

Abstract
Acute lung injury (ALI) is characterized by lung vascular endothelial cell (EC) barrier compromise resulting in increased endothelial permeability and pulmonary edema. The infection of gram-negative bacteria that produce toxins like LPS is one of the major causes of ALI. LPS activates Toll-like receptor 4, leading to cytoskeleton reorganization, resulting in lung endothelial barrier disruption and pulmonary edema in ALI. However, the signaling pathways that lead to the cytoskeleton reorganization and lung microvascular EC barrier disruption remain largely unexplored. Here we show that LPS induces calpain activation and talin cleavage into head and rod domains and that inhibition of calpain attenuates talin cleavage, RhoA activation, and pulmonary EC barrier disruption in LPS-treated human lung microvascular ECs in vitro and lung EC barrier disruption and pulmonary edema induced by LPS in ALI in vivo. Moreover, overexpression of calpain causes talin cleavage and RhoA activation, myosin light chain (MLC) phosphorylation, and increases in actin stress fiber formation. Furthermore, knockdown of talin attenuates LPS-induced RhoA activation and MLC phosphorylation and increased stress fiber formation and mitigates LPS-induced lung microvascular endothelial barrier disruption. Additionally, overexpression of talin head and rod domains increases RhoA activation, MLC phosphorylation, and stress fiber formation and enhances lung endothelial barrier disruption. Finally, overexpression of cleavage-resistant talin mutant reduces LPS-induced increases in MLC phosphorylation in human lung microvascular ECs and attenuates LPS-induced lung microvascular endothelial barrier disruption. These results provide the first evidence that calpain mediates LPS-induced lung microvascular endothelial barrier disruption in ALI via cleavage of talin.
Keywords: acute lung injury, pulmonary edema, vascular endothelial cells, ARDS, myosin light chain
Clinical Relevance
The mechanisms involved in LPS-induced acute lung injury (ALI) is not known. This study confirms the mediating role of calpain in LPS-induced lung microvascular endothelial barrier disruption. The results indicate that LPS-induced activation of calpain causes talin cleavage leading to RhoA activation, myosin light chain phosphorylation, actin stress fiber formation and lung microvascular endothelial hyperpermeability. The data from multidisciplinary approaches provides rationale for manipulating calpain in the treatment of ALI.
Acute lung injury (ALI) is characterized by lung vascular endothelial cell (EC) barrier compromise resulting in increased endothelial permeability and pulmonary edema (1, 2). The infection of gram-negative bacteria that produce toxins like lipopolysaccharides (LPS) is one of the major causes of ALI (3). Despite the use of potent antibiotics and aggressive intensive care support, the mortality rate associated with ALI is still high, and an understanding of the molecular mechanisms involved in LPS-induced ALI is urgently needed.
LPS, a proinflammatory mediator and a constituent of gram-negative bacteria cell walls, compromises EC barrier function in vitro and in vivo (4, 5). LPS activates Toll-like receptor 4, leading to RhoA activation and phosphorylation of myosin light chain (MLC) phosphatase (MLCP) targeting subunit 1 (MYPT1) in human lung microvascular ECs (HLMVECs), resulting in MLCP inhibition, MLC phosphorylation, and the increased formation of actin stress fibers, reflecting increases in EC contractility and barrier dysfunction (6, 7). Nevertheless, the signaling pathways that lead to cytoskeleton reorganization and lung microvascular EC barrier disruption remain largely unexplored.
Calpain represents a family of calcium-dependent nonlysosomal neutral cysteine endopeptidases that act via limited proteolysis of substrate proteins in mammalian cells, including HLMVECs (8). There are 15 isozymes in the family. Calpain-1 and calpain-2 are two major typical calpains and are responsible for calpain activity in lung ECs (9). Calpain-1 and calpain-2 isoforms consist of a distinct large subunit (80 kD) and a common small subunit (also named calpain-4; 30 kD) that supports calpain activities (8). Activation of calpain contributes to many physiological and pathological processes such as cell migration and proliferation, angiogenesis, and vascular remodeling (9–11). In the present study, we found that LPS activates calpain in HLMVECs and that the specific calpain inhibitor MDL28170 prevents barrier disruption of HLMVECs induced by LPS and LPS-induced ALI in vivo, suggesting that calpain activation is involved in LPS-induced ALI. It has been reported that calpain cleaves talin, and talin cleavage separates the head domain from the rod domain, thereby removing autoinhibition and stimulating talin head domain binding to integrin, inducing focal adhesion (FA) activation (12, 13). Talin-integrin-actin binding and formation of FAs are critical for RhoA activation, MLCP inhibition, MLC phosphorylation, and increased permeability in human lung ECs stimulated by edemagenic agents (14, 15). In the present study, we found for the first time that LPS induces talin cleavage into the head and rod domains in HLMVECs. These observations led us to hypothesize that calpain activation regulates talin cleavage and activation, leading to endothelial barrier disruption in ALI. Here we demonstrate that overexpression of calpain causes talin cleavage, RhoA activation, and MLCP phosphorylation, resulting in MLCP inhibition and MLC phosphorylation. We also found that depletion of talin abolishes an LPS-induced increase in stress fibers and attenuates an LPS-induced increase in MLC phosphorylation, and that overexpression of talin head and rod domains increases RhoA activation and MLC phosphorylation and enhances lung endothelial barrier dysfunction, indicating that calpain mediates LPS-induced EC permeability response via talin cleavage and activation in ALI.
Methods
An extended materials and methods section is included in the data supplement.
Conditional Endothelial-Specific Calpain-4–Knockout Mice
A conditional endothelial-specific calpain-4–knockout (KO) Capn4flox/flox;end-SCL-Cre-ERT/+;R26R/+ mouse line was obtained by crossing Capn4flox/flox with end-SCL-Cre-ERT and Gt (ROSA) 26Sortm1Sor/J mice. Deletion of calpain-4 was induced by tamoxifen (20 mg/kg/day, i.p.) for 5 days. Control mice were littermate Capn4flox/flox or Capn4flox/+ mice treated with the same tamoxifen regimen.
Mouse ALI Model
Endothelial-specific calpain-4–KO mice and their littermate control mice (8–10 wk) were anesthetized. LPS (1 mg/ml) or saline solution (control) was instilled intratracheally. Lung injuries were assessed 18–24 hours after LPS treatment. For the calpain inhibitor MDL28170 experiment, male and female C57BL6 mice 8 weeks of age were injected with MDL28170 or DMSO (20 mg/kg, i.p.) 30 minutes before and 8 hours after LPS (1 mg/kg) treatment. Lung injuries were assessed 18–24 hours after LPS treatment.
HLMVEC Culture
HLMVECs was purchased from Lonza Group Ltd. (CC-2527). Cells were cultured with 5% CO2 at 37°C using microvascular EC growth medium-2. HLMVECs from passages 3–5 were used in the in vitro experiments.
siRNA and Plasmid DNA Transfection
HLMVECs were transfected with calpain-1, calpain-2, and talin1 siRNA with Lipofectamine RNAiMAX Reagent. Cells were treated with LPS 48 hours after transfection. For plasmid transfection, talin1 head domain, talin1 rod domain, wild-type talin1, mutant talin-L432G, and C1 plasmid DNA were purified. HLMVECs were transfected with plasmid DNA using X-tremeGenE HP DNA transfection reagent. The cells were treated with LPS 48 hours after transfection.
Transwell Permeability Assay
HLMVECs were planted in the inserts (0.4‐μm pore size) with a density of 1 × 105 cells per insert and cultured to form a monolayer. Forty-eight hours after siRNA transfection, the cells were exposed to LPS for 12 hours, and then the culture medium in the inserts was replaced with 100 μl medium containing FITC-dextran (70 kD, 1 mg/ml) for 1 hour. Culture medium from the upper and lower chambers were then assayed for fluorescence.
Transendothelial Electrical Resistance Measurements
HLMVECs were planted into electric cell substrate impedance sensing array chambers (8W10E; Applied BioPhysics). Experiments were begun when the resistance was stable between 2,000 and 3,000 Ω, and the cells were then exposed to LPS to initiate changes in barrier function. Resistance was normalized to each well’s value at time 0.
Immunofluorescence Staining of HLMVECs
HLMVECs were incubated with LPS (0.2 μg/ml) for 12 hours and then stained with rhodamine phalloidin for 30 minutes. Cells were mounted and then scanned using a Zeiss LSM780 laser confocal microscope.
RhoA Activity Pull-Down Assay
RhoA activation was assessed by a pull-down assay using a RhoA Activation Assay Biochem Kit (BK036; Cytoskeleton).
Study Approval
All animal experiments were approved by the institutional animal care and use committee of Augusta University and Charlie Norwood Veterans Affairs Medical Center.
Statistical Analysis
Results are shown as the mean ± SE for n experiments. One-way ANOVA and t test analyses were used to determine the significance of differences between groups. P < 0.05 was considered significant.
Results
Inhibition of Calpain Attenuates Lung EC Barrier Disruption and Pulmonary Edema Induced by LPS in ALI in vivo
To investigate the role of calpain in lung microvascular barrier disruption induced by LPS, calpain activity was inhibited by the specific calpain inhibitor MDL28170 in an LPS-induced ALI mouse model. MDL28170 was given (20 mg/kg i.p.) 30 minutes before and 8 hours after LPS (1 mg/kg), or sterile saline solution was instilled intratracheally into mouse lungs. After 18 hours, lung vascular leakage was assessed based on Evans blue dye (EBD)–conjugated albumin flux, wet/dry lung ratio, and BAL fluid protein content. The results showed that MDL28170 significantly attenuated pulmonary edema and alveolar wall thickening (Figures 1A and E1A in the data supplement) and the increases in lung EBD levels, wet/dry lung ratio, and BAL fluid protein levels (Figures 1B–1D and see Figures E1B–E1D), suggesting that calpain is involved lung EC barrier dysfunction in LPS-induced ALI.
Figure 1.
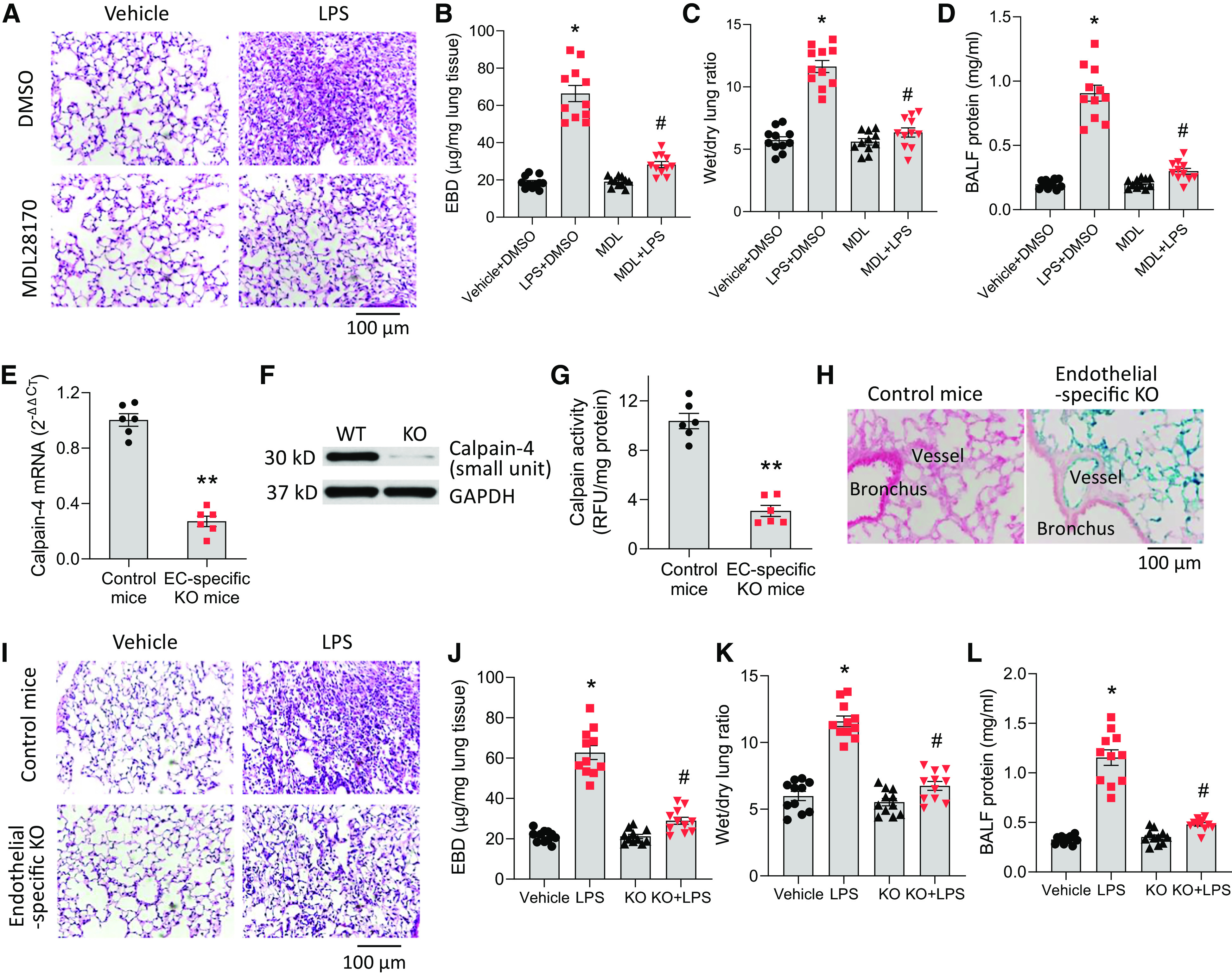
Inhibition of calpain attenuates lung endothelial cell (EC) barrier disruption and pulmonary edema induced by LPS in acute lung injury in vivo. (A–D) Eight-week-old mice (eight male, three female) were injected with the specific calpain inhibitor MDL28170 (20 mg/kg, in 50 μl DMSO, i.p.) or DMSO (50 μl) 30 minutes before and 8 hours after LPS (1 mg/kg), or sterile saline solution was instilled intratracheally into mouse lungs. After 18 hours, lung vascular leakage was assessed by using Evans blue dye–conjugated albumin flux (B), wet/dry lung ratio (C), and BAL fluid protein content (D). (A) Representative images of lung sections of control mice and mice treated with LPS and/or MDL28170. Results are expressed as mean ± SE (n = 11). *P < 0.05 versus vehicle+ DMSO; #P < 0.05 versus LPS + DMSO. (E–L) Capn4flox/flox;end-SCL-Cre-ERT;R26R/+ mice (age 8 wk; eight male and three female) were injected with tamoxifen (20 mg/kg/d, i.p.) for 5 days. Control mice were littermate Capn4flox/flox or Capn4flox/+ mice (age 8 wk, eight male and three female) with the same tamoxifen treatment. After 10 days, the mouse lung microvascular ECs were isolated from lung tissues for the measurement of calpain-4 mRNA (E), protein (F), and calpain activity (G). (H) β-Gal staining of the lung tissues. LPS (1 mg/kg) or sterile saline solution was instilled intratracheally into mouse lungs. After 18 hours, lung vascular leakage was assessed by using Evans blue dye–conjugated albumin flux (J), wet/dry lung ratio (K), and BAL fluid protein content (L). (I) Representative images of lung sections of control mice and Capn4flox/flox;end-SCL-Cre-ERT;R26R/+ mice treated with LPS. Results are expressed as mean ± SE (n = 11); *P < 0.05 versus vehicle; **P < 0.01 versus control mice; #P < 0.05 versus LPS. BALF = BAL fluid; EBD = Evans blue dye; KO = knockout; MDL = MDL28170; RFU = relative fluorescence unit; WT = wild-type. Scale bars, 100 µm.
To further study the specific role of calpain in lung microvascular EC barrier disruption in ALI, calpain was inhibited in a mouse line of EC-specific KO of calpain-4. Because the activities of calpain-1 and -2 require calpain-4 (small unit), KO of calpain-4 prevents activation of calpain-1 and -2. KO of calpain-4 was induced by injecting tamoxifen (20 mg/kg/d, i.p.) for 5 days. Control mice were littermate Capn4flox/flox or Capn4flox/+ mice that received the same tamoxifen treatment. The mouse lung microvascular ECs isolated from lung tissues of Capn4flox/flox;end-SCL-Cre-ERT;R26R/+ mice exhibit robustly lower calpain-4 mRNA, protein, and calpain activity (Figures 1E–1G). The β-gal staining of the lungs showed that strong Cre activities (blue color) were present in lung vascular endothelium (Figure 1H). There was no Cre activity in airway epithelium or airway and vascular smooth muscle, indicating that the induction of Cre was specifically achieved in the endothelium. Ten days after tamoxifen induction, LPS (1 mg/kg) or sterile saline solution was instilled intratracheally into mouse lungs. The data showed that LPS-induced pulmonary edema and alveolar wall thickening (Figure 1I) and increases in lung EBD levels, wet/dry lung ratio, and BAL fluid protein levels were attenuated in Capn4flox/flox; end-SCL-Cre-ERT;R26R/+ mice (Figures 1J–1L). Taken together, these results show that inhibition of calpain attenuates lung EC barrier disruption and pulmonary edema induced by LPS in ALI.
LPS Stimulates Calpain Activity, and Inhibition of ERK or p38 MAPK Attenuates LPS-induced Calpain Activation
HLMVECs were incubated with LPS (0.1–0.5 μg/ml) for 4–24 hours, and calpain activity was measured by detecting the calpain-mediated cleavage of the fluorogenic peptide Suc-Leu-Leu-Val-Tyr-AMC as previously described by our group (10, 16). As shown in Figures 2A and 2B, LPS treatment significantly increased calpain activity in HLMVECs in dose-dependent (LPS treatment for 24 h; Figure 2A) and time-dependent manners (LPS 0.2 μg/ml; Figure 2B). The LPS-induced increases were not accompanied by changes in protein levels of calpain-1 and -2 (Figure E2A). We and others have reported that ERK-mediated phosphorylation modulates calpain activity and increases its Ca2+ sensitivity (10, 17). Phosphorylation and activation of calpain-2 can be prevented by inhibition of p38 MAPK (mitogen-activated protein kinase) (18). Thus, we determined the effects of ERK and p38 MAPK inhibition on LPS-induced increases in calpain activity in HLMVECs. The results showed that the ERK inhibitor PD98059 and the p38 MAPK inhibitor SB203580 reduced the LPS-induced increases in calpain activity in HLMVECs (see Figures E2B and E2C), suggesting that LPS increases calpain activities via ERK and p38 MAPK.
Figure 2.
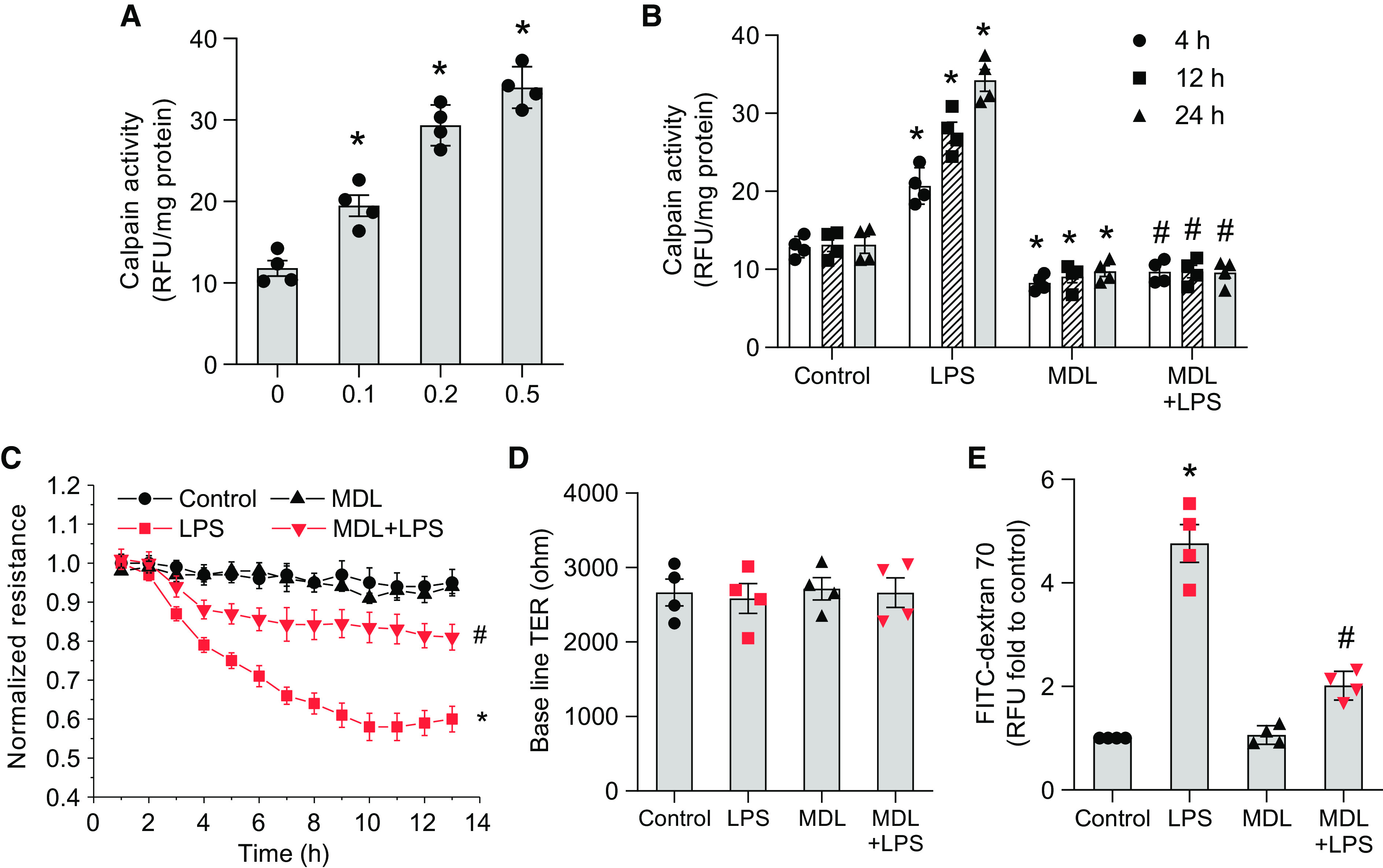
LPS stimulates calpain activity, and calpain inhibition attenuates pulmonary EC barrier disruption in vitro. Human lung microvascular ECs (HLMVECs) were incubated with LPS (0.1–0.5 μg/ml) for 4–24 hours, and then calpain activities were measured by detecting the calpain-mediated cleavage of the fluorogenic peptide Suc-Leu-Leu-Val-Tyr-AMC. (A) LPS treatment for 24 hours. (B) LPS 0.2 μg/ml treatment for 4–24 hours. (C–E) HLMVEC monolayers were incubated with MDL28170 (20 μM) for 30 minutes before LPS (0.2 μg/ml) was added, and then the transendothelial electrical resistance (TER) value was monitored in real time as a measurement of barrier integrity of the HLMVEC monolayer (C and D). The HLMVEC monolayer permeability to FITC-dextran 70 was measured by using a Transwell system (E). Results are expressed as mean ± SE (n = 4); *P < 0.05 versus control; #P < 0.05 versus LPS.
Calpain Inhibition Attenuates Pulmonary EC Barrier Disruption in vitro
HLMVEC monolayers were incubated with MDL28170 (20 μM) for 30 minutes before LPS (0.2 μg/ml) was added, and the transendothelial electrical resistance (TER) value was then monitored in real time as a measurement of the barrier integrity of the HLMVEC monolayer. The HLMVEC monolayer’s permeability to FITC-dextran 70 was measured by using a Transwell system. The endothelial cadherin junction assembly was assessed by immunostaining of VE-cadherin. The data showed that the inhibition of calpain using MDL28170 attenuated the decreases in TER value (Figures 2C and 2D) and the hyperpermeability to FITC-dextran 70 (Figure 2E) in HLMVECs treated with LPS and prevented LPS-induced endothelial cadherin junction disassembly (Figure E3). Altogether, these results indicate that LPS stimulates calpain activity and that calpain inhibition attenuates pulmonary EC barrier disruption in vitro.
Knockdown of Calpain-1 and Calpain-2 Attenuates Lung Microvascular Endothelial Barrier Disruption Induced by LPS
To further investigate whether it is calpain-1 or -2 that contributes to LPS-induced calpain activation and endothelial barrier compromise in HLMVECs, calpain-1 and -2 were knocked down. As shown in Figures 3A and 3B, knockdown of calpain-1 slightly reduced, but knockdown of calpain-2 markedly inhibited, calpain activation induced by LPS. Correspondingly, knockdown of calpain-1 slightly attenuated, but knockdown of calpain-2 greatly inhibited, LPS-induced decreases in the TER value (Figures 3D–3G) and the hyperpermeability to FITC-dextran 70 (Figure 3C) in HLMVECs treated with LPS. These data show that it is calpain-2, rather than calpain-1, that is mainly responsible for LPS-induced calpain activation and lung microvascular endothelial barrier disruption.
Figure 3.
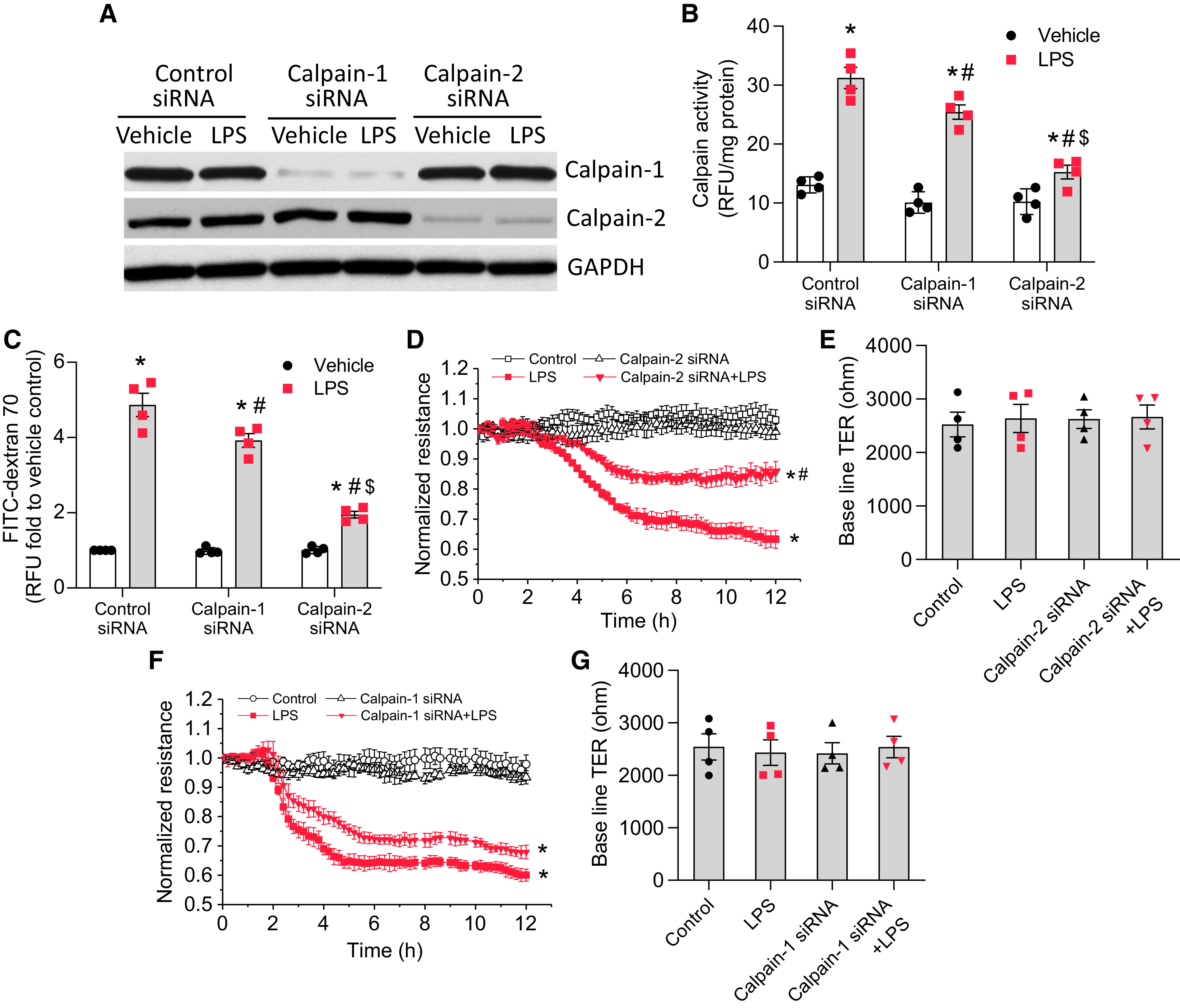
Knockdown of calpain-1 and calpain-2 attenuates lung microvascular endothelial barrier disruption induced by LPS. HLMVECs were transfected with siRNAs against calpain-1 or -2 or control siRNA. After 72 hours, the cells were incubated with LPS (0.25 μg/ml) for 12 hours, after which calpain activity (B) and the HLMVEC monolayer permeability to FITC-dextran 70 (C) were measured and during which the TER value (D–G) was monitored. (A) The blots are representative immunoblots of four independent experiments. Results are expressed as mean ± SE (n = 4); *P < 0.05 versus vehicle; #P < 0.05 versus LPS in control siRNA; $P < 0.05 versus LPS in calpain-1 siRNA.
Overexpression of Calpain-2 Promotes Talin Cleavage and Increases Actin Stress Fiber Formation, RhoA Activation, and Phosphorylation of MYPT1 and MLC in HLMVECs
HLMVECs were transfected with empty plasmids and pcDNA3.1/V5-His plasmids containing human calpain-2 for 3 days, after which p-Thr853-MYPT1, total MYPT1, pp-MLC, total MLC, and RhoA were analyzed. Talin protein was assayed by Western blot using antibodies against the N-terminal and C-terminal domains. As shown in Figure 4, calpain-2 overexpression increased calpain activity (Figure 4A) and significantly increased talin cleavage into the 45-kD head domain and the 190-kD rod domain (Figures 4B and 4C), phosphorylation of MYPT1 and MLC, and RhoA activation (Figures 4D–4G), as well as stress fiber formation (Figure 4H), in HLMVECs. These data indicate that the increases in calpain activity cause talin cleavage/activation, RhoA activation, and cytoskeletal reorganization.
Figure 4.
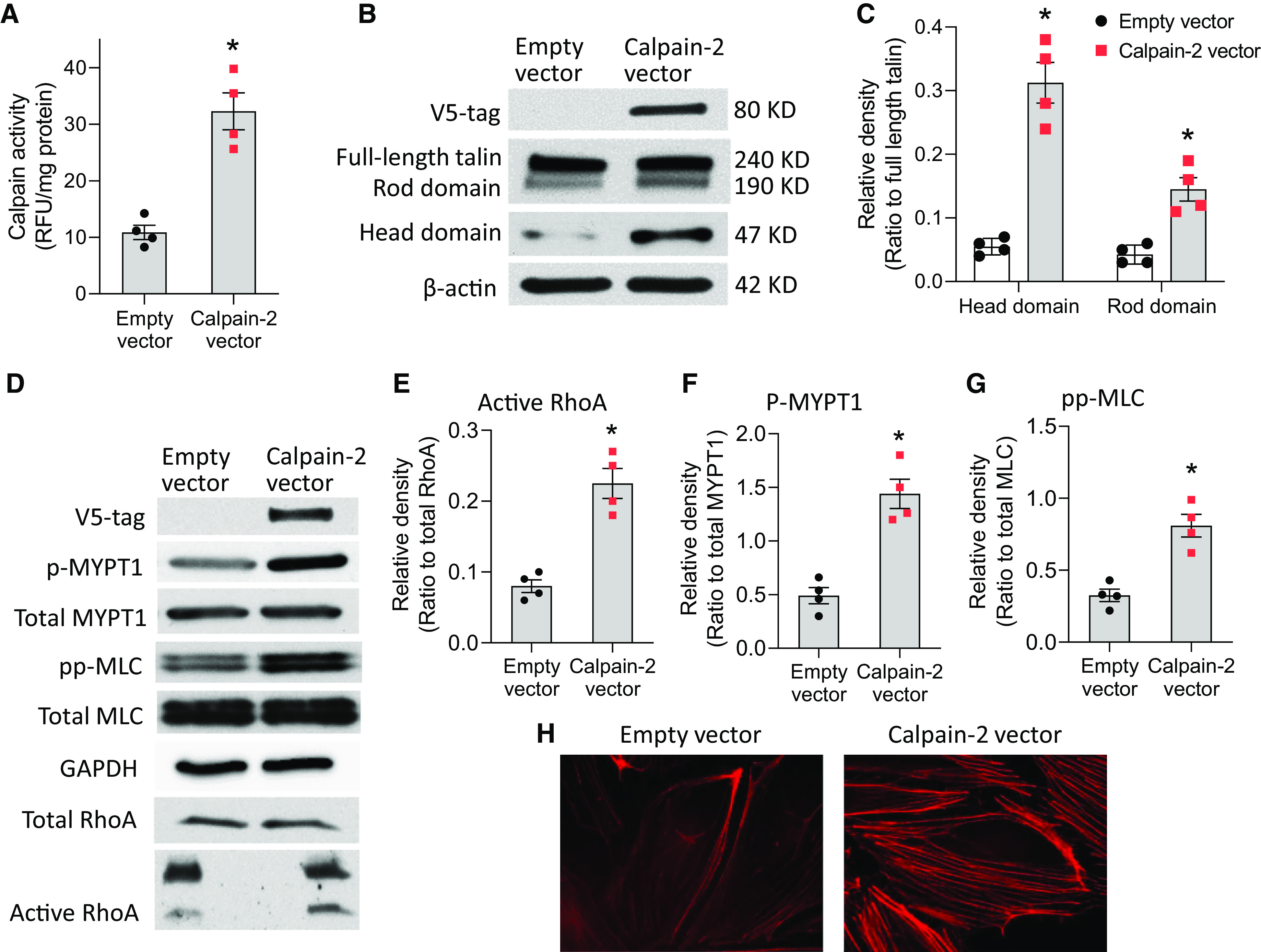
Overexpression of calpain-2 promotes talin cleavage and increases actin stress fiber formation, RhoA activation, and phosphorylation of MYPT1 (myosin light chain [MLC] phosphatase targeting subunit 1) and MLC in HLMVECs. HLMVECs were transfected with empty plasmids and pcDNA3.1/V5-His plasmids containing human calpain-2 for 3 days, after which calpain activities were assayed (A); p-Thr853-MYPT1, total MYPT1, pp-MLC, total MLC, and RhoA activation were measured (B–G); and cells were stained using rhodamine phalloidin (H). Talin protein was assayed by Western blot using antibodies against the N-terminal and C-terminal domains. Results are expressed as mean ± SE (n = 4). *P < 0.05 versus empty vector.
Knockdown of Calpain-2 Attenuates Talin Cleavage, RhoA Activation, MLC Phosphorylation, and Stress Fiber Formation in LPS-treated HLMVECs
We found that the incubation of HLMVECs with LPS resulted in talin cleavage into the head and rod domains (Figure 5A). To further investigate the role of calpain-2 in LPS-induced endothelial barrier compromise in HLMVECs, calpain-2 was knocked down. As shown in Figure 5, knockdown of calpain-2 reduced talin cleavage into the head and rod domains, RhoA activation, and MLC phosphorylation, as well as stress fiber formation, induced by LPS in HLMVECs. These results indicate that calpain may promote LPS-induced lung endothelial barrier dysfunction via a talin cleavage– related process.
Figure 5.

Knockdown of calpain-2 attenuates talin cleavage, RhoA activation, MLC phosphorylation, and stress fiber formation in HLMVECs. HLMVECs were transfected with siRNAs against calpain-2 or control siRNA. After 72 hours, the cells were incubated with LPS (0.25 μg/ml) for 12 hours, after which the talin head and rod domains, pp-MLC, total MLC, and RhoA activation were measured (A–D) and cells were stained using rhodamine phalloidin (E). Results are expressed as mean ± SE; n = 3. *P < 0.05 vs. Vehicle + Control siRNA; #P < 0.05 vs. LPS + Control siRNA.
Knockdown of Talin Reduces LPS-induced Increases in MLC Phosphorylation and Stress Fiber Formation in HLMVECs and Mitigates LPS-induced Lung Microvascular Endothelial Barrier Disruption
To study the role of talin in LPS-induced RhoA activation, MLC phosphorylation, stress fiber formation, and EC barrier compromise in HLMVECs, talin was knocked down in HLMVECs. As shown in Figures 6A–6E, knockdown of talin reduced the levels of cleaved head and rod domains and attenuated the LPS-induced RhoA activation and MLC phosphorylation as well as stress fiber formation in HLMVECs. Moreover, knockdown of talin attenuated the LPS-induced decreases in TER value (Figures 6F and 6G) and hyperpermeability to FITC-dextran 70 (Figure 6H) in HLMVECs. Collectively, these data strongly suggest that talin cleavage and activation are involved in LPS-induced cytoskeletal reorganization, leading to lung microvascular endothelial hyperpermeability.
Figure 6.
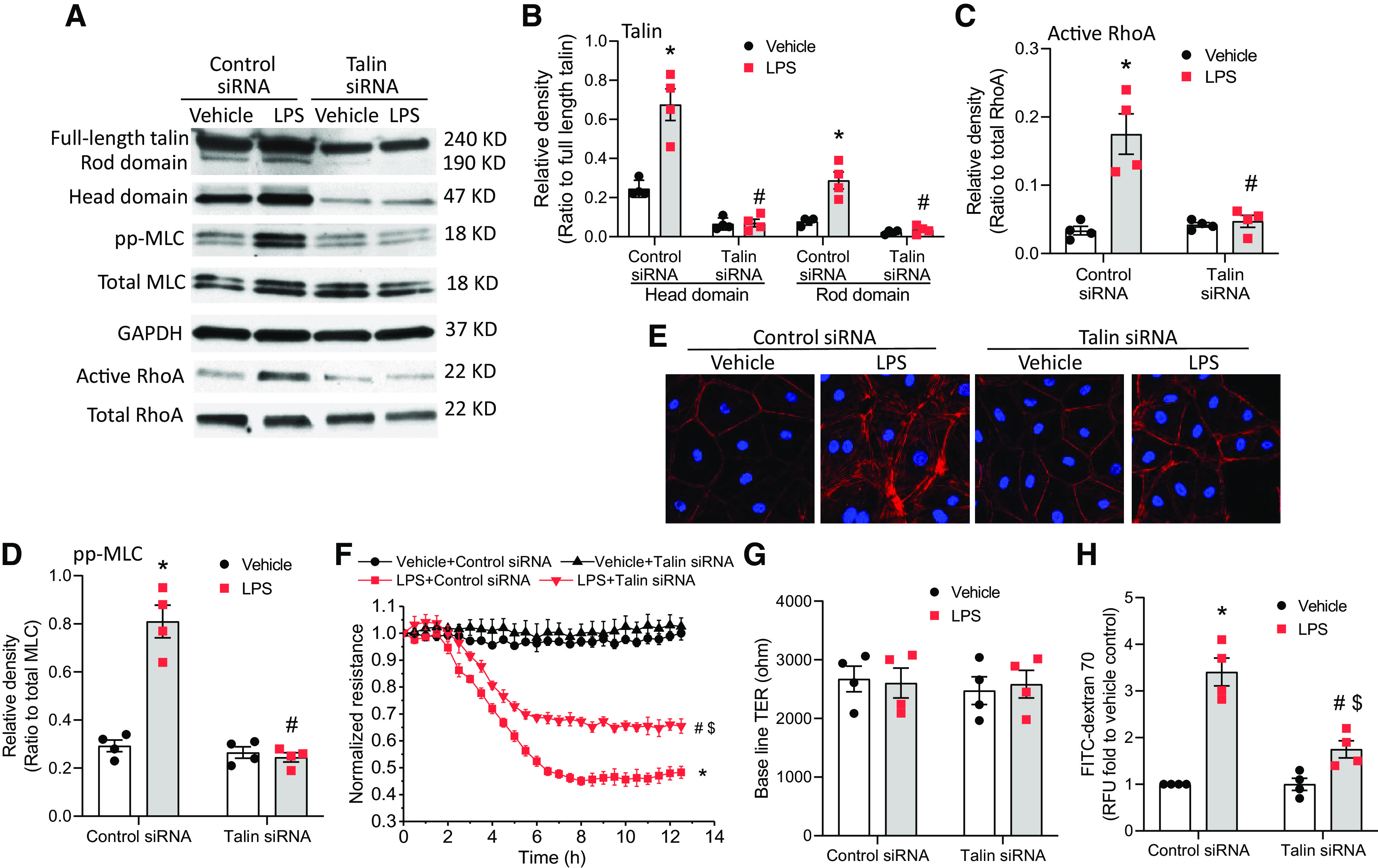
Knockdown of talin reduces LPS-induced increases in MLC phosphorylation and stress fiber formation in HLMVECs and mitigates LPS-induced lung microvascular endothelial barrier disruption. HLMVECs were transfected with siRNAs against talin or control siRNA. After 72 hours, the cells were incubated with LPS (0.25 μg/ml) for 12 hours, after which the talin head and rod domains, pp-MLC, total MLC, and RhoA activation were measured (A–D); cells were stained using rhodamine phalloidin (E); and HLMVEC monolayer permeability to FITC-dextran 70 (H) was measured and during which the TER value (F and G) was monitored. The blots and images are representative of four independent experiments. Results are expressed as mean ± SE; n = 4. *P < 0.05 versus vehicle + control siRNA; #P < 0.05 versus LPS + control siRNA; $P < 0.05 versus vehicle + talin siRNA.
Overexpression of Talin Head and Rod Domains Increases MLC Phosphorylation and Enhances Lung Microvascular Endothelial Barrier Disruption
To study the role of cleaved talin (head and rod domains) in LPS-induced cytoskeletal responses and lung microvascular endothelial barrier dysfunction, the talin head domain and rod domain were overexpressed in HLMVECs. As shown in Figures 7A–7E, overexpression of the talin head domain increased LPS-induced RhoA activation and MLC phosphorylation as well as stress fiber formation in HLMVECs. Moreover, overexpression of talin head domain exaggerated the LPS-induced decreases in the TER value (Figures 7F and 7G) and hyperpermeability to FITC-dextran 70 (Figure 7H) in HLMVECs. Furthermore, overexpression of the talin rod domain increased LPS-induced RhoA activation and MLC phosphorylation as well as stress fiber formation in HLMVECs (Figures 4A–4E). Correspondingly, LPS-induced decreases in the TER value (Figures E4F and E4G) and hyperpermeability to FITC-dextran 70 (see Figure E4H) are potentiated in HLMVECs with talin rod domain overexpression. These data confirm that cleaved talin (head and rod domains) promotes actin cytoskeletal reorganization and lung microvascular endothelial barrier dysfunction induced by LPS.
Figure 7.
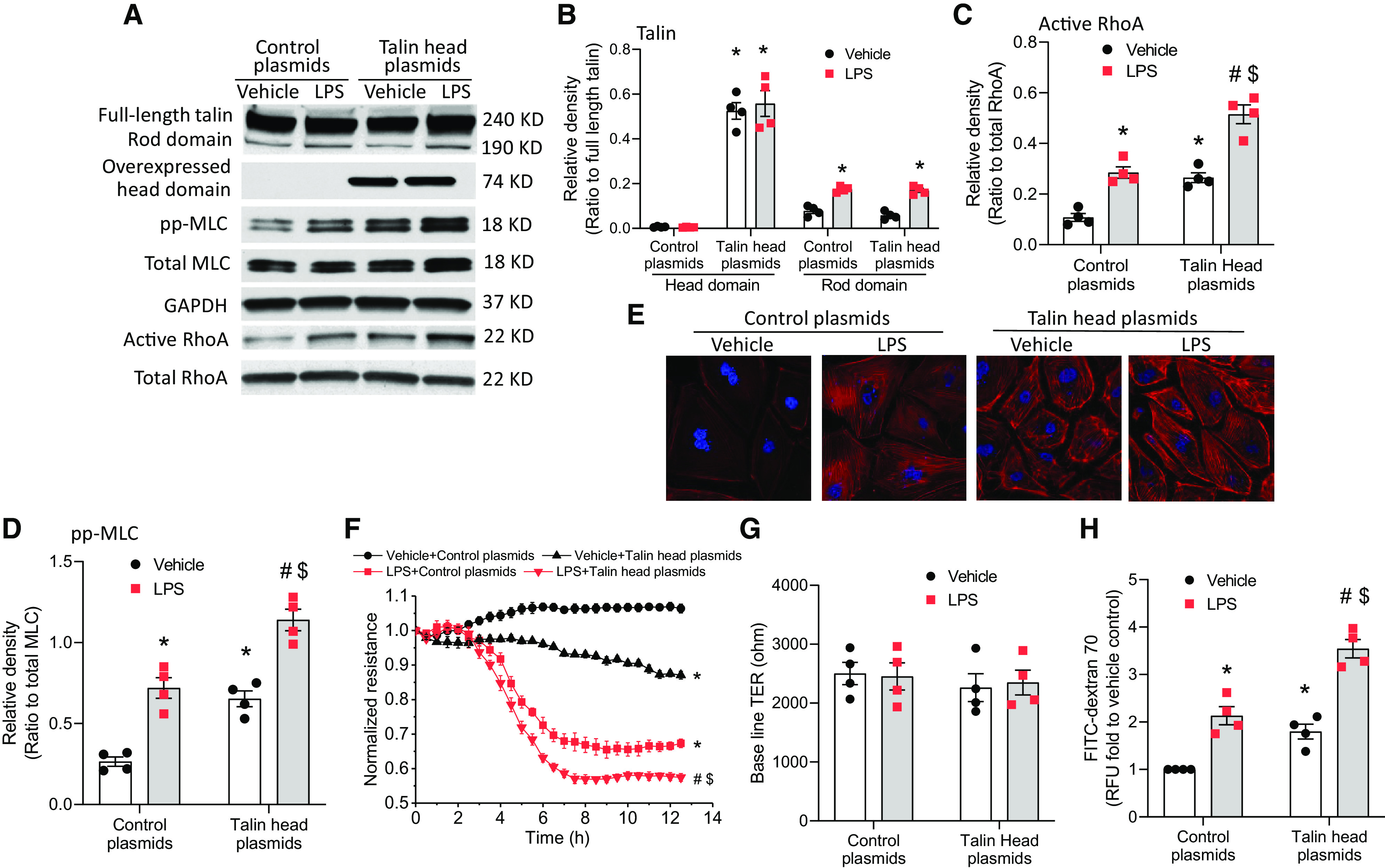
Overexpression of the talin head domain increases MLC phosphorylation and enhances lung microvascular endothelial barrier disruption. HLMVECs were transfected with talin1 head plasmids and C1 plasmids for 3 days and then incubated with LPS (0.25 μg/ml) for 12 hours, after which the talin head and rod domains, pp-MLC, total MLC, and RhoA activation were measured (A–D); cells were stained using rhodamine phalloidin (E); and the HLMVEC monolayer permeability to FITC-dextran 70 (H) was measured and during which TER value (F and G) was monitored. The blots and images are representative of four independent experiments. Results are expressed as mean ± SE; n = 4. *P < 0.05 versus vehicle + control plasmids; #P < 0.05 versus LPS + control plasmids; $P < 0.05 versus vehicle + talin head plasmids.
Overexpression of Talin Mutant Reduces LPS-induced Increases in MLC Phosphorylation in HLMVECs, and Attenuates LPS-induced Lung Microvascular Endothelial Barrier Disruption
The calpain cleavage site on talin has been identified (19). Mutating the leucine at position 432 of talin to a glycine blocked the ability of calpain to cleave talin (19). We overexpressed the calpain cleavage–resistant talin mutant in HLMVECs and found that overexpression of the talin mutant reduced LPS-induced RhoA activation and MLC phosphorylation in HLMVECs (Figures E5A–E5D) and attenuated hyperpermeability to FITC-dextran 70 in LPS-treated HLMVECs (see Figure E5E). These results confirm that calpain promotes LPS-induced lung endothelial barrier disruption via talin cleavage.
Inhibition of Calpain and Talin Attenuates the Expression of ICAM and E-Selectin Induced by LPS in HLMECs
To investigate whether calpain/talin signaling promotes endothelial inflammation in LPS-induced ALI, we determined the effects of calpain/talin inhibition on the expression of ICAM (intercellular adhesion molecule) and E-selectin induced by LPS in HLMVECs. The results showed that inhibition of calpain using MDL28170 or by knockdown of calpain-2 significantly attenuated LPS-induced increases in mRNA levels of ICAM and E-selectin in cells and protein levels of ICAM and E-selectin in cells and culture medium (Figure E6). Surprisingly, knockdown of talin markedly inhibited the increases in mRNA levels of ICAM and E-selectin in cells and protein levels of ICAM and E-selectin in cells and culture medium (Figure E7).
Discussion
In this study, we examined the role of calpain in LPS-induced lung microvascular endothelial barrier disruption in ALI. We found that LPS stimulates calpain activity and that inhibition of calpain attenuates pulmonary EC barrier disruption in vitro and lung EC barrier disruption and pulmonary edema induced by LPS in ALI in vivo. Our results indicate that calpain mediates LPS-induced lung microvascular EC barrier disruption.
Blumenthal and Malkinson first reported that calpain-like activity and limited proteolysis are involved in butylated hydroxytoluene–induced ALI (20). Other studies described positive effects of calpain inhibitors in various rodent ALI models, including scald burn–, ventilator-, and air pollution–induced lung injuries (21–25). However, the signaling mechanisms involved in calpain-mediated ALI induced by LPS have been poorly understood. In the present study, we have shown that inhibition of calpain activity by MDL28170 attenuates pulmonary EC barrier disruption in vitro and significantly prevents lung EC barrier dysfunction in LPS-induced ALI in vivo. Moreover, EC-specific KO of calpain attenuates the lung EC barrier disruption and pulmonary edema induced by LPS in ALI. Furthermore, knockdown of calpain-1 slightly reduced, but knockdown of calpain-2 markedly inhibited, the calpain activation and HLMVEC hyperpermeability induced by LPS. These data indicate that it is calpain-2, rather than calpain-1, that is mainly responsible for the calpain activation and lung microvascular endothelial barrier disruption induced by LPS.
LPS acts on lung ECs and causes RhoA activation and actin stress fiber formation, resulting in the compromise of EC barrier function in vitro and pulmonary edema in ALI in vivo (26, 27). However, the signaling pathways involved in cytoskeleton reorganization induced by LPS remain unclear. We found that LPS-induced calpain activation is not accompanied by the changes in protein levels of calpain-1 and calpain-2 in HLMVECs, suggesting that LPS regulates calpain activity via posttranslational mechanisms. We and others have reported that phosphorylation modulates calpain activity and increases its Ca2+ sensitivity (10, 17). Ser-50 phosphorylation of calpain-2 by ERK induces calpain activation in PDGF-treated lung vascular cells (10). Phosphorylation and activation of calpain-2 can be prevented by inhibition of p38 MAPK (18). Our data showed that the ERK inhibitor PD98059 and the p38 MAPK inhibitor SB203580 reduce LPS-induced increases in calpain activity in HLMVECs, suggesting that LPS induces calpain activation via ERK and p38 MAPK.
RhoA is a major regulator for actin cytoskeleton (28, 29). The downstream events for RhoA activation include the activation of Rho-associated kinase and the increased formation of actin stress fibers. RhoA and Rho-associated kinase directly catalyze MLC phosphorylation and also act indirectly via phosphorylation and inactivation of MLCP, leading to cell contraction and EC barrier disruption (28, 29). Calpain plays an important role in the activation of the Rho signaling pathway (30, 31). Several reports claimed that the inhibition of calpain activity inhibits actin stress fiber formation (32, 33). We previously reported that it is calpain-2, not calpain-1, that contributes to the increase in stress fiber formation in lung ECs (9). Interestingly, in the present study, we found that overexpression of calpain-2 induces RhoA activation and MLC phosphorylation and increases actin stress fiber formation in HLMVECs. Further, calpain-2 inhibition prevented LPS-induced RhoA activation, MLC phosphorylation, and actin stress formation in HLMVECs, suggesting that calpain-2 mediates LPS-induced RhoA activation, actin stress formation, and hyperpermeability of lung microvascular ECs.
It is unknown how calpain activates RhoA and induces actin stress formation, leading to hyperpermeability of lung microvascular ECs in ALI. Calpain acts via limited proteolysis of substrate proteins in cells (8). Among the calpain substrates, talin is the most prominent one that regulates actin cytoskeleton reorganization (19, 34). Talin links actin cytoskeleton to FAs (35). It contains distinct binding sites for actin and integrins and transduces mechanical forces from actomyosin to extracellular matrix through interactions with FA proteins (35, 36). Talin comprises two functionally different head and rod domains (37, 38). The N-terminal talin head domain (47 kD) is also known as FERM domain and includes subdomains 0–3. The F3 subdomain binds to the cytoplasmic integrins and is critical for integrin activation (37–39). The C-terminal rod (or tail) domain (190 kD) is primarily responsible for binding with other cytoskeletal proteins like actin and is also involved in integrin binding (40, 41). In quiescent cells, the C-terminal rod domain of talin self-masks a key integrin-binding site on the F3 subdomain, providing autoinhibition (42). Calpain induces talin cleavage (12), which separates the head from the rod domain, thus removing autoinhibition and stimulating talin head binding to FAs and inducing integrin activation (12, 13, 43). Several studies demonstrated that talin-integrin-actin binding and formation of FAs is critical for RhoA activation and increased contractile responses and permeability in human lung ECs stimulated by edemagenic agents (14, 15). In the present study, we found for the first time that LPS induces talin cleavage into the head and rod domains in HLMVECs and that overexpression of calpain-2 causes talin cleavage and RhoA activation in HLMVECs, resulting in MLC phosphorylation and an increase in actin stress formation. Consistent with these results, talin knockdown leads to disassembly of LPS-induced stress fibers and a decrease in LPS-induced MLC phosphorylation, reflecting inhibition of EC contractility and barrier dysfunction. Further, we demonstrated that overexpression of head talin and rod talin increases RhoA activation and MLC phosphorylation and enhances lung microvascular endothelial barrier disruption. These findings indicate that cleaved talin (i.e., head and rod domains) produced by calpain-mediated cleavage promotes RhoA activation, MLC phosphorylation, and actin stress formation in HLMVECs and enhances the lung endothelial barrier dysfunction induced by LPS. In addition, overexpression of a calpain cleavage–resistant talin mutant reduces the LPS-induced increases in MLC phosphorylation in HLMVECs and attenuates the LPS-induced lung microvascular endothelial barrier disruption. These data provide clear evidence confirming that calpain-mediated talin cleavage promotes LPS-induced actin cytoskeletal responses and lung microvascular endothelial barrier dysfunction.
In our experiments, we showed that the inhibition of calpain prevented LPS-induced endothelial adherens junction disassembly. In ALI, EC contraction caused by actin cytoskeletal reorganization leads to disruption of intercellular adherens junctions (28, 29). Several proteins, including Piezo1, β-catenin, and p120-catenin, participate in the stabilization of pulmonary endothelial adherens junctions (25, 44). It has been reported that calpain-mediated degradation/inhibition of these adherens proteins leads to ventilation/pressure– induced endothelial hyperpermeability (22, 25, 44). Thus, calpain-mediated lung endothelial barrier dysfunction may also be contributed to the disassembly of endothelial adherens junctions in addition to the actin cytoskeletal reorganization caused by talin cleavage.
It has been shown that LPS-induced apoptosis contributes to lung endothelial barrier disruption in ALI (45, 46). Notably calpain participates in apoptosis in various pathological conditions (47). Therefore, LPS-induced activation of calpain may cause endothelial apoptosis, leading to lung endothelial barrier dysfunction. Nevertheless, increased calpain activity to the level seen in LPS treatment by overexpression of calpain-2 does not compromise HLMVEC viability (Figure E8). It has been reported that overexpression of calpain-2 by >50% in mouse NR6 fibroblasts does not induce cell injury (48). Thus, it is less likely that calpain promotes LPS-induced lung endothelial barrier dysfunction via apoptosis in the present experimental condition.
Calpain has been shown to promote inflammation through degradation of I kappa B (49, 50). Endothelium-derived inflammatory factors such as ICAM and E-selectin play important roles in ALI (51, 52). Our data indicate that inhibitions of calpain attenuate LPS-induced increases in the expression of ICAM and E-selectin in HLMVECs, which might also contribute to the alleviating effect of calpain inhibition on LPS-induced lung EC barrier disruption in ALI. Furthermore, we observed unexpectedly that knockdown of talin inhibits the expression of ICAM and E-selectin induced by LPS in HLMVECs. Many processes in the signaling pathway of cytokine expression induced by LPS could be affected by talin-RhoA-MLC-cytoskeleton. RhoA and cytoskeletal reorganization may influence mRNA stability of the inflammatory cytokines (53). Actin polymerization and depolymerization may impact transcription factor recruitment into nuclei (54). Further studies are needed to clarify these possibilities.
In summary, our findings provide evidence supporting the idea that calpain mediates LPS-induced RhoA activation, MLC phosphorylation, actin stress fiber formation, and lung microvascular endothelial hyperpermeability via cleavage of talin.
Footnotes
Supported by National Institutes of Health grant HL158909 to Y.S., and A.D.V., National Heart, Lung, and Blood Institute grant HL134934 to Y.S., Augusta University intramural grant IGPCT00023 to Y.S., and U.S. Department of Veterans Affairs grant BX005350 to Y.S.
Author Contributions: Conception, design, and experiment: L.S., X.S., L.K., and W.H. Analysis and interpretation: L.S., X.S., W.H., L.K., and Y.S. Drafting the manuscript for important intellectual content: L.S., X.S., L.K., W.H., J.J., S.A.B., Z.D., R.L., D.J.R.F., A.D.V., and Y.S.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2023-0009OC on August 28, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv . 2010;23:243–252. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med . 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med . 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 4. Kovacs-Kasa A, Gorshkov BA, Kim KM, Kumar S, Black SM, Fulton DJ, et al. The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Sci Rep . 2016;6:39018. doi: 10.1038/srep39018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogatcheva NV, Zemskova MA, Poirier C, Mirzapoiazova T, Kolosova I, Bresnick AR, et al. The suppression of myosin light chain (MLC) phosphorylation during the response to lipopolysaccharide (LPS): beneficial or detrimental to endothelial barrier? J Cell Physiol . 2011;226:3132–3146. doi: 10.1002/jcp.22669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol . 2014;50:170–179. doi: 10.1165/rcmb.2012-0496OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev . 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 9. Su Y, Cui Z, Li Z, Block ER. Calpain-2 regulation of VEGF-mediated angiogenesis. FASEB J . 2006;20:1443–1451. doi: 10.1096/fj.05-5354com. [DOI] [PubMed] [Google Scholar]
- 10. Kovacs L, Han W, Rafikov R, Bagi Z, Offermanns S, Saido TC, et al. Activation of calpain-2 by mediators in pulmonary vascular remodeling of pulmonary arterial hypertension. Am J Respir Cell Mol Biol . 2016;54:384–393. doi: 10.1165/rcmb.2015-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KK, et al. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J Clin Invest . 2011;121:4548–4566. doi: 10.1172/JCI57734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan B, Calderwood DA, Yaspan B, Ginsberg MH. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem . 2001;276:28164–28170. doi: 10.1074/jbc.M104161200. [DOI] [PubMed] [Google Scholar]
- 13. Anthis NJ, Campbell ID. The tail of integrin activation. Trends Biochem Sci . 2011;36:191–198. doi: 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Nieuw Amerongen GP, Natarajan K, Yin G, Hoefen RJ, Osawa M, Haendeler J, et al. GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ Res . 2004;94:1041–1049. doi: 10.1161/01.RES.0000125627.77235.0C. [DOI] [PubMed] [Google Scholar]
- 15. Guo M, Daines D, Tang J, Shen Q, Perrin RM, Takada Y, et al. Fibrinogen-gamma C-terminal fragments induce endothelial barrier dysfunction and microvascular leak via integrin-mediated and RhoA-dependent mechanism. Arterioscler Thromb Vasc Biol . 2009;29:394–400. doi: 10.1161/ATVBAHA.108.180950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cai P, Kovacs L, Dong S, Wu G, Su Y. BMP4 inhibits PDGF-induced proliferation and collagen synthesis via PKA-mediated inhibition of calpain-2 in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol . 2017;312:L638–L648. doi: 10.1152/ajplung.00260.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem . 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- 18. Qin Q, Liao G, Baudry M, Bi X. Role of calpain-mediated p53 truncation in semaphorin 3A-induced axonal growth regulation. Proc Natl Acad Sci USA . 2010;107:13883–13887. doi: 10.1073/pnas.1008652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, et al. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol . 2004;6:977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 20. Blumenthal EJ, Malkinson AM. Changes in pulmonary calpain activity following treatment of mice with butylated hydroxytoluene. Arch Biochem Biophys . 1987;256:19–28. doi: 10.1016/0003-9861(87)90421-8. [DOI] [PubMed] [Google Scholar]
- 21. Du PR, Lu HT, Lin XX, Wang LF, Wang YX, Gu XM, et al. Calpain inhibition ameliorates scald burn-induced acute lung injury in rats. Burns Trauma . 2018;6:28. doi: 10.1186/s41038-018-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang L, Zhang Y, Lu D, Huang T, Yan K, Yang W, et al. Mechanosensitive Piezo1 channel activation promotes ventilator-induced lung injury via disruption of endothelial junctions in ARDS rats. Biochem Biophys Res Commun . 2021;556:79–86. doi: 10.1016/j.bbrc.2021.03.163. [DOI] [PubMed] [Google Scholar]
- 23. Wang T, Wang L, Moreno-Vinasco L, Lang GD, Siegler JH, Mathew B, et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part Fibre Toxicol . 2012;9:35. doi: 10.1186/1743-8977-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Minshall RD, Schwartz DE, Hu G. Cyclic stretch induces alveolar epithelial barrier dysfunction via calpain-mediated degradation of p120-catenin. Am J Physiol Lung Cell Mol Physiol . 2011;301:L197–L206. doi: 10.1152/ajplung.00048.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhong M, Wu W, Kang H, Hong Z, Xiong S, Gao X, et al. Alveolar stretch activation of endothelial piezo1 protects adherens junctions and lung vascular barrier. Am J Respir Cell Mol Biol . 2020;62:168–177. doi: 10.1165/rcmb.2019-0024OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu YC, Hsu SP, Hu MC, Lan YT, Yeh ETH, Yang FM. Pep-snasp peptide alleviates lps-induced acute lung injury through the tlr4/traf6 axis. Front Med (Lausanne) . 2022;9:832713. doi: 10.3389/fmed.2022.832713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, et al. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem . 2014;289:4710–4722. doi: 10.1074/jbc.M114.547596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cerutti C, Ridley AJ. Endothelial cell-cell adhesion and signaling. Exp Cell Res . 2017;358:31–38. doi: 10.1016/j.yexcr.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol . 2013;92:303–315. doi: 10.1016/j.ejcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 30. Kulkarni S, Saido TC, Suzuki K, Fox JEB. Calpain mediates integrin-induced signaling at a point upstream of Rho family members. J Biol Chem . 1999;274:21265–21275. doi: 10.1074/jbc.274.30.21265. [DOI] [PubMed] [Google Scholar]
- 31. Fox JE. On the role of calpain and Rho proteins in regulating integrin-induced signaling. Thromb Haemost . 1999;82:385–391. [PubMed] [Google Scholar]
- 32. Dedieu S, Poussard S, Mazères G, Grise F, Dargelos E, Cottin P, et al. Myoblast migration is regulated by calpain through its involvement in cell attachment and cytoskeletal organization. Exp Cell Res . 2004;292:187–200. doi: 10.1016/j.yexcr.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 33. Dourdin N, Bhatt AK, Dutt P, Greer PA, Arthur JS, Elce JS, et al. Reduced cell migration and disruption of the actin cytoskeleton in calpain-deficient embryonic fibroblasts. J Biol Chem . 2001;276:48382–48388. doi: 10.1074/jbc.M108893200. [DOI] [PubMed] [Google Scholar]
- 34. Fong KP, Molnar KS, Agard N, Litvinov RI, Kim OV, Wells JA, et al. Cleavage of talin by calpain promotes platelet-mediated fibrin clot contraction. Blood Adv . 2021;5:4901–4909. doi: 10.1182/bloodadvances.2021004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klapholz B, Brown NH. Talin – the master of integrin adhesions. J Cell Sci . 2017;130:2435–2446. doi: 10.1242/jcs.190991. [DOI] [PubMed] [Google Scholar]
- 36. Sun Z, Guo SS, Fässler R. Integrin-mediated mechanotransduction. J Cell Biol . 2016;215:445–456. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberts GC, Critchley DR. Structural and biophysical properties of the integrin-associated cytoskeletal protein talin. Biophys Rev . 2009;1:61–69. doi: 10.1007/s12551-009-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang JH. Pull and push: talin activation for integrin signaling. Cell Res . 2012;22:1512–1514. doi: 10.1038/cr.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. García-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, et al. Structural determinants of integrin recognition by talin. Mol Cell . 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 40. Gingras AR, Ziegler WH, Bobkov AA, Joyce MG, Fasci D, Himmel M, et al. Structural determinants of integrin binding to the talin rod. J Biol Chem . 2009;284:8866–8876. doi: 10.1074/jbc.M805937200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gingras AR, Bate N, Goult BT, Patel B, Kopp PM, Emsley J, et al. Central region of talin has a unique fold that binds vinculin and actin. J Biol Chem . 2010;285:29577–29587. doi: 10.1074/jbc.M109.095455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song X, Yang J, Hirbawi J, Ye S, Perera HD, Goksoy E, et al. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res . 2012;22:1533–1545. doi: 10.1038/cr.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hayashi M, Suzuki H, Kawashima S, Saido TC, Inomata M. The behavior of calpain-generated N- and C-terminal fragments of talin in integrin-mediated signaling pathways. Arch Biochem Biophys . 1999;371:133–141. doi: 10.1006/abbi.1999.1427. [DOI] [PubMed] [Google Scholar]
- 44. Friedrich EE, Hong Z, Xiong S, Zhong M, Di A, Rehman J, et al. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc Natl Acad Sci USA . 2019;116:12980–12985. doi: 10.1073/pnas.1902165116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu Y, Wang Y, Gong S, Tang J, Zhang J, Li F, et al. Ruscogenin alleviates LPS-induced pulmonary endothelial cell apoptosis by suppressing TLR4 signaling. Biomed Pharmacother . 2020;125:109868. doi: 10.1016/j.biopha.2020.109868. [DOI] [PubMed] [Google Scholar]
- 46. Chen L, Li W, Qi D, Wang D. Lycium barbarum polysaccharide protects against LPS-induced ARDS by inhibiting apoptosis, oxidative stress, and inflammation in pulmonary endothelial cells. Free Radic Res . 2018;52:480–490. doi: 10.1080/10715762.2018.1447105. [DOI] [PubMed] [Google Scholar]
- 47. Momeni HR. Role of calpain in apoptosis. Cell J . 2011;13:65–72. [PMC free article] [PubMed] [Google Scholar]
- 48. Shao H, Chou J, Baty CJ, Burke NA, Watkins SC, Stolz DB, et al. Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Mol Cell Biol . 2006;26:5481–5496. doi: 10.1128/MCB.02243-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruetten H, Thiemermann C. Effect of calpain inhibitor I, an inhibitor of the proteolysis of I kappa B, on the circulatory failure and multiple organ dysfunction caused by endotoxin in the rat. Br J Pharmacol . 1997;121:695–704. doi: 10.1038/sj.bjp.0701180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hamelet J, Couty JP, Crain AM, Noll C, Postic C, Paul JL, et al. Calpain activation is required for homocysteine-mediated hepatic degradation of inhibitor I kappa B alpha. Mol Genet Metab . 2009;97:114–120. doi: 10.1016/j.ymgme.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 51. Zhang N, Wang G, Sun G. Actin-binding protein, IQGAP1, regulates LPS-induced RPMVECs hyperpermeability and ICAM-1 upregulation via Rap1/Src signalling pathway. Cell Signal . 2021;85:110067. doi: 10.1016/j.cellsig.2021.110067. [DOI] [PubMed] [Google Scholar]
- 52. Goldfarb RD, Bennett CF, Butler M, Condon T, Parrillo JE. Targeting host E-selectin expression by antisense oligodeoxynucleotides as potential antiendotoxin therapy in vivo. Oligonucleotides . 2010;20:253–261. doi: 10.1089/oli.2010.0229. [DOI] [PubMed] [Google Scholar]
- 53. Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest . 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pawłowski R, Rajakylä EK, Vartiainen MK, Treisman R. An actin-regulated importin α/β-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J . 2010;29:3448–3458. doi: 10.1038/emboj.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]