

Abstract

Herein we present the first method for the synthesis of bicyclo[1.1.1]pentyl (BCP) alkyl ethers from alcohols. The reaction uses BCP–thianthrenium reagents and is catalyzed by a dual copper/photoredox catalyst system. Unlike known alkylations of tertiary alcohols via carbocation intermediates, our Cu-mediated radical process circumvents the labile BCP carbocations. The approach demonstrates a broad tolerance for functional groups when applied to primary, secondary, and even tertiary alcohols. In addition, we highlight the utility of this method in late-stage functionalizations of both natural products and pharmaceuticals as well as in the rapid construction of BCP analogs of known pharmaceuticals that would otherwise be difficult to access.
Approximately 45% of marketed small-molecule pharmaceuticals contain phenyl rings.1 In modern medicinal chemistry, replacement of planar phenyl rings with sp3-rich bioisosteres can lead to increased metabolic stability, membrane permeability, and increased solubility.2 Since the first study of the BCP analogue of (S)-(4-carboxyphenyl)glycine was described by Pellicciari and co-workers in 1996,3 the rigid three-dimensional 1,3-disubstituted BCPs have emerged as promising bioisosteres for para-substituted benzenes in drug development that maintain the exit vectors in a 180° dihedral angle.4 Over the last 15 years, numerous efforts have been devoted to the development of substituted BCPs, such as BCP amines,5 BCP arenes,5h,6 BCP alkanes,6f,7 and BCP aryl ethers5h via the functionalization of [1.1.1]propellane8 or cross-coupling reactions of BCP-based reagents.5h,6c,6d,7b,9However, alkylation of alcohols with BCP scaffolds has remained elusive. Such a new method would enable access to BCP analogs of alkyl aryl ethers that are prevalent in pharmaceuticals, such as in the selective serotonin reuptake inhibitor fluoxetine and the Parkinson’s medication safinamide. Here we report the first bicyclopentylation of alcohols using BCP–thianthrenium (TT+) reagents. Distinct from well-established alkylation of alcohols with tertiary electrophiles such as tert-butyl bromide via carbocation intermediates,10 our method proceeds through a metal-mediated radical process that can bypass the unstable BCP cations. The favorable high reduction potential and rapid mesolytic cleavage rate of BCP–TT+ allow the reaction to proceed under mild conditions with a wide variety of functional groups present and alcohols used as limiting reagents. Mechanistic studies imply that TT may potentially act as an electron-transfer mediator between the photoredox catalyst and copper catalyst. The substituted BCP–TT+ reagents can be used as structural linkers or end groups, which allows for rapid preparation of promising BCP analogs of pharmaceutically relevant ethers. To the best of our knowledge, this study represents the first example of directing-group-free transition-metal (TM)-catalyzed alkylation of tertiary alcohols that does not involve classic carbocation intermediates.
Alkyl aryl ethers are important structural motifs in pharmaceutically active compounds,11 such as in fluoxetine,12 butoxycaine,13 safinamide,14 and pranlukast.15 The current approach to BCP alkyl ethers involves the alkylation of a hydroxyl-substituted BCP with reactive alkylating reagents. Therefore, the approach is limited to reactive primary electrophiles, such as benzyl bromide, in the presence of a strong base for a conventional Williamson ether synthesis (Figure 1A, left).6c Alternatively, a conceptually distinct approach is the bicyclopentylation of alcohols with BCP-based reagents, which remains challenging due to the sluggish development of syntheses of tertiary alkyl ethers (Figure 1A, right). The Baran16a and Ohmiya16b groups reported elegant syntheses of hindered dialkyl ethers from tertiary alkyl carboxylic acids and alcohols via electrochemistry and organophotoredox catalysis, respectively. Key to the success lies in the generation of tertiary carbocations or carbocation-like intermediates under nonacidic conditions, which can be subsequently trapped by alcohol nucleophiles. However, the BCP carboxylic acid, as a bridgehead 3° carbocation precursor, does not afford the corresponding product under these conditions (Figure 1B),16a possibly because of the low stability of BCP cation intermediates (energy barrier to ring opening: ∼19 kcal/mol).17 The use of transition metals to catalyze the bicyclopentylation of alcohols involving kinetically stable BCP radical intermediates (energy barrier to ring opening: ∼26 kcal/mol),18 may avoid the skeletal rearrangement of the BCP intermediates. To date, BCP metals,6b BCP halides,6d BCP boronates,6c,7b,9 BCP amines,5a−5d and BCP carboxylate derivatives6a,6e are the most used reagents for the construction of BCPs. However, none of them have been reported for the functionalization of alcohols to construct ether bonds. Considering the potential of BCP alkyl ethers in drug discovery, we sought to develop the first TM-catalyzed bicyclopentylation of alcohols using BCP–TT+ reagents (Figure 1C).
Figure 1.

Synthesis of BCP alkyl ethers. LG, leaving group; PC, photocatalyst.
We have previously demonstrated that the cationic BCP–TT+ reagents can serve as BCP radical sources, which can engage in TM-mediated functionalization of phenols, various N-nucleophiles, and (het)aryl bromides.5h However, these conditions failed to deliver the desired product when alcohols were used as substrates due to the inherent reactivity differences between alcohols and the other, softer nucleophiles, like phenols and amines. For instance, metal alkoxides, crucial intermediates in many ether syntheses, exhibit both higher basicity and stronger reducing power compared to metal phenoxides,19,20 and alcohols are less nucleophilic than many N-nucleophiles. Consequently, in many of the TM-catalyzed C–O cross-coupling reactions involving alcohols, excess or even solvent amounts of alcohol are required.21 As part of our ongoing endeavors on BCP and thianthrenium chemistry and encouraged by recent Cu-catalyzed C–O cross-coupling reactions of alcohols,22,23 we sought the functionalization of alcohols with BCP–TT+ salts. The reaction could proceed by the reductive generation of BCP radical A (Figure 1C). Subsequently, the BCP radical would be intercepted by metal alkoxide intermediate B to form Cu(III) intermediate C, which undergoes reductive elimination for C–O bond formation.
Based on our working hypothesis, we found that the reaction between 4-methoxyphenethyl alcohol (1a) and trifluoromethyl bicyclopentyl thianthrenium salt (CF3-BCP–TT+BF4–, 1b) occurred with Cu(acac)2 as the catalyst, Na2CO3 as the base, and Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst under 460 nm irradiation to give 1 in 85% yield (Figure 2A). The use of Cu(acac)2 was crucial for the high yield, possibly because the β-diketonate ligand may facilitate the oxidative ligation of BCP radical A or stabilize the high-valent metal alkoxides C (entries 1–4).5d,23a,24,25 While Cu(TMHD)2 (TMHD = 2,2,6,6-tetramethyl-3,5-heptanedionate) can afford higher yields, we only opted for this more expensive copper source when Cu(acac)2 afforded less than 60% yield (entry 5). Control experiments showed that the copper catalyst, iridium photocatalyst, 3 Å molecular sieves, and blue LED light irradiation are necessary for efficient reaction (entries 9–15).26 The addition of 2.0 equiv of the radical trapping reagent 2,2,6,6,-tetramethylpiperidin-1-oxyl (TEMPO) suppressed the formation of 1, and a TEMPO–BCP radical adduct was observed (entry 16), consistent with the formation of BCP radicals.
Figure 2.
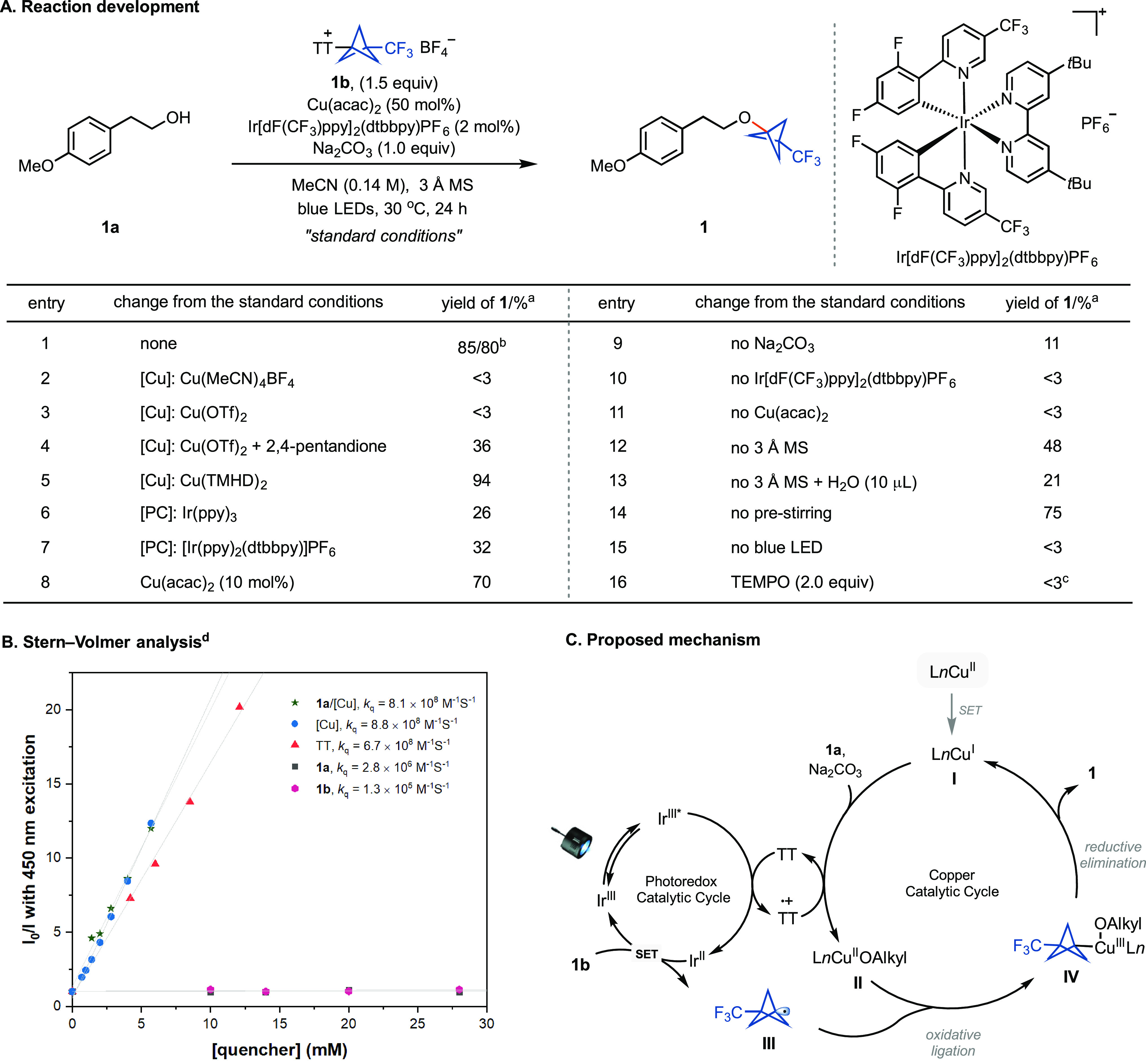
Reaction development and a mechanistic investigation. aYields were determined by 19F NMR. bIsolated yield. cThe TEMPO–BCP adduct was observed by HRMS. dThe Stern–Volmer plot of Cu(acac)2 was corrected due to the inner filter effect.
A Stern–Volmer analysis revealed an effective quenching of the photoexcited sensitizer’s (Ir[dF(CF3)ppy]2(dtbbpy)PF6) luminescence by Cu(acac)2 and thianthrene (TT) (Figure 2B). We propose that Cu(I) may act as an active catalyst formed in situ by initial single electron transfer (SET) to the Cu(II) precatalyst from the excited photoredoxcatalyst (IrIV/*IrIII = −0.9 V, E1/2 = −1.0 V for Cu(acac)2, both versus SCE in MeCN).5d,27,28 After induction, the reaction may proceed by reductive quenching of the excited Ir(III) photoredoxcatalyst by TT (*IrIII/IrII = +1.21 V, E1/2 = +1.26 V for TT, both versus SCE in MeCN).29,30 Based on this analysis and previous literature,5,28,31 a plausible catalytic cycle is shown in Figure 2C. After quenching of the excited Ir(III) photoreodoxcatalyst, the ensuing Ir(II) species can reduce the BCP–TT+ salt (IrIII/IrII = −1.4 V, E1/2 = −1.4 V for 1b, versus SCE in MeCN),5h generating BCP radical III. The TT radical cation can oxidize the Cu(I) catalyst to generate Cu(II) (supported by EPR analysis; Figure S8), which then undergoes ligand exchange with the O-nucleophile. Oxidative ligation with intermediate III affords the Cu(III) complex IV. Finally, the desired product is formed via reductive elimination from IV. All experimental observables are consistent with the proposed mechanism.32
Due to the mild conditions, a large variety of alcohols can now be bicyclopentylated to afford BCP alkyl ethers that would otherwise be challenging to synthesize by other methods (Figure 3). For example, no synthetic procedure for secondary alkyl BCP ethers is currently documented. Overall, the reaction shows broad scope and proceeds efficiently with primary (for example, 5, 8, and 15), cyclic (for example, 17, 19, and 20) and acyclic (for example, 22 and 25) secondary, and tertiary (18) alcohols. Various synthetically useful functional groups, such as esters (2, 17, 20, 22), alkenes (4, 11, 19), alkyne (6), amides (9, 12, 13), halides (8, 15, 25), ketones (19), and lactam (25) are tolerated, providing the desired products in 61–90% yield. Coordinating groups such as pyridines (8, 9), pyrimidine (21), and tertiary amine (10), which may cause catalyst poisoning, do not inhibit the desired C–O cross-coupling reactions. Notably, exclusive chemoselectivity was observed when the substrate contained an N-nucleophile (e.g., 13; vide infra). The high functional group tolerance renders this method applicable for late-stage functionalizations (LSFs) of natural and medicinal molecules such as (−)-nopol (4), tropicamide (9), perphenazin (10), pregnenolone (19), podophyllotoxin (20), and ezetimibe (25). Aldols (22), which are prone to dehydration under both acidic and basic conditions, can also be functionalized under the conditions reported here. Replacing Cu(acac)2 with Cu(TMHD)2 is crucial for the efficient bicyclopentalytion of secondary and tertiary alcohols (19). The reactivity of CN-BCP–TT+ (1c) is usually lower than for CF3-BCP–TT+ reagent 1b (compare, for example, 16 and 26 as well as 13 and 27). This difference in reactivity may be a consequence of electronic through-space interactions of the more electron-withdrawing cyano substituent, which lower the nucleophilicity of the bridgehead carbon radical toward the copper center. Although copper can be used as a catalyst (turnover number (TON) up to 7; Figure 2A, entry 8), 0.5 equiv of copper results in higher yields. Considering the complex starting materials typical for LSFs, the use of a simple copper salt in substoichiometric amounts may be acceptable.
Figure 3.
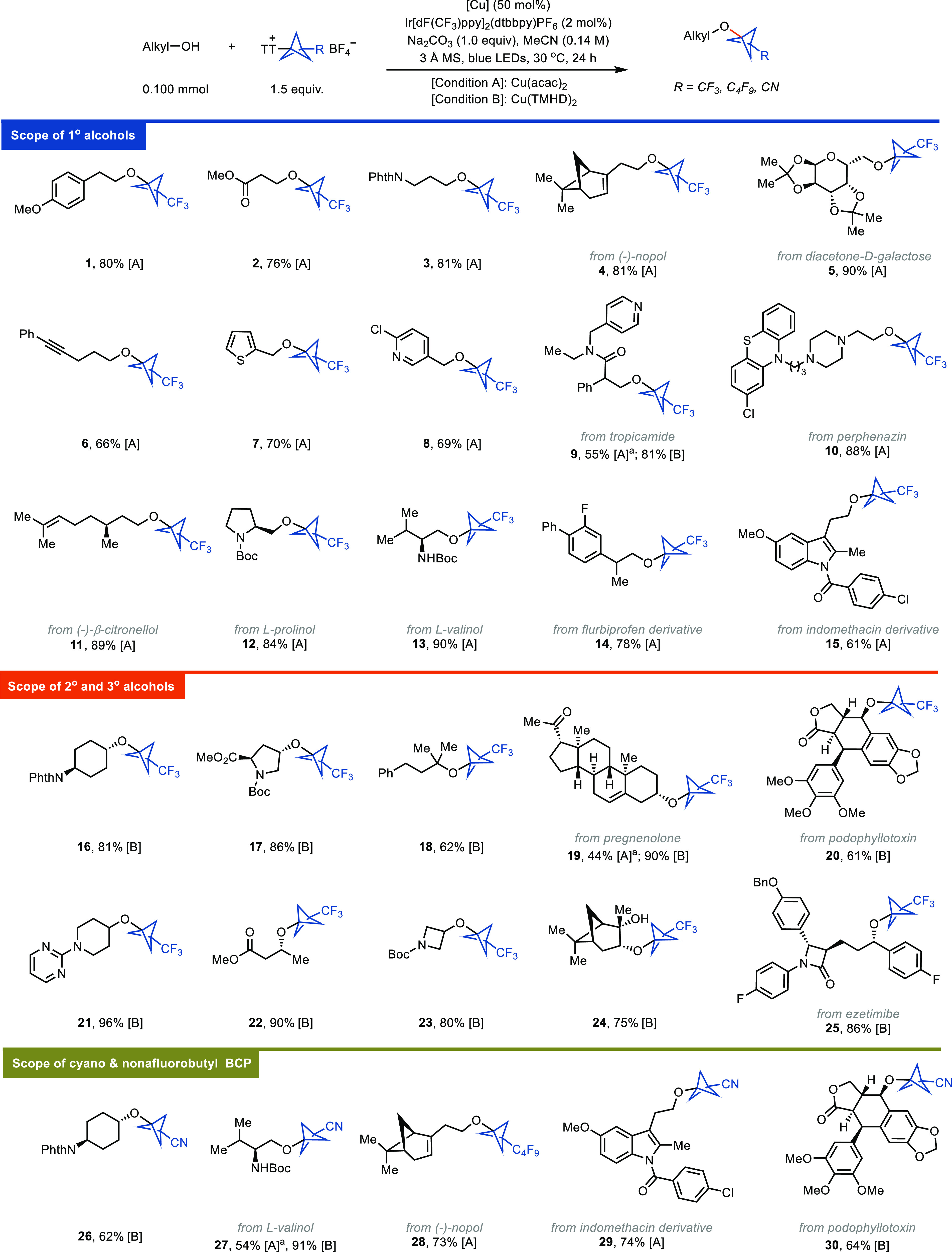
Substrate scope. aYields were determined by 19F NMR or 1H NMR.
While the scope for the alcohol is large, the scope of the BCP–TT+ reagents is limited to a rather small subset. Two aspects are conceptually challenging to expand the diversity of these reagents, at least in the chemistry presented here. Based on the data, it appears most likely that the reaction proceeds through a radical chain transfer, in which after photochemical initiation a TT radical cation is transferred from the starting material to the product (Figure 4A). For chain initiation and propagation, homolytic cleavage of the C–S bond in CF3-TT+ (31) is facile, which is the case for a rather stabilized CF3 radical. However, other substituents such as the methyl substituent in Me-TT+ (33) are bound more strongly, as supported by DFT calculations (Figure 4B). Second, other compounds that also feature a small bond dissociation energy (BDE) and would possibly engage in productive chain transfer could not be synthesized based on their lack of stability. For example, the difluoromethyl analogue of 31 (32; Figure 4 C) is unstable, presumably due to fast elimination from the cationic sulfonium salt. The cyano-substituted BCP compound 1c was synthesized through a different procedure5h and displays a unique synthesis that could not be extended to other nucleophiles, presumably due to competitive redox chemistry with oxidation of nucleophiles other than cyanide by the thianthrenium radical cation.30,33
Figure 4.

Mechanistic analysis of the synthesis of BCP–TT+ reagents. (A) Chain process of synthesis of BCP–TT+ reagents. (B) BDEs of different TT+ reagents. (C) Comparison of CF3-TT+ (31) and CF2H-TT+ (32). Ar = 3-fluoro-4-methoxyphenyl.
While the scope of the BCP–TT+ reagents is limited to only a few cases, we have highlighted the synthetic utility of our methodology to drug discovery with the few but relevant substituents in four BCP analogs of known pharmaceuticals (Figure 5A–D). The BCP analogue of fluoxetine hydrochloride, 36, was synthesized in only two steps and 85% overall yield from 1b and readily available starting material 35a. The cyano group of BCP reagent 1c can serve as a linchpin for the synthesis of other 1,3-disubstituted BCPs. For instance, cyano-BCP butyl ether (37) was obtained in 71% yield. Subsequently, hydrolysis of the cyano group to the corresponding carboxylic acid followed by alkylation afforded BCP-butoxycaine (38). Similarly, 41 underwent hydrolysis and amide condensation to give BCP-pranlukast (42). The cyano-BCP can also be selectively reduced to a BCP aldehyde, enabling reductive amination for the synthesis of BCP-safinamide (40). To the best of our knowledge, none of these analogs have been reported before. In addition, the cyano group in the BCP scaffold gives access to methylcarboxylate, methylamino, amide, and amino substituents (Figure 5E).
Figure 5.
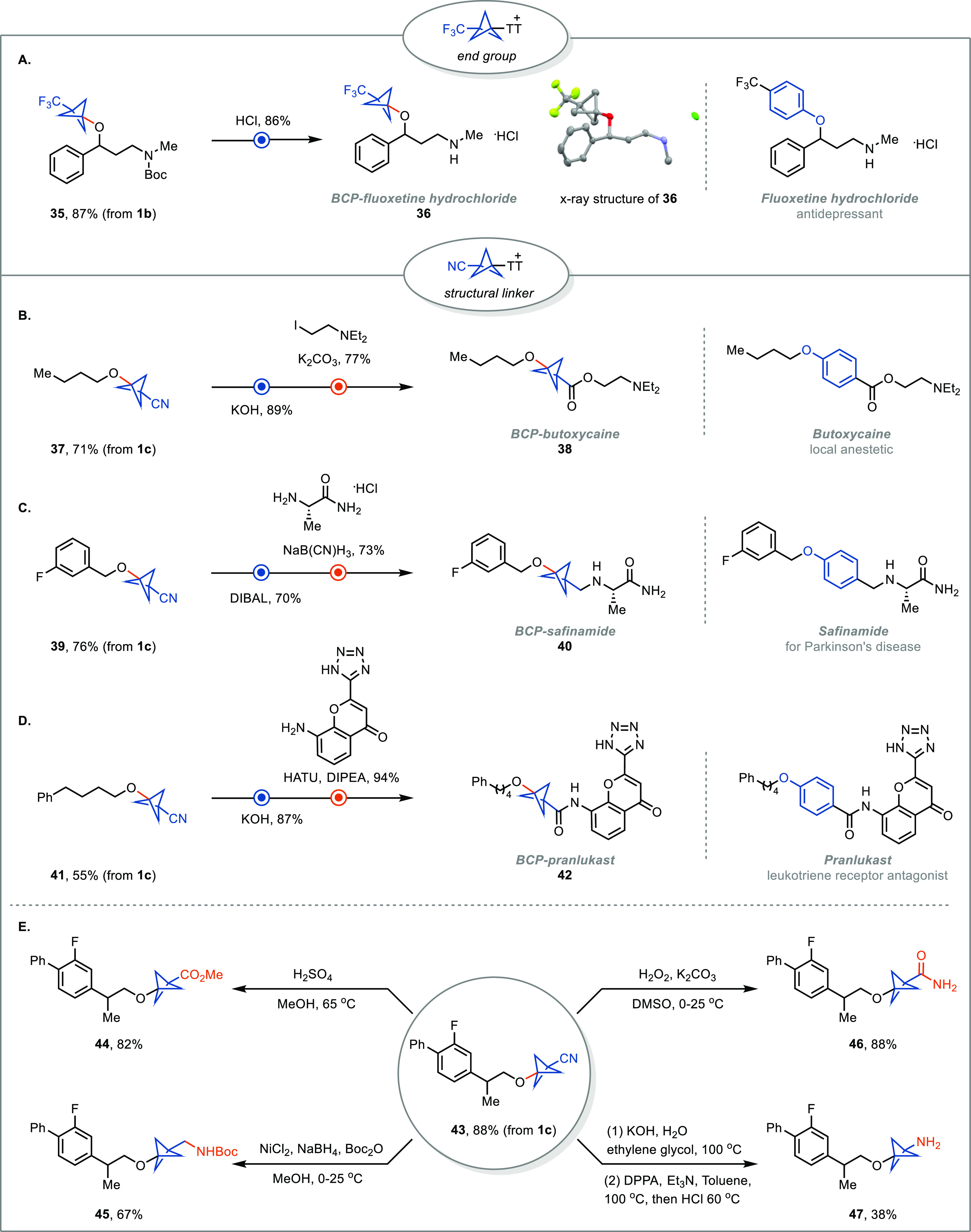
Synthesis of BCP pharmaceutical analogs. (A) Synthesis of BCP-fluoxetine hydrochloride. (B) Synthesis of BCP-butoxycaine. (C) Synthesis of BCP-safinamide. (D) Synthesis of BCP-pranlukast. (E) Further transformations of 43.
In conclusion, we have described an efficient bicyclopentylation of alcohols with BCP–thianthrenium reagents, providing a variety of BCP alkyl ether products, even at a late stage. Importantly, several bioisosteric replacements of aryl rings in small-molecule drugs were realized, which would otherwise be difficult to access currently by other methods. We anticipate that our approach can facilitate the development of saturated analogs of alkyl aryl ether drugs in the pharmaceutical industry.
Acknowledgments
We thank C. Li, S. Pandit, W. G. Whitehurst, and J. Kim (all from MPI KOFO) for helpful discussion. We thank F. Kohler and N. Haupt for mass spectrometry analysis, M. Leutzsch and C. Wirtz for NMR spectroscopy analysis, J. Rust for X-ray analysis, M. Meyer for Stern–Volmer analysis, and E. Reijerse for EPR analysis. We thank the MPI für Kohlenforschung for funding. Z.B. acknowledges the Alexander von Humboldt Foundation for a Humboldt Research Fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c10024.
Experimental procedures, spectroscopic data, and NMR spectra of all products (PDF)
Open access funded by Max Planck Society.
The authors declare the following competing financial interest(s): T.R. and Z.B. may benefit from thianthrene compound-based sales.
Supplementary Material
References
- Nilova A.; Campeau L.-C.; Sherer E. C.; Stuart D. R. Analysis of benzenoid substitution patterns in small molecule active pharmaceutical ingredients. J. Med. Chem. 2020, 63, 13389–13396. 10.1021/acs.jmedchem.0c00915. [DOI] [PubMed] [Google Scholar]
- Subbaiah M. A. M.; Meanwell N. A. Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. J. Med. Chem. 2021, 64, 14046–14128. 10.1021/acs.jmedchem.1c01215. [DOI] [PubMed] [Google Scholar]
- a Pellicciari R.; Raimondo M.; Marinozzi M.; Natalini B.; Costantino G.; Thomsen C. (S)-(+)-2-(3′-Carboxybicyclo[1.1.1]pentyl)-glycine, a Structurally New Group I Metabotropic Glutamate Receptor Antagonist. J. Med. Chem. 1996, 39, 2874–2876. 10.1021/jm960254o. [DOI] [PubMed] [Google Scholar]; b Filosa R.; Carmela Fulco M.; Marinozzi M.; Giacchè N.; Macchiarulo A.; Peduto A.; Massa A.; de Caprariis P.; Thomsen C.; Christoffersen C. T.; Pellicciari R. Design, synthesis and biological evaluation of novel bicyclo[1.1.1]pentane-based ω-acidic amino acids as glutamate receptors ligands. Bioorg. Med. Chem. 2009, 17, 242–250. 10.1016/j.bmc.2008.11.015. [DOI] [PubMed] [Google Scholar]
- a Stepan A. F.; Subramanyam C.; Efremov I. V.; Dutra J. K.; O’Sullivan T. J.; DiRico K. J.; McDonald W. S.; Won A.; Dorff P. H.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Riddell D. R.; Kauffman G. W.; Kormos B. L.; Zhang L.; Lu Y.; Capetta S. H.; Green M. E.; Karki K.; Sibley E.; Atchison K. P.; Hallgren A. J.; Oborski C. E.; Robshaw A. E.; Sneed B.; O’Donnell C. J. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active gamma-secretase inhibitor. J. Med. Chem. 2012, 55, 3414–3424. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]; b Auberson Y. P.; Brocklehurst C.; Furegati M.; Fessard T. C.; Koch G.; Decker A.; La Vecchia L.; Briard E. Improving Nonspecific Binding and Solubility: Bicycloalkyl Groups and Cubanes as para-Phenyl Bioisosteres. ChemMedChem 2017, 12, 590–598. 10.1002/cmdc.201700082. [DOI] [PubMed] [Google Scholar]; c Goh Y. L.; Cui Y. T.; Pendharkar V.; Adsool V. A. Toward Resolving the Resveratrol Conundrum: Synthesis and in Vivo Pharmacokinetic Evaluation of BCP-Resveratrol. ACS Med. Chem. Lett. 2017, 8, 516–520. 10.1021/acsmedchemlett.7b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Measom N. D.; Down K. D.; Hirst D. J.; Jamieson C.; Manas E. S.; Patel V. K.; Somers D. O. Investigation of a Bicyclo[1.1.1]pentane as a Phenyl Replacement within a LpPLA2 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 43–48. 10.1021/acsmedchemlett.6b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gianatassio R.; Lopchuk J. M.; Wang J.; Pan C.-M.; Malins L. R.; Prieto L.; Brandt T. A.; Collins M. R.; Gallego G. M.; Sach N. W.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-release amination. Science 2016, 351, 241–246. 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kanazawa J.; Maeda K.; Uchiyama M. Radical Multicomponent Carboamination of [1.1.1]Propellane. J. Am. Chem. Soc. 2017, 139, 17791–17794. 10.1021/jacs.7b11865. [DOI] [PubMed] [Google Scholar]; c Hughes J. M. E.; Scarlata D. A.; Chen A. C.; Burch J. D.; Gleason J. L. Aminoalkylation of [1.1.1]Propellane Enables Direct Access to High-Value 3-Alkylbicyclo[1.1.1]pentan-1-amines. Org. Lett. 2019, 21, 6800–6804. 10.1021/acs.orglett.9b02426. [DOI] [PubMed] [Google Scholar]; d Zhang X.; Smith R. T.; Le C.; McCarver S. J.; Shireman B. T.; Carruthers N. I.; MacMillan D. W. C. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 2020, 580, 220–226. 10.1038/s41586-020-2060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Pickford H. D.; Nugent J.; Owen B.; Mousseau J. J.; Smith R. C.; Anderson E. A. Twofold Radical-Based Synthesis of N,C-Difunctionalized Bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2021, 143, 9729–9736. 10.1021/jacs.1c04180. [DOI] [PubMed] [Google Scholar]; f Shin S.; Lee S.; Choi W.; Kim N.; Hong S. Visible-Light-Induced 1,3-Aminopyridylation of [1.1.1]Propellane with N-Aminopyridinium Salts. Angew. Chem., Int. Ed. 2021, 60, 7873–7879. 10.1002/anie.202016156. [DOI] [PubMed] [Google Scholar]; g Livesley S.; Sterling A. J.; Robertson C. M.; Goundry W. R. F.; Morris J. A.; Duarte F.; Aissa C. Electrophilic Activation of [1.1.1]Propellane for the Synthesis of Nitrogen-Substituted Bicyclo[1.1.1]pentanes. Angew. Chem., Int. Ed. 2022, 61, e202111291 10.1002/anie.202111291. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Alvarez E. M.; Bai Z.; Pandit S.; Frank N.; Torkowski L.; Ritter T. O-, N- and C-bicyclopentylation using thianthrenium reagents. Nat. Synth. 2023, 2, 548–556. 10.1038/s44160-023-00277-8. [DOI] [Google Scholar]
- a Toriyama F.; Cornella J.; Wimmer L.; Chen T. G.; Dixon D. D.; Creech G.; Baran P. S. Redox-Active Esters in Fe-Catalyzed C–C Coupling. J. Am. Chem. Soc. 2016, 138, 11132–11135. 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Makarov I. S.; Brocklehurst C. E.; Karaghiosoff K.; Koch G.; Knochel P. Synthesis of Bicyclo[1.1.1]pentane Bioisosteres of Internal Alkynes and para-Disubstituted Benzenes from [1.1.1]Propellane. Angew. Chem., Int. Ed. 2017, 56, 12774–12777. 10.1002/anie.201706799. [DOI] [PubMed] [Google Scholar]; c Kondo M.; Kanazawa J.; Ichikawa T.; Shimokawa T.; Nagashima Y.; Miyamoto K.; Uchiyama M. Silaboration of [1.1.1]Propellane: A Storable Feedstock for Bicyclo[1.1.1]pentane Derivatives. Angew. Chem., Int. Ed. 2020, 59, 1970–1974. 10.1002/anie.201909655. [DOI] [PubMed] [Google Scholar]; d Nugent J.; Shire B. R.; Caputo D. F. J.; Pickford H. D.; Nightingale F.; Houlsby I. T. T.; Mousseau J. J.; Anderson E. A. Synthesis of All-Carbon Disubstituted Bicyclo[1.1.1]pentanes by Iron-Catalyzed Kumada Cross-Coupling. Angew. Chem., Int. Ed. 2020, 59, 11866–11870. 10.1002/anie.202004090. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Polites V. C.; Badir S. O.; Keess S.; Jolit A.; Molander G. A. Nickel-Catalyzed Decarboxylative Cross-Coupling of Bicyclo[1.1.1]pentyl Radicals Enabled by Electron Donor-Acceptor Complex Photoactivation. Org. Lett. 2021, 23, 4828–4833. 10.1021/acs.orglett.1c01558. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Nugent J.; Arroniz C.; Shire B. R.; Sterling A. J.; Pickford H. D.; Wong M. L. J.; Mansfield S. J.; Caputo D. F. J.; Owen B.; Mousseau J. J.; Duarte F.; Anderson E. A. A General Route to Bicyclo[1.1.1]pentanes through Photoredox Catalysis. ACS Catal. 2019, 9, 9568–9574. 10.1021/acscatal.9b03190. [DOI] [Google Scholar]; g Yu S.; Jing C.; Noble A.; Aggarwal V. K. 1,3-Difunctionalizations of [1.1.1]Propellane via 1,2-Metallate Rearrangements of Boronate Complexes. Angew. Chem., Int. Ed. 2020, 59, 3917–3921. 10.1002/anie.201914875. [DOI] [PubMed] [Google Scholar]
- a Wong M. L. J.; Sterling A. J.; Mousseau J. J.; Duarte F.; Anderson E. A. Direct catalytic asymmetric synthesis of alpha-chiral bicyclo[1.1.1]pentanes. Nat. Commun. 2021, 12, 1644. 10.1038/s41467-021-21936-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dong W.; Yen-Pon E.; Li L.; Bhattacharjee A.; Jolit A.; Molander G. A. Exploiting the sp2 character of bicyclo[1.1.1]pentyl radicals in the transition-metal-free multi-component difunctionalization of [1.1.1]propellane. Nat. Chem. 2022, 14, 1068–1077. 10.1038/s41557-022-00979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang W.; Keess S.; Molander G. A. Dicarbofunctionalization of [1.1.1]Propellane Enabled by Nickel/Photoredox Dual Catalysis: One-Step Multicomponent Strategy for the Synthesis of BCP-Aryl Derivatives. J. Am. Chem. Soc. 2022, 144, 12961–12969. 10.1021/jacs.2c05304. [DOI] [PubMed] [Google Scholar]; d Cuadros S.; Goti G.; Barison G.; Raulli A.; Bortolato T.; Pelosi G.; Costa P.; Dell’Amico L. A general organophotoredox strategy to difluoroalkyl bicyclo-alkane (CF2-BCA) hybrid bioisosteres. Angew. Chem., Int. Ed. 2023, 62, e202303585. 10.1002/anie.202303585. [DOI] [PubMed] [Google Scholar]; e Dong W.; Keess S.; Molander G. A. Nickel-mediated alkyl-, acyl-, and sulfonylcyanation of [1.1.1]propellane. Chem. Catal. 2023, 3, 100608 10.1016/j.checat.2023.100608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Uchiyama M.; Kanazawa J. Recent Advances in the Synthetic Chemistry of Bicyclo[1.1.1]pentane. Synlett 2019, 30, 1–11. 10.1055/s-0037-1610314. [DOI] [Google Scholar]; b Pramanik M. M. D.; Qian H.; Xiao W.-J.; Chen J.-R. Photoinduced strategies towards strained molecules. Org. Chem. Front. 2020, 7, 2531–2537. 10.1039/D0QO00460J. [DOI] [Google Scholar]
- a Yu I. F.; Manske J. L.; Dieguez-Vazquez A.; Misale A.; Pashenko A. E.; Mykhailiuk P. K.; Ryabukhin S. V.; Volochnyuk D. M.; Hartwig J. F. Catalytic undirected borylation of tertiary C–H bonds in bicyclo[1.1.1]pentanes and bicyclo[2.1.1]hexanes. Nat. Chem. 2023, 15, 685–693. 10.1038/s41557-023-01159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; b VanHeyst M. D.; Qi J.; Roecker A. J.; Hughes J. M. E.; Cheng L.; Zhao Z.; Yin J. Continuous Flow-enabled Synthesis of Bench-stable Bicyclo[1.1.1]pentane Trifluoroborate Salts and Their Utilization in Metallaphotoredox Cross-couplings. Org. Lett. 2020, 22, 1648–1654. 10.1021/acs.orglett.0c00242. [DOI] [PubMed] [Google Scholar]
- Gong Y.; Zhu Z.; Qian Q.; Tong W.; Gong H. Zn- and Cu-Catalyzed Coupling of Tertiary Alkyl Bromides and Oxalates to Forge Challenging C–O, C–S, and C–N Bonds. Org. Lett. 2021, 23, 1005–1010. 10.1021/acs.orglett.0c04206. [DOI] [PubMed] [Google Scholar]
- a Enthaler S.; Company A. Palladium-catalysed hydroxylation and alkoxylation. Chem. Soc. Rev. 2011, 40, 4912–4924. 10.1039/c1cs15085e. [DOI] [PubMed] [Google Scholar]; b Evano G.; Wang J.; Nitelet A. Metal-mediated C–O bond forming reactions in natural product synthesis. Org. Chem. Front. 2017, 4, 2480–2499. 10.1039/C7QO00671C. [DOI] [Google Scholar]; c Schutyser W.; Renders T.; Van den Bosch S.; Koelewijn S. F.; Beckham G. T.; Sels B. F. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. 10.1039/C7CS00566K. [DOI] [PubMed] [Google Scholar]
- Wong D. T.; Perry K. W.; Bymaster F. P. The discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discovery 2005, 4, 764–774. 10.1038/nrd1821. [DOI] [PubMed] [Google Scholar]
- The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 12th ed.; CRC Press, 1996; p 254, compound 1566. [Google Scholar]
- Stocchi F.; Arnold G.; Onofrj M.; Kwiecinski H.; Szczudlik A.; Thomas A.; Bonuccelli U.; Van Dijk A.; Cattaneo C.; Sala P.; Fariello R. G. Improvement of Motor Function in Early Parkinson Disease by Safinamide. Neurology 2004, 63, 746–748. 10.1212/01.WNL.0000134672.44217.F7. [DOI] [PubMed] [Google Scholar]
- Keam S. J.; Lyseng-Williamson K. A.; Goa K. L. Pranlukast: a review of its use in the management of asthma. Drugs 2003, 63, 991–1019. 10.2165/00003495-200363100-00005. [DOI] [PubMed] [Google Scholar]
- a Xiang J.; Shang M.; Kawamata Y.; Lundberg H.; Reisberg S. H.; Chen M.; Mykhailiuk P.; Beutner G.; Collins M. R.; Davies A.; Del Bel M.; Gallego G. M.; Spangler J. E.; Starr J.; Yang S.; Blackmond D. G.; Baran P. S. Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 2019, 573, 398–402. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shibutani S.; Kodo T.; Takeda M.; Nagao K.; Tokunaga N.; Sasaki Y.; Ohmiya H. Organophotoredox-Catalyzed Decarboxylative C(sp3)–O Bond Formation. J. Am. Chem. Soc. 2020, 142, 1211–1216. 10.1021/jacs.9b12335. [DOI] [PubMed] [Google Scholar]
- a Wiberg K. B.; Walker F. H. [1.1.1]Propellane. J. Am. Chem. Soc. 1982, 104, 5239–5240. 10.1021/ja00383a046. [DOI] [Google Scholar]; b Wiberg K. B.; McMurdie N. Formation and reactions of bicyclo[1.1.1]pentyl-1 cations. J. Am. Chem. Soc. 1994, 116, 11990–11998. 10.1021/ja00105a046. [DOI] [Google Scholar]
- Della E. W.; Pigou P. E.; Schiesser C. H.; Taylor D. K. Experimental and calculated activation parameters for ring opening of the 1-bicyclo[1.1.1]pentyl radical: the effect of bridgehead substituents. J. Org. Chem. 1991, 56, 4659–4664. 10.1021/jo00015a018. [DOI] [Google Scholar]
- Whitesides G. M.; Sadowski J. S.; Lilburn J. Copper(I) alkoxides. Synthesis, reactions, and thermal decomposition. J. Am. Chem. Soc. 1974, 96, 2829–2835. 10.1021/ja00816a027. [DOI] [Google Scholar]
- a Vorogushin A. V.; Huang X.; Buchwald S. L. Use of Tunable Ligands Allows for Intermolecular Pd-Catalyzed C–O Bond Formation. J. Am. Chem. Soc. 2005, 127, 8146–8149. 10.1021/ja050471r. [DOI] [PubMed] [Google Scholar]; b Gowrisankar S.; Sergeev A. G.; Anbarasan P.; Spannenberg A.; Neumann H.; Beller M. A General and Efficient Catalyst for Palladium-Catalyzed C–O Coupling Reactions of Aryl Halides with Primary Alcohols. J. Am. Chem. Soc. 2010, 132, 11592–11598. 10.1021/ja103248d. [DOI] [PubMed] [Google Scholar]
- a Yu X.-Y.; Chen J.; Chen H.-W.; Xiao W.-J.; Chen J.-R. Visible-Light-Driven Copper-Catalyzed C(sp3)–O Cross-Coupling of Benzylic Radicals with Phenols. Org. Lett. 2020, 22, 2333–2338. 10.1021/acs.orglett.0c00532. [DOI] [PubMed] [Google Scholar]; b Niu J.; Guo P.; Kang J.; Li Z.; Xu J.; Hu S. Copper(I)-Catalyzed Aryl Bromides to Form Intermolecular and IntramolecularCarbon-Oxygen Bonds. J. Org. Chem. 2009, 74, 5075–5078. 10.1021/jo900600m. [DOI] [PubMed] [Google Scholar]; c Zhang L.; Israel E. M.; Yan J.; Ritter T. Copper-Mediated Etherification via Aryl Radicals Generated from Triplet States. Nat. Syn. 2022, 1, 376–381. 10.1038/s44160-022-00061-0. [DOI] [Google Scholar]; d Yang L.; Lu H. H.; Lai C. H.; Li G.; Zhang W.; Cao R.; Liu F.; Wang C.; Xiao J.; Xue D. Light-Promoted Nickel Catalysis: Etherification of Aryl Electrophiles with Alcohols Catalyzed by a Ni(II)-Aryl Complex. Angew. Chem., Int. Ed. 2020, 59, 12714–12719. 10.1002/anie.202003359. [DOI] [PubMed] [Google Scholar]
- a Chen C.; Fu G. C. Copper-Catalyzed Enantioconvergent Alkylation of Oxygen Nucleophiles. Nature 2023, 618, 301–307. 10.1038/s41586-023-06001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou Z.; Behnke N. E.; Kürti L. Copper-Catalyzed Synthesis of Hindered Ethers from α-Bromo Carbonyl Compounds. Org. Lett. 2018, 20, 5452–5456. 10.1021/acs.orglett.8b02371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Buck E.; Song Z. J.; Tschaen D.; Dormer P. G.; Volante R. P.; Reider P. J. Ullmann Diaryl Ether Synthesis: Rate Acceleration by 2,2,6,6-Tetramethylheptane-3,5-Dione. Org. Lett. 2002, 4, 1623–1626. 10.1021/ol025839t. [DOI] [PubMed] [Google Scholar]; b Chen Z.; Jiang Y.; Zhang L.; Guo Y.; Ma D. Oxalic Diamidesand Tert Butoxide: Two Types of Ligands Enabling Practical Access to Alkyl Aryl Ethers via Cu-Catalyzed Coupling Reaction. J. Am. Chem. Soc. 2019, 141, 3541–3549. 10.1021/jacs.8b12142. [DOI] [PubMed] [Google Scholar]; c Ray R.; Hartwig J. F. Oxalohydrazide Ligands for Copper Catalyzed C–O Coupling Reactions with High Turnover Numbers. Angew. Chem., Int. Ed. 2021, 60, 8203–8211. 10.1002/anie.202015654. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang Q.; Zhao Y.; Ma D. Cu-Mediated Ullmann-Type Cross-Coupling and Industrial Applicationsin Route Design, Process Development, and Scale-up of Pharmaceutical and Agrochemical Processes. Org. Process Res. Dev. 2022, 26, 1690–1750. 10.1021/acs.oprd.2c00050. [DOI] [Google Scholar]; e Shafir A.; Lichtor P. A.; Buchwald S. L. N- versus O-Arylation of Amino alcohols: Orthogonal Selectivity in Copper-Based Catalysts. J. Am. Chem. Soc. 2007, 129, 3490–3491. 10.1021/ja068926f. [DOI] [PubMed] [Google Scholar]
- a Motornov V.; Klepetářová B.; Beier P. Cu(III) Trifluoromethyl Complexes with 1,3-Diketonate Ligands and Their Versatile Reactivity in C–H Trifluoromethylation. Adv. Synth. Catal. 2023, 365, 2858–2864. 10.1002/adsc.202300695. [DOI] [Google Scholar]; b Yuan M.; Song Z.; Badir S. O.; Molander G. A.; Gutierrez O. On the Nature of C(sp3)–C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc. 2020, 142, 7225–7234. 10.1021/jacs.0c02355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We propose that the low yield is due to the inefficiency of Cu(acac)2 formation from Cu(OTf)2 and 2,4-pentanedione under the current conditions.
- The reaction is moisture-sensitive, and we speculate that prestirring the reactants with molecular sieves is helpful to remove the water from the reagents before irradiation.
- Chiyindiko E.; Conradie J. Redox behaviour of bis(β-diketonato) copper(II) complexes. Chiyindiko, E.; Conradie, J. Redox Behaviour of Bis (β-diketonato) Copper (II) Complexes. J. Electroanal. Chem. 2019, 837, 76–85. 10.1016/j.jelechem.2019.02.011. [DOI] [Google Scholar]
- Examples involving reduction of a Cu(II) precatalyst to a Cu(I) active catalyst by a photocatalyst:; a Le C. C.; Chen T. Q.; Liang T.; Zhang P.; MacMillan D. W. C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. 10.1126/science.aat4133. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dow N. W.; Cabré A.; MacMillan D. W. C. A general N-alkylation platform via copper metallaphotoredox and silyl radical activation of alkyl halides. Chem. 2021, 7, 1827–1842. 10.1016/j.chempr.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sarver P. J.; Bacauanu V.; Schultz D. M.; DiRocco D. A.; Lam Y.-h.; Sherer E. C.; MacMillan D. W. C. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem. 2020, 12, 459–467. 10.1038/s41557-020-0436-1. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerich O.; Parker V. D. The reversible oxidation of aromatic cation radicals to dications. Solvents of low nucleophilicity. Electrochim. Acta 1973, 18, 537–541. 10.1016/0013-4686(73)85015-7. [DOI] [Google Scholar]
- Li J.; Chen J.; Sang R.; Ham W.-S.; Plutschack M. B.; Berger F.; Chabbra S.; Schnegg A.; Genicot C.; Ritter T. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat. Chem. 2020, 12, 56–62. 10.1038/s41557-019-0353-3. [DOI] [PubMed] [Google Scholar]
- Given the similar rates of quenching by TT and Cu(I), the current data do not allow us to exclude similar but distinct mechanisms that do not involve quenching by TT (Figure S11).
- Murata Y.; Shine H. J. Reactions of Thianthrenium Perchlorate and Thianthrenium Trichlorodiiodide. J. Org. Chem. 1969, 34, 3368–3372. 10.1021/jo01263a034. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.