

Abstract

Decoration of semiconductor photocatalysts with cocatalysts is generally done by a step-by-step assembly process. Here, we describe the self-assembling and self-activating nature of a photocatalytic system that forms under illumination of reduced anatase TiO2 nanoparticles in an aqueous Ni2+ solution. UV illumination creates in situ a Ni+/TiO2/Ti3+ photocatalyst that self-activates and, over time, produces H2 at a higher rate. In situ X-ray absorption spectroscopy and electron paramagnetic resonance spectroscopy show that key to self-assembly and self-activation is the light-induced formation of defects in the semiconductor, which enables the formation of monovalent nickel (Ni+) surface states. Metallic nickel states, i.e., Ni0, do not form under the dark (resting state) or under illumination (active state). Once the catalyst is assembled, the Ni+ surface states act as electron relay for electron transfer to form H2 from water, in the absence of sacrificial species or noble metal cocatalysts.
Introduction
In materials science, phenomena of self-assembly can be observed on a wide range of length scales, and many examples are relevant to create specific functional materials.1 Some important photoinduced processes are based on self-aggregation, -formation, and -organization.2 Such self-assembly processes that occur by “simmering in one pot for sufficient time” represent a stark contrast to the general synthesis strategies used in a chemical or materials science laboratory. In classic synthesis approaches, complex functionality is achieved by a careful step-by-step procedure that targets an optimization of each step. For example, photocatalysts are typically designed from a semiconductor that allows for light harvesting and charge carrier generation, while cocatalysts are decorated onto the semiconductor to overcome the intrinsic kinetic hindrance of many important photocatalytic reactions. In fact, from a purely thermodynamic standpoint, the position of band edges in TiO2 can enable the reaction of H2O into H2, O2, and •OH radicals as well as H2O2.3−6 Nonetheless, even with the thermodynamic advantages for water splitting into H2 and O2, unaltered (stoichiometric) phases of TiO2 exhibit unsatisfactory rates of H2 evolution and undetected O2 evolution from pure water, as well documented in the literature.7,8 The observation that O2 generation is less than stoichiometric or entirely absent can be ascribed not only to the sluggish kinetics of the oxygen evolution reaction (OER) in the absence of any catalysts but also to the fact that photoreduced TiO2 tends to strongly adsorb oxygen as O2– or as O22–.9
More generally, for many semiconductors without cocatalysts, O2 and H2 evolution reactions are kinetically hampered.10−12 To overcome this limitation, noble metals such as Pt, Pd, Rh, among others, are decorated in the form of cocatalyst nanoparticles on the semiconductor surface for H2 evolution.13−16 To promote hole-transfer toward O2 evolution, catalysts such as IrO2 or RuO217,18 are typically used. Electron and hole transfer processes can also be facilitated using sacrificial agents.19,20
In recent years, research on cocatalysts increasingly focused on replacing “costly” noble metals with more economic alternatives, such as sulfides or phosphides21−24 of various transition metals, or their alloys.25−29 Among the latter, particularly Ni and its alloys have attracted wide interest, particularly for the surface modification of TiO2 and SrTiO3 photocatalysts.30−34 The CB and VB edges of TiO2 are positioned at −0.5 and +2.5 eV, respectively,4 while the CB and VB edges of SrTiO3 lay at about −0.8 and +2.5 eV, respectively35 (vs SHE, pH 7). The electrochemical potential of the Ni2+/Ni redox couple is −0.64 V (vs SHE, pH 7). In this framework, Ni has been found to be bifunctional, i.e., it can serve in the metallic form as cocatalyst for H2 evolution and in the oxidized form (oxyhydroxide) as cocatalyst for O2 generation.35 Some reports suggest that Ni oxide or hydroxide placed on various semiconductors can be reduced to metallic Ni by photogenerated conduction band (CB) electrons.35 However, there are controversial results on the photocatalytic reduction of Ni2+ species from solution to metallic Ni on a photocatalyst surface. Reduction of Ni2+ to metallic Ni has been reported for cases where holes in the illuminated semiconductor are rapidly consumed by a hole scavenger (e.g., methanol, ethanol, etc.).36,37 We confirmed this in previous work using in situ X-ray absorption spectroscopy (XAS) to study Ni-, Cu-, and NiCu-modified TiO2.38 For Ni-modified SrTiO3, in pure water, it has been reported that photocarrier transfer may lead to disproportionation reactions forming both Ni and NiOx sites35 or Ni/NiOx core–shell NPs.39 Such NiOx compounds or Ni/NiOx core–shell nanostructures are then able to form a NiOx cocatalyst for water oxidation to O2.
In contrast to SrTiO3, and some other complex semiconductors, for titanium dioxide (TiO2), these types of reactions are not observed in plain water. That is, to achieve facile electron and hole transfer from TiO2 to water, a noble metal cocatalyst as electron transfer mediator and an efficient hole-capturing species (such as methanol) are typically required.19,20 However, in such cases, holes react with the sacrificial agent, and methanol photoreforming replaces the water oxidation reaction.20 In plain water, on TiO2, the hole transfer reaction to water and the formation of O2 are strongly hampered.20,40 Interestingly, some works reported that on Pt-decorated titania surfaces, a two-hole transfer to water is kinetically favored, leading to H2O2 rather than O2 as reaction product.41−44 However, also this two-hole pathway to H2O2 is extremely slow on a pristine TiO2 surface, and thus the hole transfer reaction represents the real bottleneck in TiO2 photocatalytic water splitting. In a previous study, we reported that so-called gray TiO2 can provide long-lived holes that enable this two-hole pathway to H2O2.4Gray anatase is a form of reduced anatase TiO2, produced by thermal hydrogenation at 500 °C, possessing defective surface structure that can promote electron and hole transfer.45 As a result, gray titania decorated with metallic Ni particles was found to be an effective photocatalyst for the splitting of plain water into H2 and H2O2.4
Here, we demonstrate a more remarkable use of gray anatase, that is, the in situ formation of a metastable photocatalyst for H2 evolution from an aqueous Ni2+ solution. This takes place in the absence of sacrificial species or noble metal cocatalysts and without the formation of metallic Ni nanoparticles by photoreduction on the surface of gray TiO2. In situ XAS and electron paramagnetic resonance (EPR) spectroscopy show light-induced formation of defects in the semiconductor combined with the formation of surface trapped monovalent Ni+ states. We propose that such Ni+ states act as a relay for electron transfer to form H2 from water. This property is distinct to TiO2 powders hydrogenated under optimal conditions, as both nonreduced white and over-reduced black titania show negligible H2 evolution under comparable photocatalytic conditions.
Results and Discussion
In a simple gastight photoreactor (quartz tube), mildly reduced anatase titania nanopowders (gray titania45) are dispersed in an aqueous 0.4 mM NiSO4 solution and illuminated with UV light (UV LED, 365 nm, 100 mW cm–2). Under these experimental conditions, in a few hours, an increasingly effective photocatalytic system self-assembles and a metastable state is established that produces H2 from water. This self-activation process is illustrated in Figure 1 (see the Supplementary Information for additional experimental details and Figure S1 for further characterization of gray TiO2). After switching on UV illumination, during the first few hours of illumination, only negligible amounts of H2 can be detected (such amounts are in the range of or below the detection limit of the gas chromatograph, GC). Then, the H2 evolution becomes evident, and after 10 h of illumination it keeps accelerating, e.g., until 72 h of illumination (at this time the UV light was turned off and the experiment was stopped). Interestingly, these self-activation and amplification phenomena take place in the absence of any sacrificial agent. We observed a similar effect in previous work where we hypothesized the formation of a monovalent nickel cocatalyst mediator.46 Here, we prove our hypothesis by in situ XAS and EPR spectroscopy.
Figure 1.

H2 evolution performance: comparison of gray TiO2 nanoparticles with white and black TiO2 nanoparticles in 0.4 mM NiSO4·6H2O aqueous solution under illumination.
This effect is unique to gray anatase and cannot be observed for white TiO2 (untreated anatase) or black TiO2 (Figure 1). Black TiO2 is a form of over-reduced (highly defective) titania produced by hydrogenation under harsh conditions (700 °C in this case).47−50Black and gray TiO2 differ in the reduction treatment used to produce the materials from white (untreated) anatase. Consequently, the three forms of titania herein studied exhibit different density and nature of oxygen vacancies (Ti3+-OV) and, thus, different electronic properties.45 We characterized these materials in view of their electronic and defect structure in previous work.4,45,50−56 Unlike low temperature treatments (e.g., 500 °C) that maintain anatase as the only crystalline phase, hydrogenation at 700 °C (to form the over-reduced black TiO2) results in a mixed anatase–rutile composition.45 Among different “shades” of gray titania, i.e., thermally treated in H2 at different reduction temperatures (300–700 °C), the powder treated at 500 °C for 1 h (gray TiO2) shows the highest photocatalytic activity (shown in our previous work46).
Figure 1 shows that an incubation period is observed until H2 evolution is detected in significant amounts. It is known that illumination of anatase in aqueous methanolic solutions57 forms Ti3+ sites that are catalytically active for the generation of H2.58 Differently, in the present work, we show how UV light can form Ti3+ sites that generate H2 from plain water, i.e., in the absence of any hole scavenger, due to the presence of Ni2+ and via the formation of a metastable Ni+/TiO2/Ti3+ photocatalyst (discussed below). The key role of Ni ions is evident from data in Figure 1, i.e., when comparing the H2 evolution measured in an aqueous NiSO4 solution vs Ni2+ free solution. Additional data on the effect of pH, temperature, nickel salt precursor, and illumination wavelength and power density on the photocatalytic H2 evolution rate can be found in Figure S2.
A first assumption may be that photocatalytic reduction of Ni2+ to Ni° would occur under UV irradiation. In this case, over time, the titania would be decorated with Ni° nanoparticles that act as cocatalyst for H2 evolution. However, we find that after illumination (even up to 10 days), no morphological trace of particle deposition can be observed on gray titania, neither by extensive investigations by scanning electron microscopy (SEM, Figure S3) nor by high-resolution transmission electron microscopy (HR-TEM, Figure 2a,b). Nevertheless, the presence of Ni species can be clearly detected by TEM–energy dispersive spectroscopy (TEM–EDS, Figure 2c,d), which indicates that Ni is distributed uniformly on the nanopowder. The absence of particle deposition along with the evidence of a homogeneous Ni distribution hints to atomically dispersed Ni species. Similar species are commonly encountered in single atom catalysis.59−61
Figure 2.

(a–d) Gray TiO2 nanoparticles after illumination for 10 days in a 0.4 mM NiSO4·6H2O aqueous solution: (a) High-resolution TEM image. (b) High-angle annular dark-field imaging (HAADF). TEM–EDS elemental mappings of (c) Ni and (d) Ni, O, and Ti. (e) Peak fitting of the high resolution XPS spectrum in the Ni 2p region for the sputtered anatase TiO2 layer (hydrogenated at 500 °C) illuminated with a 365 nm LED (100 mW cm–2) for 24 h in a 0.4 mM NiSO4·6H2O aqueous solution.
For powder samples, the amount of Ni is below the detection limit of X-ray photoelectron spectroscopy (XPS). However, if compact, polycrystalline anatase layers produced by magnetron sputtering (on SiO2/Si) and subsequently reduced (hydrogenation at 500 °C, 1 h) are illuminated (24 h, UV light) in a 0.4 mM NiSO4·6H2O aqueous solution, XPS can reveal the presence of Ni species (Figure 2e). These layers show an onset of H2 evolution in analogy to the powder (Figure S4), and no morphological trace of photodeposited Ni particles can be observed (Figure S5). The XPS data reveal a Ni surface content of ca. 0.5 at. %, and a Ni:Ti ratio of ca. 0.04:1, indicating a low loading of Ni species (see Table S1).
The fitting of the Ni 2p XPS peak suggests the presence of Ni in various oxidations states. First, the Ni 2p consists of the Ni 2p3/2 and Ni 2p1/2 peaks at 855.9 and 873.5 eV, respectively, and with the corresponding satellite peaks at 861.3 and 879.9 eV (consistent with literature data).62 To evaluate the chemical states of Ni, peak fitting of the Ni 2p spectrum in Figure 2e was performed. For the fitting, we used empirical fits obtained from parameters derived from standard samples typically found in literature.63−66 The detected species include Niδ+, typical of surface-coordinated Ni states, and Ni2+. Consistent with SEM and TEM, no metallic Ni was detected (typical binding energy of 852.6 eV). The binding energy, full width half-maximum and percentage of the peak area of the fitted peaks comprising the 2p3/2 peak and corresponding satellite are listed in Table S2 in the Supporting Information and are consistent with literature data.62−64 The fitted spectrum with an error below 0.5% includes the 2p3/2, 2p1/3 and satellite peaks for Niδ+ (72.4%) and Ni2+ (27.6%). However, the ex situ XPS data may be affected by exposure of the sample to air due to sample removal from the electrolyte and transfer to the XPS setup. In general, this issue may affect ex situ characterization results.
Therefore, to clarify the self-activation mechanism and the role of Ni in the photocatalytic system, we performed in situ XAS experiments. Experimental details are provided in the Supporting Information. In a preliminary experiment, we studied the electrochemical reduction of a Ni(II) complex [Ni(cyclam)]2+ in an acetonitrile-based electrolyte to obtain a reference spectrum for in situ formed Ni(I) species; [Ni(cyclam)]2+ is in fact known to undergo reduction to formal Ni(I) species.67,68 Experimental details on the synthesis and characterization of the Ni(II) complex [Ni(cyclam)]2+ are provided in the SI. The X-ray absorption near-edge structure (XANES) spectrum of the complex at the Ni K-edge is shown in Figure 3.
Figure 3.

XANES spectra of the [Ni(cyclam)]2+ complex at the OCP and −2 V. Ni and NiO spectra are shown as a reference for Ni(0) and Ni(II) oxidation states, shifted along the y axis for better clarity. Inset: pre-edge region for the [Ni(cyclam)]2+ complex at the OCP and −2 V.
We measured the spectra of the complex under open circuit potential (OCP) conditions. As a reference for Ni(II), we use here a Ni K-edge XANES spectrum of NiO. The close correspondence in energy of the edge position and the overall similarity of the spectral shapes clearly demonstrate that the [Ni(cyclam)]2+ complex features divalent nickel centers. We then polarized the working electrode (a transparent conductive oxide-coated glass slide, FTO) at −2 V (vs Ag pseudoreference electrode), since preliminary voltametric experiments showed a one-electron reduction peak to Ni(I) at about −1.8 V69 (see CV in Figure S6). In the Ni K-edge spectrum, a new structure at ca. 8337.1 eV appears (green arrow in Figure 3, highlighted in the inset). We can refer to this feature as the signature of Ni(I). In addition, the spectrum at −2 V clearly indicates that Ni0, i.e., metallic nickel, does not form.
We then performed in situ XAS experiments studying the different TiO2 photocatalysts in aqueous suspensions in the presence of NiSO4, both under dark conditions and under UV light illumination (i.e., under photocatalytic H2 evolution conditions). A picture of the setup along with experimental details and can be found in Methods and Figure S7, respectively. The XANES spectra at the Ni K-edge of gray TiO2 (in a 0.4 mM solution of NiSO4) under different conditions are shown in Figure 4a. In the dark, the spectrum resembles that of Ni(II) hydrated ions. Figure S8 shows the Ni K-edge XANES spectrum of a 0.4 mM aqueous solution of NiSO4, compared to the spectra of the same solution in the presence of black, gray, and white TiO2, all under dark conditions. The close similarity of the absorption edge position for all of the spectra confirms that the oxidation state is Ni(II) in every case. Minor differences are visible when the solution is put in contact with the different TiO2 powders, which may be indicative of the formation of different Ni(II) species adsorbed onto the TiO2 nanoparticles.
Figure 4.

XANES spectra of gray (a), white (b), and black (c) TiO2 in a 0.4 mM solution of NiSO4 under intermittent UV light illumination. Right panels: magnified pre-edge regions.
Upon illumination, a new broad feature appears at ca. 8336–8338 eV, and this is indicative of the formation of Ni(I). We note that this structure in the presence of TiO2 is broader when compared to the signal obtained by electrochemical reduction of the [Ni(cyclam)]2+ complex (Figure S9). This may be related to the fact that the Ni(I) sites are adsorbed on the TiO2 nanoparticle surface and therefore are expected to experience significant local disorder. Note that no Ni0, i.e., metallic nickel, could be detected, neither in the dark (resting state) nor under illumination (active state).
When the light is switched off, this structure decreases in intensity, thus confirming that the formation of Ni(I) is driven by illumination and is reversible. The fact that the Ni(I) feature at ca. 8337 eV disappears shortly after illumination indicates that such Ni(I) species are highly reactive, i.e., are short lived, according to
Ni2++ e– → Ni+surface and 2Ni+surface + 2H+ → H2 + 2Ni2+
The XANES spectra at the Ni K-edge of white TiO2 in a 0.4 mM solution of NiSO4 under different conditions are shown in Figure 4b. In the dark, the spectrum resembles that of Ni(II) hydrated ions, and under illumination, no difference can be detected. The same is valid for this sample in the dark after illumination. As shown in Figure 1, white TiO2 shows negligible photocatalytic activity, which is now further substantiated by the fact that no Ni(I) species form, neither in the dark nor under illumination, in the case of white titania.
The XANES spectra at the Ni K-edge of black TiO2 in a 0.4 mM solution of NiSO4 under different conditions are shown in Figure 4c. In the dark, the spectrum shows a spectral weight in the energy region of Ni(I), and this increases upon illumination. After illumination, the original spectral shape is recovered only after a long time in the dark (ca. 3 h). These conclusions are, however, made less straightforward by the fact that, in the energy region characteristic of N(I), also Ni(0) has an increased spectral weight. Nevertheless, these results seem to indicate that defective Ti3+-OV sites in black TiO2 are highly active also in the dark, and a reaction with Ni(II) aqueous ions to form surface Ni(I) species takes place also without photogeneration of charge carriers in TiO2. Note that black TiO2, as shown in Figure 1, shows negligible photocatalytic activity.
In situ time-resolved XAS data help highlight the spectral differences between the three TiO2 samples, as shown in Figure 5. The value of the absorption intensity at 8337.1 eV, after normalization for the intensity at the main edge peak, is plotted as a function of time during the dark–light–dark cycles.
Figure 5.

In situ time-resolved XAS data showing the trend of the absorption intensity at 8337.1 eV vs time for the different TiO2 samples.
To sum up, the behavior of the three studied samples is very different. White TiO2 (center) is the simplest case: both under dark conditions and under UV illumination, the formation of Ni(I) can be excluded. For gray TiO2 (left) the formation of Ni(I) species requires UV illumination. The concentration of Ni(I) increases after an induction period, and decreases immediately when the illumination is switched off. The induction period resembles that observed during the photocatalytic H2 evolution experiments (see Figure 1). Black TiO2 (right), in contrast, shows the presence of Ni(I) species both in the dark and under illumination. Upon illumination, the concentration of Ni(I) increases only slightly and reaches soon a plateau. After switching off the illumination, it returns to its initial value.
Summarizing the results of photocatalytic H2 evolution experiments and in situ XAS, we observe that:
Common to all parameter variations in photocatalytic H2 evolution experiments with gray TiO2 in aqueous NiSO4 solutions (Figures 1 and S2) is that, after starting UV illumination, an incubation period is observed until H2 is produced in detectable amounts. On the contrary, in the absence of Ni(II) salt, the H2 generation remains negligible.
No Ni0, i.e., metallic nickel, can be detected, neither in the dark (resting state) nor under illumination (active state) for the tested photocatalyst.
The XAS signal ascribed to Ni(I) species (Figure 5) increases over time under UV illumination, and so does the amount of generated H2 detected by GC analysis (Figures 1 and S2), suggesting that the two redox processes (Ni+ formation and H2 evolution) are linked, and likely the formation of Ni(I) species is the first redox step, followed by subsequent H2 generation.
Results from our previous work46 suggest a strong correlation between the concentration of Ni(II) salt and the H2 generation (optimum of Ni(II) concentration 0.4 mM, as used in the present study). This can due to a trade-off between an increase in H2 generation activity, due to a higher concentration of photoformed Ni(I) states, and a decrease in effective reaction volume, due to a higher light absorbance of the aqueous phase with increasing the Ni(II) salt concentration.
Therefore, we conclude that
Ni(I) species, formed on gray TiO2 but not on white TiO2, are key to catalyze the H2 evolution in the absence of a sacrificial agent and noble metal cocatalyst.
The formation of Ni(I) species can be related to the presence of surface Ti3+ sites on TiO2, (confirmed by in situ EPR, below) which enable the formation of monovalent nickel (Ni(I)) species. The H2 evolution rate appears to correlate with the concentration of such surface Ni(I) species.
To initiate the redox chain, i.e., the formation of Ni+ active species that enable H2 generation, needed are not only available Ni2+ ions in the solution but also Ti3+ sites at the TiO2 surface to react with Ni2+ ions. It is therefore expected that the highest H2 generation rates are achieved when the system is optimized in view of light absorption and availability of surface Ti3+ sites on TiO2 and Ni2+ ions in the reaction phase.
Different hydrogenation treatments (different temperatures) form in TiO2 Ti3+-OV sites of different density, distribution, and energy, in line with in situ EPR results discussed below and in our previous work.70
Defective Ti3+-OV structures in black TiO2 are highly reactive and form Ni(I) species already in the dark. This process, however, is not reversible, indicating that Ni(I) species formed on black anatase are stabilized by a strong interaction with the defective oxide surface and, hence, are less reactive. Hence, such Ni(I) species are significantly less reactive toward H2 evolution.
Note that gray titania provides not only an adequate surface density of Ti3+-OV sites, but also a sufficient hole transfer ability to plain water, forming hydrogen peroxide, as shown in previous own work,4 in preliminary results in Figure S10, and discussed below along with in situ EPR results. This is crucial since, in the absence of efficient hole transfer, photoholes could oxidize the monovalent nickel (to Ni(II)), and this would impede the electron transfer to water for hydrogen evolution.
To validate our interpretation, we carried out in situ EPR experiments to investigate the effects of UV illumination on gray titania nanoparticles in Ni2+ aqueous solutions. Figure 6a (3D plot) and Figure 6d (2D plot) show in situ EPR resonance spectra (T = 80 K) of gray titania dispersed in a Ni2+ aqueous solution (in the absence of hole scavenger, e.g., MeOH). The sequential spectra were acquired under fast scanning conditions of the magnetic field (30 s) during a dark-light sequence (2.5 min dark followed by 27.5 min of irradiation at 325 nm, HeCd laser, power 200 mW). Under dark conditions, the recorded EPR spectra show a broad resonance signature developing in the 330–360 mT magnetic field range (gavg ≈ 1.92, see Figure 6c, blue spectrum) and two resonance signals at lower magnetic field, at g = 1.98 and g = 2.00. These signals originate from Ti3+ states located in different lattice positions.50,55 The signals at g = 2.00 corresponds to electrons trapped into oxygen vacancies, Ti3+-OV, and the resonance at g = 1.98 corresponds to Ti3+ sites formed in bulk lattice positions. The broad signature at gavg ≈ 1.92 originates from surface exposed Ti3+ sites.50,54,55 Weaker resonances are detected also in the lower magnetic field region (higher g-values), around g = 2.02–2.01; these resonances arise from trapped holes in the form of oxygen-based radicals (e.g., Ti–O• species).
Figure 6.

X-band (∼9.07–9.09 GHz) light-induced electron paramagnetic resonance (LEPR) spectra recorded at T = 80 K of gray TiO2 nanoparticles dispersed in an aqueous NiSO4 solution. Panels a–f: spectra were recorded in (oxygen containing) water, with and without light irradiation. Experimental conditions: X-band frequency 9.072 GHz, 0.4 mW applied power, 1.0 mT modulation width, 30 s sweep time for each sequential spectrum, and 0.03 s time constant. Panels g–i: spectra recorded in a 20 vol % MeOH/H2O mixture (nitrogen saturated), with and without light irradiation. Experimental conditions: X-band frequency: 9.086 GHz, 1.6 mW applied power, 0.6 mT modulation width, 60 s sweep time for each sequential spectrum, and 0.03 s time constant.
Soon after UV illumination, several changes become evident, as shown in the 3D (Figure 6a,d) and 2D plots (Figure 6b,e): (i) the appearance of the strong hole signature at g1 = 2.04 and g2 = 2.01 (see Figure 6c, red spectrum), (ii) the small increase of the signal of lattice embedded Ti3+ (gavg = 1.92), best resolved in the 2D plot shown in Figure 6b, and importantly (iii) the formation of a new resonance feature at g ≈ 2.24 that can be assigned to the development of photoactive Ni+ sites (S = 1/2, 3d84s1) (see Figure 6d,e).
The g ≈ 2.24 signal (see Figure 6f, red spectrum) is broad and very weak. This is due to the low concentration of Ni centers on the surface of gray titania along with different Ni coordination environments due to local disorder – this can explain the increase of the resonance line width. Note that before UV irradiation in the spectral range where the resonance at g ∼ 2.24 develops, another broad signal is also detected at g ∼ 2.19 (see the 2D plot in Figure 6e and the blue spectrum in Figure 6f). This resonance cannot arise from the presence of Ni2+ cations, because Ni2+ usually adopts the low spin state and is thus EPR silent (S = 0, 3d8). High spin state Ni2+, a non-Kamer system (S = 1), has such large zero-field-splitting that becomes undetectable at X-band frequencies.65 Ni2+ cations in water solution can be oxidized to Ni3+ giving an S = 1/2 configuration (3d7) upon illumination at 325 nm, which produces EPR spectral features (Figure S11, gz = 2.32 and gx = gy = 2.06 that are different than those observed here with gray titania in the dark or upon illumination (see Figure 6f, blue and red spectra, respectivley). For example, a Ni3+-EDTA complex (S = 1/2) formed in aqueous solution upon irradiation of Ni2+-EDTA solutions was reported to exhibit EPR signatures at gz = 2.336 and gx = gy = 2.139 (spectrum recorded at T = 77 K, 60Co γ-source).66 Therefore, the weak and broad resonance we observe under dark conditions at g ∼ 2.19 can be associated with the presence on the gray titania of a small fraction of less reactive Ni+ species that are strongly interacting with the defective oxide surface; such sites are already present before UV light irradiation, and form when the titania powder is suspended in the Ni salt solution. We propose that these Ni+ species form by electron transfer from surface exposed Ti3+ sites to Ni2+ cations. It should be noted that the concentration of such Ni+ species is rather low and just above the detection limit of EPR (a spin sensitive technique).
To gain further knowledge of the resonance signals developing in the g = 2.2 region and investigate the reversibility of photogeneration of Ni+ species, light-induced electron paramagnetic resonance (LEPR) experiments were performed at T = 80 K for the Ni2+-gray TiO2 water suspension upon addition of a small amount of MeOH as hole scavenger (20 vol % MeOH/H2O) and by using nitrogen saturated solutions. We applied a dark–light–dark sequence (5 min dark followed by 10 min of irradiation at 325 nm and then 33 min dark). Results are shown in Figure 6g (3D plot), Figure 6h (2D plot) and Figure 6i. In dark conditions at t = 0 min, the Ni+ signal at g = 2.19 (around 300 mT) becomes more resolved (Figure 6i, olive line), showing the presence of a shoulder at a lower magnetic field (275–290 mT). This shoulder, a distinct signal at g = 2.24 (see Figure 6g,h, time window 5–15 min, and Figure 6i, pink spectrum), belongs to highly photoactive Ni+ sites that are generated under UV irradiation of the sample in the EPR tube before freezing in liquid N2. When shutting down the UV light, the signal at g = 2.24 slowly decays at 80 K (see Figure 6g,h, time window 16–48 min, and Figure 6i, blue spectrum) while the signal at g = 2.19 does not. Therefore, we conclude that the signal at g = 2.24 belongs to the metastable Ni+ species, confirming the XANES results. We propose that, besides the fraction of Ni+ centers already formed in the dark in the as prepared materials (less reactive sites), surface adsorbed Ni2+ cations can act as (reversible) electron acceptors as soon as UV light is switched on, forming EPR active Ni+ species that are responsible for the photocatalytic H2 evolution activity of gray TiO2. The data point toward an electronic rearrangement that takes place between Ti3+ sites and adsorbed Ni2+ ions. Such reaction can be expressed in the form of: Ti3+ + Ni2+ → Ti4+–Ni+, and therefore the Ni2+ centers must be either located nearby a surface exposed Ti3+ site or trapped in a Ti3+–Ni2+ configuration.
We also monitored by in situ LEPR experiments the magnetic field region where photogenerated holes and Ti3+ sites evolve (310–350 mT), using gray TiO2 in Ni–H2O/MeOH solutions. Spectra were recorded under identical conditions (5 min underthe dark followed by 10 min UV and then 33 min dark). Results are shown in Figure 7a,b (3D and 2D plots, respectively). Without the presence of dissolved oxygen, the spectra resolution is increased and allows to observe the formation of a new Ti resonance signature under light irradiation (g = 1.997, time 5–15 min) accompanied by the disappearance of the signal at g = 1.987 (Ti3+). The disappearance of this signal exactly correlates with the formation of Ni+ observed at g = 2.24.
Figure 7.

X-band LEPR spectra recorded at T = 80 K for gray TiO2 nanoparticles dispersed in NiSO4 solution (20 vol % MeOH/H2O, nitrogen saturated), with and without light irradiation. Sequence: 5 min dark, 10 min UV, and 33 min dark. Experimental conditions: X-band frequency: 9.085 GHz, 1.6 mW applied power, 0.6 mT modulation width, 60 s sweep time for each sequential spectrum, and 0.03 s time constant.
Note that due to the presence of MeOH, the resonance associated with hole centers becomes much less dominant here, if compared to that observed under UV light in the absence of hole scavenger (shown in Figure 6a–c). The spectra in Figure 7a,b also show a small increment in the amount of surface exposed Ti3+ sites under UV light. As soon as the UV light is switched off (from t = 16 to 48 min), the signal at g = 2.012 slowly decreases (the hole centers), and so does the signal at g = 1.997. The signal at g = 1.987 (Ti3+) reforms while the excess of surface exposed Ti3+ sites generated under irradiation appears to slowly decrease over time.
By comparing the in situ EPR findings with the results of the photocatalytic experiments (Figure 1), and by considering the experimental differences between the photocatalytic and in situ EPR set-ups (temperature, illumination), we deduce that the activation toward H2 evolution occurs by a combination of a fast and a slow process. The fast process is the formation of metastable Ni+-gray titania complex, while the slow process is a reorganization of the Ti3+ defect centers.46 Hence, the ability of gray titania to evolve H2 in the absence of hole scavenger can be linked to the formation of Ni+-TiO2–Ti3+ sites; notably, and as shown in the in situ EPR spectra in Figure S12 (see inset), such active state is only metastable, i.e., when the sample is exposed to ambient conditions (room temperature, UV light switched off), the distribution of spin species formed under light (active state) relaxes back to the starting distribution seen under dark conditions (resting state), and H2 evolution ceases. In other words, the active form of the catalyst (Ni(I)-TiO2-x) is present as such only in operando, and it returns to a state close to the initial situation, i.e., inactive, once the light is switched off.
In previous work,46in situ EPR experiments showed that illumination of gray titania for several days in plain H2O in the absence of Ni2+ leads to a significant increase in the gavg ≈ 1.93 signature, i.e., a strong increase in the amount of Ti3+ surface states occurs in the absence of Ni2+. This happens because swift electron transfer (to Ni cations in the solution) is not provided. In other words, TiO2 conduction band electrons are not transferred to the environment, and hence undergo trapping at Ti3+ surface states. In contrast, in the presence of Ni2+, even after 7 days of illumination, the intensity of the EPR signal at gavg ≈ 1.92 remains almost unaltered. In other words, in the absence of Ni2+ in solution, under illumination, electron accumulation takes place, leading to a steady self-reduction of the gray anatase. In the presence of Ni2+, the electron transfer to the electrolyte (via the Ni+ surface relay) is sufficiently fast that self-reduction is limited.
On the other hand, the important role of gray titania
as hole transfer mediator4 is herein illustrated
by comparing the behavior of white versus gray TiO2 in Ni2+-containing solutions
in the absence or presence of methanol. In contrast to the inactive
white form, Gray TiO2 in 0.4 mM NiSO4 aqueous solution not only evolves H2 (via 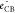 transfer
to water) but also generates H2O2 after prolonged
UV illumination (via
transfer
to water) but also generates H2O2 after prolonged
UV illumination (via 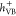 transfer to water, see H2O2 strip test in Figure S10). Instead, white TiO2 is found to evolve H2 from
Ni2+-containing solutions only in the presence of MeOH
(Figure 8); here, under
illumination, methanol acts as the hole capturing agent, and hole-transfer
is, thus, no longer the limiting factor. Note that in this case, H2 evolution takes place with gray and white TiO2 at comparable rate.
transfer to water, see H2O2 strip test in Figure S10). Instead, white TiO2 is found to evolve H2 from
Ni2+-containing solutions only in the presence of MeOH
(Figure 8); here, under
illumination, methanol acts as the hole capturing agent, and hole-transfer
is, thus, no longer the limiting factor. Note that in this case, H2 evolution takes place with gray and white TiO2 at comparable rate.
Figure 8.

Comparison of H2 production rates of gray TiO2 nanoparticles vs white anatase TiO2 nanoparticles in a 0.4 mM NiSO4 methanol–water solution (50 vol % MeOH) under UV light irradiation (365 nm LED, 100 mW cm–2).
Conclusions
In summary, in this work we demonstrated the light-induced self-assembly of a Ni+/TiO2/Ti3+ photocatalyst that is active for H2 generation from water in the absence of hole scavengers, e.g., methanol. This catalyst forms in situ when illuminating a suspension of reduced (gray) titania in a Ni2+ aqueous solution. No deposition of metallic Ni cocatalytic nanoparticles could be observed. Instead, we show that key to self-activation toward H2 evolution is the formation of monovalent Ni+ species on the TiO2 surface, combined with a light-induced rearrangement of defects in the semiconductor over time. Due to the metastable nature of the Ni+/TiO2/Ti3+ photocatalyst, in situ techniques (XAS and EPR) were used to prove the formation of monovalent Ni+ and the occurrence of defect reorganization. These self-assembly and activation processes enabling H2 evolution can be observed only for gray titania (and not for white or for black TiO2), as only gray titania provides surface Ti3+-OV states with adequate energy to form active, metastable monovalent nickel species. Gray titania, in addition, provides the ability to transfer photoholes to plain water.
In a wider context, the present work demonstrates a self-assembly process where illumination is the synthesis tool for an active metastable entity, as well as the energy provider for this entity to achieve photocatalytic H2 evolution from water. Self-amplifying reaction schemes as observed in the present work may have considerable potential for simple one-pot synthesis and use of photocatalysts.
Acknowledgments
The authors would like to acknowledge DFG and the Operational Program Research, Development and Education (European Regional Development Fund, project no. CZ.02.1.01/0.0/0.0/15_003/0000416 of the Ministry of Education, Youth and Sports of the Czech Republic) for financial support. We acknowledge the European Synchrotron Radiation Facility for provision of beamtime at ID26 beamline (DOI: 10.15151/ESRF-ES-561460108). M.A. acknowledges the Deutsche Forschungsgemeinschaft DFG (project no. DFG-grant AL 2479/1-1 30965319) and the Nederlandse Organisatie voor Wetenschappelijk Onderzoek NWO (project no. ECCM.TT.ECCM.005) for financial support. G.Z. and Z.B. acknowledge the project APPROACH No 101120397, HORIZON-WIDERA-2022-TALENTS, and the European Union under the REFRESH - Research Excellence For Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c08199.
Methods; additional SEM, XRD, photocatalytic, XPS, cyclic voltammetry, XAS, H2O2 test strips, and EPR data; in situ XAS setup pictures; additional references (PDF)
Author Contributions
All authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Whitesides G. M.; Grzybowski B. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- Eigen M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Nosaka Y. Mechanism of the OH Radical Generation in Photocatalysis with TiO2 of Different Crystalline Types. J. Phys. Chem. C 2014, 118, 10824–10832. 10.1021/jp501214m. [DOI] [Google Scholar]
- Liu N.; Mohajernia S.; Nguyen N. T.; Hejazi S.; Plass F.; Kahnt A.; Yokosawa T.; Osvet A.; Spiecker E.; Guldi D. M.; et al. Long-Living Holes in Grey Anatase TiO2 Enable Noble-Metal-Free and Sacrificial-Agent-Free Water Splitting. ChemSusChem 2020, 13, 4937–4944. 10.1002/cssc.202001045. [DOI] [Google Scholar]
- Nosaka Y.; Nosaka A. Understanding Hydroxyl Radical (•OH) Generation Processes in Photocatalysis. ACS Energy Lett. 2016, 1, 356–359. 10.1021/acsenergylett.6b00174. [DOI] [Google Scholar]
- Diesen V.; Jonsson M. Formation of H2O2 in TiO2 Photocatalysis of Oxygenated and Deoxygenated Aqueous Systems: A Probe for Photocatalytically Produced Hydroxyl Radicals. J. Phys. Chem. C 2014, 118, 10083–10087. 10.1021/jp500315u. [DOI] [Google Scholar]
- Ge M.; Cai J.; Iocozzia J.; Cao C.; Huang J.; Zhang X.; Shen J.; Wang S.; Zhang S.; Zhang K.-Q.; et al. A review of TiO2 nanostructured catalysts for sustainable H2 generation. Int. J. Hydrogen Energy 2017, 42, 8418–8449. 10.1016/j.ijhydene.2016.12.052. [DOI] [Google Scholar]
- Ni M.; Leung M. K. H.; Leung D. Y. C.; Sumathy K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renewable and Sustainable Energy Rev. 2007, 11, 401–425. 10.1016/j.rser.2005.01.009. [DOI] [Google Scholar]
- Nakata K.; Fujishima A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol., C 2012, 13, 169–189. 10.1016/j.jphotochemrev.2012.06.001. [DOI] [Google Scholar]; Osterloh F. E. Inorganic Materials as Catalysts for Photochemical Splitting of Water. Chem. Mater. 2008, 20 (1), 35–54. 10.1021/cm7024203. [DOI] [Google Scholar]
- Kudo A.; Miseki Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. 10.1039/B800489G. [DOI] [PubMed] [Google Scholar]
- Fujishima A.; Zhang X.; Tryk D. A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. 10.1016/j.surfrep.2008.10.001. [DOI] [Google Scholar]
- Schneider J.; Matsuoka M.; Takeuchi M.; Zhang J.; Horiuchi Y.; Anpo M.; Bahnemann D. W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. 10.1021/cr5001892. [DOI] [PubMed] [Google Scholar]
- Chen S.; Takata T.; Domen K. Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2017, 2, 17050. 10.1038/natrevmats.2017.50. [DOI] [Google Scholar]
- Yoshida M.; Yamakata A.; Takanabe K.; Kubota J.; Osawa M.; Domen K. ATR-SEIRAS Investigation of the Fermi Level of Pt Cocatalyst on a GaN Photocatalyst for Hydrogen Evolution under Irradiation. J. Am. Chem. Soc. 2009, 131, 13218–13219. 10.1021/ja904991p. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Teramura K.; Lu D.; Saito N.; Inoue Y.; Domen K. Noble-Metal/Cr2O3 Core/Shell Nanoparticles as a Cocatalyst for Photocatalytic Overall Water Splitting. Angew. Chem., Int. Ed. 2006, 45, 7806–7809. 10.1002/anie.200602473. [DOI] [PubMed] [Google Scholar]
- Gomes Silva C.; Juárez R.; Marino T.; Molinari R.; García H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. 10.1021/ja1086358. [DOI] [PubMed] [Google Scholar]
- Chen X.; Shen S.; Guo L.; Mao S. S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Domen K. Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies. Chem. Rev. 2020, 120, 919–985. 10.1021/acs.chemrev.9b00201. [DOI] [PubMed] [Google Scholar]
- Kamat P. V.; Jin S. Semiconductor Photocatalysis: “Tell Us the Complete Story!”. ACS Energy Lett. 2018, 3, 622–623. 10.1021/acsenergylett.8b00196. [DOI] [Google Scholar]
- Schneider J.; Bahnemann D. W. Undesired Role of Sacrificial Reagents in Photocatalysis. J. Phys. Chem. Lett. 2013, 4, 3479–3483. 10.1021/jz4018199. [DOI] [Google Scholar]
- Yuan J.; Wen J.; Zhong Y.; Li X.; Fang Y.; Zhang S.; Liu W. Enhanced photocatalytic H2 evolution over noble-metal-free NiS cocatalyst modified CdS nanorods/g-C3N4 heterojunctions. J. Mater. Chem. A 2015, 3, 18244–18255. 10.1039/C5TA04573H. [DOI] [Google Scholar]
- Zong X.; Han J.; Ma G.; Yan H.; Wu G.; Li C. Photocatalytic H2 Evolution on CdS Loaded with WS2 as Cocatalyst under Visible Light Irradiation. J. Phys. Chem. C 2011, 115, 12202–12208. 10.1021/jp2006777. [DOI] [Google Scholar]
- Sun Z.; Zheng H.; Li J.; Du P. Extraordinarily efficient photocatalytic hydrogen evolution in water using semiconductor nanorods integrated with crystalline Ni2P cocatalysts. Energy Environ. Sci. 2015, 8, 2668–2676. 10.1039/C5EE01310K. [DOI] [Google Scholar]
- Zong X.; Yan H.; Wu G.; Ma G.; Wen F.; Wang L.; Li C. Enhancement of Photocatalytic H2 Evolution on CdS by Loading MoS2 as Cocatalyst under Visible Light Irradiation. J. Am. Chem. Soc. 2008, 130, 7176–7177. 10.1021/ja8007825. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Fu A.; Chen X.; Liu L.; Ren L.; Tong L.; Ye J. Highly efficient Cu induced photocatalysis for visible-light hydrogen evolution. Catal. Today 2019, 335, 166–172. 10.1016/j.cattod.2018.11.002. [DOI] [Google Scholar]
- Peng Y.; Jiang K.; Hill W.; Lu Z.; Yao H.; Wang H. Large-Scale, Low-Cost, and High-Efficiency Water-Splitting System for Clean H2 Generation. ACS Appl. Mater. Interfaces 2019, 11, 3971–3977. 10.1021/acsami.8b19251. [DOI] [PubMed] [Google Scholar]
- Spanu D.; Recchia S.; Mohajernia S.; Tomanec O.; Kment Š.; Zboril R.; Schmuki P.; Altomare M. Templated Dewetting–Alloying of NiCu Bilayers on TiO2 Nanotubes Enables Efficient Noble-Metal-Free Photocatalytic H2 Evolution. ACS Catal. 2018, 8, 5298–5305. 10.1021/acscatal.8b01190. [DOI] [Google Scholar]
- Shahvaranfard F.; Ghigna P.; Minguzzi A.; Wierzbicka E.; Schmuki P.; Altomare M. Dewetting of PtCu Nanoalloys on TiO2 Nanocavities Provides a Synergistic Photocatalytic Enhancement for Efficient H2 Evolution. ACS Appl. Mater. Interfaces 2020, 12, 38211–38221. 10.1021/acsami.0c10968. [DOI] [PubMed] [Google Scholar]
- Pinna M.; Wei A. W. W.; Spanu D.; Will J.; Yokosawa T.; Spiecker E.; Recchia S.; Schmuki P.; Altomare M. Amorphous NiCu Thin Films Sputtered on TiO2 Nanotube Arrays: A Noble-Metal Free Photocatalyst for Hydrogen Evolution. ChemCatChem 2022, 14, e202201052 10.1002/cctc.202201052. [DOI] [Google Scholar]
- Chen W.-T.; Chan A.; Sun-Waterhouse D.; Llorca J.; Idriss H.; Waterhouse G. I. N. Performance comparison of Ni/TiO2 and Au/TiO2 photocatalysts for H2 production in different alcohol-water mixtures. J. Catal. 2018, 367, 27–42. 10.1016/j.jcat.2018.08.015. [DOI] [Google Scholar]
- Melián E. P.; Suárez M. N.; Jardiel T.; Rodríguez J. M. D.; Caballero A. C.; Araña J.; Calatayud D. G.; Díaz O. G. Influence of nickel in the hydrogen production activity of TiO2. Appl. Catal., B 2014, 152–153, 192–201. 10.1016/j.apcatb.2014.01.039. [DOI] [Google Scholar]
- Xu Y.; Xu R. Nickel-based cocatalysts for photocatalytic hydrogen production. Appl. Surf. Sci. 2015, 351, 779–793. 10.1016/j.apsusc.2015.05.171. [DOI] [Google Scholar]
- Chen W.-T.; Chan A.; Sun-Waterhouse D.; Moriga T.; Idriss H.; Waterhouse G. I. N. Ni/TiO2: A promising low-cost photocatalytic system for solar H2 production from ethanol–water mixtures. J. Catal. 2015, 326, 43–53. 10.1016/j.jcat.2015.03.008. [DOI] [Google Scholar]
- Wang J.; Mao S.; Liu Z.; Wei Z.; Wang H.; Chen Y.; Wang Y. Dominating Role of Ni0 on the Interface of Ni/NiO for Enhanced Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2017, 9, 7139–7147. 10.1021/acsami.6b15377. [DOI] [PubMed] [Google Scholar]
- Townsend T. K.; Browning N. D.; Osterloh F. E. Overall photocatalytic water splitting with NiOx–SrTiO3 – a revised mechanism. Energy Environ. Sci. 2012, 5, 9543–9550. 10.1039/c2ee22665k. [DOI] [Google Scholar]
- Wang W.; Liu S.; Nie L.; Cheng B.; Yu J. Enhanced photocatalytic H2-production activity of TiO2 using Ni(NO3)2 as an additive. Phys. Chem. Chem. Phys. 2013, 15, 12033–12039. 10.1039/C2CP43628K. [DOI] [PubMed] [Google Scholar]
- Forouzan F.; Richards T. C.; Bard A. J. Photoinduced Reaction at TiO2 Particles. Photodeposition from NiII Solutions with Oxalate. J. Chem. Phys. 1996, 100, 18123–18127. 10.1021/jp953241f. [DOI] [Google Scholar]
- Spanu D.; Minguzzi A.; Recchia S.; Shahvardanfard F.; Tomanec O.; Zboril R.; Schmuki P.; Ghigna P.; Altomare M. An Operando X-ray Absorption Spectroscopy Study of a NiCu–TiO2 Photocatalyst for H2 Evolution. ACS Catal. 2020, 10, 8293–8302. 10.1021/acscatal.0c01373. [DOI] [Google Scholar]
- Han K.; Kreuger T.; Mei B.; Mul G. Transient Behavior of Ni@NiOx Functionalized SrTiO3 in Overall Water Splitting. ACS Catal. 2017, 7, 1610–1614. 10.1021/acscatal.6b03662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosaka Y.; Nosaka A. Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. 10.1021/acs.chemrev.7b00161. [DOI] [PubMed] [Google Scholar]
- Kiwi J.; Graetzel M. Optimization of conditions for photochemical water cleavage. Aqueous platinum/TiO2 (anatase) dispersions under ultraviolet light. J. Chem. Phys. 1984, 88, 1302–1307. 10.1021/j150651a012. [DOI] [Google Scholar]
- Kiwi J.; Graetzel M. Specific analysis of surface-bound peroxides formed during photoinduced water cleavage in titanium dioxide-based microheterogeneous systems. J. Mol. Catal. 1987, 39, 63–70. 10.1016/0304-5102(87)80047-0. [DOI] [Google Scholar]
- Munuera G.; Espinós J. P.; Fernández A.; Malet P.; González-Elipe A. R. TiO2 corrosion during water photocleavage using Rh/TiO2 suspensions. J. Chem. Soc., Faraday Trans. 1990, 86, 3441–3445. 10.1039/FT9908603441. [DOI] [Google Scholar]
- Siahrostami S.; Li G.-L.; Viswanathan V.; Nørskov J. K. One- or Two-Electron Water Oxidation, Hydroxyl Radical, or H2O2 Evolution. J. Phys. Chem. Lett. 2017, 8, 1157–1160. 10.1021/acs.jpclett.6b02924. [DOI] [PubMed] [Google Scholar]
- Liu N.; Zhou X.; Nguyen N. T.; Peters K.; Zoller F.; Hwang I.; Schneider C.; Miehlich M. E.; Freitag D.; Meyer K.; et al. Black Magic in Gray Titania: Noble-Metal-Free Photocatalytic H2 Evolution from Hydrogenated Anatase. ChemSusChem 2017, 10, 62–67. 10.1002/cssc.201601264. [DOI] [PubMed] [Google Scholar]
- Qin S.; Badura Z.; Denisov N.; Tomanec O.; Mohajernia S.; Liu N.; Kment S.; Zoppellaro G.; Schmuki P. Self-assembly of a Ni(I)-photocatalyst for plain water splitting without sacrificial agents. Electrochem. Commun. 2021, 122, 106909. 10.1016/j.elecom.2020.106909. [DOI] [Google Scholar]
- Chen X.; Liu L.; Yu P. Y.; Mao S. S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746–750. 10.1126/science.1200448. [DOI] [PubMed] [Google Scholar]
- Chen X.; Liu L.; Huang F. Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 2015, 44, 1861–1885. 10.1039/C4CS00330F. [DOI] [PubMed] [Google Scholar]
- Liu N.; Schneider C.; Freitag D.; Hartmann M.; Venkatesan U.; Müller J.; Spiecker E.; Schmuki P. Black TiO2 Nanotubes: Cocatalyst-Free Open-Circuit Hydrogen Generation. Nano Lett. 2014, 14, 3309–3313. 10.1021/nl500710j. [DOI] [PubMed] [Google Scholar]
- Naldoni A.; Altomare M.; Zoppellaro G.; Liu N.; Kment Š; Zbořil R.; Schmuki P. Photocatalysis with Reduced TiO2: From Black TiO2 to Cocatalyst-Free Hydrogen Production. ACS Catal. 2019, 9, 345–364. 10.1021/acscatal.8b04068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicka E.; Altomare M.; Wu M.; Liu N.; Yokosawa T.; Fehn D.; Qin S.; Meyer K.; Unruh T.; Spiecker E.; et al. Reduced grey brookite for noble metal free photocatalytic H2 evolution. J. Mater. Chem. A 2021, 9, 1168–1179. 10.1039/D0TA09066B. [DOI] [Google Scholar]
- Will J.; Wierzbicka E.; Wu M.; Götz K.; Yokosawa T.; Liu N.; Tesler A. B.; Stiller M.; Unruh T.; Altomare M.; et al. Hydrogenated anatase TiO2 single crystals: Defects formation and structural changes as microscopic origin of co-catalyst free photocatalytic H2 evolution activity. J. Mater. Chem. A 2021, 9, 24932–24942. 10.1039/D1TA04809K. [DOI] [Google Scholar]
- Qin S.; Kim H.; Denisov N.; Fehn D.; Schmidt J.; Meyer K.; Schmuki P. Grey facet-controlled anatase nanosheets for photocatalytic H2 evolution without co-catalyst. J. Phys.: Energy 2021, 3, 034003. 10.1088/2515-7655/abd5a8. [DOI] [Google Scholar]
- Liu N.; Schneider C.; Freitag D.; Venkatesan U.; Marthala V. R. R.; Hartmann M.; Winter B.; Spiecker E.; Osvet A.; Zolnhofer E. M.; et al. Hydrogenated Anatase: Strong Photocatalytic Dihydrogen Evolution without the Use of a Co-Catalyst. Angew. Chem., Int. Ed. 2014, 53, 14201–14205. 10.1002/anie.201408493. [DOI] [PubMed] [Google Scholar]
- Mohajernia S.; Andryskova P.; Zoppellaro G.; Hejazi S.; Kment S.; Zboril R.; Schmidt J.; Schmuki P. Influence of Ti3+ defect-type on heterogeneous photocatalytic H2 evolution activity of TiO2. J. Mater. Chem. A 2020, 8, 1432–1442. 10.1039/C9TA10855F. [DOI] [Google Scholar]
- Qin S.; Denisov N.; Zhou X.; Zdražil L.; Fehn D.; Hwang I.; Bruns M.; Kim H.; Meyer K.; Schmuki P. Critical factors for photoelectrochemical and photocatalytic H2 evolution from gray anatase (001) nanosheets. J. Phys.: Energy 2022, 4, 044004. 10.1088/2515-7655/ac8ed3. [DOI] [Google Scholar]
- Wierzbicka E.; Zhou X.; Denisov N.; Yoo J.; Fehn D.; Liu N.; Meyer K.; Schmuki P. Self-Enhancing H2 Evolution from TiO2 Nanostructures under Illumination. ChemSusChem 2019, 12, 1900–1905. 10.1002/cssc.201900192. [DOI] [PubMed] [Google Scholar]
- Lo W. J.; Chung Y. W.; Somorjai G. A. Electron spectroscopy studies of the chemisorption of O2, H2 and H2O on the TiO2(100) surfaces with varied stoichiometry: Evidence for the photogeneration of Ti+3 and for its importance in chemisorption. Surf. Sci. 1978, 71, 199–219. 10.1016/0039-6028(78)90328-X. [DOI] [Google Scholar]
- Millet M.-M.; Algara-Siller G.; Wrabetz S.; Mazheika A.; Girgsdies F.; Teschner D.; Seitz F.; Tarasov A.; Levchenko S. V.; Schlögl R.; et al. Ni Single Atom Catalysts for CO2 Activation. J. Am. Chem. Soc. 2019, 141, 2451–2461. 10.1021/jacs.8b11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Gao C.; Low J.; Long R.; Kong T.; Zhu J.; Xiong Y. Heterogeneous Single-Atom Photocatalysts: Fundamentals and Applications. Chem. Rev. 2020, 120, 12175–12216. 10.1021/acs.chemrev.9b00840. [DOI] [PubMed] [Google Scholar]
- Moulder J. F.; Stickle W. F.; Sobol P. E.; Bomben K. D.. Handbook of X-ray Photoelectron Spectroscopy; Perkin Elmer Corp.,: Eden Prairie/USA, 1992. [Google Scholar]
- Grosvenor A. P.; Biesinger M. C.; Smart R. S. C.; McIntyre N. S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. 10.1016/j.susc.2006.01.041. [DOI] [Google Scholar]
- Biesinger M. C.; Payne B. P.; Lau L. W. M.; Gerson A.; Smart R. S. C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 2009, 41, 324–332. 10.1002/sia.3026. [DOI] [Google Scholar]
- Dubey P.; Kaurav N.; Devan R. S.; Okram G. S.; Kuo Y. K. The effect of stoichiometry on the structural, thermal and electronic properties of thermally decomposed nickel oxide. RSC Adv. 2018, 8, 5882–5890. 10.1039/C8RA00157J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt H. W.; Legrand D.; Bancroft G. M. Interpretation of Ni2p XPS spectra of Ni conductors and Ni insulators. Phys. Chem. Miner. 2000, 27, 357–366. 10.1007/s002690050265. [DOI] [Google Scholar]
- Abba F.; De Santis G.; Fabbrizzi L.; Licchelli M.; Manotti Lanfredi A. M.; Pallavicini P.; Poggi A.; Ugozzoli F. Nickel(II) Complexes of Azacyclams: Oxidation and Reduction Behavior and Catalytic Effects in the Electroreduction of Carbon Dioxide. Inorg. Chem. 1994, 33, 1366–1375. 10.1021/ic00085a026. [DOI] [Google Scholar]
- Di Casa M.; Fabbrizzi L.; Licchelli M.; Poggi A.; Sacchi D.; Zema M. A novel fluorescence redox switch based on the formal NiII/NiI couple. J. Chem. Soc., Dalton Trans. 2001, 1671–1675. 10.1039/b101310f. [DOI] [Google Scholar]
- Lovecchio F. V.; Gore E. S.; Busch D. H. Oxidation and reduction behavior of macrocyclic complexes of nickel. Electrochemical and electron spin resonance studies. J. Am. Chem. Soc. 1974, 96, 3109–3118. 10.1021/ja00817a016. [DOI] [Google Scholar]
- Bad’ura Z.; Naldoni A.; Qin S.; Bakandritsos A.; Kment Š.; Schmuki P.; Zoppellaro G. Light-Induced Migration of Spin Defects in TiO2 Nanosystems and their Contribution to the H2 Evolution Catalysis from Water. ChemSusChem 2021, 14, 4408–4414. 10.1002/cssc.202101218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.