

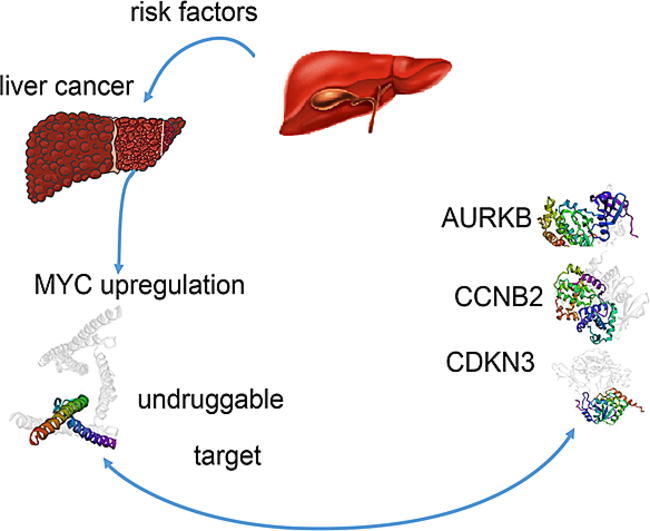
Graphical abstract
Keywords: LIHC, Tumor, Treatment, Biomarker, Therapy
Highlights
-
•
Scientific research of MYC associated liver hepatocellular carcinoma (LIHC) was to be the dominant force responsible for poor prognosis.
-
•
The possible risk factors attributed to LIHC include hepatitis B or C virus infection, excessive alcohol intake, inactivity, and a high-lipid diet associated with fatty liver. Serveral traditional clinical management options for LIHC, treatment is still limited due to toxic side effects.
-
•
MYC is difficult to target because of its undruggable structure. We aimed to uncover MYC-associated molecular targets to provide new strategies for LIHC treatment.
-
•
Several biomarkers of potential and promising utility for managing liver cancer therapy were identified in our work.
Abstract
Background
Aberrations in MYC underlie a large proportion of liver hepatocellular carcinoma (LIHC) cases; however, MYC is difficult to target because of its undruggable structure. We aimed to uncover MYC-associated molecular targets to provide new strategies for LIHC treatment.
Methods
LIHC transcriptome datasets and clinical information were obtained from The Cancer Genome Atlas. A series of bioinformatics analyses were performed for 370 patients who were stratified based on the median MYC expression level (high-MYC group and low-MYC group). Correlation analysis was performed to determine relationships between the expression of key MYC-associated genes and prognosis, DNA promotor methylation, and immune cell infiltration. Gene ontology and Kyoto Encyclopedia of Genes and Genomes Pathway enrichment analyses were performed to elucidate the functions of these genes in LIHC. Their expression and functions in LIHC were further verified using transgenic mice overexpressing c-Myc under control of the hepatocyte-specific promoter (Alb-Cre).
Results
AURKB, CCNB2, and CDKN3 were overexpressed in LIHC patients with high MYC expression and were associated with poor prognosis. Upregulation of these 3 genes was significantly correlated with hypomethylated promoter status, advanced T stage, metastasis, and immune cell infiltration in LIHC patients. Functional enrichment analyses indicated that these genes participate in the “p53 signaling pathway” and “cell cycle”. Furthermore, RT-PCR and IHC analysis revealed that their mRNA and protein expression levels were upregulated in an Alb-Cre;cMYClsl/- mouse model. Drugs that target these 3 MYC-related genes were identified.
Conclusion
Taken together, our results identify biomarkers of potential utility for managing liver cancer therapy owing to their significance in tumorigenesis, proliferation, and tumor immunity.
Introduction
As one of the most lethal tumors, liver hepatocellular carcinoma (LIHC) has caused millions of deaths over the past several decades, accompanied by increasing incidence [[1], [2], [3]]. In 2020, LIHC was ranked as the 7th most fatal tumor with > 905,000 new cases, accounting for up to 4.7 % of cancer in 185 countries with an 8.3 % overall mortality rate [4]. Developing counties have especially carried a heavy disease-related economic burden due to outdated medical options [[5], [6]]. Reported risk factors attributed to LIHC include hepatitis B or C virus infection, excessive alcohol intake, inactivity, and a high-lipid diet associated with fatty liver [[7], [8], [9]]. Although a variety of treatment options have been implemented for LIHC, the overall survival rates are still low due to underdiagnosis in the early stage and latency of distal metastases [[1], [10]]. Therefore, traditional options for LIHC therapy, including surgical removal, local ablation, and oral dosing with chemotherapy agents, represent poor treatment modalities with severe adverse side effects [[1], [11]]. CAR-T cell treatment has shown success in treating hematologic carcinomas, but treatment of solid tumors is hindered by the tumor microenvironment [[12], [13]]. As an alternative, molecular targeted therapies have shown promise as a strategy for improving cancer symptoms with less side effects [[8], [14], [15]]. Thus, the identification of novel core genes associated with LIHC holds immense potential value in the development of new approaches for diagnosis and treatment.
The MYC family of transcription factors, which includes C-MYC, N-MYC and l-MYC, functions as oncogenes in many human cancers [[16], [17], [18]]. Though the expression of MYC genes is generally elevated in LIHC, their underlying mechanisms are poorly understood [[19], [20]]. In this study, we collected mRNA expression datasets of 370 LIHC patients from The Cancer Genome Atlas (TCGA) and classified them into 2 groups based on median expression levels (high-MYC and low-MYC groups). We screened for tumor-specific differentially expressed genes (DEGs) associated with MYC expression by Gene Expression Profiling Interactive Analysis (GEPIA) [21]. We also conducted exploratory analyses of the pairwise relationships between expression of these genes and survival, DNA promotor methylation, and tumor related immune cell infiltration. Furthermore, we performed functional enrichment and protein–protein interaction (PPI) analyses to reveal the roles of MYC-associated hub genes in LIHC. Finally, we validated the expression levels of these genes by quantitative polymerase chain reaction (qPCR) and immunostaining of genetically modified mouse liver tissues. Taken together, our results elucidate candidate biomarkers and potential therapeutic targets to advance the understanding and treatment of LIHC.
Materials and methods
Data collection and processing
The mRNA-sequencing data and clinical information for LIHC and normal tissues were acquired from The Cancer Genome Atlas database (TCGA, https://www.cancer.gov/tcga). The dataset contained 370 LIHC samples and 50 paired normal samples that were evaluated on the Illumina Hiseq platform. Raw data were standardized according to the Fragments Per Kilobase of transcript per Million mapped reads. MYC over-expression in LIHC specimens compared with normal tissues was also evaluated at the protein level by extracting data from the Human Protein Atlas (HPA, https://www.proteinatlas.org/). Mutation sequence data were collected from cBioportal (https://www.cbioportal.org/) and OncoVar (https://oncovar.org/welcome/index).
DEG analysis, functional enrichment and survival analysis
The LIHC samples were divided into 2 groups, with the median expression of MYC as the cutoff value. Differentially expressed genes (DEGs) between the high-MYC and low-MYC groups were filtered using thresholds of logFC > 2 and p < 0.05, and up-regulated genes in LIHC from the TCGA datasets were identified by GEPIA (https://gepia.cancer-pku.cn/) using the same thresholds. Volcano plots of DEGs were visualized by GraphPad Prism (version 9.3.0), and Venn diagrams were generated by R software (version 3.6.3) using packages “ggplot2” and “VennDiagram” to evaluate overlapping DEGs. Differences in the expression levels of AURKB, CCNB2 and CDKN3 between LIHC and normal healthy tissues were analyzed and visualized using R software. The genes were also identified in pan-cancers and corresponding normal tissues using TCGA and GTEx datasets.
Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DEGs were conducted in Metascape (https://metascape.org/gp/index.html). The top 100 genes related to LIHC were acquired by GEPIA, and functional enrichment was implemented using DAVID (https://david.ncifcrf.gov/) and visualized by R package “ggplot2” (version 3.3.3).
Clinical data from LIHC patients, including overall survival (OS), disease specific survival (DSS), and progress free interval (PFI), were downloaded from TCGA and processed in R package using “survmine” (version0.4.9) and “survival” (version3.2.10) with the median gene expression as a cutoff value. Logrank p value < 0.05 was considered statistically significant. To evaluate predictive performance, receiver operating characteristic (ROC) curves were plotted using R package “pROC” (version 1.17.0.1). For increased robustness, the relationship between gene expression and 7 clinical traits, including T stage, N stage, distant metastasis, pathologic stage, gender, age and weight, was investigated using R software.
DNA methylation and mutation analysis
The methylation module of the UALCAN (https://ualcan.path.uab.edu/) portal was used to verify the methylation degree of promoter regions in LIHC and adjacent normal liver tissue. Heatmaps of the methylation patterns and methylation-related survival analysis accompanied by covariate adjustment were acquired in MethSurv (https://biit.cs.ut.ee/methsurv/). CpG islands were evaluated using MEXPRESS (https://mexpress.be/). P values < 0.05 were considered statistically significant.
The online tool OncoVar [22] was used to identify somatic mutations and driver events in LIHC. A total of 379 liver hepatocellular carcinoma samples (TCGA, Firehose Legacy) were acquired in cBioportal to explore genetic alterations.
Immune cell infiltration analysis
The online TIMER (https://timer.cistrome.org/) server was used to systematically analyze immune cell infiltration and immune-related survival. The R package “GSVA” (version 1.34.0) was applied to generate lollipop diagrams for visualization of the correlations between gene expression and 24 immune cell types across the TCGA database, including activated DCs; B cells; CD8 T cells; cytotoxic cells; DCs; eosinophils; immature DC; macrophages; mast cells; neutrophils; NK CD56 bright cells; NK CD56 dim cells; NK cells; plasmacytoid DCs; T cells; T helper cells; T central memory cells; T effector memory cells; T follicular helper cells; T gamma delta cells; Th1 cells; Th17 cells; Th2 cells; and Treg cells. The relationship between the gene expression levels and immune cells across 30 cancers was estimated using TISIDB (https://cis.hku.hk/TISIDB/index.php).
Functional enrichment analysis and protein–protein interaction (PPI) network construction
The “similar gene” module of GEPIA was used to acquire data on MYC and MYC-associated genes from LIHC samples in TCGA. R packages “clusterProfiler” (version3.14.3) and “ggplot2” were used to plot histograms and predict GO annotation, which consisted of biological processes (BP), molecular function (MF), and cellular component (CC) categories. The top 100 genes obtained were uploaded to the DAVID functional annotation tool (https://david.ncifcrf.gov/tools.jsp), and gene annotation lists were generated. Then, R package “ggplot2” was used to decipher KEGG pathway enrichment bubble maps. To predict the activation or inhibition function of signaling pathways, we used the web tool GSCALite (https://bioinfo.life.hust.edu.cn/web/GSCALite/) with “TCGA-LIHC” and “Pathway Activity” settings to elucidate the expression of hub genes in common pathways, including apoptosis, cell cycle, DNA damage response, EMT, hormone AR, hormone ER, PI3K/AKT, RAS/MAPK, and RTK pathways (activation and inhibition). Finally, we used the STRING (https://string-db.org/) website to assess protein–protein correlations using default parameters.
Real-time polymerase chain reaction (RT-PCR) and immunohistochemistry (IHC)
Tumor tissues from Alb-Cre+/Myc+ mice were dissected, and RT-PCR and immunohistochemical staining (IHC) were performed to compare mRNA and protein expression levels to those in normal tissues of double negative littermates. For the RT-PCR, the following primers were used: mGAPDH F, GGTTGTCTCCTGCGACTTCA; mGAPDH R, TGGTCCAGGGTTTCTTACTCC; mAURKB F, GGGTGCTCTGCTATGAACTGATGG; mAURKB R, CCACCTTGACAATCCGACGATACG; mCCNB2 F, ACAAATGTCAACAAGCAGCCGAAAC; mCCNB2 R, GCAGAGCAGAGCATCAGAGAAAGC; mCDKN3 F, GCTGCTGGGAAATCATGGAGGAG; mCDKN3 R, TGGCTTGCTGTGGTGAGATTGAG; mTNF-α F, GCTACGACGTGGGCTACAG; mTNF-α R, CCCTCACACTCAGATCATCTTCT; miNOS F, TGGCTCGCTTTGCCACGGACGAGACGGA; miNOS R, GGAGCTGCGACAGCAGGAAGGCAGCGGG; mIL-6F, TTGGTCCTTAGCCACTCCTCC; mIL-6 R, TAGTCCTTCCTACCCCAATTTCC. For IHC [23], polyclonal rabbit anti-AURKB (D151481, BBI, China), rabbit monoclonal anti-Cyclin B2/CCNB2 (ab185622, Abcam, UK), and rabbit polyclonal anti-CDKN3 (bs-5743R, Bioss, China) antibodies were diluted 1:200.
Drug sensitivity analysis
To investigate potential therapeutic targets for AURKB, CCNB2 and CDKN3, datasets from Therapeutics Response Portal (CTRP) were analyzed using the GSCALite tool. A novel dataset from HERB (https://herb.ac.cn/), an integrated high-throughput assay- and literature-guided database for Chinese medicine analysis[24], was also used to access traditional Chinese medicine targets.
Statistical analysis
All statistical analyses were processed using R software (version 3.6.3). The Student’s t-test was used for comparison of differences between two groups. Correlations among AURKB, CCNB2, CDKN3 and MYC expression were analyzed with a Pearson correlation. Kaplan-Meier curves and the log-rank test were used to assess the overall survival (OS), disease specific survival (DSS), and progress free interval (PFI) of LIHC patients. Data is presented as mean ± standard deviation and a P-value < 0.05 was considered statistically significant.
Results
MYC is upregulated in LIHC within a network of differentially regulated mRNAs that includes AURKB, CCNB2, and CDKN3
To verify the role of MYC in a human LIHC cohort from TCGA, we evaluated its distribution in 369 LIHC and 160 normal control liver samples by GEPIA. As expected, MYC was overexpressed in LIHC specimens compared to normal hepatic tissues (Figure S1A and B). To further investigate features that may impact MYC expression in LIHC, we analyzed the relationship between MYC expression and DNA methylation, copy number, and genetic alteration. The expression of MYC was significant negatively correlated with DNA methylation and positively correlated with copy number (Figure S1C, D). Additional analysis demonstrated that the MYC gene is altered in seventeen percent of patients with LIHC, with gene amplification being the most common genetic alteration (Figure S1E-G). Therefore, these data suggest that MYC upregulation is associated with DNA hypomethylation, gene amplification and other genetic alterations within the LIHC cohort.
Next, to identify differentially expressed genes (DEGs) that are associated with MYC activation in LIHC, we compared gene expression levels in the 370 LIHC versus 50 control normal adjacent samples by GEPIA. A total of 118 genes were upregulated in LIHC samples compared to adjacent tissues using p < 0.05 and log fold change [FC] > 2 as thresholds (Fig. 1A). We performed Gene ontology (GO) analysis and Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of these 118 differential genes by Metascape and determined that LIHC was associated with pathways related to “cell cycle”, “Mitotic G1 phase and G1/S transition” and “DNA replication” (Fig. 1B, C).
Fig. 1.

Identification of differentially expressed genes (DEGs) in LIHC. (A) Volcano plot of DEGs between LIHC tissues and normal liver tissues using GEPIA. KEGG (B) and GO (C) functional enrichment analysis of DEGs in LIHC versus adjacent tissues. (D) Volcano plot of high-MYC and low-MYC LIHC groups using datasets from TCGA. (E) Venn diagram of overlapping DEGs associated with high MYC expression and LIHC-specific expression.
To identify key genes that mediate the MYC response, we sorted the tumor samples into high-MYC and low-MYC groups according the median levels of MYC expression and identified 5442 genes that were differentially expressed according to the MYC level (Fig. 1D). Next, we evaluated the overlap between LIHC-associated DEGs and MYC-high DEGs, which yielded 6 candidate mRNAs, including ALG1L, AURKB, CCNB2, CDKN3, EPS8L3, and FABP5 (Fig. 1E). These findings are consistent with potential roles for AURKB,CCNB2, andCDKN3 in MYC-dependent LIHC.
AURKB, CCNB2, and CDKN3 are co-expressed in LIHC and are predictive of clinical parameters
To further evaluate the relationship between the expression of MYC and AURKB, CCNB2, and CDKN3 in LIHC, we performed co-expression analyses. Surprisingly, no obvious co-expression between MYC expression and the levels of these genes was detected (Fig. 2A). However, in paired analysis of high-MYC and low-MYC groups, all three genes had statistically higher expression levels in the high-MYC expression group (Fig. 2B). Additionally, AURKB, CCNB2, and CDKN3 showed high correlation levels among each other (Fig. 2C).
Fig. 2.

Distribution of expression for MYC and MYC-associated genes in LIHC. (A) Correlation analysis of the expression of AURKB, CCNB2 and CDKN3 genes and the expression of MYC in LIHC samples. (B) Distribution of AURKB, CCNB2 and CDKN3 genes in low-MYC and high-MYC groups. (C) Correlation analysis of the reciprocal association of AURKB, CCNB2 and CDKN3.
To determine whether the levels of AURKB, CCNB2 and CDKN3 affect LIHC outcomes, we performed Kaplan-Meier survival analysis according to the median levels of these genes. The results suggest that overexpression of each of the 3 genes is associated with shorter overall survival (OS), disease specific survival (DSS), and progress free interval (PFI) in LIHC patients (Fig. 3A-C). To further explore the predictive values of these genes in LIHC, we performed receiver operating characteristic (ROC) analysis. The adjusted area under the curve values of 1-, 3-, and 5-year of overall survival for AURKB (0.672, 0.622, and 0.617, respectively), CCNB2 (0.682, 0.618, and 0.599, respectively) and CDKN3 (0.664, 0.622, and 0.592, respectively) (Fig. 3D) suggest that these genes have moderate prognostic relevance in LIHC.
Fig. 3.

Survival and predictive analysis for MYC-associated genes in LIHC. (A-C) Kaplan-Meier analysis according to high and low expression of AURKB, CCNB2, and CDKN3 of overall survival (A), disease specific survival (B) and progress free interval (C). (D) ROC curve analysis of the predictive capabilities of AURKB, CCNB2, and CDKN3 for the survival of LIHC patients.
To better understand the importance of AURKB, CCNB2 and CDKN3, we evaluated their expression according to clinicopathological characteristics, including T stage, lymph node metastasis (N), distal metastasis (M), age, gender, weight, and histopathology. For all 3 genes, expression was predominantly associated with advanced T stage (T2 + T3) compared to early-stage (T1) of LIHC tumors (Figure S4A-B). No significance was observed between T1 and T4 stage, potentially explained by the limited sample size (p > 0.01). Beside, no significance was observed between Stage Ⅰ and Stage Ⅳ (Figure S4B). Furthermore, higher expression was observed according to both the M and N stages (Figure S4C-D). Interestingly, for all three genes, expression was also associated with age and weight, though no significant differences between genders were observed (Figure S5A-C). Therefore, these results support the potential relevance of AURKB, CCNB2 and CDKN3 expression in LIHC.
DNA methylation and gene alteration statuses of AURKB, CCNB2 and CDKN3 promoters are altered in LIHC
To determine whether the differential expression of AURKB, CCNB2 and CDKN3 in LIHC is accompanied by differences in promoter methylation levels, we evaluated their methylation statuses in LIHC and adjacent para-carcinoma tissues using the methylation module of UALCAN. The overall promoter methylation levels of AURKB and CCNB2 were significantly downregulated in LIHC, whereas the levels of CDKN3 were not significantly altered (Fig. 4A-C). Next, we used MEXPRESS [28] and MethSurv [29] to identify candidate methylation sites in LIHC in the 3 gene promoters. Heatmaps obtained from MethSurv showed that cg09132923 and cg18576335 of AURKB, and cg00649500 and cg24032252 of CDKN3 were highly methylated in LIHC tissues, whereas there were no candidate methylation sites identified for CCNB2 (Fig. 4D-F). Notably, in survival analysis, high levels of cg09132923 and cg00649500 were associated with a favorable prognosis, while cg18576335 and cg24032252 did not show consistent trends (Fig. 4G-J). To further evaluate the relationship between methylation and gene transcriptional profiles, we implemented MEXPRESS, which detected 9 sites for AURKB, 8 sites for CCNB2, and 6 sites for CDKN3 that were hypermethylated in LIHC (Figure S6A-C). This included the cg09132923, cg00649500 and cg24032252 sites, which is consistent with the MethSurv analysis.
Fig. 4.

Methylation analysis of the AURKB, CCNB2 and CDKN3 promoters in LIHC. Methylation was assessed using UALCAN (A-C) and MethSurv (D-F). (G-J) Survival analysis of methylation sites cg09132923 and cg18576335 of AURKB, and cg00649500 and cg24032252 of CDKN3.
Gene alteration, especially oncogenic alteration in human tumoral tissues, frequently influences primary tumor formation, development and metastases [[30], [31]]. Therefore, we visualized the gene alteration patterns in LIHC using the online tool OncoVar. Seven classification types of variants were detected in tumor livers and ranked from high to low frequency (Figure S7A and B). Three subgroups of variants were further explored, including single nucleotide polymorphisms (SNP), insertion mutations (INS) and deletion mutations (DEL) (Figure S7C). Among them, SNPs accounted for a relatively large proportion of variation. We calculated the numbers of single nucleotide variation (SNV) and base substitution types in LIHC patients (Figure S7D) and concurrently arranged them in decreasing order (Figure S7E), which was visualized in a waterfall plot (Figure S7F). Transitions (Ti) were rarely observed in LIHC compared with transversions (Tv) (Figure S7G). Finally, mutation events of AURKB, CCNB2 and CDKN3 derived from LIHC tissue and pan-cancer in TCGA were determined (Figure S8). The results indicate that AURKB was deep depleted in 2.5 % of LIHC cases, while CCNB2 and CDKN3 were significant amplified in 1.1 % and 1.4 % of LIHC, respectively. These results indicate that AURKB, CCNB2 and CDKN3 expression may be regulated by both promoter methylation and gene alterations in LIHC.
Immune infiltration status correlates with AURKB, CCNB2 and CDKN3 expression in LIHC
To evaluate potential effects of AURKB, CCNB2 and CDKN3 on the immune environment in LIHC, we quantified immune infiltration signatures in LIHC from TCGA using the “Gene module” option in TIMER. Multiple immune cells exhibited a significantly positive relationship with AURKB expression, including B cells (p = 2.02e − 22, cor = 0.493), CD8+ T cells (p = 1.60e − 10, cor = 0.337), CD4+ T cells (p = 1.14e − 05, cor = 0.234), macrophages (p = 1.29e − 12, cor = 0.372), neutrophils (p = 1.43e − 06, cor = 0.256), and dendritic cells (p = 1.64e − 15, cor = 0.414) (Fig. 5A). Similarly, the expression of CCNB2 was positively associated with B cells (p = 6.57e − 22, cor = 0.487), CD8+ T cells (p = 2.78e − 11, cor = 0.35), CD4+ T cells (p = 7.86e − 09, cor = 0.305), macrophages (p = 6.27e − 18, cor = 0.444), neutrophils (p = 1.49e − 09, cor = 0.318), and dendritic cells (p = 6.57e − 19, cor = 0.457) (Fig. 5B). The CDKN3 expression levels were also positively associated with B cells (p = 1.63e − 18, cor = 0.45), CD8+ T cells (p = 2.19e − 08, cor = 0.297), CD4+ T cells (p = 3.18e − 06, cor = 0.248), macrophages (p = 2.33e − 13, cor = 0.383), neutrophils (p = 3.28e − 07, cor = 0.271), and dendritic cells (p = 6.65e − 13, cor = 0.377) (Fig. 5C).
Fig. 5.

Immune infiltration analysis in LIHC and the association with AURKB, CCNB2 and CDKN3 expression. The correlation between AURKB, CCNB2 and CDKN3 expression and different immune cell types by TIMER (A-C), R software (D-F), and Immune cell-related survival analysis (G) in LIHC. aDC, activated dendritic cells; iDC, immature dendritic cells; pDC, plasmacytoid dendritic cells; Tcm, T central memory cells; Tem, T effector memory cells; TFH, T follicular helper cells; Tgd, T gamma delta cells.
For confirmation, we analyzed 24 types of immune cell infiltration in LIHC using R package “GSVA” (Fig. 5D-F). Immune cell-related survival analysis indicated that enhanced CD4+ T cells, macrophages and neutrophil expansion induce invasive properties in LIHC and lead to poor prognosis (Fig. 5G). To further investigate the roles of AURKB, CCNB2 and CDKN3 in tumor immune responses, we employed TISIDB in order to evaluate the importance of the 3 genes across 30 cancer types. As shown in Figure S9A-C, the heatmap obtained from TISIDB supports the positive correlation of activated CD4+ T cell and Th2 cell infiltration in tumor lesions with the expression of AURKB, CCNB2 and CDKN3 in most cancer types.
Functional enrichment analysis of AURKB, CCNB2, CDKN3 and MYC in LIHC
To elucidate the underlying molecular mechanisms of AURKB, CCNB2, CDKN3 and MYC in LIHC, we conducted GO and KEGG enrichment analysis. GO analysis revealed that AURKB, CCNB2 and CDKN3 concurrently participate in the “regulation of nuclear division”, “sister chromatid segregation”, “mitotic nuclear division”, and “cell cycle” pathways (Fig. 6). Furthermore, MYC was central in its association with “snoRNA binding”, “catalytic activity” and “acting on RNA” (Figure S10A) and was also linked with the “cell cycle” pathway (Figure S10B). Notably, AURKB, CCNB2 and CDKN3 were involved in the “p53 signaling” pathway, and CCNB2 and CDKN3 were also master regulators of “DNA replication” (Fig. 6B, D, F).
Fig. 6.

Enrichment analysis of AURKB, CCNB2, and CDKN3-associated biological pathways. GO (A, C, E) and KEGG (B, D, F) analyses of AURKB, CCNB2 and CDKN3.
Next, we used the GSCALite website to explore the inhibition or activation roles of AURKB, CCNB2, CDKN3 and MYC in major signaling pathways. The expression of all 4 genes was positively correlated with “cell cycle”, “apoptosis” and “endothelial mesenchymal transition (EMT)” signal pathway activation, each of which are core functions in tumor initiation and development (Fig. 7A, B). Finally, we predicted protein–protein interactions (PPI) of AURKB, CCNB2 and CDKN3 using the STRING bioinformatics tool. The top 10 core genes with highest connectivity degree are visualized in Figure S11A-C, and the interrelationship between AURKB, CCNB2, CDKN3 and MYC was predicted (Figure S11D). These results support the interconnection of AURKB, CCNB2, CDKN3 and MYC in LIHC.
Fig. 7.

The correlations between AURKB, CCNB2, CDKN3 and MYC expression and common tumor-associated pathways. The correlations were predicted using the GSCALite website.
Overexpression of AURKB, CCNB2 and CDKN3 promotes tumorigenesis in Alb-cMyc mice
For better understanding of the potential mechanisms of AURKB, CCNB2 and CDKN3 in high-MYC LIHC, we established an LIHC mouse model. First, Alb + mice were crossbred with cMyc+ mice to generate Alb+/cMyc+ mice (Fig. 8A, B). These mice gained a gross tumor burden by 4 months after birth (Fig. 8C). Using traditional HE staining, we demonstrated that livers of WT mice had an orderly arrangement, while those of Alb+/cMyc+ mice had a disordered cell arrangement with signs of degeneration, karyomegaly, and multiple nuclei (Fig. 8D, E). Notably, significant upregulation of AURKB, CCNB2 and CDKN3 transcriptional levels was verified in resultant LIHC tumors, in which cMyc was highly enhanced (Fig. 8F). Moreover, the protein expression levels of AURKB, CCNB2 and CDKN3 were significantly increased in the LIHC of Alb+/cMYC + mice as compared to in normal liver tissue as evaluated by IHC (Fig. 8G-L). Besides, the expression of Ki67 was elevated in high-MYC LIHC tissue versus normal tissue, consistent with the role for cell cycle pathways in MYC-dependent LIHC as suggested by our enrichment analyses (Fig. 8M, N). Finally, qPCR analysis results of the expressions (Fig. 8O) and correlations (Fig. 8P) of three inflammatory marker genes (interleukin-6 (IL-6), tumor necrosis factor-α (TNFα) and inducible nitric oxide synthase (iNOS)) verified our hypothesis about the infiltration of immune cells in mice liver cancer.
Fig. 8.

AURKB, CCNB2, CDKN3 and MYC expression in Alb-cMYC mice with LIHC. (A) Scheme for intercrossing Myc and Alb-Cre mice to generate Alb-cMYC mice. (B) Genotyping for validation of Alb and cMYC in the Alb-cMYC mice. (C) LIHC formed in the Alb-cMYC mouse model. (D-E) Representative HE staining images of WT mouse normal tissues and Alb-cMYC mouse LIHC tissues. (F) qRT-PCR validation of AURKB, CCNB2, and CDKN3 levels in WT mice and the Alb-cMYC mouse model. (G-N) IHC Protein detection of AURKB (G-H), CCNB2 (I-J), CDKN3 (K-L) and Ki67 (M−N) in WT mice and the LIHC mouse model (all 400 × ). (O—P) qPCR analysis results of the expressions (O) and correlations (P) of three inflammatory marker genes (IL-6, TNFα and iNOS).
Prediction of drugs that target AURKB, CCNB2 and CDKN3
Given the predicted oncogenic significance of AURKB, CCNB2, and CDKN3, we sought to identify drugs that target these proteins. According to CTRP datasets, CDKN3 was predicted to be severely antagonized by 12 drugs or small molecules, AURKB by 33 drugs, and CCNB2 by 9 drugs (Figure S12A). We also detected the molecular effects of traditional Chinese medicines (herbs/ingredients) for AURKB, CCNB2 and CDKN3 using HERB. The results identified 3, 4, and 16 candidates for these proteins that may serve as potential therapeutic targets (Figure S12B). Notably, all 3 of the genes were predicted to be sensitive to CAO MEI CHE ZHOU CAO, namely Trifolium fragiferum (Figure S12B). Therefore, these results could assist in the identification of new potential therapies for MYC-associated LIHC.
Discussion
LIHC remains a major challenge for cancer therapy owing to its local regional recurrence [32], aggressiveness [33] and high worldwide mortal rate [34]. Although there are numerous traditional clinical management options for LIHC, treatment is still limited due to toxic side effects. Thus, there is an urgent need to develop useful biomarkers for LIHC patients. Accumulating evidence suggests that deregulated MYC plays a vital role in human cancer tumorigenesis via its transcriptional regulatory motifs downstream or upstream of pathways associated with cell proliferation [35], with well-established roles for MYC in lung cancer and hematologic malignancies [[36], [37]]. However, efforts to directly target MYC have not been successful thus far, partly because of limitations in its protein structure [18]. Accordingly, we sought to identify novel druggable targets that are associated with MYC to overcome this challenge in LIHC.
To identify new MYC-related targets, we screened 118 mRNAs that were upregulated in LIHC as evaluated by GEPIA. Enrichment analysis revealed that these mRNAs were concentrated in “cell cycle”, “DNA replication” and the “p53 signaling pathway”. Next, we identified 5440 mRNAs that were differentially expressed in high-MYC compared with low-MYC LIHC groups and performed intersection analysis, which yielded 6 mRNAs. We chose 3 of them (AURKB, CCNB2 and CDKN3) for further exploration due to their established roles in tumorigenesis [[25], [26], [27]] and PPI relationship with MYC. To confirm the functional roles of these genes, we repeated GO and KEGG analysis, which verified that these three identified MYC-related genes are also enriched in the “cell cycle” signaling pathway. Because cell-cycle deregulation accounts for the overwhelming majority of uncontrolled proliferation of tumor cells [38], these results are consistent with functional roles for these genes in LIHC. Correlation analysis verified potential roles for their high expression in LIHC prognosis. Taken together, these results suggest that AURKB, CCNB2 and CDKN3 may serve as biomarkers and new therapeutic targets for MYC-driven LIHC.
Aurora kinase B (AURKB) is a member of mitotic protein kinase family that functions as a unique regulator in chromosome segregation and cell mitosis [[39], [40]]. Consistently, previous studies have revealed that aberrant AURKB expression is associated with tumorigenesis and growth of many cancers. For example, Long et al. [41] performed bioinformatics analysis, indicating that AURKB is a key tumorigenic factor in cholangiocarcinoma. Nie et al. [42] demonstrate that AURKB directly triggers histone phosphorylation of CCND1 in gastric cancer cell lines at Serine 10. Furthermore, Jiang et al. [37] revealed that AURKB reciprocally activates MYC in T-cell lymphoma, stabilizing MYC via phosphorylation at Serine 67. Additionally, Chen et al. [43] demonstrated that overexpressed MYC activates BOP1 ribosomal biogenesis factor, which increases AURKB activity and results in chromosomal segregation mistakes in colorectal cancer cell lines. Nevertheless, the underlying mechanism of AURKB in LIHC remained to be clarified. In our study, we detected increased AURKB in multiple LIHC and paired samples using the data from TCGA and GTEx datasets. AURKB was associated with clinical characteristics of LIHC, including T stage, age, pathologic stage, and weight. More importantly, ROC analysis results demonstrate that high AURKB expression levels are a relatively accurate predictor for prognosis in LIHC patients, with high AURKB in LIHC associated with a shorter OS, DSS and PFI.
As a member of the cyclin family, CCNB2 has a crucial role in the G2 to M phase transition [44]. Many studies have verified that CCNB2 has a role in cell cycle regulation [[45], [46]], which is consistent with our KEGG enrichment results. Wang et al. [47] determined that CCNB2 triggers cell senescence by activating SASP/Cathepsin B and PGE2, which mediates malignant transformation in glioma. Qian et al. [44] revealed that CCNB2 is significantly overexpressed in non-small cell lung cancer compared to adjacent normal lung tissues. Furthermore, Xia et al. [48] reported that increased expression of CCNB2 is associated with rhabdomyosarcoma tumor volumes when the diameter of tumor > 5 cm. CCNB2 overexpression has also been shown to contribute to poor survival in hepatocellular carcinoma [49], which is supported by the results of our study. Additionally, using Alb-cMYC mice with liver cancer, we verified that CCNB2 was significantly upregulated, thus supporting its association with unfavorable survival in LIHC.
Cyclin-dependent kinase inhibitor-3 (CDKN3) is a CDK2-associated dual specificity phosphatase that functions as a regulator of the mitotic phase of the cell cycle [50]. Consistently, CDKN3 expression fluctuates during different cell cycle phases and is especially increased in M phase [50]. Cen et al. [51] demonstrated that an upregulated CDKN3/E2F1 axis in renal cell carcinoma promotes tumor cell proliferation and metastasis. Aberrant expression of CDKN3 reduces survival times in various human cancers [50], including esophageal cancer [52], ovarian cancer [53], and gastric cancer [54]. In our study, immunohistochemical experiments suggest that AURKB, CCNB2 and CDKN3 are highly increased in Alb-cMYC liver cancer specimens compared to WT normal liver tissue, thus supporting their roles in LIHC.
In this study, we also evaluated the role of methylation in the regulation of AURKB, CCNB2 and CDKN3. Previous studies have reported that high methylation levels are correlated with gene expression [[55], [56], [57], [58]]. Aberrant DNA methylation is detected in various cancer types, though the understanding of its role in LIHC is limited [[59], [60], [61]]. Therefore, we studied DNA methylation patterns in LIHC using UALCAN,MEXPRESS, and MethSurv databases. AURKB and CCNB2 were demonstrated to be hypomethylated in LIHC specimens, with associated increases in expression levels in LIHC. Furthermore, we demonstrated a negative correlation between gene expression and promoter methylation at specific sites in the AURKB and CCNB2 promoters in LIHC that are associated with prognosis. These findings support the possibility that aberrant methylation contributes to the development of LIHC.
Increasing evidence indicates that immune cell infiltration in tumor tissues affects cancer progression and patient prognosis [[62], [63], [64], [65], [66]]. AURKB [67], CCNB2 [26] and CDKN3 [68] have each been evaluated as immune intervenors in various cancers. Therefore, we used the online web servers TISIDB and TIMER to demonstrate potential roles for these genes in tumor immunity in LIHC. Our survival analysis results suggest that high levels of CD4+ T cell, macrophage, and neutrophil infiltration are related to unfavorable prognosis in LIHC. However, the functions of immune cells in tumorigenesis remains controversial. LIHC patients with high T cell levels have been demonstrated to have a lower recurrence and better prognosis [69]. By contrast, Fu et al. [70] demonstrated that excessive immune cell infiltration increases immunity tolerance in LIHC owing to activation of multiple immune suppressor mechanisms. Additionally, Ma et al. [71] demonstrated that CD8+ T cells are highly enriched in malignant neoplasm, which is consistent with our results. Taken together, these findings indicate that the MYC-related genes identified in this study (AURKB, CCNB2 and CDKN3) may have an influence on immunity in LIHC.
Though most of the results of our research were performed by web database analysis, the relevance of the findings were further verified in a mouse liver cancer model using Alb-cMYC transgenic mice. These results provide additional evidence of the potential integral roles of AURKB, CCNB2 and CDKN3 in MYC-driven LIHC. Notably, we demonstrated that these genes are predicted drug targets, which suggest therapeutic potential. A comprehensive exploration of drug function in LIHC, as well as targeted assays for ablating gene expression, may further elucidate the mechanisms of these genes in promoting MYC-driven LIHC.
Conclusion
In summary, we identified AURKB, CCNB2 and CDKN3 as oncogenic factors and potential therapeutic targets for MYC-driven LIHC. Our study suggested that their expression levels are related to worse prognosis, proliferation, and immune cell infiltration. Therefore, these genes may be robust biomarkers for predicting prognosis in LIHC patients with highly expressed MYC.
Ethics statement
All experiments involving animals were conducted according to the ethical policies and procedures approved by Institutional Animal Care and Use Committee (IACUC) guidelines at Tongji University School of Medicine(SYDW-19–215). Patient data originated from TCGA database and HPA did not require ethics committee approval, and this study complied with the publication guidelines provided by TCGA.
Consent for publication
Not applicable.
Availability of data and material
The datasets supporting the conclusions of this article are included within the article.
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
CRediT authorship contribution statement
Sha Li: Conceptualization, Writing – review & editing. Pei Xue: . Xun Diao: . Qi-Yu Fan: . Kun Ye: . Xiao-Mei Tang: . Jia Liu: . Zhong-Yan Huang: . Qing-Hai Tang: . Cheng-You Jia: . Rui Xin: . Zhong-Wei Lv: . Ji-Bin Liu: Conceptualization, Writing – review & editing. Yu-Shui Ma: Conceptualization, Writing – review & editing, Supervision. Da Fu: Conceptualization, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This study was supported partly by grants from the National Natural Science Foundation of China (81772932 and 81972214), Natural Science Foundation of Hunan Province (2021JJ30060); Key Research and Development Program of Hunan Province (2021NK2026), Key Program of Hunan Provincial Department of Science and Technology (2020WK2020), Shanghai Committee of Science and Technology (21140903500), Shanghai Natural Science Foundation (20ZR1472400), Construction of Clinical Medical Centre for Tumor Biological Samples in Nantong (HS2016004), Science and Technology Project of Nantong (JC2018010) and the Jiangsu Provincial University Natural Science Funding (19KJB180021).
We would like to thank the Laboratory Animal Center of Tongji University (Shanghai, China) for providing the animal experimental platform.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2023.01.010.
Contributor Information
Ji-Bin Liu, Email: tians2008@ntu.edu.cn.
Yu-Shui Ma, Email: mayushui@tongji.edu.cn.
Da Fu, Email: fu80da90@163.com.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Anwanwan D., Singh S.K., Singh S., Saikam V., Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1) doi: 10.1016/j.bbcan.2019.188314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cocker F., Chien Yee K., Palmer A.J., de Graaff B. Increasing incidence and mortality related to liver cancer in Australia: time to turn the tide. Aust N Z J Public Health. 2019;43(3):267–273. doi: 10.1111/1753-6405.12889. [DOI] [PubMed] [Google Scholar]
- 3.Chen S., Cao Q., Wen W., Wang H. Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett. 2019;460:1–9. doi: 10.1016/j.canlet.2019.114428. [DOI] [PubMed] [Google Scholar]
- 4.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 5.Liu S., Li Y., Zeng X., Wang H., Yin P., Wang L., et al. Burden of Cardiovascular Diseases in China, 1990–2016: Findings From the 2016 Global Burden of Disease Study. JAMA Cardiol. 2019;4(4):342–352. doi: 10.1001/jamacardio.2019.0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Axelsson U., Rydén L., Johnsson P., Edén P., Månsson J., Hallberg I.R., et al. A multicenter study investigating the molecular fingerprint of psychological resilience in breast cancer patients: study protocol of the SCAN-B resilience study. BMC Cancer. 2018;18(1):789. doi: 10.1186/s12885-018-4669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrick J.L., Florio A.A., Znaor A., Ruggieri D., Laversanne M., Alvarez C.S., et al. International trends in hepatocellular carcinoma incidence, 1978–2012. Int J Cancer. 2020;147(2):317–330. doi: 10.1002/ijc.32723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang D.-D., Wang W.-E., Ma Y.-S., Shi Y., Yin J., Liu J.-B., et al. A miR-212-3p/SLC6A1 Regulatory Sub-Network for the Prognosis of Hepatocellular Carcinoma. Cancer Manag Res. 2021;13:5063–5075. doi: 10.2147/CMAR.S308986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniai M. Alcohol and hepatocarcinogenesis. Clin Mol Hepatol. 2020;26(4):736–741. doi: 10.3350/cmh.2020.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 11.Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 12.Grosser R., Cherkassky L., Chintala N., Adusumilli P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell. 2019;36(5):471–482. doi: 10.1016/j.ccell.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hont A.B., Cruz C.R., Ulrey R., O'Brien B., Stanojevic M., Datar A., et al. Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J Clin Oncol. 2019;37(26):2349–2359. doi: 10.1200/JCO.19.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keenan T.E., Tolaney S.M. Role of Immunotherapy in Triple-Negative Breast Cancer. J Natl Compr Canc Netw. 2020;18(4):479–489. doi: 10.6004/jnccn.2020.7554. [DOI] [PubMed] [Google Scholar]
- 15.Bedard P.L., Hyman D.M., Davids M.S., Siu L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395(10229):1078–1088. doi: 10.1016/S0140-6736(20)30164-1. [DOI] [PubMed] [Google Scholar]
- 16.Beaulieu M.-E., Jauset T., Massó-Vallés D., Martínez-Martín S., Rahl P., Maltais L., et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med. 2019;11(484) doi: 10.1126/scitranslmed.aar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoellein A., Fallahi M., Schoeffmann S., Steidle S., Schaub F.X., Rudelius M., et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 2014;124(13):2081–2090. doi: 10.1182/blood-2014-06-584524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H., Liu H., Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung J.H., Lee H.-J., Kim J.-H., Sim D.Y., Im E., Kim S., et al. Colocalization of MID1IP1 and c-Myc is Critically Involved in Liver Cancer Growth via Regulation of Ribosomal Protein L5 and L11 and CNOT2. Cells. 2020;9(4) doi: 10.3390/cells9040985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buendia M.-A., Bourre L., Cairo S. Myc target miRs and liver cancer: small molecules to get Myc sick. Gastroenterology. 2012;142(2):214–218. doi: 10.1053/j.gastro.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 21.Li C., Tang Z., Zhang W., Ye Z., Liu F. GEPIA2021: integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021;49(W1):W242–W246. doi: 10.1093/nar/gkab418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T., Ruan S., Zhao X., Shi X., Teng H., Zhong J., et al. OncoVar: an integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res. 2021;49(D1):D1289–D1301. doi: 10.1093/nar/gkaa1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao C., Zhou Y., Wang H., Deng F., Chen Y., Zhu X., et al. Adipose-derived stem cells alleviate radiation-induced dermatitis by suppressing apoptosis and downregulating cathepsin F expression. Stem Cell Res Ther. 2021;12(1):447. doi: 10.1186/s13287-021-02516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang S., Dong L., Liu L., Guo J., Zhao L., Zhang J., et al. HERB: a high-throughput experiment- and reference-guided database of traditional Chinese medicine. Nucleic Acids Res. 2021;49(D1):D1197–D1206. doi: 10.1093/nar/gkaa1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka K., Yu H.A., Yang S., Han S., Selcuklu S.D., Kim K., et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 2021;39(9) doi: 10.1016/j.ccell.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pabla S., Conroy J.M., Nesline M.K., Glenn S.T., Papanicolau-Sengos A., Burgher B., et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J Immunother Cancer. 2019;7(1):27. doi: 10.1186/s40425-019-0506-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berumen J., Espinosa A.M., Medina I. Targeting CDKN3 in cervical cancer. Expert Opin Ther Targets. 2014;18(10):1149–1162. doi: 10.1517/14728222.2014.941808. [DOI] [PubMed] [Google Scholar]
- 28.Li Y., Gu J., Xu F., Zhu Q., Ge D., Lu C. Novel methylation-driven genes identified as prognostic indicators for lung squamous cell carcinoma. Am J Transl Res. 2019;11(4):1997–2012. [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C., Hao X., Zhang S., Hu W., Li J., Sun J., et al. Characterization of the prognostic values of the family in gastric cancer. Therap Adv Gastroenterol. 2019;12 doi: 10.1177/1756284819858507. 1756284819858507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timar J., Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020;39(4):1029–1038. doi: 10.1007/s10555-020-09915-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garnis C., Buys T.P.H., Lam W.L. Genetic alteration and gene expression modulation during cancer progression. Mol Cancer. 2004;3:9. doi: 10.1186/1476-4598-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding X., He M., Chan A.W.H., Song Q.X., Sze S.C., Chen H., et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology. 2019;157(6) doi: 10.1053/j.gastro.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Craig A.J., von Felden J., Garcia-Lezana T., Sarcognato S., Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17(3):139–152. doi: 10.1038/s41575-019-0229-4. [DOI] [PubMed] [Google Scholar]
- 34.Affo S., Yu L.-X., Schwabe R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dang C.V. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massó-Vallés D., Beaulieu M.-E., Soucek L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin Ther Targets. 2020;24(2):101–114. doi: 10.1080/14728222.2020.1723548. [DOI] [PubMed] [Google Scholar]
- 37.Jiang J., Wang J., Yue M., Cai X., Wang T., Wu C., et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell. 2020;37(2) doi: 10.1016/j.ccell.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng K., He Z., Kitazato K., Wang Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics. 2019;9(1):104–125. doi: 10.7150/thno.30308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z., Yu Z., Wang G.-H., Zhou Y.-M., Deng J.-P., Feng Y., et al. Promotes the Metastasis of Gastric Cancer, Possibly by Inducing EMT. Cancer Manag Res. 2020;12:6947–6958. doi: 10.2147/CMAR.S254250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Q., Ding L., Zi Z., Gao S., Wang C., Wang Y., et al. Viral-Mediated AURKB Cleavage Promotes Cell Segregation and Tumorigenesis. Cell Rep. 2019;26(13) doi: 10.1016/j.celrep.2019.02.106. [DOI] [PubMed] [Google Scholar]
- 41.Long J., Huang S., Bai Y., Mao J., Wang A., Lin Y., et al. Transcriptional landscape of cholangiocarcinoma revealed by weighted gene coexpression network analysis. Brief Bioinform. 2021;22(4) doi: 10.1093/bib/bbaa224. [DOI] [PubMed] [Google Scholar]
- 42.Nie M., Wang Y., Yu Z., Li X., Deng Y., Wang Y., et al. AURKB promotes gastric cancer progression via activation of expression. Aging (Albany NY) 2020;12(2):1304–1321. doi: 10.18632/aging.102684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen B., Dragomir M.P., Fabris L., Bayraktar R., Knutsen E., Liu X., et al. The Long Noncoding RNA CCAT2 Induces Chromosomal Instability Through BOP1-AURKB Signaling. Gastroenterology. 2020;159(6) doi: 10.1053/j.gastro.2020.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian X., Song X., He Y., Yang Z., Sun T., Wang J., et al. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed Pharmacother. 2015;74:222–227. doi: 10.1016/j.biopha.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Li J., Tang J.-X., Cheng J.-M., Hu B., Wang Y.-Q., Aalia B., et al. Cyclin B2 can compensate for Cyclin B1 in oocyte meiosis I. J Cell Biol. 2018;217(11):3901–3911. doi: 10.1083/jcb.201802077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daldello E.M., Luong X.G., Yang C.-R., Kuhn J., Conti M. Cyclin B2 is required for progression through meiosis in mouse oocytes. Development. 2019;146(8) doi: 10.1242/dev.172734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Zhang H., Wang M., He J., Guo H., Li L., et al. CCNB2/SASP/Cathepsin B & PGE2 Axis Induce Cell Senescence Mediated Malignant Transformation. Int J Biol Sci. 2021;17(13):3538–3553. doi: 10.7150/ijbs.63430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia T., Meng L., Zhao Z., Li Y., Wen H., Sun H., et al. Bioinformatics prediction and experimental verification identify MAD2L1 and CCNB2 as diagnostic biomarkers of rhabdomyosarcoma. Cancer Cell Int. 2021;21(1):634. doi: 10.1186/s12935-021-02347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao X., Wang X., Zhang S. Bioinformatics identification of crucial genes and pathways associated with hepatocellular carcinoma. Biosci Rep. 2018;38(6) doi: 10.1042/BSR20181441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cress W.D., Yu P., Wu J. Expression and alternative splicing of the cyclin-dependent kinase inhibitor-3 gene in human cancer. Int J Biochem Cell Biol. 2017;91(Pt B) doi: 10.1016/j.biocel.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cen J., Liang Y., Huang Y., Pan Y., Shu G., Zheng Z., et al. Circular RNA circSDHC serves as a sponge for miR-127-3p to promote the proliferation and metastasis of renal cell carcinoma via the CDKN3/E2F1 axis. Mol Cancer. 2021;20(1):19. doi: 10.1186/s12943-021-01314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J., Che W., Wang W., Su G., Zhen T., Jiang Z. CDKN3 promotes tumor progression and confers cisplatin resistance via RAD51 in esophageal cancer. Cancer Manag Res. 2019;11:3253–3264. doi: 10.2147/CMAR.S193793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li T., Xue H., Guo Y., Guo K. CDKN3 is an independent prognostic factor and promotes ovarian carcinoma cell proliferation in ovarian cancer. Oncol Rep. 2014;31(4):1825–1831. doi: 10.3892/or.2014.3045. [DOI] [PubMed] [Google Scholar]
- 54.Liu X., Wu J., Zhang D., Bing Z., Tian J., Ni M., et al. Identification of Potential Key Genes Associated With the Pathogenesis and Prognosis of Gastric Cancer Based on Integrated Bioinformatics Analysis. Front Genet. 2018;9:265. doi: 10.3389/fgene.2018.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai J.Y., Wang X., Wang B., Sun W., Jordahl K.M., Kolb S., et al. DNA methylation and cis-regulation of gene expression by prostate cancer risk SNPs. PLoS Genet. 2020;16(3):e1008667. doi: 10.1371/journal.pgen.1008667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y., Liu C., Cheng H., Tian S., Liu Y., Wang S., et al. DNA methylation and its effects on gene expression during primary to secondary growth in poplar stems. BMC Genomics. 2020;21(1):498. doi: 10.1186/s12864-020-06902-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klupczyńska E.A., Ratajczak E. Can Forest Trees Cope with Climate Change?-Effects of DNA Methylation on Gene Expression and Adaptation to Environmental Change. Int J Mol Sci. 2021;22(24) doi: 10.3390/ijms222413524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen M., Wong C.-M. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer. 2020;19(1):44. doi: 10.1186/s12943-020-01172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klutstein M., Nejman D., Greenfield R., Cedar H. DNA Methylation in Cancer and Aging. Cancer Res. 2016;76(12):3446–3450. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- 60.Yi L., Wu G., Guo L., Zou X., Huang P. Comprehensive Analysis of the PD-L1 and Immune Infiltrates of mA RNA Methylation Regulators in Head and Neck Squamous Cell Carcinoma. Mol Ther Nucleic Acids. 2020;21:299–314. doi: 10.1016/j.omtn.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X., Xu M., Xu X., Zeng K., Liu X., Sun L., et al. METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol Ther. 2020;28(2):599–612. doi: 10.1016/j.ymthe.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Fujii S.-I., Shimizu K. Immune Networks and Therapeutic Targeting of iNKT Cells in Cancer. Trends Immunol. 2019;40(11):984–997. doi: 10.1016/j.it.2019.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Lhuillier C., Rudqvist N.-P., Yamazaki T., Zhang T., Charpentier M., Galluzzi L., et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J Clin Invest. 2021;131(5) doi: 10.1172/JCI138740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mondino A., Manzo T. To Remember or to Forget: The Role of Good and Bad Memories in Adoptive T Cell Therapy for Tumors. Front Immunol. 2020;11:1915. doi: 10.3389/fimmu.2020.01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang Y., Zhang A.X.J., Chen G., Wu Y., Gu W. Prognostic and therapeutic TILs of cervical cancer-Current advances and future perspectives. Mol Ther Oncolytics. 2021;22:410–430. doi: 10.1016/j.omto.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu C., Sui S., Shang Y., Yu Z., Han J., Zhang G., et al. The landscape of immune cell infiltration and its clinical implications of pancreatic ductal adenocarcinoma. J Adv Res. 2020;24:139–148. doi: 10.1016/j.jare.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Punt S., Malu S., McKenzie J.A., Manrique S.Z., Doorduijn E.M., Mbofung R.M., et al. Aurora kinase inhibition sensitizes melanoma cells to T-cell-mediated cytotoxicity. Cancer Immunol Immunother. 2021;70(4):1101–1113. doi: 10.1007/s00262-020-02748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye Z.-S., Zheng M., Liu Q.-Y., Zeng Y., Wei S.-H., Wang Y., et al. Survival-associated alternative splicing events interact with the immune microenvironment in stomach adenocarcinoma. World J Gastroenterol. 2021;27(21):2871–2894. doi: 10.3748/wjg.v27.i21.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mizukoshi E., Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol. 2019;12(1):52. doi: 10.1186/s13045-019-0742-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fu Y., Liu S., Zeng S., Shen H. From bench to bed: the tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):396. doi: 10.1186/s13046-019-1396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma J., Zheng B., Goswami S., Meng L., Zhang D., Cao C., et al. PD1 CD8 T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):331. doi: 10.1186/s40425-019-0814-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.