitor, 500 nM of WEE1 inhibitor AZD1775 or 10 μM of CDC7 inhibitor PHA‐767491.
- The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV3B.
- The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann‐Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV3C.
- HeLa S3 was sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min with or without 10 μM of CDC7 inhibitor PHA‐767491. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV3D.
- Western‐blot analysis of the indicated proteins upon treatment with 5 mM HU for 240 min with or without 10 μM of CDC7 inhibitor PHA‐767491.
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min with or without 10 μM of CDC7 inhibitor PHA‐767491. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. 100 individual DNA fibers were counted for each condition, andbiological replicates are shown in Fig EV3G.
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min with or without 20 μM of Roscovitine. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV3H.
- HeLa S3 cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV3J.
- Control U‐2 OS cells (U‐2 OS) or U‐2 OS cells that overexpress the three RPA subunits (SuperRPA) were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, and the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. ns, not significant. 100 individual DNA fibers were counted for each condition, biological replicates are shown in Fig EV3L.
Source data are available online for this figure.
Figure EV3. DNA resection in the absence of GNL3 is a consequence of increased origin firing (related to Fig 3).
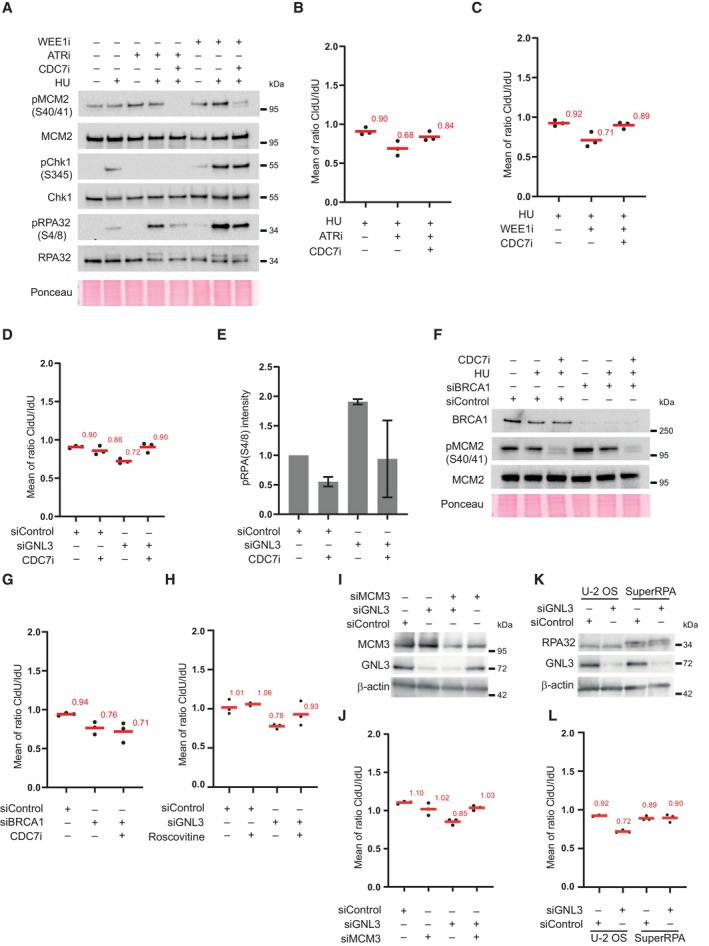
- Western‐blot analysis upon treatment with HU and inhibition of CDC7, WEE1, or ATR.
- Graphic representation (related to Fig 3B) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Graphic representation (related to Fig 3C) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Graphic representation (related to Fig 3D) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Quantification of RPA phosphorylation (S4/8) upon GNL3 depletion and CDC7 inhibition. The error bars represent the standard deviation between three biological replicates.
- Western‐blot analysis of HeLa S3 cells depleted or not for BRCA1 upon treatment with CDC7 inhibitor.
- Graphic representation (related to Fig 3F) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Graphic representation (related to Fig 3G) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of HeLa S3 cells.
- Graphic representation (related to Fig 3H) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of control U‐2 OS cells (U‐2 OS) and U‐2 OS cells that express the three RPA subunits (SuperRPA).
- Graphic representation (related to Fig 3I) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
BRCA1 is recruited to HU‐stalled forks (Dungrawala et al, 2015) and its depletion increases DNA resection induced by HU (Schlacher et al, 2012). BRCA1 is thought to protect stalled forks from resection by directly blocking nucleases. If this is the case, inhibition of CDC7 should have no effect on protection by BRCA1. To test this prediction, we depleted cells of BRCA1 and measured the level of resection in the absence or presence of the CDC7 inhibitor. As expected, depletion of BRCA1 increased resection; treatment with CDC7 inhibitor, however, did not decrease the level of resection (Figs 3F and EV3F and G). These data strongly suggests that fork protection by GNL3 differs mechanistically from fork protection by BRCA1.
To ensure that the observations were not due to possible side effects of the CDC7 inhibitor, we inhibited origin firing with roscovitine, a CDK inhibitor (Petermann et al, 2010; Toledo et al, 2013). We sequentially labeled cells with IdU and CldU for 30 min each and then treated them with HU for 4 h in the presence of roscovitine. Resection was significantly decreased in the presence of roscovitine, confirming that the resection occurring upon GNL3 depletion in the presence of HU is due to an excess of origin firing (Figs 3G and EV3H). Finally, we reduced the number of available origins by depleting MCM3, a subunit of the MCM complex. Partial depletion of MCM3 significantly decreased the level of resection upon GNL3 depletion in response to HU (Figs 3H and EV3I and J). We conclude that the resection occurring in absence of GNL3 in response to HU is due to an excess of replication origins.
One attractive hypothesis to explain how an excess of fired replication origins would increase resection in response to HU is the exhaustion of the pool of RPA due to the excessive activation of replication origins in a GNL3‐depleted background. To test this hypothesis, we took advantage of the Super RPA cell line that expresses 2‐ to 3‐fold excess of all three RPA subunits (Toledo et al, 2013). We sequentially labeled cells with IdU and CldU for 30 min each and then treated them with HU for 4 h in the control U‐2 OS cell line and in the SuperRPA cell line. Depletion of GNL3 induced resection upon HU treatment in the U‐2 OS cell line as predicted (Figs 3I and EV3K and L). In contrast, the depletion of GNL3 in the SuperRPA cell line did not induce significant resection upon HU treatment. We conclude that the enhanced resection observed upon GNL3 depletion and HU treatment is a consequence of increased origin firing that induces RPA exhaustion.
GNL3 interacts with ORC2 in the nucleolus
To understand how GNL3 might influence replication origin firing, we used proximity‐dependent biotinylation identification (BioID; Roux et al, 2012) to identify the proteins in proximity to GNL3 by mass spectrometry. We established a Flp‐In T‐Rex HEK293 cell line expressing a DOX‐inducible GNL3 cDNA fused to the biotin ligase BirA and FLAG. Upon induction with DOX for 16 h, we observed by immunofluorescence microscopy GNL3‐BirA‐FLAG mainly in the nucleolus (Fig EV4A). Moreover, by using streptavidin conjugated to Alexa Fluor 488 to detect exogenous biotin, we observed a strong signal (Fig EV4A) demonstrating that GNL3‐BirA‐FLAG is well localized and can biotinylate proteins in its proximity. In four independent experiments, we induced expression of GNL3‐BirA‐FLAG with DOX for 16 h and labeled proteins in its proximity with exogenous biotin for 4 h. Then we purified the biotinylated proteins on streptavidin beads and analyzed them by mass spectrometry. We calculated the LogRatio of the peptides detected upon addition of DOX and biotin compared to the peptides detected in the negative controls (treatment with either DOX or biotin alone) and represented the data in a Volcano plot (Fig 4A). As expected, GNL3 was highly enriched as well as several nucleolar proteins that are known to be in proximity (e.g., GNL3L, GNL2, DDX21, Ki67 or NPM1). Notably, enrichment of ORC2, one of the components of the origin recognition complex, suggested a possible mechanism in the regulation of replication origin firing by GNL3. To confirm the association of ORC2 with GNL3, we immunoprecipitated each of the proteins and analyzed the immunoprecipitates by western blotting; we found GNL3 in immunoprecipitates of ORC2 and vice versa (Fig 4B). Mass spectrometry analysis of the proteins that co‐immunoprecipitated when using a specific antibody against ORC2 confirmed the presence of GNL3 and most of the ORC subunits, whereas immunoprecipitation with an irrelevant control IgG contained neither GNL3 nor ORC subunits. Moreover, there was a significant overlap between the co‐immunoprecipitated proteins and those found by BioID of GNL3: among the 88 proteins significantly enriched by BioID, 35 were found by coimmunoprecipitation with ORC2 (Fig EV4B) and most of them (24/35) are proteins localized in the nucleolus. This suggests that at least a subset of ORC2 might be localized in the nucleolus and that the interaction between ORC2 and GNL3 is likely to occur in this compartment. The association of GNL3 with chromatin (Fig 2E), however, suggests that GNL3 and ORC2 may also interact at or near replication origins. To test this, we performed GNL3 chromatin immunoprecipitation followed by deep sequencing (ChIP‐seq) and found 3,412 binding sites for GNL3. We compared these binding sites with ORC2‐binding sites (Miotto et al, 2016), but found no significant overlap (Figs 4C and EV4C), indicating that the GNL3–ORC2 interaction may occur mostly in the nucleolus rather than on vicinity of replication origins. To test this, we analyzed the GNL3‐ORC2 interaction using proximity ligation assay (PLA) and found most foci at the border of regions that stained lightly with DAPI and that correspond to nucleoli (Fig EV4D), thus supporting our hypothesis. The PLA signal was strongly decreased upon depletion of GNL3, validating its specificity. To validate that the interaction between GNL3 and ORC2 is occurring in the nucleolus, we labeled the nucleolus using an antibody directed against NOP1, a nucleolar protein, before performing PLA between ORC2 and GNL3 (Fig 4D). We could observe a good colocalization between GNL3‐ORC2 PLA signal and NOP1, confirming that GNL3 and ORC2 interact mainly in the nucleolus.
Figure EV4. GNL3 interacts with ORC2 in the nucleolus (related to Fig 4).
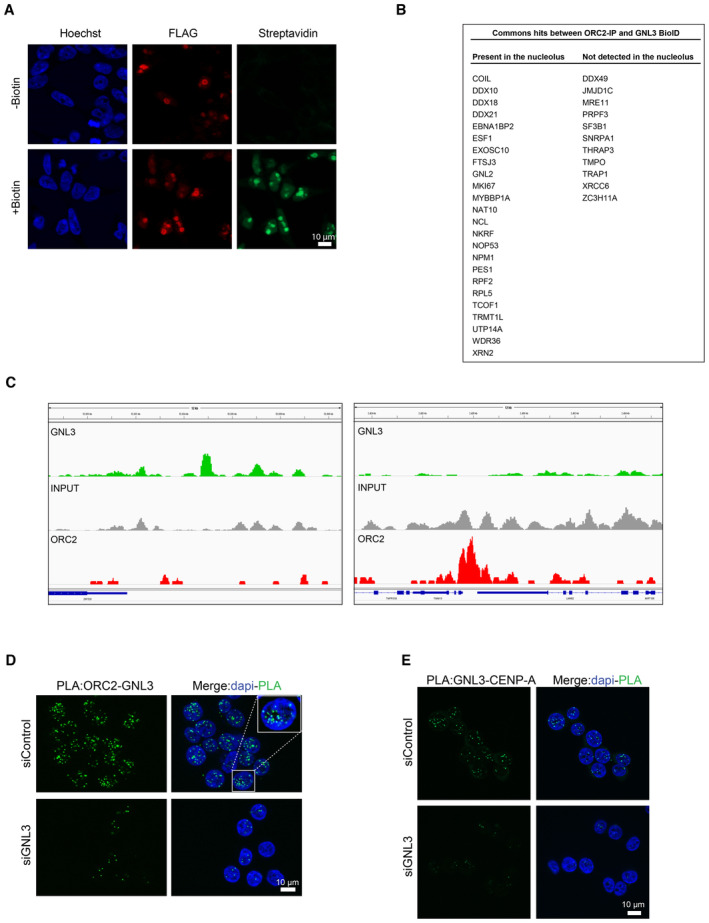
- Immunofluorescence experiment in Flp‐In T‐Rex HEK293 cells showing the expression of GNL3‐BirA‐FLAG and the biotinylation by BirA (revealed by streptavidin coupled with Alexa‐488) upon addition of biotin.
- Common hits found by mass spectrometry between ORC2 immunoprecipitation and GNL3 BioID. The localization was determined using The Human Protein Atlas database (https://www.proteinatlas.org/).
- Examples of GNL3 peaks on chromosome 19 obtained by ChIP‐seq of GNL3, INPUT is shown as negative control. The ORC2 ChIP‐seq data are obtained from (Miotto et al, 2016).
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3 in HeLa S3 cells.
- PLA (proximity ligation assay) analyzing the proximity between GNL3 and CENP‐A in HeLa S3 cells.
Figure 4. GNL3 interacts with ORC2 in the nucleolus.
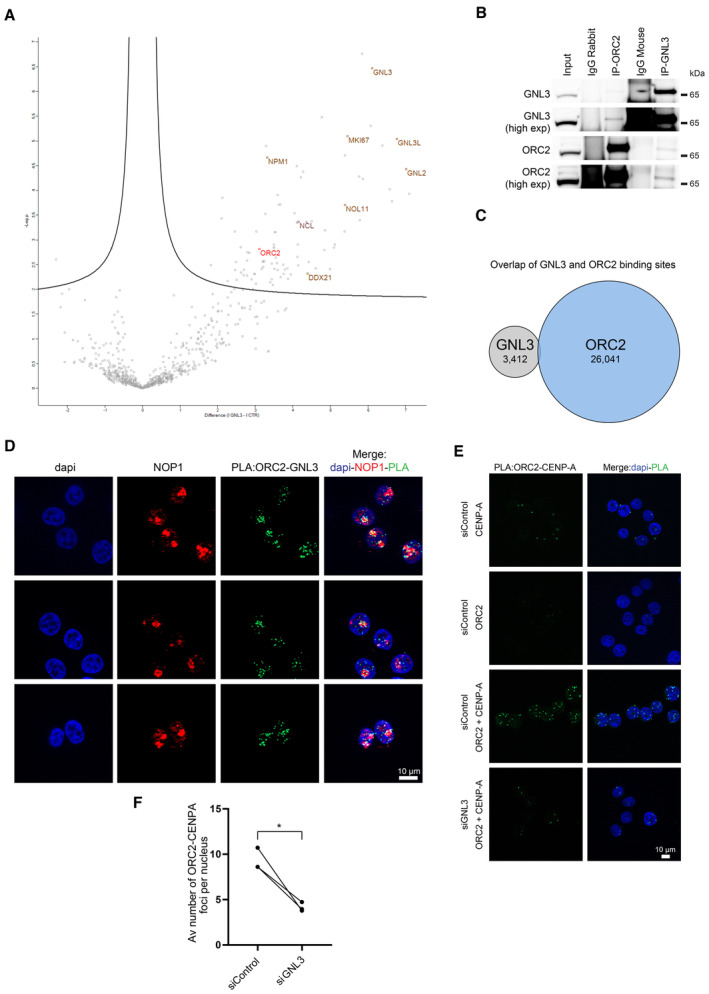
- GNL3‐BioID experiment analyzed by mass spectrometry. Expression of GNL3‐BirA‐FLAG in HEK293 Flp‐in cells was induced with doxycycline for 16 h then biotin was added for 4 h. For negative controls cells were treated 16 h with doxycycline alone or 4 h with biotin alone. Four biological replicates were analyzed by mass spectrometry. Label‐free quantification was performed using MaxQuant (Cox & Mann, 2008) and statistical analysis using Perseus (Tyanova et al, 2016). The volcano plot shows the proteins that are significantly (two‐tailed t‐test, false discovery rate = 0.05) enriched upon induction of GNL3‐BirA‐FLAG and addition of biotin. The full list of proteins is available in Dataset EV2.
- Western‐blot analysis of GNL3 and ORC2 immunoprecipitates in K562 cells.
- Comparison of the genomic location of GNL3 and ORC2. Chromatin immunoprecipitation of GNL3 followed by deep sequencing was performed in HeLa S3. GNL3‐binding sites were compared to ORC2‐binding sites obtained from Miotto et al (2016).
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3 in HeLa S3 cells that are stained with an antibody directed against NOP1.
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and CENP‐A in HeLa S3 cells using the indicated antibodies.
- Graphic representation of the average number of PLA ORC2‐CENP‐A foci in three biological replicates. For statistical analysis, paired t‐test was used; *P < 0.05.
Source data are available online for this figure.
Our data strongly suggest the presence of a subset of ORC2 into the nucleolus. This is consistent with previous results regarding ORC2 role at centromeres independently of its function in the ORC complex (Prasanth et al, 2004; Huang et al, 2016; Bauwens et al, 2021) since centromeres are often localized in the vicinity of the nucleolus (Wong et al, 2007; Padeken et al, 2013; Peng et al, 2023). In support of this, we observed an interaction between GNL3 and the centromere‐specific histone H3 variant CENP‐A using PLA (Fig EV4E). We hypothesized that GNL3 may be required for the recruitment of ORC2 at centromeres, a possible readout of its nucleolar localization. To test this, we performed PLA between ORC2 and CENP‐A. As expected, many PLA foci of ORC2 and CENP‐A were found in normal cells when compared to controls treated with only the antibody against ORC2 or that against CENP‐A (Fig 4E). When the cells were depleted of GNL3, however, the average number of PLA foci per cell was reduced by about two‐fold (Fig 4F), indicating that ORC2 recruitment at centromeres depends in part on the availability of GNL3. We conclude that the presence of ORC2 at centromeres may reflect its nucleolar localization and could be regulated by GNL3 suggesting that GNL3 may regulate ORC2 subnuclear localization.
Accumulation of GNL3 into the nucleolus limits origin firing
GNL3 may use its long residency time in the nucleolus (Meng et al, 2007) to regulate ORC2 subnuclear localization and limits replication origin firing. To test this idea, we took advantage of a mutant of GNL3 (GNL3‐dB) that has a shorter residency time in the nucleolus and diffuses in the nucleoplasm (Fig 5A) (Tsai & McKay, 2002, 2005). We depleted endogenous GNL3 with a specific siRNA and expressed siRNA‐resistant, DOX‐inducible GNL3‐dB fused with FLAG in Flp‐In T‐Rex HeLa cells. GNL3‐dB had a level of expression comparable to GNL3‐WT (Fig 5B). We next checked GNL3‐WT and GNL3‐dB localization using immunofluorescence with an antibody directed against FLAG (Fig 5C) and confirmed that GNL3‐dB localization is not restricted to the nucleolus, as GNL3‐WT (Tsai & McKay, 2002, 2005). To test if this change in the nuclear distribution of GNL3 is affecting its interaction with ORC2, we performed PLA. Consistent with our previous observations (Figs 4D and EV4D), we found that exogenous GNL3‐WT interacts with ORC2 mostly in proximity of the nucleolus (Figs 5D and EV5A). Surprisingly, we observed that the interaction between GNL3‐dB and ORC2 occurred more frequently (Fig 5E) and was mainly in the nucleoplasm consistent with its localization (Fig EV5B). We conclude that when GNL3 is diffusing in the nucleoplasm, it increases its ability to interact with ORC2. Interestingly, by performing immunofluorescence experiment in the presence of cytoskeletal buffer (CSK) to remove soluble proteins, we observed that the signal corresponding to GNL3‐dB in the nucleoplasm was strongly reduced (Fig 5F). Since ORC2 is mostly associated with chromatin (Ohta et al, 2003), we postulate that GNL3‐dB interacts with a fraction of ORC2 that may not be localized to chromatin and could reflect a change in ORC2 subnuclear localization. To test if this putative change in ORC2 subnuclear localization is related with the regulation of replication origin firing, we measured the GIFD using DNA combing (Fig 5G). As shown before (Fig 2D), depletion of GNL3 increased GIFD. Expression of GNL3‐WT decreased GIFD to the level of the siControl as expected. In contrast, expression of GNL‐dB failed to decrease GIFD (Figs 5G and EV5C). From this experiment, we conclude that accumulation of GNL3 in the nucleolus is required to limit replication origin firing. We propose that the inability of GNL3‐dB to accumulate in the nucleolus may perturb the subnuclear localization of ORC2, which may impact the licensing of replication origins.
Figure 5. Accumulation of GNL3 into the nucleolus limits origin firing.
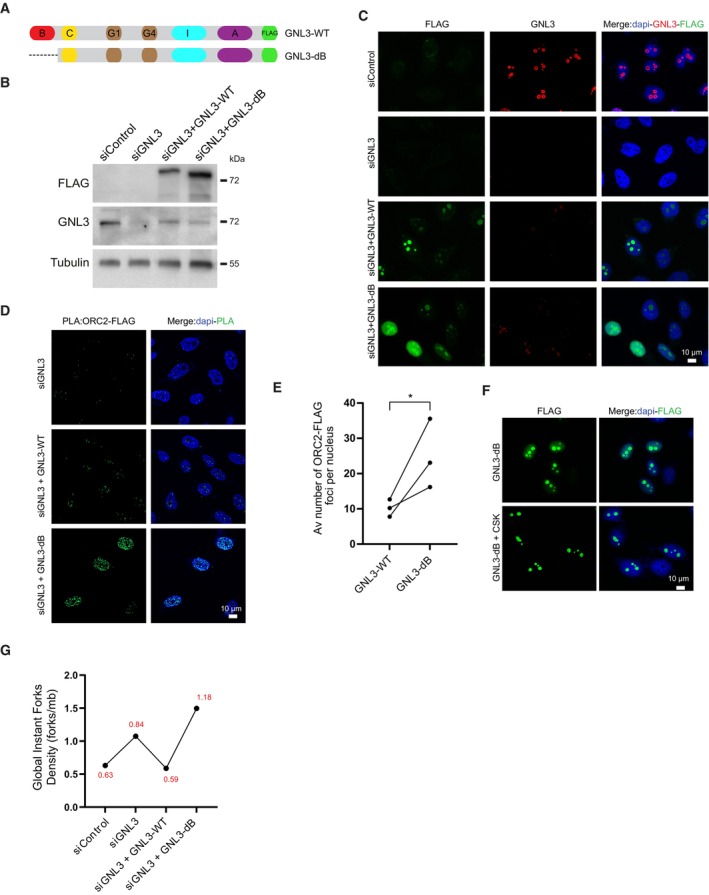
- Schematic representation of human GNL3 protein with its associated domains (B: basic domain; C: coiled‐coil domain; G1: GTP‐binding motif 1; G4: GTP‐binding motif 4; I: intermediate domain; A: acidic domain). GNL3‐WT and GNL3‐dB are fused with FLAG.
- Western‐blot analysis of Flp‐In T‐Rex HeLa cells expressing exogenous GNL3‐WT or GNL3‐dB. Cells were transfected with siControl or siGNL3 for 48 h then expression of exogenous GNL3‐FLAG (resistant to the siRNA against GNL3) was induced using 10 μg/ml of doxycycline for 16 h.
- Immunofluorescence analysis of Flp‐in T‐Rex HeLa cells expressing exogenous GNL3‐WT or GNL3‐dB.
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3‐FLAG or GNL3‐dB‐FLAG in HeLa Flp‐In cells upon doxycycline induction.
- Graphic representation of the average number of PLA ORC2‐FLAG foci in three biological replicates. For statistical analysis paired t‐test was used; *P < 0.05.
- Immunofluorescence experiment of HeLa Flp‐In cells expressing GNL3‐dB with or without pre‐extraction with cytoskeletal buffer (CSK).
- Analysis of GIFD (Global Instant Fork Density) by DNA combing in HeLa cells (n = 1; a biological replicate is shown in Fig EV5C). GIFD value is indicated in red.
Source data are available online for this figure.
Figure EV5. Accumulation of GNL3 into the nucleolus limits origin firing (related to Figs 5 and 6).
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3‐WT‐FLAG or GNL3‐dB‐FLAG in HeLa Flp‐In cells upon doxycycline induction using the indicated antibodies.
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3‐dB‐FLAG in HeLa Flp‐In cells upon doxycycline induction using the indicated antibodies.
- Biological replicate of the GIFD (Global Instant Fork Density) analysis performed in Fig 5G. GIFD value is indicated in red.
- Graphic representation (related to Fig 6A) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Graphic representation (related to Fig 6B) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Western‐blot analysis of Flp‐In T‐Rex HeLa cells expressing exogenous GNL3‐WT or GNL3‐dB mutants. Cells were transfected with siControl or siGNL3 for 48 h then the expression of GNL3‐WT and GNL3‐dB (resistant to the siRNA against GNL3) was induced using the indicated doses of doxycycline for 16 h.
- Immunofluorescence analysis of Flp‐in T‐Rex HeLa cells expressing GNL3‐dB at the indicated doses of doxycycline.
- Graphic representation (related to Fig 6C) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
- Immunofluorescence experiment using the indicated antibodies of Flp‐in T‐Rex HeLa cells expressing or not exogenous FLAG‐ORC2.
- Graphic representation (related to Fig 6E) of the medians of CldU/IdU ratios in three biological replicates, the average is indicated in red.
The control of ORC2 subnuclear localization limits DNA resection
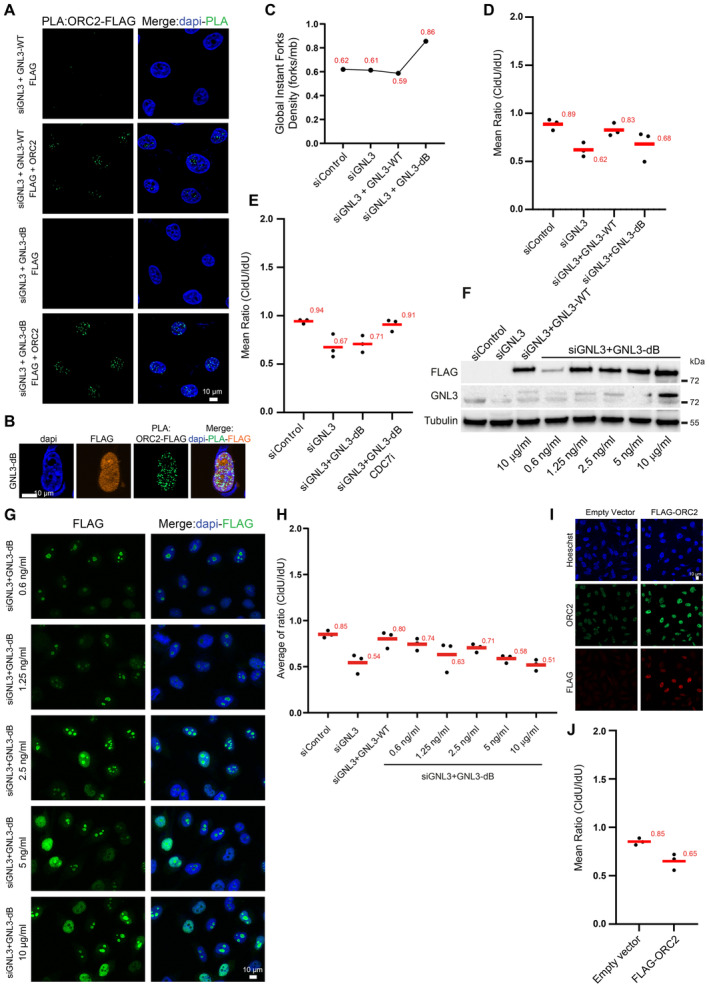
We showed that the major cause of DNA resection upon GNL3 depletion is due to increased origin firing. Since origin firing is augmented upon GNL3‐dB expression, DNA resection in response to HU should also increase. As already shown (Fig 1F), expression of GNL3‐WT almost completely suppressed the increased resection induced by GNL3 depletion (Figs 6A and EV5C). In contrast, GNL3‐dB, did not fully complement the increased resection due to GNL3 depletion (Figs 6A and EV5D). If the reason for increased resection upon GNL3‐dB expression is the upregulation of origin firing, its inhibition should decrease resection. Consistent with this hypothesis, inhibition of origin firing using CDC7 inhibitor reduced significantly the resection occurring upon GNL3‐dB expression and treatment with HU (Figs 6B and EV5E). We conclude that the ability of GNL3‐dB to increase origin firing is responsible for the increased resection upon HU treatment. We then wanted to check if this is due to a change in ORC2 subnuclear localization. To this purpose we modulated GNL3‐dB expression using different concentrations of DOX (Fig EV5F) and observed that the diffusion in the nucleoplasm was dependent on DOX concentration (Fig EV5G). Strikingly, we observed that the amount of resection was largely correlated with the level of expression of GNL3‐dB (Figs 6C and EV5H). Importantly, we observed that the level of interaction between GNL3‐dB and ORC2 was also largely dependent on the level of expression of GNL3‐dB (Fig 6D). We propose that the diffusion of GNL3 in the nucleoplasm perturbs the function of ORC2, a phenomenon that increases origin firing and induces DNA resection in response to HU. If this model is true, ORC2 overexpression by itself should also induce DNA resection in response to HU since its level in the nucleoplasm would be increased. To test this hypothesis, we transfected a plasmid containing ORC2 tagged with FLAG (Fig EV5I) and measured the level of DNA resection upon treatment with HU. Strikingly, we observed that overexpression of ORC2 by itself increases nascent DNA resection (Figs 6E and EV5J). This demonstrates that an excessive amount of ORC2 in the nucleoplasm induces DNA resection, a situation that may phenocopy the depletion of GNL3 or the expression of GNL3‐dB. We conclude that nucleolar localization of GNL3 is required to prevent excessive DNA resection in response to exogenous replication stress by possibly ensuring the correct subnuclear localization of ORC2.
Figure 6. The control of ORC2 subnuclear localization limits DNA resection.
- Flp‐in T‐Rex HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, the red line indicates the median. For statistical analysis Mann–Whitney test was used; ****P < 0.0001; ***P < 0.001; ns non‐significant. 100 individual DNA fibers were counted for each condition, biological replicates are shown in Fig EV5D.
- Flp‐in T‐Rex HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min with or without 10 μM of CDC7 inhibitor PHA‐767491. The ratio between CldU and IdU is plotted, the red line indicates the median. For statistical analysis Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, biological replicates are shown in Fig EV5E.
- Flp‐in T‐Rex HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, the red line indicates the median. For statistical analysis Mann–Whitney test was used; ****P < 0.0001; ***P < 0.001; ns non‐significant. 100 individual DNA fibers were counted for each condition, biological replicates are shown in Fig EV5H.
- PLA (proximity ligation assay) analyzing the proximity between ORC2 and GNL3‐dB‐FLAG in HeLa Flp‐In cells treated with indicated doses of doxycycline and stained with an antibody directed against NOP1.
- Flp‐in T‐Rex HeLa cells were sequentially labeled for 30 min with IdU and for 30 min with CldU then treated with 5 mM HU for 240 min. The ratio between CldU and IdU is plotted, the red line indicates the median. For statistical analysis, Mann–Whitney test was used; ****P < 0.0001. 100 individual DNA fibers were counted for each condition, and biological replicates are shown in Fig EV5J.
Source data are available online for this figure.
Discussion
GNL3/nucleostemin was first described as a nucleolar protein required for cell proliferation (Tsai & McKay, 2002), and several studies have highlighted its role(s) in maintaining genome integrity (Tsai, 2014). Here, we investigate the role of GNL3 in response to exogenous replication stress. We demonstrate that GNL3 protects stalled replication forks from resection by exonucleases and this protection depends on the number of replication origins that fire. We show that the long residency time of GNL3 in the nucleolus is required to protect stalled replication forks from resection by possibly regulating the sub nuclear localization of ORC2. We propose a model in which an excess of fired replication origins due to the absence of GNL3 induces DNA resection upon treatment with exogenous replication stress due to RPA exhaustion (Fig 7A).
Figure 7. GNL3 prevents resection of stalled replication forks by sequestrating ORC2 in the nucleolus.
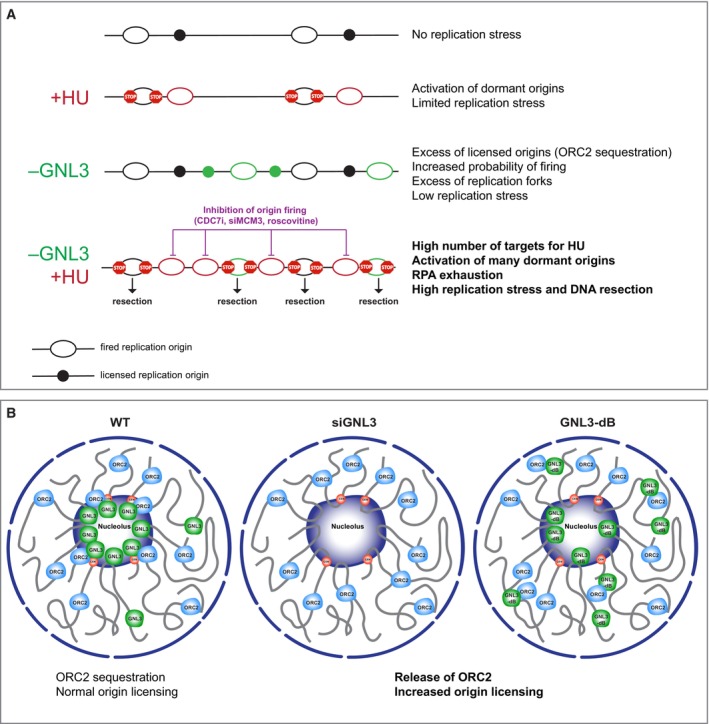
- HU‐treatment stalls active replication forks and activate dormant origins. When GNL3 is impaired, more origins may be licensed increasing the probability of replication origin firing. This could lead to over replication and replication stress. When GNL3‐deficient cells are treated with HU, the number of targets for HU is increased leading to activation of more dormant origins, a situation that leads to the exhaustion of the pool of available RPA that may explain the resection of nascent DNA. Inhibition of origin firing with CDC7 inhibitor, roscovitine or depletion of MCM3 counteracts this effect.
- Thanks to its ability to accumulate into the nucleolus, GNL3 may sequester a subset of ORC2 to precisely regulate origin licensing. When GNL3 is depleted (siGNL3) or does not accumulate in the nucleolus, ORC2 is released into the nucleoplasm leading to more licensed origins and DNA resection upon HU treatment.
GNL3 limits the firing of replication origins
GNL3 depletion induces activation of the DNA damage response during S phase and γH2A.X phosphorylation (Meng et al, 2013; Lin et al, 2014) indicating a strong level of replication stress. Consistent with this and as previously observed (Lin et al, 2014), we noticed a weak enrichment in cells in G2/M cells (Figs 2A and EV2B) that may account for incomplete replication due to replication stress (Harrigan et al, 2011). However, the reason for the endogenous replication stress upon GNL3 depletion was largely unknown. Here we propose that the increased replication origin firing caused by GNL3 depletion may be the major cause of endogenous replicative stress. The regulation of replication origin firing may be less constrained in absence of GNL3 leading to regions under‐ or over‐replicated (Blow et al, 2011) that may induce the accumulation of cells in G2/M. This is supported by the fact that replication timing is not affected upon GNL3 depletion, a measurement that does not represent stochastic variations between individual cells. In addition, recent data suggest that the firing of replication origins is more stochastic than previously thought (Klein et al, 2021; Wang et al, 2021). In addition, the reduced fork velocity observed upon GNL3 depletion may also impair the replication of specific regions. We detected GNL3 in the vicinity of replication forks using iPOND coupled with mass spectrometry (Lebdy et al, 2023) and Western‐blot (Fig 1F) suggesting that its depletion may directly impair fork speed. Yet, the recruitment of GNL3 at replication forks is low, and it was not detected by other iPOND screens performed in basal conditions (Lopez‐Contreras et al, 2013; Sirbu et al, 2013). In fact, GNL3 was found at nascent DNA only in FANCJ‐knockout cells that tend to heterochromatinize their genome (Schwab et al, 2013; Peng et al, 2018), suggesting that GNL3 may be in proximity of nascent DNA only in specific regions. Interestingly, GNL3 is localized in the vicinity of centromeres and in the proximity of nucleolar‐associated domains (NADs) that are both heterochromatic regions (Bersaglieri et al, 2022; Peng et al, 2023). Therefore, the weak association of GNL3 to nascent DNA may simply reflect its localization in proximity of heterochromatic regions undergoing DNA replication without participating in the replication process per se. This is supported by our recent findings showing that iPOND efficacy is biased by genome organization (Lebdy et al, 2023). Based on this, we propose that the slow replication fork speed observed upon GNL3 depletion is more likely a compensation mechanism of the increased replication origin firing rather than a direct impact on replication fork speed. This is supported by the fact that inhibition of replication origin firing suppresses the slower replication fork speed upon GNL3 depletion (Fig 2G). This model is also largely supported by the abundance of GNL3 in the nucleolus as well as its interaction with ORC2.
Excessive replication origin firing induces DNA resection in response to replication stress
We show that GNL3 protects nascent DNA at stalled replication forks from resection by nucleases. Yet GNL3 is not recruited to HU‐stalled forks, suggesting that it is not able to directly protect nascent DNA. In addition, the recruitment of RAD51, BRCA1, or RIF1, which are known forks protectors (Hashimoto et al, 2010; Schlacher et al, 2011; Mukherjee et al, 2019), was not impacted by GNL3 impairment. Finally, we conclude that the increased resection seen upon GNL3 depletion is related to the increased replication origin firing because it is suppressed by inhibition of regulators (CDC7 and CDK) of origin firing or by the depletion of MCM3. This conclusion is consistent with data showing that CDC7 inhibition prevents nascent strand resection (Sasi et al, 2018; Jones et al, 2021). We also found that inhibition of ATR or WEE1, both of which increase origin firing (Beck et al, 2012; Moiseeva et al, 2017, 2019), increases the resection of nascent DNA in a CDC7‐dependent manner. Collectively, these data suggest that when cells with an excess of fired replication origins are challenged with replication stress, it induces nascent DNA resection. Having more replication origins that fire means more replication forks that may be targeted by exogenous replication stress like HU, which in turn activates more dormant origins (Fig 7A). This situation may lead to the exhaustion of RPA causing resection as previously proposed for the occurrence of DNA lesions in the case of ATR or WEE1 inhibition (Toledo et al, 2013). Consistent with this, increased expression of the three RPA subunits significantly decreased the nascent DNA resection occurring in the absence of GNL3 upon HU treatment. We propose that DNA resection may be largely induced by excessive replication origin firing that causes the exhaustion of RPA. However, the nascent DNA resection that occurs in the absence of BRCA1 was not suppressed by CDC7 inhibition. This confirms a direct role for BRCA1 in protecting nascent DNA. Thus, we conclude that nascent DNA resection can be promoted either by loss of a protein that protects the DNA directly, like BRCA1, or by loss of proteins such as GNL3 that act indirectly and may induce RPA exhaustion due to excessive replication origin firing. BRCA1, FANCD2, and RAD51 were the first proteins found to act as fork protectors (Hashimoto et al, 2010; Schlacher et al, 2011, 2012). Since then, several other proteins have been found to protect stalled forks from resection by nucleases (Liao et al, 2018; Rickman & Smogorzewska, 2019; Berti et al, 2020). Given our findings here, it would be interesting to investigate whether the numerous proteins that protect stalled replication are acting directly or indirectly.
Nucleolar concentration of GNL3 is required to prevent DNA resection
To test if GNL3 localization has a role in preventing DNA resection, we expressed a mutant of GNL3, GNL3‐dB, that is diffusing in the nucleoplasm (Tsai & McKay, 2002, 2005). We conclude that the concentration of GNL3 in the nucleolus prevents DNA resection in response to replication stress. Since resection occurring upon GNL3‐dB expression is dependent on origin firing and expression of GNL3‐dB increases replication origin firing, it appears that the accumulation of GNL3 in the nucleolus limits origin firing, which in turns limits DNA resection in response to replication stress. One of the main features of GNL3 is its strong residency time in the nucleolus (Meng et al, 2007), this ability may allow GNL3 to regulate proteins involved in replication origin firing by nucleolar sequestration for instance (Wang et al, 2019). In addition to known nucleolar proteins, our GNL3 BioID screen uncovered ORC2 as one of the best hits. ORC2 is one of the components of the origin recognition complex and therefore is required for the licensing of replication origins. Therefore, in the absence of ORC2, less replication origins fire, consequently the inter‐origin distance is increased, and fork speed is increased (Shibata et al, 2016). We hypothesized that GNL3 may be able to regulate ORC2 subnuclear distribution to limit licensing, something that may explain the increase in replication origin firing we observed upon GNL3 depletion. GNL3 could also regulate ORC2 functions that are not directly linked with the ORC complex but that may influence licensing such as its role at centromeres (Prasanth et al, 2004; Huang et al, 2016; Bauwens et al, 2021), at nuclear pores (Richards et al, 2022) or in sister chromatid cohesion (Shimada & Gasser, 2007; MacAlpine et al, 2010). In support to this, we found that GNL3 depletion decreases ORC2 recruitment to centromeres. ORC2 SUMOylation is required to prevent re‐replication of pericentromeric heterochromatin (Huang et al, 2016), suggesting that GNL3 depletion may impair centromeres replication through ORC2 regulation. Since centromeres and replication origins may derive from a common ancestor (Hu & Stillman, 2023), it may also be possible that ORC2 localized at centromeres has a broader role in the control of DNA replication. We have found that GNL3‐dB interacts more frequently with ORC2 than GNL3‐WT and that this interaction occurs mostly in the nucleoplasm. GNL3‐dB may interact with ORC2 from the ORC complex localized on chromatin, however since GNL3‐dB is not associated with chromatin in the nucleoplasm we do not favor this possibility. We rather envision that the interaction between ORC2 and GNL3‐dB in the nucleoplasm may be due to a release of ORC2 from the nucleolus or from the chromatin. This is supported by the fact that the level of resection increases with the level of GNL3‐dB expression, something that could gradually increase the release of ORC2 from the nucleolus. Consistent with this, overexpression of ORC2 by itself increases DNA resection upon HU treatment. Based on our findings, our model is a sequestration of ORC2 by GNL3 to prevent excessive licensing, a possible cause of DNA resection in response to replication stress (Fig 7B). This may explain why both GNL3 depletion and expression of GNL3‐dB lead to nascent strand resection, since in both cases ORC2 may not be properly sequestrated in the nucleolus. More work is obviously required to fully demonstrate this model by analyzing for example the distribution of ORC2 on chromatin upon GNL3 loss at genome‐wide level.
The ability of GNL3 to prevent excessive firing of replication origins may also be caused by a more global role of GNL3 on nuclear organization possibly related to compartmentation. Although GNL3 is found only in chordates, it belongs to the family of YlqF‐related GTPases that is conserved in Eukarya, Bacteria and Archea (Reynaud et al, 2005; Mier et al, 2017; Quiroga‐Artigas et al, 2022). GNL3 is the more recent member of the family and seemed to have co‐evolved with sub compartments of the nucleolus. Growing evidence indicates that the nucleolus is involved in the 3D organization of the genome (Iarovaia et al, 2019) and particularly of centromeric DNA and heterochromatin (Wong et al, 2007; Padeken et al, 2013; Bersaglieri et al, 2022; Peng et al, 2023). Therefore, GNL3 may play a larger role in the organization of centromeres, or other regions of heterochromatin, by keeping them in proximity to the nucleolus thanks to its long residency time (Meng et al, 2006). For example, GNL3 may mediate interactions between nucleolus and heterochromatin as proposed for Ki‐67 (Sobecki et al, 2016; van Schaik et al, 2022) and NPM1 (Holmberg Olausson et al, 2014), two proteins localized to the nucleolar rim like GNL3 (Stenstrom et al, 2020). It has been recently shown that genome organization is a determinant of the locations of replication origins (Emerson et al, 2022), therefore GNL3 may have a broader role in the regulation of replication origin firing. GNL3 is an essential gene in mice (Beekman et al, 2006; Zhu et al, 2006) and GNL3‐null HeLa S3 clones we obtained using CRISPR‐Cas9 grew poorly preventing us from performing any experiments, this is consistent with a broader role for GNL3. Future studies will need to rely on inducible systems such as degrons and/or point mutants to fully understand the role(s) of GNL3 during replication.
Materials and Methods
Cell lines
HeLa S3 (ATCC CCL‐2.2), Flp‐In T‐Rex 293 (ThermoFisher R78007), HeLa Flp‐In T‐Rex (authenticated with Eurofins, gift from Jean‐Hugues Guervilly and Pierre‐Henri Gaillard, Centre de Recherche en Cancérologie de Marseille, France), and U‐2 OS cells (gift from Jiri Lukas, Novo Nordisk Foundation Center for Protein Research) were cultured in Dulbecco's modified Eagle's media (DMEM). HCT116 (obtained from SIRIC Montpellier Cancer) and K562 (authenticated with Eurofins) cells were cultured in Roswell Park Memorial Institute medium (RPMI). Culture media was supplemented with 10% fetal bovine serum (Biowest) and penicillin/streptomycin (Sigma‐Aldrich). Cells were incubated in a 5% CO2 at 37°C in in level 2 laboratory. Selection of integrated clones in Flp‐In cells was done using hygromycin and blasticidin.
Inhibitors, drugs, and antibiotics
The following reagents were used: etoposide (Sigma‐Aldrich E1383), camptothecin (Sigma‐Aldrich C9911), hydroxyurea (Sigma‐Aldrich H8627), doxycycline (Clontech 631311), hygromycin B Gold (InvioGen), zeocin (Invitrogen 46‐0509), blasticidin (InvivoGen), ATR inhibitor VE‐821 (TINIB‐TOOLS), WEE1 inhibitor AZD1775 (Selleckchem), CDC7 inhibitor PHA‐767491 (Selleckchem), and roscovitine (Sigma‐Aldrich R7772).
Plasmids construction
GNL3 cDNA cloned in pDONR223 (obtained from Montpellier Genomic Collection) was introduced using Gateway method in pDEST_pcDNA5_FLAG_C‐term and pDEST_pcDNA5_BirA‐FLAG_C‐term (gifts from Anne‐Claude Gingras, Lunenfeld‐Tanenbaum Research Institute at Mount Sinai Hospital, Toronto, Canada). GNL3‐dB was created by gene synthesis (Invitrogen GeneArt Gene Synthesis) and introduced using Gateway method into pDEST_pcDNA5_FLAG_C‐term. ORC2 cDNA (a gift from Eric Julien, Institut de Recherche en Cancérologie de Montpellier) was introduced using Gateway method into pDEST_pcDNA5_FLAG_N‐term (gift from Anne‐Claude Gingras, Lunenfeld‐Tanenbaum Research Institute at Mount Sinai Hospital, Toronto, Canada).
Gene silencing
siRNAs against MRE11, CtIP, and EXO1 were provided by Yea‐Lih Lin (Institut de Génétique Humaine, Montpellier) and are described in Coquel et al (2018). For GNL3 depletion, siGENOME SMARTpool (M‐016319‐00; GGACAUACAUGAAGAAUUG; GUGGACAGGUGCCUCAUUA; CCAGGAAACUGUUGAUGAA; CAUCGUAUCUCCACUUAAU), and individual siRNA oligonucleotide (D‐016319‐01; GGACAUACAUGAAGAAUUG) were purchased from Dharmacon. For MCM3 depletion, siGENOME SMARTpool (M‐003274‐02; GGACAUCAAUAUUCUUCUA; GCAGGUAUGACCAGUAUAA; GGAAAUGCCUCAAGUACAC; GACCAUAGAGCGACGUUAU) was purchased from Dharmacon. For RIF1 depletion, siGENOME SMARTpool (D‐027983‐00; UCACGUAGOCCUAAAUUUA; AGACGGUGCUCUAUUGUUA; UGAGGAGAUCUAAAIGGUU; CAGAAGAGUCCAUUGCAUA) was purchased from Dharmacon. siRNAs were transfected using INTERFERin (Polypus transfection).
Western‐blot
Cellular extracts were resuspended in Laemmli buffer (65.8 mM Tris, 26.3% glycerol, 2.1% SDS, and Bromophenol blue) and boiled at 95°C for 5 min. Proteins were separated by SDS‐PAGE using home‐made or precast gels (Bio‐Rad) with suitable percentage then transferred onto nitrocellulose membranes (GE Healthcare or Bio‐Rad). Membranes were blocked with 5% nonfat milk in TBS‐T (10 mM Tris pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 1 h then incubated with the primary antibodies overnight. Membranes were washed three times with TBS‐T then incubated with the corresponding secondary antibody. Finally, membranes were developed with Clarity Western ECL Blotting Substrate (Bio‐Rad), and images were acquired using a ChemiDoc System (Bio‐Rad). Antibodies against the following proteins were used: Ser345 Phospho‐Chk1 (Cell Signaling Technology 2348), Chk1 (Santa Cruz sc‐8408), PCNA (Sigma‐Aldrich P8825), Ser4/8 Phospho‐RPA32 (Bethyl A300‐245A), RPA32 (Calbiochem NA18), histone H3 (Abcam ab62642), GNL3 (Bethyl A300‐600A, Santa Cruz sc‐166460 or Sigma‐Aldrich SAB1407312), Ser33 Phospho‐RPA32 (Bethyl A300‐246), Tubulin (Sigma Aldrich T5168), CDC45 (Santa Cruz sc‐20685), Ser40 Phospho‐MCM2 (Abcam ab133243), MRE11 (Novus NB100‐142), BRCA1 (Santa Cruz sc‐642), CtIP (Abcam ab70163), RAD51 (Santa Cruz sc‐8349), MCM3 (Abcam ab4460), EXO1 (Bethyl A302‐639), RIF1 (A300‐568A‐M; Bethyl), β‐actin (Sigma‐Aldrich A1978), and FLAG (Sigma Aldrich F1804).
esiRNA screening
The 25 esiRNA (Sigma‐Aldrich) corresponding to 24 candidates plus 1 negative control (EGFP) are described in Dataset EV1. HCT116 were seeded in 96‐wells plates and transfected with esiRNAs using Oligofectamine (ThermoFisher). After 48 h, transfected cells were subjected to 4 h treatment with 1 μM camptothecin then fixed for 15 min using 4% paraformaldehyde (PFA). Cells were permeabilized with 75% EtOH for 30 min on ice. 96‐wells plate was incubated with primary antibody against Ser139 Phospho‐H2A.X (Millipore 05‐636) for 60 min then with secondary antibody antimouse coupled with Alexa568 (ThermoFisher A‐11011) and finally with DAPI for 30 min. All the washes were performed with PBS‐BSA 1%. 96 wells were scanned using a Nexcelom Celigo, and images were analyzed using Celigo software. DAPI staining was used to measure the level of Ser139 Phospho‐H2A.X in the nucleus for each esiRNA.
Proximity ligation assay (PLA)
Cells were grown on coverslips to reach 70–80% confluency then fixed with 2% paraformaldehyde (PFA) and 0.02% sucrose in PBS for 20 min at room temperature. Cells were permeabilized with 0.5% Triton X100‐PBS for 20 min then washed PBS‐3% BSA. Coverslips were incubated with primary antibodies in PLA blocking solution (Sigma‐Aldrich) overnight at 4°C then washed with PBS. PLA probes (antimouse minus DUO92004 and antirabbit plus DUO92002, Sigma‐Aldrich) were incubated together in PLA blocking solution for 20 min then added on the coverslips for 1 h at 37°C then washed two times with buffer A (150 mM NaCl, 10 mM Tris, 0.5% Tween). PLA kit was used (DUO92014, Sigma‐Aldrich) for the following steps. Coverslips were incubated with ligase (1/40 dilution in ligase buffer) for 30 min at 37°C. Coverslips were washed two times with buffer A and incubated with polymerase (1/80 dilution in amplification buffer) for 100 min at 37°C. Coverslips were washed two times with buffer B (200 mM NaCl, 400 mM Tris‐Base), dried, and then mounted on glass slides with DAPI‐containing mounting medium (DUO82040 Sigma‐Aldrich). Cells were analyzed by fluorescence microscopy, and quantification of the number of foci was performed using Fiji software. Antibodies against the following proteins were used: ORC2 (Bethyl A302‐734A), CENP‐A (Thermo Fisher MA1‐20832), FLAG (Sigma Aldrich F1804), and GNL3 (Bethyl A300‐600A and Santa Cruz sc‐166460).
Flow cytometry
When indicated cells were first labeled with 20 μM IdU for 10 min and then fixed with ice‐cold 70% ethanol. Then cells were treated with RNase during 60 min and then for 30 min with 2 M HCl. Next, the cells were incubated with a BrdU/IdU antibody from BD Biosciences (347580) for 60 min or with an anti‐pH3S10 (Cell Signaling 9701) overnight, and then with an Alexa 488 conjugated antimouse IgG (Invitrogen) at room temperature for 30 min. Finally, the cells were stained with 5 μg/ml of propidium iodide in PBS and analyzed using a MACSquant analyzer (Miltenyi Biotec). Results were analyzed using Flowjo (https://www.flowjo.com).
Replication analysis by DNA combing
Asynchronous cells were labeled 20 min with IdU, 20 min with CldU and then chased 90 min with thymidine. Purification of HMW gDNA, DNA combing, and replication analysis were performed as in Bialic et al (2015) with the following modifications. Agarose plugs containing gDNA were washed in TNE50 containing 100 mM NaCl, digested O/N at 42°C with 3 U β‐agarase (New England Biolabs) and again for 2 h with 2 U β‐agarase. DNA was combed in MES buffer also containing 100 mM NaCl. Briefly, genomic DNA was combed on silanized coverslips, denatured with NaOH, and sites of DNA synthesis revealed using anti‐IdU (red), anti‐CldU (green), and anti‐ssDNA (blue) antibody pairs. Primary antibodies were rat anti‐BrdU (clone BU1/75, Abcam ab6326) for CldU, mouse anti‐BrdU (clone B44, Becton Dickinson), for IdU and mouse autoanti‐ssDNA (from DSHB) for DNA. Washes were performed with PBS‐T containing 0.05% Triton X100. Secondary antibodies were Alexa488 Goat anti‐rat IgG, Alexa546 Goat anti‐mouse IgG, Alexa647 Goat anti‐Mouse IgG2a (Life Technologies). Imaging was performed on a Zeiss AxioImager Z1 microscope with YFP, Cy3, and Cy5 filter blocks, equipped with a 40× objective (EC Plan Neofluar 1.3 NA oil) and scMOS ZYLA 4.2 MP camera (2048*2048 pixels, 6.5 μm pixel size). Red‐to‐green signals show fork direction (yellow arrow). Fork velocity (FV) is calculated by dividing the length of the green tract by the pulse time (in kb/min). Global instant fork density (GIFD) was calculated using the formula that accounts for the doubling of DNA during S phase:
where Nf is the number of bicolor forks, DNA the total length of DNA measured (in Mb), and G1%, S%, and G2M% the fraction of cells in G1, S, and G2 or M phases, respectively, calculated from flow cytometry profiles using the same cells as for DNA combing.
Isolation of proteins on nascent DNA (iPOND)
iPOND was performed largely as previously described (Lossaint et al, 2013; Ribeyre et al, 2016). HeLa S3 cells were pulse labeled with 10 μM EdU for indicated times and chases were performed with 10 μM thymidine. Cells were fixed with 1% formaldehyde for 5 min or 2% for 15 min followed or not by quenching of formaldehyde by 5 min incubation with 0.125 M glycine. Fixed samples were collected by centrifugation at 1,000 g for 3 min, washed three times with PBS and stored at ‐80°C. Cells were permeabilized with 0.5% triton for 30 min and click chemistry was used to conjugate biotin‐TEG‐azide (Eurogentec) to EdU‐labeled DNA in PBS containing 10 mM sodium ascorbate, 10 μM biotin‐TEG‐azide, and 2 mM CuSO4. Cells were re‐suspended in lysis buffer (10 mM Hepes‐NaOH; 100 mM NaCl; 2 mM EDTA PH8; 1 mM EGTA; 1 mM PMSF; 0.2% SDS; 0.1% Sarkozyl), and sonication was performed using a Qsonica sonicator with the following settings: 30% power, 20 s constant pulse and 50 s pause for a total sonication time of 5 min on ice with water. Lysates were centrifuged at 15,000 g for 10 min at room temperature. Supernatants were normalized by DNA quantification using a nanodrop device. Biotin conjugated DNA–protein complexes were captured using overnight incubation with magnetic beads coated with streptavidin (Ademtech). Captured complexes were washed with lysis buffer and 500 mM NaCl. Proteins associated with nascent DNA were eluted under reducing conditions by boiling into SDS sample buffer for 30 min at 95°C and analyzed by Western blot.
DNA fibers labeling
DNA fibers labeling was performed as previously described (Lossaint et al, 2013; Ribeyre et al, 2016). Cells were labeled with 25 μM IdU, washed with warm media and exposed to 50 μM CldU. Cells were lysed and DNA fibers were stretched onto glass slides are left to air dry then fixed in methanol/acetic acid (3:1) for 10 min. The DNA fibers were denatured with 2.5 M HCl for 60 min, washed with PBS, and blocked with 2% BSA in PBS‐Tween for 60 min. IdU replication tracks were revealed with a mouse anti‐BrdU/IdU antibody from BD Biosciences (347580) and CldU tracks with a rat anti‐BrdU/CldU antibody from Eurobio (ABC117‐7513). The following secondary antibodies were used: Alexa fluor 488 anti‐mouse antibody (Life A21241) and Cy3 anti‐rat antibody (Jackson Immunoresearch 712‐166‐153). Fibers were visualized and imaged by Carl Zeiss Axio Imager Apotome using 40× Plan Apo 1.4 NA oil immersion objective. Replication tracks lengths were analyzed using ImageJ software. Statistical analysis was performed using Graphpad Prism software, and at least 80 individual DNA fibers were counted for each individual experiment.
Immunofluorescence
Cells were grown on coverslips to reach 70–80% confluency then directly with 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature. When indicated, incubation with cytoskeletal (CSK) buffer (10 mM PIPES pH 6.8, NaCl 100 mM, sucrose 300 mM, MgCl2 3 mM, EGTA 1 mM, Triton X‐100 0.5%) was performed. Cells were permeabilized by with 0.2% Triton X100‐PBS for 10 min then transferred into 0.1% Tween‐PBS for 5 min. Coverslips were then incubated with primary antibodies in 0.1% Tween‐5% BSA‐PBS for 1–2 h, washed with 0.1% Tween‐PBS, then incubated with secondary antibodies in Tween 0.1%‐BSA 5%‐PBS for 1 h. All the incubations were carried out in darkness in a humidified chamber at room temperature. Finally, coverslips are washed again with 0.1% Tween‐PBS, incubated with Hoechst to label DNA for 5 min, and then mounted on glass slides with Prolong (Life). Cells were analyzed by fluorescence microscopy. Antibodies against the following proteins were used: FLAG (Sigma Aldrich F1804), Streptavidin‐Alexa Fluor 488 (Life S32354), NOP1 (Novus NBP2‐46881), and GNL3 (Bethyl A300‐600A).
Replication timing experiments and microarrays
Cells were incubated with 50 μM of BrdU for 90 min and collected, washed three times with PBS and then fixed in ethanol 75%. Cells were re‐suspended in PBS with RNAse (0.5 mg/ml) and then with propidium iodide (50 μg/ml) followed by incubation in the dark at room temperature for 30 min with low agitation. Two fractions of 150,000 cells, S1 and S2 corresponding to Early and Late S‐phase fractions, respectively, were sorted by flow cytometry using a Becton Dickinson FACS Melody. Whole DNA was extracted with lysis buffer (50 mM Tris pH 8, 10 mM EDTA, 300 mM NaCl, 0.5% SDS) and 0.2 mg/ml of Proteinase K for 2 h at 65°C. Neo‐synthesized DNA were immunoprecipitated with BrdU antibodies (Anti‐BrdU Pure, BD Biosciences, #347580) as previously described (Fernandez‐Vidal et al, 2014). To control the quality of enrichment of early and late fractions in S1 and S2, qPCR was performed with BMP1 oligonucleotides (early control) and with Dppa2 oligonucleotides (late control; (Hiratani et al, 2008)). Microarray hybridization requires a minimum of 1,000 ng of DNA. To obtain sufficient specific immunoprecipitated DNA for this hybridization step, whole genome amplification was conducted (WGA, Sigma) on immunoprecipitated DNA. A post WGA qPCR was performed to preserve specific enrichment in both S1 and S2 fractions. Early and late amplified neo‐synthesized DNA were then labeled with Cy3 and Cy5 ULS molecules, respectively (Genomic DNA labeling Kit, Agilent). The hybridization was performed according to the manufacturer instructions on 4 × 180 K mouse microarrays (SurePrint G3 Mouse CGH Microarray Kit, 4 × 180 K, AGILENT Technologies, reference genome: mm9). Microarrays were scanned with an Agilent High‐Resolution C Scanner using a resolution of 3 μm and the autofocus option. Feature extraction was performed with the Feature Extraction 9.1 software (Agilent Technologies). For each experiment, the raw datasets were automatically normalized by the Feature extraction software. Analysis was performed using the STAR‐R software described in Hadjadj et al (2020). The statistical comparison was conducted between early and late domains from both cell lines in order to determine segments where replication timing changes. Graphical representation was generated with START‐R suit.
Chromatin immunoprecipitation and deep sequencing (ChIP‐seq)
About 20.106 of Hela S3 cells per sample were prepared for sonication following the True‐ChIP chromatin shearing kit protocol for high cell concentration from Covaris. Cells were cross‐linked in 1% methanol‐free formaldehyde during 5 min before cell lysis and nuclei preparation. Washed nuclei were sonicated for 15 min at 6°C to obtain DNA fragments of 100–800 pb using the E220evolution Covaris machine following parameters indicated in the provided protocol. After dilution with one volume of immunoprecipitation dilution buffer (Covaris), sonicated samples were precleared with 3 μl/ml of protein G magnetic beads (Ademtech) during 1 h at 4°C. Each sample was then normalized to an equal amount of protein (associated to precleared chromatin), and input samples were collected after this step. Normalized samples were then incubated with 1 μg of GNL3 antibody (Bethyl A300‐600A) overnight at 4°C, before incubation with 20 μl/ml of protein G magnetic beads (previously blocked overnight at 4°C in immunoprecipitation dilution buffer with 1% BSA) during 4 h at 4°C. Chromatin bound to beads was then washed 5 min at room temperature in each following buffers: low salt buffer (150 mM NaCl, 20 mM Tris HCl pH = 8, 2 mM EDTA, 1% Triton, 0.1% SDS); high salt buffer (500 mM NaCl, 20 mM Tris HCl pH = 8, 2 mM EDTA, 1% Triton, 0.1% SDS); LiCl buffer (0.25 M LiCl, 10 mM Tris–HCl pH = 8, 1 mM EDTA, 1% sodium deoxycholate, 1% NP‐40); TE buffer (10 mM Tris–HCl pH = 8, 1 mM EDTA). Washed beads were eluted in 200 μl of elution buffer (100 mM NaHCO3, 1% SDS) during 15 min at 30°C with shaking. Eluted chromatin and input samples were reverse‐cross‐linked overnight at 65°C with 0.2 M NaCl and 0.02 mg/ml of RNAse A and incubated 1 h with Proteinase K (400 μg/ml final concentration). DNA was purified using the ChIP DNA Prep Adem kit (Ademtech) following the provided protocol. DNA bound to beads was eluted in 50 μL of elution buffer. Quantity of DNA was measured with the Qubit 1× dsDNA HS Assay kit (Invitrogen), using a Qubit 2.0 fluorometer (Thermofisher scientific). GNL3 ChIP was repeated three times and 10 ng of each ChIP and each corresponding input were pooled together and send to the MGX sequencing platform of Montpellier, France (https://www.mgx.cnrs.fr/). DNA banks were sequenced using the Illumina‐Novaseq‐6000 machine to obtain 150 bp paired‐end reads. Sequencing data were processed and analyzed using the online Galaxy platform (https://usegalaxy.org/). Reads were aligned on the February 2009 human reference genome (GRCh37/Hg19) using Bowtie2 tool with default parameters. GNL3 Peaks were discovered using MACS2 callpeak tool using input as control file with a q‐value < 0.005. ORC2 peaks file was taken from Miotto et al (2016).
Chromatin fractionation
Cells were seeded at 80% confluency and collected by trypsinization followed by centrifugation for 3 min (1,200 g) at room temperature. The pellets were washed with PBS then resuspended with CSK buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 300 mM Sucrose, 1 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 0.1% Triton X‐100, 1 mM ATP, 1× protease inhibitor) and kept for 10 min on ice. Lysed cells were then centrifuged for 3 min (3,000 g) at 4°C. The resulting supernatant presenting the soluble protein fraction was transferred to another Eppendorf tube, and the pellet was washed with CSK buffer for 10 min on ice followed by centrifugation for 3 min (3,000 g) at 4°C. The resulting pellet that represents the in‐soluble fraction of proteins was then resuspended in 2× Laemmli buffer and incubated at 95°C for 10 min before western blot analysis.
BioID
Flp‐In T‐Rex 293 cell lines were stably transfected with Flag‐BirA‐GNL3. Cells seeded at 75% confluency were incubated with 10 μg/ml of doxycycline for 16 h and then with 50 μM biotin for 4 h. Cells were washed once with PBS and lysed with RIPA/SDS buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP‐40, 0.2% SDS, 0.5% sodium deoxycholate) complemented with 1× complete protease inhibitor and 250 U benzonase (Sigma‐Aldrich, CE1014). Lysed cells were incubated on a rotating wheel for 1 h at 4°C followed by sonication on ice with 30% amplitude for 3 cycles of 10 s (2 s ON‐2 s resting) separated with 10 s of resting. Sonicated lysate was next centrifuged for 30 min (7,750 g) at 4°C, the cleared supernatant was transferred to a new tube, and protein concentration was quantified using Bradford protein assay. For each condition, 500 μg of proteins were incubated with 30 μl of Streptavidin‐Agarose beads (Sigma‐Aldrich, CS1638) on a rotating wheel for 3 h at 4°C. Beads were next washed sequentially with 1 ml of each buffer starting with lysis buffer, wash buffer 1 (2% SDS in H2O), wash buffer 2 (0.2% sodium deoxycholate, 1% Triton X‐100, 500 mM NaCl, 1 mM EDTA, and 50 mM Hepes pH 7.5), wash buffer 3 (250 mM LiCl, 0.5% NP‐40, 0.5% sodium deoxycholate, 1 mM EDTA, 500 mM NaCl and 10 mM Tris pH 8), and finally wash buffer 4 (50 mM Tris pH 7.5 and 50 mM NaCl). Biotinylated proteins were eluted from the magnetic beads using 40 μl of 2× Laemmli buffer and incubated at 95°C for 10 min.
Proteomics analysis of BioID samples
Biotinylated proteins were migrated on SDS PAGE for a short migration. After reduction (DTT 1 M, 30 min at 60°C), an alkylation (IAA 0.5 M, 30 min RT) proteins were digested using trypsin (Gold, Promega, 1 μg/sample, overnight at 37°C). For LC MSMS analysis, samples were loaded onto a 50 cm reversed‐phase column (75 mm inner diameter; Acclaim PepMap 100 C18; Thermo Fisher Scientific) and separated with an UltiMate 3000 RSLC system (Thermo Fisher Scientific) coupled to a QExactive HF system (Thermo Fisher Scientific). Separation of the peptides was performed following a gradient from 2 to 25% buffer B (0.1% AF in 80% ACN) for 100 min at a flow rate 300 nl/min, then 25–40% in 20 min and finally 40–90% in 3 min. Tandem mass spectrometry analyses were performed in a data‐dependent mode. Full scans (350–1,500 m/z) were acquired in the Orbitrap mass analyzer with a resolution of 60,000 at 200 m/z. For MS scans, 3e6 ions were accumulated within a maximum injection time of 60 ms. The 12 most intense ions with charge states ≥ 2 were sequentially isolated (1e5) with a maximum injection time of 100 ms and fragmented by higher‐energy collisional dissociation (normalized collision energy of 28) and detected in the Orbitrap analyzer at a resolution of 30,000. Raw spectra were processed with MaxQuant v 1.6.5.0 (Cox & Mann, 2008) using standard parameters with match between runs option. Spectra were matched against the UniProt reference proteome (release 2019_06; http://www.uniprot.org) of Homo sapiens and 250 frequently observed contaminants, as well as reversed sequences of all entries. The maximum false discovery rate (FDR) for peptides and proteins was set to 0.01. Representative protein ID in each protein group was automatically selected using the in‐house developed Leading tool (Raynaud et al, 2018).
Immunoprecipitation
Whole‐cell extracts of K562 cells were prepared using lysis buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 5 mM EDTA pH8, 0.5% NP40) supplemented with protease inhibitor cocktail (Roche), 1 mM PMSF, 1 mM MgCl2 and Benzonase Nuclease 250 units/10 millions of cells (E1014‐25KU, Sigma). Immunoprecipitations were performed overnight at 4°C with protein G Dynabeads (Thermo Fisher Scientific) coupled to either rabbit immunoglobulin G (IgG) (P120‐201, Bethyl Laboratories), rabbit GNL3 (sc‐166460, Santa Cruz Biotechnology) or rabbit ORC2 antibody (A302‐734A, Bethyl Laboratories). Beads were washed four times with lysis buffer, then washed three times with 50 mM Tris HCl pH8. The immunoprecipitated complexes were eluted in 50 mM Tris HCl pH8 containing 1% SDS for 15 min à 56°C with agitation. IP samples were mixed with 1× Bolt Sample Reducing agent (Thermo Fisher Scientific) and 1× Bolt LDS Sample Buffer (Thermo Fisher Scientific), loaded and resolved on pre‐cas Bolt Bis‐Tris gels (Thermo Fisher Scientific), then transferred onto nitrocellulose membrane (GE Healthcare). Membranes were blocked in 5% fat‐free milk in PBS, incubated overnight at 4°C with primary antibodies directed against ORC2 (A302‐734A, Bethyl Laboratories) and GNL3 (sc‐166460, Santa Cruz Biotechnology). A cognate secondary antibody coupled to horseradish peroxidase was used and revealed with the Super Signal West Dura Extended Duration Substrate kit (Thermo Fisher Scientific). Acquisition was performed using the Fusion FX (Vilber), and image analysis was performed using ImageJ (https://imagej.nih.gov/ij/).
Proteomics analysis of immunoprecipitation
Sample preparation: Tryptic peptides from the immunoprecipitated complexes (=eluate) were obtained by Strap Micro Spin Column according to the manufacturer's protocol (Protifi, NY, USA). Briefly, proteins from 140 μL of the eluate were diluted 1:1 with 2× reducing‐alkylating buffer (20 mM TCEP, 100 mM chloroacetamide in 400 mM TEAB pH 8.5 and 4% SDS) and left 5 min at 95°C to allow reduction and alkylation in one step. Strap‐binding buffer was applied to precipitate proteins on quartz and proteolysis took place during 14 h at 37°C with 1 μg Trypsin‐sequencing grade (Promega). After speed‐vacuum drying of eluted peptides, these were solubilized in 0.1% trifluoroacetic acid (TFA) in 10% acetonitrile (ACN). Liquid chromatography‐coupled mass spectrometry analysis (LC–MS): LC–MS analyses were performed on a Dionex U3000 HPLC nanoflow system coupled to a TIMS‐TOF Pro mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany). One μl was loaded, concentrated, and washed for 3 min on a C18 reverse‐phase precolumn (3 μm particle size, 100 Å pore size, 75 μm inner diameter, 2 cm length, from Thermo Fisher Scientific). Peptides were separated on an Aurora C18 reverse phase resin (1.6 μm particle size, 100 Å pore size, 75 μm inner diameter, 25 cm length mounted onto the Captive nanoSpray Ionization module, IonOpticks, Middle Camberwell Australia) with a 60 min overall run‐time gradient ranging from 99% of solvent A containing 0.1% formic acid in milliQ‐grade H2O to 40% of solvent B containing 80% acetonitrile, 0.085% formic acid in mQH2O. The mass spectrometer acquired data throughout the elution process and operated in DDA PASEF mode with a 1.1 s/cycle, with timed ion mobility spectrometry (TIMS) mode enabled and a data‐dependent scheme with full MS scans in PASEF mode. This enabled a recurrent loop analysis of a maximum of the 120 most intense nLC‐eluting peptides which were CID‐fragmented between each full scan every 1.1 s. Ion accumulation and ramp time in the dual TIMS analyzer were set to 50 ms each and the ion mobility range was set from 1/K0 = 0.6 Vs cm‐2 to 1.6 Vs cm‐2. Precursor ions for MS/MS analysis were isolated in positive mode with the PASEF mode set to « on » in the 100–1.700 m/z range by synchronizing quadrupole switching events with the precursor elution profile from the TIMS device. The cycle duty time was set to 100%, accommodating as many MSMS in the PASEF frame as possible. Singly charged precursor ions were excluded from the TIMS stage by tuning the TIMS using the otof control software, (Bruker Daltonik GmbH). Precursors for MS/MS were picked from an intensity threshold of 2,500 arbitrary units (a.u.) and resequenced until reaching a “target value” of 20,000 a.u taking into account a dynamic exclusion of 0.40 s elution gap. Protein quantification and comparison: The mass spectrometry data were analyzed using Mascot version 2.5.1 (http://www.matrixscience.com/). The database used was a concatenation of Homo sapiens sequences from the Swissprot databases (release June 2020: 563,972 sequences; 203,185,243 residues) and an in‐house list of frequently found contaminant protein sequences. The enzyme specificity was trypsin's. The precursor and fragment mass tolerances were set to 20 ppm. Oxidation of methionines was set as variable modifications, while carbamidomethylation of cysteines was considered complete. FDR was kept below 1% on both peptides and proteins. For comparative analysis, peptide count results from Mascot were assembled with the MyPROMS (Poullet et al, 2007) software (version 3.1).
Author contributions
Rana Lebdy: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Marine Canut: Data curation; formal analysis; investigation; methodology. Julie Patouillard: Methodology. Jean‐Charles Cadoret: Data curation; formal analysis; funding acquisition; investigation; methodology; writing – review and editing. Anne Letessier: Data curation; investigation; methodology; writing – review and editing. Josiane Ammar: Methodology. Jihane Basbous: Writing – review and editing. Serge Urbach: Methodology. Benoit Miotto: Funding acquisition; writing – review and editing. Angelos Constantinou: Funding acquisition; writing – review and editing. Raghida Abou Merhi: Supervision; writing – review and editing. Cyril Ribeyre: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
PDF+
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgments
We thank all the present and former lab members for comments and suggestions on the project and the manuscript. We are grateful to Pierre‐Henry Gaillard, Jean‐Hugues Guervilly, Maud de Dieuleveult, Anne‐Claude Gingras, Yea‐Lih Lin, Claudia Lukas, Jiri Lukas, Domenico Maiorano, Eric Julien, and Montpellier Genomic Collection for reagents. We thank Armelle Lengronne, Antoine Aze, Domenico Maiorano, Eric Julien, Sébastien Britton, Olivier Ganier, and Joelle Nassar for discussions and comments as well as Marie‐Pierre Blanchard and Amélie Sarazzin from the Montpellier Imaging Platform for their support. We acknowledge Carol Featherstone of Plume Scientific Communication Services for professional scientific editing during the preparation of the manuscript. We are grateful to Montpellier Combing Facility (Etienne Schwob and Marjorie Drac) and Montpellier GenomiX facility (Hugues Paranello). Mass spectrometry analysis of BioID samples was carried out using the facilities of the Montpellier Proteomics Platform (PPM, BioCampus). We thank Céline Gongora, Nadia Vie, and Naoill Abdellaoui for their help with the use of the Celligo. We thank the 3P5 proteom'IC facility (Johanna Bruce, Cedric Broussard and François Guillonneau) at Institut Cochin, which is supported by the DIM Thérapie Génique Région Ile‐de‐France, IBiSA, and the Labex GR‐Ex. This work was supported by a Jeunes Chercheurs Jeunes Chercheuses grant from the Agence National de la Recherche (REPLIBLOCK ANR‐17‐CE12‐0034‐01), and an Emergence grant from Cancéropole Grand Sud‐Ouest to Cyril Ribeyre as well as a grant from Programme labellisé Fondation ARC to Angelos Constantinou. Rana Lebdy was funded by fellowships from Azm & Saade Association and Fondation ARC pour la recherche sur le cancer. Benoit Miotto and Anne Letessier are partners of Labex “Who am I?” (ANR‐11‐LABX‐0071 and ANR‐11‐IDEX‐005‐02) and are supported by Fondation pour la Recherche Medicale (AJE20151234749). Jean‐Charles Cadoret thanks the IdEx Université Paris Cité (ANR‐18‐IDEX‐0001).
EMBO reports (2023) 24: e57585
Contributor Information
Raghida Abou Merhi, Email: raboumerhi@ul.edu.lb.
Cyril Ribeyre, Email: cyril.ribeyre@cnrs.fr.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD045389 (http://www.ebi.ac.uk/pride/archive/projects/PXD045389) and 10.6019/PXD045389. ChIP‐seq sequencing data and replication timing data for this study have been submitted to the NCBI Gene Expression Omnibus and are available under accession number GSE190253 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190253) and GSE180865 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180865), respectively.
References
- Bauwens S, Lototska L, Koundrioukoff S, Debatisse M, Ye J, Gilson E, Mendez‐Bermudez A (2021) The telomeric protein TRF2 regulates replication origin activity within pericentromeric heterochromatin. Life (Basel) 11: 267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Nähse‐Kumpf V, Larsen MSY, O'Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuåsen RG et al (2012) Cyclin‐dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 32: 4226–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman C, Nichane M, De Clercq S, Maetens M, Floss T, Wurst W, Bellefroid E, Marine JC (2006) Evolutionarily conserved role of nucleostemin: controlling proliferation of stem/progenitor cells during early vertebrate development. Mol Cell Biol 26: 9291–9301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersaglieri C, Kresoja‐Rakic J, Gupta S, Bar D, Kuzyakiv R, Panatta M, Santoro R (2022) Genome‐wide maps of nucleolus interactions reveal distinct layers of repressive chromatin domains. Nat Commun 13: 1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Cortez D, Lopes M (2020) The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21: 633–651 [DOI] [PubMed] [Google Scholar]
- Bialic M, Coulon V, Drac M, Gostan T, Schwob E (2015) Analyzing the dynamics of DNA replication in Mammalian cells using DNA combing. Methods Mol Biol 1300: 67–78 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Ge XQ, Jackson DA (2011) How dormant origins promote complete genome replication. Trends Biochem Sci 36: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, Bensimon A (2007) Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 18: 3059–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL et al (2018) SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61 [DOI] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella‐Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB (2012) Mouse Rif1 is a key regulator of the replication‐timing programme in mammalian cells. EMBO J 31: 3678–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtot L, Hoffmann JS, Bergoglio V (2018) The protective role of dormant origins in response to replicative stress. Int J Mol Sci 19: 3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaek CR, Petrosius V, Benada J, Erichsen L, Damgaard RB, Sorensen CS (2022) WEE1 kinase protects the stability of stalled DNA replication forks by limiting CDK2 activity. Cell Rep 38: 110261 [DOI] [PubMed] [Google Scholar]
- Emerson DJ, Zhao PA, Cook AL, Barnett RJ, Klein KN, Saulebekova D, Ge C, Zhou L, Simandi Z, Minsk MK et al (2022) Cohesin‐mediated loop anchors confine the locations of human replication origins. Nature 606: 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Vidal A, Guitton‐Sert L, Cadoret J‐C, Drac M, Schwob E, Baldacci G, Cazaux C, Hoffmann J‐S (2014) A role for DNA polymerase θ in the timing of DNA replication. Nat Commun 5: 4285 [DOI] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, Mechali M (2015) DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16: 360–374 [DOI] [PubMed] [Google Scholar]
- Hadjadj D, Denecker T, Guérin E, Kim S‐J, Fauchereau F, Baldacci G, Maric C, Cadoret J‐C (2020) Efficient, quick and easy‐to‐use DNA replication timing analysis with START‐R suite. NAR Genom Bioinform 2: lqaa045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP (2011) Replication stress induces 53BP1‐containing OPT domains in G1 cells. J Cell Biol 193: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C‐W, Lyou Y, Townes TM, Schübeler D, Gilbert DM (2008) Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg Olausson K, Nistér M, Lindström MS (2014) Loss of nucleolar histone chaperone NPM1 triggers rearrangement of heterochromatin and synergizes with a deficiency in DNA methyltransferase DNMT3A to drive ribosomal DNA transcription. J Biol Chem 289: 34601–34619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Stillman B (2023) Origins of DNA replication in eukaryotes. Mol Cell 83: 352–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Cheng J, Bawa‐Khalfe T, Yao X, Chin YE, Yeh ETH (2016) SUMOylated ORC2 recruits a histone demethylase to regulate centromeric histone modification and genomic stability. Cell Rep 15: 147–157 [DOI] [PubMed] [Google Scholar]
- Iarovaia OV, Minina EP, Sheval EV, Onichtchouk D, Dokudovskaya S, Razin SV, Vassetzky YS (2019) Nucleolus: a central hub for nuclear functions. Trends Cell Biol 29: 647–659 [DOI] [PubMed] [Google Scholar]
- Jones MJK, Gelot C, Munk S, Koren A, Kawasoe Y, George KA, Santos RE, Olsen JV, McCarroll SA, Frattini MG et al (2021) Human DDK rescues stalled forks and counteracts checkpoint inhibition at unfired origins to complete DNA replication. Mol Cell 81: 426–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein KN, Zhao PA, Lyu X, Sasaki T, Bartlett DA, Singh AM, Tasan I, Zhang M, Watts LP, Hiraga SI et al (2021) Replication timing maintains the global epigenetic state in human cells. Science 372: 371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Carr AM (2013) Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 122: 33–45 [DOI] [PubMed] [Google Scholar]
- Lebdy R, Patouillard J, Larroque M, Urbach S, Abou Merhi R, Larroque C, Ribeyre C (2023) The organizer of chromatin topology RIF1 ensures cellular resilience to DNA replication stress. Life Sci Alliance 6: e202101186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung W, Simoneau A, Saxena S, Jackson J, Patel PS, Limbu M, Vindigni A, Zou L (2023) ATR protects ongoing and newly assembled DNA replication forks through distinct mechanisms. Cell Rep 42: 112792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Ji F, Helleday T, Ying S (2018) Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 19: e46263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Ibrahim W, Peng CY, Finegold MJ, Tsai RY (2013) A novel role of nucleostemin in maintaining the genome integrity of dividing hepatocytes during mouse liver development and regeneration. Hepatology 58: 2176–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Meng L, Lin TC, Wu LJ, Pederson T, Tsai RY (2014) Nucleostemin and GNL3L exercise distinct functions in genome protection and ribosome synthesis, respectively. J Cell Sci 127: 2302–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Contreras AJ, Ruppen I, Nieto‐Soler M, Murga M, Rodriguez‐Acebes S, Remeseiro S, Rodrigo‐Perez S, Rojas AM, Mendez J, Munoz J et al (2013) A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep 3: 1105–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A (2013) FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 51: 678–690 [DOI] [PubMed] [Google Scholar]
- MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM (2010) Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10: 425–448 [DOI] [PubMed] [Google Scholar]
- Mechali M (2010) Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol 11: 728–738 [DOI] [PubMed] [Google Scholar]
- Meng L, Yasumoto H, Tsai RY (2006) Multiple controls regulate nucleostemin partitioning between nucleolus and nucleoplasm. J Cell Sci 119: 5124–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Zhu Q, Tsai RY (2007) Nucleolar trafficking of nucleostemin family proteins: common versus protein‐specific mechanisms. Mol Cell Biol 27: 8670–8682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Lin T, Peng G, Hsu JK, Lee S, Lin SY, Tsai RY (2013) Nucleostemin deletion reveals an essential mechanism that maintains the genomic stability of stem and progenitor cells. Proc Natl Acad Sci U S A 110: 11415–11420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mier P, Perez‐Pulido AJ, Reynaud EG, Andrade‐Navarro MA (2017) Reading the evolution of compartmentalization in the ribosome assembly toolbox: the YRG protein family. PloS One 12: e0169750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Ji Z, Struhl K (2016) Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc Natl Acad Sci U S A 113: E4810–E4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva T, Hood B, Schamus S, O'Connor MJ, Conrads TP, Bakkenist CJ (2017) ATR kinase inhibition induces unscheduled origin firing through a Cdc7‐dependent association between GINS and And‐1. Nat Commun 8: 1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, Bakkenist CJ (2019) WEE1 kinase inhibitor AZD1775 induces CDK1 kinase‐dependent origin firing in unperturbed G1‐ and S‐phase cells. Proc Natl Acad Sci U S A 116: 23891–23893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C et al (2008) A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4: 357–365 [DOI] [PubMed] [Google Scholar]
- Mukherjee C, Tripathi V, Manolika EM, Heijink AM, Ricci G, Merzouk S, de Boer HR, Demmers J, van Vugt MATM, Ray Chaudhuri A (2019) RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat Commun 10: 3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, Tatsumi Y, Fujita M, Tsurimoto T, Obuse C (2003) The ORC1 cycle in human cells: II. Dynamic changes in the human ORC complex during the cell cycle. J Biol Chem 278: 41535–41540 [DOI] [PubMed] [Google Scholar]
- Padeken J, Mendiburo MJ, Chlamydas S, Schwarz HJ, Kremmer E, Heun P (2013) The nucleoplasmin homolog NLP mediates centromere clustering and anchoring to the nucleolus. Mol Cell 50: 236–249 [DOI] [PubMed] [Google Scholar]
- Peng M, Cong K, Panzarino NJ, Nayak S, Calvo J, Deng B, Zhu LJ, Morocz M, Hegedus L, Haracska L et al (2018) Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep 24: 3251–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Hou Y, Meng H, Cao Y, Wang X, Jia L, Chen Q, Zheng Y, Sun Y, Chen H et al (2023) Mapping nucleolus‐associated chromatin interactions using nucleolus Hi‐C reveals pattern of heterochromatin interactions. Nat Commun 14: 350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, Helleday T (2010) Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 107: 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poullet P, Carpentier S, Barillot E (2007) myProMS, a web server for management and validation of mass spectrometry‐based proteomic data. Proteomics 7: 2553–2556 [DOI] [PubMed] [Google Scholar]
- Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B (2004) Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J 23: 2651–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent C, Dimitrov S (2003) Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 116: 3677–3685 [DOI] [PubMed] [Google Scholar]
- Quiroga‐Artigas G, de Jong D, Schnitzler CE (2022) GNL3 is an evolutionarily conserved stem cell gene influencing cell proliferation, animal growth and regeneration in the hydrozoan Hydractinia. Open Biol 12: 220120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud F, Homburger V, Seveno M, Vigy O, Moutin E, Fagni L, Perroy J (2018) SNAP23‐Kif5 complex controls mGlu1 receptor trafficking. J Mol Cell Biol 10: 423–436 [DOI] [PubMed] [Google Scholar]
- Reynaud EG, Andrade MA, Bonneau F, Ly TB, Knop M, Scheffzek K, Pepperkok R (2005) Human Lsg1 defines a family of essential GTPases that correlates with the evolution of compartmentalization. BMC Biol 3: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeyre C, Zellweger R, Chauvin M, Bec N, Larroque C, Lopes M, Constantinou A (2016) Nascent DNA proteomics reveals a chromatin remodeler required for topoisomerase i loading at replication forks. Cell Rep 15: 300–309 [DOI] [PubMed] [Google Scholar]
- Richards L, Das S, Nordman JT (2022) Rif1‐dependent control of replication timing. Genes (Basel) 13: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, Smogorzewska A (2019) Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol 218: 1096–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Acebes S, Mouron S, Mendez J (2018) Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J Biol Chem 293: 12855–12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi NK, Coquel F, Lin YL, MacKeigan JP, Pasero P, Weinreich M (2018) DDK has a primary role in processing stalled replication forks to initiate downstream checkpoint signaling. Neoplasia 20: 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schaik T, Manzo SG, Vouzas AE, Liu NQ, Teunissen H, de Wit E, Gilbert DM, van Steensel B (2022) Dynamic chromosomal interactions and control of heterochromatin positioning by Ki‐67. EMBO Rep 23: e55782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51‐BRCA1/2. Cancer Cell 22: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ (2021) The central role of DNA damage in the ageing process. Nature 592: 695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shin‐ya K, Niedzwiedz W (2013) FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J Cell Biol 201: 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata E, Kiran M, Shibata Y, Singh S, Kiran S, Dutta A (2016) Two subunits of human ORC are dispensable for DNA replication and proliferation. Elife 5: e19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Gasser SM (2007) The origin recognition complex functions in sister‐chromatid cohesion in Saccharomyces cerevisiae . Cell 128: 85–99 [DOI] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu‐Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288: 31458–31467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobecki M, Mrouj K, Camasses A, Parisis N, Nicolas E, Llères D, Gerbe F, Prieto S, Krasinska L, David A et al (2016) The cell proliferation antigen Ki‐67 organises heterochromatin. Elife 5: e13722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soniat MM, Myler LR, Kuo HC, Paull TT, Finkelstein IJ (2019) RPA phosphorylation inhibits DNA resection. Mol Cell 75: 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenstrom L, Mahdessian D, Gnann C, Cesnik AJ, Ouyang W, Leonetti MD, Uhlen M, Cuylen‐Haering S, Thul PJ, Lundberg E (2020) Mapping the nucleolar proteome reveals a spatiotemporal organization related to intrinsic protein disorder. Mol Syst Biol 16: e9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- Tsai RY (2014) Turning a new page on nucleostemin and self‐renewal. J Cell Sci 127: 3885–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RY, McKay RD (2002) A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev 16: 2991–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RY, McKay RD (2005) A multistep, GTP‐driven mechanism controlling the dynamic cycling of nucleostemin. J Cell Biol 168: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Wang M, Bokros M, Theodoridis PR, Lee S (2019) Nucleolar sequestration: remodeling nucleoli into amyloid bodies. Front Genet 10: 1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Klein KN, Proesmans K, Yang H, Marchal C, Zhu X, Borrman T, Hastie A, Weng Z, Bechhoefer J et al (2021) Genome‐wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell 81: 2975–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LH, Brettingham‐Moore KH, Chan L, Quach JM, Anderson MA, Northrop EL, Hannan R, Saffery R, Shaw ML, Williams E et al (2007) Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res 17: 1146–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Nitta E, Nagamatsu G, Ikushima YM, Hosokawa K, Arai F, Suda T (2013) Nucleostemin is indispensable for the maintenance and genetic stability of hematopoietic stem cells. Biochem Biophys Res Commun 441: 196–201 [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H (2012) Rif1 regulates the replication timing domains on the human genome. EMBO J 31: 3667–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Yasumoto H, Tsai RY (2006) Nucleostemin delays cellular senescence and negatively regulates TRF1 protein stability. Mol Cell Biol 26: 9279–9290 [DOI] [PMC free article] [PubMed] [Google Scholar]