

Abstract
Ufmylation plays a crucial role in various cellular processes including DNA damage response, protein translation, and ER homeostasis. To date, little is known about how the enzymes responsible for ufmylation coordinate their action. Here, we study the details of UFL1 (E3) activity, its binding to UFC1 (E2), and its relation to UBA5 (E1), using a combination of structural modeling, X‐ray crystallography, NMR, and biochemical assays. Guided by Alphafold2 models, we generate an active UFL1 fusion construct that includes its partner DDRGK1 and solve the crystal structure of this critical interaction. This fusion construct also unveiled the importance of the UFL1 N‐terminal helix for binding to UFC1. The binding site suggested by our UFL1‐UFC1 model reveals a conserved interface, and competition between UFL1 and UBA5 for binding to UFC1. This competition changes in the favor of UFL1 following UFM1 charging of UFC1. Altogether, our study reveals a novel, terminal helix‐mediated regulatory mechanism, which coordinates the cascade of E1‐E2‐E3‐mediated transfer of UFM1 to its substrate and provides new leads to target this modification.
Keywords: AlphaFold2, DDRGK1, UFC1, UFL1, ufmylation
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology
A combination of AlphaFold2 modeling, X‐ray crystallography, NMR, and biochemical experiments reveals a crucial role for the UFL1 N‐terminal helix in binding to UFC1 and productive ufmylation.
Introduction
Protein modifications by UFM1 (ufmylation) play a crucial role in many cellular processes, including DNA damage repair, anti‐viral responses, and unfolded protein responses (Banerjee et al, 2020). Therefore, not surprisingly, UFM1 is expressed in most tissues, including, brain, kidney, and liver (Lemaire et al, 2011). UFM1 is initially translated as a precursor of 85 amino acids, which is then cleaved during maturation by UFM1‐specific proteases, removing the last two amino acids and exposing a VG sequence at the C‐terminus (Kang et al, 2007). This mature UFM1 is subjected to a three‐enzyme cascade involving E1‐UBA5, E2‐UFC1, and E3‐UFL1, which is responsible for the attachment of UFM1 to target proteins. First, UBA5 activates UFM1 in an ATP‐dependent process. Next, UFM1 is transferred from UBA5 to the active site cysteine of UFC1, forming a thioester bond. Finally, with the assistance of UFL1, UFM1 is transferred from UFC1 to the target protein (Komatsu et al, 2004; Tatsumi et al, 2010). Overall, similar to ubiquitin and other ubiquitin‐like proteins, UFM1 undergoes a maturation process that enables its conjugation to target proteins via an E1, E2, and E3 enzymatic cascade (Cappadocia & Lima, 2018).
Surprisingly, UFL1 lacks structural elements that are common to other E3 ligase enzymes, namely, a RING domain, a HECT‐type catalytic domain, or an RBR structure (Wenzel et al, 2011; Berndsen & Wolberger, 2014; Zheng & Shabek, 2017). In addition, while some atypical E3 enzymes possess a motif required for interaction with their ubiquitin‐like protein (Reverter & Lima, 2005; Cappadocia et al, 2015), whether UFL1 has a UFM1‐interacting motif is uncertain. Therefore, it remains to be determined whether UFL1 functions in a novel mechanism that does not exist in other E3 ligases. It was shown previously that ufmylation by UFL1 of the nuclear receptor coactivator, ASC1, requires DDRGK1 (also known as UFBP1) (Yoo et al, 2014). Moreover, a recent model of the interaction between UFL1 and DDRGK1 generated by AlphaFold2 has revealed structural complementation between the two proteins (Peter et al, 2022). Besides binding to DDRGK1, UFL1 interacts with LZAP (also known as the adapter protein CDK5RAP3), forming a ternary complex (Wu et al, 2010). The latter has been suggested to possess a motif allowing UFM1 binding (Picchianti et al, 2023). Currently, structural data on UFL1 are still missing, and it is unclear how UFL1 binds UFC1 to promote ufmylation.
Deep learning of modeling of protein structures is revolutionizing the field of structural biology, spearheaded by AlphaFold2 developed by DeepMind (Jumper et al, 2021; Tunyasuvunakool et al, 2021; Varadi et al, 2022). As models are either available or can be generated within a short time on platforms such as ColabFold (Mirdita et al, 2022), they will accelerate studies that previously depended on the expression, purification, and crystallization of one or more proteins, a process that could take years, if successful at all. Structural models provide guidelines for the generation of stable proteins, by identifying disordered regions that hamper protein expression and could be truncated for improved expression. Beyond the study of protein monomers, the structures of many protein complexes can now be modeled (preprint: Evans et al, 2021; Humphreys et al, 2021; Bryant et al, 2022), including interactions mediated by short motifs (Johansson‐Åkhe & Wallner, 2022; Tsaban et al, 2022). It is now possible to also study the interaction of regions that adopt a stable structure only upon interaction, as for example the interaction of motifs located within disordered regions with their partners, as well as complementation of a full domain by two proteins.
With these tools in hand, we set out to study the ufmylation system and reveal yet unsolved challenges in our understanding of this complex regulatory pathway. We report here on two major advances: (i) The design of a functional fusion protein that is composed of truncated parts of the UFL1‐DDRGK1 complex. This construct allows to significantly simplify the study of UFL1 activity and, importantly, has enabled us to solve, for the first time, a crystal structure of UFL1 bound to DDRGK1. (ii) The identification of the critical role of the UFL1 N‐terminal helix in UFC1 binding and ufmylation. Our model suggests that E2 UFC1 uses the same site to bind both E1 UBA5 as well as E3 UFL1 helices, which we confirm by NMR studies. Of note, this helix‐mediated interaction was revealed in a model generated without any prior information, highlighting the contribution of AlphaFold2 to the revelation of new interaction details and regulation.
Results
AlphaFold2‐assisted engineering of an active UFM1 E3‐ligase
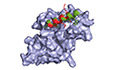
UFL1 has been suggested to be only active in the presence of DDRGK1 (Witting & Mulder, 2021). In line with a parallel recent study (Peter et al, 2022), our starting point was a model of the UFL1‐DDRGK1 interaction (see Figs 1A and EV1A and B) that we generated using AlphaFold2 (see Materials and Methods). The model shows the crucial contribution of DDRGK1 to complement the first winged helix (WH) domain repeat of UFL1 (residues 27–58) and explains why neither DDRGK1 nor UFL1 are folded when expressed alone. This model, as well as additional information about the importance of different regions (Tatsumi et al, 2010), assisted us in the design of a fusion construct encompassing DDRGK1‐UFL1 (Fig 1B), in which we removed the N‐terminal region of DDRGK1 and the C‐terminal region of UFL1 (i.e., DDRGK1:207–314 ‐ UFL1:1–200). Furthermore, we noted the predicted low confidence of the N‐terminal helix of UFL1 (average AlphaFold2 pLDDT < 70) (Fig EV1B) and therefore generated a second construct in which we also truncated this N‐terminal UFL1 helix, DDRGK1‐UFL1ΔN (DDRGK1:207–305 ‐ UFL1:27–200). To test the AlphaFold2‐based design of the above fusion proteins comprising UFL1‐DDRGK1, we purified the fusion proteins (Fig EV1C) and tested their molecular weights in solution using SEC‐MALS analysis. Both proteins behave as monomers with a molecular weight corresponding to their amino acid sequence (Fig 1C). Overall, our results imply that the fusion protein is soluble, similar to the co‐expression of UFL1 with DDRGK1 that allows purification of a soluble UFL1‐DDRGK1 complex (Peter et al, 2022).
Figure 1. AlphaFold2‐assisted generation of an active fusion protein for ufmylation.
-
AAlphaFold2 structural model of the DDRGK1‐UFL1 complex (the first 206 residues of DDRGK1 are not shown); DDRGK1 is colored in orange; UFL1 colored in green. A blowup shows the winged helix domain shared by DDRGK1 and UFL1.
-
BDetails of the fusion constructs. DDRGK1 was connected to UFL1, removing the N‐terminal region of DDRGK1 and the C‐terminal region of UFL1. In a second construct, we also removed the N‐terminal helix of UFL1 (DDRGK1‐UFL1ΔN), due to its flexibility suggested by AlphaFold2 (see Text). Regions removed from the parent proteins are shown in gray (WH: winged helix domains, PCI: proteasome‐COP9‐initiation factor 3 domain, W and H indicate a partial WH domain).
-
CSEC‐MALS profiles of fusion proteins.
-
DCrystal structure of the DDRGK1‐UFL1ΔN fusion. The four winged helix (WH) domains are colored as in (A).
-
E–HIn vitro ufmylation assays. (E, F) Western blots show that in the presence of DDRGK1‐UFL1 an additional band appears. This band corresponds to ufmylated DDRGK1‐UFL1 and is absent without any fusion protein or in the presence of DDRGK1‐UFL1ΔN (E—blotting with anti‐FLAG, since UFM1 has a FLAG‐tag; F—blotting with anti‐Myc since DDRGK1‐UFL1 has a Myc‐tag); *Indicates nonspecific bands that exist also in the absence of ATP. (G, H) Western blots show a band corresponding to ufmylated UFL1 only in the presence of the ternary complex containing the UFL1 N‐terminus (same corresponding anti‐FLAG and anti‐Myc were used); *Indicates degraded UFL1/UFL1ΔN. See Fig EV1D–H for protein loading controls.
Source data are available online for this figure.
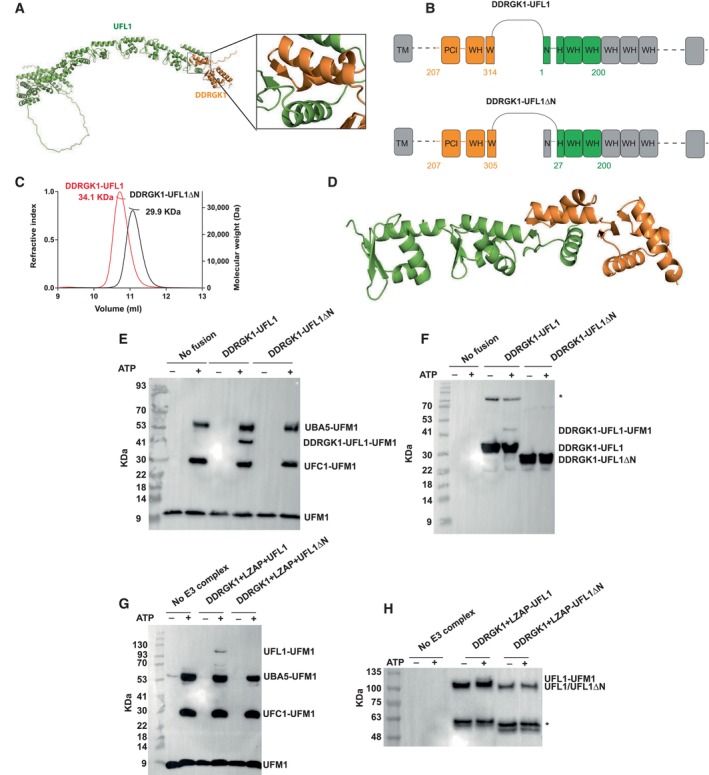
Figure EV1. The UFL1‐DDRGK1 complex.
- A
-
BModel of the DDRGK1‐UFL1 complex, colored according to pLDDT, highlighting the lower confidence in the structure of the N‐terminal helix.
-
CSDS–PAGE showing the purity of the indicated fusion proteins.
-
D–HLoading controls of in vitro ufmylation assays (accompanies Fig 1E–H). (D, E) fusion constructs, (F, G) ternary complex. (H) Presence of UFL1/UFL1ΔN in the membrane: Since we hardly see UFL1/UFL1ΔN in the Ponceau staining (F), we performed Western blot analysis with anti‐Myc. All reactions were loaded on bis‐Tris‐PAGE (except the reaction of Fig 1G, which was loaded on 8–16% Tris gel).
To date, structural data on DDRGK1‐UFL1 complexes are based on AlphaFold2 models (Peter et al, 2022). This motivated us to exploit our fusion proteins for the determination of their crystal structure. We successfully solved the crystal structure of DDRGK1‐UFL1ΔN to 3.1 Å resolution (Table EV1). The structure reveals four repeats of WH domains: the first is contributed by DDRGK1, and the second is formed partially by DDRGK1 and partially by UFL1, while the last two are from UFL1 (Fig 1D). This structure is very similar (backbone RMSD = 1.4 Å) to the corresponding AlphaFold2 model. In the crystal structure, the last 18 amino acids (UFL1 amino acids 183–200) are not detected, suggesting a flexible region. Overall, our crystal structure suggests that the fusion protein maintains the overall architecture of the UFL1‐DDRGK1 complex as observed in the AlphaFold2 model.
With the above fusion proteins in hand, we tested their functionality as E3 ligases. To that end, we incubated pure UBA5, UFC1, and UFM1 with and without fusion proteins and analyzed the ufmylation pattern. As shown in Fig 1E, in the absence of a fusion protein or in the presence of DDRGK1‐UFL1ΔN, the ufmylation pattern is unchanged, showing only ufmylated UBA5 and ufmylated UFC1. However, in the presence of DDRGK1‐UFL1, an additional band corresponding to ufmylated DDRGK1‐UFL1 appears. Moreover, to support that indeed this band corresponds to ufmylated DDRGK1‐UFL1, we tagged the fusion protein with a Myc‐tag and used this tag for western blot analysis. As expected, only in the presence of DDRGK1‐UFL1 an extra band corresponding to ufmylated DDRGK1‐UFL1 appeared (Fig 1F). Overall, our data suggest that in the context of the fusion protein, the N‐terminal helix is essential for E3 ligase activity.
Next, we demonstrated the importance of the UFL1 N‐terminal helix in the context of the native complex (rather than the fusion above). Since in cells UFL1 and DDRGK1 are found in a complex with an additional protein named LZAP, we tested the importance of the UFL1 N‐terminal helix within the context of this ternary complex, comprising UFL1, DDRGK1, and LZAP. To this end, we overexpressed these three proteins in 293T cells and purified the complex. As expected, we observed a band corresponding to ufmylated UFL1 only in the presence of a complex containing the UFL1 N‐terminus (Fig 1G and H). Overall, our data suggest that the UFL1‐N‐terminal helix is critical for ligase activity not only in the fusion protein but also in the native complex containing all three proteins.
A structural model of the UFC1‐UFL1 interaction reveals the critical role of the helix in the N‐terminal tail of UFL1
Why is the UFL1 N‐terminal helix critical to its ligase activity? Motivated by the contribution of the AlphaFold2 model to the successful design of a fusion protein with E3 ligase activity, we modeled the binding of UFC1 to the UFL1‐DDRGK1 complex (Figs 2A and EV2A). This model revealed a critical role of the UFL1 N‐terminal helix in UFC1 binding (Figs 2B and EV2B). This helix occupies a pocket on the side opposite to the UFC1 active site cysteine at position 116, leaving the latter exposed for aminolysis. UFC1 residues located in the interface with the UFL1 N‐terminus, Y36, I40, K47, and R55 contribute significantly to binding (as identified using computational alanine scanning, see Table EV2). In the UFL1 N‐terminal helix, the residues W5, I8, L11, and F15 that are involved in UFC1 binding are highly conserved (Fig 2C). Interestingly, the UFL1 N‐terminus was modeled as a helix with high confidence when in complex with UFC1 (in contrast to the lower confidence of this helix in the model of the free UFL1 structure), indicating that it might be disordered and only fold into a helix upon binding to UFC1 (Fig 2D). Of note, while we have previously demonstrated the ability of AlphaFold2 to model peptide–protein interactions at high confidence (Tsaban et al, 2022), it was still surprising that AlphaFold2 could identify the binding region on UFL1 in the full‐length protein (i.e., generate a model of this interaction using the full UFL1 protein, in contrast to modeling the interaction between only the N‐terminal helix of UFL1 and UFC1), which is not always trivial (preprint: Bret et al, 2023; preprint: Lee et al, 2023).
Figure 2. The N‐terminal helix of UFL1 is crucial for binding of UFC1 and activity.
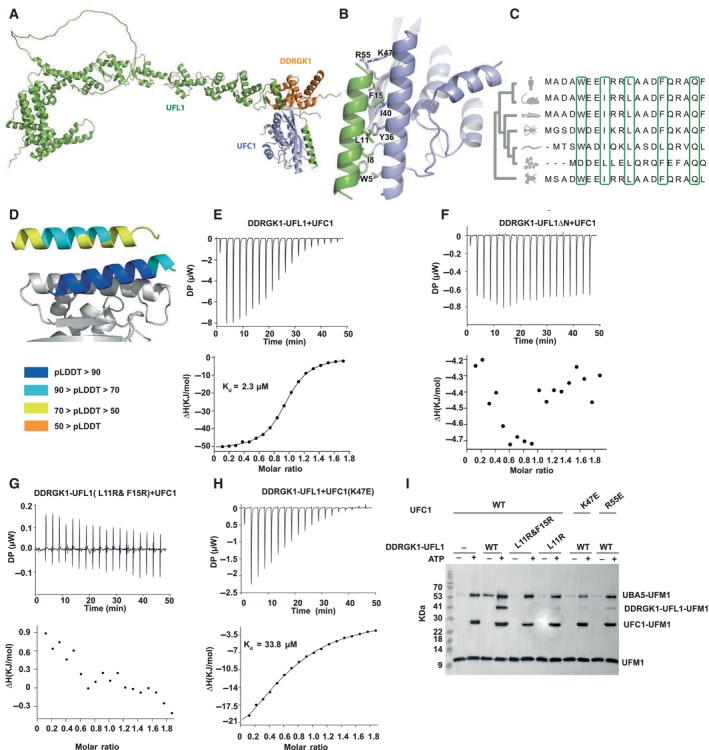
-
AOverall view of the UFC1‐DDRGK1‐UFL1 ternary complex (UFC1 colored in violet).
-
BDetails of the interaction: UFL1 N‐terminal helix bound to UFC1.
-
CAnalysis of conservation of UFL1 N‐terminal helix shows evolutionary conservation of the residues predicted to be involved in binding.
-
DThe N‐terminal helix (colored according to pLDDT) is modeled with high confidence when bound to UFC1 (in gray), in contrast to the low confidence for this helix in the unbound structure (shown on top).
-
E–HITC experiments of UFC1 binding to fusion proteins: (E) UFC1 binding to DDRGK1‐UFL1; (F) UFC1 binding to DDRGK1‐UFL1ΔN (G) UFC1 binding to DDRGK1‐UFL1 double mutant (L11R & F15R); (H) mutant UFC1 (K47E) binding to DDRGK1‐UFL1. In all the experiments, top graphs represent raw data of heat flow versus time. The area under the peaks of the upper panel was integrated and plotted as kJ per mole of UFC1 as a function of binding stoichiometry in the bottom panel. Thermodynamic parameters are summarized in Table EV3.
-
IWestern blot showing the effect of UFC1 mutants or DDRGK1‐UFL1 mutants on ufmylation; blot with anti‐FLAG since UFM1 has FLAG‐tag. See Fig EV2D for protein loading control.
Source data are available online for this figure.
Figure EV2. UFC1 interaction with UFL1 N‐terminal helix.
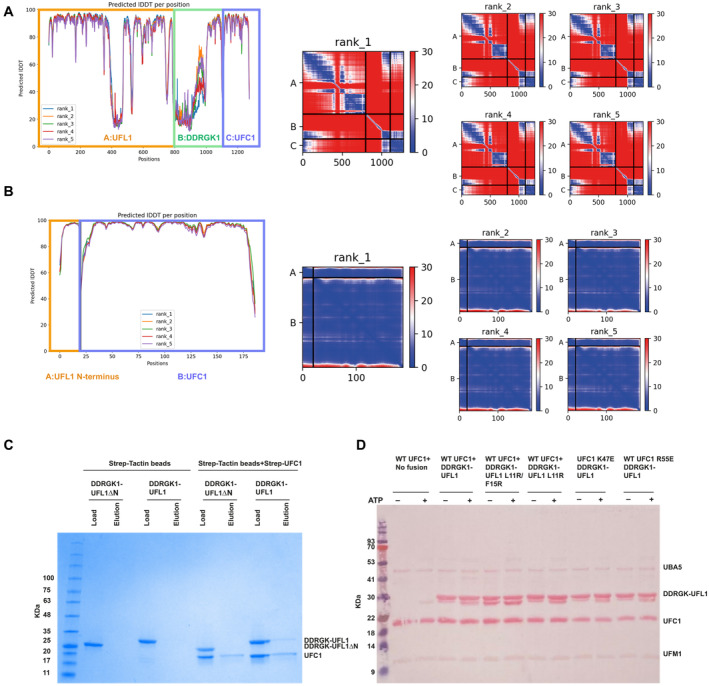
-
A, BModel confidence of the AlphaFold2 complex prediction of A UFL1‐DDRGK1‐UFC1 and B UFL1 N‐terminal helix‐UFC1. Left: Predicted local distance difference test (pLDDT); Right: predicted Align Error (pAE) plots of model ranked 1, and models ranked 2–5.
-
CCoomassie stain gel shows that UFC1 binds to DDRGK1‐UFL1 but not DDRGK1‐UFL1ΔN, demonstrating that this interaction depends on the presence of the N‐terminal helix of UFL1.
-
DLoading control of in vitro ufmylation assay with mutants of DDRGK1‐UFL1 and UFC1 (accompanies Fig 2I).
To validate the AlphaFold2 model, we first tested the binding of UFC1 to DDRGK1‐UFL1 or DDRGK1‐UFL1ΔN using ITC. In line with the model, only the fusion protein possessing the UFL1 N‐terminal helix binds to UFC1 (Kd = 2.3 μM; Fig 2E and F and Table EV3). We reconfirmed this also by pulldown experiments (Fig EV2C). Next, we performed binding experiments with DDRGK1‐UFL1 fusion proteins possessing mutations in UFL1 residues that are involved in UFC1 binding. As shown in Fig 2G, DDRGK1‐UFL1 with the double mutations L11R and F15R completely abolished the binding to UFC1. Similarly, mutation in UFC1 K47E significantly increased the Kd to 33.8 μM (Fig 2H). Accordingly, functional assays with these mutants showed significant reduction in ufmylated DDRGK1‐UFL1, highlighting the importance of the UFL1 N‐terminal helix for ufmylation (Fig 2I).
Figure EV3. UBA5 versus UFL1 computational competition assay.
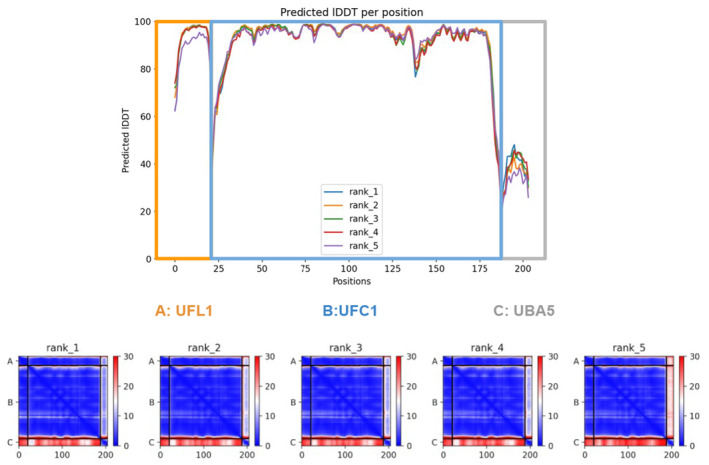
Model confidence of the AlphaFold2 complex prediction of UFL1 N‐terminal‐UFC1‐UBA5 C‐terminal. Top: predicted local distance difference test (pLDDT); Bottom: predicted Align Error (pAE) plots of all models (accompanies Fig 4C).
To further support our model of the interaction of the UFL1 N‐terminus with UFC1, we used NMR spectroscopy to define the changes in UFC1 chemical shifts upon binding to DDRGK1‐UFL1 or DDRGK1‐UFL1ΔN. To that end, we exploited the reported assigned (1H, 15N)‐HSQC NMR spectra for UFC1 (Liu et al, 2005). As expected from our activity and ITC experiments (Figs 1E–H and 2E–H), the addition of DDRGK1‐UFL1, but not DDRGK1‐UFL1ΔN, to 15N‐labeled UFC1 caused strong attenuations (Fig 3A and B). In line with the AlphaFold2 model, these attenuations include residues from UFC1 ⍺‐helix I (residues 26–48) and from β‐strand I (residues 54–58), that directly interact with the UFL1 N‐terminus (Fig 3C). Interestingly, besides the above residues, the N‐terminal half of UFC1 ⍺‐helix II (residues 135–145) also showed chemical perturbations. This region is located on the other side according to our AlphaFold2 model and not directly involved in UFL1 N‐terminus binding, raising the possibility that these residues are allosterically regulated by binding of the UFL1 N‐terminus to UFC1.
Figure 3. Characterization of UFC1 binding to DDRGK1‐UFL1 using NMR.
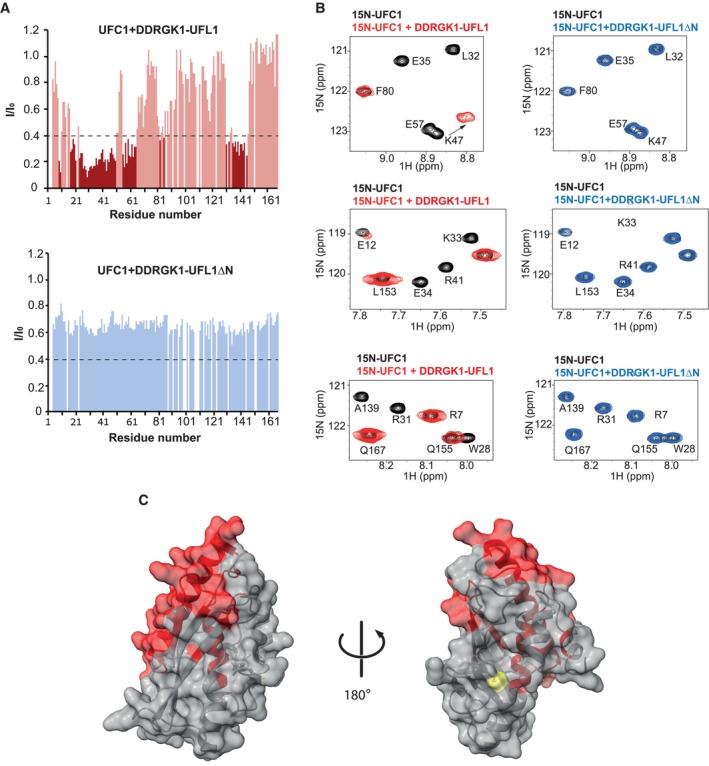
- Intensity changes of UFC1 residue peaks upon addition of 1.5‐fold excess (300 μM) of DDRGK1‐UFL1 (upper graph) or 2‐fold excess (400 μM) of DDRGK1‐UFL1ΔN (lower graph). Dark colors indicate shifted residues (I/I0 ≤ 0.4), and light colors indicate unshifted residues (I/I0 > 0.4). Removal of the N‐terminal UFL1 regions significantly impairs UFC1‐DDRGK1‐UFL1 complex formation. The dashed line indicates I/I0 = 0.4.
- Selected regions of the 1H–15N HSQC spectrum: 0.2 mM UFC1 alone (black) and in the presence of twofold excess of DDRGK1‐UFL1 (red) or DDRGK1‐UFL1ΔN (blue).
- Structure of UFC1 [PDB ID: 7NW1 (Kumar et al, 2021)] with residues displaying significant intensity changes (I/IO ≤ 0.4) upon addition of DDRGK1‐UFL1 colored in red; UFC1 active site cysteine labeled in yellow.
Source data are available online for this figure.
According to our model and NMR data (Figs 2B and 3), UFC1 binds the N‐terminal helix of UFL1 using the same binding pocket with which it also binds to the C‐terminal helix of UBA5 (Fig 4A), as shown by a crystal structure solved previously by us (Kumar et al, 2021). This is in line with previous reports that in ubiquitin‐conjugating enzymes, the same binding site on the E2 that is responsible for E1 binding is also required for E3 binding (Eletr et al, 2005). These data suggest a competition between UFL1 and UBA5 for binding to UFC1. To verify this, we performed an NMR based competition experiment between 15N‐labeled UBA5 C‐terminus (UBA5 347–404) bound to UFC1, and UFC1+DDRGK1‐UFL1 (Fig 4B). As expected, addition of UFC1 to 15N UBA5 C‐terminus caused large changes to the UBA5 1H–15N HSQC spectrum, in the form of chemical shift perturbations (Fig 4B, center panel). Addition of DDRGK1‐UFL1 to the UBA5‐UFC1 complex, however, caused shifting of NMR cross‐peaks in UBA5 to their unbound position, confirming that UFL1 and UBA5 bind to the same surface of UFC1 (Fig 4B, right panel). To summarize, these results demonstrate that both E1 UBA5 and E3 UFL1 use the same pocket on E2 UFC1 for binding, revealing important details of the regulation of ufmylation.
Figure 4. UBA5 and UFL1 compete for binding to UFC1.
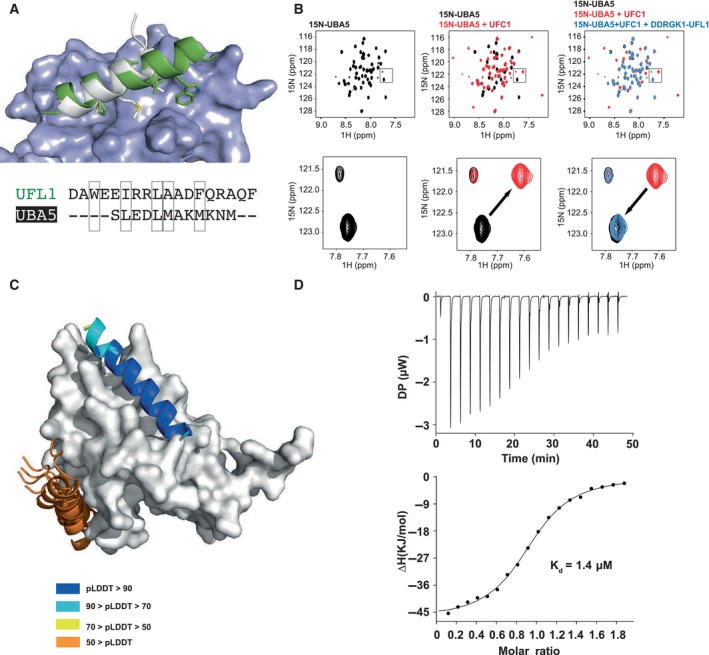
- Superposition of the model of the UFL1 N‐terminal helix‐UFC1 complex onto the solved structure of the UBA5 N‐terminal helix‐UFC1 complex (PDB ID: 7NW1) suggests that both bind to the same binding site; UFC1 is shown in surface representation, UFL1 (green), UBA5 (white).
- NMR 1H–15N HSQC spectra revealing competitive binding: Left: 1H–15N HSQC spectrum of 15N UBA5 alone (150 μM, black); Center: Overlay of spectrum after addition of UFC1 (300 μM, red); Right: Overlay of spectrum after addition of both UFC1 (300 μM) and DDRGK1‐UFL1 (700 μM, blue), showing return to the unbound spectra. Upper panel: Full spectrum; Lower panel: Inset focusing on a specific peak, showing its shift in the presence of UFC1, which is reverted after addition of competing UFL1.
- Computational competition assay using AlphaFold2 modeling of the structure of UFC1 in the presence of both helices (N‐terminal helix of UFL1 and C‐terminal helix of UBA5) identifies UFL1 as the stronger binder, forcing UBA5 to another, incorrect location. Note the high pLDDT for the UFL1 helix, compared to the corresponding pLDDT values for UBA5. See also Fig EV3.
- ITC experiment of UFC1 binding to UFL1 N‐terminal helix (1–21). The top graph represents raw data of heat flow versus time. The area under the peaks of the upper panel was integrated and plotted as kJ per mole of UFC1 as a function of binding stoichiometry in the bottom panel. Thermodynamic parameters are summarized in Table EV3.
Source data are available online for this figure.
To simulate the ability of the two specific helices (i.e., of E1 UBA5 residues 389–404 and E3 UFL1 residues 1–21) to compete for E2 UFC1, we ran AlphaFold2 on a sequence that contains both peptides, assuming that the stronger binding peptide will bind to the receptor binding site (an approach suggested by Chang & Perez, 2023). In that experiment, the UFL1 helix invariably outcompeted the UBA5 helix in the binding site and was modeled with significantly higher confidence (Fig 4C). This is anticipated since UFL1 binds as a longer helix (20 residues vs. 12 residues of UBA5) and generates a larger hydrophobic interaction surface. Indeed, the E3 UFL1 helix binds to UFC1 with Kd = 1.4 μM affinity (measured using ITC, Fig 4D), which is stronger than the corresponding affinity of the UBA5 C‐terminus to UFC1 (Kd = 2.5 μM, reported by us previously; Kumar et al, 2021). Within the natural context, the relative binding affinities might however change.
In the conjugation pathway, UFL1‐DDRGK1 has to bind the charged E2 (UFC1~UFM1). In the case of ubiquitin, several of its E3 ligases bind to the charged E2s, and this, in turn, enhances the discharge of ubiquitin from the E2s (Metzger et al, 2014). To test whether the binding of UFL1‐DDRGK1 to charged UFC1 (UFC1~UFM1) enhances UFC1 discharge, we performed a single turnover discharge assay. Initially, we charged UFC1 using UFM1 and UBA5. We then treated the reaction with EDTA to block UBA5 activity. Finally, we added free lysine in the presence or absence of DDRGK1‐UFL1. As shown in Fig 5A, the discharge of UFC1 was stimulated in the presence of DDRGK1‐UFL1, suggesting that in the presence of UFL1, the thioester bond is more susceptible to the attacking lysine.
Figure 5. DDRGK1‐UFL1 exhibits higher affinity for charged UFC1 and enhances destabilization of the latter.
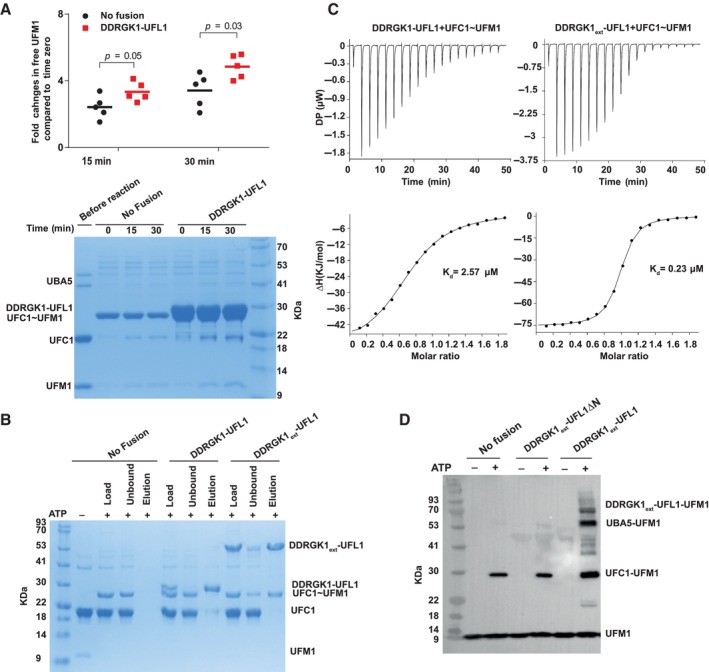
- Single turnover lysine discharge assay showing the effect of DDRGK1‐UFL1 on discharge of UFC1 by free lysine. Five independent experiments were performed to quantify the effect of DDRGK1‐UFL1 on UFC1 discharge. The amount of free UFM1 was quantified at each time point and was normalized against time zero (for 15 min time point pval = 0.05, for 30 min time point pval = 0.03, two‐tailed unpaired Student's t‐test, data as mean ± SD, n = 5 technical replicates). Lower panel: A representative gel showing UCF1~UFM1 discharge upon addition of lysine in the presence and absence of DDRGK1‐UFL1 fusion protein.
- Coomassie stain gel shows that in a pulldown assay DDRGK1ext‐UFL1, but not DDRGK1‐UFL1, preferentially binds to charged UFC1 compared to uncharged UFC1. Specifically, only charged UFC1 is detected in the elution of the pulldown with DDRGK1ext‐UFL1 (last lane).
- ITC experiments of charged UFC1 binding to DDRGK1‐UFL1 (left panel) and DDRGK1ext‐UFL1 (right panel). The top graph represents raw data of heat flow versus time. The area under the peaks of the upper panel was integrated and plotted as kJ per mole of UFC1 as a function of binding stoichiometry in the bottom panel. Thermodynamic parameters are summarized in Table EV3.
- In vitro ufmylation assay. Western blot shows that changes in the ufmylation pattern are only observed in the presence of DDRGK1ext‐UFL1 possessing the UFL1 N‐terminus (blotting with anti‐FLAG). See Fig EV4B for protein loading controls.
Source data are available online for this figure.
Next, we asked if the fusion protein prefers binding to charged UFC1. To that end, we generated a mixture of charged and uncharged UFC1 with an excess of uncharged UFC1 (see Materials and Methods section for more details). We used this mixture for a pulldown experiment with DDRGK1‐UFL1 to evaluate binding preferences to charged UFC1. The DDRGK1‐UFL1 fusion construct did not show any preferential binding to the charged UFC1 (Fig 5B). A recent study reported that DDRGK1 contains a UFM1 binding site located at positions 116–224 (Ishimura et al, 2022), which are missing in this fusion construct (Fig 1B). This raises the possibility that preferential binding of DDRGK1‐UFL1 to charged UFC1 requires this region. Therefore, we generated an extended fusion protein, DDRGK1ext‐UFL1 (DDRGK1:87–314 ‐ UFL1:1–200). Indeed, this extended fusion protein shows preferential binding to the charged UFC1 (Fig 5B): while the reaction input contains more uncharged than charged UFC1, after binding to the extended DDRGK1‐UFL1, this ratio is inversed, showing mainly charged UFC1. To complement this experiment, we also measured the affinity of the fusion proteins for charged UFC1 using ITC. As anticipated, DDRGK1‐UFL1 that lacks the UFM1 binding site in DDRGK1 exhibited a Kd of 2.57 μM, similar to that observed with uncharged UFC1 (Fig 5C). However, the binding of charged UFC1 to DDRGK1ext‐UFL1 resulted in a 10‐fold increase in binding affinity (Kd = 0.23 μM). Of note, DDRGK1ext‐UFL1 does not show higher affinity to uncharged UFC1 compared to DDRGK1‐UFL1 lacking the UFM1‐binding site of DDRGK1 (Fig EV4A). Collectively, our data suggest that the UFM1 binding site on DDRGK1, previously demonstrated to bind free UFM1, is also capable of binding UFM1 once it is charged on UFC1, thereby facilitating preferential binding to the charged form.
Figure EV4. Binding and activity characterization of DDRGK1ext‐UFL1.
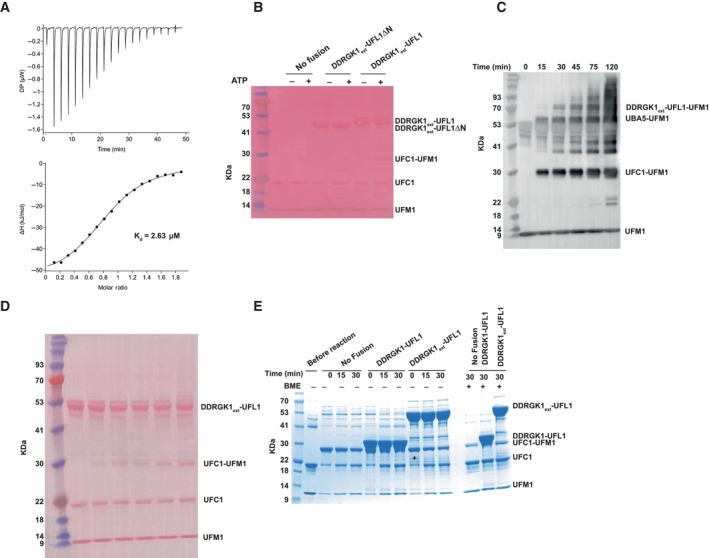
- ITC experiment of UFC1 binding to DDRGK1ext‐UFL1. The top graph represents raw data of heat flow versus time. The area under the peaks of the upper panel was integrated and plotted as kJ per mole of UFC1 as a function of binding stoichiometry in the bottom panel. Thermodynamic parameters are summarized in Table EV3.
- Loading control of in vitro ufmylation assay (accompanies Fig 5D).
- In vitro ufmylation assay in the presence of DDRGK1ext‐UFL1. Western blot analysis of ufmylated products as function of time (blotting with anti‐FLAG, since UFM1 has an FLAG‐tag).
- Loading control of (C).
- Single turnover lysine discharge assay showing the effect of DDRGK1‐UFL1 or DDRGK1ext‐UFL1 on discharge of UFC1 by free lysine.
To study the necessity of the UFL1 N‐terminus for ufmylation activity when DDRGK1 contains the UFM1 binding site, we conducted a ufmylation assay on DDRGK1ext‐UFL1 possessing or lacking the UFL1 N‐terminus. As depicted in Fig 5D, only DDRGK1ext‐UFL1, which includes the N‐terminus, exhibited a distinct alternation in the ufmylation pattern when compared to the pattern observed without the fusion protein. Moreover, we showed an increase over time in ufmylated products in the presence of DDRGK1ext‐UFL1 (Fig EV4C and D). Next, we investigated whether the presence of the UFM1‐binding site on DDRGK1 would result in a more significant stimulation of discharge. To that end, we compared the discharge of UFC1~UFM1 in the presence of DDRGK1‐UFL1 or DDRGK1ext‐UFL1 that includes the UFM1 binding site. However, as shown in Fig EV4E, we did not observe any improvement in the stimulation upon the introduction of DDRGK1ext‐UFL1. Furthermore, we did not detect an increase in ufmylated UFC1 (UFM1 that is attached to UFC1 via an isopeptide bond) in the presence of UFL1 fragments. In summary, our findings highlight the crucial role of the UFL1 N‐terminus in ufmylation activity, even in the presence of the UFM1 binding site within DDRGK1.
Discussion
Transient interactions between E1, E2, and E3 are indispensable for protein modifications by ubiquitin and ubiquitin‐like proteins (UBLs); therefore, understanding their binding mechanisms is of high interest. Here, we show that the UFL1 N‐terminal helix that resides outside the WH domains is responsible for the binding to UFC1 (Figs 1 and 2). This helix binds UFC1 on the side opposite to the active site cysteine at position 116 (Fig 3), leaving the latter exposed for aminolysis. Intriguingly, this mode of binding overlaps with the binding site of UBA5 C‐terminal helix on UFC1, suggesting competition between UBA5 and UFL1 on UFC1 (Fig 4). This competition allows UFC1 to execute its dual role in the conjugation process. Initially, UFC1 has to bind the charged UBA5 and participate in the trans‐thiolation reaction where UFM1 is transferred to the UFC1 active site cysteine. Then, UFL1 outcompetes UBA5, leaving the UFC1 active site cysteine exposed and amenable to a substrate's nucleophilic attack.
In a previous study, we reported a Kd value of ~1 μM for the affinity between UFC1 and UBA5 (Soudah et al, 2019), which is similar to the Kd obtained here for DDRGK1‐UFL1 and UFC1. This similarity raises questions about how the binding competition between these enzymes ensures productive ufmylation. Our discovery that DDRGK1‐UFL1 exhibits a higher affinity for charged UFC1 can explain how the former can outcompete UBA5. However, as observed with ubiquitin and other UBL enzymes, conformational changes in E1 and E2 enzymes during the transthiolation process further affect the affinity between these enzymes (Yuan et al, 2021). This therefore can impact the competition with E3. Another parameter that can affect this competition is the cellular localization of UBA5, UFC1, and UFL1. While UFC1 and UBA5 are freely present in the cytosol, UFL1 is associated with the ER membrane through its interaction with DDRGK1 (Liang et al, 2020). Additionally, recent findings indicate that LZAP, another partner of the UFL1‐DDRGK1 complex, also possesses a UFM1 binding site (Ishimura et al, 2023). This further assists in bringing UFC1 ~ UFM1 in proximity to UFL1, enhancing its ability to outcompete UBA5. In addition, it is also important to note that when UFC1 is no longer charged, it has an increased likelihood of leaving the ER environment. Consequently, it becomes available for interaction with UBA5, thereby initiating another cycle of charging.
Currently, the role of preferential binding of DDRGK1‐UFL1 to charged UFC1 in catalysis is not fully understood. It is still uncertain whether this binding induces a closed conformation in charged UFC1, akin to what is observed with ubiquitin E2 enzymes, and subsequently facilitates catalysis (Huang et al, 2007; Lee & Schindelin, 2008; Olsen & Lima, 2013; Stewart et al, 2016). Recently, Peter et al (2022) demonstrated that the presence of the complex comprising UFL1 and DDRGK1 enhances the discharge of charged UFC1 by free lysine. This observation suggests that the binding of the UFL1‐DDRGK1 complex to the charged UFC1 assists in making the thioester bond more favorable for transfer. In the context of our fusion protein, we observed a slight increase in UFC1 discharge when the fusion protein is present, and this effect was independent of whether DDRGK1 possesses or lacks its UFM1 binding site. The relatively weak stimulation of discharge observed with the fusion proteins, in comparison with the native complex, suggests that there may be additional regions in UFL1 or DDRGK1 that are absent in the fusion proteins, which are necessary for this stimulation.
While both free lysine discharge experiments and DDRGK1‐UFL1 auto‐ufmylation assays are valuable for elucidating the modification mechanism, it is essential to acknowledge that these assays employ non‐native substrates. Consequently, the lower yield observed in the auto‐ufmylation assay may be attributed to the use of non‐native substrates. Similar to the above‐mentioned assays, in the context of a native substrate a lysine residue functions as the attacking moiety. However, within the native substrate, this lysine resides within its real environment, which can influence its physicochemical properties and, consequently, impact the yield of the ufmylation reaction. Specifically, whether the substrate itself has a catalytic role that contributes to the reaction is still unclear and requires further investigation.
Many of the advances in this study were possible thanks to accurate structural models, which highlight the major role played by interactions that involve significant stabilization of the monomer structure(s) upon binding. The model of the UFL1‐DDRGK1 complex was crucial to realize the formation of a full, stable WH domain from two unstable parts contributed by UFL1 and DDRGK1 (Fig 1), and suggested that addition of DDRGK1 would solve the long‐time challenge of successful expression and purification of active UFL1. The model of the UFL1‐UFC1 interaction contributed to our understanding of the transfer of UFM1 via the E1‐E2‐E3 cascade onto the substrate, by highlighting the roles played by terminal helices in binding to UFC1, by both E1 (UBA5) and E3 (UFL1) (Fig 4A). This study underlines the significant contribution of deep learning methods such as Alphafold2 to our advanced ability to research, and ultimately to understand, the complexity of regulatory interactions.
Materials and Methods
AlphaFold2 predictions
In general, structural models of individual proteins and complexes were generated using ColabFold (Mirdita et al, 2022). Unless indicated otherwise, the predictions were run with all five models and default seed, default multiple sequence alignment generation using the MMSeqs2 server and with three recycles, without linkers between the monomers. The “computational competition assay” was run as suggested by Chang and Perez (2023), providing both competing peptides in a single prediction run, provided before and after the receptor UFC1 sequence. All structure visualizations were created with PyMOL (Schrodinger LLC, 2010).
Calculation of sequence conservation
Conservation of UFL1 was calculated with the ConSurf server (Yariv et al, 2023), using default parameters. Alignment of the human and model animal sequences of UFL1 was performed using ClustalOmega (Sievers et al, 2011) on the UniProt server (UniProt Consortium, 2021).
Computational alanine scanning
To estimate the contribution of different residues to the binding of UFL1 N‐terminal helix to UFC1, we applied alanine scanning using the Robetta server (Kortemme et al, 2004). Residues with predicted effect of ΔΔGbinding > 1.0 kcal/mol were retained as hotspot residues.
Cloning and mutagenesis
The fusion constructs DDRGK1207–314‐UFL11–200 (corresponding to DDRGK1‐UFL1 in the main text), DDRGK1207–305‐UFL127–200 (corresponding to DDRGK1‐UFL1ΔN in the main text), DDRGK187–314‐UFL11–200 (corresponding to DDRGK1ext‐UFL1 in the main text), and DDRGK187–305‐UFL127–200 (corresponding to DDRGK1ext‐UFL1ΔN in the main text) were generated using Gibson assembly (Gibson assembly master mix, New England Biolabs) according to the manufacturer's protocol and are cloned in pET15b. The point mutants of UFC1 were generated by PfuUltra II Fusion HS DNA polymerase, by Agilent. The point mutants of DDRGK1‐UFL1 were generated by PCR and Gibson assembly. A list of all primers is provided in Table EV4. All of the constructs were verified by DNA sequencing.
UFL1 1–794 and UFL1 27–794 were cloned in pLVX with an N‐terminal 3xMyc tag and a C‐terminal Strep tag in a BamH1/Xba1 site. LZAP with C‐terminal His tag and DDRGK1 50–314 were cloned in pCDNA 3.4 using Gibson assembly.
Cell culture and transfections
293T cells (American Type Culture Collection) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, and incubated at 37°C in the presence of 5% CO2. 293T cells were grown to 70% confluence in T175 flasks and transiently co‐transfected with 12 μg of 3xMyc_UFL1 1–794_Strep_pLVX/3xMyc_UFL1 27–794_Strep_pLVX, 24 μg of LZAP‐His_pCDNA 3.4 and 24 μg of DDRGK1 50–314_ pCDNA 3.4 (total 60 μg of plasmid DNA), and 120 μl Transporter™ 5 transfection reagent per flask.
Protein expression and purification
UBA5, UFC1, and UFM1 were expressed and purified as previously described (Oweis et al, 2016). All the fusion constructs and point mutants were expressed in E. coli T7 express (New England Biolabs). The transformed cells were grown in 2xYT and induced at 16°C overnight with 0.3 mM isopropyl‐β‐thio‐galactoside (IPTG). For the NMR experiments UFC1 and UBA5 (347–404), transformed cells were grown in standard M9 minimal media supplemented with 15NH4Cl and induced at 20°C overnight with 0.3 mM IPTG. The induced cells were harvested by centrifugation at 8,000 g for 15 min. Pellets were resuspended in lysis buffer (50 mM NaH2PO4 pH 8.0, 400 mM NaCl, 10 mM imidazole, and 5 mM β‐mercaptoethanol), supplemented with 1 mM phenyl‐methyl sulfonyl fluoride (PMSF) and DNase. Cells were disrupted using a microfluidizer (Microfluidics). Lysate was cleared by centrifugation at 29,000 g for 45 min and was subjected to 5 ml His‐Trap columns (GE Healthcare). The protein was eluted with a linear imidazole gradient of 15–300 mM in 20 column volumes. Fractions containing the purified protein were pooled and dialyzed overnight at 4°C against dialysis buffer (25 mM NaH2PO4 pH 8.0, 300 mM NaCl, and 5 mM β‐mercaptoethanol) in the presence of TEV protease. Cleaved protein was then subjected to a second round of His‐Trap column, and flow‐through containing the cleaved protein was collected. Furthermore, purification was done using 16/60 Superdex 75 pg or 16/60 Superdex 200 pg size exclusion chromatography as applicable, equilibrated in buffer containing Tris–Cl pH 7.5 (20 mM), NaCl (200 mM), and DTT (2 mM).
To purify the UFL1 ligase complex from mammalian cells, 48 h after transfection, 293T cells were scraped from T175 flasks, transferred to 50‐ml conical tubes, and centrifuged at 1,000 g for 5 min. Cell pellets were resuspended in ice‐cold lysis buffer (50 mM Tris–Cl, pH 8, 600 mM NaCl, I mM DTT, and 1× protease inhibitor cocktail). The cells were disrupted using microfluidizer and incubated in ice for 30 min with 10 μg/ml avidin, followed by centrifugation at 20,000 g for 20 min at 4°C. The supernatant was loaded to a 5 ml Strep‐tactin superflow HC column. The protein was eluted with 50 mM Tris–Cl, pH 8, 500 mM NaCl, and 7.5 mM Desthiobiotin in 10 column volumes. Furthermore, purification was done using 16/60 Superdex 200 pg equilibrated in a buffer containing Tris–Cl pH 7.5 (20 mM), NaCl (400 mM), and DTT (1 mM). The purified proteins were concentrated and flash‐frozen in liquid N2 and stored at −80°C.
In vitro ufmylation assay
UBA5 (0.5 μM), FLAG‐UFM1 (7.5 μM), UFC1 (3 μM), and fusion fragments (3 μM each) were mixed together in a buffer containing HEPES (50 mM pH 8.0), NaCl (100 mM), and MgCl2 (10 mM). For assay with native protein, 0.5 μM of ternary complex was used. The concentration of the proteins was the same for all reactions involving point mutants unless specified. Reactions were initiated by the addition of ATP (5 mM) and were incubated at 30°C for 45 min. The negative control sample was incubated without ATP. After incubation, the reactions were stopped by adding a 5× SDS‐sample buffer containing β‐mercaptoethanol. The samples along with the control were then loaded on 12% Bis‐Tris PAGE followed by immunoblot with anti‐Myc (UFL1) (Santa Cruz, sc‐40) or anti‐FLAG (UFM1) (Merck, F1804) antibody.
In vitro pulldown assay
Recombinant purified strep‐UFC1 (5 μM) and fusion fragments (5 μM each) were mixed in PBS in total volume of 50 μl for 1 h at room temperature (RT) and subsequently incubated with Strep‐Tactin beads (Iba Lifesciences). The mixtures were washed twice with 1xPBS. The bound proteins were eluted using 7.5 mM Desthiobiotin in 50 mM HEPES, pH 8.0, and 300 mM NaCl buffer. Then, the samples were analyzed by 4–15% SDS–PAGE followed by Coomassie Brilliant Blue staining.
Discharge assay
For the charging, 10 μM of UFC1 was incubated with 0.5 μM UBA5 and 10 μM UFM1 in reaction buffer (50 mM HEPES pH 8.0, 100 mM NaCl, and 10 mM MgCl2) with 5 mM ATP for 10 min at 30°C. The reaction was stopped by 10‐min incubation at RT with 50 mM EDTA (pH 8.0). In order to start the discharge, the reaction was incubated at 37°C with 50 mM lysine (pH 8.0) and with or without 20 μM of DDRGK1‐UFL1 fusion protein. The discharge reaction was stopped by addition of SDS‐sample buffer without any reducing agent, at different time points (0, 15, and 30 min) and loaded on 12% Bis‐Tris PAGE under non‐reducing conditions followed by Coomassie staining.
Preparation of charged UFC1
UFC1 T106S/C116K (25 μM) was incubated at 37°C along with UBA5 (10 μM) and Strep‐tagged UFM1 (40 μM) for 4 h. The 5‐ml reaction was initiated by ATP (5 mM) and conducted at sodium bicarbonate buffer (50 mM pH 9.8), NaCl (100 mM), and MgCl2 (10 mM). The formation of isopeptide‐linked UFC1‐UFM1 covalent complex was verified by SDS–PAGE. After the incubation, the reaction mixture was loaded on the strep‐tactin superflow column. UFC1‐Strep‐tagged UFM1 covalent complex and unreacted residual Strep‐tagged UFM1 bound to the column, while UBA5 and unreacted UFC1 were separated in the flow‐through. The covalent complex was eluted using Desthiobiotin. The purified complex was verified in SDS–PAGE. Furthermore, purification was done using a Superdex 75 analytical column to separate UFC1‐Strep‐tagged UFM1 covalent complex and the unreacted Strep‐tagged UFM1. The purified complex was concentrated and stored at −80°C.
Binding of DDRGK1‐UFL1 fusion proteins to charged/uncharged UFC1
A mixture of charged and uncharged UFC1 with an excess of uncharged UFC1 was generated by adding UBA5 (0.5 μM), UFC1 (20 μM), and UFM1 (5 μM) in a buffer containing HEPES (50 mM pH 8.0), NaCl (100 mM), and MgCl2 (10 mM). Reactions were initiated by the addition of ATP (5 mM) and were incubated at 30°C for 30 min. The negative control sample was incubated without ATP. Then, fusion fragments DDRGK1‐UFL1 or DDRGK1ext‐UFL1 (5 μM each) that contain a C‐terminal Strep tag were added to the reaction and incubated with Strep‐Tactin beads (Iba Lifesciences) for 1 h in 4°C. The mixtures were washed twice with 1xPBS. The bound proteins were eluted using 7.5 mM Desthiobiotin in 50 mM HEPES, pH 8.0, and 300 mM NaCl buffer. Then, the samples were analyzed by 12% Bis‐Tris non‐reducing PAGE followed by Coomassie Brilliant Blue staining.
Size exclusion chromatography with multi‐angle light scattering analysis
For the SEC‐MALS experiments, the fusion fragments were loaded on an analytical SEC column (Superdex 75 10/300 GL) equilibrated with a buffer containing 20 mM Tris (pH 7.5), 200 mM NaCl, and 1 mM DTT. Molecular mass within the chromatographic peak was calculated using ASTRA software, version 7 (Wyatt Technologies).
Crystallography
Crystals of DDRGK1‐UFL1ΔN were grown at 20°C using the hanging drop vapor diffusion method. Protein was concentrated to 100 mg/ml and crystalized in a solution containing 0.7 M Ammonium tartrate dibasic and 0.1 M sodium acetate trihydrate, at pH 4.6. The crystals were cryoprotected using a reservoir solution containing 25% glycerol and flash‐frozen in liquid nitrogen.
Diffraction data for the DDRGK1‐UFL1ΔN crystals were collected on beamline ESRF ID30A‐3 at 100°K. Data were processed using Dials (Waterman et al, 2016) and scaled using Aimless (Evans, 2011). The putative space group was determined to be hexagonal P62 or its enantiomorph. The structure was solved by molecular replacement (MR) with MOLREP (Vagin & Teplyakov, 2010) using the AlphaFold2 model of the UFL1‐DDRGK1 heterodimer (Fig 1A). The translation function confirmed the space group to be P64. The asymmetric unit contains two chains of the chimeric protein. The MR model was refined in REFMAC5 (Murshudov et al, 2011) and BUSTER. The electron density was subject to density modification with NCS averaging using Parrot (Cowtan, 2010; Emsley et al, 2010). The model was further refined using REFMAC5 with input density modification phases (Pannu et al, 1998). The model was rebuilt using COOT (Emsley et al, 2010) and ISOLDE (Croll, 2018) implemented in ChimeraX (Pettersen et al, 2021). Details of the quality of the refined model are presented in Table EV1.
NMR spectroscopy
All NMR experiments were carried out at 25°C on a 23.5T (1,000 MHz) Bruker spectrometer equipped with triple resonance (x,y,z) gradient cryoprobe. The experiments were processed with NMRPipe58 and analyzed with NMRFAM‐SPARKY59. The interaction of UFC1 with DDRGK1‐UFL1 fragments was monitored by 2D 1H–15N HSQC experiments with the assignments for UFC1 transferred from the BMRB [entry 6546 (Liu et al, 2005)]. DDRGK1‐UFL1 (100–400 μM) and DDRGK1‐UFL1ΔN (400 μM) were titrated into 200 μM of 15N‐labeled UFC1 in 20 mM Tris pH 7.6. For competition experiments, we used 15N‐labeled UBA5 (residues 347–404; 150 μM), UFC1 (300 μM), and DDRGK1‐UFL1 (700 μM).
Isothermal titration calorimetric experiments (ITC)
Isothermal titration calorimetric experiments were performed using PEAQ‐ITC (Malvern Instruments, Malvern) at 25°C. The binding experiments between WT UFC1 and DDRGK1‐UFL1 (n = 2), DDRGK1‐UFL1ΔN or point mutants of DDRGK1‐UFL1 (L11R, L11R + F15R) and between point mutants UFC1 (K47E, R55E) and DDRGK1‐UFL1, were performed in buffer containing 20 mM Tris–Cl, 150 mM NaCl, and 1 mM DTT, at pH 7.5, where fusion constructs (0.8 mM) were titrated into 80 μM UFC1. The binding experiments between charged UFC1 (UFC1~UFM1) and DDRGK1‐UFL1 or DDRGK1ext‐UFL1 (n = 2) were performed in buffer containing 20 mM Tris–Cl, 200 mM NaCl, and 1 mM DTT, at pH 7.5, where fusion constructs (0.24 mM) were titrated into 24 μM charged UFC1. The experiment between WT UFC1 and DDRGK1extUFL1 was performed in the same buffer where fusion constructs (0.2 mM) were titrated into 20 μM UFC1. The ITC experiment between WT UFC1 and UFL1 N‐terminal helix (1–21 aa) (n = 2) was done in a buffer containing 20 mM Tris–Cl, 200 mM NaCl, 1 mM DTT, and 4% DMSO at pH 7.5 where N‐terminal peptide (0.26 mM) was titrated into 26 μM UFC1. All the titrations were done via 19 individual injections of 2 μl volume each, following the first injection (0.4 μl) that was disregarded. An interval of 150 s was allowed between each injection, and the stirring speed was set at 750 rpm to achieve complete thermodynamic equilibration. The experiment data were analyzed with MicroCal PEAQ‐ITC analysis software, and “1 set of sites” models were used for data fitting.
Author contributions
Sayanika Banerjee: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Julia K Varga: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Manoj Kumar: Investigation; methodology. Guy Zoltsman: Investigation; methodology. Shahar Rotem‐Bamberger: Investigation; methodology; writing – review and editing. Einav Cohen‐Kfir: Methodology. Michail N Isupov: Resources; methodology. Rina Rosenzweig: Supervision; funding acquisition; investigation; methodology; writing – review and editing. Ora Schueler‐Furman: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Reuven Wiener: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported, in whole or in part, by the Israel Science Foundation, founded by the Israel Academy of Science and Humanities (grant number 491/2021 to RW and grant number 301/2021 to OS‐F) and by the Israel Cancer Research Fund (award ID 21‐113‐PG to RW). JKV is supported by a Marie Sklodowska‐Curie European Training Network Grant #860517 (Ubimotif). We thank the staff of ESRF beamline ID30‐A for the help with data collection.
EMBO reports (2023) 24: e56920
Contributor Information
Ora Schueler‐Furman, Email: ora.furman-schueler@mail.huji.ac.il.
Reuven Wiener, Email: reuvenw@ekmd.huji.ac.il.
Data availability
The crystal structure of the UFL1‐DDRGK1ΔN fusion structure has been deposited to the Protein Data Bank with PDB ID: 8BX9 (http://www.rcsb.org/pdb/explore/explore.do?structureId=8BX9). The AlphaFold2 models have been deposited to ModelArchive and assigned the following identifiers: ma‐8k8ml (https://www.modelarchive.org/doi/10.5452/ma‐8k8ml), ma‐uzjo7 (https://www.modelarchive.org/doi/10.5452/ma‐uzjo7), ma‐enci1 (https://www.modelarchive.org/doi/10.5452/ma‐enci1), and ma‐vte0s (https://www.modelarchive.org/doi/10.5452/ma‐vte0s).
References
- Banerjee S, Kumar M, Wiener R (2020) Decrypting UFMylation: how proteins are modified with UFM1. Biomolecules 10: 1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen CE, Wolberger C (2014) New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 21: 301–307 [DOI] [PubMed] [Google Scholar]
- Bret H, Andreani J, Guerois R (2023) From interaction networks to interfaces: scanning intrinsically disordered regions using AlphaFold2. bioRxiv 10.1101/2023.05.25.542287 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Bryant P, Pozzati G, Elofsson A (2022) Improved prediction of protein‐protein interactions using AlphaFold2. Nat Commun 13: 1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappadocia L, Lima CD (2018) Ubiquitin‐like protein conjugation: structures, chemistry, and mechanism. Chem Rev 118: 889–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappadocia L, Pichler A, Lima CD (2015) Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat Struct Mol Biol 22: 968–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Perez A (2023) Ranking peptide binders by affinity with AlphaFold. Angew Chem Int Ed 62: e202213362 [DOI] [PubMed] [Google Scholar]
- Cowtan K (2010) Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr 66: 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI (2018) ISOLDE: a physically realistic environment for model building into low‐resolution electron‐density maps. Acta Crystallogr D Struct Biol 74: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletr ZM, Huang DT, Duda DM, Schulman BA, Kuhlman B (2005) E2 conjugating enzymes must disengage from their E1 enzymes before E3‐dependent ubiquitin and ubiquitin‐like transfer. Nat Struct Mol Biol 12: 933–934 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR (2011) An introduction to data reduction: space‐group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 67: 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, O'Neill M, Pritzel A, Antropova N, Senior AW, Green T, Žídek A, Bates R, Blackwell S, Yim J et al (2021) Protein complex prediction with AlphaFold‐Multimer. bioRxiv 10.1101/2023.07.04.547638 [PREPRINT] [DOI]
- Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA (2007) Basis for a ubiquitin‐like protein thioester switch toggling E1‐E2 affinity. Nature 445: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys IR, Pei J, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, Zhang J, Ness TJ, Banjade S, Bagde SR et al (2021) Computed structures of core eukaryotic protein complexes. Science 374: eabm4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, El‐Gowily AH, Noshiro D, Komatsu‐Hirota S, Ono Y, Shindo M, Hatta T, Abe M, Uemura T, Lee‐Okada H‐C et al (2022) The UFM1 system regulates ER‐phagy through the ufmylation of CYB5R3. Nat Commun 13: 7857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Ito S, Mao G, Komatsu‐Hirota S, Inada T, Noda NN, Komatsu M (2023) Mechanistic insights into the roles of the UFM1 E3 ligase complex in ufmylation and ribosome‐associated protein quality control. Sci Adv 9: eadh3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson‐Åkhe I, Wallner B (2022) Improving peptide‐protein docking with AlphaFold‐Multimer using forced sampling. Front Bioinform 2: 959160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K et al (2007) Two novel ubiquitin‐fold modifier 1 (Ufm1)‐specific proteases, UfSP1 and UfSP2. J Biol Chem 282: 5256–5262 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K (2004) A novel protein‐conjugating system for Ufm1, a ubiquitin‐fold modifier. EMBO J 23: 1977–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T, Kim DE, Baker D (2004) Computational alanine scanning of protein‐protein interfaces. Sci STKE 2004: pl2 [DOI] [PubMed] [Google Scholar]
- Kumar M, Padala P, Fahoum J, Hassouna F, Tsaban T, Zoltsman G, Banerjee S, Cohen‐Kfir E, Dessau M, Rosenzweig R et al (2021) Structural basis for UFM1 transfer from UBA5 to UFC1. Nat Commun 12: 5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Schindelin H (2008) Structural insights into E1‐catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 134: 268–278 [DOI] [PubMed] [Google Scholar]
- Lee CY, Hubrich D, Varga JK, Schäfer C, Welzel M, Schumbera E, Đokić M, Strom JM, Schönfeld J, Geist JL et al (2023) Systematic discovery of protein interaction interfaces using AlphaFold and experimental validation. bioRxiv 10.1101/2023.08.07.552219 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Lemaire K, Moura RF, Granvik M, Igoillo‐Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E, Cnop M, Schuit F (2011) Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress‐induced apoptosis. PLoS One 6: e18517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, Corn JE (2020) A genome‐wide ER‐phagy screen highlights key roles of mitochondrial metabolism and ER‐resident UFMylation. Cell 180: 1160–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Aramini J, Atreya HS, Eletsky A, Xiao R, Acton T, Ma L, Montelione GT, Szyperski T (2005) GFT NMR based resonance assignment for the 21 kDa human protein UFC1. J Biomol NMR 32: 261 [DOI] [PubMed] [Google Scholar]
- Metzger MB, Pruneda JN, Klevit RE, Weissman AM (2014) RING‐type E3 ligases: master manipulators of E2 ubiquitin‐conjugating enzymes and ubiquitination. Biochim Biophys Acta 1843: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M (2022) ColabFold: making protein folding accessible to all. Nat Methods 19: 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Lima CD (2013) Structure of a ubiquitin E1‐E2 complex: insights to E1‐E2 thioester transfer. Mol Cell 49: 884–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oweis W, Padala P, Hassouna F, Cohen‐Kfir E, Gibbs DR, Todd EA, Berndsen CE, Wiener R (2016) Trans‐binding mechanism of ubiquitin‐like protein activation revealed by a UBA5‐UFM1 complex. Cell Rep 16: 3113–3120 [DOI] [PubMed] [Google Scholar]
- Pannu NS, Murshudov GN, Dodson EJ, Read RJ (1998) Incorporation of prior phase information strengthens maximum‐likelihood structure refinement. Acta Crystallogr D Biol Crystallogr 54: 1285–1294 [DOI] [PubMed] [Google Scholar]
- Peter JJ, Magnussen HM, DaRosa PA, Millrine D, Matthews SP, Lamoliatte F, Sundaramoorthy R, Kopito RR, Kulathu Y (2022) A non‐canonical scaffold‐type E3 ligase complex mediates protein UFMylation. EMBO J 41: e111015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picchianti L, Sánchez de Medina Hernández V, Zhan N, Irwin NA, Groh R, Stephani M, Hornegger H, Beveridge R, Sawa‐Makarska J, Lendl T et al (2023) Shuffled ATG8 interacting motifs form an ancestral bridge between UFMylation and autophagy. EMBO J 42: e112053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverter D, Lima CD (2005) Insights into E3 ligase activity revealed by a SUMO‐RanGAP1‐Ubc9‐Nup358 complex. Nature 435: 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC (2010) The PyMOL molecular graphics system.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J et al (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudah N, Padala P, Hassouna F, Kumar M, Mashahreh B, Lebedev AA, Isupov MN, Cohen‐Kfir E, Wiener R (2019) An N‐terminal extension to UBA5 adenylation domain boosts UFM1 activation: isoform‐specific differences in ubiquitin‐like protein activation. J Mol Biol 431: 463–478 [DOI] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: more than just middle men. Cell Res 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi K, Sou Y, Tada N, Nakamura E, Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E et al (2010) A novel type of E3 ligase for the Ufm1 conjugation system. J Biol Chem 285: 5417–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaban T, Varga JK, Avraham O, Ben‐Aharon Z, Khramushin A, Schueler‐Furman O (2022) Harnessing protein folding neural networks for peptide‐protein docking. Nat Commun 13: 176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A et al (2021) Highly accurate protein structure prediction for the human proteome. Nature 596: 590–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480–D489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66: 22–25 [DOI] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A et al (2022) AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res 50: D439–D444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman DG, Winter G, Gildea RJ, Parkhurst JM, Brewster AS, Sauter NK, Evans G (2016) Diffraction‐geometry refinement in the DIALS framework. Acta Crystallogr D Struct Biol 72: 558–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474: 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witting KF, Mulder MPC (2021) Highly specialized ubiquitin‐like modifications: shedding light into the UFM1 enigma. Biomolecules 11: 255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lei G, Mei M, Tang Y, Li H (2010) A novel C53/LZAP‐interacting protein regulates stability of C53/LZAP and DDRGK domain‐containing Protein 1 (DDRGK1) and modulates NF‐kappaB signaling. J Biol Chem 285: 15126–15136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariv B, Yariv E, Kessel A, Masrati G, Chorin AB, Martz E, Mayrose I, Pupko T, Ben‐Tal N (2023) Using evolutionary data to make sense of macromolecules with a “face‐lifted” ConSurf. Protein Sci 32: e4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW, Ka SH, Sou Y‐S, Komatsu M, Tanaka K et al (2014) Modification of ASC1 by UFM1 is crucial for ERα transactivation and breast cancer development. Mol Cell 56: 261–274 [DOI] [PubMed] [Google Scholar]
- Yuan L, Lv Z, Adams MJ, Olsen SK (2021) Crystal structures of an E1‐E2‐ubiquitin thioester mimetic reveal molecular mechanisms of transthioesterification. Nat Commun 12: 2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Shabek N (2017) Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem 86: 129–157 [DOI] [PubMed] [Google Scholar]