

Abstract
Ricin toxin A subunit (RTA) is the catalytic subunit of ricin, which depurinates an adenine from the sarcin/ricin loop in eukaryotic ribosomes. There are no approved inhibitors against ricin. We used a new strategy to disrupt RTA–ribosome interactions by fragment screening using surface plasmon resonance. Here, using a structure-guided approach, we improved the affinity and inhibitory activity of small-molecular-weight lead compounds and obtained improved compounds with over an order of magnitude higher efficiency. Four advanced compounds were characterized by X-ray crystallography. They bind at the RTA–ribosome binding site as the original compound but in a distinctive manner. These inhibitors bind remotely from the catalytic site and cause local conformational changes with no alteration of the catalytic site geometry. Yet they inhibit depurination by ricin holotoxin and inhibit the cytotoxicity of ricin in mammalian cells. They are the first agents that protect against ricin holotoxin by acting directly on RTA.
Graphical Abstract

INTRODUCTION
Ricin is one of the most potent toxins known and is classified as a category B threat due to its accessibility, stability, and toxicity.1 It is easily extracted from castor beans and is processed worldwide on an industrial scale for biodiesel and lubricant production as castor oil. Escherichia coli (STEC) and Shigella dysenteriae, which produce the related Shiga toxins (Stxs), are potentially fatal, foodborne pathogens.2–4 Ricin and Stxs are ribosome inactivating proteins (RIPs) that catalyze the removal of the same adenine base from the sarcin/ricin loop (SRL) of the large rRNA, thereby inhibiting protein synthesis.5 Ricin contains an enzymatically active A subunit (RTA) and a cell binding B subunit (RTB) linked by a disulfide bond. The B subunit is a galactose/N-acetyl galactosamine-binding lectin, which promotes endocytosis and retrograde transport to the endoplasmic reticulum (ER).6 RTA is released from RTB in the ER and is retrotranslocated into the cytoplasm where it depurinates the SRL, inhibits protein synthesis, and causes cell death.7 RTA–antibody complexes have been explored as immunotoxins against leukemia and lymphoma. However, off-target effects, including vascular leak syndrome, have limited their utility.8–10 Development of inhibitors against the action of ricin on human ribosomes could provide an antidote against cases of ricin poisoning and ameliorate the side effects of RTA complexes in cancer therapy.
The active site of RTA has been extensively explored as a potential target for inhibitors by a structure-based design.11,12 However, these compounds had poor solubility and did not exhibit potent inhibitory activity due to the large and highly polar active site, which is not readily druggable.13–15 Pterin-based molecules that fit into the active site were poorly soluble and incompatible with cell and animal models of toxicity.11,12 Transition state analogs of the catalytic reaction inhibited the depurination of small stem-loop RNA substrates at low pH but did not protect eukaryotic ribosomes at physiological pH.16 High-throughput, cell-based screens identified compounds that targeted host proteins rather than the toxin itself.17–21 Only one small molecule, Retro-2, has been shown to have activity in protecting mice against ricin by blocking retrograde trafficking.18
We described that RTA binds to a protein component of the ribosome, the P stalk distinct from the site of depurination (Figure 1A).22,23 In eukaryotes, the P stalk is a pentameric protein complex formed by a P0 protein and two P1–P2 heterodimers.24,25 The C-terminal domain (CTD) of P0, P1, and P2 proteins, especially their last 11 residues [SDDDMGFGLFD], are highly conserved among the eukaryotes.26 Peptides corresponding to CTD of P proteins interact with RTA27 and several other RIPs, including trichosanthin (TCS),28 maize RIP (MOD),29 and Stx, allowing RIPs to access the SRL.30–32 The elongation factors bind to the CTD of the P proteins, which deliver them to the SRL.33,34
Figure 1.
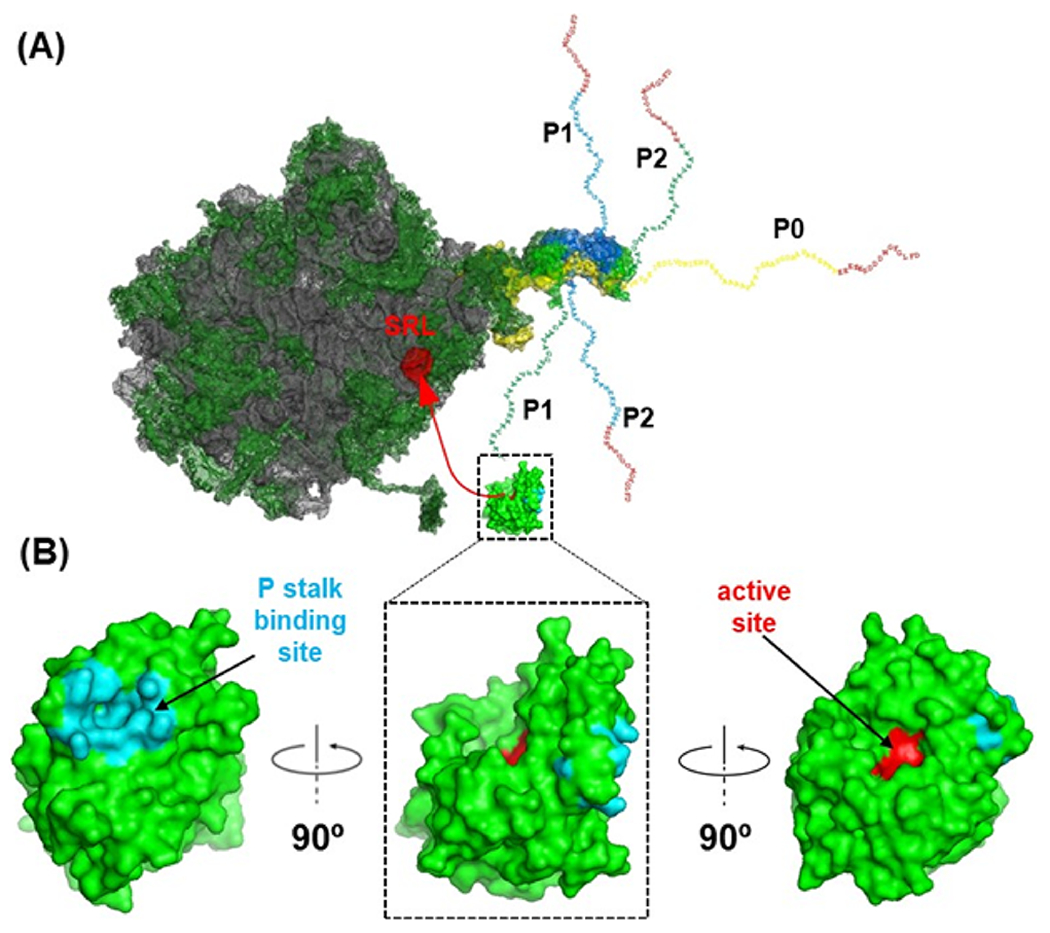
Model showing the interaction of RTA with the P stalk to access the SRL. (A) S. cerevisiae 26S rRNA (PDB ID: 3U5H) and the 60S subunit (PDB ID: 3U5I) interacting with RTA (PDB ID: 6URX) are shown. The rRNA is shown in gray, ribosomal proteins are shown in green, and SRL is shown in red. The fitted schematic structure shows the P0 fragment complexed with the N-terminal domain of P proteins from Archaea (PDB ID: 3AIY). The flexible extensions of the C-terminal tails of P1, P2, and P0 proteins are shown. The red C-terminal ends indicate the identical 16 amino acids at the C-termini of the five P proteins. (B) RTA is shown in green in three different orientations. The active site (Tyr80, Tyr123, Glu177, Arg180, and Trp211) is shown in red, and the P stalk binding site (Tyr183, Ser203, Leu207, Leu232, Gln233, Arg234, Arg235, Phe240, Ile247, Leu248, Pro250, and Ile251) is shown in cyan. The interaction of the P stalk binding site of RTA with the P protein C-termini positions the active site toward the SRL.
RTA binds the ribosome via a two-step mechanism, which involves first slow and non-stalk specific interactions with the ribosome followed by fast interactions with the P stalk.35–37 RTA cannot depurinate the naked rRNA or an RNA mimic of the SRL at physiological pH because ribosome binding is required for activity.16 The P stalk binding site of RTA is on the opposite face of the active site (Figure 1B).36 P stalk binding stimulates the depurination of the SRL by a conformational change and by positioning the active site of RTA toward the SRL.36,38 The P stalk binding site is a hydrophobic pocket on RTA formed by Tyr183, Leu207, Leu232, Phe240, Val242, Ile247, P250, and Ile251 and binds only to the last six residues [GFGLFD] of an 11-mer peptide (P11) mimicking the conserved CTD of P proteins.39,40 Arg235 just outside the hydrophobic pocket is the most critical arginine at the P stalk binding site.36,41 Arg235 forms hydrogen bonds with the last two residues (Phe114 and Asp115) of P11.39,40 We reported that Leu232, Tyr183, and Phe240 of the hydrophobic pocket contribute cumulatively to the toxicity of RTA.38 A quadruple mutant, which combined alanine mutations in these residues with the R235A mutation, completely abolished the activity of RTA in mammalian cells, establishing the ribosome binding site as a new target for drug discovery.38 We recently reported the first X-ray crystal structure of Stx2a with P11 (PDB ID: 6X6H) and showed that, as observed with RTA, only the last six residues of P11 inserted into a shallow pocket on Stx2A1.42 However, Stx2a formed a distinct P11-binding mode within a different surface pocket relative to RTA and TCS, suggesting different ribosome recognition mechanisms for each RIP.42
Peptides mimicking the last 4 to 11 amino acids of P proteins bind to the P stalk binding site and inhibit depurination activity by disrupting RTA–ribosome interactions.27 The P stalk binding site of RTA has not been previously targeted by small molecule inhibitors. We used a surface plasmon resonance (SPR) based fragment screen and discovered two small molecule fragments, CC10501 and CC70601, which bind the hydrophobic pocket at the P stalk binding site away from the active site and inhibit the activity of RTA.43 Here, we used a structure-based design exploring the CC10501 chemical scaffold as the starting point for optimization and constructed a focused library. Structure activity relationships (SARs) of the compounds in the library were examined by measuring their binding affinity and inhibitory activity on eukaryotic ribosomes. A new group of compounds was identified with improved affinity and ribosome protection activity. Four leading compounds in this group were co-crystallized with RTA. All four occupied the same binding site as CC10501 but in a distinctive manner. The best compounds protected cells against the toxicity of ricin holotoxin over an order of magnitude more effectively than CC10501. This is the first family of agents capable of protecting against ricin by acting directly on the ribosome/RTA interaction rather than on cellular trafficking or catalytic site interactions and provides a new path for further optimization.
RESULTS
Compound Design for Fragment Optimization.
The crystal structure of RTA in complex with CC10501 (PDB ID: 6URX) (Figure 2)43 revealed that the inhibitor binds in the same deep hydrophobic pocket of RTA as Leu9 and Phe10 of the P11 peptide (SDDDMGFGLFD) (5GU4).40 Leu9 makes several hydrophobic interactions with Ile251, while the aromatic side chain of Phe10 forms an offsetting π-stack with Tyr183 and Phe240.43 The aromatic ring of CC10501 similarly establishes π-stacking interactions with Tyr183 and Phe240 and comparable hydrophobic associations with Ile251.43 The carboxylate moiety on CC10501 and the carboxylate of C-terminal Asp11 form similar hydrogen bond interactions with the guanidine group of Arg234 and Arg235 just outside the hydrophobic pocket (Figure 2).43 CC10501 binds RTA with a dissociation constant (KD) of 270 μM and inhibits its ribosome depurination activity with an IC50 of 181 μM, supporting the biological function for this interaction.43
Figure 2.

Peptide and inhibitor binding to the RTA P stalk pocket. Superpositioned stereoview of the P11 peptide (green, 5GU4) and CC10501 (yellow, 6URX) bound at the P stalk binding pocket of RTA. The stick model of the RTA binding site is shown together with the electrostatic surface. The hydrogen bond interactions of CC10501 with RTA are highlighted (black dotted lines).
To optimize CC10501, we assembled a library of 37 compounds from Maybridge bearing a carboxylic acid group, as well as libraries containing carboxylic acid analogs (amides, esters, hydroxamic acids and esters) that contain a carbonyl group. These libraries were screened by measuring their binding affinity (KD) and inhibitory activity on eukaryotic ribosomes. Based on the compounds with the highest affinity, we synthesized 97 compounds to probe unique interactions with Phe240, Tyr183, Ile251, and the surface residues Arg234 and Arg235. The medicinal chemistry effort toward the improvement of CC10501 is summarized in Figure 3. Using CC10501 as a starting point, we attempted to get hydrogen bonding with Tyr183 with strategically positioned heteroatoms. However, none of them showed better inhibitory activities. Subsequently, we explored bioisosteric groups to replace the carboxylic acid moiety with boronic acid, hydroxamic acid, amide, and tetrazoles. We also tried to incorporate different cyclic/heterocyclic hydrophobic fragments at the thiophene 5-position as well as several thiophene core replacements to enhance the hydrophobic interaction, including phenyl picolinic acids, phenyl nicotinic acids, and phenyl furoic acids. However, the 2-carboxy-5-phenylthiophene proved optimal. We followed only the compounds with improved affinity and inhibitory activity against RTA, and we refer to those compounds as the ”advanced compounds”. These compounds contained an ionizable carboxylate functional group (Table 1).
Figure 3.

Medicinal chemistry synthetic strategy toward the improvement of CC10501. The red circles represent where additional improvements in binding interactions were sought by altering the volume and additional hydrophobicity. The green box was varied to explore the effects of other heteroaromatic thiophene core replacements. Carboxyl group replacements (the fuchsia oval) were explored by substitution with carboxylic acid bioisosteres.
Table 1.
The Chemical Structures, Affinity, and Inhibitory Activity of CC10501 Analogs Using Yeast Ribosomes

|
The affinities were determined by steady-state fitting with the free Rmax model using the Biacore Insight Evaluation Software.
The affinity of these compounds could not be determined by steady-state fitting with the free Rmax model.
The synthesis of RU-NT-57 is described in Scheme 1. Initial attempts of coupling thiophene-2-carbonitrile with 1-bromo-4-methylbenzene catalyzed with palladium acetate gave no product. We speculated that a higher temperature was needed to activate the C–H bond of thiophene-2-carbonitrile. Increasing the reaction temperature to 150 °C was needed to obtain compound 1 in 45% yield. Cycloaddition of 1 with sodium azide in the presence of ammonium chloride afforded RU-NT-57.
Scheme 1.

Synthesis of 5-(5-(p-Tolyl)thiophen-2-yl)-1H-tetrazole (RU-NT-57)
The preparation of RU-NT-128 is reported in Scheme 2. Suzuki coupling of 5-bromothiophene-2-carbonitrile with (2,6-dimethylphenyl)boronic acid gave compound 2 in 70% yield. Cycloaddition of compound 2 with sodium azide in the presence of ammonia chloride afforded RU-NT-128 in 85% yield.
Scheme 2.

Synthesis of 5-(5-(2,6-Dimethylphenyl)thiophen-2-yl)-1H-tetrazole (RU-NT-128)
The synthesis of RU-NT-70, 74, 75, 93, 102, 105, 110, 111, 113, 115, and 124 started from methyl 5-bromothiophene-2-carboxylate as shown in Scheme 3. Suzuki coupling of methyl 5-bromothiophene-2-carboxylate with the corresponding aryl boronic acid followed by ester hydrolysis yielded the corresponding carboxylic acids.
Scheme 3.

Synthesis of RU-NT-70, 74, 75, 93, 102, 105, 110, 111, 113, 115, and 124
The percent inhibition of the depurination of yeast ribosomes and the equilibrium dissociation constants (KD’s) for the 15 advanced compounds determined using Biacore 8K+ are shown in Table 1. The KD values were determined using steady-state fitting with the free maximal binding (Rmax) model. Using the free Rmax model, we could not determine the affinity of CC10501 because the sensorgram displayed a slope during binding suggesting that secondary binding occurred as the compound concentration increased (Figure S1A). The KD for CC10501 was 584 μM using steady-state fitting with the constant Rmax model when RU-NT-93 was used as a positive control. This value is closer to the actual KD than the previously published value of 270 μM43 obtained using the P11 peptide as a positive control since the molecular weight (MW) of CC10501 (204 Da) is closer to that of RU-NT-93 (232 Da) than P11 (1218 Da). Even when steady-state fitting with the constant Rmax model was used, the highest concentration of CC10501 did not reach equilibrium.43 Similarly, we could not determine the KD value of RU-NT-57 using steady-state fitting with the free Rmax model possibly because secondary binding occurred as the compound concentration increased (Figure S1B). We were able to obtain the KD values for all other analogs using steady-state fitting with the free Rmax model (Table 1). The inhibitory activity on eukaryotic ribosomes was determined by qRT-PCR using each fragment at 100 μM (Table 1).44 The qRT-PCR method measures the depurination level directly after isolation of the depurinated rRNA44 and is subject to less interference by the fragments than other reporter-based assays.45 CC10501 showed 45% protection of yeast ribosomes at 100 μM (Table 1).
Replacement of the carboxylic acid moiety of CC10501 and RU-NT-93 afforded tetrazole analogs RU-NT-57 and RU-NT-128 (Table 1). Although they were similar in structure, RU-NT-128 had a higher affinity (Figure S1C,D). RU-NT-128 showed 93% protection from depurination, while RU-NT-57 showed 52% protection at 100 μM (Table 1). The phenyl rings in these two compounds are different in their orientation due to the ortho substitution. A rotation toward an orthogonal conformation of the thiophene and phenyl in RU-NT-128 positions the H-bond acceptor/donor in the proximity of the Tyr183 hydroxy-aryl ring.
The design of compounds (Table 1) included consideration of the conformational preferences about the thiophene-phenyl bond of CC10501. Thiophene is thought to be isosteric with phenyl, so the combination is somewhat analogous to biphenyl. Generally, biphenyl shows a dihedral angle skewed 45°, while 2,6-dimethylbiphenyl would, a priori, prefer an orthogonal relationship. This conformational binding preference of the inhibitor causes the thiophene-carboxyl plane to be rotated from 40° toward 90° off the plane of the thiophene ring. RU-NT-70, RU-NT-74, RU-NT-75, RU-NT-93, RU-NT-102, and RU-NT-124 served as conformational analogs where the thiophene-carboxyl would vary from a rotation of the thiophene plane of 40–45° (as in 70 or 102) to being driven to adopt a near orthogonal relationship (as in 75 and 93). The four advanced compounds, RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102, were at equilibrium when the affinity was calculated (Figure 4). RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 showed higher affinity than CC10501. RU-NT-93 showed the highest affinity among the four advanced compounds (Table 1). These compounds showed 81–91% protection against depurination at 100 μM (Table 1). RU-NT-74 and RU-NT-75 differ by only one ortho methyl group substituent, making a small difference in the sterics of the ring. RU-NT-74 showed lower affinity than RU-NT-75 (Figure S2). RU-NT-124 showed a high nonspecific interaction, the lowest affinity (Figure S2), and the lowest inhibition of depurination at 100 μM among the conformational analogs (Table 1). The decreased activity of RU-NT-124 may be due to the clash of extended isopropyl with Ser203 or Leu232.
Figure 4.

Interaction of advanced compounds with RTA by SPR. The binding sensorgrams of RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 (upper) and the steady-state fitting (lower) with free Rmax are shown. Biacore 8K+ was used to measure the interaction and determine the equilibrium dissociation constant (KD). RTA was immobilized on flow cell 2 (Fc2) of a CM5 chip up to 5800–6000 RU using amine coupling. Fc1 was activated and blocked without RTA as reference. Fragments at 0.7, 2.1, 6.2, 18.5, 55.6, 166.7, and 500 μM were passed over both surfaces at 30 μL/min for 60 s and dissociated for 60 s. The running buffer was PBS-P (20 mM phosphate, 2.7 mM KCl, 137 mM NaCl, and 0.05% surfactant P20, pH 7.4) with 2% DMSO. Buffer mismatch was corrected by reference (Fc1) subtraction and DMSO correction. We did not observe any nonspecific interaction of any of the fragments alone on the reference cell using these conditions. All fragments were run on four different channels as replicates. Data were analyzed using the Biacore Insight Evaluation Software. The vertical blue line indicates that the highest concentration used is high enough for a reliable KD measurement.
In consideration of the interaction between the thiophene acid and Arg234 and Arg235 that determines fragment location, we reasoned that the orientation of the phenyl ring, by virtue of ortho substitution, would place it closer to Tyr183 if it were orthogonal to the thiophene. In this way, RU-NT-110 was designed to pick up a π-stacking interaction with Tyr183, where the orthogonal ortho fluorophenyl might be projected toward Tyr183. The oxygen-containing systems appended to the ortho phenyl position: (a) a H-bond acceptor as the methyl ether in RU-NT-111, (b) a −OH group as a H-bond donor or acceptor in RU-NT-113, (c) an oxygen locked in a tetrahydrofuran (THF) ring to lend added basicity/H-bond acceptor capability to the THF oxygen in RU-NT-105, or (d) a polyethylene glycol (PEG) group at the ortho phenyl position in RU-NT-115 as potential H-bond acceptors from Tyr183. Both RU-NT-111 and RU-NT-113 would be expected to adopt an orthogonal relationship with the thiophene ring since they both carry a second ortho substituent. The near orthogonal orientation of the phenyl and thiophene rings may place the ortho substituents, o-F (110), o-OMe (111), and o-OH (113), close to Tyr183 such that they engage in H-bonding.
RU-NT-110, RU-NT-111, and RU-NT-113 had similar affinity, while RU-NT-105 and RU-NT-115 had lower affinity (Figure S3). RU-NT-111 and 113 displayed stronger inhibition of depurination (84 and 82% protection at 100 μM, respectively), while RU-NT-105 and RU-NT-115 showed lower inhibitory activity (Table 1). The lower affinity and lower binding levels of RU-NT-105 and RU-NT-115 (Figure S3) suggested that the PEG group of RU-NT-115 might be too large to fully engage in the pocket. The fused THF ring associated with 105 was also not active (Table 1).
RTA Structure and Binding of Inhibitors.
We obtained the X-ray crystal structure of the leading four compounds bound to RTA by co-crystallization. The crystallizations of RTA in complex with RU-NT-70, RU-NT-75, RU-NT-93, or RU-NT-102 were performed by sitting drop vapor diffusion, summarized in Table S1. The structures were solved using PHASER in different space groups (Figure 5 and Table S2). All polypeptide residues are clearly observed in the electron density map except for a few terminal residues and side chains of surface residues. The electron densities for the inhibitors were well resolved in the structures (Figure S4).
Figure 5.

The crystal structure of RTA–inhibitor complexes. (A) The structure and the active site of RTA. The active site is highlighted with the black circle. (B) The RTA–inhibitor complex structures are superimposed, and the binding of the RTA structures with RU-NT-70 (green, 7MLN), RU-NT-75 (cyan, 7MLO), RU-NT-93 (pink, 7MLP), and RU-NT-102 (yellow, 7MLT) is highlighted in the black rectangle. All inhibitors are bound in the ribosome binding pocket, which is on the opposite face of the active site. The RTA structure is rotated 180° counterclockwise in (B).
The crystal structures of RTA and full-length ricin have been determined previously,46,47 and the structures determined here with all four inhibitors are similar to the previously reported RTA structures. The RMSD deviation of these structures are 0.293–0.323 Å. Five amino acid residues including Tyr80, Tyr123, Glu177, Arg180, and Trp211 form the active site pocket of RTA (Figure 5A). In ricin holotoxin, the Cys4 of ricin toxin B chain (RTB) forms a disulfide bond with Cys259 of RTA. The C-terminal residue (Phe262) of RTB interacts with a hydrophobic pocket of RTA formed by Tyr183, Leu207, Leu232, Phe240, Val242, Ile247, Leu248, and Ile251. This hydrophobic interaction and the disulfide bond stabilize the RTA–RTB heterodimer and maintain the heterodimeric state of ricin. This hydrophobic pocket is opposite to the active site of RTA and provides the binding space for the current inhibitors (Figure 5B).
The structure of RTA with RU-NT-70 (1.52 Å resolution) shows the inhibitor bound into the hydrophobic P stalk binding pocket of RTA (Figure 5B). The aromatic ring of RU-NT-70 forms a π–π stacking interaction with Tyr183 and a π–T stacking with Phe240. Residues Leu232 and Ile251 also have hydrophobic interactions with the aromatic ring of RU-NT-70, and the inhibitor carboxylate shares hydrogen bond interactions with two waters, the Arg234 guanidinium and a backbone nitrogen of Arg235 (Figure 6A).
Figure 6.

Stereoview of inhibitor-bound RTA structures. Inhibitor complexes of RU-NT-70 (7MLN) (A), RU-NT-75 (7MLO) (B), RU-NT-93 (7MLP) (C), and RU-NT-102 (7MLT) (D) are shown. All panels are shown in the same structural orientation. Amino acids interacting with the inhibitors are highlighted. Selected hydrogen bond interactions are shown in black dotted lines. The RTA residues interacting with the inhibitors within 4 Å are highlighted. Electrostatic views of these panels are shown in Figure S5.
The other RTA complex structures with RU-NT-75 and RU-NT-93 were determined in space group P1211, while the structure with RU-NT-102 was determined in space group P6322. These inhibitors bind in the same hydrophobic ribosome binding pocket of RTA that accommodates RU-NT-70. The binding of these inhibitors is stabilized by H-bonds and hydrophobic amino acids in the binding pocket. The distinct alkyl substituents of the phenyl rings cause distinct binding site interactions as summarized in Table 2.
Table 2.
RTA Amino Acid Residues in Contact with the Inhibitors within 4.0 Å of the Binding Pocket
| inhibitors | amino acid residues |
|---|---|
| RU-NT-70 | Tyr183, Ser203, Leu207, Leu232, Gln233, Arg234, Arg235, Phe240, Ile247, Leu248, Ile251 |
| RU-NT-75 | Tyr183, Ser203, Leu232, Gln233, Arg234, Arg235, Phe240, Ile247, Leu248, Ile251 |
| RU-NT-93 | Tyr183, Ser203, Leu207, Leu232, Gln233, Arg234, Arg235, Phe240, Ile247, Leu248, Ile251 |
| RU-NT-102 | Tyr183, Leu207, Leu232, Gln233, Arg234, Arg235, Phe240, Ile247 |
In RU-NT-75, the thiophene-carboxylate plane is rotated 86° (near orthogonal) off the plane of the phenyl ring (Figure 7 and Figure S5) and is sandwiched between the two Arg residues: 5.15 Å from Arg234 and 3.80 Å from Arg235. The carboxylate of RU-NT-75 has two H-bond interactions including one with a water molecule and one with a backbone nitrogen of Arg235 (Figure 6B). The phenyl and thiophene rings are stabilized by interactions with Arg234/Arg235.
Figure 7.

Stereoview superposition of the inhibitors. RU-NT-70 (green, 7MLN), RU-NT-75 (cyan, 7MLO), RU-NT-93 (pink, 7MLP), and RU-NT-102 (yellow, 7MLT). The binding of RU-NT-75 (cyan) and RU-NT-93 (pink) has the thiophene-carboxylate plane perpendicular to that of the other inhibitors because of steric hindrance in these inhibitors. The common geometry of the substituted phenyl group suggests it is the major determinant for inhibitor binding. Key amino acid residues involved in the hydrophobic interactions are highlighted in blue.
In RU-NT-93, the thiophene-carboxylate plane is perpendicular to the disubstituted 2,6-dimethyl phenyl ring (Figure 7 and Figure S5). The carboxylate of RU-NT-93 has two H-bond interactions including one with a water molecule and one with a backbone nitrogen of Arg235 (Figure 6C). A different mode of binding was observed for the thiophene carboxylate in RU-NT-93 relative to RU-NT-70, 75, and 102. The new binding pose involved the thiophene sandwiched between the Arg235 guanidine and Tyr183 instead of the H-bond interactions generated by the carboxylate and Arg234/235 in the similar structures. The carboxylate makes a 2.84 Å interaction with the backbone NH of Arg235. The phenyl ring in RU-NT-93 could potentially capture a stronger π-stacking interaction with the Phe240 or Tyr183 phenyl ring. The hindered rotation about the thiophene-phenyl C–C bond provides a conformational preference on the molecule due to the 2,6-methyl groups. This conformation places the phenyl ring orthogonal to the plane of thiophene that projects the 2- and 6-methyl of phenyl substituents toward Tyr183 and Phe240 where the inhibitor can engage in a hydrophobic interaction and/or a π-stacking interaction, while the carbonyl is closer to Arg235, increasing the H-bonding. The improved KD of RU-NT-93 describes the improved stacking and hydrophobic interactions with the aromatic ring of the inhibitor due to differences in the binding mode (Figure 7 and Table 2).
In RU-NT-102, the thiophene carboxylate is 44.3° off the plane of the ortho ethyl phenyl ring (Figure 7 and Figure S5) and is sandwiched between the two Arg residues: 3.23 Å from Arg234 and 3.40 Å from Arg235. The carboxylate of RU-NT-102 has two H-bond interactions including one with a water molecule and one with a backbone nitrogen of Arg235. The position of the inhibitor phenyl and thiophene rings is stabilized by the H-bond interaction with Arg235 along with π-stacking interactions of the phenyl ring with Phe240 (T-shaped stack) on one side and Tyr183 (offset rings) on the other side48 (Figure 6D).
Inhibitory Activities of the Advanced Compounds.
The 50% depurination inhibitory activity (IC50) of RTA on yeast ribosomal RNA in vitro was determined by qRT-PCR.44 The data for the percent inhibition at different fragment concentrations were fitted with Michaelis–Menten kinetics using the OriginPro software. All compounds had improved IC50 compared to CC10501. RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 gave 10-, 4-, 13-, and 12-fold improved IC50 values relative to CC10501, respectively (Table 3 and Figure 8).
Table 3.
The IC50 Values of the Advanced Compounds
| inhibitors | molecular weight | IC50 (μM)a |
|---|---|---|
| CC10501 | 204 | 181 ± 40 |
| RU-NT-70 | 281 | 19 ± 5 |
| RU-NT-75 | 246 | 48 ± 11 |
| RU-NT-93 | 232 | 14 ± 3 |
| RU-NT-102 | 232 | 15 ± 3 |
The IC50 values were determined in vitro by qRT-PCR using yeast ribosomes as described in Figure 8.
Figure 8.

Depurination inhibition in vitro using yeast ribosomes. The compounds were incubated with RTA for 5 min, and yeast ribosomes were added to start the reaction. The reactions were set at room temperature for 5 min, and the RNA extraction buffer was added to stop the reaction. The RNA was purified, and the depurination levels were determined using qRT-PCR. The percent depurination relative to the reaction without compound is shown. Data are presented as the average of three technical replicates from three to nine biological replicates. Each color represents one biological replicate. The data were fit using the OriginPro software with the Michaelis–Menten model.
Depurination Inhibition and Protection against Ricin Holotoxin in Cell-Based Assays.
Structure-based optimization led to inhibitors with low micromolar RTA binding affinity and similar potency for inhibition against RTA depurination of yeast ribosomes. To test their cellular protection activity, we used cultured Vero cells to determine if pretreatment with the fragments protected cells from exposure to ricin holotoxin. One hour of pretreatment was sufficient to allow for maximal protection. The cells were deprived of serum for pretreatment and toxin exposure to reduce the effect of fragment binding to serum proteins. In the presence of 200 pM ricin, cellular ribosome depurination increased linearly for about 4 h. We selected 1 h of pretreatment by each compound and 2 h of depurination by ricin to compare the fragments. Two hours after toxin addition, cellular RNA was isolated and used for qRT-PCR to determine the level of depurination.
RNA from cells without pretreatment was used as the control to represent full depurination (100%). The compounds did not cause any cytotoxicity to Vero cells by themselves even at 500 μM. Pretreating Vero cells with the parent compound, CC10501, showed very little inhibition of ribosome depurination (Figure 9). In contrast, cells treated with the lowest concentration of each inhibitor (250 μM) resulted in ~40–75% reduction in ribosome depurination with increased protection at 500 μM. The greatest protection was observed for RU-NT-93, exhibiting 71% reduction at 250 μM and 85% at 500 μM, and for RU-NT-102, which showed 73% reduction at 250 μM and 82% at 500 μM. There was little effect of the serum on the ability of the compounds to protect ribosomes against depurination by ricin.
Figure 9.

Inhibition of depurination by ricin holotoxin in Vero cells. Vero cells were plated at 1 × 105/mL and grown for 24 h. Cells were treated with each compound followed by 200 pM ricin as described in the methods. Percent depurination was measured by qRT-PCR compared to DMSO-treated cells at 2 h. Data are presented as the average of three technical replicates from three biological replicates with error bars showing the standard error. The Dunnett test was used to compare each compound to control, *** P < 0.001.
Next, we tested if the reduced ribosome depurination observed would translate into protection for cell growth and cell viability. The same conditions of pre-exposure in serum deprived cells prior to ricin holotoxin treatment were used. Preincubation with 250 and 500 μM compound was compared with controls (all at 0.5% DMSO final concentration) to a titration series of ricin holotoxin up to 400 pM. At 24 h after toxin addition, viability was determined with the Cell Titer-Glo luminescent viability assay. The parent compound CC10501 barely shifted the EC50 by ricin (Figure 10). RU-NT-70 shifted the EC50 twofold at 500 μM. RU-NT-93 and RU-NT-102 offered the best protection against ricin, shifting the EC50 values approximately 10-fold relative to CC10501 at 500 μM, in good agreement with their similar potencies for depurination inhibition in the Vero cell assay. Similar trends were also obtained with the MTT viability assay. RU-NT-75 at 500 μM interfered with both the Cell Titer Glo and the MTT assays.
Figure 10.

Cell protection against ricin holotoxin. Vero cells were treated with each fragment and a dilution series of ricin in triplicate in 96-well format as described in the methods. At 24 h, the Cell Titer-Glo reagent was added according to manufacturer’s instructions and luminescence was measured. Viability was expressed as a percentage of control cells treated with 0.5% DMSO.
We examined the effect of the four advanced compounds on depurination and toxicity of Stx2a holotoxin in Vero cells. In the presence of 1 nM Stx2a, cellular depurination increased linearly for 4 h. We treated cells for 1 h by each compound, and 2 h after toxin addition, cellular RNA was isolated and used for qRT-PCR. The compounds did not inhibit depurination by Stx2a in Vero cells (Figure S6) and had only minimal effects in viability assays using Stx2a holotoxin over a dose range causing substantial loss of viability (Figure S7). RU-NT-93 and RU-NT-102 did not alter the toxicity of an unrelated cytotoxin, cisplatin. These results showed the specificity of the compounds for viability protection against ricin consistent with their ability to inhibit ricin depurination.
DISCUSSION
Advanced Compounds Targeting RTA–ribosome Interactions Inhibit the Toxicity of Ricin Holotoxin.
Effective inhibitors are needed to protect against ricin, which remains a global biothreat agent.1 During cytotoxic ricin action, the C-terminal residues of ribosomal P proteins bind the hydrophobic pocket of RTA to allow the catalytic site to depurinate the SRL (Figure 1A). Despite significant efforts toward the identification of compounds that directly target RTA, very few successes have been reported to date. This is because high-throughput screening (HTS) assays that detect ribosome depurination are not available for screening large libraries of small molecules. Disruption of ribosome interactions of RIPs such as ricin and Stxs by small molecules is even more difficult since their ribosome binding site contains five P proteins with identical C-termini. We used a new strategy to identify small molecules that target the ribosome binding of RTA by screening fragment libraries. Fragment libraries require much fewer compounds than HTS to explore the chemical space with a relatively high hit rate due to the small size of the fragments.43 We screened a 1000-compound Maybridge core fragment library by SPR and identified about 70 fragments with dose-dependent binding to RTA.43
Fragment hits have low affinity because of their small size. In most cases, it is not possible to determine the affinity of a fragment hit accurately because of the difficulty in reaching the concentration range required (up to 10-fold above the KD) for the steady-state affinity analysis. Hence, KD values of the fragments from the primary screen serve to provide approximate values. We identified two fragments, CC10501 and CC70601, which bound RTA at the ribosome binding site with mid micromolar affinity and inhibitory activity.43 Here, we aimed for improvement in both the affinity and inhibitory activity of CC10501 to guide our optimization efforts. We optimized CC10501 using a structure-based design and obtained a new group of compounds with over an order of magnitude higher efficiency. Four inhibitors with improved protection against ricin were characterized kinetically and structurally by high-resolution X-ray crystallography. They all bound in the hydrophobic P protein binding pocket at the CTD of RTA. They inhibited the cytotoxicity of ricin holotoxin in mammalian cells without showing any cytotoxicity by themselves. These results demonstrate the feasibility of inhibiting ricin by disrupting the ribosome–RTA interaction by small molecules.
The Ribosome Binding Site Is a Better Target for Inhibitors than the Active Site of RTA.
We observed no major changes in the crystal structure of the RTA active site upon inhibitor binding at the ribosome binding pocket. Yet the advanced compounds inhibited the ability of RTA to inactivate eukaryotic ribosomes. The IC50 values of the inhibitors from the yeast ribosome assay were approximately an order of magnitude lower than the KD values measured by SPR, a difference that we have also observed with peptides that bind in the hydrophobic pocket.27 The small molecules and peptides bind to the P stalk binding site that is on the opposite face of the active site.36 The inhibitors do not compete with the SRL for binding RTA but compete with binding of the P protein CTD to RTA. The interaction of the P protein CTD with the hydrophobic pocket on RTA stimulates the depurination activity of RTA by promoting a conformational change.36,41 Therefore, the inhibition of P stalk binding causes a decrease in ribosome inactivation. Small molecule binding at the hydrophobic pocket may stabilize RTA at a particular conformation preventing the binding of the P protein CTD and the stimulation of depurination, thereby enhancing the inhibition of depurination.
The dissociation constants measured by SPR for the four compounds emphasized here improved relative to the original compound. We were able to detect inhibition of toxicity in mammalian cells in the same relative order as the affinity of the advanced compounds, while the original compound did not show any inhibition. Of the entire set of CC10501 analogs tested, RU-NT-93 and RU-NT-102 were the best compounds with 13- and 12-fold improvement, respectively, in IC50 against yeast ribosomes (Table 3). Both compounds protected Vero cells against depurination by ricin holotoxin and caused over an order of magnitude increase in the viability of Vero cells exposed to ricin. They did not protect Vero cells against depurination by Stx2a or inhibit the cytotoxicity of Stx2a in Vero cells, indicating their specificity for ricin. Both compounds are sufficiently hydrophobic to cross the cellular membrane. The octanol–water coefficient (cLogP) and the topological polar surface area (tPSA) are predictors for absorption across cellular membranes. RU-NT-93 with a cLogP of 4.03 and a tPSA of 37.3 and RU-NT-102 with a cLogP of 4.36 and a tPSA of 37.3 possess properties consistent with absorption into mammalian cells.
The SPR affinity data correlated with the Vero cell protection data, indicating similar thermodynamic and biological functions, suggesting that the affinity is predictive of the effect in cells. None of the hits targeted the active site of RTA, indicating that the active site of RTA is a more difficult target for small molecule inhibitors.43 This might also be true for other RIPs, such as Stxs, that interact with the P stalk to depurinate the SRL.
RU-NT-93 Binds RTA in a Distinctive Manner.
The structural analysis established that the advanced compounds bind in the same hydrophobic pocket of RTA that accommodates the CTD of P proteins. The binding interactions of RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 are primarily hydrophobic but also include hydrogen bonding between the common carboxylate group of the inhibitors with a backbone carbonyl of Arg235, the Arg234 guanidinium, and structural waters (Figure 6). While the carboxylates of RU-NT-75, RU-NT-93, and RU-NT-102 have two H-bond interactions, the carboxylate of RU-NT-70 is stabilized by four hydrogen bonds (Figure 6). The binding pose of RU-NT-75 and RU-NT-93 showed that the thiophene carboxylate groups were perpendicular to those of the other inhibitors because of steric hindrance in these inhibitors. The common geometry of the substituted phenyl group in all four compounds indicated that it is the major determinant for inhibitor binding (Figure 7).
The crystal structure analysis revealed a new binding pose for RU-NT-93 relative to the similar structures. The thiophene carboxylate was sandwiched between Arg235 and Tyr183 instead of Arg234/235. RU-NT-93 has a cLogP of 4.02, while RU-NT-75 has the 2,6-dimethyl of RU-NT-93 but an additional 4-methyl group that raises the cLogP to 4.52. The binding of RU-NT-93 was 0.7 Å deeper in the binding pocket compared to RU-NT-75. In the RU-NT-93 bound structure, Leu207 participated in the inhibitor binding (Figure 6C), but this interaction was missing in the RU-NT-75 bound structure (Figure 6B). This pose of RU-NT-93 has a more favorable binding in the RTA hydrophobic pocket. RU-NT-93 may have better affinity and inhibition against RTA due to the improved stacking and hydrophobic interactions with the aromatic ring of RU-NT-93. The interaction with Leu207 identifies a new location within the hydrophobic pocket, which may define new interactions for inhibitor optimization. The hydrophobic pocket in the phenyl binding region of the inhibitors is sufficiently open to provide potentially new binding interactions with this site. The RU-NT-93 chemical scaffold will serve as a positive control for future inhibitor development, as well as a probe for understanding ricin/ribosome interactions.
To determine if we can corroborate the X-ray structure analysis by molecular docking, we used the MOE (2019.0102) software to dock the compounds into the available X-ray structures (PDB IDs: 6URX, 7MLN, 7MLO, 7MLP, and 7MLT). We found that the docking mostly recapitulated the structures we observed by X-ray crystallography (Figure S8). Our initial lead series were structurally similar, and the docking score did not clearly differentiate between them due to small differences in the binding energy (Table S3). These studies provided evidence that by using the X-ray crystal structure of the bound ligand and molecular docking, we can design better inhibitors.
Ricin is a difficult target for therapeutic intervention because poisoning symptoms appear within 4–12 h after inhalation. The inhibitors reported here can inhibit the toxicity of ricin holotoxin after it is internalized and transported to the ER, where RTA is released from RTB, and after RTA enters the cytosol.6 Hence, inhibitors that target the ribosome binding site of RTA have a longer therapeutic window than inhibitors targeting the holotoxin or the B subunit. To our knowledge, this is the first example of small molecule inhibitors binding directly at the ribosome binding site of RTA and inhibiting the cytotoxicity of ricin holotoxin without affecting ricin trafficking or ribotoxic stress pathways associated with ricin-induced cell death. The low micromolar potency observed in cell-based assays suggests that further improvements in inhibitor binding are possible. Small molecule selective inhibitors against RTA may have potential as antidotes against ricin toxicity and might also rescue normal cells after RTA-immunotoxin cancer therapy.16
CONCLUSIONS
The CC10501 chemical scaffold was used as a template to improve small molecule inhibitor binding against RTA. We have characterized the advanced inhibitors chemically and biochemically and found that these inhibitors have over 10-fold improved inhibitory activity relative to CC10501. Crystal structure studies of the complexes with RTA showed binding of these inhibitors in the peripheral hydrophobic pocket that interacts with the C-termini of ribosomal P proteins. The inhibitors characterized here are the first small molecule chemical agents that act directly on the ribosome interaction site of RTA and show efficacy against ricin holotoxin in cells. This research provides a starting point for optimizing the next-generation small molecule inhibitors against ricin toxin.
EXPERIMENTAL SECTION
General Procedures.
The reagents were purchased and used without additional purification. LC–MS was performed on an Agilent 1100 system with a Waters Micromass ZQ spectrometer using a 5 μL injection on an XBridge C18 (3.5 μm, 4.6 × 50 mm) column at a temperature of 40 °C with a 4 min gradient from 5% A to 95% B (solvent A: 10 mM ammonium formate in water; solvent B: acetonitrile) at a flow rate of 2 mL/min. The detection used a diode array scanning from 190 to 600 nm or with dual wavelength detectors at 220 and 254 nm (mass detection cone voltage: 30 V). NMR was obtained on a Varian VNMRS 300 MHz in chloroform-d (1H: δ7.26) or DMSO-d6 (1H: δ2.50). Microwave reactions were run on a Biotage Initiator system. Flash column chromatography purifications were performed using Biotage Isolera systems. Unless otherwise stated, the purities of the final compounds were equal to or greater than 95% by LC–MS analysis. The purities were confirmed with 1H NMR to look for residual solvents or non-UV active impurities.
5-(p-Tolyl)thiophene-2-carbonitrile (1).
Palladium (II) acetate (0.22 mg, 0.001 mmol) was added to a solution of thiophene-2-carbonitrile (93 μL, 1 mmol), 1-bromo-4-methylbenzene (246 μL, 2 mmol), and potassium acetate (196 mg, 2 mmol) in dimethylacetamide (5 mL) under nitrogen, and the reaction mixture was heated to 150 °C for 20 h. After cooling to room temperature, the mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to give the product (43 mg, 45%) as a yellow solid. ESI-MS: 200.1 (M + H)+.
5-(5-(p-Tolyl)thiophen-2-yl)-1H-tetrazole (RU-NT-57).
A mixture of 5-(p-tolyl)thiophene-2-carbonitrile (43 mg, 0.22 mmol), sodium azide (140 mg, 2.16 mmol), and ammonium chloride (116 mg, 10 mmol) was suspended in DMF, and the resulting mixture was stirred at 120 °C under nitrogen overnight. After cooling to room temperature, the mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to give the product (36 mg, 70%) as a yellow solid. ESI-MS: 243.0 (M + H)+. 1H NMR (300 MHz, DMSO-d6) δ 7.69 (d, J = 3.9 Hz, 1H), 7.48 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.9 Hz, 1H), 7.17 (d, J = 7.9 Hz, 2H), 2.30 (s, 3H).
General Procedure for Suzuki Coupling and Ester Hydrolysis.
A mixture of methyl 5-bromothiophene-2-carboxylate (1 equiv), aryl boronic acid (2 equiv), Pd(PPh3)4 (0.1 equiv), and K2CO3 (3 equiv) was dissolved in dioxane (10 mL) and water (1 mL). The mixture was flushed with nitrogen for 3 min and then refluxed under nitrogen overnight. After cooling to room temperature, the reaction mixture was partitioned between water and ethyl acetate. The organic layer was washed with water and brine and dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to give the corresponding ester. NaOH (10%, 4 mL) was added to a stirred solution of the ester in MeOH (3 mL) and THF (3 mL). The mixture was stirred at room temperature for 2 h. After the completion of the reaction by LC–MS, the reaction mixture was partitioned between 1 N HCl and ethyl acetate. The organic layer was washed with water and brine and dried over anhydrous Na2SO4, filtered, and concentrated to afford the corresponding carboxylic acid. RU-NT-70, 74–75, 93, 102, 105, 110–111, 113, 115, and 124 were prepared using this general method.
5-(o-Tolyl)thiophene-2-carboxylic Acid (RU-NT-70).
ESI-MS: 217.2 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.68–7.61 (m, 1H), 7.32 (d, J = 7.0 Hz, 1H), 7.26–7.11 (m, 3H), 7.00 (d, J = 3.8 Hz, 1H), 2.36 (s, 3H).
5-(2,4-Dimethylphenyl)thiophene-2-carboxylic Acid (RU-NT-74).
ESI-MS: 231.3 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.69–7.55 (m, 1H), 7.26–7.16 (m, 1H), 7.08–6.91 (m, 3H), 2.31 (s, 3H), 2.27 (s, 3H).
5-Mesitylthiophene-2-carboxylic Acid (RU-NT-75).
ESI-MS: 245(M – H)−. 1H NMR (300 MHz, CDCl3) δ 7.62 (d, J = 3 Hz, 1H), 6.79 (d, J = 0.5 Hz, 2H), 6.65 (d, J = 3 Hz, 1H), 2.17 (s, 3H), 1.97 (s, 6H).
5-(2,6-Dimethylphenyl)thiophene-2-carboxylic Acid (RU-NT-93).
ESI-MS: 231.3 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.67 (d, J = 3 Hz, 1H), 7.17–6.97 (m, 3H), 6.75 (d, J = 3 Hz, 1H), 2.07 (s, 6H).
5-(2-Ethylphenyl)thiophene-2-carboxylic Acid (RU-NT-102).
ESI-MS: 231.3 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.65 (d, J = 3.8 Hz, 1H), 7.30–7.14 (m, 4H), 6.96 (d, J = 3.8 Hz, 1H), 2.67 (q, J = 7.5 Hz, 2H), 1.11 (t, J = 7.5 Hz, 3H).
5-(2,3-Dihydrobenzofuran-7-yl)thiophene-2-carboxylic Acid (RU-NT-105).
ESI-MS: 245.4 (M – H)−. 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 3.0 Hz, 1H), 7.48 (d, J = 3.0 Hz, 1H), 7.36 (d, J = 9.0 Hz, 1H), 7.11 (d, J = 9.0 Hz, 1H), 6.83 (t, J = 9.0 Hz, 1H), 4.64 (t, J = 9.0 Hz, 2H), 3.21 (t, J = 9.0 Hz, 2H).
5-(2-Fluoro-6-methylphenyl)thiophene-2-carboxylic Acid (RU-NT-110).
ESI-MS: 235.3 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.70 (d, J = 3.8 Hz, 1H), 7.27–7.15 (m, 1H), 7.02 (d, J = 7.7 Hz, 1H), 6.98–6.86 (m, 2H), 2.23 (s, 3H).
5-(2-Methoxy-6-methylphenyl)thiophene-2-carboxylic Acid (RU-NT-111).
ESI-MS: 247.3 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.62 (m, 1H), 7.20 (t, J = 7.9 Hz, 1H), 6.88–6.67 (m, 3H), 3.68 (s, 3H), 2.13 (s, 3H).
5-(2-Hydroxy-6-methylphenyl)thiophene-2-carboxylic Acid (RU-NT-113).
ESI-MS: 233.3 (M – H)−. 1H NMR (300 MHz, CDCl3) δ 7.53 (d, J = 3.0 Hz, 1H), 6.88 (t, J = 7.8 Hz, 1H), 6.72 (d, J = 3.0 Hz, 1H), 6.61–6,53 (m, 2H), 1.96 (s, 3H).
5-(2-((2-Methoxyethoxy)methyl)phenyl)thiophene-2-carboxylic Acid (RU-NT-115).
ESI-MS: 291.4 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.65 (d, J = 3.5 Hz, 1H), 7.54–7.24 (m, 4H), 7.14 (d, J = 3.5 Hz, 1H), 4.47 (s, 2H), 3.62–3.43 (m, 4H), 3.29 (s, 3H).
5-(2,6-Diisopropylphenyl)thiophene-2-carboxylic Acid (RU-NT-124).
ESI-MS: 287.4 (M – H)−. 1H NMR (300 MHz, DMSO-d6) δ 7.68 (d, J = 3.6 Hz, 1H), 7.51 (d, J = 4.7 Hz, 1H), 7.29 (t, J = 6.0 Hz, 1H), 7.13 (d, J = 3.6 Hz, 1H), 6.78 (d, J = 3.6 Hz, 1H), 2.72–2.63 (m, 2H), 1.06 (t, J = 6.0 Hz, 12H).
5-(2,6-Dimethylphenyl)thiophene-2-carbonitrile (2).
A mixture of methyl 5-bromothiophene-2-carbonitrile (141 mg, 0.75 mmol), (2,6-dimethylphenyl)boronic acid (135 mg, 0.9 mmol), Pd(PPh3)4 (87 mg, 0.075 mmol), and K2CO3 (310 mg, 2.25 mmol) was dissolved in dioxane (10 mL) and water (1 mL). The mixture was flushed with nitrogen for 3 min and then refluxed under nitrogen overnight. After cooling to room temperature, the reaction mixture was partitioned between water and ethyl acetate. The organic layer was washed with water and brine and dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to give 5-(2,6-dimethylphenyl)thiophene-2-carbonitrile (112 mg, 70%). ESI-MS: 214.1 (M + H)+.
5-(5-(2,6-Dimethylphenyl)thiophen-2-yl)-1H-tetrazole (RU-NT-128).
A mixture of 5-(2,6-dimethylphenyl)thiophene-2-carbonitrile (55 mg, 0.26 mmol), sodium azide (168 mg, 2.60 mmol), and ammonium chloride (140 mg, 2.60 mmol) was suspended in DMF, and the mixture was stirred at 120 °C under nitrogen overnight. After cooling to room temperature, the mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to give the product (56 mg, 85%) as a yellow solid. ESI-MS: 257.1 (M + H)+. 1H NMR (300 MHz, DMSO-d6) δ 7.75 (d, J = 3.1 Hz, 1H), 7.23–6.95 (m, 3H), 6.82 (d, J = 3.1 Hz, 1H), 2.10 (s, 6H).
RTA Binding and Affinity Measurement Using SPR.
The affinity of all compounds in the RTA focused library was measured using Biacore 8K+ using the same conditions as previously reported.43 Biacore 8K+ has eight channels with two flow cells (Fc) in each channel. RTA was immobilized on Fc2 of a CM5 chip at 5800 to 6000 RU using the amine coupling method. Fc1 was activated and blocked without RTA using the same method as Fc2 as reference. Compounds were passed over Fc1 and Fc2 in all eight channels in parallel. Data were analyzed using the Biacore Insight Evaluation Software with DMSO correction. The binding sensorgrams of Fc1 were subtracted from Fc2 to correct for the buffer mismatch. The buffer alone was subtracted from all sensorgrams at different concentrations to correct for any baseline drift. The running buffer was PBS-P (20 mM phosphate, 2.7 mM KCl, 137 mM NaCl, and 0.05% surfactant P20, pH 7.4) with 2% DMSO. Fragments at 0.7, 2.1, 6.2, 18.5, 55.6, 166.7, and 500 μM were passed over both surfaces at 30 μL/min for 60 s and dissociation for another 60 s. There was no nonspecific binding with any of the compounds tested.
In Vitro Depurination Inhibition of Yeast Ribosomes.
Yeast ribosome purification and depurination inhibition were conducted using the qRT-PCR method as previously reported.43 Compounds were diluted in twofold series to concentrations ranging from 10 to 200 μM in the RIP buffer and DMSO (25 mM KCl, 20 mM Tris–HCl pH 7.5, 5 mM MgCl2, and 1% DMSO). RTA was diluted to 4 nM and yeast ribosomes to 200 nM in RIP buffer–DMSO. Final concentrations for each component in the 100 μL reaction were 1 nM RTA, 50 nM ribosomes, and each compound ranging from 5 to 100 μM. Reactions were carried out by mixing 25 μL of 4 nM RTA with 50 μL of the diluted compound and incubated at room temperature for 5 min. Then, 25 μL of 200 nM yeast ribosomes was added and the reaction was incubated at room temperature for an additional 5 min. The reaction was stopped by the addition of 100 μL 2× extraction buffer (240 mM NaCl, 50 mM Tris–HCl pH 8.8, 20 mM EDTA, and 2% SDS). RNA was prepared by phenol and chloroform extraction followed by ethanol precipitation. Precipitated RNA was washed twice with 70% ethanol, dried, and resuspended in 30 μL of RNase-free water. RNA concentration was determined by absorbance at 260 nm.
Depurination Assay in Mammalian Cells.
Vero cells were maintained in Dulbecco’s modified Eagle medium (DMEM) with penicillin and streptomycin and 10% fetal calf serum supplements. For depurination, cells were plated in this medium at 1 × 105/mL in 24-well tissue culture treated plates at 500 μL per well and grown for 24 h. Compounds were added to 500 μL of the medium lacking serum supplementation from 100% DMSO stocks to give the designated final concentrations for each treatment and vortexed to dissolve. The final concentration of DMSO in controls and treatments was 0.5%. The medium was removed from each well and replaced with the vortexed fragment solution. The plate was incubated at 37 °C, 5% CO2 for 1 h. Ricin holotoxin was added from the stock diluted in phosphate-buffered saline to a final concentration of 200 pM. Wells were further incubated for 2 h prior to harvest. The medium was removed, and cells were collected in 350 μL of the lysis buffer from the Qiagen Plus RNA Mammalian Cell kit. Total RNA was extracted from Vero cells using the RNeasy Plus Mini Kit (Qiagen) either immediately or after storage at −80 °C. The High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) was used for cDNA conversion of ~375 ng of total RNA in a 20 μL reaction.
qRT-PCR and Data Analysis.
The qRT-PCR assays were performed on a StepOnePlus Real Time PCR System (Thermo Fisher Scientific) using the Power SYBR Green Master Mix (Thermo Fisher Scientific) in a total volume of 20 μL with 5 μL of cDNA diluted 1:50 and forward and reverse primers at a final concentration of 250 nM each. The PCR primer pair sequences in 5′ to 3′ direction were T G C C A T G G T A A T C C T G C T C A G T A and TCTGAACCTGCGGTTCCACA for the depurinated human rRNA and GATGTCGGCTCTTCCTATCATTGT and CCAGCTCACGTTCCCTATTAGTG for the human 28S rRNA as the endogenous control. Quantification by the comparative CT method was done using the StepOnePlus Software V2.3 with depurination in the absence of an inhibitor (control) representing 100% depurination. Data are presented as the average of three technical replicates from three biological replicates with error bars showing the standard error.
Viability Analysis.
Protection was performed in 96-well white plates at the same seeding density (1 × 105 mL). At 24 h, the medium was removed and replaced by 100 μL of the serum free medium with fragment or DMSO added and vortexed. One hour later, ricin was added from a dilution series to give final concentrations as noted. Viability was measured at 24 h using the Cell Titer-Glo 3D Cell Viability Assay (Promega) according to the manufacturer’s conditions. Luminescence was measured with a BioTek 4 Synergy plate reader. Viability was expressed as a percentage of control cells treated with 0.5% DMSO.
Co-crystallization of RTA Inhibitor Complexes.
The co-crystallization of RTA with inhibitors RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 was done by the sitting drop vapor diffusion method at 22 °C. RTA (5 mg/mL) was mixed with inhibitors in a 1:5 molar ratio and incubated for 2 h on ice. The incubated samples were screened by using the Microlytic (MCSG1-4) and Hampton (crystal screenHT) crystallization conditions. The crystallization was set up in 96-well INTELLI plates (ART ROBBINS) using the CRYSTAL-GRYPHON crystallization robot (ART ROBBINS). Each crystallization drop contained 0.5 μL of the RTA inhibitor mixture and 0.5 μL of the well solution. The volume of the well solution was 70 μL. Good quality crystals were obtained in 1–2 weeks. The crystallization and crystal handling processes are summarized in Table S1.
Data Collection and Processing.
The high-resolution diffraction data were collected from 1.52 to 1.80 Å resolutions at the LRL-CAT beam line (Argonne National Laboratory, Argonne, IL) at 0.97931 Å wavelength. All the diffraction data were processed using the iMOSFLM and scaled by the AIMLESS program of the CCP4 suite in different space groups as summarized in Table S2.49 The quality of the data was analyzed using the SFCHECK and XTRIAGE.50,51 The Matthews coefficient (Vm) calculations were done to calculate the number of monomer molecules present in the asymmetric unit.50 The data collection and processing statistics are summarized in Table S2.
Structure Determination and Refinement.
The crystal structures of RTA in complex with inhibitors were solved by molecular replacement using PHASER.52 The chain-A of the wild-type RTA (PDB ID: 1RTC) structure was used as the initial phasing model. The model obtained from PHASER was manually adjusted and completed using the graphics program COOT.53 The structure refinement was performed by the REFMAC5 program using standard protocols for the NCS refinement.54 The inhibitor molecules were left out from the models in the beginning of the refinement. After building all the waters into the structures, inhibitor molecules were fitted in their respective electron densities. The final refinement statistics of the structure are summarized in Table S2.
Structure Analysis.
All structural superimpositions were done using the SSM protocol of COOT. The geometry analyses of the final model were done using MolProbity.55 Further structure analyses, including the calculation of the B-factor profiles, was done using the BAVERAGE program of the CCP4 suite.50 The figures were made using the molecular graphics program PyMOL. For RTA structures, subunit-A was used for all the structural analyses and comparisons.
Molecular Modeling and Docking.
The compounds CC10501, RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 were docked into the active site of RTA using the MOE 2019.0102 software (University-Wide License). The ”QuickPrep” feature with standard settings was used to optimize the protein. Each compound was docked in each of the resolved protein structures. The docking process was performed by using the ”Induced Fit” protocol. In this simulation, the ”Triangle Matcher/London dG” and “Forcefield/GBVI-WSA dG” parameters were chosen as the placement and refinement methods, respectively. Fifty ligand conformations were generated and scored. The best docking score was reported. In this mode, the protein residues were allowed to move, thereby allowing ligands to fit the conformation of the binding site.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stephanie P. Delatola and Arly Volmar for help with the in vitro depurination assay used in screening. We thank Dr. Chhaya Dharia for purifying RTA, Dr. John McLaughlin for statistical analysis, and Dr. Przemek Grela for Figure 1A. Research support was provided by NIH research grant AI072425 to Nilgun E. Tumer and NIH research grant GM041916 to Vern L. Schramm, Albert Einstein College of Medicine. We thank V.L.S. for advice and editing the manuscript. The School of Environmental and Biological Sciences (SEBS) Biomolecular Interaction Analysis Core Facility is supported by NIH Shared Instrumentation Grant S10 OD026750, which we gratefully acknowledge. The Einstein Crystallographic Core X-Ray diffraction facility is supported by NIH Shared Instrumentation Grant S10 OD020068, which we gratefully acknowledge. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
ABBREVIATIONS USED
- DMF
dimethyl formamide
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- HTS
high-throughput screening
- PEG
polyethylene glycol
- RIP
ribosome inactivating protein
- RTA
ricin toxin A subunit
- RTB
ricin toxin B subunit
- Stx
Shiga toxin
- SPR
surface plasmon resonance
- SRL
sarcin/ricin loop
- THF
tetrahydrofuran
- TCS
trichosanthin
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01370.
Tables S1–S3; Figures S1–S8; analytical LC–MS traces; NMR spectra; and PDB validation report for the structures of RU-NT-70, RU-NT-75, RU-NT-93, and RU-NT-102 (PDF)
Molecular formula strings (CSV)
PDB docking structure file: RU-NT-70 (PDB)
PDB docking structure file: RU-NT-75 (PDB)
PDB docking structure file: RU-NT-93 (PDB)
PDB docking structure file: RU-NT-102 (PDB)
Accession Codes
RU-NT-70 (7MLN), RU-NT-75 (7MLO), RU-NT-93 (7MLP), and RU-NT-102 (7MLT). The authors will release the atomic coordinates and experimental data upon article publication. PDB IDs have been provided in the figure legends.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c01370
The authors declare no competing financial interest.
Contributor Information
Xiao-Ping Li, Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08901, United States.
Rajesh K. Harijan, Department of Biochemistry, Albert Einstein College of Medicine, Jack and Pearl Resnick Campus, Bronx, New York 10461, United States.
Bin Cao, Molecular Design and Synthesis Core, Rutgers University Biomolecular Innovations Cores, Office for Research, Rutgers University, Piscataway, New Jersey 08854, United States; Present Address: Eternity Bioscience Inc. 6 Cedarbrook Drive, Cranbury, NJ 08512, United States.
Jennifer N. Kahn, Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08901, United States
Michael Pierce, Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08901, United States.
Anastasiia M. Tsymbal, Molecular Design and Synthesis Core, Rutgers University Biomolecular Innovations Cores, Office for Research, Rutgers University, Piscataway, New Jersey 08854, United States
Jacques Y. Roberge, Molecular Design and Synthesis Core, Rutgers University Biomolecular Innovations Cores, Office for Research, Rutgers University, Piscataway, New Jersey 08854, United States
David Augeri, Molecular Design and Synthesis Core, Rutgers University Biomolecular Innovations Cores, Office for Research, Rutgers University, Piscataway, New Jersey 08854, United States; Present Address: DJ Augeri Pharma Consulting, LLC Princeton, NJ 08540, United States.
Nilgun E. Tumer, Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08901, United States
REFERENCES
- (1).Audi J; Belson M; Patel M; Schier J; Osterloh J Ricin Poisoning: A Comprehensive Review. JAMA 2005, 294, 2342–2351. [DOI] [PubMed] [Google Scholar]
- (2).Paton JC; Paton AW Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev 1998, 11, 450–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Boerlin P; McEwen SA; Boerlin-Petzold F; Wilson JB; Johnson RP; Gyles CL Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol 1999, 37, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Law D Virulence factors of Escherichia coli O157 and other Shiga toxin-producing E.coli. J. Appl. Microbiol 2000, 88, 729–745. [DOI] [PubMed] [Google Scholar]
- (5).Endo Y; Mitsui K; Motizuki M; Tsurugi K The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem 1987, 262, 5908–5912. [PubMed] [Google Scholar]
- (6).Sowa-Rogozinska N; Sominka H; Nowakowska-Golacka J; Sandvig K; Slominska-Wojewodzka M Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins 2019, 11, 350. (Published: June 18, 2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Spooner RA; Lord JM How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol 2011, 357, 19–40. [DOI] [PubMed] [Google Scholar]
- (8).Liu XY; Pop LM; Schindler J; Vitetta ES Immunotoxins constructed with chimeric, short-lived anti-CD22 monoclonal antibodies induce less vascular leak without loss of cytotoxicity. mAbs 2012, 4, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Weidle UH; Tiefenthaler G; Schiller C; Weiss EH; Georges G; Brinkmann U Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics 2014, 11, 25–38. [PubMed] [Google Scholar]
- (10).Polito L; Djemil A; Bortolotti M Plant toxin-based immunotoxins for cancer therapy: A short overview. Biomedicines 2016, 4, 12. (Published: June 1, 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Robertus JD; Monzingo AF The structure of ribosome inactivating proteins. Mini. Rev. Med. Chem 2004, 4, 477–486. [DOI] [PubMed] [Google Scholar]
- (12).Wahome PG; Robertus JD; Mantis NJ Small-molecule inhibitors of ricin and Shiga toxins. Curr. Top. Microbiol 2012, 357, 179–207. [DOI] [PubMed] [Google Scholar]
- (13).Pruet JM; Saito R; Manzano LA; Jasheway KR; Wiget PA; Kamat I; Anslyn EV; Robertus JD Optimized 5-membered heterocycle-linked pterins for the inhibition of Ricin Toxin A. ACS Med. Chem. Lett 2012, 3, 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saito R; Pruet JM; Manzano LA; Jasheway K; Monzingo AF; Wiget PA; Kamat I; Anslyn EV; Robertus JD Peptideconjugated pterins as inhibitors of ricin toxin A. J. Med. Chem 2013, 56, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jasheway K; Pruet J; Anslyn EV; Robertus JD Structure-based design of ricin inhibitors. Toxins 2011, 3, 1233–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ho MC; Sturm MB; Almo SC; Schramm VL Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci U. S. A 2009, 106, 20276–20281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Saenz JB; Doggett TA; Haslam DB Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun 2007, 75, 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stechmann B; Bai SK; Gobbo E; Lopez R; Merer G; Pinchard S; Panigai L; Tenza D; Raposo G; Beaumelle B; Sauvaire D; Gillet D; Johannes L; Barbier J Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [DOI] [PubMed] [Google Scholar]
- (19).Wahome PG; Bai Y; Neal LM; Robertus JD; Mantis NJ Identification of small-molecule inhibitors of ricin and Shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wahome PG; Ahlawat S; Mantis NJ Identification of small molecules that suppress ricin-induced stress-activated signaling pathways. PLoS One 2012, 7, No. e49075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Redmann V; Gardner T; Lau Z; Morohashi K; Felsenfeld D; Tortorella D Novel class of potential therapeutics that target ricin retrograde translocation. Toxins 2014, 6, 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chiou J-C; Li X-P; Remacha M; Ballesta JP; Tumer NE The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol 2008, 70, 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Grela P; Li XP; Horbowicz P; Dzwierzynska M; Tchorzewski M; Tumer NE Human ribosomal P1-P2 heterodimer represents an optimal docking site for ricin A chain with a prominent role for P1 C-terminus. Sci. Rep 2017, 7, 5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guarinos E; Santos C; Sanchez A; Qiu DY; Remacha M; Ballesta JP Tag-mediated fractionation of yeast ribosome populations proves the monomeric organization of the eukaryotic ribosomal stalk structure. Mol. Microbiol 2003, 50, 703–712. [DOI] [PubMed] [Google Scholar]
- (25).Grela P; Krokowski D; Gordiyenko Y; Krowarsch D; Robinson CV; Otlewski J; Grankowski N; Tchorzewski M Biophysical properties of the eukaryotic ribosomal stalk. Biochemistry 2010, 49, 924–933. [DOI] [PubMed] [Google Scholar]
- (26).Tchorzewski M The acidic ribosomal P proteins. Int. J. Biochem. Cell Biol 2002, 34, 911–915. [DOI] [PubMed] [Google Scholar]
- (27).Li X-P; Kahn JN; Tumer NE Peptide mimics of the ribosomal P stalk inhibit the activity of ricin A chain by preventing ribosome binding. Toxins 2018, 10, 371. (Published: Sept. 13, 2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chan DS; Chu L-O; Lee K-M; Too PH; Ma K-W; Sze KH; Zhu G; Shaw P-C; Wong K-B Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acids Res. 2007, 35, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang Y; Mak AN-S; Shaw PC; Sze KH Solution structure of an active mutant of maize ribosome-inactivating protein (MOD) and its interaction with the ribosomal stalk protein P2. J. Mol. Biol 2010, 395, 897–907. [DOI] [PubMed] [Google Scholar]
- (30).McCluskey AJ; Poon GM; Bolewska-Pedyczak E; Srikumar T; Jeram SM; Raught B; Gariepy J The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol 2008, 378, 375–386. [DOI] [PubMed] [Google Scholar]
- (31).McCluskey AJ; Bolewska-Pedyczak E; Jarvik N; Chen G; Sidhu SS; Gariépy J Charged and hydrophobic surfaces on the A chain of Shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PLoS One 2012, 7, No. e31191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chiou J-C; Li X-P; Remacha M; Ballesta JPG; Tumer NE Shiga toxin 1 is more dependent on the P proteins of the ribosomal stalk for depurination activity than Shiga toxin 2. Int. J. Biochem. Cell Biol 2011, 43, 1792–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bargis-Surgey P; Lavergne JP; Gonzalo P; Vard C; Filhol-Cochet O; Reboud JP Interaction of elongation factor eEF-2 with ribosomal P proteins. Eur. J. Biochem 1999, 262, 606–611. [DOI] [PubMed] [Google Scholar]
- (34).Carlson MA; Haddad BG; Weis AJ; Blackwood CS; Shelton CD; Wuerth ME; Walter JD; Spiegel PC Ribosomal protein L7/L12 is required for GTPase translation factors EF-G, RF3 and IF2 to bind in their GTP state to 70S ribosomes. FEBS J. 2017, 284, 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li X-P; Chiou J-C; Remacha M; Ballesta JP; Tumer NE A two-step binding model proposed for the electrostatic interactions of ricin A chain with ribosomes. Biochemistry 2009, 48, 3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li XP; Kahn PC; Kahn JN; Grela P; Tumer NE Arginine residues on the opposite side of the active site stimulate the catalysis of ribosome depurination by ricin A chain by interacting with the P-protein stalk. J. Biol. Chem 2013, 288, 30270–30284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li XP; Grela P; Krokowski D; Tchorzewski M; Tumer NE Pentameric organization of the ribosomal stalk accelerates recruitment of ricin a chain to the ribosome for depurination. J. Biol. Chem 2010, 285, 41463–41471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhou Y; Li XP; Kahn JN; McLaughlin JE; Tumer NE Leucine 232 and hydrophobic residues at the ribosomal P stalk binding site are critical for biological activity of ricin. Biosci. Rep 2019, 39, BSR20192022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Fan X; Zhu Y; Wang C; Niu L; Teng M; Li X Structural insights into the interaction of the ribosomal P stalk protein P2 with a type II ribosome-inactivating protein ricin. Sci. Rep 2016, 6, 37803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shi WW; Tang YS; Sze SY; Zhu ZN; Wong KB; Shaw PC Crystal structure of ribosome-inactivating protein ricin A chain in complex with the C-terminal peptide of the ribosomal stalk protein P2. Toxins 2016, 8, 296. (Published: Oct 13, 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou Y; Li XP; Chen BY; Tumer NE Ricin uses arginine 235 as an anchor residue to bind to P-proteins of the ribosomal stalk. Sci. Rep 2017, 7, 42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rudolph MJ; Davis SA; Tumer NE; Li XP Structural basis for the interaction of Shiga toxin 2a with a C-terminal peptide of ribosomal P stalk proteins. J. Biol. Chem 2020, 295, 15588–15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li XP; Harijan RK; Kahn JN; Schramm VL; Tumer NE Small molecule inhibitors targeting the interaction of ricin toxin A subunit with ribosomes. ACS Infect Dis. 2020, 6, 1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pierce M; Kahn JN; Chiou J; Tumer NE Development of a quantitative RT-PCR assay to examine the kinetics of ribosome depurination by ribosome inactivating proteins using Saccharomyces cerevisiae as a model. RNA 2011, 17, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhou Y; Li XP; Kahn JN; Tumer NE Functional assays for measuring the catalytic activity of ribosome inactivating proteins. Toxins 2018, 10, 240. (Published: June 14, 2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Rutenber E; Katzin BJ; Ernst S; Collins EJ; Mlsna D; Ready MP; Robertus JD Crystallographic refinement of ricin to 2.5 Å. Proteins 1991, 10, 240–250. [DOI] [PubMed] [Google Scholar]
- (47).Mlsna D; Monzingo AF; Katzin BJ; Ernst S; Robertus JD Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993, 2, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McGaughey GB; Gagne M; Rappe AK pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem 1998, 273, 15458–15463. [DOI] [PubMed] [Google Scholar]
- (49).Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG iMOSFLM: a new graphical interface for diffractionimage processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- (51).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (55).Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.