

Abstract
Glioblastoma is a very aggressive tumor and represents the most common primary brain malignancy. Key characteristics include its high resistance against conventional treatments, such as radio‐ and chemotherapy and its diffuse tissue infiltration, preventing complete surgical resection. The analysis of migration and invasion processes in a physiological microenvironment allows for enhanced understanding of these phenomena and can lead to improved therapeutic approaches. Here, we combine two state‐of‐the‐art techniques, adult organotypic brain tissue slice culture (OTC) and light‐sheet fluorescence microscopy (LSFM) of cleared tissues in a combined method termed OTCxLSFM. Using this methodology, we can show that glioblastoma tissue infiltration can be effectively blocked through treatment with arsenic trioxide or WP1066, as well as genetic depletion of the tetraspanin, transmembrane receptor CD9, or signal transducer and activator of transcription 3 (STAT3). With our analysis pipeline, we gain single‐cell level, three‐dimensional information, as well as insights into the morphological appearance of the tumor cells.
Keywords: glioblastoma, glioma stem‐like cells, invasion, light‐sheet microscopy
Subject Categories: Cancer, Methods & Resources, Signal Transduction
Glioblastoma, an aggressive brain tumor, is resistant to standard treatments and infiltrates brain tissue. OTCxLSFM reveals that blocking infiltration is possible through pharmacological and genetic interventions, providing insights into tumor cell behavior.
Introduction
Tumors arising in the central nervous system are particularly devastating considering that the brain is one of the most delicate organs with a very special physiology including the blood–brain barrier, its specialized cell types, and its vital importance preventing excessive surgery. The most common and aggressive primary brain tumors are glioblastomas (GBM), which are graded as a grade IV tumor according to the current WHO classification system (Louis et al, 2021). Key characteristics of GBM are their particularly high resistance against conventional chemotherapeutics and radiotherapy and their fast and highly infiltrative growth into the surrounding brain tissue, which frequently leads to tumor dispersal across the corpus callosum (Becker & Yu, 2012), further complicating surgical removal and localized therapies such as radiation treatment. Despite drastic therapy consisting of maximally possible surgical resection followed by combined radio‐chemotherapy with temozolomide (TMZ), the median survival barely exceeds 1 year and the 5‐year‐survival rate is below 5% (Stupp et al, 2005, 2007, 2009; Luwor et al, 2013). Although novel treatment approaches such as tumor treating fields show some success (Toms et al, 2019; Rominiyi et al, 2020), recurrences are common and overall survival is still particularly low. In fact, patients suffering from GBM have one of the worst prognoses compared to other primary cancers (Quaresma et al, 2015).
One major reason for failure of glioma treatment is the infiltration of tumor cells into the surrounding brain parenchyma. This inevitably excludes a total resection of the tumor and recurrence is virtually unavoidable due to the presence of treatment‐resistant glioma stem‐like cells (GSCs). It is known for many years that glioma cells migrate along existing anatomic brain structures like white matter tracts, blood vessels, and the extracellular matrix via interaction of multiple transmembraneous receptors (Ariza et al, 1995; Wiranowska et al, 2006; Krusche et al, 2016). Recently, it has been reported that particularly white matter tract invasion of GSCs is regulated by a Notch1 > Sox9 > Sox2 feedback loop (Wang et al, 2019). Compared to the majority of tumor cells, these GSCs have, among other features, a higher differentiation potential, allowing them to replenish a tumor, usually reside within specialized niches (Gilbertson & Rich, 2007; Soeda et al, 2009; Hide et al, 2019), and express various marker proteins associated with stemness (e.g., OLIG2, SOX2, SOX9) while lacking marker proteins of terminal differentiation like NeuN or MAP2 GSCs (Boumahdi et al, 2014; Trepant et al, 2015; Yoshimura et al, 2015; Bradshaw et al, 2016; Garros‐Regulez et al, 2016; Tirosh et al, 2016; Voronkova et al, 2017). Similarly, the signal transducer and activator of transcription 3 (STAT3) pathway is frequently upregulated in GBM and GSCs and is known to regulate migration and invasion (Kim et al, 2014; Yu et al, 2014; Galoczova et al, 2018). Currently, in a phase II clinical trial against recurrent malignant glioma (NCT05879250), the JAK2/STAT3 inhibitor WP1066, an analog of caffeic acid, effectively induces apoptosis in gliomas and reduces VEGF production in solid malignancies, suggesting modulation of downstream transcriptional products regulated by p‐STAT3 (Hussain et al, 2007; Groot et al, 2022).
Current approaches to study migration and invasion of GBM cells are either restricted to “classical” cell cultures of cells grown in serum‐supplemented medium and consist of highly artificial scratch (wound healing) assays or invasion assays such as Boyden chamber assay (de Gooijer et al, 2018). For once, these assays might have value in a very basic question, but they fail to provide the three‐dimensional complexity and composition of a brain parenchyma. As such, even a Boyden chamber assay, which employs Matrigel as a surrogate for an extracellular matrix (ECM), is more suited for epithelial cancers considering the composition of the ECM in the brain compared to epithelia. On the other hand, complex model systems such as orthotopically transplanted animals represent the most physiological models, but the study of invasion is usually restricted to staining of sequential sections and therefore very cumbersome, time‐consuming, and error prone given that the sections need to be perfectly aligned. A rarely used, but powerful method consists of placing fluorescently labeled tumor cells onto murine brain slices and monitoring tumor development for several days to weeks. We have successfully applied these so‐called organotypic tissue slice cultures (OTCs) in previous studies and could show that this method has potential for further development (Remy et al, 2018; Linder et al, 2019a, 2022; Gerstmeier et al, 2021). So far, we have analyzed tumor growth using an epifluorescence microscope and measured the tumor area on top of the brain slices. However, since GBM are known for their infiltrative phenotype and our current method lacks an efficient way to study invasion, we combined the OTC model with state‐of‐the‐art multicolor fluorescence light‐sheet microscopy (LSFM) (Hotte et al, 2019).
Light‐sheet fluorescence microscopy (LSFM) is a three‐dimensional imaging technique particularly suited for the analysis of large specimens, such as tumor biopsies, in their entirety (so‐called in toto imaging) (Pampaloni et al, 2013). In LSFM, optical sectioning is achieved by illuminating individual planes of a specimen with a laser light sheet. The fluorescence emitted at each plane is collected by an objective lens placed perpendicularly to the optical axis of the light sheet. By translating the specimen through the light sheet, a three‐dimensional data set is recorded. At variance with confocal microscopy, in LSFM, only the plane that is imaged is excited by the light sheet. Thus, photobleaching and phototoxicity in the specimen are reduced by several orders of magnitude compared to confocal microscopy. Moreover, the acquisition time is much faster compared to a standard confocal microscope (Stelzer et al, 2021).
The combination of LSFM and optical clearing is particularly powerful for digital pathology (Pampaloni et al, 2015). Optical clearing is obtained by homogenizing the refractive index in the specimen by using organic or aqueous solvents as index‐matching media. With optical clearing, whole organs (including the human brain) become highly transparent. Therefore, even deep tissue regions are accessible to investigation with light microscopy (Hof et al, 2021). In general, optical clearing only works with chemically fixed samples. Current efforts aim to increase the throughput of the specimens that can be analyzed with LSFM and optical clearing and to simplify specimen preparation. For instance, Glaser et al (2022) developed an open‐top LSFM for the fast imaging of very large, cleared organs. We recently introduced thermoformed fluorinated ethylene propylene (FEP)‐foil cuvettes for the multiscale microscopy analysis of live and optically cleared large tissue specimens (Hof et al, 2021). The FEP‐foil cuvettes allow for a straightforward and fast specimen preparation, which increases and improves the throughput of LSFM.
Here, we provide first data from our newly developed method OTCxLSFM, by combining a physiological tumor growth assay using OTCs with a microscope technique (LSFM) that is suited to measure large samples with high resolution after tissue clearing using CUBIC2 (Richardson & Lichtman, 2015). As a model substance, we employ arsenic trioxide (As2O3, ATO), which we recently employed to inhibit stemness in GSCs, when combined with the natural anticancer agent (−)‐gossypol (Gos, also known as AT‐101) (Voss et al, 2010; Meyer et al, 2018; Linder et al, 2019a). In addition, we implemented the usage of the WP1066 against the JAK/STAT3 pathway. We further provide evidence that this model system is also suited for genetically modified tumor cells by investigating the role of the tetraspanin, transmembrane receptor CD9 (Podergajs et al, 2016; Linder et al, 2019a), and STAT3 (Kim et al, 2014) for migration and invasion.
Results
Arsenic trioxide inhibits migration in vitro and tissue infiltration ex vivo
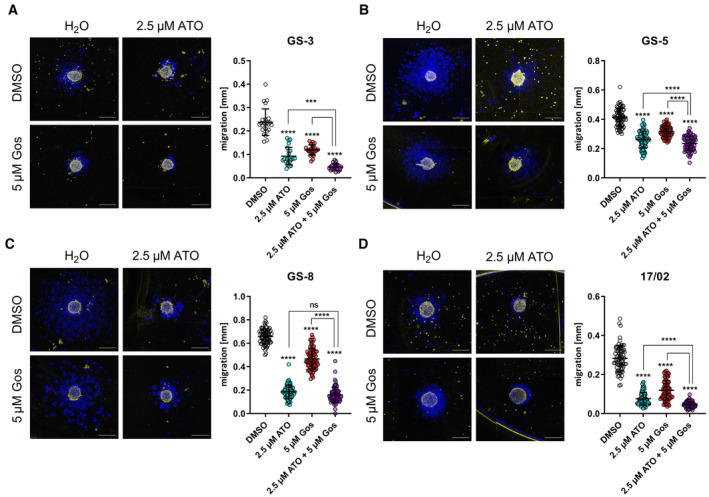
In our previous work, we showed that the combination of arsenic trioxide (ATO) + gossypol (Gos) reduces stemness of GSCs and regulates many proteins that can be clustered under the term “movement” (Linder et al, 2019a; Data ref: Linder et al, 2019b). Based on these data we hypothesized that ATO (±Gos) might be able to inhibit the migratory capacity of GSCs. In order to validate this hypothesis, we developed an assay that employs the capacity of GSCs to grow adherently on laminin‐coated plates, without changing their stem‐like phenotype (Pollard et al, 2009) enabling us to analyze migration using free‐floating GSC spheres. Using this assay, we could show that within 30 min after transferring spheres onto coated plates, the sphere attaches to the plates and the cells start to migrate. Therefore, we applied ATO, Gos, and the combination of both to three GSC lines, GS‐3 (Fig 1A), GS‐5 (Fig 1B), and GS‐8 (Fig 1C) (Gunther et al, 2008), and one primary culture, 17/02 (Fig 1D) (Linder et al, 2019a), which are all sensitive to ATO treatment (Linder et al, 2019a). This experiment confirmed our hypothesis and newly provided information that both single treatments effectively block migration in all four cell lines, whereas ATO is more effective than Gos. The combination treatment is more effective than ATO treatment in GS‐3 (Fig 1A), GS‐5 (Fig 1B), and 17/02 (Fig 1D) indicative of the synergism between the two drugs that we previously determined.
Figure 1. Arsenic trioxide (ATO) alone or in combination with (−)‐gossypol (Gos) effectively blocks migration of GSCs.
-
A–DSphere migration assays of GS‐3 (A), GS‐5 (B), and GS‐8 (C) and of the primary culture 17/02 (D), treated with sublethal concentrations of ATO (2.5 μM), Gos (5 μM), or solvent (H2O and DMSO, respectively). The experiment was performed at least in three biological replicates performed in triplicate (n ≥ 9). Migration distance was measured after 0 h (false color, yellow) and 24 h (false color, blue). Scale bars: 500 μm.
Data information: In (A–D), data are presented as mean ± SEM. ns, not significant; ***P < 0.001; ****P < 0.0001; against DMSO/H2O‐treated cells (two‐way ANOVA with Tukey's multiple‐comparison test; GraphPad Prism 9).
Source data are available online for this figure.
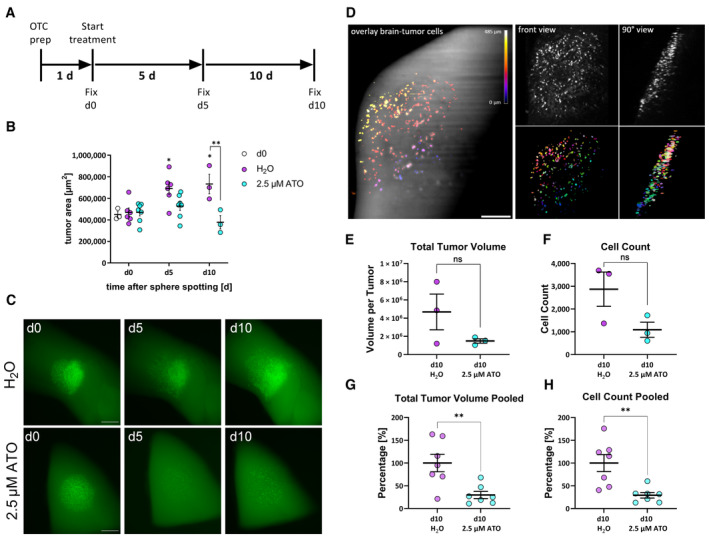
In order to apply our newly developed method and to test its capacity for three‐dimensional analyses, we performed a proof‐of‐concept experiment by treating GS‐5 GFP/Luc tumors (Linder et al, 2019a) using ATO. We chose only ATO treatment because it exerts a very strong migration inhibition on its own and we applied GS‐5 because in our experience this cell culture has the most consistent growth pattern ex vivo. Based on our previous data (Linder et al, 2019a) we anticipated that after 5 days of treatment, a reduced tumor growth will occur and that after 10 days a potent reduction in tumor size will be apparent (experimental setup Fig EV1A). Accordingly, we chose three time points to test the OTCxLSFM method (Fig EV1A–C): 1 day after tumor sphere spotting (treatment d0 “untreated”), 5 days after the initial treatment, and 10 days after the initial treatment (Fig 2A). For each time point, we analyzed at least three slices. First, we measured changes in the tumor area over time (see Materials and Methods and Remy et al, 2018; Linder et al, 2019a) for each time point (Fig 2B) and could confirm our previous findings (Linder et al, 2019a). Before treatment (d0), all tumors are of comparable size, whereas after 5 days, the solvent‐treated tumors are significantly larger, while ATO‐treated tumors are not, indicating growth inhibition as anticipated. After 10 days, the solvent‐treated tumors further increased in size, while ATO‐treated tumors were smaller compared to the initial tumor sizes. In fact, the tumors after ATO treatment are significantly smaller compared to solvent treatment at the same time point. An example for each treatment and time point is depicted in Fig 2C. The microscopic evaluation further showed that tumor cells begin to invade the surrounding parenchyma very quickly. This is especially true for solvent‐treated tumors (compare H2O d5 with d0). In addition, the measurement of the tumor projection area likely underestimates the true effect size of ATO treatment, since the tumor area does not significantly change the intensity of the GFP signal, however, it is greatly reduced after ATO treatment, indicating that the tumor cells are effectively depleted. In order to address this discrepancy, we applied a three‐dimensional analysis (Materials and Methods and assembly for light‐sheet imaging Fig EV1A–C) of the tumor with light‐sheet microscopy to obtain a more complete picture, using Fiji as an open‐source image processing software based on ImageJ2 (Schindelin et al, 2012), as well as generated macros (https://doi.org/10.5281/zenodo.8332648) for processing the image stacks. Figure 2D shows the three‐dimensional rendering of a representative organotypic tumor culture (a control specimen treated with solvent) recorded with the light‐sheet microscope. The GFP fluorescence signal emitted by the tumor cells is nearly one order of magnitude stronger compared to the background autofluorescence emission of the surrounding brain. The background fluorescence is indeed particularly useful for locating the spreading of the tumor cells in the anatomy of the surrounding brain tissue. The rendered image stack can be rotated and viewed under different angles (a frontal and a lateral view of the tumor are shown in Fig 2D). The migration of the tumor cell into the underlying tissue can be readily visualized by color coding the depth (Fig 2D). The high signal‐to‐noise ratio of the fluorescence images allows an efficient segmentation of the individual tumor cells and the quantitative analysis of multiple cell parameters. Placing the tumor sphere onto specific areas of the brain leads to difference in overall migration potential. However, all regions of the tumor hemisphere are used in all conditions to compensate for potential outliers (Fig EV1D). The effect of the ATO treatment on tumor volume and cell count, as obtained from the light‐sheet 3D image stacks is shown in Fig 2E and F. This analysis revealed a strong tendency toward decreased total tumor volume (Fig 2E) as well as a reduced total cell count per tumor (Fig 2F). Notably, neither difference reached statistical significance likely due to low sample size (n = 3 tumors). To address this issue, we repeated the experiment and observed a similar trend (Appendix Fig S1). Normalizing and pooling the data revealed that both the total tumor volume (Fig 2G) as well as total cell count (Fig 2H) are significantly reduced.
Figure EV1. Generating OTCs.
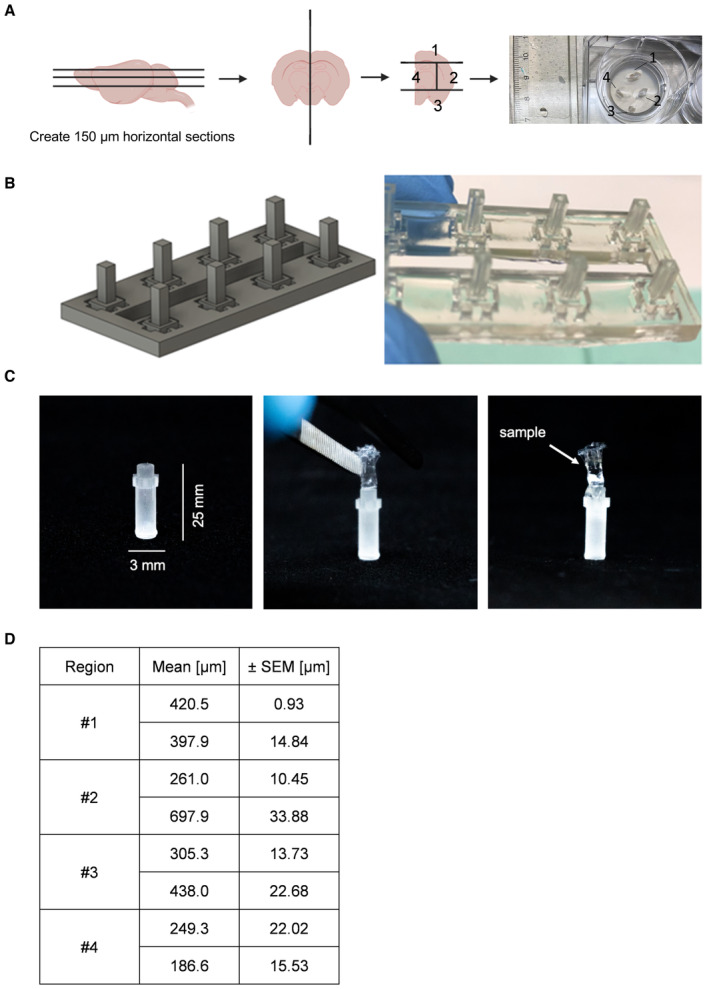
- Schematic setup of generating 150‐μm‐thick mouse brain slices (OTC) and cutting them along the hemisphere into four intersections and placing them on a Millicell culture insert. Scheme created with BioRender.com.
- 3D rendering of the CAD design of the thermoforming positive molds for the LSFM holders. An array of eight positive molds is shown. The molds have a square 2 × 2 mm cross‐section and a height of 5 mm. On the right panel, the real 3D‐printed molds array is displayed.
- One thermoformed FEP‐foil LSFM holder (3 × 25 mm) glued to a sample holder, followed by an OTC is placed inside (right panel).
- Table depicting the mean and SEM of the tumor cell dispersion of one biological replicate (n = 2 tumors per region) of spheres placed onto different brain regions depicted in (A) using GraphPad Prism 9.
Figure 2. Arsenic trioxide effectively reduces tumor growth in adult organotypic tissue slice culture based on epifluorescence microscopy and OTCxLSFM‐based single‐cell analysis.
-
ASchematic timeframe of the OTC experiments.
-
BQuantification of tumor area before treatment (d0) and 5 (d5) and 10 days (d10) after treatment with 2.5 μM ATO or solvent (H2O) [measured in 3 to 7 biological replicates (n ≥ 3)].
-
CEpifluorescence microscopic pictures of the same tumor after treatment with solvent (H2O) or 2.5 μM ATO. Scale bars 500 μm.
-
DLeft panel: Overlay of brain slice and tumor cells. The image results from overlaying the maximum projection of the autofluorescence signal from the brain with the GFP signal emitted by the tumor cells. The color coding represents the depth in the image stack. Right panel, top: the front and 90° views of the tumor cells are shown (both maximum projections of the original image stack). Right panel, bottom: front and 90° views of the corresponding segmented and labeled 3D data set. Scale bar 200 μm.
-
E, FThe total tumor volume (E) and cell count (F) parameters are extracted from the segmented 3D data set (n = 3 tumors, being biological replicates).
-
G, HPooled data of the total tumor volume (G) and cell count (H) (n = 7 tumors) reveal efficient depletion after ATO treatment. The values presented in (E, F), as well as Appendix Fig S1 (n = 4 tumors, being biological replicates), were first normalized to H2O‐treated tumor per experiment and subsequently pooled to perform statistical analyses.
Data information: In (B, E–H), data are presented as mean ± SEM. ns, not significant; *P < 0.05; **P < 0.01, against H20‐treated cells (unpaired T‐test with Welch's test; GraphPad Prism 9).
Source data are available online for this figure.
Next, to obtain a more complete understanding of the cellular alterations following the ATO treatment, we performed a comprehensive cell shape analysis (Fig 3), where single cells were extracted from three tumors. This method revealed that the overall cellular volume of ATO‐treated tumors is prominently and significantly reduced (Fig 3A). Additionally, we performed a 3D analysis of tumor cell dispersion from the calculated center of the tumor mass (Fig 3B). This readout is indicative of overall migratory/invasive capacity. As expected from our previous report showing a reduction of many movement‐related proteins (Linder et al, 2019a) and our in vitro data display ATO‐treated tumor cells an overall reduced distance from the centroid. In line with these findings, we observed a decrease in cellular sphericity, although this difference was only minor (Fig 3C), while the elongation of ATO‐treated tumor cells was increased upon treatment (Fig 3D). Similarly, we observed that the compactness of the cells is diminished (Fig 3E).
Figure 3. Morphological parameters extracted from the segmented 3D data set.
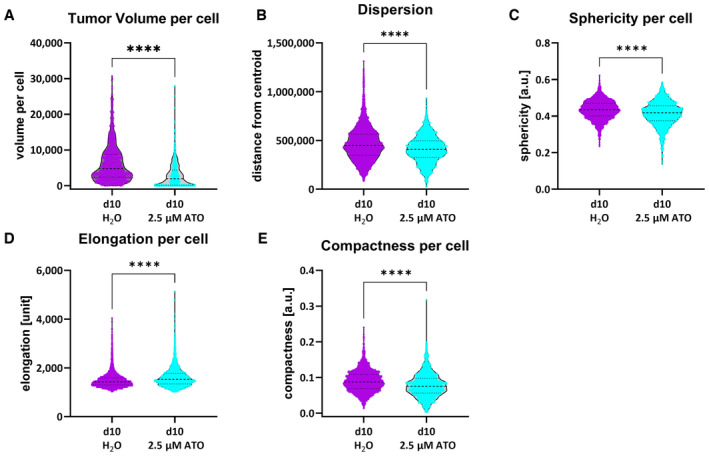
-
A–EThe tumor volume per cell (A), the tumor dispersion, that is, the distance to the tumor centroid (B), the sphericity per cell (C), the elongation per cell (D), and the compactness per cell (E) were generated from GSC line GS‐5 treated with 2.5 μM ATO or solvent (H2O). Single cells from the GSC line GS‐5 were measured from three tumors as biological replicates for each condition (H2O: n = 2,164 single cells; ATO: n = 1,365 single cells).
Data information: In (A–E), data are presented as mean ± SEM. ****P < 0.0001, against H20‐treated cells (unpaired T‐test with Mann–Whitney test; GraphPad Prism 9).
Source data are available online for this figure.
Stable depletion of CD9 prevents migration in vitro and reduces tissue infiltration ex vivo
Hereafter, we wanted to challenge our method by investigating migration and invasion using a genetic knockdown model. We chose to deplete the tetraspanin CD9 because it has been previously shown to be important for GBM migration and invasion (Podergajs et al, 2016) and the CD9 protein was among the most decreased hits after ATO + Gos treatment from our previous report (Linder et al, 2019a).
For this purpose, we first depleted CD9 via stable shRNA‐mediated knockdown (CD9‐KD) in GS‐5 as well as two cell lines that have previously been described to contain high levels of CD9 protein, NCH644, and NCH421k (Podergajs et al, 2016). Using GS‐5 cells, we successfully confirmed efficient depletion of CD9 protein expression (Fig EV2A). With these cells, we first assessed migration in vitro using the sphere migration assay with GS‐5 cells (Fig EV2B and C). This approach revealed that GS‐5 CD9‐KD cells migrate slower compared to their parental cell line, showing a significantly lower distance covered after 24, 48, and 72 h. After 96 h, likely reflecting the maximal migration distance, no more difference is apparent. For NCH644 (Fig 4A) and NCH421k (Fig 4B), we could also confirm CD9 depletion via qPCR. Unfortunately, these two cell lines only weakly adhere to laminin‐coated surfaces and are therefore not suitable for sphere migration assays. Hence, we performed modified Boyden chamber assay to investigate invasion processes. This approach revealed that for NCH644 (Fig 4C and D) almost no difference in the number of invaded cells could be determined for the cells that invaded through the matrix (Fig 4C and D, upper half), whereas significantly fewer spheres could be determined in the bottom well (Fig 4C and D, lower half), indicating that NCH644 CD9‐KD cells are hindered in their invasive potential after 72 h. Similarly, NCH421k CD9‐KD cells (Fig 4E and F) invaded through the matrigel matrix to a reduced extent (Fig 4E and F, upper half), which is reflected in fewer spheres in the bottom well after 72 h (Fig 4E and F, lower half). Hereafter, we analyzed changes in stemness marker expression as well as stemness features in both GSCs. This approach revealed a potent reduction in CXCR4, OLIG2 and SOX2 expression in both lines (Appendix Fig S2A and B), as well as blockade of sphere‐forming potential after 7 days of incubation, as shown by fewer and smaller spheres upon CD9 depletion (Appendix Fig S2C and D).
Figure EV2. CD9 depletion leads to reduced migration in GS‐5 GSCs.
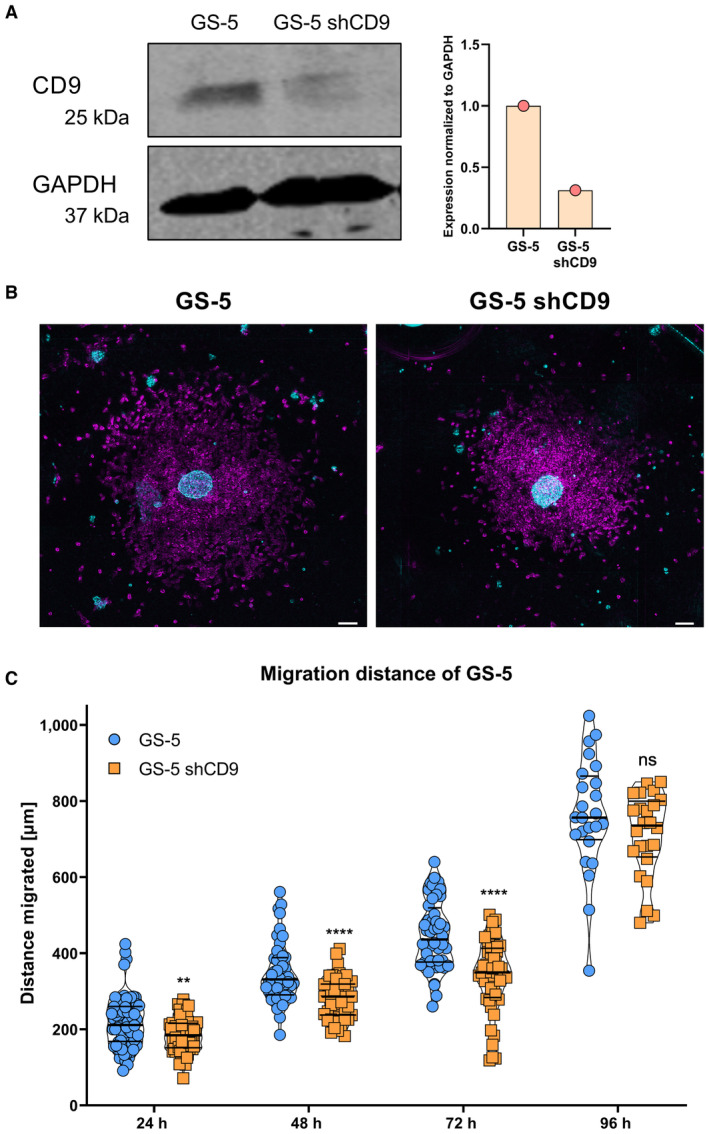
- The expression of CD9 in GS‐5 GSCs after lentiviral transduction of a shCD9 plasmid was analyzed via Western blot and quantified in relation to GAPDH expression (n = 1).
- False‐color images of the sphere migration assay of GS‐5 and GS‐5 shCD9 GSCs. Time points are displayed for 0 h (false color, cyan) and 48 h (false color, magenta). Scale bars: 100 μm.
- Quantification of the sphere migration assay of GS‐5 and GS‐5 shCD9 GSCs showed difference in migration upon CD9 depletion. The experiment was performed independently at least three times with eight replicates (n ≥ 24 biological replicates).
Data information: In (C), data are presented as mean ± SEM. ns, not significant; **P < 0.01; ****P < 0.0001 against GS‐5 (unpaired T‐test with Mann–Whitney test, GraphPad Prism 9).
Figure 4. Boyden chamber invasion assay of NCH644 and NCH421k GSCs after CD9 depletion.
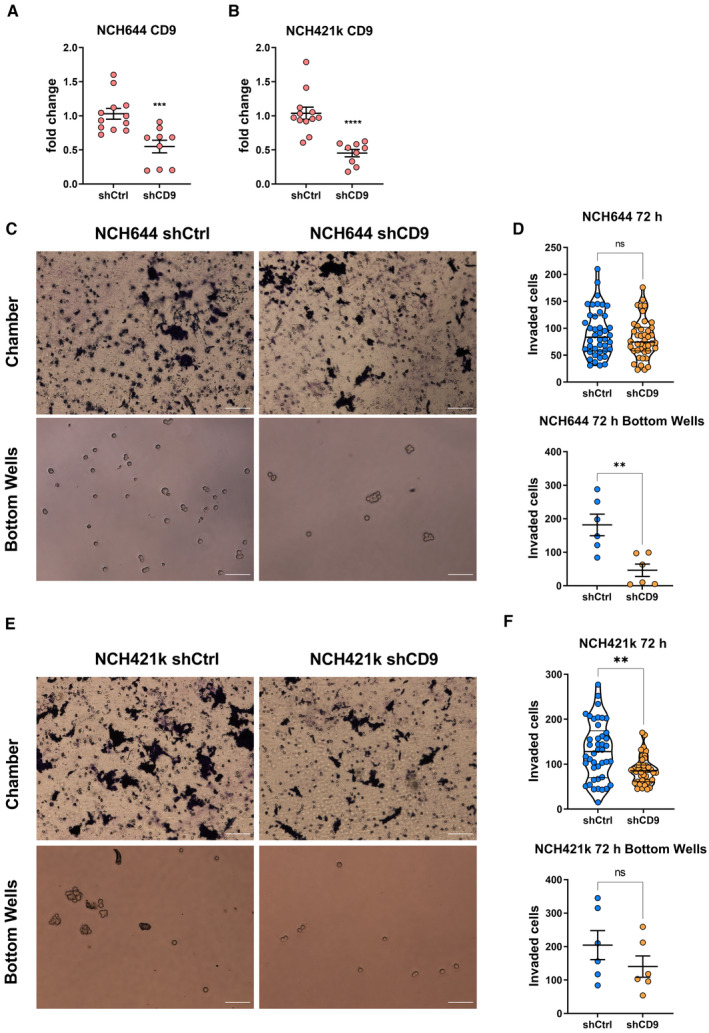
-
A, BKnockdown validation of NCH644 (A) and NCH421k (B) cells shows significant decrease in CD9 expression after depletion. GSCs were measured three to four times in biological triplicate (n ≥ 9).
-
CBright‐field images of GSC NCH644 performing Boyden chambers after 72 h of incubation. Visualizing the chamber, as well as cells migrated through the Matrigel and collecting in the bottom well. Scale bars 100 μm.
-
DQuantification of GSC NCH644 (n = 6, being biological replicates). Quantifying the Boyden chamber GSCs were counted in seven vision fields at 20× magnification. Quantification of the bottom wells, being the total number of cells in the bottom well.
-
EBright‐field images of GSC NCH421k performing Boyden chambers after 72 h of incubation. Visualizing the chamber, as well as cells migrated through the Matrigel and collecting in the bottom well. Scale bars 100 μm.
-
FQuantification of GSC NCH421k (n = 6, being biological replicates). Quantifying the Boyden chamber GSCs was counted in seven vision fields at 20× magnification. Quantification of the bottom wells, being the total number of cells in the bottom well.
Data information: In (A, B, D, F), data are presented as mean ± SEM. ns, not significant; **P < 0.01; ***P < 0.001; ****P < 0.0001 against shCtrl cells (unpaired T‐test with Mann–Whitney test; GraphPad Prism 9).
Source data are available online for this figure.
Hereafter, we performed OTCs with the NCH644 and NCH421k shCtrl and shCD9 cells to analyze if CD9 depletion affects tumor growth and tumor cell infiltration. For NCH644GFP+ tumors, we observed that over the 7‐day observation period, CD9‐depleted tumors showed a tendency for slower growth compared to CD9‐proficient tumors (Fig 5A). Figure 5A shows the epifluorescence images on the left part, where the overall tumor area can be visualized in a 2D image. The montage (right part) of the 3D reconstruction light‐sheet images displays the same tumor on the latest time point (d7) at 30° angles to visualize tissue invasion/migration of the tumor cells. The epifluorescence quantification shows, at the latest analyzed time point (d7, Fig 5B, upper half) only a slight tendency toward a decreased tumor area in CD9‐depleted tumors. However, by applying our three‐dimensional analysis pipeline after image acquisition using OTCxLSFM, the tumor cell dispersion (Fig 5B, lower left) is significantly reduced, indicating reduced invasion after CD9‐KD. Similarly, the frequency distribution (Fig 5B, lower right) showed that more than 20% of tumor cells with CD9‐KD only migrate up to 100 μm, and also display a skewed frequency distribution. In contrast, CD9‐proficient cells display a more bell‐shaped distribution with a mean migration distance of approximately 250 μm. Additionally, CD9‐proficient cells migrate up to over 800 μm, whereas CD9‐KD cells only migrate to a maximum of approximately 600 μm. For NCH421k, very similar results were obtained (Fig 5C and Appendix Fig S3). As depicted in the representative epifluorescence images, as well as the montages of the 3D reconstruction after OTCxLSFM (Appendix Fig S3), it is apparent that the NCH421kGFP+ shCD9 tumors are smaller and less invasive. This can also be quantified in the covered area according to the epifluorescence analysis (Fig 5C upper half). Our three‐dimensional approach revealed a reduced tumor cell dispersion (Fig 5C, lower left), indicative of reduced invasiveness, as well as a left‐skewed frequency distribution after CD9 depletion (Fig 5C, lower right), similar to NCH644 tumors. Having a mean migration distance of 300 μm in NCH421k CD9‐proficient cells and only a mean migration distance of around 210 μm in CD9‐KD cells.
Figure 5. CD9 depletion affects tumor growth in ex vivo organotypic tissue culture tumor growth kinetics in NCH644 and NCH421k GSCs.
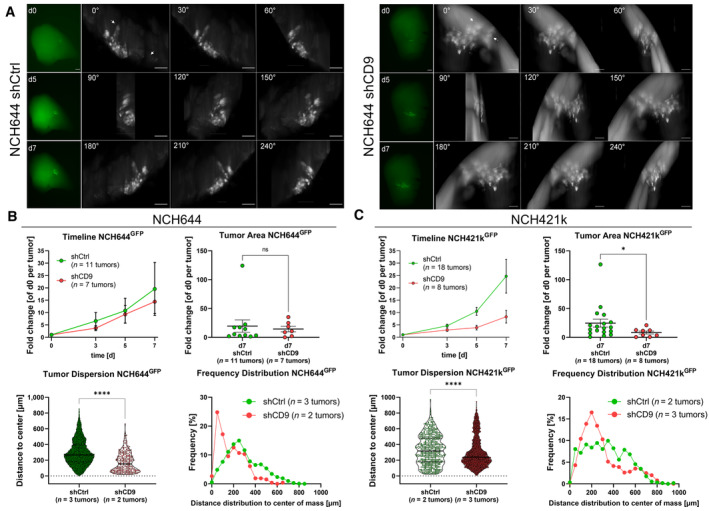
- Epifluorescence microscopic pictures (left panels, green) of the same tumor of NCH644 shCtrl or shCD9 after 0, 5, and 7 days of incubation. Scale bars 200 μm. The montage of 3D reconstruction LSFM image displays the same tumor at the latest time point (d7) of incubation for NCH644 shCtrl or shCD9 in 30° angles (right panels, gray). Arrows display cells that reached their overall maximal migration distance. Scale bars 100 μm. 360° videos of the LSFM images are provided as source data.
- Evaluation of the epifluorescence pictures for NCH644 (shCtrl n = 11 tumors and shCD9 n = 7 tumors; being biological replicates), the summarized growth kinetic over the entire observation time (timeline), and point plot depicting individual fold‐changes tumor sizes (tumor area) at the latest time point (d7). For the evaluation of the light‐sheet images, single cells were measured from biological replicates of n = 3 tumors of shCtrl (n = 9,604 single cells) and n = 2 tumors of shCD9 (n = 262 single cells) NCH644 GSCs. Violin plot depicting tumor dispersion; each point represents one tumor cell and frequency distribution showing the relative amount of migration distance in 50 μm bins.
- Evaluation of the epifluorescence pictures for NCH421k (shCtrl n = 18 tumors and shCD9 n = 8 tumors; being biological replicates), the summarized growth kinetic over the entire observation time (timeline), and point plot depicting individual fold‐changes tumor sizes (tumor area) at the latest time point (d7). For the evaluation of the light‐sheet images, single cells were measured from biological replicates of n = 2 tumors of NCH421k shCtrl (n = 572 single cells) and n = 3 tumors of NCH421k shCD9 (n = 6,047 single cells) GSCs. Violin plot depicting tumor dispersion, each point represents one tumor cell and frequency distribution showing the relative amount of migration distance in 50 μm bins.
Data information: In (B, C), data are presented as mean ± SEM. ns, not significant; *P < 0.05; ****P < 0.0001 against shCtrl cells (unpaired T‐test with Mann–Whitney test, GraphPad Prism 9).
Source data are available online for this figure.
To further test and validate our new pipeline of quantitative analysis of tumor cell invasion and migration, we established another genetically and pharmacological model system using GBM MZ‐54 cells (Hetschko et al, 2008). We genetically depleted STAT3, a known driver for glioma invasion, using CRISPR‐KO in MZ‐54 cells (Kim et al, 2014; Remy et al, 2022). To pharmacologically inhibit migration and invasion, we applied the JAK2/STAT3 inhibitor WP1066 (Groot et al, 2022), which is currently investigated in phase II clinical trial (NCT05879250) for treating recurrent glioma. 3D light‐sheet imaging (Fig EV3A) confirmed significantly decreased tumor areas (Fig EV3B) and less tumor dispersion (Fig EV3C) in STAT3 knockouts (KO) (mean migrated distance: 282 μm) and WP1066‐treated MZ‐54 wt cultures (mean migrated distance: 267 μm) versus controls (mean migrated distance: 342 μm) after 5 days of incubation. Therefore, our pipeline could successfully be adapted to another genetic as well as pharmacological model system, using a second GBM cell model and an additional target for therapeutic intervention.
Figure EV3. Genetically STAT3 depletion and pharmacological JAK2/STAT3 inhibition affect tumor growth in ex vivo organotypic tissue culture in MZ‐54 cells.
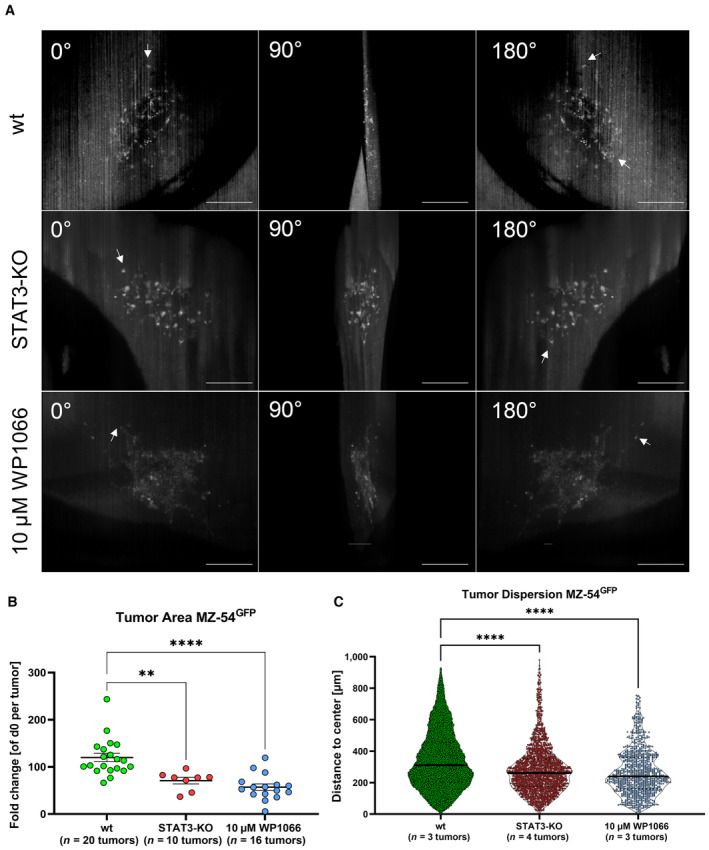
- Montages of 3D reconstruction of light‐sheet images displayed in 90° angles of the same tumor at the latest time point (d5) of MZ‐54 wt, MZ‐54 STAT3‐KO or MZ‐54 wt treated with 10 μM WP1066. Arrows display cells which reached their overall maximal migration distance. Scale bars 200 μm.
- Point plot depicting individual fold‐changes tumor sizes at the latest time point (d5) generated from epifluorescence microscopy of MZ‐54 wt (n = 20 tumors), MZ‐54 STAT3‐KO (n = 10 tumors), and MZ‐54 wt treated with 10 μM WP1066 (n = 16 tumors), being biological replicates for each condition.
- Violin plot depicting single tumor cell dispersion at the latest time point (d5) generated from light‐sheet images of MZ‐ 54. wt n = 3 tumors (n = 103,314 single cells), MZ‐54 STAT3‐KO n = 4 tumors (n = 2040 single cells), and MZ‐54 treated with 10 μM WP1066 n = 3 tumors (n = 932 single cells), each point represents one tumor cell.
Data information: In (B, C), data are presented as mean ± SEM. **P < 0.01; ****P < 0.0001 against MZ‐54 wt (unpaired T‐test with Mann–Whitney test, GraphPad Prism 9).
Lastly, and in contrast by analyzing the same samples observed in Fig 5 using “classical” confocal microscopy (Fig EV4), we could not confirm our findings obtained in NCH644. Hence, no difference in tumor cell dispersion was found and the frequency distribution showed for both, shCtrl and shCD9‐cells, a left‐skewed distribution. Notably, the farthest migration distance was 400 μm in both conditions and likely represents a technical limitation (Fig EV4A). For NCH421k shCtrl and shCD9 cells, a decreased tumor cell dispersion could be validated, but, again, the maximum migrated distance was only approximately 450 μm (Fig EV4B). We therefore concluded that only the acquisition of the entire tumor using OTCxLSFM outcompetes the time‐consuming analysis of a fraction of the tumors, thereby overcoming the limitations and false interpretations resulting from the analyses of large specimens using confocal microscopy.
Figure EV4. Quantification of confocal generated pictures observing CD9 depletion performing ex vivo organotypic tissue culture using GSC line NCH644 and NCH421k shCtrl and shCD9.
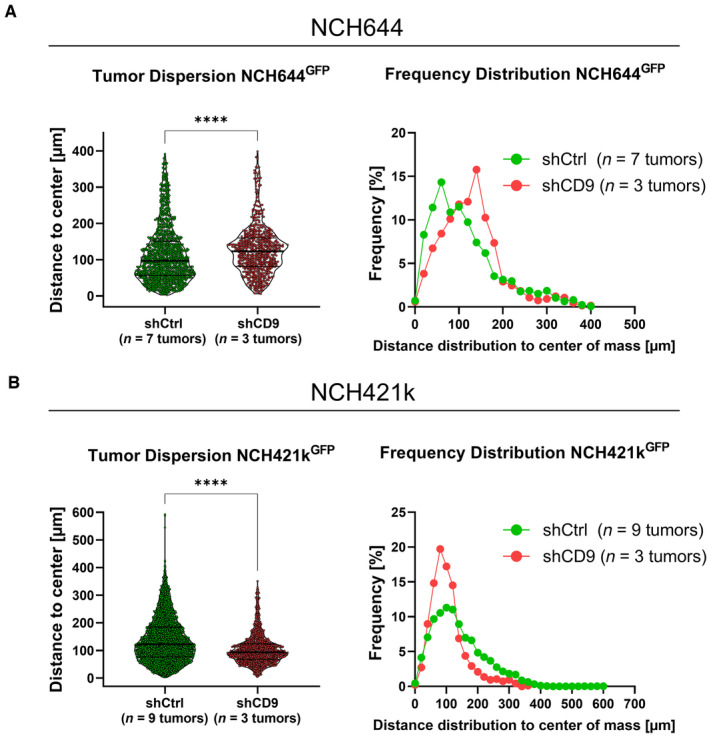
- Violin plot depicting single tumor cell dispersion, each point represents one tumor cell, for NCH644 shCtrl (n = 7 tumors and n = 1,247 single cells) and shCD9 (n = 3 tumors and n = 671 single cells). Frequency distribution showing the relative amount of migration distance in 25 μm bins of NCH644 tumors.
- Violin plot depicting single tumor cell dispersion, each point represents one tumor cell for NCH421k shCtrl (n = 9 tumors and n = 3,345 single cells) and shCD9 (n = 3 tumors and n = 958 single cells). Frequency distribution showing the relative amount of migration distance in 25 μm bins of NCH421k tumors.
Data information: In (A, B), data are presented as mean ± SEM. ****P < 0.0001 against shCtrl cells (unpaired T‐test with Mann–Whitney test, GraphPad Prism 9).
Discussion
Here, we present a novel approach to analyze and quantify GBM migration and invasion in a physiological environment. By combining two state‐of‐the‐art methods, (i) murine, adult organotypic (OTC) brain slices to model GBM growth and (ii) advanced fluorescence light‐sheet microscopy of cleared tissues (LSFM), we developed OTCxLSFM and provided examples of its application to the quantitative analysis of GBM invasion. One of the major novelties of the OTCxLSFM assay is that it provides access to the three‐dimensional migratory behavior of the cells. The three‐dimensional data sets obtained with OTCxLSFM allow us to avoid biases arising from neglecting the depth in 2D images. In fact, in 2D images, the invasion of the brain tissue by the GBM cells cannot be quantified, whereas the ability of tumor cells to spread into the tissue's extracellular matrix rather than on the surface is a key factor determining the lethality of the tumor and its response to the pharmacological treatment. Another important benefit of the OTCxLSFM assay consists of the much faster imaging speed of LSFM compared to the confocal microscope. This allows to measure > 10 specimens in their entirety in one imaging session without compromises in terms of depth acquisition.
The development of suitable, complex preclinical models is a necessity to reflect the complexity of a tumor such as GBM. GBM is characterized by its profound heterogeneity, both between patients (intertumoral) and within the same tumor (intratumoral) (Robertson et al, 2019). Complicating matters further is the diffuse infiltration, as well as tissue penetration, mediated via diverse mechanisms such as collective invasion, perineuronal satellitosis, diffuse infiltration, and vessel co‐option (Chouleur et al, 2020; Seano & Jain, 2020). Many approaches exist to tackle this complexity, each carrying its own advantages and disadvantages (reviewed in Souberan & Tchoghandjian, 2020), and we present the next‐level development of OTCs. One potential drawback of using OTCs is that, depending on the brain region used, the tumor growth can be different, as we showed here (Fig EV1). In previous studies (Remy et al, 2018; Linder et al, 2019a, 2022; Gerstmeier et al, 2021), we harnessed this as a further layer of complexity nicely reflecting the known intertumoral heterogeneity of GBM and addressed this by using sufficiently high samples sizes. Similarly, by harnessing the acquisition speed of LSFM, intrinsic heterogeneity can be accounted for. The concept of studying GBM growth using brain slices has been applied in multiple studies (de Bouard et al, 2007; Parker et al, 2017, 2018) mainly employing post‐natal brain slices derived not only from mice but also from rats grown under high‐serum, species‐unmatched serum supplementation (Ren et al, 2015; Ghoochani et al, 2016; Sidorcenco et al, 2020), mainly focusing on tumor growth. Some reports show applicability toward analysis of angiogenesis and tumor microenvironment. However, these studies mainly rely on further sectioning the slices thereby further increasing the workload. More recent studies, including our own, developed an OTC platform based on adult murine brain slices grown under serum‐free conditions (Marques‐Torrejon et al, 2018; Meyer et al, 2018; Remy et al, 2018; Linder et al, 2019a; Gerstmeier et al, 2021), thereby ensuring that tumors grow in a more authentic environment because GBM is mostly a tumor of adult patients. One major drawback of current approaches is the lack of thorough investigation of the three‐dimensional tissue invasion of GBM cells within these slices. Although this problem can be addressed by further performing fine sections of these slices or by employing tissue clearing and confocal microscopy, these approaches are either very labor intensive and time‐consuming or, in the case of confocal microscopy, usually restricted to a small fraction of the entire tumor. Meaning, not observing the whole entity of the brain slice, which can be the case for confocal microscopy, false quantification can be observed (Fig EV4). In contrast, with LSFM, it is possible to image very large specimens, while maintaining single‐cell resolution, thereby greatly reducing the needed acquisition time, as well as reducing photobleaching and phototoxicity of the sample, by simultaneously increasing the possible sample size. Nonetheless, most studies, as well as our own, mostly rely on antibody‐based stainings, which usually have to be laboriously optimized and in case of thick tissue section, as in our case, require prolonged incubation times. This incubation may further lessen tissue integrity, which likely results in suboptimal analyses. Hence, we propose that future applications of OTCxLSFM and similar approaches should use either fluorescently labeled proteins of interest or reporter for different cell types/cell states. One promising example is the recently described reporter set by Schmitt et al (2021). They developed reporter (so‐called genetic tracing cassettes) based on the transcriptional profile of proneural, mesenchymal, and classical GBM cells based on the classification by Verhaak et al (2010). Using different fluorescent proteins, for example, proneural and mesenchymal GBM cells, they were able to show the proneural‐to‐mesenchymal shift, recapitulating cellular heterogeneity and plasticity (Schmitt et al, 2021).
As a proof‐of‐concept approach for interfering with tumor invasion in OTCxLSFM, we chose ATO or WP1066 as pharmacological agents and CD9 or STAT3 as targets for the genetic depletion models. Firstly, we have recently shown that the combination of ATO and Gos can effectively block GSC stemness, and proteomic analysis revealed that almost 40% of all decreased proteins were related to cell movement and CD9 as one the most strongly depleted proteins (Data ref: Linder et al, 2019b). We confirmed these data using four GSCs lines showing that, in particular, ATO and the combination of ATO and Gos can block migration. Since ATO alone was already highly effective we chose to only apply the ATO single treatment. Using an OTC approach we first confirmed our previous results (Linder et al, 2019a), showing that ATO leads to continuous reduction in tumor areas and can, after 10 days of treatment, also reduce the area below its original size. The three‐dimensional analysis using OTCxLSFM revealed that not only the area on top of the slices is reduced but also tissue invasion is greatly disturbed while inducing morphological changes in the cells indicative of reduced invasiveness. Next, we wanted to show the applicability of our novel tool using a transgenic approach. For this purpose, we depleted CD9 in GS‐5, as well as in two additional cell lines NCH644 and NCH421k (Campos et al, 2014), which have been previously shown to express high levels of CD9 and displayed reduced migration/invasion after CD9 depletion (Podergajs et al, 2016). After confirmation of reduced migration potentials in all three GSC lines, we next performed OTC experiments with NCH644 and NCH421k GSCs and showed that NCH644 CD9 KD cells do not display reduced tumor areas, which contrasts with the previously described improved survival of tumor‐bearing mice (Podergajs et al, 2016), while NCH421k CD9 KD displayed smaller tumors in line with the literature (Podergajs et al, 2016). However, after quantification of the light‐sheet images, it could be proven that both GSC lines showed reduced migration of tumor cells into the surrounding brain tissue, indicating the superior informative value of OTCxLSFM. When analyzing a subset of the entire tumor using confocal microscopy, we again could only validate these results in one of the two cell lines, by only receiving maximum migration distances of around 400 μm, compared to its full migration potential, being 800 μm using light‐sheet imaging. These comparative analyses demonstrate that it is an absolute necessity to analyze the complete tumor including distant migration of individual tumor cells. The analysis of a randomly selected subset brings in an unnecessary bias, which will lead to false interpretations. In addition, the acquisition via OTCxLSFM is much faster and reduces photobleaching and phototoxicity, which is also a crucial factor, when considering increasing the throughput. One notable advantage of confocal microscopy versus LSFM is the improved image quality and lack of artifacts that frequently occur using light‐sheet approaches, which eases sample processing. To further underscore the suitability of the OTCxLSFM model as a robust, yet comparatively less labor‐intensive tool to study GBM invasion, we targeted the JAK/STAT3‐signaling pathway as a key player of infiltrative tumor growth in this tumor entity (Weissenberger et al, 2010; Priester et al, 2013; Linder et al, 2019c). In line with this well‐known function of JAK2/STAT3 signaling, pharmacological blockade of JAK2 activity with WP‐1066 (Hussain et al, 2007) (ClinicalTrials.gov ID: NCT01904123) and CRISPR‐Cas‐mediated ablation of STAT3 led to robust and very similar inhibitory effects on tumor dispersion in the OTCxLSFM model, further underscoring the usefulness and reproducibility of this novel approach. We and others (Kim et al, 2014; Zheng et al, 2014; Yu et al, 2020) extensively studied the role of this pathway (Weissenberger et al, 2010; Priester et al, 2013; Linder et al, 2019c), allowing a direct comparison of the OTCxLSFM data obtained in the present study. Overall, the congruence of obtained data is very high, (Weissenberger et al, 2010; Priester et al, 2013; Linder et al, 2019c) and are faithfully recapitulated in OTCxLSFM, demonstrating that our new model system allows quick and reproducible analysis of GBM cell infiltration in an authentic brain environment.
In summary, we present a novel analysis tool by combining two state‐of‐the‐art techniques; adult brain slices to assay tumor growth and multifluorescent LSFM to analyze large and complex samples on a single cell level in 3D. We anticipate that this system will provide novel valuable insights into GBM migration/invasion processes and that our analytical pipelines are transferable to other complex model systems.
Materials and Methods
Cells and cell culture
Human glioblastoma stem‐like cells GS‐5 GFP/Luc have been described previously (Linder et al, 2019a) and are derived from GS‐5 (Gunther et al, 2008), which were kindly provided by Katrin Lamszus (UKE Hamburg, Germany). NCH421k and NCH644 (Campos et al, 2010) were kindly provided by Christel Herold‐Mende (University Hospital Heidelberg, Germany). The cells were cultured in neurobasal medium (Gibco, Darmstadt, Germany). The medium was supplemented with 1× B27, 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco), 1× GlutaMAX (Gibco), 20 ng/ml epidermal growth factor (EGF, Peprotech, Hamburg, Germany), and 20 ng/ml fibroblast growth factor (FGF, Peprotech). HEK293T (ATCC #CRL‐3216) were cultured in Dulbecco's modified Eagle's medium (DMEM GlutaMAX) supplied with heat‐inactivated 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco). Cell line MZ‐54 was first described in Hetschko et al (2008). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM GlutaMAX) supplied with heat‐inactivated 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco). Arsenic trioxide (As2O3, ATO; Sigma‐Aldrich) was solved in 1 M NaOH, diluted with PBS (Gibco) to 0.5 M and solved at 80°C while stirring. The solution was then sterile filtrated and kept for long‐term storage. An intermediate dilution of 15 mM was used for all consecutive dilutions. AT‐101 ((−)‐gossypol, Bio‐Techne GmbH, Wiesbaden‐Nordenstadt, Germany) and WP1066 (Merck, #573097) were dissolved in DMSO (Carl Roth GmbH, Karlsruhe, Germany) and used at a concentration of 10 μM. Staurosporine (STS) (Enzo Life Science, #ALX‐380) was diluted in DMSO to a 3 mM stock solution. No data were excluded during the performance of the experiments.
Synthesis of lentiviral supernatant and transduction
The laboratory where this study was performed has S1 and S2 safety authorization. HEK293T cells were used to generate lentiviral particles. Therefore, the cells were seeded in six‐well plates at a cell density of 300,000 cells and incubated overnight at 37°C. Next, medium of the HEK293T cells was changed to transfection medium containing DMEM and 10% FCS (Gibco), incubating the cells for 30 min at 37°C. Afterward, transfection with the plasmid DNA was performed. For this, the DNA mix (1.34 μg shRNA‐plasmid DNA (TRCN0000296954; TRCN0000291711, Sigma‐Aldrich, Taufkirchen, Germany) or pLV[Exp]‐EGFP:T2A:Bsd‐CMV > ORF_Stuffer (VectorBuilder, Neu‐Isenburg, Germany), 0.67 μg gag/pol plasmid (psPAX2, addgene #12260), and 0.86 μg VSV‐G envelope plasmid (pMD2.G, addgene #12259)) was prepared in 110 μl Opti‐MEM, vortexed, and incubated for 10 min. At this time, the Fugene HD mix (8.6 μl Fugene HD (Promega, Madison, WI, USA) in 110 μl Opti‐MEM) was prepared and mixed thoroughly. After incubation, 110 μl of the Fugene HD mix was pipetted onto the DNA mix and incubated for 30 min at room temperature. Following the incubation, 220 μl of the mixture was added to the HEK293T cells. The plate was incubated for 6 to 7 h at 37°C. Afterward, the medium was aspirated and refreshed using NB‐A medium, following incubation overnight. After 16 and 40 h, the supernatants were collected and pooled with the supernatants from the day before and afterward filtered using a 0.45 μm filter and stored for a short time period at −21°C or were used directly. Next, GSCs were used to generate stable shCD9 cell lines. Similarly, GFP positive cell lines from the corresponding shCD9 cells were generated after confirming efficient depletion of CD9 via western blot or qPCR. In addition, MZ‐54 wt and MZ‐54 STAT3‐KO cells (Remy et al, 2022), inheriting the following sequences to generate STAT3‐KO: sgSTAT3_A_sense: 3′‐aaacCAGTGGCTGCAGTCTGTAGAC‐5′, sgSTAT3_A_antisense: 3′‐CACCGTCTACAGACTGCAGCCACTG‐5′; sgSTAT3_C_sense: 3′‐aaacGTGGGAAGAATCACGCCTTC‐5′, sgSTAT3_C_antisense: 3′‐CACCGAAGGCGTGATTCTTCCCAC‐5′, were used for generating GFP‐positive cells as well. For this purpose, the corresponding virus was produced using pLV[Exp]‐EGFP:T2A:Bsd‐CMV > ORF_Stuffer. Prior to seeding the cells in a 24‐well plate with a cell density of 500,000 cells per well, wells were treated with protamine sulfate to allow better adhesion onto the plate. Afterward, the viral supernatants were added in a 1:1 dilution onto the corresponding cells. The cells were incubated for 72 h at 37°C. After incubation, the media were changed to medium containing half the concentration of the selection antibiotic puromycin (Santa Cruz Biotechnology, Heidelberg, Germany)/blasticidin (Thermo Fisher Scientific, Waltham, MA, USA). After 24 h of incubation, the full concentration of the selection antibiotic (puromycin 0.5–2 μg/ml and blasticidin 1.25–2.5 μg/ml) was added at the cell line‐dependent concentration. NCH421k and NCH644 were transduced with the virus made with the shCD9 constructs TRCN0000296954 and TRCN0000291711, respectively. The viability of the cells was checked every day for 4 days, containing a washing step each day. After four washing steps, the cells were able to be transferred into S1 laboratories, with stable GFP transduction for OTCxLSFM measurement.
Quantitative real‐time polymerase chain reaction
To analyze differential gene expression in GSCs, quantitative real‐time polymerase chain reaction (qRT–PCR) was performed. For this purpose, 300,000 cells were seeded in a six‐well plate in triplicate and were incubated for 48 h. Afterward, cells were pelleted by centrifugation and washed with PBS. After this, the RNA was isolated directly. The isolation was performed according to the manufacturer's protocol from the ExtractMe Total RNA Kit (7Bioscience, Hartheim, Germany; https://blirt.eu/wp‐content/uploads/2018/08/TOTAL‐RNA_protocol_en_29012018_druk_v2‐min.pdf). After isolation, the RNA concentration was measured using the Tecan Spark plate reader. The RNA samples were used directly for cDNA synthesis, using 1 μg of the sample. After this, the corresponding amount of DEPC‐H2O was added. Further, 1 μl of 50 μM Oligo(dT)20, 1 μl random hexamer primer (3 μg/μl), and 1 μl 10 mM dNTP mix was added to each of the RNA mixtures. The total volume was now set up to 14 μl. After pipetting, the mixture was transferred into an Eppendorf cycler (Eppendorf AG, Hamburg, Germany) and incubated for 5 min at 65°C, followed by being cooled down on ice for 1 min. Followed by adding a mixture of the enzyme Superscript III (200 U; Invitrogen, Carlsbad, USA), DEPC‐H2O, 0.1 M DTT, and 5× first‐strand buffer, samples were heated up again for 5 min at 25°C, followed by a 1 h incubation at 50°C and lastly a 15 min incubation at 70°C. The resulting cDNA was diluted with 180 μl of DEPC‐H2O and used directly for qRT–PCR. For each target gene, the corresponding primer was added [TaqMan probes were bought from Thermo Fischer; TBP (Hs00427620_m1), CD9 (Hs01124022_m1), SOX2 (Hs01053049_s1), OLIG2 (Hs00300164_s1), and CXCR4 (Hs00607978_s1)]. To each master mix of the gene of interest, FastStart master mix and DEPC‐H2O were added. This mixture was pipetted on a micro‐AMP fast 96‐well reaction plate, as well as the desired cDNA. Afterward, the plate was sealed with an adhesive film and was analyzed using the StepOnePlus qPCR machine (Applied Biosciences, Foster City, CA, USA). Gene expression quantification using the ΔΔC T method was performed using Graphpad Prism 9 (GraphPad Software, Inc., La Jolla, CA, USA). Therefore, the samples were baseline corrected by subtracting the housekeeper gene (TBP). Following another baseline correction by normalizing the samples toward the shCtrl cells, the fold change of each gene was generated by performing the equation: Y = 2−Y. The generated data were visualized, and ordinary one‐way ANOVA was performed to provide statistical information about the gene expression of each.
SDS–PAGE and Western blot
For protein lysate preparation, SDS–PAGE, and Western blotting, we followed established procedures from Linder et al (2019c). Briefly, membranes were blocked with 5% BSA in TBS‐Tween20 (TBS‐T) for 1 h at room temperature. Primary antibodies, CD9 (Abcam, #ab236630) and GAPDH (Calbiochem, #CD1001), were diluted in 5% BSA/TBS‐T and incubated at 4°C overnight. Secondary antibodies (goat anti‐mouse or goat anti‐rabbit, Licor) were applied at room temperature for 1 h. Detection was carried out using a LI‐COR Odyssey reader (LI‐COR Biosciences).
Sphere formation assay
To determine changes in the sphere area/number and stemness character of GSCs after CD9 depletion, cells were seeded at a density of 500 cells per well in a 96‐well plate. Each condition was seeded in 5 to 10 replicates. Next, the cells were incubated in an incubator at 37°C for 7 days for the NCH cell lines. Pictures of the spheres were obtained with a Tecan Spark plate reader (Tecan, Männedorf, Austria), which was equipped with a camera. After this, the pictures were evaluated using Fiji (Schindelin et al, 2012). The spheres were counted with a self‐made macro for Fiji. The mean sphere area as well as the number of spheres were obtained.
Sphere migration assay
To analyze migration using GSC tumor, spheres 2,000 to 3,000 cells were seeded into U‐shaped 96‐well plates 1–2 days prior to the experiments. Additionally, conventional 96‐well plates (flat wells) were coated using 10 μg/ml laminin (Sigma‐Aldrich) either overnight at 4°C or for 3 h at 37°C. Shortly before the experiments, the plates were prewarmed in case of overnight coating and laminin was removed. Two wells of the U‐shaped 96‐well plates were pooled into one well of the coated plate, thereby ensuring that at least one sphere was placed into every well. The plates are immediately placed into an incubator for 30 min. During that time, the spheres will start to adhere to the plate. Hereafter, the treatment medium is added and images of the entire wells are taken using a Tecan Spark plate reader tempered at 37°C (Tecan). After the indicated time points, additional pictures are acquired. To analyze cellular migration out of the spheres, the corresponding pictures are analyzed using Fiji (Schindelin et al, 2012). After applying the “find edges” function, the pictures are merged in suitable false‐color channels. Then, the most migrated cell is manually selected using the line tool starting from the sphere at the first time point (t0) to the time point of interest (tx). Starting from this cell, three additional cells at 90°C angle are measured. By applying the correct pixel/size ratio using the “set scale” function, the migration distance in μm was obtained.
Sphere invasion assay
This assay was performed to analyze the invasive potential of GSCs. Corning® Bio‐Coat® Matrigel® Invasion Chambers with 8.0 μm PET membranes were rehydrated for 2 h at 37°C using NB‐A medium with only 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco) added. During incubation, the cells were prepared in NB‐A medium containing only penicillin and streptomycin at a cell density of 80,000 cells per chamber (Hira et al, 2020). This supports the invasive behavior of the cells by generating a growth factor‐dependent gradient and by placing the chambers in NB‐A complete medium (including growth factors). After rehydration of the chambers, the medium was removed and cells were pipetted in the corresponding chambers alongside adding complete medium under the chambers, guaranteeing the growth factor gradient. After this step, the chambers were incubated for 72 h at 37°C. After incubation, the medium including the seeded cells was removed and the cells were fixed using 1 ml of methanol (Sigma‐Aldrich) incubated for 2 min. After removing the chambers from the methanol, the chambers were stained with 1 ml of 0.1% crystal violet (Sigma‐Aldrich) for 2 min. The chambers were washed with distilled H2O and placed in a new 24‐well plate waiting to dry. During the drying process, cells that did not migrate through the Matrigel were removed using a cotton swab. The chambers were then analyzed with a fluorescent microscope (Nikon Eclipse TE2000‐S) using only bright‐field setting. The invaded cells were counted individually using seven vision fields and a 20× objective. An overview picture was generated with a 10× objective.
Adult organotypic slice cultures and ex vivo tumor growth assay
Adult organotypic tissue slice culture (OTC) was carried out as described previously (Remy et al, 2018; Linder et al, 2019a). Mouse brains were dissected and the dura mater was removed. Subsequently, mouse brains were placed in warm (35–37°C) 2% low‐melting agarose (Carl Roth). Using a VT1000 Vibratome (Leica, Wetzlar, Germany), 150‐ to 200‐μm‐thick transverse sections were obtained. The sections were transferred to parafilm and kept moist using a drop of PBS. Hereafter, the brain slices were cut into pieces of approximately 2 × 5 mm to ensure that they fit into FEP‐foil container for subsequent microscopy (Fig EV1A and C). These sections were placed on Millicell cell culture inserts (Merck KGaA, Darmstadt, Germany) and cultured in six‐well plates using FCS‐free medium consisting of DMEM/F12 supplied with 1× B27, 1× N2 supplement, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco). One day later, one tumor sphere was placed into the center of each brain slice snippet (day 0). Adequate spheres were prepared by seeding 2,000–3,000 cells/well in U‐shaped 96‐well plates using 200 μl medium. They were allowed to grow for 1–3 days to aggregate into one large sphere. One day after sphere spotting into the brain slices, the treatment was started and refreshed three times per week. Tumor growth was evaluated after pictures were taken regularly by Nikon SMZ25 stereomicroscope equipped with a P2‐SHR Plan Apo 2× objective operated by NIS elements software. Tissue integrity of mouse brain slices over time was validated by propidium iodide [Sigma‐Aldrich, #P4864 (PI)] staining of tumor‐free cultures, revealing small rims of PI‐positive (dead) cells at the cutting edge of the slices already on d0, while inner regions of the slices remain fully intact (Fig EV5A and B, left panel). Only minor integrity loss at the maximum incubation time of 10 days, as indicated by single PI‐positive cells in the inner region of slices, is visible (Fig EV5A and B, middle panel). Treatment with 5 μM STS on d9 for 16 h (Mewes et al, 2012) served as a positive control for cell death (Fig EV5A and B, right panel). The experiments in this study using mouse brains, from C57BL6j mice, are a killing for scientific purposes according to the German animal protection law and do not require prior approval, but reporting to the respective local authority (Regierungspräsidium Darmstadt). C57BL6j are kept in type II long cages, bought via Envigo, and observed via the “Tierschutzbeauftragte und zentrale Forschungseinrichtung” of the Goethe University Hospital.
Figure EV5. Integrity of mouse brain slices.

- PI staining of mouse brain slice pictured after d0 and maximum incubation time d10. Slices were incubated on day 9 for 16 h with 5 μM staurosporine (STS) as indicator for cell death. Scale bars 200 μm.
- Close‐up of pictures depicted in white boxes in (A) for better visualization. Scale bars 100 μm.
Specimen preparation and mounting in the light‐sheet microscope
Specimen holder production and assembly
A new LSFM specimen holder suitable for the size of the brain OTC used in the work was designed with CAD software (the open‐source parametric CAD software FreeCAD or Fusion360 by Autocad). Next, the model was converted to stereolithographic format (STL) for 3D printing. 3D printing was performed with an LCD stereolithographic printer (Anycubic Photon S) (Fig EV1B). FEP‐foil custom cuvettes were realized following the procedure described in Hotte et al (2019). Briefly, 3D‐printed positive molds containing a 4 × 4 array of square pillars with a cross‐section of 2 × 2 mm and a length of 4 mm were used as vacuum thermoforming tools. After heating a roughly 10 × 10 cm 50‐μm‐thick FEP foil in the thermoforming machine, vacuum was applied and the shape of the positive mold was impressed in the foil. Next, the individual FEP‐foil cuvettes were cut out of the array and glued with UV‐sensitive resin (Anycubic) or two‐component epoxidic glue (Uhu) onto the top of the holder (Fig EV1B and C).
Mounting the OTC specimen in the holder
Individual PFA fixed and with CUBIC‐2 optically cleared OTC snippets were picked up from the multiwell plate with a fine‐tip brush and carefully deposited in the FEP‐foil cuvette glued on the specimen holder (Fig EV1C). The cuvette was previously partially filled up with CUBIC‐2 solution to fully embed the OTC specimen in the clearing solution.
LSFM imaging
LSFM is a three‐dimensional fluorescence microscopy that allows a fast acquisition of image stacks in large and highly scattering samples. Optical sectioning of the sample is achieved through a laser light sheet, which selectively excites fluorescence in single plane of the sample at one time. The emitted light in each plane is collected by an objective lens arranged perpendicular to the optical axis of the light source and detected by a sensitive camera (CCD or CMOS). By axially translating the sample through the light sheet, a three‐dimensional data set is recorded. An advantage of using LSFM is that only the imaged plane is excited by the light sheet. Thus, photobleaching and phototoxicity of the sample are reduced compared to confocal microscopy. Furthermore, the acquisition time is decreased and LSFM provides a new opportunity to investigate the biology of cells and other microorganisms with high spatial and temporal resolution (Pampaloni et al, 2015).
For LSFM imaging, a custom‐built “monolithic digitally‐scanned light sheet microscope” (mDSLM) was used (Pampaloni et al, 2013). The mDSLM features a motorized xyzϑ‐stage placed below the specimen chamber. Light sheet imaging was performed with an Epiplan‐Neofluar 2.5×/0.06 illumination objective (Carl Zeiss), an N‐Achroplan 10×/0.3 detection objective (Carl Zeiss), and a Neo CCD camera (ANDOR Technology, Ireland). Laser wavelength and bandpass filter sets (center wavelength/full‐width at half maximum): 561 nm, 607/70; 488, 525/50; and 405, 447/50 nm. Prior to imaging, the mounted OTC specimen was carefully inserted in the mDSLM chamber, also filled with CUBIC‐2 solution. After correct positioning of the specimen to visualize the tumor in its entirety, the image stack recording was started.
Image processing pipeline
The open‐source Fiji (Schindelin et al, 2012) (ImageJ version 1.53q, Java version 1.8.0_172, 64 bit) was used to process the raw image stack recorded with the light‐sheet microscope, as well as the confocal microscope images. The raw data were all saved as uncompressed tif stacks.
Preprocessing
After uploading in Fiji, the stacks were first cropped to the region of interest (ROI). Next, the data were denoised with the ImageJ plugin “SNR V2 Variational Stationary Noise Remover” (Escande et al, 2017) (Macro: https://doi.org/10.5281/zenodo.8332648) to suppress stripe artifacts deriving from the light‐sheet illumination. Thereafter, the background intensity was subtracted from every slice using the “Subtract Background” function (Fiji, ball radius of 50 pixels). Next, a median filter was applied, via “process” and “filters,” to the image stack.
Segmentation
For segmentation of the preprocessed data, the Fiji/ImageJ plugins 3D ImageJ Suite (Ollion et al, 2013) and MorphoLibJ (Legland et al, 2016) (Macro: https://doi.org/10.5281/zenodo.8332648) were used. First, the seeds corresponding to each cell in the 3D stack were determined with the “3D Maxima Finder” function (3D ImageJ Suite). Next, the “3D Spot Segmentation” function (3D ImageJ Suite) was applied, to determine the size of each cell, using integrated watershed from the segmentation. The resulting segmented 3D data set was post‐processed by using MorphoLibJ tools “Labels Size Filtering,” which allows for the removal of segmented debris particles, and “Label Edition,” which allows for manually correcting further segmentation artifacts.
Data extraction
Finally, the 3D cell centroids and the various 3D morphological parameters shown in Figs 3 and 5, as well as in Figs EV3 and EV4, were extracted by using the 3D ImageJ Suite functions “3D Centroid” by Fiji, calculating the center of mass of the tumor and calculating the single‐cell migration distance in x‐, y‐, and z‐coordinates of the previous segmented tumor cells. Resulting in the tumor dispersion by generating the median of the migrated distance of all cells from each x‐, y‐, and z‐coordinate, followed by calculating the overall migrated distance for each individual cell. “3D Shape Measure” achieves information about the compactness, sphericity, and elongation of the segmented tumor cells in a 3D manner as well.
Visualization
The z‐stacks were processed into 3D maxima projections using the “3D Projection” function and followed by generating a montage in desired angles, via “Image Stacks” and “Montage”. Using the 3D projection, black and white or even false‐color images can be generated via “merge channels”. The depth along the Z‐axis is color coded by using the ImageJ plug‐in “Z‐stack Depth Colorcode 0.0.2” (https://github.com/UU‐cellbiology/ZstackDepthColorCode, Eugene A. Katrukha, Utrecht University).
Statistics
All statistical analyses applied are either unpaired T‐test with Welch's test or unpaired T‐test with Mann–Whitney test or one‐way ANOVA with Tukey's multiple‐comparison or Two‐Way ANOVA with Tukey's multiple‐comparison test using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA), always indicated in the figure legends.
Author contributions
Alicia Haydo: Data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Andrej Wehle: Formal analysis; investigation; visualization. Christel Herold‐Mende: Resources. Donat Kögel: Resources; supervision; funding acquisition; methodology; writing – review and editing. Francesco Pampaloni: Conceptualization; resources; data curation; software; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft. Benedikt Linder: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
The authors would like to thank Hildegard König for ongoing excellent technical assistance. The authors would like to thank Dr. Stefan Liebner and the Edinger Institute for assistance with the epifluorescence microscopy analysis. The graphical abstract and the schematic setup of the OTCs in Fig EV1A were created with BioRender.com. This study was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Council) to BL (LI 3687/2‐1) and DK (Project‐ID 259130777—SFB 1177). Research in the group of FP is supported by EU FET‐Open (828931), EU Transition Open (01 101057894), Wilhelm Sander‐Stiftung (2020.008.1), and the German Aerospace Center (DLR) (IMMUNO3D‐SHAPE (grant number 50WB2019)). Open access funding enabled and organized by Projekt DEAL.
EMBO reports (2023) 24: e56964
Data availability
Fiji Macros are published via zenodo.org and accessible over the link: https://doi.org/10.5281/zenodo.8332648.
References
- Ariza A, Lopez D, Mate JL, Isamat M, Musulen E, Pujol M, Ley A, Navas‐Palacios JJ (1995) Role of CD44 in the invasiveness of glioblastoma multiforme and the noninvasiveness of meningioma: an immunohistochemistry study. Hum Pathol 26: 1144–1147 [DOI] [PubMed] [Google Scholar]
- Becker KP, Yu J (2012) Status Quo—standard‐of‐care medical and radiation therapy for glioblastoma. Cancer J 18: 12–19 [DOI] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E et al (2014) SOX2 controls tumour initiation and cancer stem‐cell functions in squamous‐cell carcinoma. Nature 511: 246–250 [DOI] [PubMed] [Google Scholar]
- Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T (2016) Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg 3: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE, Ahmadi R, Lohr J, Dictus C, Gdynia G et al (2010) Differentiation therapy exerts antitumor effects on stem‐like glioma cells. Clin Cancer Res 16: 2715–2728 [DOI] [PubMed] [Google Scholar]
- Campos B, Gal Z, Baader A, Schneider T, Sliwinski C, Gassel K, Bageritz J, Grabe N, von Deimling A, Beckhove P et al (2014) Aberrant self‐renewal and quiescence contribute to the aggressiveness of glioblastoma. J Pathol 234: 23–33 [DOI] [PubMed] [Google Scholar]
- Chouleur T, Tremblay ML, Bikfalvi A (2020) Mechanisms of invasion in glioblastoma. Curr Opin Oncol 32: 631–639 [DOI] [PubMed] [Google Scholar]
- de Bouard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, Guillamo JS (2007) Antiangiogenic and anti‐invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol 9: 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gooijer MC, Guillen Navarro M, Bernards R, Wurdinger T, van Tellingen O (2018) An experimenter's guide to glioblastoma invasion pathways. Trends Mol Med 24: 763–780 [DOI] [PubMed] [Google Scholar]
- Escande P, Weiss P, Zhang WX (2017) A variational model for multiplicative structured noise removal. J Math Imaging Vis 57: 43–55 [Google Scholar]
- Galoczova M, Coates P, Vojtesek B (2018) STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett 23: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garros‐Regulez L, Aldaz P, Arrizabalaga O, Moncho‐Amor V, Carrasco‐Garcia E, Manterola L, Moreno‐Cugnon L, Barrena C, Villanua J, Ruiz I et al (2016) mTOR inhibition decreases SOX2‐SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets 20: 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstmeier J, Possmayer AL, Bozkurt S, Hoffmann ME, Dikic I, Herold‐Mende C, Burger MC, Munch C, Kogel D, Linder B (2021) Calcitriol promotes differentiation of glioma stem‐like cells and increases their susceptibility to temozolomide. Cancers (Basel) 13: 3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoochani A, Yakubov E, Sehm T, Fan Z, Hock S, Buchfelder M, Eyupoglu IY, Savaskan NE (2016) A versatile ex vivo technique for assaying tumor angiogenesis and microglia in the brain. Oncotarget 7: 1838–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7: 733–736 [DOI] [PubMed] [Google Scholar]
- Glaser AK, Bishop KW, Barner LA, Susaki EA, Kubota SI, Gao G, Serafin RB, Balaram P, Turschak E, Nicovich PR et al (2022) A hybrid open‐top light‐sheet microscope for versatile multi‐scale imaging of cleared tissues. Nat Methods 19: 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot J, Ott M, Wei J, Kassab C, Fang D, Najem H, O'Brien B, Weathers SP, Matsouka CK, Majd NK et al (2022) A first‐in‐human Phase I trial of the oral p‐STAT3 inhibitor WP1066 in patients with recurrent malignant glioma. CNS Oncol 11: CNS87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, Modrusan Z, Meissner H, Westphal M, Lamszus K (2008) Glioblastoma‐derived stem cell‐enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27: 2897–2909 [DOI] [PubMed] [Google Scholar]
- Hetschko H, Voss V, Horn S, Seifert V, Prehn JH, Kogel D (2008) Pharmacological inhibition of Bcl‐2 family members reactivates TRAIL‐induced apoptosis in malignant glioma. J Neurooncol 86: 265–272 [DOI] [PubMed] [Google Scholar]
- Hide T, Shibahara I, Kumabe T (2019) Novel concept of the border niche: glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell‐like features. Brain Tumor Pathol 36: 63–73 [DOI] [PubMed] [Google Scholar]
- Hira VV, Breznik B, Van Noorden CJ, Lah T, Molenaar RJ (2020) 2D and 3D in vitro assays to quantify the invasive behavior of glioblastoma stem cells in response to SDF‐1alpha. Biotechniques 69: 339–346 [DOI] [PubMed] [Google Scholar]
- Hof L, Moreth T, Koch M, Liebisch T, Kurtz M, Tarnick J, Lissek SM, Verstegen MMA, van der Laan LJW, Huch M et al (2021) Long‐term live imaging and multiscale analysis identify heterogeneity and core principles of epithelial organoid morphogenesis. BMC Biol 19: 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotte K, Koch M, Hof L, Tuppi M, Moreth T, Verstegen MMA, van der Laan LJW, Stelzer EHK, Pampaloni F (2019) Ultra‐thin fluorocarbon foils optimise multiscale imaging of three‐dimensional native and optically cleared specimens. Sci Rep 9: 17292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB (2007) A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res 67: 9630–9636 [DOI] [PubMed] [Google Scholar]
- Kim JE, Patel M, Ruzevick J, Jackson CM, Lim M (2014) STAT3 activation in glioblastoma: biochemical and therapeutic implications. Cancers (Basel) 6: 376–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusche B, Ottone C, Clements MP, Johnstone ER, Goetsch K, Lieven H, Mota SG, Singh P, Khadayate S, Ashraf A et al (2016) EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem‐like cells. Elife 5: e14845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda‐Carreras I, Andrey P (2016) MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32: 3532–3534 [DOI] [PubMed] [Google Scholar]
- Linder B, Wehle A, Hehlgans S, Bonn F, Dikic I, Rodel F, Seifert V, Kogel D (2019a) Arsenic trioxide and (−)‐gossypol synergistically target glioma stem‐like cells via inhibition of Hedgehog and Notch signaling. Cancers (Basel) 11: 350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Wehle A, Hehlgans S, Bonn F, Dikic I, Rodel F, Seifert V, Kogel D (2019b) Arsenic trioxide and (−)‐gossypol synergistically target glioma stem‐like cells via inhibition of Hedgehog and Notch signaling. Cancers (Basel) 11: 350 (https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD009249) [DATASET] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Weirauch U, Ewe A, Uhmann A, Seifert V, Mittelbronn M, Harter PN, Aigner A, Kogel D (2019c) Therapeutic targeting of Stat3 using lipopolyplex nanoparticle‐formulated siRNA in a syngeneic orthotopic mouse glioma model. Cancers (Basel) 11: 333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Zoldakova M, Kornyei Z, Kohler LHF, Seibt S, Menger D, Wetzel A, Madarasz E, Schobert R, Kogel D et al (2022) Antitumor effects of a new retinoate of the fungal cytotoxin illudin M in brain tumor models. Int J Mol Sci 23: 9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella‐Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23: 1231–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor RB, Baradaran B, Taylor LE, Iaria J, Nheu TV, Amiry N, Hovens CM, Wang B, Kaye AH, Zhu HJ (2013) Targeting Stat3 and Smad7 to restore TGF‐beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene 32: 2433–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques‐Torrejon MA, Gangoso E, Pollard SM (2018) Modelling glioblastoma tumour‐host cell interactions using adult brain organotypic slice co‐culture. Dis Model Mech 11: dmm031435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes A, Franke H, Singer D (2012) Organotypic brain slice cultures of adult transgenic P301S mice—a model for tauopathy studies. PLoS ONE 7: e45017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Zielke S, Michaelis JB, Linder B, Warnsmann V, Rakel S, Osiewacz HD, Fulda S, Mittelbronn M, Munch C et al (2018) AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 14: 1693–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escude C, Boudier T (2013) TANGO: a generic tool for high‐throughput 3D image analysis for studying nuclear organization. Bioinformatics 29: 1840–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampaloni F, Ansari N, Stelzer EH (2013) High‐resolution deep imaging of live cellular spheroids with light‐sheet‐based fluorescence microscopy. Cell Tissue Res 352: 161–177 [DOI] [PubMed] [Google Scholar]
- Pampaloni F, Chang BJ, Stelzer EH (2015) Light sheet‐based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissues. Cell Tissue Res 360: 129–141 [DOI] [PubMed] [Google Scholar]
- Parker JJ, Lizarraga M, Waziri A, Foshay KM (2017) A human glioblastoma organotypic slice culture model for study of tumor cell migration and patient‐specific effects of anti‐invasive drugs. J Vis Exp 53557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JJ, Canoll P, Niswander L, Kleinschmidt‐DeMasters BK, Foshay K, Waziri A (2018) Intratumoral heterogeneity of endogenous tumor cell invasive behavior in human glioblastoma. Sci Rep 8: 18002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podergajs N, Motaln H, Rajcevic U, Verbovsek U, Korsic M, Obad N, Espedal H, Vittori M, Herold‐Mende C, Miletic H et al (2016) Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 7: 593–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M et al (2009) Glioma stem cell lines expanded in adherent culture have tumor‐specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4: 568–580 [DOI] [PubMed] [Google Scholar]
- Priester M, Copanaki E, Vafaizadeh V, Hensel S, Bernreuther C, Glatzel M, Seifert V, Groner B, Kogel D, Weissenberger J (2013) STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro Oncol 15: 840–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaresma M, Coleman MP, Rachet B (2015) 40‐year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: a population‐based study. Lancet 385: 1206–1218 [DOI] [PubMed] [Google Scholar]
- Remy J, Linder B, Weirauch U, Konovalova J, Marschalek R, Aigner A, Kogel D (2018) Inhibition of PIM1 blocks the autophagic flux to sensitize glioblastoma cells to ABT‐737‐induced apoptosis. Biochim Biophys Acta Mol Cell Res 1866: 175–189 [DOI] [PubMed] [Google Scholar]
- Remy J, Linder B, Weirauch U, Day BW, Stringer BW, Herold‐Mende C, Aigner A, Krohn K, Kögel D (2022) STAT3 enhances sensitivity of glioblastoma to drug‐induced autophagy‐dependent cell death. Cancers (Basel) 14: 339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yu S, Chen C, Wang L, Liu Z, Wu Q, Wang L, Zhao K, Yang X (2015) Invasion and anti‐invasion research of glioma cells in an improved model of organotypic brain slice culture. Tumori 101: 390–397 [DOI] [PubMed] [Google Scholar]
- Richardson DS, Lichtman JW (2015) Clarifying tissue clearing. Cell 162: 246–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson FL, Marques‐Torrejon MA, Morrison GM, Pollard SM (2019) Experimental models and tools to tackle glioblastoma. Dis Model Mech 12: dmm040386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rominiyi O, Vanderlinden A, Clenton SJ, Bridgewater C, Al‐Tamimi Y, Collis SJ (2020) Tumour treating fields therapy for glioblastoma: current advances and future directions. Br J Cancer 124: 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MJ, Company C, Dramaretska Y, Barozzi I, Göhrig A, Kertalli S, Großmann M, Naumann H, Sanchez‐Bailon MP, Hulsman D et al (2021) Phenotypic mapping of pathologic cross‐talk between glioblastoma and innate immune cells by synthetic genetic tracing. Cancer Discov 11: 754–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seano G, Jain RK (2020) Vessel co‐option in glioblastoma: emerging insights and opportunities. Angiogenesis 23: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorcenco V, Krahnen L, Schulz M, Remy J, Kogel D, Temme A, Krugel U, Franke H, Aigner A (2020) Glioblastoma tissue slice tandem‐cultures for quantitative evaluation of inhibitory effects on invasion and growth. Cancers (Basel) 12: 2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda A, Park M, Lee D, Mintz A, Androutsellis‐Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB et al (2009) Hypoxia promotes expansion of the CD133‐positive glioma stem cells through activation of HIF‐1alpha. Oncogene 28: 3949–3959 [DOI] [PubMed] [Google Scholar]
- Souberan A, Tchoghandjian A (2020) Practical review on preclinical human 3D glioblastoma models: advances and challenges for clinical translation. Cancers (Basel) 12: 2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer EHK, Strobl F, Chang B‐J, Preusser F, Preibisch S, McDole K, Fiolka R (2021) Light sheet fluorescence microscopy. Nat Rev Methods Primers 1: 73 [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996 [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Gilbert MR, Chakravarti A (2007) Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol 25: 4127–4136 [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 10: 459–466 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG et al (2016) Single‐cell RNA‐seq supports a developmental hierarchy in human oligodendroglioma. Nature 539: 309–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toms SA, Kim CY, Nicholas G, Ram Z (2019) Increased compliance with tumor treating fields therapy is prognostic for improved survival in the treatment of glioblastoma: a subgroup analysis of the EF‐14 phase III trial. J Neurooncol 141: 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepant AL, Bouchart C, Rorive S, Sauvage S, Decaestecker C, Demetter P, Salmon I (2015) Identification of OLIG2 as the most specific glioblastoma stem cell marker starting from comparative analysis of data from similar DNA chip microarray platforms. Tumour Biol 36: 1943–1953 [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronkova MA, Luanpitpong S, Rojanasakul LW, Castranova V, Dinu CZ, Riedel H, Rojanasakul Y (2017) SOX9 regulates cancer stem‐like properties and metastatic potential of single‐walled carbon nanotube‐exposed cells. Sci Rep 7: 11653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss V, Senft C, Lang V, Ronellenfitsch MW, Steinbach JP, Seifert V, Kogel D (2010) The pan‐Bcl‐2 inhibitor (−)‐gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res 8: 1002–1016 [DOI] [PubMed] [Google Scholar]
- Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, Li L, Yang ZY, Liao XM, Cai J et al (2019) Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1‐SOX2 positive‐feedback loop. Nat Neurosci 22: 91–105 [DOI] [PubMed] [Google Scholar]
- Weissenberger J, Priester M, Bernreuther C, Rakel S, Glatzel M, Seifert V, Kogel D (2010) Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin Cancer Res 16: 5781–5795 [DOI] [PubMed] [Google Scholar]
- Wiranowska M, Ladd S, Smith SR, Gottschall PE (2006) CD44 adhesion molecule and neuro‐glial proteoglycan NG2 as invasive markers of glioma. Brain Cell Biol 35: 159–172 [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Shiino A, Muraki K, Fukami T, Yamada S, Satow T, Fukuda M, Saiki M, Hojo M, Miyamoto S et al (2015) Arsenic trioxide sensitizes glioblastoma to a myc inhibitor. PLoS ONE 10: e0128288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14: 736–746 [DOI] [PubMed] [Google Scholar]
- Yu S, Yu X, Sun L, Zheng Y, Chen L, Xu H, Jin J, Lan Q, Chen CC, Li M (2020) GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene 39: 5042–5055 [DOI] [PubMed] [Google Scholar]
- Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, Jia X, Jiang T, Zhang J, Li X et al (2014) JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII‐expressing glioblastoma. Neuro Oncol 16: 1229–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]