

Abstract
Stimulator of interferon (IFN) genes (STING, also named MITA, ERIS, MPYS, or TMEM173) plays an essential role in DNA virus‐ or cytosolic DNA‐triggered innate immune responses. Here, we demonstrate that the RING‐in‐between RING (RBR) E3 ubiquitin ligase family member RING‐finger protein (RNF) 144A interacts with STING and promotes its K6‐linked ubiquitination at K236, thereby enhancing STING translocation from the ER to the Golgi and downstream signaling pathways. The K236R mutant of STING displays reduced activity in promoting innate immune signal transduction. Overexpression of RNF144A upregulates HSV‐1‐ or cytosolic DNA‐induced immune responses, while knockdown of RNF144A expression has the opposite effect. In addition, Rnf144a‐deficient cells exhibit impaired DNA virus‐ or cytosolic DNA‐triggered signaling, and RNF144A protects mice from DNA virus infection. In contrast, RNF144A does not affect RNA virus‐ or cytosolic RNA‐triggered innate immune responses. Taken together, our findings identify a new positive regulator of DNA virus‐ or cytosolic DNA‐triggered signaling pathways and a critical ubiquitination site important for fully functional STING during antiviral responses.
Keywords: antiviral innate immune responses, DNA virus, posttranslational modification, signal transduction, ubiquitination
Subject Categories: Immunology, Post-translational Modifications & Proteolysis, Signal Transduction
RBR family E3 ubiquitin ligase RNF144A promotes DNA virus‐ or cytosolic DNA‐triggered signaling. RNF144A interacts with STING and increases its K6‐linked ubiquitination, thereby enhancing STING translocation from the ER to the Golgi.
Introduction
Upon viral infection, innate immune responses are initiated by pattern recognition receptors (PRRs), which recognize pathogen‐associated molecular patterns (PAMPs) from viruses, including nucleic acids, proteins, and carbohydrates (Carty et al, 2021). The recognition of intracellular viral DNA relies on a series of PRRs called DNA sensors, with cGAS and IFI16 as bona fide intracellular viral DNA receptors (Chan & Gack, 2016). Based on the genetic evidence that STING‐deficient cells fail to produce interferons (IFNs) in response to dsDNA stimulation, the adaptor protein STING is considered to be essential for the DNA sensor‐triggered antiviral signaling (Cai et al, 2014). Additionally, STING‐deficient mice exhibited impaired production of type I IFNs in response to DNA virus infection and were susceptible to lethal infection with DNA viruses, including HSV‐1, vaccinia virus, and murine gammaherpesvirus 68 (MHV68) (Ni et al, 2018).
Posttranslational modification (PTM) with ubiquitin is critical to the tight control and adjustment of the strength and duration of the antiviral immune responses by affecting the activities, stabilities, or localizations of target proteins (Liu et al, 2016; Zhou et al, 2017). Ubiquitin, the conserved 76‐amino acid polypeptide, is universally expressed in most tissues of eukaryotic organisms and fulfills essential functions through its conjugation with other intracellular proteins (Pickart, 2001). All seven Lys residues (K6, K11, K27, K29, K33, K48, and K63) in ubiquitin can be linked to another ubiquitin, leading to the generation of different types of ubiquitin chains with distinct functions (Park et al, 2014). While K48‐ and K63‐linked chains are the best‐known ubiquitination types, accumulating evidence has suggested that the other types of chains assembled through K6, K11, K27, K29, and K33 residues deserve equal attention in the regulation of multiple biological processes (Tracz & Bialek, 2021). The modification of ubiquitination is mediated by three coordinated enzymes, E1 ubiquitin‐activating enzyme, E2 ubiquitin‐conjugating enzyme, and E3 ubiquitin‐protein ligase (Zinngrebe et al, 2014). Till now, in the human genome, more than 600 E3 ubiquitin‐protein ligases have been identified, contributing to the high substrate specificity for protein ubiquitination (Schapira et al, 2019). Several E3 ubiquitin‐protein ligases have been demonstrated in the regulation of STING trafficking, signaling, and degradation. For example, mitochondrial E3 ubiquitin protein ligase 1 (MUL1) catalyzes the ubiquitination of STING and facilitates optimal STING trafficking (Ni et al, 2017). TRIM32 and AMFR target STING for K63‐linked or K27‐linked ubiquitination, respectively, and positively modulate type I IFN production (Zhang et al, 2012; Wang et al, 2014). Our previous research has indicated that RNF90 regulates antiviral responses by targeting STING for K48‐linked ubiquitination and subsequent degradation after viral infection (Yang et al, 2020a). RNF5 and Trim30α also regulate STING ubiquitination and degradation (Zhong et al, 2009; Wang et al, 2015).
E3 ubiquitin‐protein ligases have been structurally classified into four main classes, RING‐, U‐Box‐, Cullin‐RING‐, and HECT‐type (Uchida & Kitagawa, 2016). Recently, the RBR (RING‐in‐between RING) E3 ubiquitin ligases have been considered a novel group of E3 ligases that transfer ubiquitin from the activated E2 by a RING/HECT hybrid mechanism with RING and RING‐like domains (Wang et al, 2020). Among the 12 human RBR family members, Parkin and HOIP are relatively well‐studied E3 ligases, known for their roles in many cellular functions and pathogenesis of human diseases (Uchida & Kitagawa, 2016). RNF144A, which contains an RBR domain in the N‐terminus and a potential single‐transmembrane (TM) domain in the C‐terminus, remains poorly characterized (Ho et al, 2015; Uchida & Kitagawa, 2016). RNF144A is distributed in both plasma membrane and the membranes of intracellular vesicles including endosomes, lysosomes, endoplasmic reticulum (ER), perinuclear vesicles, and others, which might be caused by the presence of the TM domain (Ho et al, 2014). This special localization suggests that RNF144A has the potential to contribute to the regulation of receptors and their signaling. Till now, RNF144A has been suggested to play a role in several cellular processes, including the regulation of epidermal growth factor receptor (EGFR) transport in intracellular vesicles, the suppression of breast cancer growth and metastasis, the enhancement of DNA damage‐induced cell apoptosis targeting DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), and the induction of cell death under oxidative stress (Ho et al, 2014; Uchida & Kitagawa, 2016; Ho & Lin, 2018; Han & Kim, 2020; Yang et al, 2020b). However, the functional and mechanistic role of RNF144A in antiviral innate immune responses remains unclear.
In this study, we demonstrate that RNF144A is induced by HSV‐1 infection and positively regulated HSV‐1‐triggered antiviral innate immune responses. Rnf144a‐deficient bone marrow‐derived macrophages (BMDMs), mouse embryonic fibroblasts (MEFs), and peritoneal macrophages (PMs) exhibit impaired DNA virus‐ or cytosolic DNA‐triggered signaling and RNF144A prevents HSV‐1 infection in mice. Further studies on the mechanism by which RNF144A enhances STING‐mediated host defense reveal that RNF144A promotes K6‐linked ubiquitination of STING at K236. Additionally, the K236R mutant of STING displays reduced activity in promoting innate immune signal transduction. Taken together, our findings identify a new positive regulator of DNA virus‐ or cytosolic DNA‐triggered signaling pathways and a critical ubiquitination site necessary for fully functional STING during antiviral responses.
Results
RNF144A positively regulates DNA virus‐ or exogenous cytosolic DNA‐triggered innate immune responses
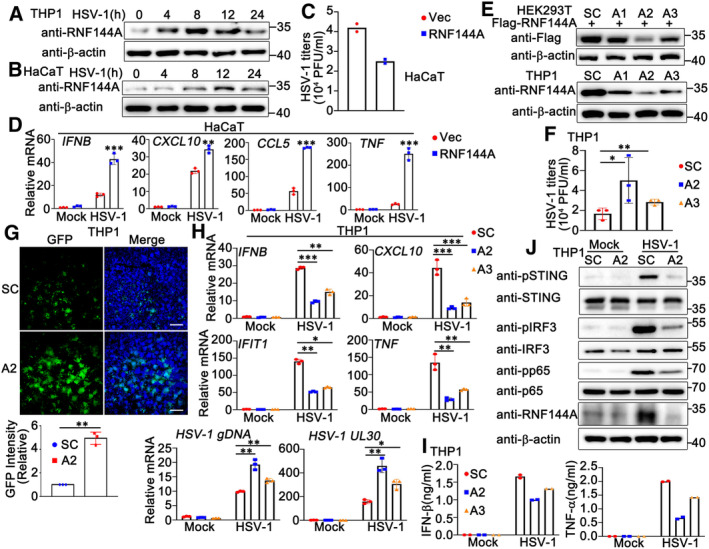
To explore the potential function of RNF144A in DNA virus infection, we first determined whether the expression of RNF144A was associated to DNA virus infection. Phorbol‐12‐myristate‐13‐acetate (PMA)‐differentiated THP1 (PMA‐THP1, a human macrophage‐like cell line) cells or HaCaT cells (human keratinocytes) were infected with DNA virus HSV‐1 and the expression of RNF144A was evaluated by immunoblot assays. As shown in Fig 1A and B, RNF144A expression was induced by HSV‐1 infection and peaked around 8–12 h after infection, suggesting that RNF144A might have a role during DNA virus invasion. Next, we examined the effect of RNF144A overexpression on DNA virus infection. HaCaT cells were transfected with RNF144A and then infected with HSV‐1. Plaque assay results indicated that RNF144A overexpression inhibited HSV‐1 infection, which motivated us to explore the involvement of RNF144A in HSV‐1‐triggered antiviral innate immune responses (Fig 1C). The effect of RNF144A on HSV‐1 infection‐induced transcription of downstream genes such as IFNB, CXCL10, CCL5, and TNF was evaluated in HaCaT cells by real‐time PCR assays. A significant increase in the production of these proteins at the mRNA level was noticed in RNF144A‐transfected cells compared to control cells after HSV‐1 infection (Fig 1D), suggesting a positive regulatory role of RNF144A in innate immune responses to DNA virus infection. Next, we investigated RNF144A function in innate immune responses that were induced by the transfection of exogenous DNA, such as HSV60 (a 60 bp oligonucleotide containing viral DNA motifs derived from the HSV‐1 genome) and HT DNA (herring testis DNA). As shown in Fig EV1A, overexpression of RNF144A in HaCaT cells upregulated the production of IFNB, CXCL10, and IL‐6 in response to HSV60 and HT DNA transfection.
Figure 1. RNF144A promotes DNA virus‐ or exogenous cytosolic DNA‐triggered innate immune responses.
-
A, BPMA‐THP1 cells (A) or HaCaT keratinocytes (B) were stimulated with HSV‐1 (MOI = 1) for the indicated periods. Afterward, the cells were lysed for immunoblot assays.
-
CHaCaT keratinocytes were transfected with the empty vector (Vec) or the RNF144A expressing plasmid for 24 h and then infected with HSV‐1 for 24 h. The titers of HSV‐1 were determined by standard plaque assay.
-
DHaCaT keratinocytes were transfected with the empty vector (Vec) or the RNF144A expressing plasmid and then infected with HSV‐1 (MOI = 1) for 8 h. The cells were then lysed for real‐time PCR assays.
-
EHEK293T cells were transfected with Flag‐RNF144A and then transfected with control siRNA (SC) or RNF144A‐specific siRNA (A1, A2, and A3). At 24 h after transfection, the cells were lysed for immunoblot assays (top). PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A1, A2, and A3). At 24 h after transfection, the cells were infected with HSV‐1 (MOI = 1) for 8 h and then immunoblot assays were performed (bottom).
-
FPMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2 and A3). At 24 h after transfection, the cells were infected with HSV‐1 (MOI = 1) for 24 h. The titers of HSV‐1 were determined by standard plaque assays.
-
GPMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2). At 24 h after transfection, the cells were infected with GFP‐HSV‐1 (MOI = 1) for 24 h and then immunofluorescence assays were performed. Nuclei were stained with DAPI. Scale bar, 500 μm.
-
HPMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2 and A3). At 24 h after transfection, the cells were infected with HSV‐1 (MOI = 1) for 8 h. The cells were then lysed for real‐time PCR assays.
-
IPMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2 and A3). At 24 h after transfection, the cells were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were collected and subjected to ELISA assays.
-
JPMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with HSV‐1 for 8 h. The cells were lysed for immunoblot assays.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01, ***P < 0.001. Data shown are from two (C, I) or at least three independent biological replicates (A, B, D–H, J). In (D, H), each data point represents a technical replicate. In (C, F, I), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
Figure EV1. RNF144A promotes DNA virus‐ or exogenous cytosolic DNA‐triggered innate immune responses.
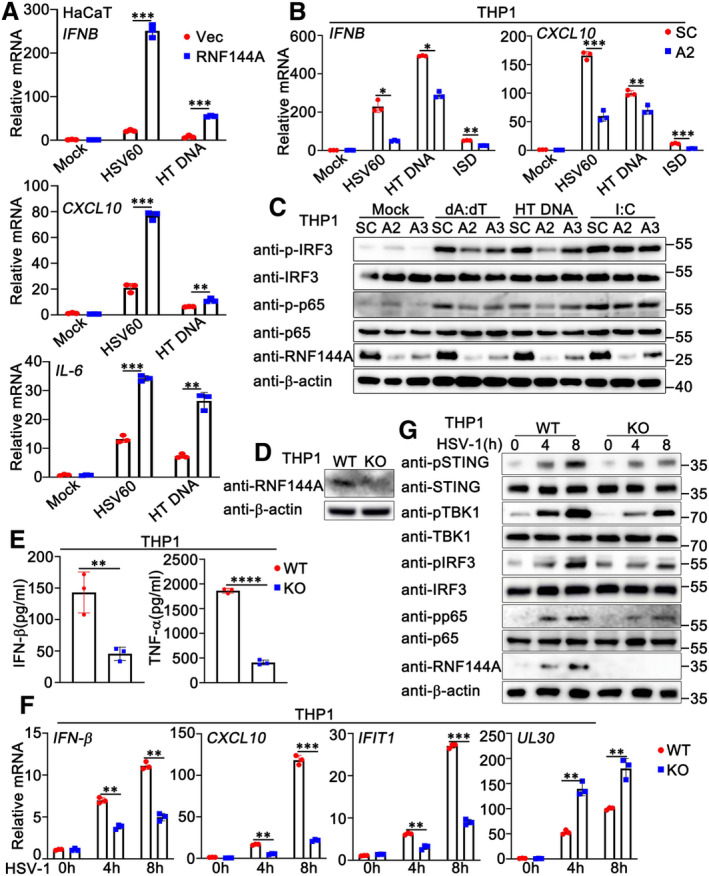
- HaCaT keratinocytes were transfected with the empty vector (Vec) or the RNF144A plasmid for 24 h and then treated with HSV60 (1 μg/ml), HT‐DNA (1 μg/ml), or left untreated for 8 h. The cells were lysed for real‐time PCR assays.
- PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2). At 24 h after transfection, the cells were transfected with HSV60 (1 μg/ml), HT‐DNA (1 μg/ml), or ISD (1 μg/ml) for 8 h. The cells were lysed for real‐time PCR assays.
- PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2, A3). At 24 h after transfection, the cells were transfected with poly(dA:dT) (1 μg/ml), HT‐DNA (1 μg/ml), or poly(I: C) (2.5 μg/ml) for 8 h. The cells were lysed for immunoblot assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) PMA‐THP1 cells were infected with HSV‐1 (MOI = 1) for 8 h. The cells were lysed for immunoblot assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) PMA‐THP1 cells were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) PMA‐THP1 cells were infected with HSV‐1 (MOI = 1) for the indicated periods. The cells were lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) PMA‐THP1 cells were infected with HSV‐1 (MOI = 1) for the indicated periods. The immunoblot assays were then performed.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01 and ***P < 0.001. Data shown are representative of at least three independent biological replicates. In (A, B, F), each data point represents a technical replicate. In (E), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
To further investigate the role of RNF144A in DNA virus infection, we purchased three pairs of siRNA oligonucleotides specific for RNF144A (A1, A2, and A3) and used the knockdown approach to evaluate the role of endogenous RNF144A in HSV‐1 infection. As shown in Fig 1E, both A2 and A3 efficiently inhibited exogenous and endogenous RNF144A expression, and A2 exhibited higher efficiency than A3 in the inhibition of RNF144A expression. Therefore, A2 and A3 were used to investigate the effect of RNF144A on HSV‐1 infection in PMA‐THP1 cells. As shown in Fig 1F, both A2 and A3 promoted HSV‐1 infection compared to control siRNA, and the increase in viral infection by A2 transfection was more significant than A3, consistent with their efficiency in the inhibition of RNF144A expression. Further, fluorescence microscopy images of GFP‐HSV‐1 also indicated that the knockdown of RNF144A enhanced HSV‐1 infection (Fig 1G). Then, we examined the role of RNF144A in HSV‐1‐induced antiviral responses. The results suggested that RNF144A knockdown inhibited the production of IFNB, CXCL10, IFIT1, and TNF at the mRNA levels (Fig 1H) and the induction of IFN‐β and TNF‐α at the protein levels (Fig 1I) upon HSV‐1 infection. Additionally, real‐time PCR assays suggested that RNF144A knockdown promoted HSV‐1 infection (Fig 1H). In addition, consistent with their inhibitory efficiency on RNF144A expression, the effects of A2 on these host defense responses and HSV‐1 infection were more significant than A3 (Fig 1H and I). Correspondingly, RNF144A knockdown decreased HSV‐1‐induced phosphorylation of STING, IRF3, and p65 significantly (Fig 1J). Similarly, knockdown of RNF144A also downregulated the innate immune responses that were triggered by exogenous cytosolic DNA (Fig EV1B and C). However, we found that knockdown of RNF144A did not make significant differences in the activation of IRF3 and p65 upon the transfection of poly (I:C), suggesting that RNF144A might only play a role in host defense against cytosolic DNA, but not cytosolic RNA (Fig EV1C). Then, Rnf144a‐deficient THP1 cells were generated by CRISPR and the role of RNF144A in the regulation of antiviral immune responses was explored further. The expression of RNF144A could not be detected in Rnf144a‐deficient PMA‐THP1 cells, as suggested by immunoblot assays (Fig EV1D). Compared to wild‐type PMA‐THP1 cells, upon HSV‐1 infection, Rnf144a‐deficient cells exhibited decreased antiviral innate immune responses (Fig EV1E–G). Taken together, our findings suggest a positive regulatory role of RNF144A in innate immune responses against DNA virus infection or exogenous DNA transfection.
RNF144A restricts DNA virus infection and promotes antiviral innate immune responses in a mouse model
To confirm the role of RNF144A in the regulation of host defense against DNA virus, Rnf144a‐deficient mice were generated by CRISPR/Cas9 strategy. As shown in Fig 2A, RNF144A expression was knocked out successfully in protein levels in both cells and organs from Rnf144a‐deficient mice. Then wild‐type and Rnf144a‐deficient mice were intraperitoneally injected with HSV‐1, and the in vivo effect of RNF144A on DNA virus infection was evaluated. Greater body weight loss and lower survival rate after HSV‐1 infection were observed in Rnf144a‐deficient mice compared to control wild‐type groups (Fig 2B and C). Further, HSV‐1‐induced destruction in the lungs was more severe in Rnf144a‐deficient mice than in wild‐type mice (Fig 2D). Next, HSV‐1 infection and antiviral responses were examined in a variety range of organs from wild‐type and Rnf144a‐deficient mice. In the lung, liver, and spleen, 24 h after HSV‐1 infection, compared to wild‐type mice, Rnf144a‐deficient mice exhibited reduced production of Ifnb, Cxcl10, Ifit1, and Il‐6 and elevated genomic DNA copies of HSV‐1 (HSV‐1 gDNA) (Fig 2E). Consistently, RNF144A deficiency resulted in impaired antiviral responses and enhanced HSV‐1 infection in the brains of mice 4 days after HSV‐1 infection (Fig 2F). In addition, ELISA results indicated that the protein levels of IFN‐β, TNF‐α, and IL‐6 in the serum were much lower in Rnf144a‐deficient mice than in wild‐type mice 6 h after intravenous infection with HSV‐1 (Fig 2G). Finally, wild‐type and Rnf144a‐deficient mice were intranasally infected with HSV‐1 for 24 h, and then cytokine secretion was examined in the bronchoalveolar lavage fluids (BALFs). As shown in Fig 2H, after HSV‐1 infection, RNF144A deficiency caused reduced secretion of IFN‐β, TNF‐α, and IL‐6 in the BALFs of mice. In all, our findings suggest that RNF144A protects mice from DNA virus infection by the manipulation of innate immune responses.
Figure 2. RNF144A restricts DNA virus infection and promotes antiviral innate immune responses in a mouse model.
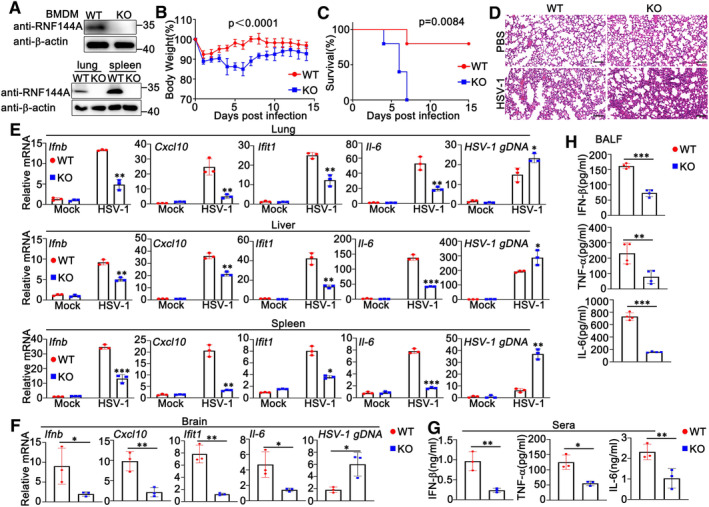
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for 8 h. The cells were lysed for immunoblot assays (top). wild‐type (WT) and Rnf144a‐deficient (KO) mice were intravenously infected with HSV‐1 (1 × 107 PFU) for 24 h and then lung and spleen sections were analyzed by immunoblot assays (bottom).
- Sex and age‐matched wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 4, 8‐week‐old) were intravenously infected with HSV‐1 (1 × 107 PFU). The body weight loss was monitored for 14 days.
- Sex and age‐matched wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 5, 8‐week‐old) were intravenously infected with HSV‐1 (2 × 107 PFU). The survival of these mice was monitored for 15 days.
- Sex and age‐matched wild‐type (WT) and Rnf144a‐deficient (KO) mice were intravenously infected with HSV‐1 (1 × 107 PFU) for 24 h and lung sections were analyzed by H&E staining. Scale bar, 100 μm.
- Wild‐type (WT) and Rnf144a‐deficient (KO) mice were intravenously infected with HSV‐1 (1 × 107 PFU) for 24 h and then the lungs, livers, and spleens of the mice were subjected to real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 3, 8‐week‐old) were intravenously infected with HSV‐1 (1 × 107 PFU) for 4 days and then the brains of the mice were subjected to real‐time PCR assays.
- ELISA of IFN‐β and TNF‐α in serum of wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 3, 8‐week‐old) 6 h after intravenous infection with HSV‐1 (1 × 107 PFU).
- Wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 4, 8‐week‐old) were intranasally infected with HSV‐1 (1 × 107 PFU) for 24 h. Bronchoalveolar lavage fluid (BALF) was collected and ELISA assays were performed.
Data information: Unpaired t‐test (B, E–H), or log‐rank (Mantel–Cox) test (C), *P < 0.05, **P < 0.01 and ***P < 0.001. Data shown are representative of at least three independent biological replicates, with each data point representing an independent biological experiment. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A deficiency impairs DNA virus‐ or exogenous cytosolic DNA‐triggered innate immune responses in BMDMs and PMs
Given that RNF144A promoted antiviral responses and inhibited HSV‐1 infection in mouse models, we further investigated the effect of RNF144A on DNA virus‐induced host defense at the cellular level. BMDMs and PMs were isolated from Rnf144a‐deficient and control wild‐type mice, respectively, and then infected with HSV‐1, followed by the evaluation of viral infection and antiviral innate immune responses. As shown in Figs 3A and B, and EV2A, compared to wild‐type cells, Rnf144a‐deficient BMDMs or PMs displayed a significant increase in HSV‐1 infection. Accordingly, HSV‐1‐induced production of Ifnb, Il‐6, Cxcl10 and Ifit1 at the mRNA level was much lower in Rnf144a‐deficient BMDMs than in control wild‐type cells (Fig 3C). Similar results were obtained in PMs (Fig EV2B). Next, we investigated the function of RNF144A in innate immune responses evoked by exogenous cytosolic DNA, including HSV60, VACV70 (a 70 bp dsDNA oligonucleotide containing vaccinia virus motifs), HT DNA, and the cytosolic DNA‐analog poly (dA:dT). Similar to the results from HSV‐1 infection, the production of Ifnb, Il‐6, Cxcl10 and Ifit1 in response to stimulation with HSV60, HT DNA, or poly (dA:dT) was significantly lower in Rnf144a‐deficient BMDMs compared to control wild‐type cells (Fig 3D). Consistently, following transfection with HSV60, IFN‐stimulatory DNA (ISD), VACV70, or HT DNA, Rnf144a‐deficient PMs displayed decreased production of Ifnb and Il‐6 compared to wild‐type PMs (Fig EV2C). Notably, innate immune responses triggered by cyclic GMP‐AMP (cGAMP), an endogenous second messenger produced by cGAS that activates STING to generate type I IFNs and subsequent innate immune responses, were also decreased by RNF144A deficiency in BMDMs (Fig 3D). Then, we evaluated the effect of RNF144A on DNA virus or exogenous cytosolic DNA‐triggered secretion of type I IFNs and proinflammatory cytokines by ELISA. In accordance with the findings at the mRNA level, inhibited production of IFN‐β, TNF‐α, and IL‐6 upon stimulation with HSV‐1, HSV60, VACV70, poly (dA:dT), or HT DNA was observed in Rnf144a‐deficient BMDMs compared to control wild‐type cells (Fig 3E and F). Similarly, RNF144A deficiency in PMs resulted in inhibited secretion of IFN‐β and TNF‐α in response to HSV‐1 infection (Fig EV2D). Consistently, RNF144A deficiency caused the restricted formation of IRF3 dimers and decreased phosphorylation of STING, IRF3, TBK1, and p65 in responses to HSV‐1 infection or HSV60 stimulation as suggested by immunoblot assays in both BMDMs and PMs (Figs 3G and H, and EV2E and F). Further, after HSV‐1 infection, impaired formation of STING dimers was observed in Rnf144a‐deficient PMs compared to wild‐type PMs (Fig EV2G). Taken together, our research indicates that RNF144A deficiency impairs innate immune responses against DNA viruses or exogenous cytosolic DNA in BMDMs and PMs.
Figure 3. RNF144A deficiency impairs DNA virus or exogenous cytosolic DNA‐triggered innate immune responses in BMDMs.
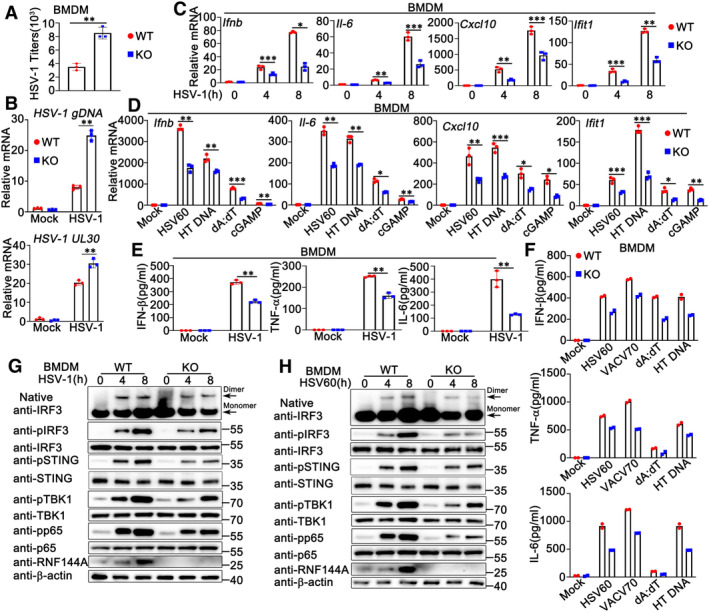
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for 24 h. The titers of HSV‐1 were determined by standard plaque assay.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for 8 h. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for the indicated periods. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were transfected with HSV60 (1 μg/ml), HT‐DNA (1 μg/ml), poly(dA:dT) (1 μg/ml) and cGAMP (1 μg/ml) for 8 h. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were then collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were transfected with HSV60 (1 μg/ml), VACV70 (1 μg/ml), poly(dA:dT) (1 μg/ml) and HT‐DNA (1 μg/ml) for 24 h. The supernatants were then collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were infected with HSV‐1 (MOI = 1) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were transfected with HSV60 (1 μg/ml) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01 and ***P < 0.001. Data shown are from two (F) or at least three independent biological replicates (A–E, G, H). In (B–D), each data point represents a technical replicate. In (A, E, F), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
Figure EV2. RNF144A deficiency impairs DNA virus or exogenous cytosolic DNA‐triggered innate immune responses in PMs.
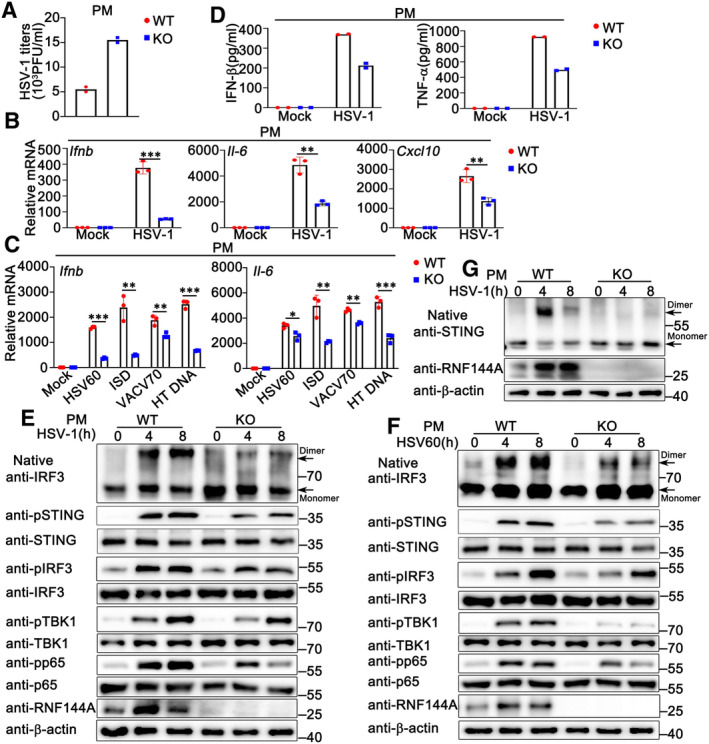
-
AWild‐type (WT) and Rnf144a‐deficient (KO) PMs were infected with HSV‐1 (MOI = 1) for 24 h. The titers of HSV‐1 were determined by standard plaque assay.
-
BWild‐type (WT) and Rnf144a‐deficient (KO) PMs were infected with HSV‐1 (MOI = 1) for 8 h. The cells were lysed for real‐time PCR assays.
-
CWild‐type (WT) and Rnf144a‐deficient (KO) PMs were transfected with HSV60 (1 μg/ml), ISD (1 μg/ml), VACV70 (1 μg/ml), or HT‐DNA (1 μg/ml) for 8 h. The cells were lysed for real‐time PCR assays.
-
DWild‐type (WT) and Rnf144a‐deficient (KO) PMs were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were collected and subjected to ELISA assays.
-
E, FWild‐type (WT) and Rnf144a‐deficient (KO) PMs were infected with HSV‐1 (MOI = 1) (E) or transfected with HSV60 (1 μg/ml) (F) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
-
GWild‐type (WT) and Rnf144a‐deficient (KO) PMs were infected with HSV‐1 (MOI = 1) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01, ***P < 0.001. Data shown are from two (A, D) or at least three independent biological replicates (B, C, E–G). In (B, C), each data point represents a technical replicate. In (A, D), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A deficiency impairs DNA virus or exogenous cytosolic DNA‐triggered innate immune responses in MEFs
Next, we investigated the role of RNF144A in HSV‐1 infection and innate immune responses in non‐immune cells. MEFs were obtained from wild‐type and Rnf144a‐deficient mice and infected with HSV‐1 or stimulated with exogenous cytosolic DNA. Compared to wild‐type MEFs, Rnf144a‐deficient MEFs displayed an increase in HSV‐1 infection as suggested by plaque assays (Fig 4A), real‐time PCR assays (Fig 4B), fluorescence microscopy (Fig 4C), and immunoblot assays (Fig 4D). Then, the effect of RNF144A on antiviral innate immune responses was evaluated. As shown in Fig 4E, upon HSV‐1 infection or HSV60 transfection, lower levels of mRNA expression of Ifnb, Il‐6, Ccl5 and Ifit1 were observed in Rnf144a‐deficient MEFs than in control wild‐type cells. Accordingly, ELISA assays suggested that RNF144A deficiency restricted the secretion of IFN‐β, IL‐6, and TNF‐α in response to HSV‐1 infection (Fig 4F) or exogenous cytosolic DNA stimulation (Fig 4G) in MEFs. In addition, compared to control wild‐type MEFs, Rnf144a‐deficient MEFs exhibited a significant decrease in antiviral signal transduction after HSV‐1 infection or HSV60 transfection, including the phosphorylation of STING, TBK1, IRF3, and p65, the formation of IRF3 dimers, and the nuclear translocation of IRF3 and p65 (Fig 4H–J). Finally, after HSV60 transfection, the culture supernatants from wild‐type and Rnf144a‐deficient BMDMs were collected and used to protect MEFs against HSV‐1 infection. As shown in Fig 4K, in plaque assays, RNF144A deficiency in BMDMs resulted in more HSV‐1 infection in MEFs, suggesting RNF144A deficiency impaired antiviral immune responses upon HSV60 transfection. In all, our findings suggest RNF144A deficiency restricts antiviral innate immune responses upon DNA virus infection or exogenous cytosolic DNA stimulation in MEFs.
Figure 4. RNF144A deficiency impairs DNA virus‐ or exogenous cytosolic DNA‐triggered innate immune responses in MEFs.
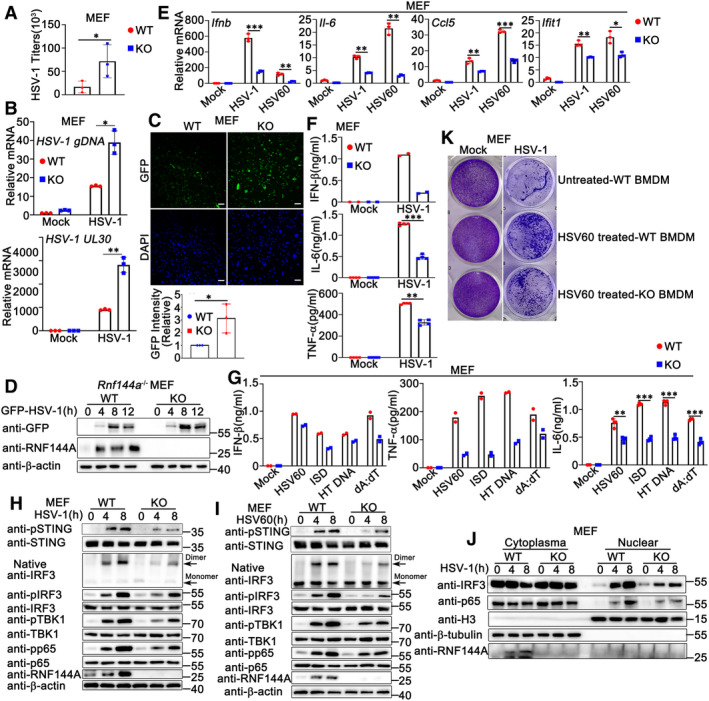
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for 24 h. The titers of HSV‐1 were determined by standard plaque assay. n = 3 biological replicates.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for 8 h. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with GFP‐HSV‐1 (MOI = 1) for 24 h and immunofluorescence assays were then performed. Nuclei were stained with DAPI. Scale bar, 100 μm. The GFP fluorescence intensity was determined by ImageJ (bottom). n = 3 biological replicates.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with GFP‐HSV‐1 (MOI = 1) for the indicated periods. Immunoblot assays were then performed.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) or transfected with HSV60 (1 μg/ml) for 8 h. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were then collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were transfected with HSV60 (1 μg/ml), ISD (1 μg/ml), HT‐DNA (1 μg/ml) and poly(dA:dT) (1 μg/ml) for 24 h. The supernatants were then collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were transfected with HSV60 (1 μg/ml) for the indicated periods. Native‐PAGE and SDS–PAGE assays were then performed.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for the indicated periods and then fractionated into cytosolic and nuclear subfractions. Immunoblot assays were then performed as indicated.
- Wild‐type (WT) and Rnf144a‐deficient (KO) BMDMs were transfected with mock or HSV60 (1 μg/ml). Equal volumes of culture supernatants from these treatments were applied to fresh MEFs, followed by HSV‐1 infection. The proliferation of cells was examined by crystal violet staining.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01 and ***P < 0.001. Data shown are from two (G left and middle panels) or at least three independent biological replicates (A–F, G right panel, H–K). In (B, E), each data point represents a technical replicate. In (A, F, G), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A does not affect RNA virus‐ or cytosolic RNA‐triggered innate immune responses
To further investigate the role of RNF144A in antiviral innate immune responses, we examined the effect of RNF144A on RNA virus infection. Unlike the phenomenon observed in HSV‐1‐infected mice, after vesicular stomatitis virus (VSV) infection, Rnf144a‐deficient mice did not exhibit significant differences from control wild‐type mice in the body weight loss, as well as the production of IFN‐β, proinflammatory cytokines, and antiviral proteins in live, lung, spleen, serum, and BALFs (Fig EV3A–E). Consistently, RNF144A deficiency did not affect Sendai virus (SeV)‐triggered secretion of IFN‐β and IL‐6 in the serum of mice (Fig EV3C). At the cellular level, no significant differences were observed between wild‐type and Rnf144a‐deficient MEFs in the innate immune responses against RNA virus or cytosolic RNA (Fig EV3F–H). In all, our observations indicate RNF144A does not regulate RNA virus‐ or cytosolic RNA‐induced innate immune responses.
Figure EV3. RNF144A does not affect RNA virus‐ or cytosolic RNA‐triggered innate immune responses.
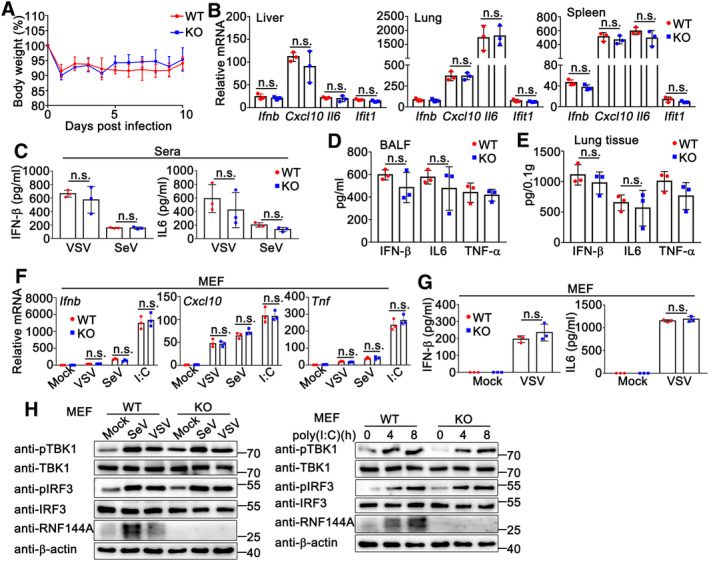
- Sex and age‐matched wild‐type (WT) and Rnf144a‐deficient (KO) mice (n = 7, 8‐week‐old) were intravenously infected with VSV (5 × 107PFU). The body weight loss was monitored for 10 days.
- Wild‐type (WT) (n = 3) and Rnf144a‐deficient (KO) (n = 3) mice were intravenously infected with VSV (5 × 107 PFU) for 24 h and then the lungs, livers, and spleens of the mice were subjected to real‐time PCR assays.
- Serum IFN‐β and IL‐6 were determined by ELISA in wild‐type (WT) (n = 3) and Rnf144a‐deficient (KO) (n = 3) mice 6 h after intravenous infection with VSV or SeV.
- Wild‐type (WT) (n = 3) and Rnf144a‐deficient (KO) mice (n = 3) were intranasally infected with VSV (5 × 107 PFU) for 24 h. Bronchoalveolar lavage fluid (BALF) was collected and ELISA assays were performed.
- Following homogenization, the lung suspensions in (D) were centrifuged, and the supernatants were subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with VSV (MOI = 1), SeV (5 HA units/ml), or transfected with poly(I: C) (1 μg/ml) for 8 h. The cells were then lysed for real‐time PCR assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with VSV (MOI = 1) for 24 h. The supernatants were collected and subjected to ELISA assays.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with VSV (MOI = 1), SeV (5 HA units/ml), or transfected with poly(I: C) (1 μg/ml) for the indicated periods and then the cells were lysed for immunoblot assays.
Data information: Two‐tailed unpaired Student's t‐test, n.s., not significant (P > 0.05). Data are representative of at least three independent biological replicates. In (F), each data point represents a technical replicate. In (B–E, G), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A deficiency impairs the expression of type I IFNs, proinflammatory cytokines and ISGs
We further performed global RNA‐sequencing (RNA‐seq) analysis to identify genes and pathways regulated by RNF144A. Differentially expression analysis identified 1,062 up‐regulated genes (log2 > 1) and 1,797 down‐regulated genes (log2 < −1) in response to RNF144A deficiency in HSV‐1 infected MEFs (Appendix Fig S1A and B). Gene‐set‐enrichment analysis (GSEA) was performed, and the results demonstrated that the core‐enriched genes were related to the cytosolic DNA sensing and HSV‐1 infection‐related pathways (Appendix Fig S1C). Additionally, Gene ontology (GO) analysis further demonstrated that these genes were involved in cellular antiviral immune responses against HSV‐1 infection (Appendix Fig S1D). Differential expression analysis further displayed that RNF144A deficiency resulted in the down‐regulation of many type I IFN‐induced IFN‐stimulated genes (ISGs) and proinflammatory cytokines upon HSV‐1 infection (log2 ≥ 1) (Appendix Fig S1E). In all, our findings suggest that RNF144A positively regulates antiviral responses by controlling the expression of type I IFNs, proinflammatory cytokines, and ISGs.
RNF144A interacts with STING
To explore the underlying mechanism by which RNF144A regulated host antiviral responses, firstly, we tried to identify the potential target protein of RNF144A in DNA sensor‐triggered signaling pathways, containing the generation of cGAMP by cGAS, the production of type I IFNs by the activation of STING‐TBK1‐IRF3 signaling pathway and the induction of antiviral state by type I IFNs. Thus, we examined the effect of RNF144A deficiency on these processes. As shown in Fig 5A, after IFN‐β stimulation, wild‐type and Rnf144a‐deficient MEFs produced equal amounts of transcripts of Cxcl10 and Ifit1. In both BMDMs and MEFs, after HSV‐1 infection, RNF144A deficiency did not make a significant difference in cGAMP generation (Fig 5B). Similar results were obtained in HSV60‐ or HT DNA‐transfected MEFs (Fig EV4A). Further, in cGAS‐deficient HeLa cells, RNF144A knockdown decreased cGAMP‐triggered activation of TBK1 and IRF3 (Fig EV4B and C). Meantime, in HSV‐1‐infected IFI16‐deficient PMA‐THP1 cells, RNF144A knockdown remained to inhibit the production of IFNB and CXCL10, and the activation of TBK1 and IRF3, suggesting the function of RNF144A was independent on IFI16 (Fig EV4D–G). These findings suggest the function of RNF144A was independent of the generation of cGAMP, IFN‐β‐triggered signaling, and the existence of cGAS and IFI16. Next, selective STING antagonist C‐176 was administered and the role of RNF144A in STING‐mediated antiviral innate immune responses was evaluated. As shown in Fig EV4H and I, after the treatment of C‐176, RNF144A deficiency did not affect HSV‐1‐triggered innate immune responses, including the production of Ifnb, Cxcl10, Ifit1, Tnf and Il‐6, and the phosphorylation of TBK1, IRF3, and p65. Further, in STING‐deficient THP1 cells, RNF144A knockdown did not affect HSV‐1 or SeV‐induced production of IFNB, CXCL10, IFIT1 and IL‐6 (Fig EV4J). Additionally, in STING‐deficient THP1 cells, RNF144A knockdown did not affect HSV‐1‐induced production of IFNB, CXCL10, and IFIT1 (Fig EV4K), and HSV‐1 infection (Fig EV4K–M), indicating that the effect of RNF144A on HSV‐1 infection was dependent on the existence of STING and innate immune responses, not by affecting virus directly. These findings suggest that there is a bigger chance to find the target protein of RNF144A in the STING‐TBK1‐IRF3 signaling pathway than in cGAMP generation and IFN‐β‐induced signal transduction. Thus, co‐immunoprecipitation assays were performed between exogenously expressed RNF144A and cGAS, STING, TBK1, or IRF3, and the results indicated that RNF144A might be associated with STING (Fig 5C and D). Then, this association was confirmed by co‐immunoprecipitation between endogenous RNF144A and STING (Fig 5E and F). Additionally, the RNF144A‐STING interaction was enhanced by HSV‐1 infection or HT‐DNA stimulation, which might be caused at least partly by the increase of RNF144A expression (Fig 5E and F). To further validate the RNF144A‐STING interaction, we performed in situ‐proximity ligation assay (PLA) using anti‐Flag and anti‐STING antibodies. Rnf144a‐deficient MEFs were transfected with Flag‐RNF144A and then stimulated with HSV‐1, HSV60, or left untreated. As shown in Fig 5G, the red dots, which depicted the co‐localization of RNF144A and STING, were detected in untreated cells and were increased after HSV‐1 infection or HSV60 transfection. Further, the co‐localization of RNF144A and STING was also suggested by confocal images (Figs 5H and EV5A). Next, we tried to explore the regions responsible for the RNF144A‐STING interaction. Surprisingly, although a series of truncation mutants of both proteins were tested, co‐immunoprecipitation assays indicated that only the full‐length RNF144A interacted with the full‐length STING (Fig 5I and J), suggesting the integrity of both proteins was required for their interaction. Notably, the two reported E3‐ligase defective mutants of RNF144A, C20/23A and C198A, with destroyed RING domain (Ho & Lin, 2018; Yang et al, 2020b), exhibited a much weaker association with STING (Fig 5K), indicating the integrity of the RING domain was also essential to the RNF144A‐STING interaction. In all, our findings suggest RNF144A interacts with STING.
Figure 5. RNF144A interacts with STING.
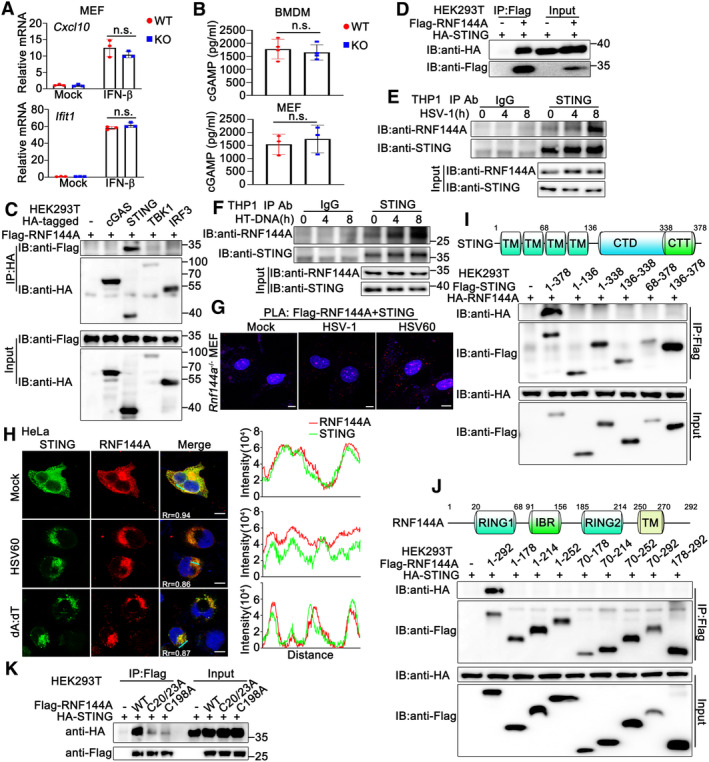
-
AWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were left untreated or treated with IFN‐β (100 ng/ml) for 4 h before real‐time PCR assays.
-
BWild‐type (WT) and Rnf144a‐deficient (KO) BMDMs (top) or MEFs (bottom) were infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were then collected and subjected to ELISA assays.
-
C, DHEK293T cells were transfected with indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) assays were performed as indicated.
-
E, FPMA‐THP1 cells were infected with HSV‐1 (MOI = 1) (E) or transfected with HT‐DNA (1 μg/ml) (F) for the indicated periods and then the cell lysates were subjected to immunoprecipitation (IP) and immunoblot (IB) assays as indicated.
-
GRnf144a‐deficient (KO) MEFs were transfected with Flag‐RNF144A. At 24 h after transfection, the cells were treated with HSV‐1 (MOI = 1), HSV60 (1 μg/ml), or left untreated for 8 h, and then in situ PLA assays were performed to examine the colocalization of RNF144A and STING. The RNF144A‐STING complex is visualized in red; nuclei are shown in blue. Scale bar, 10 μm.
-
HHeLa cells were transfected with plasmids expressing HA‐STING and Flag‐RNF144A. At 24 h after transfection, HeLa cells were transfected with HSV60 (1 μg/ml), poly(dA:dT) (1 μg/ml), or left untreated for 8 h. Immunofluorescence assays were performed using anti‐HA (green) and anti‐Flag (red). Nuclei were stained with DAPI. Scale bar, 10 μm. The plot of pixel intensity along the green line is shown in the right panel. Pearson's correlation coefficient was calculated using ImageJ software. Rr, Pearson's correlation coefficient.
-
IA schematic diagram of full‐length STING (top). TM, transmembrane; CTD, carboxy‐terminal domain; CTT, carboxy‐terminal tail. HEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) assays were performed as indicated (bottom).
-
JA schematic diagram of full‐length RNF144A (top). RING, really interesting new gene; IBR, in between RING; TM, transmembrane. HEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) assays were performed as indicated (bottom).
-
KHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, immunoprecipitation (IP) and immunoblot (IB) assays were performed as indicated.
Data information: Two‐tailed unpaired Student's t‐test, n.s., not significant (P > 0.05). Data shown are representative of at least three independent biological replicates. In (A), each data point represents a technical replicate. In (B), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
Figure EV4. RNF144A regulates STING‐mediated signaling pathways.
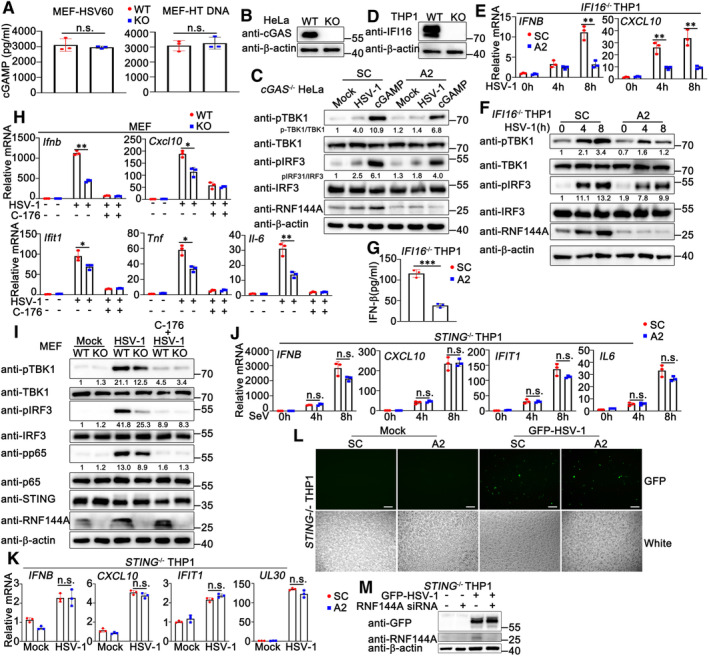
-
AWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were transfected with HSV60 (1 μg/ml) or HT DNA (1 μg/ml) for 24 h. The supernatants were collected and subjected to ELISA assays.
-
BWild‐type (WT) and cGAS‐deficient (KO) HeLa cells were lysed for immunoblot assays.
-
CcGAS‐deficient (KO) HeLa cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with HSV‐1 or transfected with cGAMP (1 μg/ml) for 8 h. The cells were lysed for immunoblot assays.
-
DWild‐type (WT) and IFI16‐deficient (KO) PMA‐THP1 cells were lysed for immunoblot assays.
-
E, FIFI16‐deficient (KO) PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with HSV‐1 (MOI = 1) for the indicated periods. Afterward, the cells were lysed for real‐time PCR (E) or immunoblot assays (F).
-
GIFI16‐deficient (KO) PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with HSV‐1 (MOI = 1) for 24 h. The supernatants were collected and subjected to ELISA analysis.
-
H, IWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were treated with C‐176 (1 μM) or left untreated for 24 h and then infected with HSV‐1 (MOI = 1) for 8 h. Afterward, the cells were lysed for real‐time PCR (H) or immunoblot assays (I).
-
JSTING‐deficient (KO) PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with SeV for the indicated periods. Afterward, the cells were lysed for real‐time PCR assays.
-
KSTING‐deficient (KO) PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with HSV‐1 for 8 h. Afterward, the cells were lysed for real‐time PCR assays.
-
L, MSTING‐deficient (KO) PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2) for 24 h and then infected with GFP‐HSV‐1 for 24 h. Afterward, the cells were subjected to image acquisition (L) or immunoblot assays (M). Scale bar: 200 μm (L). White: bright field microscopy (L).
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01, n.s., not significant (P > 0.05). Data shown are representative of at least three independent biological replicates. In (E, H, J, K), each data point represents a technical replicate. In (A, G), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
Figure EV5. RNF144A promotes the ubiquitination of STING and regulates its translocation.
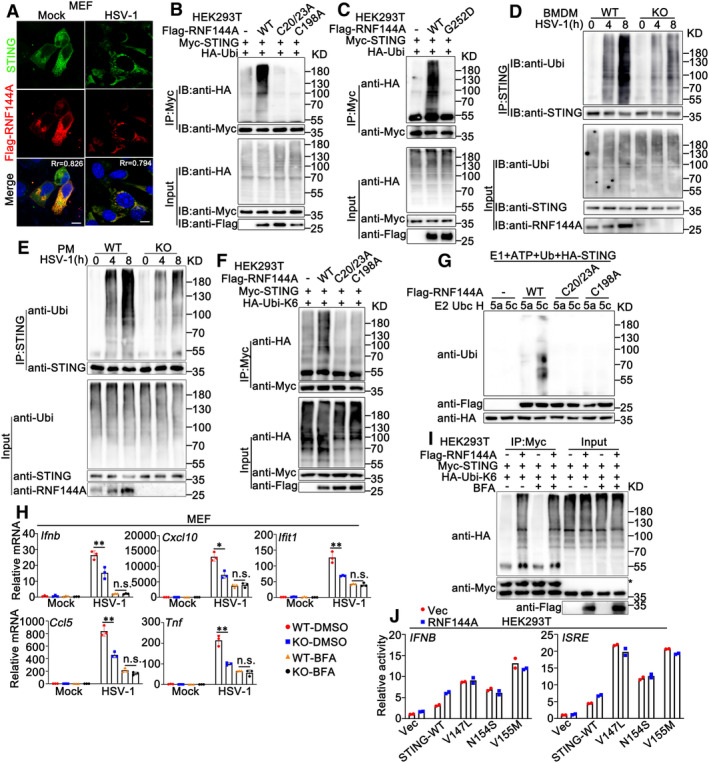
-
AMEFs were transfected with expressing plasmids for Flag‐RNF144A. At 24 h after transfection, MEFs were infected with HSV‐1 (MOI = 1) or left uninfected for 8 h. Immunofluorescence assays were performed using anti‐STING (green) and anti‐Flag (red). Nuclei were stained with DAPI. Scale bar: 10 μm. Pearson's correlation coefficient was calculated using ImageJ software. Rr, Pearson's correlation coefficient.
-
B, CHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
D, EWild‐type (WT) and Rnf144a‐deficient (KO) BMDMs (D) or PMs (E) were infected with HSV‐1 (MOI = 1) for the indicated periods. Afterward, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
FHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
GImmunoblot analysis of STING ubiquitination in vitro. STING, wild‐type RNF144A, and its mutants were quickly translated in vitro. Then, the biotin‐ubiquitin E1 and indicated E2s were added for the in vitro ubiquitination assays.
-
HWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were treated with BFA (5 μg/ml) or left untreated for 3 h and then infected with HSV‐1 (MOI = 1) for 8 h. Afterward, the cells were lysed for real‐time PCR assays.
-
IHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were treated with BFA (5 μg/ml) or left untreated for 3 h. Afterward, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays. *Heavy chain of the antibody.
-
JLuciferase activity in HEK293 cells transfected with an IFNB or ISRE luciferase reporter, together with plasmids as indicated.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01, n.s., not significant (P > 0.05). Data shown are from two (J) or at least three independent biological replicates (A–I). In (H), each data point represents a technical replicate. In (J), each data point represents an independent biological replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A promotes the ubiquitination of STING
Given that the E3 ligase RNF144A interacted with STING and regulated STING‐mediated host defense, we investigated whether RNF144A regulated the function of STING by manipulating its ubiquitination. To determine the effect of RNF144A on the ubiquitination of STING, we transfected HEK293T cells with increasing amounts of RNF144A together with STING and ubiquitin, followed by co‐immunoprecipitation assays. As shown in Fig 6A, exogenously expressed RNF144A promoted the ubiquitination of STING in a dose‐dependent pattern. However, the C20/23A and C198A mutants of RNF144A failed to enhance the ubiquitination of exogenously expressed STING in HEK293T cells (Fig EV5B), indicating that the intact catalytic activity of RNF144A is required for its effect on STING ubiquitination. Surprisingly, the G252D mutant of RNF144A found in human cancers, which preserves its ubiquitin ligase activity but loses its membrane localization, barely promoted the ubiquitination of STING (Fig EV5C), indicating the membrane localization was essential to the effect of RNF144A on STING. To exclude the possible interference by endogenous RNF144A, wild‐type RNF144A and its mutants were transfected into Rnf144a‐deficient MEFs and the effect of these ligase‐dead mutants was examined. As expected, the C20/23A and C198A mutants of RNF144A lost the ability to increase the ubiquitination of endogenous STING in MEFs (Fig 6B). Additionally, neither the C20/23A nor the C198A mutant of RNF144A increased HSV60‐induced production of Ifnb and Cxcl10 to the same extent as wild‐type RNF144A did, indicating that the integrity of RING domain was required for the role of RNF144A in STING ubiquitination and STING‐mediated signaling pathway (Fig 6C). Furthermore, knockdown of RNF144A in PMA‐THP1 cells decreased HSV‐1‐induced ubiquitination of STING (Fig 6D). Next, we evaluated the effects of RNF144A deficiency on the ubiquitination of STING. Wild‐type and Rnf144a‐deficient MEFs were infected with HSV‐1 and then the ubiquitination of STING was investigated. As shown in Fig 6E, compared to control wild‐type cells, Rnf144a‐deficient MEFs displayed impairment in HSV‐1‐induced ubiquitination of STING. Similar results were observed in Rnf144a‐deficient BMDMs and PMs (Fig EV5D and E). Then, we explored the type of ubiquitin linkage that was promoted by RNF144A. Expression plasmids for ubiquitin mutants retaining only a single lysine residue, including K6, K11, K27, K29, K33, K48, and K63, were transfected into cells together with RNF144A for immunoprecipitation and immunoblot analysis. The results showed that RNF144A mainly promoted ubiquitination of STING by the K6 mutant ubiquitin (in which only the Lys residue 6 was retained), indicating that RNF144A mainly enhanced the K6‐linked ubiquitination of STING (Fig 6F and G). This phenomenon was further confirmed by the usage of the K6R (in which only the Lys residue 6 was mutated to Arg), K29R (in which only the Lys residue 29 was mutated to Arg), and K48R (in which only the Lys residue 48 was mutated to Arg) mutants of ubiquitin. Immunoprecipitation and immunoblot analysis indicated that RNF144A increased K48R or K29R‐mediated ubiquitination of STING, but not K6R, indicating the Lys residue 6 was essential for the RNF144A‐triggered linkage of STING with ubiquitin (Fig 6H). As expected, the C20/23A and C198A mutants of RNF144A did not affect K6‐linked ubiquitination of STING (Fig EV5F). Finally, to examine whether RNF144A ubiquitinated STING directly as an E3‐ligase, in vitro ubiquitination assays were performed. We observed that RNF144A enhanced the ubiquitination of STING in a cell‐free translation system with only E1 and E2 enzymes (Fig 6I). However, the C20/23A and C198A mutants of RNF144A did not affect the ubiquitination of STING in this system (Fig EV5G). Collectively, our findings demonstrate that RNF144A promotes the K6‐linked ubiquitination of STING.
Figure 6. RNF144A promotes the ubiquitination of STING.
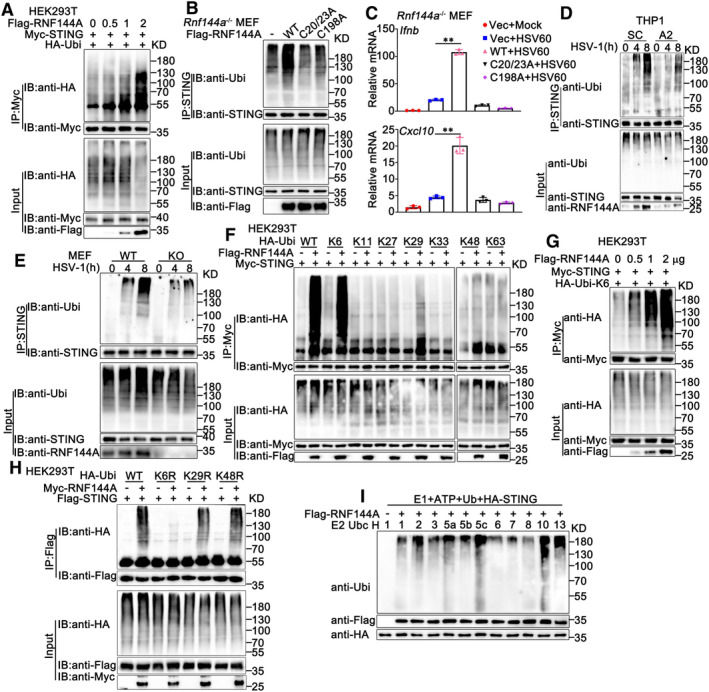
- HEK293T cells were transfected with the indicated plasmids and increasing amounts of Flag‐RNF144A (0, 0.5, 1, and 2 μg). At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
- Rnf144a‐deficient (KO) MEFs were transfected with various combinations of plasmids as indicated. 24 h later, immunoprecipitation (IP) and immunoblot (IB) assays were performed.
- Rnf144a‐deficient (KO) MEFs were transfected with an empty vector (Vec), wild‐type RNF144A plasmid, or its mutants for 24 h and then transfected with HSV60 (1 μg/ml) for 8 h. The cells were then lysed for real‐time PCR assays.
- PMA‐THP1 cells were transfected with control siRNA (SC) or RNF144A‐specific siRNA (A2). At 24 h after transfection, the cells were infected with HSV‐1 (MOI = 1) for the indicated periods. Afterward, immunoprecipitation (IP) and immunoblot (IB) assays were performed.
- Wild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for the indicated periods. Afterward, immunoprecipitation (IP) and immunoblot (IB) assays were performed.
- HEK293T cells were transfected with various combinations of plasmids as indicated. 24 h later, immunoprecipitation (IP) and immunoblot (IB) assays were performed.
- HEK293T cells were transfected with the indicated plasmids and increasing amounts of Flag‐RNF144A (0, 0.5, 1, and 2 μg). At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
- HEK293T cells were transfected with various combinations of plasmids as indicated. 24 h later, immunoprecipitation (IP) and immunoblot (IB) assays were performed.
- Immunoblot analysis of STING ubiquitination in vitro. STING and wild‐type RNF144A were quickly translated in vitro, and then the biotin‐ubiquitin E1 and indicated E2s were added for the in vitro ubiquitination assays.
Data information: Two‐tailed unpaired Student's t‐test, **P < 0.01. Data shown are representative of at least three independent biological replicates. In (C), each data point represents a technical replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A deficiency impairs the translocation, dimerization, and complex formation of STING
Next, we explored how RNF144A regulated the function of STING. As shown in Fig EV4I, RNF144A deficiency did not affect the expression of STING. It has been reported that upon binding to cGAMP, STING translocates from the ER through ERGIC to the Golgi, where STING recruits TBK1 to activate the type I IFN and proinflammatory responses through IRF3 and NF‐κB (Taguchi et al, 2021). Therefore, we examined whether RNF144A affected STING trafficking during HSV‐1 infection. As shown in Fig 7A–C, after HSV‐1 infection, RNF144A deficiency caused an increase in ER retention of STING (indicated by ER marker calnexin), and a decrease in the redistribution of STING in ERGIC (indicated by P58) and Golgi (indicated by GM130), suggesting RNF144A was required for the translocation of STING in response to HSV‐1 infection. In addition, upon treatment with Brefeldin A (BFA), which inhibited the STING movement, no significant difference was observed in the HSV‐1‐induced production of Ifnb, Cxcl10, Ifit1, Tnf and Ifit1 between wild‐type and Rnf144a‐deficient MEFs (Fig EV5H), suggesting that the effect of RNF144A on antiviral responses might be dependent on the regulation of STING trafficking. Furthermore, K6‐linked ubiquitination of STING by RNF144A occurred even in the presence of BFA, indicating that ubiquitination of STING by RNF144A was independent of STING translocation (Fig EV5I).
Figure 7. RNF144A deficiency impairs the translocation, dimerization, and complex formation of STING.
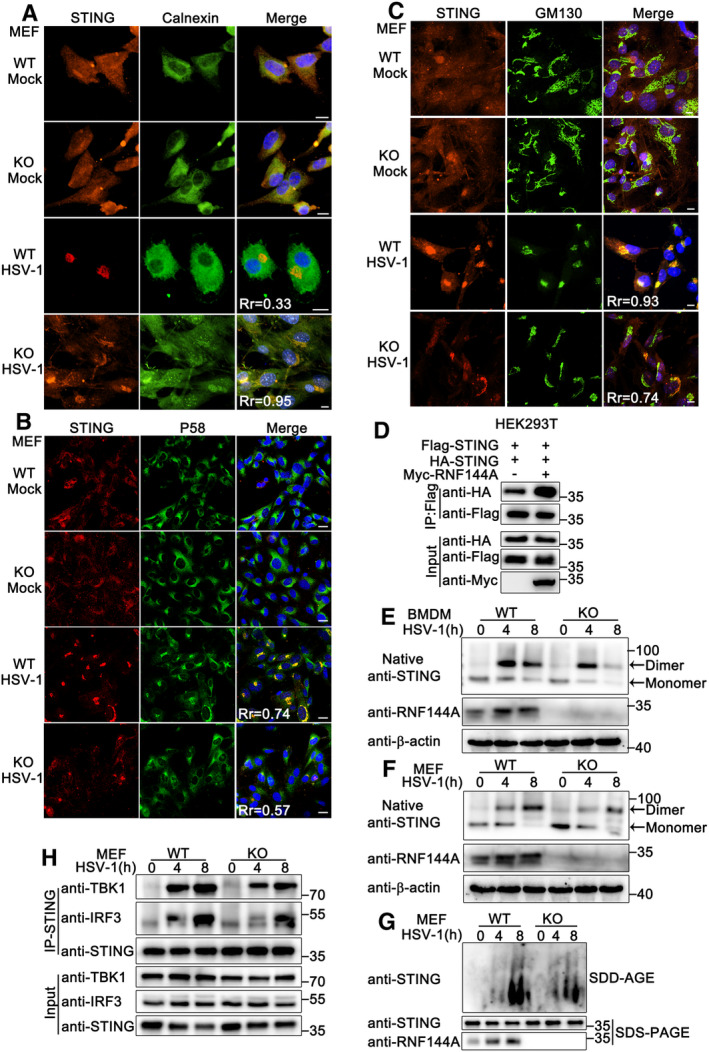
-
A–CWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for 4 h. Afterward, the cells were fixed and labeled with STING (red) and calnexin (ER marker, green) (A), P58 (ERGIC marker, green) (B), or GM130 (Golgi marker, green) (C) antibody. Scale bar: 10 μm (A, C); 20 μm (B). Pearson's correlation coefficient was calculated using ImageJ software. Rr, Pearson's correlation coefficient.
-
DHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
E, FWild‐type (WT) and Rnf144a‐deficient (KO) BMDMs (E) or MEFs (F) were infected with HSV‐1 (MOI = 1), or left uninfected for the indicated periods. The nondenatured lysates were separated in native PAGE gel and then subjected to immunoblot assays.
-
GWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for the indicated periods. Cell lysates were resolved by SDD–AGE and SDS–PAGE and subjected to immunoblot assays.
-
HWild‐type (WT) and Rnf144a‐deficient (KO) MEFs were infected with HSV‐1 (MOI = 1) for the indicated periods. Afterward, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
Data information: Data shown are representative of at least three independent biological replicates.
Source data are available online for this figure.
The binding of STING with its ligands promotes the dimerization of STING, which is necessary for STING to signal (Li et al, 2022). Therefore, we investigated the effect of RNF144A on the formation of STING dimers. Coimmunoprecipitation assays indicated that RNF144A enhanced the dimerization of overexpressed STING (Fig 7D). In wild‐type BMDMs and MEFs, HSV‐1 infection induced STING dimerization, which was decreased by RNF144A deficiency (Fig 7E and F). Furthermore, the SDD‐AGE assays indicated that RNF144A deficiency impaired the aggregation of STING in response to HSV‐1 (Fig 7G). In addition, compared to wild‐type MEFs, Rnf144a‐deficient MEFs exhibited a drop in the affinity of STING with TBK1 and IRF3 upon the infection of HSV‐1 (Fig 7H). Three gain‐of‐function mutants of STING (V147L, N154S, or V155M), which are associated with an auto‐inflammatory disease called STING‐associated vasculopathy with onset in infancy (SAVI), localize to perinuclear compartments instead of the ER (Dobbs et al, 2015; Mukai et al, 2016; Taguchi et al, 2021). Interestingly, we found that RNF144A could only enhance the activation of an IFN‐β or ISRE reporter that was induced by wild‐type STING, but not by SAVI mutants (Fig EV5J), indicating the contribution of RNF144A to the activation of STING might be mainly in the ER, instead of post‐ER compartments. In all, these results suggest RNF144A regulates the translocation, dimerization, and complex formation of STING.
RNF144A promotes ubiquitination of STING at K236
To investigate whether the RNF144A‐promoted ubiquitination of STING was crucial for its function in antiviral signaling pathways, we sought to identify the ubiquitination site at STING that was promoted by RNF144A. A series of STING mutants were generated with the potential lysine sites replaced by arginine residues individually. As shown in Fig 8A, wild‐type STING, and its mutants were transfected into HEK293T cells along with Myc‐RNF144A and HA‐Ubi, and the ubiquitination of STING and its mutants was examined. Immunoprecipitation and immunoblot analysis indicated that the K236R mutant of STING was barely ubiquitinated by RNF144A, suggesting RNF144A promoted the ubiquitination of STING at K236 (Fig 8A). Next, we investigated whether the mutation at K236 affected the function of RNF144A and STING. The wild‐type and the K236R mutant of STING were transfected into HEK293T cells, together with cGAS and an IFN‐β or ISRE reporter. Luciferase assays indicated that the K236R mutant of STING could not enhance the activity of IFN‐β and ISRE reporters as much as the wild‐type STING did (Fig 8B). In contrast, RNF144A only promoted the activity of IFN‐β and ISRE reporters that were induced by wild‐type STING, but not by the K236R mutant of STING (Fig 8B). Consistently, the K236R mutation of STING impaired its ability to activate TBK1 and IRF3 and blocked the positive regulatory role of RNF144A in the STING function (Fig 8C). In addition, the K236R mutants of STING displayed a lower affinity with TBK1 and IRF3 compared to wild‐type STING (Fig 8D and E). To exclude the effects of endogenous STING, we compared the function of the wild‐type and the K236R mutant of STING in STING‐deficient cells. STING‐deficient PMA‐THP1 cells were overexpressed with wild‐type STING or its K236R mutant, and then HSV‐1‐induced signaling pathways were evaluated. As shown in Fig 8F and H, compared to wild‐type STING, the K236R mutant of STING resulted in weaker innate immune responses. Consistent with this, compared to wild‐type STING, there was less distribution of the K236R mutant on ERGIC in HSV‐1 infected STING‐deficient PMA‐THP1 cells (Fig 8I). Taken together, our observations indicate that RNF144A promotes the ubiquitination of STING at K236, and ubiquitination at this site is critical for the function of STING in innate immune responses.
Figure 8. RNF144A promotes ubiquitination of STING at K236.
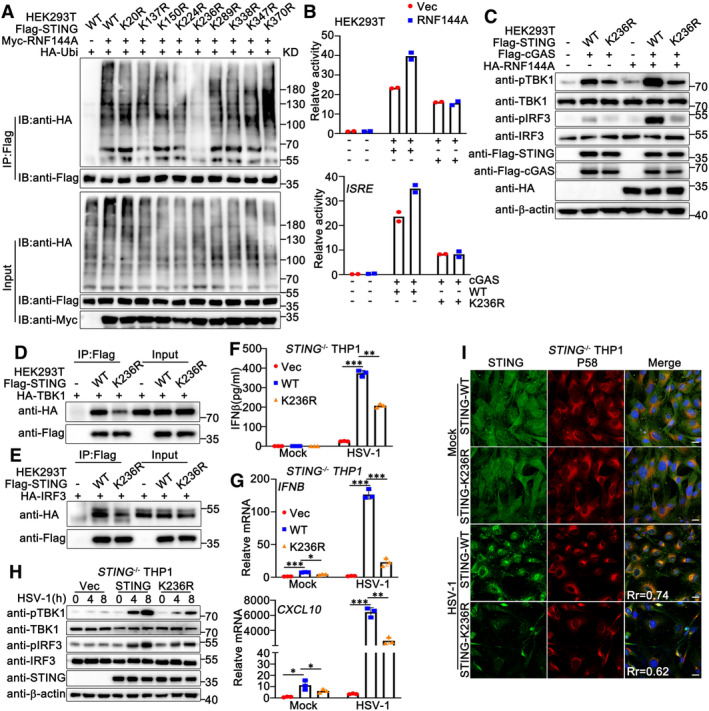
-
AHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
BLuciferase activity in HEK293 cells transfected with an IFNB or ISRE luciferase reporter, together with plasmids as indicated.
-
CHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed for immunoblot assays.
-
D, EHEK293T cells were transfected with the indicated plasmids. At 24 h after transfection, the cells were lysed and subjected to immunoprecipitation (IP) and immunoblot (IB) assays.
-
FSTING‐deficient (KO) PMA‐THP1 cells were overexpressed with the indicated plasmids. 24 h later, the cells were infected with HSV‐1 for 24 h and then lysed for ELISA assays.
-
GSTING‐deficient (KO) PMA‐THP1 cells were overexpressed with the indicated plasmids. 24 h later, the cells were infected with HSV‐1 for 8 h and then lysed for real‐time PCR assays.
-
HSTING‐deficient (KO) PMA‐THP1 cells were overexpressed with the indicated plasmids. 24 h later, the cells were infected with HSV‐1 for the indicated periods and then lysed for immunoblot assays.
-
ISTING‐deficient (KO) PMA‐THP1 cells were overexpressed with the indicated plasmids. 24 h later, the cells were infected with HSV‐1 (MOI = 1) for 4 h. Afterward, the cells were fixed and labeled with STING (green) and P58 (ERGIC marker, red) antibodies. Scale bar: 10 μm. Pearson's correlation coefficient was calculated using ImageJ software. Rr, Pearson's correlation coefficient.
Data information: Two‐tailed unpaired Student's t‐test, *P < 0.05, **P < 0.01, ***P < 0.001. Data shown are from two (B) or at least three independent biological replicates (A, C–I). In (B, F), each data point represents an independent biological replicate. In (G), each data point represents a technical replicate. Error bars are presented as mean ± SD.
Source data are available online for this figure.
RNF144A can act as a promoter of systemic lupus erythematous
STING also plays an important role in autoimmune diseases, including systemic lupus erythematosus (SLE), which is characterized by high levels of type I IFNs and pro‐inflammatory cytokines (Hasan et al, 2015; Motwani et al, 2019). Therefore, we wondered whether RNF144A was dysregulated in SLE. The involvement of RNF144A in SLE was evaluated by the analysis of two large‐scale gene expression profiles of leukocytes and peripheral blood mononuclear cells (PBMCs) of 302 SLE patients and 27 healthy donors, including GSE37356 (Data ref: Ramsey‐Goldman, 2012)and GSE45291 (Data ref: Bienkowska & Browning, 2013), and in both datasets, the results indicated that RNF144A was upregulated in SLE patients compared to healthy donors (Appendix Fig S2A). We then isolated PBMCs from both SLE patients and healthy donors and confirmed the upregulation of RNF144A in the PBMCs from SLE patients, with higher IFNB expression compared to that in healthy donors (Appendix Fig S2B). These observations prompted us to investigate the role of RNF144A in the regulation of type I IFN signaling in SLE patients. PBMCs were isolated from SLE patients and transfected with RNF144A‐specific siRNA A2 or control siRNA. As shown in Appendix Fig S2C, the mRNA levels of IFNB and IFIT1 were markedly decreased in RNF144A‐silenced PBMCs of SLE patients, and the expression levels of CXCL10 and CCL5 also exhibited a trend of downregulation by RNF144A knockdown, suggesting that knockdown of RNF144A attenuated the type I IFN signaling response in the PBMCs of SLE patients. Taken together, these data indicate that RNF144A may function as an enhancer of the autoimmune diseases like SLE.
Discussion
While the current study has uncovered several functions of RNF144A, mainly related to its role in cell proliferation, apoptosis, and cancer (Andrieu et al, 2017), the effect of RNF144A on innate immune responses remains poorly understood. To our knowledge, this is the first report to demonstrate the role of RNF144A in antiviral innate immune responses. In this study, we have mainly characterized RNF144A as a positive regulator of DNA virus‐ or exogenous DNA‐triggered signaling pathways. We used HSV‐1 to establish an infection model with mice and cells. The exogenous DNA that we selected to induce innate immune responses included viral DNA (HSV60 and VACV70), HT DNA, ISD, and the cytosolic DNA‐analog poly (dA:dT). With the treatment of HSV‐1 infection or these commonly used types of DNA ligands, we observed the following findings. Firstly, RNF144A expression was induced by HSV‐1 infection or viral DNA stimulation. Secondly, in an HSV‐1‐infected mouse model, the deficiency of RNF144A caused a decrease in body weight, survival rate, and antiviral immune responses in various organs. Thirdly, at the cellular level, overexpression, knockdown, or knockout of RNF144A in immune and non‐immune cells yielded consistent results showing that RNF144A restricted HSV‐1 infection and promoted antiviral innate immune responses. Based on these findings, we conclude that RNF144A positively regulated DNA virus‐ or exogenous DNA‐triggered innate immune signaling pathways.
The translocation of STING from the ER to perinuclear compartments, including the Golgi, endosomes, and autophagy‐related compartments, is essential for its function in innate immune signaling pathways (Dobbs et al, 2015; Mukai et al, 2016; Taguchi et al, 2021). In our study, we found that the deficiency of RNF144A caused an increase in the ER retention of STING, and a decrease in its redistribution to the ERGIC and Golgi apparatus, suggesting a regulatory role of RNF144A in innate immune responses through modulation of STING translocation. Although RNF144A also regulated the dimerization and complex formation of STING, our investigation revealed that RNF144A could not enhance the activation of an IFN‐β reporter induced by SAVI mutants, indicating that RNF144A mainly regulated the activation of STING by its effect on ER retention, rather than post‐ER compartments. However, the exact mechanisms underlying how RNF144A manipulated the redistribution of STING remain unclear. Although it is known that RNF144A has a specific location in cells, including the membranes of the ER and perinuclear vesicles, further studies are needed to clarify the mechanisms by which RNF144A regulates the translocation of STING.
Differently linked polyubiquitin chains are associated with diverse physiological roles (Hertzog & Rehwinkel, 2020). While K48‐ and K63‐linked chains have been broadly investigated in the past years, the other types of chains assembled through K6, K11, K27, K29, and K33 residues remain understudied (Tracz & Bialek, 2021). K6‐linked ubiquitination is involved in the regulation of mitophagy and the DNA damage response, by controlling the degradation, stability, and other non‐degradative processes of target proteins (Tracz & Bialek, 2021). In innate immunity, K6‐linked ubiquitination is involved in the binding of IRF3 to the promoter of genes encoding type I IFNs, but the exact mechanism and the involved E3 ligase remain unknown (Zhang et al, 2020). Upon HSV‐1 infection, TRIM13 has been reported to promote the K6‐liked ubiquitination of STING and its subsequent degradation, leading to the negative regulation of antiviral innate immune responses (Ni et al, 2018). In our study, we found that RNF144A also promoted K6‐linked ubiquitination of STING, but in a non‐degradative pattern. Instead, the K6‐linked ubiquitination of STING promoted by RNF144A was associated with the translocation of STING from the ER to the Golgi. Therefore, this is the first report to demonstrate that K6‐linked ubiquitination regulates translocation and play a positive regulatory role in antiviral innate immune responses.
Ubiquitination at distinct lysine residues of STING regulates its function. For instance, MUL1 ubiquitinates STING on K224 via K63‐linked polyubiquitination, which facilitates optimal STING trafficking and the transcription of host defense genes (Ni et al, 2017). In our research, we demonstrated that RNF144A added K6 polyubiquitin to STING at K236. Compared to wild‐type STING, the transfection of the K236R mutant of STING resulted in weaker innate immune responses. Furthermore, while RNF144A enhanced wild‐type STING‐induced signal transduction, RNF144A barely increased the activation of TBK1 and IFN‐β reporter triggered by the transfection of the K236R mutant of STING. These findings indicate that K236 of STING is important not only for its modification by RNF144A but also for the full function of STING in antiviral responses. We noticed that another two E3 ubiquitin ligases, autocrine motility factor receptor (AMFR) and TRIM32, also targeted STING for ubiquitination at lysine residues including K236. AMFR promoted the K27‐linked polyubiquitination of STING, whereas TRIM32 catalyzed the K63‐linked polyubiquitination of STING (Zhang et al, 2012; Wang et al, 2014). Interestingly, three different E3 ubiquitin ligases target the same lysine residues for ubiquitination in different types.
Indeed, it would be fascinating to clarify the relationship between RNF144A, AMFR, and TRIM32. However, despite the ability of exogenously expressed RNF144A to co‐immunoprecipitate with AMFR (Appendix Fig S3A), in HSV‐1‐infected THP1 cells, no endogenous interaction between RNF144A and AMFR or TRIM32 was observed (Appendix Fig S3B), suggesting that the exogenous interaction between RNF144A and AMFR may be artificial. Further, we did not observe a significant effect of RNF144A on the interaction between STING and AMFR or TRIM32 (Appendix Fig S3C). When transfected with cGAS and STING, RNF144A, AMFR, and Trim32 promoted cGAS‐STING‐induced IFN‐β expression independently (Appendix Fig S4D). However, RNF144A only slightly enhanced AMFR‐ or TRIM32‐induced IFN‐β expression, and not as effectively as RNF144A did by itself (Appendix Fig S3D). When AMFR or Trim32 was knocked down, RNF144A still had a strong impact on HSV‐1‐induced antiviral immune responses (Appendix Fig S3E and F). However, in Rnf144a‐deficient MEFs, AMFR or Trim32 knockdown only slightly inhibited HSV‐1‐induced antiviral immune responses (Appendix Fig S3F), indicating that RNF144A might be required for the full function of AMFR and Trim32, but further studies are needed to draw a conclusion.
In addition, Wang et al (2014) reported that using the two‐step immunoprecipitation assay, they failed to detect any ubiquitination signal of STING in the presence of Trim32 and they considered that Trim32 did not catalyze the polyubiquitination of STING per se but might promote the poly‐ubiquitination of other proteins in the STING complex. In the same article, Wang et al (2014) identified that other than K236, AMFR also promoted the ubiquitination of STING at K137, K150, and K224. These results may suggest that Trim32 and AMFR do not have to compete with RNF144A for the modification of STING at K236 to exert their roles in signal transduction. In summary, our current findings indicate that these three molecules are all necessary for STING activation, but may function through distinct mechanisms.
In addition to its essential role in DNA virus‐triggered signaling pathways, STING is involved in the immune response against RNA viruses in a virus‐ and cell‐type‐specific manner. STING‐deficient mice were defective in antiviral signaling pathways to some RNA viruses (Ni et al, 2018). Therefore, we wondered whether RNF144A regulated RNA virus infection. However, upon infection with VSV or SeV, at both the cellular and animal levels, the deficiency of RNF144A did not cause significant differences in viral infection and antiviral innate immune responses. One possible explanation is that DNA virus infection leads to rapid ubiquitination and phosphorylation of STING, whereas RNA virus infection up‐regulates the expression of STING and does not cause any post‐translational modifications nor the degradation of STING (Ni et al, 2018). However, more evidence is needed to confirm this theory.
Aberrant STING activation by self‐DNA causes elevated levels of type I IFNs, leading to inflammatory diseases such as SLE (Ahn & Barber, 2014). Therefore, the inhibition of type I IFNs is considered a promising strategy in the treatment of SLE (Khamashta et al, 2016; Furie et al, 2017). Our findings demonstrated that RNF144A was significantly up‐regulated in leukocytes and PBMCs of SLE patients through integrative analysis of large‐scale gene expression profiles of two SLE cohorts. Consistently, PBMCs isolated from SLE patients exhibited higher expression levels of RNF144A and IFN‐β. Considering RNF144A is a positive regulator of the STING‐mediated signaling pathway, RNF144A may serve as a target for SLE treatment. As expected, knockdown of RNF144A in the PBMCs from SLE patients decreased the production of type I IFNs, ISG, and other proinflammatory cytokines, suggesting RNF144A has the potential to be a target for treating aberrant type I IFN activity in SLE patients.
Except for the effects in antiviral innate immunity and auto‐inflammatory diseases, emerging evidence indicates the involvement of STING in antitumor immune response, and STING agonists are currently being extensively developed as a new strategy for cancer treatment (Sokolowska & Nowis, 2018). The expression pattern of RNF144A and its effects on STING‐mediated signaling in cancer cells remain unknown. Further clarification of the role of RNF144A in the STING pathway in cancer cells may shed light on the development of new cancer therapeutics.
In summary, our findings indicate that RNF144A acts as a new positive regulator of STING‐mediated antiviral innate immune responses triggered by DNA viruses. RNF144A interacts with STING and promotes its K6‐linked ubiquitination at K236, leading to an increase in the translocation of STING from the ER to the Golgi (Appendix Fig S4). These observations demonstrate that RNF144A and the ubiquitination at K236 of STING are crucial for the regulation of STING translocation and subsequent STING‐mediated antiviral responses.
Materials and Methods
Mice
Rnf144a‐deficient C57/BL6 mice were purchased from Shanghai Model Organisms. Mice were housed in a facility with access to food and water and were maintained under a 12‐h light/12‐h dark cycle. All animal procedures were performed according to guidelines approved by the Committee on animal care at Xinxiang Medical University, China. The age‐ and sex‐matched wild‐type and Rnf144a‐deficient C57/BL6 mice were used in the experiments.
Isolation of peripheral blood mononuclear cells from SLE patients
PBMCs were isolated from the blood of SLE patients using Ficoll‐plaque (GE, 17144002). The study was conducted according to the principles of the Declaration of Helsinki and approved by the Institutional Review Board of the Medical Ethic Committee of the First Affiliated Hospital of Xinxiang Medical University, in accordance with its guidelines for the protection of human subjects. (Approval Number [2021]‐KY‐079‐01). Informed consent was obtained from all subjects and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
cDNA constructs and reagents
Human RNF144A and its deletion mutants were amplified by PCR using cDNA from THP1 cells and were subsequently cloned into a pcDNA3 vector (Invitrogen). All RNF144A deletion mutants were constructed by PCR and also subcloned into a pcDNA3 vector. Human STING and its mutants, Ubi and its mutants, pIFN‐β‐Luc, and ISRE‐Luc were described previously (Yang et al, 2013, 2020a).
The following antibodies were used for immunoblot analysis or immunoprecipitation: anti‐Flag (F3165, Sigma‐Aldrich), anti‐HA (901515, Biolend), anti‐Myc (66004‐1‐Ig, Proteintech), anti‐IFI16 (sc‐8023, Santa Cruz), anti‐cGAS (26416‐1‐AP, Proteintech), anti‐STING (19851‐1‐AP, Proteintech), anti‐p‐TBK1 (5483T, Cell Signaling Technology), anti‐TBK1 (CSB‐PA024154LA01HU, Flarbio), anti‐p‐IRF3 (4947, Cell Signaling Technology), anti‐IRF3 (11312‐1‐AP, Proteintech), anti‐p‐p65 (3033, Cell Signaling Technology), anti‐p65 (10745‐1‐AP, Proteintech), anti‐RNF144A (26144‐1‐AP, Proteintech; sc‐393432, Santa Cruz Biotechnology), anti‐Ubi (sc‐8017, Santa Cruz Biotechnology), anti‐β‐tubulin (10068‐1‐AP, Proteintech), anti‐Histone H3 (CSB‐PA010109LA01HU, Flarbio), and anti‐β‐actin (60008‐1, Proteintech).
The PMA (S1819) was purchased from Beyotime Biotechnology. C‐176 (S6575) was obtained from Selleck. The poly(dA:dT) (tlrl‐patn), HSV60 (tlrl‐hsv60n), cGAMP (tlrl‐nacga23), and poly(I:C) (tlrl‐picw) were obtained from InvivoGen. Herring testis (HT) DNA (D6898) was purchased from Sigma. VACV70 was synthesized by GENEWIZ. The sequence was as follows: 5′‐CCATCAGAAAGAGGTTATATATTTTTGTGAGACCATGGAAGAGAGAGAAAGAGATAAAACTTTTTTACGACT‐3′.
Cell culture, transfection, and stimulation
HEK293T and THP1 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. HaCaT keratinocytes were purchased from Procell Life Science & Technology Co., Ltd., (Wuhan, China). IFI16‐deficient THP1 cells (thpd‐koifi16) and STING‐deficient THP1 cells (thpd‐kostg) were purchased from InvivoGen. cGAS‐deficient HeLa cells were kindly provided by Zhiduo Liu (Shanghai Jiao Tong University). HaCaT, HeLa, and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM). THP1 cells were grown in RPMI 1640. PMA‐THP1 cells referred to THP1 cells that were pretreated with 100 ng/ml PMA for 24 h. All cells were supplemented with 10%FBS (Gibco), 4 mM L‐glutamine, 100 μg/ml penicillin, and 100 U/ml streptomycin under humidified conditions with 5% CO2 at 37°C. Transfection of HaCaT, HeLa, HEK293T, THP1, and MEF cells was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Preparations of PMs, BMDMs, and MEFs
PMs, BMDMs, and MEFs were obtained from wild‐type or Rnf144a‐deficient female C57/BL6 mice. The procedure for generating PMs, BMDMs, and MEFs has been described previously (Yang et al, 2021).
Immunoprecipitation and immunoblot analysis
Immunoprecipitation and immunoblot analysis were performed as described previously (Wang et al, 2015).
IRF3 dimerization assay
IRF3 dimerization assay was performed as described previously (Wang et al, 2023a).
Real‐time PCR
Total RNA was extracted from the cultured cells with TRIzol reagent (Invitrogen) as described by the manufacturer. All gene transcripts were quantified by real‐time PCR with SYBR Green qPCR Master Mix using a 7500 Fast real‐time PCR system (Applied Biosystems). The relative fold induction was calculated using the method. The primers used for real‐time PCR were described previously (Wang et al, 2023b) and shown in Appendix Table S1.
ELISA
THP1 cells or HaCaT keratinocytes were infected with viruses or transfected with synthetic nucleic acids for 24 h. The supernatants were collected for measurement of IFN‐β (KE00187, Proteintech) and TNF‐α (88‐7346‐88, Thermo Fisher Scientific). 8‐week‐old wild‐type mice were infected with HSV‐1, VSV or SeV for 6 h. Then the serum of mice was collected for measurement of IFN‐β (R&D), TNF‐α (88‐7323‐88, Thermo Fisher Scientific), and IL‐6 (88‐7064‐88, Thermo Fisher Scientific) by ELISA.
RNA interference
RNF144A Stealth‐RNAi siRNA was designed by the Invitrogen BLOCKiT RNAi Designer. The small interfering RNA (siRNA) sequences were as follows:
A1: Forward, 5′‐GCAAGUGCAGUAAAGGUGACGACGA‐3′; Reverse, 5′‐UCGUCGUCACCUUUACUGCACUUGC‐3′.
A2: Forward, 5′‐GAGUACCCAGUGGAGCAGAUGACAA‐3′; Reverse, 5′‐UUGUCAUCUGCUCCACUGGGUACUC‐3′.
A3: Forward, 5′‐GCCUGAAACAGUAUGUUGAGCUCUU‐3′; Reverse, 5′‐AAGAGCUCAACAUACUGUUUCAGGC‐3′.
The Silencer Select negative control siRNA was purchased from Invitrogen (Catalog no. 4390843). PMA‐THP1 cells were transfected with siRNA using Lipofectamine 2000 according to the manufacturer's instructions. At 24 h after transfection, the cells were used for further experiments.
Viruses and infection
Cells were infected with HSV‐1 (KOS strain, multiplicity of infection of 1) for 1.5 h. Then the cells were washed with PBS and cultured in fresh media. In the mouse model, age‐ and sex‐matched groups of mice were intravenously infected with HSV‐1. HSV‐1 viral titer was determined by the plaque‐forming assay on Vero cells.
The infection of VSV was performed as described previously (Yang et al, 2021).
Luciferase reporter gene assay
Luciferase reporter gene assays were performed as described previously (Wang et al, 2017).
Confocal microscopy
After treatment, THP1, HeLa, and MEF cells were fixed with 4% PFA in PBS, permeabilized with Triton X‐100, and then blocked with 1% BSA in PBS. Nuclei were stained with 4, 6‐diamidino‐2‐phenylindole (DAPI). Images were taken using Nikon A1R microscopy.
In situ PLA
In situ PLA assay was performed as described previously (Wang et al, 2023b).
RNA‐seq analysis
After HSV‐1 stimulation for 4 h, total RNA was extracted from control and Rnf144a‐deficient MEF cells using the TRIzol reagent. Library preparation and sequencing were performed by BGI (Shenzhen, China). To take an insight into the change of phenotype, GO (http://www.geneontology.org/) and KEGG (https://www.kegg.jp/) enrichment analysis of annotated different expressed genes was performed by Dr. Tom (https://biosys.bgi.com/). The significant levels of terms and pathways were corrected by Q value with a rigorous threshold (Q value ≤ 0.05) by Bonferroni. All RNA‐seq data have been deposited in the NCBI database and are publicly available. The accession number is PRJNA972845.
Nuclear extracts
The nuclear extracts were prepared as described previously (Yang et al, 2018).
Statistics
The data are presented as the means ± SD from at least three independent experiments. The statistical comparisons between the different treatments were performed using the unpaired Student t‐test, and P < 0.05 was considered statistically significant.
Author contributions
Bo Yang: Conceptualization; resources; data curation; software; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Jinyong Pei: Conceptualization; data curation; formal analysis; validation; investigation; methodology. Chen Lu: Conceptualization; data curation; formal analysis; validation; investigation; methodology. Yi Wang: Formal analysis; validation; investigation; methodology. Mengyang Shen: Formal analysis; validation; investigation; methodology. Xiao Qin: Formal analysis; investigation; methodology. Yulu Huang: Formal analysis; investigation; methodology. Xi Yang: Validation; investigation. Xin Zhao: Validation; investigation. Shujun Ma: Validation; investigation. Zhishan Song: Validation; investigation. Yinming Liang: Resources; investigation. Hui Wang: Resources; supervision. Jie Wang: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We want to thank all the members of Henan Key Laboratory of Immunology and Targeted Drug for sharing valuable material and research support. This work was supported by the National Natural Science Foundation of China Grants (31970847, 32070949, U2004103, and 32170871), 111 Project (NO. D20036), Natural Science Foundation of Henan Province (212300410065 and 222300420064), Program for Science & Technology Innovation Talents in Higher Education of Henan Province (23HASTIT050).
EMBO reports (2023) 24: e57528
Contributor Information
Bo Yang, Email: byang94@xxmu.edu.cn.
Yinming Liang, Email: yinming.liang@foxmail.com.
Hui Wang, Email: wanghui@xxmu.edu.cn.
Jie Wang, Email: jiewang618@xxmu.edu.cn.
Data availability
RNA‐seq data: Sequence Read Archive PRJNA972845 (https://www.ncbi.nlm.nih.gov/sra/PRJNA972845).
References
- Ahn J, Barber GN (2014) Self‐DNA, STING‐dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol 31: 121–126 [DOI] [PubMed] [Google Scholar]
- Andrieu G, Ledoux A, Branka S, Bocquet M, Gilhodes J, Walzer T, Kasahara K, Inagaki M, Sabbadini RA, Cuvillier O et al (2017) Sphingosine 1‐phosphate signaling through its receptor S1P5 promotes chromosome segregation and mitotic progression. Sci Signal 10: eaah4007 [DOI] [PubMed] [Google Scholar]
- Bienkowska JAN, Browning J (2013) Expression profiling by array GSE4591 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45291). [DATASET]
- Cai X, Chiu YH, Chen ZJ (2014) The cGAS‐cGAMP‐STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54: 289–296 [DOI] [PubMed] [Google Scholar]
- Carty M, Guy C, Bowie AG (2021) Detection of viral infections by innate immunity. Biochem Pharmacol 183: 114316 [DOI] [PubMed] [Google Scholar]
- Chan YK, Gack MU (2016) Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14: 360–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N (2015) STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18: 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S et al (2017) Anifrolumab, an anti‐interferon‐alpha receptor monoclonal antibody, in moderate‐to‐severe systemic lupus erythematosus. Arthritis Rheumatol 69: 376–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Kim KT (2020) RNF144a induces ERK‐dependent cell death under oxidative stress via downregulation of vaccinia‐related kinase 3. J Cell Sci 133: jcs247304 [DOI] [PubMed] [Google Scholar]
- Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC III, Lehrman MA et al (2015) Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 43: 463–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzog J, Rehwinkel J (2020) Regulation and inhibition of the DNA sensor cGAS. EMBO Rep 21: e51345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SR, Lin WC (2018) RNF144A sustains EGFR signaling to promote EGF‐dependent cell proliferation. J Biol Chem 293: 16307–16323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SR, Mahanic CS, Lee YJ, Lin WC (2014) RNF144A, an E3 ubiquitin ligase for DNA‐PKcs, promotes apoptosis during DNA damage. Proc Natl Acad Sci USA 111: E2646–E2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SR, Lee YJ, Lin WC (2015) Regulation of RNF144A E3 ubiquitin ligase activity by self‐association through its transmembrane domain. J Biol Chem 290: 23026–23038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, Drappa J, Wang L, Greth W, CD1067 Study Investigators (2016) Sifalimumab, an anti‐interferon‐alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double‐blind, placebo‐controlled study. Ann Rheum Dis 75: 1909–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yu Z, Fang Q, Yang M, Huang J, Li Z, Wang J, Chen T (2022) The transmembrane endoplasmic reticulum‐associated E3 ubiquitin ligase TRIM13 restrains the pathogenic‐DNA‐triggered inflammatory response. Sci Adv 8: eabh0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Qian C, Cao X (2016) Post‐translational modification control of innate immunity. Immunity 45: 15–30 [DOI] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, Fitzgerald KA (2019) DNA sensing by the cGAS‐STING pathway in health and disease. Nat Rev Genet 20: 657–674 [DOI] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, Taguchi T (2016) Activation of STING requires palmitoylation at the Golgi. Nat Commun 7: 11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Konno H, Barber GN (2017) Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol 2: eaah7119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Ma Z, Damania B (2018) cGAS and STING: at the intersection of DNA and RNA virus‐sensing networks. PLoS Pathog 14: e1007148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Jin HS, Aki D, Lee J, Liu YC (2014) The ubiquitin system in immune regulation. Adv Immunol 124: 17–66 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Ramsey‐Goldman RKB (2012) Expression profiling by array GSE37356 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37356). [DATASET]
- Schapira M, Calabrese MF, Bullock AN, Crews CM (2019) Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 18: 949–963 [DOI] [PubMed] [Google Scholar]
- Sokolowska O, Nowis D (2018) STING signaling in cancer cells: important or not? Arch Immunol Ther Exp (Warsz) 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T, Mukai K, Takaya E, Shindo R (2021) STING operation at the ER/Golgi interface. Front Immunol 12: 646304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracz M, Bialek W (2021) Beyond K48 and K63: non‐canonical protein ubiquitination. Cell Mol Biol Lett 26: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida C, Kitagawa M (2016) RING‐, HECT‐, and RBR‐type E3 ubiquitin ligases: involvement in human cancer. Curr Cancer Drug Targets 16: 157–174 [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, Yu H, Pan Y, Wang C (2014) The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41: 919–933 [DOI] [PubMed] [Google Scholar]
- Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, Lin G, Lian Z, Jiang Z, Sun B (2015) TRIM30alpha is a negative‐feedback regulator of the intracellular DNA and DNA virus‐triggered response by targeting STING. PLoS Pathog 11: e1005012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yang S, Liu L, Wang H, Yang B (2017) HTLV‐1 Tax impairs K63‐linked ubiquitination of STING to evade host innate immunity. Virus Res 232: 13–21 [DOI] [PubMed] [Google Scholar]
- Wang P, Dai X, Jiang W, Li Y, Wei W (2020) RBR E3 ubiquitin ligases in tumorigenesis. Semin Cancer Biol 67: 131–144 [DOI] [PubMed] [Google Scholar]
- Wang J, Qin X, Huang Y, Zhang G, Liu Y, Cui Y, Wang Y, Pei J, Ma S, Song Z et al (2023a) Sirt1 negatively regulates cellular antiviral responses by preventing the cytoplasmic translocation of interferon‐inducible protein 16 in human cells. J Virol 97: e0197522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Qin X, Huang Y, Zhang Q, Pei J, Wang Y, Goren I, Ma S, Song Z, Liu Y et al (2023b) TRIM7/RNF90 promotes autophagy via regulation of ATG7 ubiquitination during L. monocytogenes infection. Autophagy 19: 1844–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Wang J, Wang Y, Zhou H, Wu X, Tian Z, Sun B (2013) Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J Immunol 190: 3613–3619 [DOI] [PubMed] [Google Scholar]
- Yang B, Song D, Liu Y, Cui Y, Lu G, Di W, Xing H, Ma L, Guo Z, Guan Y et al (2018) IFI16 regulates HTLV‐1 replication through promoting HTLV‐1 RTI‐induced innate immune responses. FEBS Lett 592: 1693–1704 [DOI] [PubMed] [Google Scholar]
- Yang B, Liu Y, Cui Y, Song D, Zhang G, Ma S, Liu Y, Chen M, Chen F, Wang H et al (2020a) RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog 16: e1008387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Zhang Y, Li DD, Zhang FL, Liu HY, Liao XH, Xie HY, Lu Q, Zhang L, Hong Q et al (2020b) RNF144A functions as a tumor suppressor in breast cancer through ubiquitin ligase activity‐dependent regulation of stability and oncogenic functions of HSPA2. Cell Death Differ 27: 1105–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zhang G, Qin X, Huang Y, Ren X, Sun J, Ma S, Liu Y, Song D, Liu Y et al (2021) Negative regulation of RNF90 on RNA virus‐triggered antiviral immune responses targeting MAVS. Front Immunol 12: 730483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hu MM, Wang YY, Shu HB (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63‐linked ubiquitination. J Biol Chem 287: 28646–28655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang D, Wang P, Zhao Y, You F (2020) OTUD1 negatively regulates type I IFN induction by disrupting noncanonical ubiquitination of IRF3. J Immunol 204: 1904–1918 [DOI] [PubMed] [Google Scholar]
- Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, Shu HB (2009) The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30: 397–407 [DOI] [PubMed] [Google Scholar]
- Zhou Y, He C, Wang L, Ge B (2017) Post‐translational regulation of antiviral innate signaling. Eur J Immunol 47: 1414–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinngrebe J, Montinaro A, Peltzer N, Walczak H (2014) Ubiquitin in the immune system. EMBO Rep 15: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]