

Abstract
Interferons (IFN) are crucial antiviral and immunomodulatory cytokines that exert their function through the regulation of a myriad of genes, many of which are not yet characterized. Here, we reveal that lipin‐2, a phosphatidic acid phosphatase whose mutations produce an autoinflammatory syndrome known as Majeed syndrome in humans, is regulated by IFN in a STAT‐1‐dependent manner. Lipin‐2 inhibits viral replication both in vitro and in vivo. Moreover, lipin‐2 also acts as a regulator of inflammation in a viral context by reducing the signaling through TLR3 and the generation of ROS and release of mtDNA that ultimately activate the NLRP3 inflammasome. Inhibitors of mtDNA release from mitochondria restrict IL‐1β production in lipin‐2‐deficient animals in a model of viral infection. Finally, analyses of databases from COVID‐19 patients show that LPIN2 expression levels negatively correlate with the severity of the disease. Overall, these results uncover novel regulatory mechanisms of the IFN response driven by lipin‐2 and open new perspectives for the future management of patients with LPIN2 mutations.
Keywords: COVID‐19, inflammasome, interferon, lipin‐2, MCMV
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Lipin‐2 expression is controlled by interferons, reducing cytomegalovirus replication and inflammasome activation. Patients with decreased LPIN2 expression experience increased inflammatory responses during SARS‐CoV‐2 infection.

Introduction
Interferons (IFNs) are essential cytokines in the fight against viral infections and also display immunomodulatory actions (Schneider et al, 2014). Engagement of pathogen‐associated molecular patterns (PAMPs) with pattern recognition receptors (PRRs) at the plasma and endosomal membranes and the cytosol, generate IFNs (Janeway Jr & Medzhitov, 2002; Toshchakov et al, 2002). Once synthesized and released from cells, they may act in an autocrine or paracrine fashion by binding to specific cell surface receptors. Although there are three categories of IFNs, only type I (IFNα and β) and II (IFNγ) are relevant to immune cells. They signal through different receptors, i.e. type I IFNs bind to IFNAR, while IFNγ binds to IFNGR. After recognition of their respective ligands, both receptors activate a signaling cascade initiated by Janus kinases (JAK), and TYK2 in the case of IFNAR, that culminates in the activation of several members of the signal transducer and activator of transcription (STAT) family, and the upregulation of interferon‐stimulated genes (ISGs) (Levy et al, 1988; Darnell Jr et al, 1994). Specifically, type I IFNs promote the phosphorylation of STAT1 and STAT2 that, together with IRF9 form the interferon‐stimulated gene factor 3 (ISGF3) complex. ISGF3 translocates to the nucleus and binds a DNA sequence motif known as the interferon‐stimulated response element (ISREs) present in type I ISGs (Stark & Darnell Jr, 2012). On the other hand, IFNγ promotes the phosphorylation and dimerization of STAT1, which binds to the IFNγ activation site (GAS) DNA element to initiate the transcription of type II ISGs (Decker et al, 1991; Schneider et al, 2014). This type of ISGs can also be upregulated by type I IFNs through the promotion of STAT1 and STAT3 homo and heterodimers. ISGs participate in mounting an effective antiviral state that may help to fight intracellular bacteria and parasites, and initiate an adequate adaptive immune response (Schoggins et al, 2011). However, despite many years of efforts, only a few ISGs have been characterized in detail.
Lipin‐2 is a member of a family of phosphatidic acid phosphatase enzymes which are central to lipid metabolism, as they provide the diacylglycerol (DAG) that is used within the de novo pathway for phospholipid and triacylglycerol (TAG) biosynthesis (Balboa et al, 1998; Péterfy et al, 2001; Zhang & Reue, 2017). The gene encoding for lipin‐2, LPIN2, is mutated in patients that suffer from an autoinflammatory disease known as Majeed syndrome (Ferguson et al, 2005). These patients experience recurrent flares of fever and inflammation in their joints and skin. The causes underlying these symptoms are still poorly defined (Majeed et al, 1989; Ferguson & El‐Shanti, 2021).
In general, autoinflammatory diseases develop subsequent to dysregulated activation of innate immune cells (McDermott et al, 1999; Tartey & Kanneganti, 2020). We have recently described that macrophages defective in lipin‐2 exhibit an exacerbated production of IL‐1β due to increased classical activation of the NLRP3 inflammasome (Lordén et al, 2017; Balboa et al, 2019). Inflammasomes are intracellular machineries, key in the battle against pathogens, that assemble to activate caspase‐1 (Martinon et al, 2002). Once caspase‐1 is activated, it cleaves pro‐IL‐1β and pro‐IL‐18 to produce the mature cytokines. In macrophages, two different signals are needed for IL‐1β production through the NLRP3 inflammasome (Bauernfeind et al, 2009; Franchi et al, 2009). The first signal is generated by the binding of PAMPs to Toll‐like receptors (TLRs) or cytokines like TNF‐α. This upregulates IL1B through activation of transcription factors such as NF‐κB. The second signal is generated by cellular events such K+ efflux and mitochondria or lysosome damage.
Canonical activation of the NLRP3 inflammasome in macrophages occurs through sequential exposure of the cells to bacterial lipopolysaccharide (LPS, TLR4 ligand—first signal) and ATP (P2X7 receptor ligand—second signal). The absence of lipin‐2 leads to enhanced LPS signaling that augments pro‐IL‐1β production, and increased K+ efflux due to over‐activation of P2X7 receptors (Lordén et al, 2017). While these findings establish that lipin‐2 acts as a brake to control the extent of inflammation, many questions are still open. One of these refers to the way innate immune cells, particularly macrophages, regulate the expression of lipin‐2. This is a key question to solve because it may provide important clues as to where and when does lipin‐2 exert its anti‐inflammatory and metabolic regulatory roles and, perhaps, aid in the development of strategies aimed to better manage the symptoms of Majeed disease.
In this study, we show that Lpin2 is upregulated by IFNs through STAT1, and this constitutes a key event for the latter to express their antiviral activity. We also demonstrate that lipin‐2 protects mitochondria from oxidative damage and reduces mtDNA release, thereby providing a mechanism to explain its anti‐inflammatory actions during NLRP3 activation by intracellular viral molecular patterns. Further, we show that symptomatic patients infected with SARS‐CoV‐2, the virus responsible for the COVID‐19 pandemic, can be distinguished from asymptomatic patients on the basis of their very different lipin‐2 expression levels. Collectively, our study suggests that: (i) unexplained recurrent flares of symptoms in Majeed patients, and more specifically their high IL‐1β levels, could be due to otherwise unnoticed viral infections; (ii) inhibitors of mtDNA release can be helpful in the management of Majeed disease; and (iii) measuring LPIN2 levels may be useful to identify higher‐risk COVID‐19 patients.
Results
TLRs and IFNs regulate the expression of lipin‐2 in BMDMs
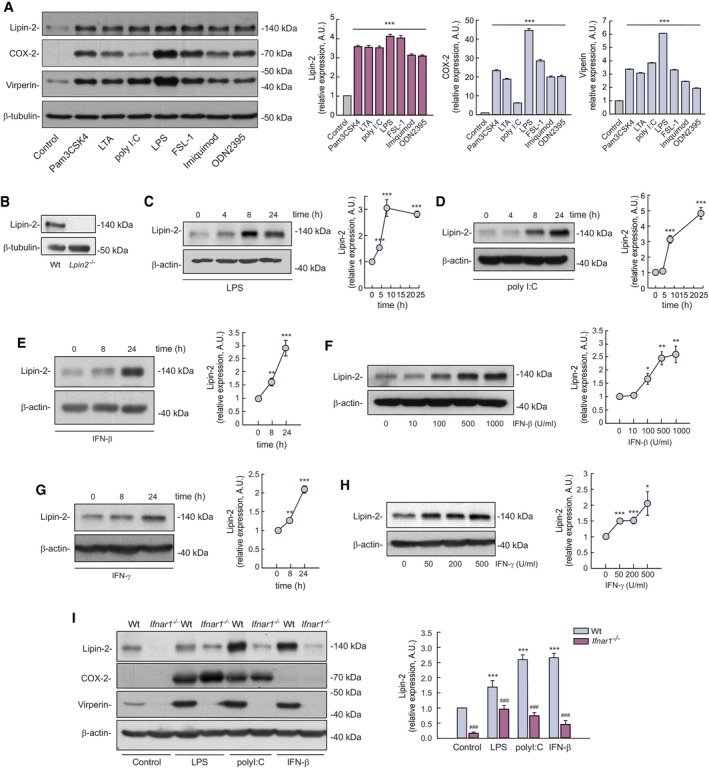
We began this study by analyzing whether the engagement of classical PRRs such as the TLRs promotes changes in the expression levels of lipin‐2 in innate immune cells. To this end, we stimulated bone marrow derived macrophages (BMDMs) with known TLR agonists and analyzed the expression of lipin‐2 by immunoblot. The antibody against lipin‐2 used in the present study recognizes a ~ 140 kDa band. This antibody has previously been used by others in murine macrophage cell lines (Watahiki et al, 2020). Lipin‐2 is heavily phosphorylated in cells. This radically changes its electrophoretic mobility from a predicted 95 kDa band to the observed ~ 140 kDa band in SDS–PAGE (Eaton et al, 2014). The results showed that agonists of TLR1/2 (Pam3CSK4), TLR2 (lipoteichoic acid, LTA), TLR3 (poly(I:C)), TLR4 (LPS), TLR5 (Flagellin), TLR2/6 (FSL‐1), TLR7 (imiquimod) and TLR9 (ODN1826) upregulated lipin‐2 expression to various degrees (Fig 1A and Appendix Fig S1). The specificity of the antibody used to detect lipin‐2 was verified by analyzing lysates from Lpin2 −/− macrophages (Fig 1B). In these cells, the antibody failed to detect a 140 kDa band (Fig 1B). Conversely, in cells overexpressing lipin‐2 the antibody recognized the expected 140 kDa band (Appendix Fig S2). As a positive control that cells were activated properly by all TLR stimuli used, we also investigated the expression of proteins known to be upregulated by TLRs, such as COX‐2 and viperin. The levels of both proteins increased in cells stimulated with all the agonists (Fig 1A). Time‐course analyses of lipin‐2 expression in cells activated with different TLR agonists indicated that the enzyme experienced measurable increases in expression at different time points after stimulation, reaching a plateau of expression between 8 and 18 h depending on the TLR activated (Fig 1C and D, and Appendix Fig S1).
Figure 1. Effect of TLR agonists and interferons on lipin‐2 expression.
-
ABMDMs were treated or not (control) with 1 μg/ml Pam3CSK4 (TLR1/2), 1 μg/ml LTA (TLR2), 25 μg/ml poly(I:C) (TLR3), 200 ng/ml LPS (TLR4), 100 μg/ml FSL‐1 (TLR6/2), 5 μg/ml Imiquimod (TLR7) or 1 μM ODN 2395 (TLR9) for 24 h. Cells lysates were analyzed by immunoblot using specific antibodies against lipin‐2, COX‐2, viperin, and β‐tubulin as a loading control. Quantifications of the bands relative to β‐tubulin and normalized to Control cell values are shown on the right.
-
BHomogenates from Wt and Lpin2 −/− BMDMs were analyzed by immunoblot using antibodies against lipin‐2 and β‐tubulin (loading control).
-
C, DBMDMs were treated with 200 ng/ml LPS (C) or 25 μg/ml poly(I:C) (D) for the indicated periods of time. Quantifications of the bands relative to β‐actin and normalized to time zero values are shown on the right.
-
E–HBMDMs were treated with 500 U/ml IFN‐β (E) or 200 U/ml IFN‐γ (G) for the indicated periods of time, or with the indicated concentrations of IFN‐β (F) or IFN‐γ (H) for 24 h. Homogenates were analyzed by immunoblot using antibodies against lipin‐2 and β‐actin (loading control). Quantifications of the bands relative to β‐actin and normalized to time zero or untreated cells are shown on the right.
-
IWt or Ifnar1 −/− BMDMs were treated or not (control) with 200 ng/ml LPS, 25 ng/ml poly(I:C), or 500 U/ml IFN‐β for 24 h. Homogenates were analyzed by immunoblot using antibodies against lipin‐2 and β‐actin (loading control). Quantifications of the bands relative to β‐actin and normalized to Wt control cells are shown on the right. Shown are representative experiments of at least two independent ones. *P < 0.05; **P < 0.01; ***P < 0.001, ### P < 0.001.
Data information: The immunoblots in (A, C, D, F, and I) are representative examples from three biological replicates. The immunoblots in (E, G, and H) are representative examples from five biological replicates. Two different experiments were performed. Data represent the mean ± SEM. P‐value of one‐way ANOVA followed by Holm‐Sidak test (A) or Student's t‐test (C–I). *P < 0.05; **P < 0.01; ***P < 0.001, ### P < 0.001. *, treated vs. control cells (unstimulated or time = 0), # Wt vs. Ifnar−/− cells.
Source data are available online for this figure.
The finding that a delay exists before lipin‐2 levels start to increase after TLR activation suggests the existence of a factor that is produced prior to and intermediates in the process. Because a number of TLR receptors, including TLR3, TLR4, TLR7, and TLR9 are strong inducers of IFN expression (Chow et al, 2015), we sought to study the possible involvement of IFN in regulating lipin‐2 expression in macrophages. As shown in Fig 1E–H, treating BMDMs with IFN‐β or IFN‐γ increased the expression of lipin‐2 in a time and dose‐dependent manner. Importantly, cells deficient in the IFN‐β receptor subunit 1 (Ifnar1) displayed reduced expression levels of lipin‐2, and their capacity to increase lipin‐2 levels after TLR activation or IFN‐β treatment was significantly reduced (Fig 1I). These results indicate that both basal and stimulated IFN levels impact on lipin‐2 expression.
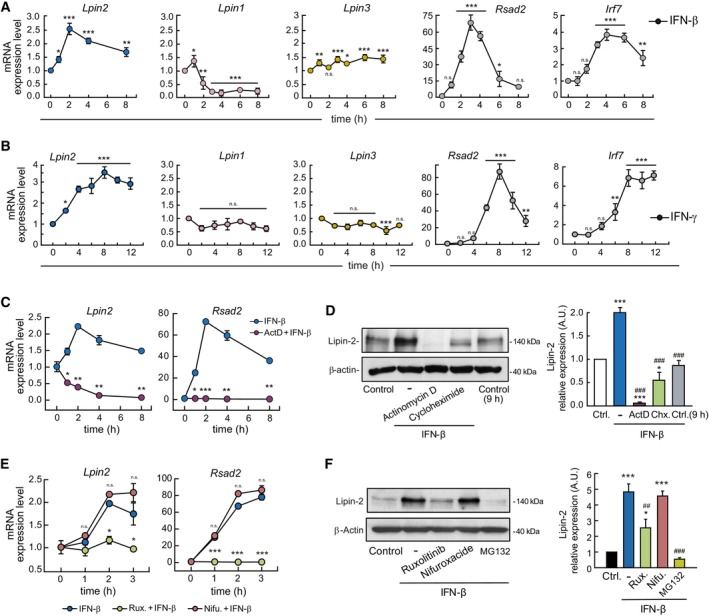
To ascertain whether lipin‐2 levels were regulated at the protein or mRNA level, we studied the levels of Lpin2 mRNA during cellular stimulation by analyzing gene expression arrays (Raza et al, 2014a; Data ref: Raza et al, 2014b). We found that LPS and IFNs increased Lpin2 mRNA levels in a time‐dependent manner, while the mRNA levels for other members of the lipin family of enzymes, i.e. Lpin1 and Lpin3, were decreased or unchanged respectively (Appendix Fig S3). We confirmed these results in IFN‐β‐ and IFN‐γ‐treated BMDMs, by evaluating by qPCR the expression levels of Lpins and also those of Rsad2/viperin and Irf7, two well‐known ISGs (Fig 2A and B). The data showed that among lipins, Lpin2 selectively increased its mRNA levels during macrophage stimulation with TLR agonists and by IFNs.
Figure 2. Lpin2 mRNA levels are controlled by interferons.
-
A, BBMDMs were treated with 500 U/ml IFN‐β (A) or 200 U/ml IFN‐γ (B) for the indicated periods of time. mRNA levels for the indicated genes were analyzed by qPCR, using Gapdh as reference and normalized to time zero.
-
CBMDMs were treated with 500 U/ml IFN‐β in the presence or absence of 1 μg/ml Actinomycin D (ActD). mRNA levels were analyzed by qPCR, using Gapdh as reference and normalized to time zero.
-
DBMDMs were left untreated (Control and Control 9 h) or treated with 500 U/ml IFN‐β in the absence or presence of 1 μg/ml Actinomycin D (ActD) or 10 μg/ml Cycloheximide (Chx.). Cellular homogenates were analyzed by immunoblot using antibodies against lipin‐2 and β‐actin (loading control). Quantifications of the bands relative to β‐actin and normalized to Control cells are shown on the right.
-
E, FBMDMs were untreated (Control) or treated with 500 U/ml IFN‐β in the presence or absence of 5 μM Ruxolitinib (Rux.), 10 μM Nifuroxazide (Nifu.) or 20 μM MG132 (only in F). mRNA levels were analyzed by qPCR, using Gapdh as reference and normalized to time zero. (E) Cellular homogenates were analyzed by immunoblot using antibodies against lipin‐2 and β‐actin (loading control) (F). Quantifications of the bands relative to β‐actin are shown on the right.
Data information: Data from (A–C and E) are shown as means ± SEM of biological triplicates. Two experiments were performed. The immunoblots in (D, F) are representative examples from three biological replicates. Data represent the mean ± SEM. P value of one‐way ANOVA followed by Holm‐Sidak test (A, B, D, F), or Student's t‐test (C, D). (A, B) *, IFN‐treated vs. time = 0. (C) *, IFN‐β vs. ActD+IFN‐β. (E) *, IFN‐β vs. Rux + IFN‐β or Nifu+IFN‐β. (D, F) *, IFN‐β treated vs. control cells; # conditions indicated vs. just IFN‐β treated. n.s., not significant, *P < 0.05; **P < 0.01; ***P < 0.001; ## P < 0.01; ### P < 0.001.
Source data are available online for this figure.
STAT1 participates in the transcriptional upregulation of Lpin2
In the next series of experiments, the events controlling Lpin2 upregulation by IFNs were investigated. We used actinomycin D and cycloheximide to inhibit transcription and translation, respectively. The results indicated that transcription is necessary to increase Lpin2 mRNA levels and that both transcription and translation events are needed for lipin‐2 expression to increase during macrophage activation by IFN‐β (Fig 2C and D). To further demonstrate the involvement of IFN in the transcriptional activation of Lpin2, we analyzed newly transcribed levels of Lpin2 mRNA by gene expression arrays (Dölken et al, 2008; Data ref: Robertson & Ghazal, 2016). The results showed that IFN‐γ induces the transcription of new mRNA for Lpin2 (Appendix Fig S4).
Next, the signaling events that participate in Lpin2 mRNA upregulation were evaluated. Engagement of IFN receptors initiates signaling events through activation of JAK1 and TYK2 (IFNAR) or JAK1 and JAK2 (IFNGR). Subsequently, IFNGR activates STAT1, while IFNAR activates STAT1 and STAT2, and both receptors can secondarily activate STAT3 (Stark & Darnell Jr, 2012). We analyzed first the effect of the selective JAK1/2 inhibitor ruxolitinib on lipin‐2 expression. Treatment of BMDMs with ruxolitinib completely abolished the induction of Lpin2 mRNA, as well as the increases in lipin‐2 protein levels by IFN‐β (Fig 2E and F). In contrast, nifuroxazide, a STAT3 inhibitor (Nelson et al, 2008), had no effect. The same behavior was found in BMDMs activated with the TLR3 agonist poly(I:C), thus suggesting the key involvement of JAKs in mediating IFN‐induced lipin‐2 expression (Appendix Fig S5).
It has previously been shown that lipin‐2 levels can be regulated by ubiquitination and proteasome degradation (Watahiki et al, 2020). We studied the effect of the proteasome inhibitor MG132 on the capacity of IFN‐β to increase lipin‐2 expression levels. We found that the inhibitor did not increase the levels of expression of lipin‐2, indicating that a decreased proteolysis by the proteasome does not play a role in the increased expression of the enzyme in this scenario.
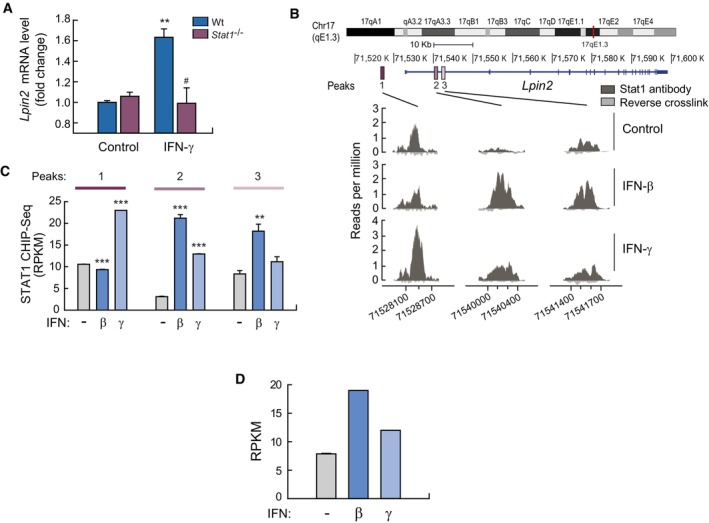
To assess the role of STAT1 in Lpin2 transcriptional regulation, we analyzed gene expression array data from STAT1−/− BMDMs stimulated with IFN‐γ (Semper et al, 2014a; Data ref: Semper et al, 2014b). The data showed that, in the absence of STAT1, the ability of IFN‐γ to increase Lpin2 mRNA levels is abolished (Fig 3A). To study whether the transcriptional activation of Lpin2 correlates with binding of STAT1 to the Lpin2 locus, we performed analyses of ChIP‐seq data from BMDMs stimulated with IFN‐β and IFN‐γ (Ng et al, 2011a; Data ref: Ng et al, 2011b). The results presented in Fig 3 indicate that STAT1 binds to three different sequences in the locus of Lpin2 (peaks 1, 2 and 3). The first peak is around 5 kb upstream of the initiation of the transcription, while the second and third peaks are located downstream, between the first and second exons, at around 7.8 and 8.2 kb respectively of the initiation of the transcription (Fig 3B). Analysis of the density of reads in each peak showed that STAT1 is already attached to peak 1 in unstimulated cells, and treatment with IFN‐γ but not IFN‐β, increases the number of reads to this region, indicating an enhanced binding of STAT1 to this sequence (Fig 3B and C). In contrast, STAT1 is almost absent of peak 2 in unstimulated cells, but both IFNs promote its binding to this region, with IFN‐β being more potent in this regard. Finally, only IFN‐β appears to significantly induce STAT1 binding to the sequence present in peak 3. Under these experimental conditions, Lpin2 mRNA levels were induced by both IFN‐β and IFN‐γ (Fig 3D).
Figure 3. STAT‐1 binds to Lpin2 locus to participate in its transcriptional upregulation during interferon stimulation.
-
AWt and STAT1−/− BMDMs were left untreated (Control) or treated with 100 U/ml IFN‐γ for 6 h, and gene expression was analyzed by microarrays. Fold change respect to Wt control cells are shown. Data are from triplicate samples and represent means ± SEM. **P < 0.01, # P < 0.05 by Student's t‐test. *, IFN‐γ treated vs. control cells; # STAT1−/− vs. Wt cells.
-
BBMDMs were left untreated (Control) or treated with IFN‐β or IFN‐γ for 6 h and Chip‐seq analysis was performed using anti‐STAT1 antibodies. Shown are the main three STAT1 peaks called by MACS associated to the Lpin2 locus (dark gray). Reads from just reverse crosslink is also shown (light gray).
-
CShown are Chip‐seq reads per kilobase per million reads (RPKM) values for STAT1‐binding peaks associated with Lpin2 relative to input controls. Statistics were performed with respect to unstimulated samples (−) for each peak. **P < 0.01; ***P < 0.001 by Student's t‐test.
-
DBMDMs were left untreated (−) or treated with IFN‐β or IFN‐γ for 6 h, and gene expression was analyzed by RNA‐seq. Data are shown as RPKM for peaks associated to Lpin2.
Source data are available online for this figure.
Analysis of the sequences of the STAT1 peaks found in the Lpin2 locus revealed the presence of GAS consensus motifs in the three peaks, and a unique ISRE consensus motif in peak 2 (Appendix Fig S6). These data suggest that IFN‐γ promotes the binding of STAT1 to GAS sequences in peaks 1 and 2, while IFN‐β induces the binding of STAT‐1 to the ISRE/GAS sequences found in peak 2, and to the GAS sequence found in peak 3. In addition to inducing the formation of ISGF3, it has been reported that IFN‐β may promote the dimerization of STAT1 (GAF) (Ivashkiv & Donlin, 2014). This event may help explain the induction of STAT1 binding to GAS sequences present in the Lpin2 locus during IFN‐β stimulation of BMDMs (Fig 3B and C). Altogether, the data illustrate the participation of STAT‐1 in the induction of Lpin2 transcription by IFN.
Lipin‐2 controls viral replication
One of the key functions of IFNs is to control viral infections through the transcriptional induction of ISGs. Given the regulation of lipin‐2 expression by IFNs, we went on to assess whether the phosphatase plays a role in the antiviral response. For these experiments, we chose murine CMV (MCMV) because IFN signaling is crucial for the control of its infection in vivo (Lio et al, 2016). Accordingly, we first evaluated whether MCMV‐infected BMDMs were able to increase Lpin2 mRNA levels (Blanc et al, 2013; Data ref: Blanc et al, 2015). Figure 4 shows that this was the case. The effect was particularly noteworthy after 3 h of infection, and occurred in parallel with well‐known ISGs like Rsad2 or Irf7. In contrast, Lpin1 and Lpin3 were not substantially increased under these conditions (Fig 4A). We confirmed by qPCR that macrophage infection by MCMV increased Lpin2 expression (Fig 4B). To assess whether lipin‐2 had any influence on virus replication, we infected BMDMs with an MCMV carrying the GFP gene (MCMV‐GFP), and GFP fluorescence was analyzed in the cultures at different times post‐infection. Infected wild type BMDMs experienced a progressive increase in fluorescence after a 3‐day infection period (Fig 4C). Importantly, lipin‐2‐deficient macrophages manifested higher increases in fluorescence than wild type cells from days 2–6 after infection. This indicated enhanced viral replication. To verify whether the increase in replication was accompanied by an augmented production of infective viral particles in Lpin2 −/− macrophages, supernatants of infected macrophages were collected and viral infectivity was analyzed on mouse embryonic fibroblast (MEFs) monolayers. Macrophage lipin‐2 deficiency caused a significant increase in fluorescence forming units (FFUs) (Fig 4D). The same results were also observed when the infectivity of the supernatants collected at different times from the infected macrophages was analyzed by standard plaque assays on MEFs (Fig 4E).
Figure 4. Lipin‐2 restricts MCMV replication in vitro and in vivo .
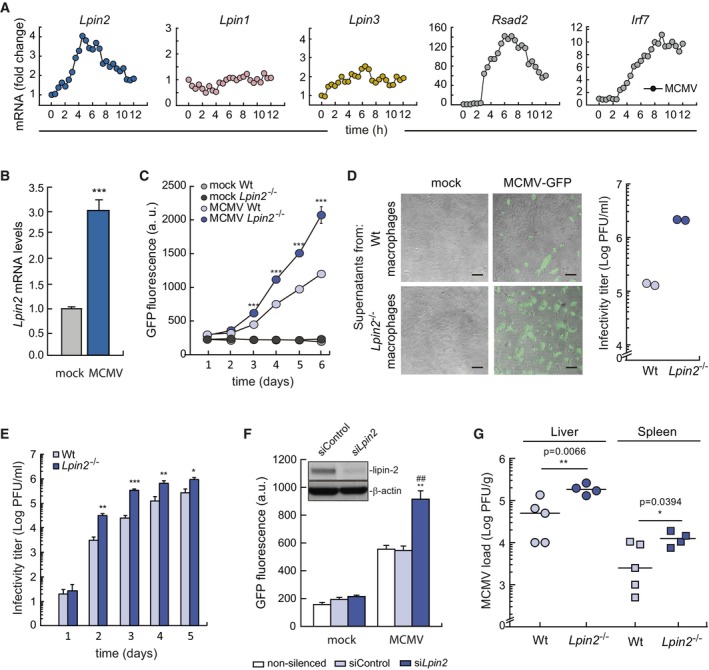
- BMDMs were infected with MCMV (MOI = 1) for the indicated time points. Gene expression was analyzed by microarrays. Data are from a single biological replicate at each time point. Data shows fold gene expression change respect to expression levels at 0 h.
- BMDMs were infected with MCMV (MOI = 1) for 4 h and mRNA levels for Lpin2 were quantified by qPCR, using Gapdh as reference. Data were normalized to mock infected cells. Data from biological triplicate samples are represented as mean ± SEM. ***P < 0.001, Student's t‐test.
- BMDMs from Wt or Lpin2 −/− animals were mock infected or infected with MCMV‐GFP (MOI = 2) for 2 h. GFP fluorescence present in cell cultures was analyzed at the indicated time points after infection. Data from biological triplicate samples are represented as means ± SD. ***P < 0.001, Student's t‐test.
- BMDMs from Wt or Lpin2 −/− animals were mock infected or infected with MCMV‐GFP (MOI = 4). Supernatants were collected at day 3 post infection and, either transferred to MEF monolayers which were examined by microscopy 24 h post‐infection (left panels) or titrated by standard plaque assays on MEFs, determining the number of FFU under the microscope (right panel). Scale bars: 100 μm.
- BMDMs from Wt or Lpin2 −/− animals were mock infected or infected with MCMV‐GFP (MOI = 2), supernatants were collected at different times after infection and titrated by standard plaque assays on MEFS, counting PFU under the microscope. Data from biological triplicate samples are represented as mean ± SD. *P < 0.5, **P < 0.01, ***P < 0.001, Student's t‐test.
- RAW264.7 cells silenced for Lpin2 or not were mock infected or infected with MCMV‐GFP (MOI = 2). GFP fluorescence present in cell cultures was analyzed 5 days after infection. Data from biological triplicate samples are represented as mean ± SEM. **P < 0.01, ## P < 0.01, Student's t‐test. * siLpin2 vs. siControl, # siLpin2 vs. unsilenced cells. The insert shows lipin‐2 expression in silenced cells by immunoblot.
- Wt (n = 5) and Lpin2 −/− (n = 4) animals were intraperitoneally injected with MCMV (3.5 × 106 PFU per mouse) and, after 4 days of infection viral titers were analyzed in the liver and the spleen of animals by standard plaque assays, counting PFUs. Statistical differences were tested by Student's t‐test, and P‐values are shown.
Source data are available online for this figure.
To further establish the involvement of lipin‐2 in MCMV replication, experiments were conducted in lipin‐2‐silenced RAW 264.7 macrophages (treated with specific siRNAs). These experiments confirmed that MCMV replicate better when lipin‐2 levels are decreased (Fig 4F). These data support the notion that acute elimination of lipin‐2 is sufficient to produce the observed effects on viral behavior.
To study whether these results could be translated to animal models of infection, mice were infected with MCMV for 5 days, and viral titers were determined in liver and spleen, two primary organs for viral replication. In agreement with the in vitro data, we observed that lipin2‐deficient animals display a higher viral load in both organs (Fig 4G). Collectively, these data suggest that the induction of Lpin2 expression during MCMV infection plays a role in the control of viral replication.
We also tested whether lipin‐2 participates in the control of viruses that are known to restrain IFN production and the anti‐viral activity promoted by several ISGs, such as Vaccinia virus (VACV) (Smith et al, 2018). As expected, replication of VACV in infected BMDMs was not affected by the phosphatase (Appendix Fig S7).
Lipin‐2 controls inflammation during virus‐derived nucleic acid recognition
Beyond their role in anti‐viral defense, IFNs have also been described as modulators of immunopathogenic events curtailing collateral damage during infection (Lee & Ashkar, 2018). Thus we evaluated whether lipin‐2, as a part of the IFN response, was involved in decreasing the inflammation during viral recognition by macrophages. IL‐1β is a key cytokine during inflammatory reactions. IL‐1β is essential for the host response to pathogens, but is also fully capable of generating damage when produced at too high levels (Masters et al, 2009). We evaluated whether the presence of lipin‐2 affected IL‐1β production by BMDMs treated sequentially with poly(I:C) (viral nucleic acid mimetic) and ATP (to activate the inflammasome). Lipin‐2‐deficient BMDMs produced much higher levels of IL‐1β than wild type cells (Fig 5A). This correlated with increased levels of Il1b mRNA during poly(I:C) stimulation in Lpin2 −/− BMDMs and silenced RAW264.7 macrophages (Fig 5B and Appendix Fig S8). Importantly, the same behavior was also found in LPIN2–silenced primary human macrophages (Fig 5C). The participation of TLR3 in the upregulation of I1b mRNA expression was confirmed by silencing the receptor in BMDMs (Fig 5D). Analysis of the signal transduction events implicated showed that NF‐κB (p65) experienced a quicker translocation to the nucleus in lipin‐2‐deficient cells compared with lipin‐2 expressing cells. This indicated an earlier activation of this factor during poly(I:C) stimulation (Fig 5E). TLR3 signaling is also known to involve MAPK activation (Janeway Jr & Medzhitov, 2002). Evaluation of ERK, JNK and p38 phosphorylation by immunoblot showed that the activation of all of these kinases was also ahead of time in lipin‐2‐deficient macrophages (Fig 5F). We also found that MAPK inhibitors (PD98059 for MEK, the kinase upstream of ERK; SP600125 for JNK, and SB203580 for p38), significantly reduced the expression levels of Il1b mRNA in macrophages expressing or not lipin‐2. The most efficient of these was the p38 inhibitor (SB203580) (Fig 5G). We next evaluated whether the expression levels of proteins that participate in inflammasome activation, the cytosolic machinery that ultimately activates caspase‐1 to proteolytically mature pro‐IL‐1β, could also be enhanced in lipin‐2‐deficient macrophages during the poly(I:C) challenge. Both NLRP3 expression (inflammasome receptor) and mRNA levels for Casp1 (caspase‐1) were increased under these circumstances (Fig 5H and I). As a consequence of the increased signaling through TLR3 that characterized cells deficient in lipin‐2, we also found that, after poly(I:C) stimulation, they displayed increased expression levels for TLR3 itself. Other ISGs such as Ifih1/MDA5, Ddx58 (RIG‐I), and Eif2ak2 (PKR), which are intracellular receptors for dsRNA, and Rsad2/viperin, a dual role factor for viral host defense (West et al, 2015), were also found to be elevated. Collectively, these results show that lipin‐2 regulates the production of IL‐1β as well as ISGs expression levels by regulating the signal transduction events that occur downstream of TLR3 engagement.
Figure 5. Lipin‐2 controls inflammasome priming by molecules that mimic viral nucleic acids.
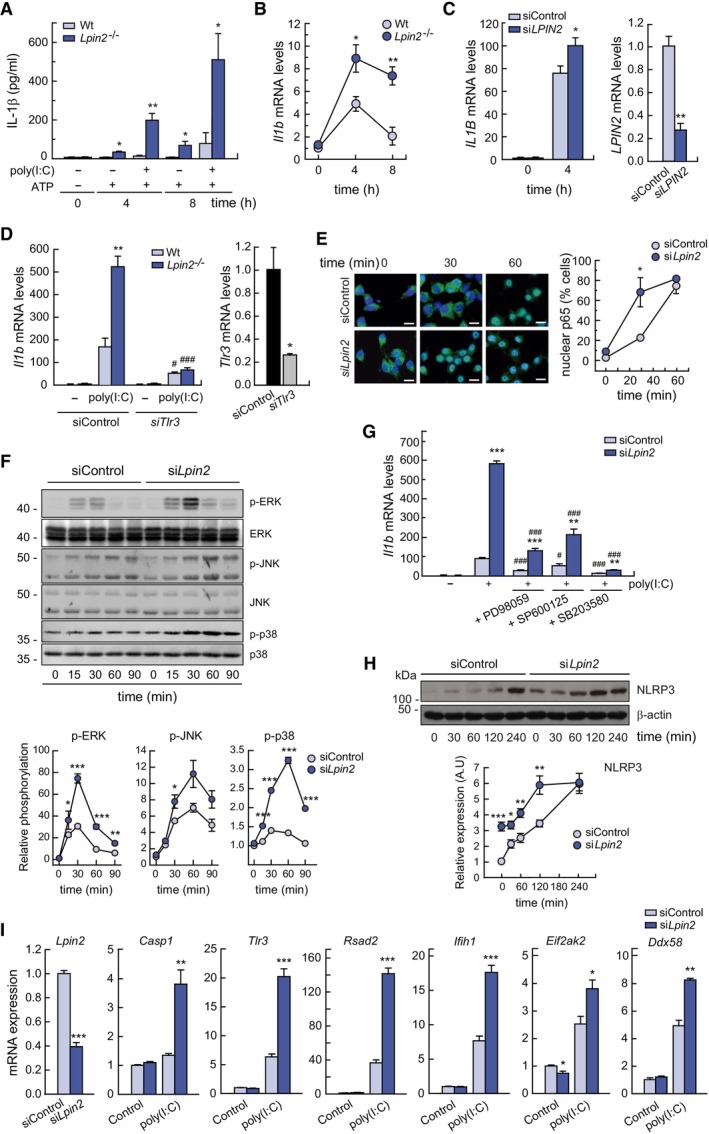
- BMDMs from Wt or Lpin2 −/− animals were left untreated, primed with 10 μg/ml of poly(I:C) for 4 h and activated or not with 2 mM ATP for 4 or 8 h, as indicated. IL‐1β levels present in cell supernatants were quantified by specific ELISAs.
- BMDMs from Wt or Lpin2 −/− animals were left untreated or treated with 10 μg/ml poly(I:C) for 4 and 8 h. mRNA levels for Il1b were analyzed by qPCR, using Gapdh as reference and normalized to time zero.
- Primary human macrophages, nucleofected with control siRNAs (siControl) or against LPIN2 (siLPIN2), were treated or not with 25 μg/ml poly(I:C) for 4 h and mRNA levels for IL1B (left panel) or LPIN2 (right panel) were analyzed by qPCR. ACTB was used as reference.
- BMDMs from Wt or Lpin2 −/− animals were transfected with control siRNAs or against TLR3 (siTLR3) and left untreated or stimulated with 10 μg/ml of poly(I:C) for 4 h. mRNA levels for Il1b (left panel) or TLR3 (right panel) were analyzed by qPCR, using Gapdh as reference.
- RAW264.7 cells silenced for Lpin2 or not were treated with 25 μg/ml of poly(I:C) for the indicated times. Cells were immunostained using specific antibodies against p65. Nuclei were stained with DAPI. Images obtained by confocal microscopy are shown (left panel). Percentage of cells with nuclear p65 translocation is represented (right panel) (n = 60). Scale bars: 10 μm.
- Homogenates from RAW264.7 cells treated as in F were analyzed by immunoblot using antibodies against phospho‐ERK, phospho‐JNK and phospho‐p38. Total ERK, JNK and p38 were used as a loading controls respectively. Densitometric quantification of the bands are represented in the lower panels.
- RAW264.7 cells silenced for Lpin2 or not were treated with 10 μM of the indicated MAPK inhibitors prior to stimulation with 25 μg/ml of poly(I:C) for 4 h. mRNA levels for Il1b were analyzed by qPCR, using Gapdh as reference.
- RAW264.7 cells silenced for Lpin2 or not were treated with 25 μg/ml of poly(I:C) for the indicated periods of time. Cell homogenates were analyzed by immunoblot using specific antibodies against NLRP3 and β‐actin. Densitometric quantification of the bands are represented in the lower panel.
- RAW264.7 cells silenced for Lpin2 or not were left untreated (Control) of stimulated with 25 μg/ml of poly(I:C) for 4 h. mRNA levels for the indicated genes were analyzed by qPCR mRNA levels for Il1b were analyzed by qPCR, using Gapdh as reference.
Data information: Data shown in (A–D, F–I) from biological triplicate samples are represented as mean ± SEM. (E) Data (n = 60) are represented as mean ± SEM. Figures show representative experiments of two independent ones. *P < 0.05; **P < 0.01; ***P < 0.001; # P < 0.05; ### P < 0.001, Student's t‐test. *, Lpin2‐deficient vs. control cells; #, in (D), poly(I:C)‐treated TLR3‐deficient vs. poly(I:C)‐treated siControl cells; #, in (G), cells treated with MAPKs inhibitors vs. just poly(I:C) treated cells.
Source data are available online for this figure.
Lipin‐2 controls NLRP3 inflammasome activation induced by transfected poly(I:C)
To investigate whether viral nucleic acid mimetics can provide the second signal for inflammasome activation, we transfected primed macrophages with poly(I:C), to make it accessible to intracellular receptors such as the inflammasomes, and studied IL‐1β production. The results showed that, when transfected with poly(I:C), primary human macrophages, BMDMs, and RAW264.7 macrophage‐like cells all increased IL‐1β levels. Importantly, the absence of lipin‐2 further enhanced the response (Fig 6A–C). IL‐1β production was dependent on the dose of transfected poly(I:C) (Fig 6D).
Figure 6. NLRP3 inflammasome activation by cytosolic poly(I:C) is regulated by lipin‐2.
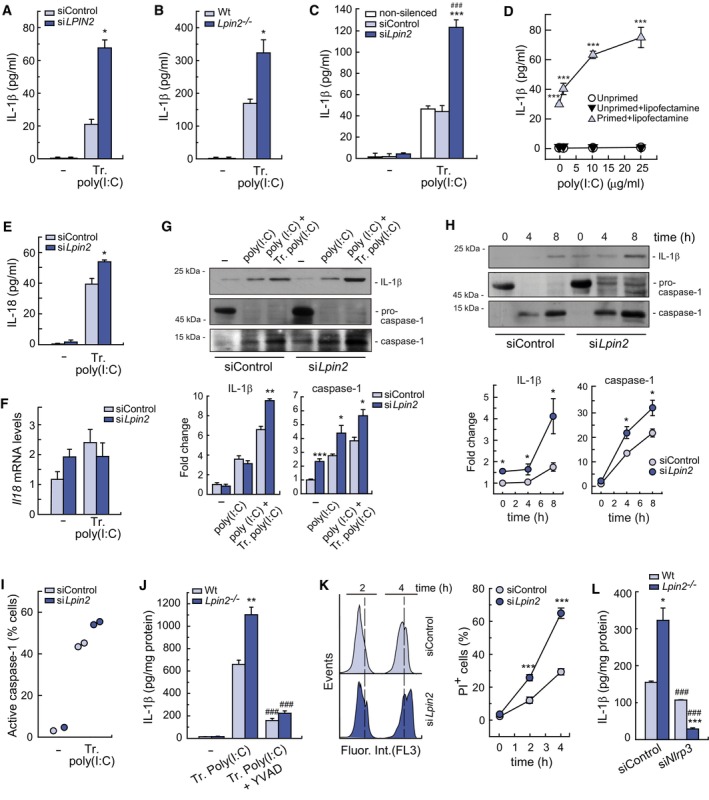
-
A–CSilenced primary human macrophages (A), Wt and Lpin2 −/− BMDMs (B) or non‐silenced/silenced RAW264.7 cells (C) were primed with 10 μg/ml poly(I:C) for 4 h and then transfected (Tr. poly(I:C) or not (−) with 10 μg/ml of poly(I:C) for 4 h. IL‐1β present in cellular supernatants were quantified by specific ELISAs.
-
DRAW264.7 cells were primed or not and then treated with different doses of poly(I:C) in the presence or absence of lipofectamine for 4 h. IL‐1β present in cellular supernatants was quantified using a specific ELISA.
-
E, FRAW264.7 cells silenced for Lpin2 or not were transfected with 10 μg/ml of poly(I:C). IL‐18 present in cellular supernatants was quantified using a specific ELISA (E), and Il18 mRNA levels were quantified by qPCR, using Gapdh as reference (F).
-
G, HBMDMs from Wt or Lpin2 −/− animals were left untreated (−), primed with 10 μg/ml of poly(I:C) for 4 h (poly(I:C) and then transfected with 10 μg/ml of poly(I:C) (poly(I:C) + Tr. poly(I:C)) for 4 h (G), or the indicated periods of time (H). IL‐1β and mature caspase‐1 present in cellular supernatants from the same number of cells were analyzed by immunoblot. Densitometric quantification of bands is represented in lower panels.
-
IRAW264.7 cells were treated as in (C). Cells were stained with FAM‐FLICA during the last 60 min of activation and fluorescence was analyzed by flow cytometry. Data are shown as the percentage of cells with active caspase‐1 (FAM‐FLICA positive cells).
-
JBMDMs were pretreated or not with 10 μM YVAD for 30 min and then treated as in (B). IL‐1β present in cellular supernatants was quantified with a specific ELISA.
-
KPrimed RAW264.7 cells were transfected with 10 μg/ml of poly(I:C) for the indicated periods of time and assayed for PI permeability by flow cytometry. Right panel shows percentage of PI+ cells.
-
LBMDMs were silenced or not for NLRP3, primed and transfected with 10 μg/ml of poly(I:C) for 4 h. IL‐1β present in cellular supernatants were quantified by specific ELISAs.
Data information: Data shown in (A–H, J–L) are from biological triplicate samples and are shown as means ± SEM. Two independent experiments were performed. Data in I are from duplicate samples. *P < 0.05; **P < 0.01; ***P < 0.001, ### P < 0.001, Student's t‐test. For figures (A–C, E, and G–L): *, lipin‐2‐deficient vs. control cells. For figure (D): *, Cells primed+lipofectamine vs. unprimed. For (C): #, lipin2‐deficient vs. unsilenced cells. For (J): #, Transfected poly(I:C) + YVAD vs. Transfected poly(I:C). For (L): #, siNLRP3 vs. siControl treated cells.
Source data are available online for this figure.
Il18 mRNA and pro‐IL‐18 levels are usually constitutive in cells (Gu et al, 1997). Conversion of pro‐IL‐18 to its mature form has been used as a reliable marker of inflammasome activation (Lordén et al, 2017). Analysis of IL‐18 levels in poly(I:C)‐transfected macrophages showed an increase in cells deficient in lipin‐2 (Fig 6E). We did not observe significant changes in Il18 mRNA levels under these circumstances (Fig 6F). Immunoblot analyses of the supernatants of these cells confirmed the increase of IL‐1β protein levels, especially in lipin‐2‐deficient cells (Fig 6G and H). Importantly, fragments of active caspase‐1 were also present at elevated amounts in the supernatants of lipin‐2‐deficient cells, and appeared earlier than IL‐1β (Fig 6G and H). Use of a fluorescent probe that specifically binds to active caspase‐1 indicated that transfection of poly(I:C) promoted the activation of the protease, with higher levels being found in cells deficient in lipin‐2 (Fig 6I). Further, inhibition of caspase‐1 activity by the specific inhibitor Y‐VAD blunted the production of IL‐1β by transfected poly(I:C), and this occurred both in wild type and Lpin2 −/− BMDMs (Fig 6J).
Caspase‐1 is involved in a type of cell death called pyroptosis. Thus, macrophage cell death was evaluated in cells that had been first primed and then transfected with poly(I:C). For these experiments we took advantage of the property of propidium iodide to permeate damaged cell membranes. Flow cytometry data showed that lipin‐2‐deficient cells experienced higher levels of death than cells normally expressing the enzyme (Fig 6K). Finally, elimination of the inflammasome receptor NLRP3 by mRNA silencing reduced the capacity of BMDMs to release IL‐1β, especially in cells from Lpin2 −/− mice (Fig 6L). Together, these data demonstrate that lipin‐2 restrains NLRP3 inflammasome activation during recognition of cytosolic viral nucleic acid mimics.
Lipin‐2 regulates mtDNA release during cytosolic poly(I:C) recognition
The presence of cytosolic mtDNA has been suggested as a key event for the activation of the NLRP3 inflammasome (Zhou et al, 2011; Zhong et al, 2018). To unveil possible mechanisms for the increased activation of the NLRP3 inflammasome by transfected poly(I:C), especially under reduced lipin‐2 levels, we focused on studying whether cytosolic mtDNA is involved in the production of IL‐1β in this model. Primed BMDMs, transfected with poly(I:C), were used to study cytosolic mtDNA levels in cytosolic fractions by analyzing the presence of the mtDNA‐encoded gene Dloop (Fig 7A). Transfection of poly(I:C) increased the presence of Dloop in the cytosol of BMDMs, and this was augmented in cells from Lpin2 −/− animals. To verify whether mtDNA was related to IL‐1β production, we eliminated mtDNA by treating immortalized wild type and Lpin2 −/− BMDMs with ethidium bromide (ρ0 phenotype) (King & Attardi, 1989). IL‐1β synthesis was then evaluated in primed macrophages transfected with poly(I:C). We found that elimination of mtDNA significantly reduced IL‐1β production, and such reduction was more pronounced in cells deficient in lipin‐2 (Fig 7B). In agreement with these data, we also observed that loss of mtDNA decreased the activation of caspase‐1 by transfected polyI:C in both wild type and lipin‐2‐deficient cells, as analyzed by flow cytometry (Fig 7C). As a consequence, cell death was also decreased in ρ0 cells from both phenotypes (Fig 7D).
Figure 7. Lipin‐2 controls inflammasome activation by reducing mtDNA release and ROS production.
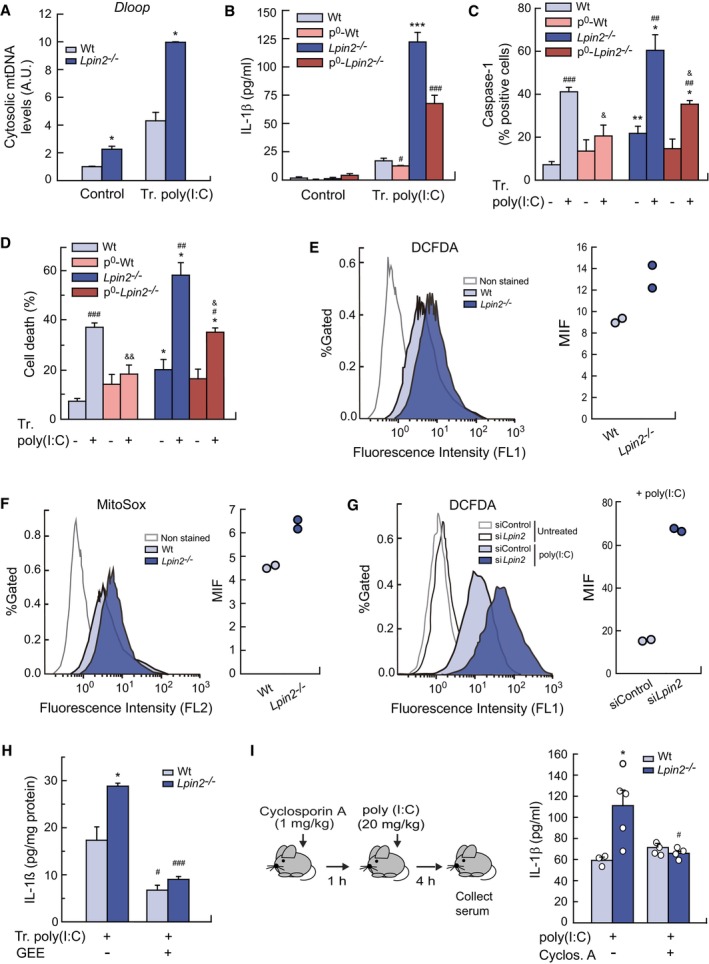
-
APrimed BMDMs from Wt and Lpin2 −/− animals were transfected or not (Control) with 10 μg/ml poly(I:C) for 4 h. Cytosol mtDNA levels were analyzed by qPCR as mentioned in M&M.
-
B–DBMDMs from Wt and Lpin2 −/− animals depleted or not of mtDNA (p0) were primed and transfected with 10 μg/ml poly(I:C) for 4 h. IL‐1β present in cellular supernatants were quantified by specific ELISAs (B). Active caspase‐1 and cell death were analyzed by flow cytometry after cell treatment with FAM‐FLICA (C) or PI (D).
-
E, FPrimed BMDMs were transfected with 10 μg/ml poly(I:C) for 4 h and labeled with 10 μM DCFA (E) or 5 μM MitoSOX (F), and fluorescence was analyzed by flow cytometry. Right panels show the mean fluorescence intensities (MIF) observed in left panels.
-
GRAW264.7 cells were silenced for Lpin2, treated with 10 μg/ml of poly(I:C) for 4 h, labeled with 10 μM DCFA, and analyzed by flow cytometry. Right panel shows the MIF observed in left panels.
-
HPrimed BMDMs from Wt and Lpin2 −/− animals were transfected or not (Control) with 10 μg/ml poly(I:C) for 4 h. During the activation, macrophages were treated or not with 5 mM GEE. IL‐1β present in cellular supernatants was quantified with a specific ELISA.
-
IMice were treated with 1 mg/kg cyclosporine A for 1 h and then injected with 20 mg/kg of poly(I:C). Four hours later, serum was collected and IL‐1β levels were analyzed with a specific ELISA.
Data information: Data shown (A–D, H) are from biological triplicate samples and are shown as mean ± SEM. Two independent experiments were performed. Data in (E–G) are from duplicate biological samples. (I) Data shown are from three Wt animals, and five Lpin2 −/− animals treated with poly(I:C) and 4 Wt and Lpin2 −/− animals treated with Cyclosporin A and poly(I:C). Data represent the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; # P < 0.05; ## P < 0.01; ### P < 0.001; & P < 0.05; && P < 0.01, Student's t‐test. *, Lpin2‐deficient vs. control cells/animals; #, Tr. poly(I:C) treated cells/animals vs. control cells/animals of the same phenotype; &, p0 vs. cells of the same phenotype and activation.
Source data are available online for this figure.
During NLRP3 inflammasome activation by mtDNA, oxidative processes appear to play an important role (Zhong et al, 2018). By using specific fluorescent probes, we measured total ROS production in cells and also specifically in mitochondria. We found that lipin‐2‐deficient cells transfected with poly(I:C) produced significantly more ROS, both total and mitochondrial, than cells expressing normal lipin‐2 levels (Fig 7E and F). The same effect in cellular ROS production was observed upon extracellular poly(I:C) treatment (priming signal for inflammasome activation through TLR3) (Fig 7G). Interestingly, treatment of macrophages with the antioxidant glutathione ethyl ester (GEE) strongly blunted the production of IL‐1β, an effect that was more marked in BMDMs from Lpin2 −/− animals (Fig 7H).
One of the mechanisms that have been described for mtDNA exit from the mitochondria is through the opening of the mitochondrial permeability transition pore, which occurs during excessive ROS production (Rottenberg & Hoek, 2017; Bahat et al, 2021). Pore opening can be inhibited by cyclosporin A, a drug that has also been shown to destabilize mitochondria and reduce IL‐1β production by a number or stimuli (Iyer et al, 2013). To evaluate whether the findings reported above could be relevant to animal models of inflammation, we analyzed IL‐1β levels in the serum of animals treated with poly(I:C), and analyzed the effect of cyclosporin A pretreatment as well. We found that cyclosporin A severely curtailed IL‐1β production in lipin‐2‐deficient animals, while having no discernible effect on wild type animals (Fig 7I).
Overall, these results suggest that lipin‐2 restrains both mtDNA release and oxidative events that play a role during inflammasome activation by viral nucleic acid mimics.
Lipin‐2 controls IL‐1β during cytomegalovirus infection
To corroborate our findings with poly(I:C) regarding IL‐1β production, experiments were also conducted with MCMV‐infected macrophages. We found that the absence of lipin‐2 increased IL‐1β levels in the supernatants of MCMV‐infected BMDMs (Fig EV1A). Accordingly, virus‐infected Lpin2 −/− animals had higher concentrations of the cytokine in serum than control ones (Fig EV1B). The same behavior was also found in HCMV‐infected human macrophages (Fig EV1C). These results confirm that lipin‐2 restricts inflammasome activation during viral encounters.
Figure EV1. Lipin‐2 effects on IL‐1β production during cytomegalovirus infection.
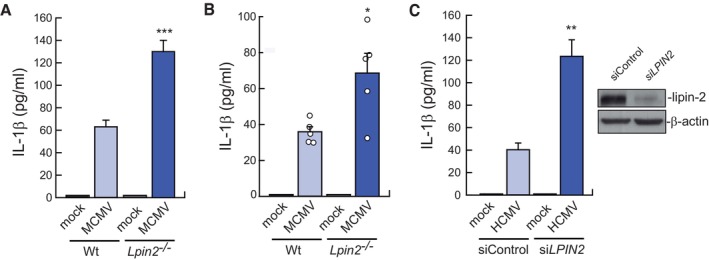
- LPS‐primed BMDMs from Wt or Lpin2 −/− animals were mock infected or infected with MCMV (MOI = 2). IL‐1β levels were measured in cell supernatants using a specific ELISA after 36 h of infection.
- Wt (n = 5) and Lpin2 −/− (n = 5) animals were intraperitoneally injected with MCMV (3.5 × 106 PFU per mouse) and IL‐1β levels were evaluated in serum using specific ELISAs after 36 h of infection.
- LPS‐primed primary human macrophages, nucleofected with control siRNAs (siControl) or against LPIN2 (siLPIN2), were mock infected or infected with HCMV (MOI = 5) for 36 h. IL‐1β levels in cell supernatants were measured in cell supernatants using a specific ELISA. Right insert: lipin‐2 expression in silenced cells by immunoblot.
Data information: Data shown are from biological triplicate samples and are expressed as means ± SEM. *P < 0.05; **P < 0.01, ***P < 0.001, by Student's t‐test.
LPIN2 expression decreases in symptomatic COVID‐19 patients and correlates with increases in damaging factors
Late in 2019, a new coronavirus, SARS‐CoV‐2, was identified as the cause of a severe acute respiratory syndrome known as COVID‐19 (Chen et al, 2020; Zhou et al, 2020). Although the majority of infected people are asymptomatic or develop mild symptoms, a significant number of patients develop a severe disease. As a consequence, around 1% of the patients die and some suffer from long‐standing sequelae.
Severity of COVID‐19 has been associated with a dysfunctional immune response characterized by low or delayed IFN responses and exaggerated production of proinflammatory cytokines (Hadjadj et al, 2020). An interesting feature of SARS‐CoV‐2 infection is that the virus modulates the immune response in a way that suppresses the IFN‐I system but induces and sustains high levels of chemokine mRNAs, resulting in an imbalanced host response (Konno et al, 2020; Lei et al, 2020; Miorin et al, 2020; Thoms et al, 2020; Xia et al, 2020). Indeed, it has been demonstrated that upregulation of IFN responses in COVID‐19 asymptomatic patients correlates with a better control of the illness (Masood et al, 2021a).
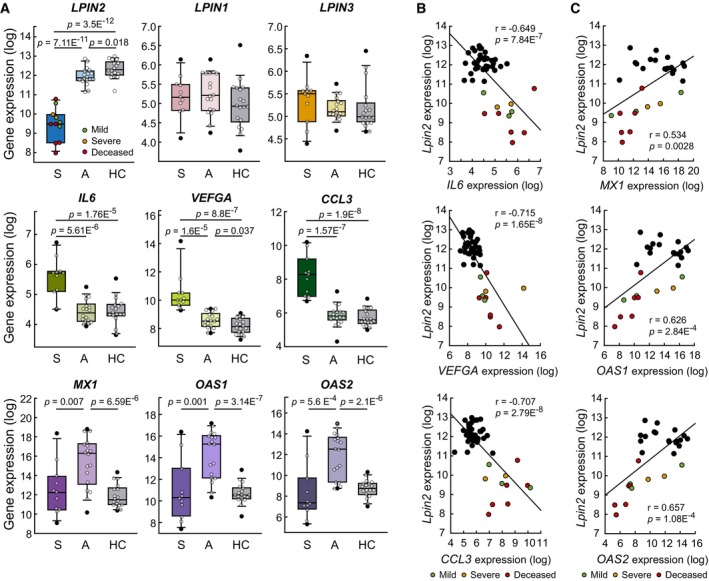
Based on the findings described above showing that lipin‐2 levels are regulated by IFN and that the expression levels of lipin‐2 inversely modulate the proinflammatory capacity of macrophages, we sought to analyze whether LPIN2 levels were altered in COVID‐19 patients. We analyzed public available databases of gene expression arrays from blood samples of healthy control subjects, asymptomatic and symptomatic patients (Masood et al, 2021a; Data ref: Masood et al, 2021b). The results are shown in Fig 8 and indicate that LPIN2 levels were significantly decreased in symptomatic compared with asymptomatic individuals or healthy control subjects (Fig 8A). Interestingly, patients with lower expression of LPIN2 experienced severe symptoms and were actually not able to recover from the illness (Fig 8A). The rest of the genes of the LPIN family, LPIN1 and LPIN3 showed no changes in their expression levels. As previously described, samples from symptomatic patients showed an increased expression of the proinflammatory genes IL6, VEFGA and CCL3, related with the cytokine storm (Chen et al, 2020), the endothelial dysfunction (Rovas et al, 2021), and the increased recruitment of innate immune cells (Chi et al, 2020) that characterize patients with severe COVID‐19 (Masood et al, 2021a). Interestingly, LPIN2 expression levels negatively correlated with the expression of these genes (Fig 8B). However, canonical ISGs such as MX1, OAS1, and OAS2 showed higher expression levels in asymptomatic patients than in symptomatic ones, as shown for LPIN2 (Fig 8A and C). Collectively, these results are highly suggestive of a correlation between LPIN2 expression levels and IFN responses and the capacity to overcome the disease in COVID‐19 patients.
Figure 8. LPIN2 expression decreases in symptomatic COVID‐19 patients.
-
A–CGene expression analyses were performed using the GSE177477 database. (A) Box plots showing the indicated gene expression analysis in samples from symptomatic (S, n = 11) and asymptomatic (A, n = 18) patients with COVID‐19, and healthy controls (HC, n = 18). The central band is the median, boxes define 25th to 75th percentiles and whiskers 10th and 90th percentiles. P as shown by Student's t‐test. (B–C) Scatter plots showing the correlation between LPIN2 and proinflammatory/damage‐related gene expression levels (n = 47, B), or between LPIN2 and canonical ISGs expression levels (COVID‐19 samples, n = 29, C). Pearson's coefficient tests were performed to assess statistical significance.
Source data are available online for this figure.
Discussion
After viral interference factors were discovered in the late 50's (Isaacs & Lindenmann, 1957), research on IFNs has demonstrated that they exert a myriad of effects on pathogen elimination, immunity and metabolism. To exert their actions, IFN receptors activate a complex array of transcription factors that participate in the regulated expression of many different genes. The identification and description of the roles of the proteins encoded by those genes is crucial to better define how IFNs impact on the aforementioned processes. Here we unveil the regulated expression of Lpin2 by IFNs, and the involvement of this phosphatase in a number of key immunoinflammatory events. These include the control of viral infections, inflammasome activation by viral‐derived PAMPs, and the negative correlation between LPIN2 levels and severity of COVID‐19.
Macrophages possess the ability to differentially reprogram their lipidome in response to TLR stimulation (Hsieh et al, 2020). In BMDMs, TLRs are able to modify the cellular levels of a number of lipid families, including phospholipids, DAG and TAG. Deletion of the IFN receptor ifnar results in reduced production of TAG (Hsieh et al, 2020). Together with our present results, these studies suggest a scenario whereby the capacity of certain TLRs to reprogram the lipidome in macrophages to generate TAG may depend on their capacity to generate IFN, and that lipin‐2, a lipid phosphatase that participates in TAG synthesis, may act as master regulator of the pathway. Of note, the other members of the lipin family, lipin‐1 and lipin‐3 are down regulated or very modestly regulated, respectively, during TLR or IFN stimulation of macrophages.
The finding that lipin‐2 acts as an antiviral effector for MCMV infection is in agreement with its regulation by IFN, both being part of the mechanisms that the cytokine puts into motion to fight the viral infection. Several metabolism‐related effects found during human cytomegalovirus infection could help to explain the antiviral role of lipin‐2. In the first place, factors such as Rsad2/viperin, an ISG that is used as part of the IFN‐induced antiviral response to inhibit the replication of many viruses, may be used by cytomegaloviruses in their own benefit to increase viral infectivity (Seo et al, 2011; Dumbrepatil et al, 2020). The mechanism appears to be related with the relocalization of viperin from the endoplasmic reticulum to mitochondria, where it inhibits β‐oxidation and ATP production. This will result in disruption of the actin cytoskeleton, and also with the accumulation of lipids that the virus needs to form the viral envelope (Seo et al, 2011; Dumbrepatil et al, 2020). Interestingly, we show here that lipin‐2 reduces Rsad2/viperin expression in macrophages during viral‐derived PAMP activation, thus suggesting a possible metabolic mechanism to restrict cytomegalovirus replication. Second, it is well known that IFN reduces cholesterol synthesis to control viral infection (Blanc et al, 2011). However, it has also been described that human cytomegalovirus promotes cholesterol efflux from infected cells through viral proteins, thus helping the virus escape immune control (Low et al, 2016). In this regard, we have previously described that lipin‐2 impacts on macrophage cholesterol levels in such a way that, in the absence of lipin‐2, the levels of cholesterol are reduced in macrophages (Lordén et al, 2017). Thus, during an infection with cytomegalovirus, the effect that lipin‐2 exerts on cholesterol levels could be disadvantageous for cytomegalovirus propagation and immune evasion.
Majeed syndrome patients that carry mutations in the LPIN2 gene develop very early in their lives symptoms such as recurrent episodes of fever and inflammation in bones and skin (Majeed et al, 1989; Ferguson et al, 2005). Macrophages defective in LPIN2 have an enhanced predisposition to activate the inflammasome NLRP3 in a classical way (Lordén et al, 2017). Interestingly, we have now discovered that viral‐associated molecular patterns exhibit the capacity to both increase signaling during the priming step of inflammasome activation and promote an increased inflammasome activation when recognized intracellularly by macrophages. These findings may be important to explain, at least in part, why Majeed syndrome patients suffer from recurrent episodes of fever without obvious infection. It seems likely that low‐load viral infections, which would not generate symptoms in healthy individuals, could induce the production of the pyrogen IL‐1β, reaching levels high enough to generate peaks of fever in Majeed patients (Dinarello et al, 1986). Also, the exaggerated production of IL‐1β by myeloid cells from these patients may influence the polarization of anti‐inflammatory/regulatory macrophages (M2), to generate M2 cells that express proinflammatory cytokines such as IL‐6 or TNFα, and display increased osteoclastogenic capacity (Bhuyan et al, 2021). The increased levels of IL‐1β promoted by low levels of viral infections could participate in the development of proinflammatory M2 macrophages. This would provide an explanation to the flares of osteomyelitis that these patients also develop.
Our results related with the mechanism through which intracellular viral nucleic acid mimics increase inflammasome activation in lipin‐2‐deficient macrophages, prompt us to consider that, similar to what we have observed in lipin‐2‐deficient mice, treatment of patients with inhibitors of mtDNA release may help reduce inflammasome activation during viral encounters. This would lower IL‐1β synthesis and ameliorate symptoms in these patients. One of these inhibitors, cyclosporin A, has been used in the clinic as an immunosuppressant during organ transplantation for decades. Due to the recent confirmation of the importance of the mitochondrial permeability transition pore in the development of many diseases, inhibitors such as cyclosporine A have now been proposed as possible treatment options for these conditions, including viral infections (Winquist & Gribkoff, 2020; Han et al, 2022).
Despite the detrimental actions of an excessive and uncontrolled Il‐1β production, the importance of inflammasomes and IL‐1β in antimicrobial defenses is demonstrated by the increased susceptibility to infections exhibited by mice and humans defective in IL‐1β receptor/inflammasomes (Man et al, 2017). It is manifested as well by the capacity of viruses and other pathogens to dampen the production and actions of the cytokine (Biolatti et al, 2018). Thus, it is tempting to speculate that a consequence of the enhanced inflammasome activation in Majeed syndrome patients would also include a more effective elimination of some viruses that are very sensitive to inflammasome activation. However, we also show in this work that viruses that developed robust mechanisms to evade innate immune responses such as cytomegaloviruses (Biolatti et al, 2018), are able to replicate better in the absence of lipin‐2. Infection of Majeed syndrome patients with this type of viruses would predictably be more severe. In this regard, the conclusions obtained from samples from SARS‐CoV‐2‐infected patients point to the idea that patients with LPIN2 mutations and/or reduced LPIN2 expression levels would be at risk for severe COVID‐19. Overall, it may seem that, depending on the type of virus, Majeed syndrome patients would be more protected, or more vulnerable to infection. Either case, they would presumably display an exacerbated inflammasome activation and IL‐1β production during the encounters.
In the current COVID‐19 pandemic only a minor portion of the patients develop a severe disease and eventually die. Most of the population is believed to be able to mount an early and strong antiviral, IFN‐dependent response that reduces viral replication and spreading from the upper airways to the lungs and other tissues (Blanco‐Melo et al, 2020). If initial IFN responses are defective, SARS‐CoV‐2 replicates without control, spread to the abovementioned organs, and inflammation exacerbates due to an exaggerated immune cell recruitment (Zhang et al, 2020a). These antiviral actions occur in spite of the SARS‐CoV‐2 capacity to decrease IFN responses (Xu et al, 2021). Thus it is important to identify in the very early steps of infection those patients who could have difficulties to mount an adequate antiviral response. We have identified that LPIN2 expression levels in blood samples from Covid‐19 patients provide a clear‐cut difference between symptomatic from asymptomatic patients. LPIN2 levels appear to perform better in this regard than canonical ISGs like MX1, OAS1 or OAS2. Whether the low levels of LPIN2 in symptomatic Covid‐19 patients are a cause or a consequence of the disease, cannot be told at this stage. We speculate that patients with constitutive low LPIN2 expression levels could be at risk of experiencing a more severe disease due to the increased inflammatory response. This idea receives support from the fact that expression levels of inflammatory and damage‐related factors such as IL6, VEFGA and CCl3 inversely correlate with LPIN2 expression. However, based on our observations that LPIN2 expression is regulated by IFNs, it is also possible that LPIN2 levels in COVID‐19 patients reflect their IFN responses.
Whatever the cause for the reduced levels of LPIN2 in symptomatic patients, we are tempted to speculate that early analysis of LPIN2 expression in COVID‐19 patients could help in the stratification of those patients. Those with very low LPIN2 levels would possibly develop a severe illness; hence they could benefit from early treatment with exogenous IFN, more specifically, IFN‐β (Vinh et al, 2021; Sodeifian et al, 2022). This treatment has proven to be efficacious in critical COVID‐19 patients with inborn errors for IFN or that carry autoantibodies against IFN (Bastard et al, 2020; Zhang et al, 2020b, 2022; Aricò et al, 2022). Clearly, future studies should validate LPIN2 as an accurate biomarker of IFN action and/or COVID‐19 severity, and its use for the design of personalized therapies.
Materials and Methods
Reagents
LPS (Escherichia coli 0111: B4), and poly(I:C) (P0913) were from Sigma‐Aldrich. N‐palmitoyl‐S‐[2,3‐bis(palmitoyloxy)‐(2R,2S)‐propyl]‐Cys‐[S]‐Ser‐[S]‐Lys(4) trihydrochloride (Pam3SCK4), lipoteichoic acid from Staphylococcus aureus (LTA), S‐(2,3‐bispalmitoyloxypropyl)‐Cys‐Gly‐Asp‐ProLys‐His‐Pro‐Ser‐Phe (FSL‐1), imiquimod, and synthetic oligodeoxynucleotide 1826 (5‐TCCATGACGTTCCTGACGTT‐3; ODN1826) were purchased from InvivoGen. Murine IFN‐β (12405‐1) was from Pbl Assay Science, and murine IFN‐γ (12343534) was from Immunotools. Actinomycin D (294940010), cycloheximide (AC357420010), and cyclosporine A (457970010) were purchased from Thermo Scientific. The antibody against lipin‐2 was obtained from Bethyl Laboratories (A303‐703A) (Montgomery, TX, USA). Antibodies against phospho‐p38 MAPK (Thr180/Tyr182) (4511), phospho‐p44/42 MAPK (p‐ERK) (Thr202/Tyr204) (9101), phospho‐SAPK/JNK (Thr183/Tyr185) (4668), p38 MAPK (9212), p42‐p44 MAPK (ERK) (4695), SAPK/JNK (9252), NF‐KB p65 (8242) and IL‐1β (12242) were from Cell Signaling. Antibodies against active caspase‐1 (sc‐515) were from Santa Cruz. Nigericine, Ac‐YVAD‐AOM, and SB203582 were from Calbiochem, Ruxolitinib was from LC Laboratories. Specific kits for the measurement of IL‐1β and IL‐18 levels were from Invivogen (#88‐7013‐88) and Sino Biologicals (#E‐EL‐M0033), respectively. All other reagents were purchased from Sigma‐Aldrich.
Animals
Mice, including Lpin2 −/− animals, were maintained as previously described (Lordén et al, 2017). 8–12‐week‐old animals were used for experimentation in sex‐matched groups. Wild type littermates were used as control animals (heterozygous animals were used for breeding). All procedures involving animals were carried out under the supervision of the Institutional Committee of Animal Care and Usage of the University of Valladolid (Approval No. 7406000), and are in accordance with the guidelines established by the Spanish Ministry of Agriculture, Food, and Environment, and the European Union.
Cells
Bone marrow‐derived macrophages (BMDMs), primary human macrophages, and RAW264.7 cells were obtained, cultured, maintained and silenced as previously described (Valdearcos et al, 2012; Rubio et al, 2015; Lordén et al, 2017). BMDMs were differentiated from precursors obtained from mice femurs by culturing for 7 days in RPMI 1640 supplemented with 10% FBS, 20% conditioned media from L929 fibroblast, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Human macrophages were differentiated from human blood monocytes present in buffy coats of healthy anonymous volunteer donors obtained from the Centro de Hemoterapia y Hemodonación de Castilla y León (Valladolid, Spain), as previously described (Rubio et al, 2015; Lordén et al, 2017). Blood cells were diluted with PBS (1:1), layered over a cushion of Ficoll‐Paque and centrifuged at 750 g for 30 min. Mononuclear cells from the interphase were collected, washed with PBS, resuspended in RPMI 1640 free of serum and allowed to adhere to sterile plastic dishes for 2 h at 37°C. Non‐adherent cells were extensively washed out. Macrophage differentiation was achieved by incubating adherent monocytes in RPMI with 5% of human serum (which contains high quantities of M‐CSF) in the absence of other cytokine sources for 10–14 days. Culture medium was replaced every 3 days.
RAW 264.7 macrophages (ATTC, TIB‐71™) were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine. MEFs derived from BALB/c mice were cultivated in DMEM supplemented 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin, and 2 mM glutamine.
All cells were maintained under mycoplasma‐free conditions. For inflammasome activation analyses, cells were primed with 10 ng/ml of poly(I:C) for 4 h. Afterward, they were transfected with 10 ng/ml poly(I:C) using 0.8 μl Lipofectamine (Thermo Fisher Scientific) per mg poly(I:C) for 4 h.
Gene silencing
Gene expression silencing was performed in murine cells using specific ON‐Target plus siRNAs obtained from Dharmacon (Thermo Scientific). Control siRNAs were ON‐Target plus Non‐targeting Control Pool siRNAs (D‐00810‐10). BMDMs and RAW 264.7 cells were silenced by introducing complexes of siRNAs (20 nM final concentration in culture) with Lipofectamine RNAiMAX (Thermo Fisher Scientific) as specified by the manufacturer (Pindado et al, 2007; Ruipérez et al, 2009). The cells were used after 2 days of transfection.
For LPIN2 silencing in human cells Silencer® Select siRNAs were obtained from Ambion (sense sequence, 5′‐GAA GUU GGG UGA UAA CGG ATT‐3′, and antisense sequence 5′‐UCC GUU AUC ACC CAA CUU CAT‐3′). Control siRNAs used were Ambion™ Silencer™ Select Negative Control No.2 siRNAs (4390847). siRNAs were introduced after 10–14‐days differentiation by Nucleofection (Amaxa Biosystems) following the manufacturer's instructions (Y‐010 program), at 20–40 nM siRNA final concentration in culture (Valdearcos et al, 2011). Macrophages were placed in culture and allowed to rest for 2 days. A 75–90% reduction of lipin‐2 levels was routinely achieved using this methodology.
Immunoblot
Cell homogenates were obtained by lysis with the buffer 20 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5 mM Na4P2O7, 50 mM β‐glycerophosphate, 270 mM sucrose, 0.1% 2‐mercaptoethanol, 1% Triton X‐100, 100 μM PMSF, 1 mM Na3VO4, 10 mM NaF and a protease inhibitor cocktail (P‐8340, Sigma). After centrifugation at 15,000 g for 10 min, 50–100 μg of cellular protein was separated by SDS–PAGE and transferred to PVDF membranes. Blocked membranes (5% defatted dry milk or 5% bovine serum albumin in PBS) were then treated with specific antibodies at 1:1,000 in PBS with 0.5% defatted dry milk and 0.1% Tween 20. The exception was anti‐ lipin‐2 (1:500) and β‐actin (1:20,000). Secondary antibodies HRP linked (GE Healthcare #NA931 and #NA9340) were used at 1:5,000 dilution. Bands were visualized using ECL chemiluminescent substrate (Amersham Biosciences).
For analysis of protein present in cellular supernatants, trichloracetic acid was added to supernatants at a final concentration of 10%, mixed vigorously and incubated at 4°C for 30 min. Samples were then centrifuged at 16,000 g 10 min at 4°C. Pellets were washed with cold acetone, dried at 63°C, resuspended in PBS and analyzed by immunoblot.
Quantitative PCR (qPCR)
RNA was obtained using the TRIzol reagent (Ambion) and cDNA was then produced using Verso cDNA Synthesis Kit (Thermo Fisher Scientific), following the manufacturer's instructions. qPCR was performed from 20 ng of cDNA using the Brilliant III Ultra‐Fast SYBR® Green QPCR Master Mix (Agilent Technologies), following the manufacturer's instructions. Relative mRNA expression was obtained using the ΔΔ C t method (Schmittgen & Livak, 2008). Quantitative real‐time RT–PCR analysis was performed in a LightCycler 480 (Roche) as previously described (Valdearcos et al, 2012; Meana et al, 2014). Genes of reference were Gapdh for murine cells, and ACTB for human cells. Primers used for murine genes were: Gapdh AGGTCGGTGTGAACGGATTTG and TGTAGACCATGTAGTTGAGGTCA; Lpin1, CTCCGCTCCCGAGAGAAAG and TCATGTGCAAATCCACGGACT; Lpin2, AGTTGACCCCATCACCGTAG and CCCAAAGCATCAGACTTGGT; Lpin3, TGGAATTGGGATGACAAGGT and CACTGCAAGTACCCCTTGGT; Il1b, GCAACTGTTCCTGAACTCAACT and TCTTTTGGGGTCCGTCAACT; Il18, CAAACCTTCCAAATCACTTCCT and TCCTTGAAGTTGACGCAAGA; Irf7, GAGACTGGCTATTGGGGGAG and GACCGAAATGCTTCCAGGG; Nlrp3, ATCAACAGGCGAGACCTCTG and GTCCTCCTGGCATACCATAG; Rsad2, TGCTGGCTGAGAATAGCATTAGG and GCTGAGTGCTGTTCCCATCT; Tlr3 GTGAGATACAACGTAGCTGACTG and TCCTGCATCCAAGATAGCAAGT; Ifih1 AGATCAACACCTGTGGTAACACC and CTCTAGGGCCTCCACGAACA; Eif2k2 ATGCACGGAGTAGCATTACG and TGACAATCCACCTTGTTTTCG T; Ddx58 AAGAGCCAGAGTGTCAGAATCT and AGCTCCAGTTGGTAATTTCTTGG. Primers used for human genes were: ACTB, ATTGCCGACAGGATGCAGAA and GCTGATCCACATCTGCTGGAA; LPIN2, CCTCTCCTCAGACCAGATCG and GGAGAATCTGTCCCAAAGCA; IL1B, ATGATGGCTTATTACAGTGGCAA and GTCGGAGATTCGTAGCTGGA.
Constructs
The plasmid mlipin‐2‐EGFP was previously described (Valdearcos et al, 2012). A plasmid expressing only lipin2 was generated by introducing of a stop codon between the lipin‐2 and EGFP sequences using the Q5 site‐directed mutagenesis Kit (New England Biolabs). The primers used were:5´‐aagtcgacggtaccgcgggcCCCGGGATCCATCG‐3´and 5′‐ aagccaggtcatccaggtcATTGGG TCTCGCCAG‐3′. Confirmation of the correct insertion of the cDNA was performed by sequencing. Plasmids were transfected using Lipofectamine™ LTX and PLUS™ reagents following the manufacturer's instructions.
Microarray expression data
To study the kinetics of mRNA expression levels of BMDMs treated with either LPS (0.5 ng/ml, 5 ng/ml, or 50 ng/ml), or IFN‐β or IFN‐γ (10 U/ml), data were obtained from GSE44292 and analyzed as previously described (Raza et al, 2014a; Data ref: Raza et al, 2014b).
Gene expression data of MCMV infected BMDMs was obtained from GSE42505 and analyzed as previously described (Blanc et al, 2013; Data ref: Blanc et al, 2015). Data to unravel the participation of STAT1 in gene expression induced by treatment of BMDMs (wild type and Stat1−/−) with 100 U/ml IFN‐γ for 6 h were obtained from GSE48970 and analyzed as previously described (Semper et al, 2014a; Data ref: Semper et al, 2014b).
Newly transcribed mRNA
Newly transcribed Lpin2 mRNA data were obtained from GSE63290. Experimental design and data analysis was described elsewhere (Dölken et al, 2008; Robertson et al, 2016; Data ref: Robertson & Ghazal, 2016). Briefly, BMDMs were left untreated or treated with 10 U/ml IFN‐γ. Newly transcribed RNA was then labeled with 200 μM 4‐thiouridine every 30 min for 8 h. For analysis, newly transcribed RNA samples and total RNA samples were hybridized to Affymetrix GeneST v1.0 arrays.
Chip‐seq analysis of STAT1 binding to Lpin2 locus
Data from Chip‐Seq experimentation using BMDMs stimulated with IFN‐β or IFN‐γ for 6 h were obtained from GSE33913. The experimental details were described elsewhere (Ng et al, 2011a; Data ref: Ng et al, 2011b). Briefly, chromatin from stimulated cells was cross‐linked and immunoprecipitated using antibodies against STAT1 (Santa Cruz). After sequencing, STAT1 peaks were defined using Model‐based analysis of ChIP‐Seq (MACS) (Zhang et al, 2008). Reads were aligned to the mouse genome and normalized to reads per kilobase per million reads sequenced (RPKM) (Ng et al, 2011a; Data ref: Ng et al, 2011b).
In vitro viral infection
For in vitro infection experiments, the MCMV‐GFP recombinant virus, a derivative of the BAC MCMV MW97.01 based on the MCMV strain (ATCC VR‐1399) that carries the GFP gene inserted in the ie2 locus, was used (Mathys et al, 2003). Macrophages were infected with MCMV‐GFP at 2–4 multiplicity of infection (MOI) during 2 h, including a centrifugal enhancement of infectivity step (Hudson, 1988). Cells were then washed with PBS and fresh culture medium was added. GFP fluorescence of the cell cultures was analyzed after infection using a Fluoroskan Ascent FL Fluorimeter (Thermo Electron Corporation) employing a wavelength of 475 nm for fluorescence excitation and 510 nm for fluorescence emission. For experiments with human macrophages, the HCMV strain TB40/E was used (Sinzger et al, 2008).
To quantify infectious virus, supernatants from infected macrophages were harvested, clarified, and transferred to MEFs immortalized by p53 to determine by standard plaque assay titrations viral plaque‐forming units (PFU) or fluorescence‐forming units (FFU) using a fluorescence microscope.
IL‐1β production was measured using BMDMs or human macrophages primed with 200 ng/ml of LPS before MCMV (MOI = 2) or HCMV infection (MOI = 5) respectively. Cell supernatants were obtained 36 h after infection, and analyzed for IL‐1β content by ELISA.
In vivo viral infection model
For in vivo viral infections, the BAC‐derived MCMV MW97.01, referred here as MCMV, was used (Wagner et al, 1999). Mice were intraperitoneally infected with 1 ml PBS containing 3.5 × 106 PFU of MCMV. After 5 days of infection, mice were anesthetized with 100 mg/kg ketamine (Merial) and 5 mg/kg xylazine (Bayer). Mice were sacrificed by cervical dislocation to harvest the organs of interest. Livers and spleens were homogenized in DMEM 3%FBS (10% weight/volume), sonicated, centrifuged and viral titers from the supernatants were determined by standard plaque assays, including a centrifugal enhancement step, following the same protocol mentioned above. For IL‐1β quantification, mice were infected for 36 h. Afterward, serum was collected and IL‐1β was analyzed by ELISA.
In vivo inflammasome activation model
Mice were intraperitoneally treated with 1 mg/kg cyclosporin A for 1 h to inhibit the mitochondrial permeability transition pore, and then injected with 20 mg/kg poly(I:C) to induce inflammasome activation. After 4 h of treatment, serum was collected as described above. Serum concentration of IL‐1β was analyzed by ELISA.
Immunostainings
Cells were seeded in glass coverslips, stimulated and washed before they were fixated with 4% PFA and 3% sucrose 10 min at room temperature. Cells were permeabilized with 0.1% Triton X‐100 for 2 min, washed three times with PBS, and incubated in 0.5% BSA, 50 mM glycine, and 4% goat serum in PBS for 30 min at room temperature. Cells were then washed three times and incubated with an anti‐p65 antibody (1:200) (Cell Signaling). diluted in 0.1% BSA (w/v) in PBS for 1 h at room temperature. After washing, cells were incubated with an Alexa Fluor®488‐goat anti rabbit (1:2,000) diluted in 0.1% BSA (w/v) in PBS for 1 h at room temperature, protected from light. Finally, nuclei were stained with 1 μg/ml DAPI in PBS for 5 min at room temperature, and washed with milli‐Q H2O. Coverslips were mounted onto slides and fluorescence was examined using an oil immersion, 63×, 1.4 NA, HCX PL APO CS objective in a Leica TCS SP5X confocal microscope. DAPI was excited using an UV laser line at 405 nm and fluorescence emission was collected between 441 and 470 nm. AF®488 was excited using a supercontinuum visible laser, at 488 nm and fluorescence emission was collected between 499 and 545 nm. To obtain the percentage of cells with p65 in the nucleus, all the nuclei in the image (DAPI) and the nuclei with green staining (p65) were counted using the Cell Counter plugin of ImageJ.
Flow cytometry
Active caspase‐1 was assayed using the probe FAM‐FLICA (FAM‐YVAD‐FMK, Immunochemistry Technologies), a fluorescent inhibitor that binds and labels active caspase‐1. The manufacturer's instructions were followed. Briefly, the cells were labeled with FAM‐FLICA during 1 h at 37°C, washed to eliminate the excess of probe, and fluorescence was analyzed by flow cytometry (FL1, Gallios, Beckman Coulter). Data analysis was performed using the software Kaluza, version 1.3.
Cellular membrane permeabilization was assayed by PI uptake. Briefly, cells were treated with 50 μg/ml of PI in PBS for 5 min, and cellular fluorescence analyzed by flow cytometry in FL3. Data was analyzed using the Kaluza software.
To measure total cellular ROS or mitochondrial ROS levels, the probe DCFDA (2′,7′‐dichlorofluorescin diacetate, Sigma‐Aldrich) or MitoSOX were used respectively. Briefly, cells were stimulated and treated with 10 μM DCFDA or 5 μM MitoSOX during the last 30 min of stimulation. Cells were then washed twice with PBS and fluorescence analyzed by flow cytometry (FL1 for DCFDA, or FL2 for MitoSOX, Gallios, Beckman Coulter). Data analysis was performed using the software Kaluza, version 1.3.
MtDNA depletion
Bone marrow–derived macrophages were cultured for 3 weeks in the presence of 100 ng/ml ethidium bromide, 100 μg/ml sodium pyruvate and 50 μg/ml uridine, as previously described (Nakahira et al, 2011). mtDNA depletion (ρ0) was confirmed by qPCR using primers for mitochondrial (Dloop) and nuclear (Tert) genes.
Cytosolic mtDNA quantification
Cytosolic mtDNA isolation and quantification was performed as previously described with slight modifications (West et al, 2015). Cells were divided into two equal aliquots. One aliquot was resuspended in Tri‐reagent® (Molecular Research Center, Inc) to isolate total cellular DNA. These extracts were used as normalization controls for total mtDNA. Cells from the second aliquot were resuspended in 150 mM NaCl, 50 mM HEPES pH7.4, containing 20 μg/ml digitonin to permeabilize the plasma membrane without disrupting the nucleus or any organelle. After 10 min incubation, the cells were centrifuged at 980 g for 3 min three times to obtain a cytosolic supernatant. This fraction was centrifuged further at 17,000 g for 10 min to completely eliminate nuclei, mitochondria, or endoplasmic reticulum. Protein was quantified from a small fraction and DNA was isolated from this cytosolic pure fractions using QIAQuick Nucleotide Removal Columns (QIAGEN) following the manufacturer's instructions. A cytosolic volume equivalent to 1 μg protein was used to analyze mtDNA content by qPCR using primers for Dloop. Ct values for total Dloop expression in total cellular DNA extract were used for normalization.
Statistics
No blinding of the data was performed. Except where indicated, data are described as means ± standard error of the mean (SEM). Statistical significance was determined by Student's t‐test, and one‐way ANOVA followed by Holm‐Sidak test conducted in SigmaPlot software, version 14.0. P < 0.05 was considered statistically significant. Statistical tests and number of samples are described in the figure legends.
Author contributions
Nagore de Pablo: Data curation; formal analysis; investigation; methodology. Clara Meana: Data curation; formal analysis; investigation; methodology. Javier Martínez‐García: Data curation; formal analysis; investigation; methodology. Pablo Martínez‐Vicente: Data curation; formal analysis; investigation; methodology. Manuel Albert: Data curation; formal analysis; investigation; methodology. Susana Guerra: Data curation; formal analysis; supervision; funding acquisition; validation. Ana Angulo: Data curation; formal analysis; supervision; funding acquisition; validation; writing – original draft. Jesús Balsinde: Resources; formal analysis; supervision; funding acquisition; validation; writing – original draft; project administration; writing – review and editing. María A Balboa: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; visualization; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interest statement
The authors state they have no competing interests or disclosures.
Supporting information
Appendix S1
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). We thank Montserrat Duque and Eva Merino for technical assistance. This work was supported by the Spanish Ministry of Economy, Industry, and Competitiveness (grant SAF2016‐80883‐R), Spanish Ministry of Science and Innovation (MICIN/AEI/10.13039/501100011033; grants PID2019‐105989RB‐I00; PID2020‐116918RB‐100; and PID2020‐117425RB‐C21), CIBERDEM‐ISCIII (grant CB07/08/0004), and the Regional Government of Castile and Leon (grant CSI141P20, cofinanced by the European Union through the European Regional Development Fund, “A Way of Making Europe”). N. de Pablo and J. Martínez‐García were supported by predoctoral fellowships from the Spanish Ministry of Science and Innovation (FPU14‐02879) and the Regional Government of Castile and Leon (E‐47‐2021‐0015435), respectively. Instituto de Biología y Genética Molecular is a research center partnered between the Spanish National Research Council (CSIC) and the University of Valladolid.
EMBO reports (2023) 24: e57238
Data availability
This study includes no data deposited in external repositories.
References
- Aricò E, Bracci L, Castiello L, Urbani F, Casanova JL, Belardelli F (2022) Exploiting natural antiviral immunity for the control of pandemics: lessons from Covid‐19. Cytokine Growth Factor Rev 63: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahat A, MacVicar T, Langer T (2021) Metabolism and innate immunity meet at the mitochondria. Front Cell Dev Biol 9: 720490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboa MA, Balsinde J, Dennis EA (1998) Involvement of phosphatidate phosphohydrolase in arachidonic acid mobilization in human amnionic WISH cells. J Biol Chem 273: 7684–7690 [DOI] [PubMed] [Google Scholar]
- Balboa MA, de Pablo N, Meana C, Balsinde J (2019) The role of lipins in innate immunity and inflammation. Biochim Biophys Acta 1864: 1328–1337 [DOI] [PubMed] [Google Scholar]
- Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V et al (2020) Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science 370: eabd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes‐Alnemri T, Wu J, Monks BG, Fitzgerald KA et al (2009) NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuyan F, de Jesus AA, Mitchell J, Leikina E, VanTries R, Herzog R, Onel KB, Oler A, Montealegre Sanchez GA, Johnson KA et al (2021) Novel Majeed syndrome‐causing LPIN2 mutations link bone inflammation to inflammatory M2 macrophages and accelerated osteoclastogenesis. Arthritis Rheumatol 73: 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biolatti M, Gugliesi F, Dell'Oste V, Landolfo S (2018) Modulation of the innate immune response by human cytomegalovirus. Infect Genet Evol 64: 105–114 [DOI] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, Khondoker M, Dickinson P, Sing G, Rodríguez‐Martín S et al (2011) Host defense against viral infection involves interferon mediated down‐regulation of sterol biosynthesis. PLoS Biol 9: e1000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ et al (2013) The transcription factor STAT‐1 couples macrophage synthesis of 25‐hydroxycholesterol to the interferon antiviral response. Immunity 38: 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ et al (2015) Gene Expression Omnibus GSE42505. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42505). [DATASET]
- Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D et al (2020) Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 181: 1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130: 2620–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Ge Y, Wu B, Zhang W, Wu T, Wen T, Liu J, Guo X, Huang C, Jiao Y et al (2020) Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J Infect Dis 222: 746–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J, Franz KM, Kagan JC (2015) PRRs are watching you: localization of innate sensing and signaling regulators. Virology 479–480: 104–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE Jr, Kerr IM, Stark GR (1994) Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421 [DOI] [PubMed] [Google Scholar]
- Decker T, Lew DJ, Mirkovitch J, Darnell JE Jr (1991) Cytoplasmic activation of GAF, an IFN‐gamma‐regulated DNA‐binding factor. EMBO J 10: 927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Cannon JG, Mier JW, Bernheim HA, LoPreste G, Lynn DL, Love RN, Webb AC, Auron PE, Reuben RC et al (1986) Multiple biological activities of human recombinant interleukin 1. J Clin Invest 77: 1734–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P et al (2008) High‐resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbrepatil AB, Zegalia KA, Sajja K, Kennedy RT, Marsh ENG (2020) Targeting viperin to the mitochondrion inhibits the thiolase activity of the trifunctional enzyme complex. J Biol Chem 295: 2839–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JM, Takkellapati S, Lawrence RT, McQueeney KE, Boroda S, Mullins GR, Sherwood SG, Finck BN, Villén J, Harris TE (2014) Lipin 2 binds phosphatidic acid by the electrostatic hydrogen bond switch mechanism independent of phosphorylation. J Biol Chem 289: 18055–18066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson PJ, El‐Shanti H (2021) Majeed Syndrome: a review of the clinical, genetic and immunologic features. Biomolecules 11: 367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, Munnich A, Lyonnet S, Majeed HA, El‐Shanti H (2005) Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 42: 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Núñez G (2009) TNF‐ mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: 792–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K et al (1997) Activation of interferon‐gamma inducing factor mediated by interleukin‐1β converting enzyme. Science 275: 206–209 [DOI] [PubMed] [Google Scholar]
- Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier‐Gobeaux C et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 369: 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee MK, Jang Y, Cho WJ, Kim M (2022) Repurposing of cyclophilin A inhibitors as broad‐spectrum antiviral agents. Drug Discov Today 27: 1895–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh WY, Zhou QD, York AG, Williams KJ, Scumpia PO, Kronenberger EB, Hoi XP, Su B, Chi X, Bui VL et al (2020) Toll‐like receptors induce signal‐specific reprogramming of the macrophage lipidome. Cell Metab 32: 128–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson JB (1988) Further studies on the mechanism of centrifugal enhancement of cytomegalovirus infectivity. J Virol Methods 19: 97–108 [DOI] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc Royal Soc Lond 147: 258–267 [Google Scholar]
- Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper‐Adrian V, Han R, Qiao L et al (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197–216 [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, Sauter D, Gifford RJ, USFQ‐COVID19 Consortium , Nakagawa S et al (2020) SARS‐CoV‐2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep 32: 108185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJ, Ashkar AA (2018) The dual nature of type I and type II interferons. Front Immunol 9: 2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L et al (2020) Activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun 11: 3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Kessler DS, Pine R, Reich N, Darnell JE Jr (1988) Interferon‐induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev 2: 383–393 [DOI] [PubMed] [Google Scholar]
- Lio CW, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, Sharma S (2016) cGAS‐STING signaling regulates initial innate control of cytomegalovirus infection. J Virol 90: 7789–7797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lordén G, Sanjuán‐García I, de Pablo N, Meana C, Alvarez‐Miguel I, Pérez‐García MT, Pelegrín P, Balsinde J, Balboa MA (2017) Lipin‐2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med 214: 511–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low H, Mukhamedova N, Cui HL, McSharry BP, Avdic S, Hoang A, Ditiatkovski M, Liu Y, Fu Y, Meikle PJ et al (2016) Cytomegalovirus restructures lipid rafts via a US28/CDC42‐mediated pathway, enhancing cholesterol efflux from host cells. Cell Rep 16: 186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, al‐Gharbawy F, Majeed SA, al‐Gazzar AH (1989) Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J Pediatr 115: 730–734 [DOI] [PubMed] [Google Scholar]
- Man SM, Karki R, Kanneganti TD (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277: 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 10: 417–426 [DOI] [PubMed] [Google Scholar]
- Masood KI, Yameen M, Ashraf J, Shahid S, Mahmood SF, Nasir A, Nasir N, Jamil B, Ghanchi NK, Khanum I et al (2021a) Upregulated type I interferon responses in asymptomatic COVID‐19 infection are associated with improved clinical outcome. Sci Rep 11: 22958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masood KI, Yameen M, Nasir A, Shahid S, Hasan Z (2021b) Gene Expression Omnibus GSE177477. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE177477). [DATASET]
- Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 27: 621–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys S, Schroeder T, Ellwart J, Koszinowski UH, Messerle M, Just U (2003) Dendritic cells under influence of mouse cytomegalovirus have a physiologic dual role: to initiate and to restrict T cell activation. J Infect Dis 187: 988–999 [DOI] [PubMed] [Google Scholar]
- McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T et al (1999) Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97: 133–144 [DOI] [PubMed] [Google Scholar]
- Meana C, Peña L, Lordén G, Esquinas E, Guijas C, Valdearcos M, Balsinde J, Balboa MA (2014) Lipin‐1 integrates lipid synthesis with proinflammatory responses during TLR activation in macrophages. J Immunol 193: 4614–4622 [DOI] [PubMed] [Google Scholar]
- Miorin L, Kehrer T, Sanchez‐Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E et al (2020) SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA 117: 28344–28354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, Chauhan D, Anderson KC, Frank DA (2008) Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 112: 5095–5102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T (2011a) IκB kinase epsilon (IKKɛ) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci USA 108: 21170–21175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T (2011b) Gene Expression Omnibus GSE33913. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33913) [DATASET]
- Péterfy M, Phan J, Xu P, Reue K (2001) Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet 27: 121–124 [DOI] [PubMed] [Google Scholar]
- Pindado J, Balsinde J, Balboa MA (2007) TLR‐3‐dependent induction of nitric oxide synthase in macrophages via a cytosolic phospholipase A2/cyclooxygenase‐2 pathway. J Immunol 179: 4821–4828 [DOI] [PubMed] [Google Scholar]
- Raza S, Barnett MW, Barnett‐Itzhaki Z, Amit I, Hume DA, Freeman TC (2014a) Analysis of the transcriptional networks underpinning the activation of murine macrophages by inflammatory mediators. J Leukoc Biol 96: 167–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza S, Freeman TC, Hume DA (2014b) Gene Expression Omnibus GSE44292. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44292) [DATASET]
- Robertson KA, Ghazal P (2016) Gene expression Omnibus GSE63290. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63290). [DATASET]
- Robertson KA, Hsieh WY, Forster T, Blanc M, Lu H, Crick PJ, Yutuc E, Watterson S, Martin K, Griffiths SJ et al (2016) An interferon regulated microRNA provides broad cell‐intrinsic antiviral immunity through multihit host‐directed targeting of the sterol pathway. PLoS Biol 14: e1002364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, Hoek JB (2017) The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16: 943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovas A, Osiaevi I, Buscher K, Sackarnd J, Tepasse PR, Fobker M, Kühn J, Braune S, Göbel U, Thölking G et al (2021) Microvascular dysfunction in COVID‐19: the MYSTIC study. Angiogenesis 24: 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio JM, Rodríguez JP, Gil‐de‐Gómez L, Guijas C, Balboa MA, Balsinde J (2015) Group V secreted phospholipase A2 is up‐regulated by interleukin‐4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J Immunol 194: 3327–3339 [DOI] [PubMed] [Google Scholar]
- Ruipérez V, Astudillo AM, Balboa MA, Balsinde J (2009) Coordinate regulation of Toll‐like receptor‐mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J Immunol 182: 3877–3883 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semper C, Leitner NR, Lassnig C, Parrini M, Mahlakõiv T, Rammerstorfer M, Lorenz K, Rigler D, Müller S, Kolbe T et al (2014a) STAT1β is not dominant negative and is capable of contributing to gamma interferon‐dependent innate immunity. Mol Cell Biol 34: 2235–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semper C, Leitner NR, Lassnig C, Parrini M, Mahlakõiv T, Rammerstorfer M, Lorenz K, Rigler D, Müller S, Kolbe T et al (2014b) Gene Expression Omnibus GSE48970. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48970). [DATASET] [DOI] [PMC free article] [PubMed]
- Seo J‐Y, Yaneva R, Hinson ER, Cresswell P (2011) Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332: 1093–1097 [DOI] [PubMed] [Google Scholar]
- Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B (2008) Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human Cytomegalovirus TB40/E. J Gen Virol 89: 359–368 [DOI] [PubMed] [Google Scholar]
- Smith GL, Talbot‐Cooper C, Lu Y (2018) How does Vaccinia virus interfere with interferon? Adv Virus Res 100: 355–378 [DOI] [PubMed] [Google Scholar]
- Sodeifian F, Nikfarjam M, Kian N, Mohamed K, Rezaei N (2022) The role of type I interferon in the treatment of COVID‐19. J Med Virol 94: 63–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Darnell JE Jr (2012) The JAK‐STAT pathway at twenty. Immunity 36: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartey S, Kanneganti TD (2020) Inflammasomes in the pathophysiology of autoinflammatory syndromes. J Leukoc Biol 107: 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens‐Kiani T, Cheng J et al (2020) Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science 369: 1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ et al (2002) TLR4, but not TLR2, mediates IFN‐β‐induced STATIα/β‐dependent gene expression in macrophages. Nat Immunol 3: 392–398 [DOI] [PubMed] [Google Scholar]
- Valdearcos M, Esquinas E, Meana C, Gil‐de‐Gómez L, Guijas C, Balsinde J, Balboa MA (2011) Subcellular localization and role of lipin‐1 in human macrophages. J Immunol 186: 6004–6013 [DOI] [PubMed] [Google Scholar]
- Valdearcos M, Esquinas E, Meana C, Peña L, Gil‐de‐Gómez L, Balsinde J, Balboa MA (2012) Lipin‐2 reduces proinflammatory signaling induced by saturated fatty acids in macrophages. J Biol Chem 287: 10894–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh DC, Abel L, Bastard P, Cheng MP, Condino‐Neto A, Gregersen PK, Haerynck F, Cicalese MP, Hagin D, Soler‐Palacín P et al (2021) Harnessing type I IFN immunity against SARS‐CoV‐2 with early administration of IFN‐β. J Clin Immunol 41: 1425–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Jonjic S, Koszinowski UH, Messerle M (1999) Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73: 7056–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watahiki A, Shimizu K, Hoshikawa S, Chiba M, Kitamura H, Egusa H, Fukumoto S, Inuzuka H (2020) Lipin‐2 degradation elicits a proinflammatory gene signature in macrophages. Biochem Biophys Res Commun 524: 477–483 [DOI] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winquist RJ, Gribkoff VK (2020) Targeting putative components of the mitochondrial permeability transition pore for novel therapeutics. Biochem Pharmacol 177: 113995 [DOI] [PubMed] [Google Scholar]
- Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY (2020) Evasion of type I interferon by SARS‐CoV‐2. Cell Rep 33: 108234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Biswal M, Neal A, Hai R (2021) Review Devil's tools: SARS‐CoV‐2 antagonists against innate immunity. Curr Res Virol Sci 2: 100013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Reue K (2017) Lipin proteins and glycerolipid metabolism: roles at the ER membrane and beyond. Biochim Biophys Acta 1859: 1583–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bastard P, Bolze A, Jouanguy E, Zhang SY, Human Genetic Effort COVID, Cobat A, Notarangelo LD, Su HC, Abel L et al (2020a) Life‐threatening COVID‐19: defective interferons unleash excessive inflammation. Med 1: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada‐Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C et al (2020b) Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science 370: eabd4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Matuozzo D, Le Pen J, Lee D, Moens L, Asano T, Bohlen J, Liu Z, Moncada‐Velez M, Kendir‐Demirkol Y et al (2022) Recessive inborn errors of type I IFN immunity in children with COVID‐19 pneumonia. J Exp Med 219: e20220131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez‐Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273 [DOI] [PMC free article] [PubMed] [Google Scholar]