

Abstract
The emergence of SARS-CoV-2 variants and drug-resistant mutants calls for additional oral antivirals. The SARS-CoV-2 papain-like protease (PLpro) is a promising but challenging drug target. In this study, we designed and synthesized 85 noncovalent PLpro inhibitors that bind to the newly discovered Val70Ub site and the known BL2 groove pocket. Potent compounds inhibited PLpro with inhibitory constant Ki values from 13.2 to 88.2 nM. The co-crystal structures of PLpro with eight leads revealed their interaction modes. The in vivo lead Jun12682 inhibited SARS-CoV-2 and its variants, including nirmatrelvir-resistant strains with EC50 from 0.44 to 2.02 μM. Oral treatment with Jun12682 significantly improved survival and reduced lung viral loads and lesions in a SARS-CoV-2 infection mouse model, suggesting PLpro inhibitors are promising oral SARS-CoV-2 antiviral candidates.
One-Sentence Summary:
Structure-guided design of SARS-CoV-2 PLpro inhibitors with in vivo antiviral efficacy in a mouse model.
The COVID-19 pandemic is a timely call for the urgent need for orally bioavailable antivirals. The FDA has approved three direct-acting antivirals, including two viral RNA-dependent RNA polymerase inhibitors, remdesivir and molnupiravir, and one viral main protease (Mpro) inhibitor, nirmatrelvir (1). Remdesivir is administered by intravenous injection, and its use is limited to hospitalized patients. The clinical efficacy of remdesivir is also controversial (2). Molnupiravir is a mutagen and should not be used in pregnant women (3). Nirmatrelvir is co-administered with ritonavir, a CYP3A4 inhibitor, to improve the in vivo half-life (4). For this reason, Paxlovid, a combination of nirmatrelvir and ritonavir, has drug-drug interaction concerns. Ensiltrelvir is a second-generation Mpro inhibitor approved in Japan (5). Although it is highly efficacious in clinical trials (6), ensitrelvir is a potent CYP3A4 inhibitor that may lead to severe adverse drug-drug interactions with other medications (7, 8). Mutant SARS-CoV-2 viruses with resistance against remdesivir or nirmatrelvir have been identified from viral passage experiments in cell culture (9–11) and drug-treated COVID-19 patients (12–15). Therefore, additional antivirals with alternative mechanisms of action are urgently needed to combat drug-resistant and emerging SARS-CoV-2 variants.
The papain-like protease (PLpro) is one of the two viral cysteine proteases encoded by SARS-CoV-2. PLpro cleaves the viral non-structural (nsp) polyproteins at the nsp1/2, nsp2/3, and nsp3/4 junctions and is pivotal for viral replication (16). In addition, PLpro suppresses the host immune response through cleaving ubiquitin and interferon-stimulated gene 15 (ISG-15) modifications from host proteins (17–19). The SARS-CoV-2 PLpro sequence is 82.9% identical to SARS-CoV-1 PLpro. PLpro is highly conserved among SARS-CoV-2 variants (20), rendering it a high-profile antiviral drug target (19, 21). However, despite decades of medicinal chemistry optimization and high-throughput screening, no drug-like PLpro inhibitor has shown in vivo antiviral efficacy in SARS-CoV-2-infected animal models (20). PLpro substrates contain a consensus motif LXGG↓(N/K/X) (16). The S1 and S2 subsites of PLpro form a narrow tunnel for binding two glycines (22). The absence of binding pockets near the catalytic cysteine Cys111 presents a challenge in designing highly potent PLpro inhibitors. In this study, we describe the design of potent PLpro inhibitors by exploiting a novel drug-binding site that accommodates the ubiquitin Val70 side chain (Val70Ub). We validate PLpro as a viable drug target by demonstrating the in vivo antiviral efficacy of a designed PLpro inhibitor Jun12682 with oral administration in SARS-CoV-2 infected mice.
Discovery of the binding site for ubiquitin Val70 as a new drug binding site on PLpro
Before our study, noncovalent and covalent PLpro inhibitors were reported (20, 23). One potent noncovalent PLpro inhibitor is XR8–24, which has an IC50 of 0.56 μM in the fluorescence resonance energy transfer (FRET) enzymatic assay and an EC50 of 1.2 μM in the antiviral assay (24). Compound 7 (Cp7) is a rationally designed covalent PLpro inhibitor with a fumarate reactive warhead that inhibits PLpro with an IC50 of 0.094 μM and SARS-CoV-2 replication with an EC50 of 1.1 μM (25). Inspired by these results, we designed a hybrid covalent PLpro inhibitor Jun11313 by converting the naphthalene in Cp7 to 3-phenylthiophene (Fig. 1A); Jun11313 potently inhibited PLpro with an IC50 of 0.12 μM.
Fig. 1. X-ray crystal structure of the covalent inhibitor Jun11313 with SARS-CoV-2 PLpro and structure-based design of biarylphenyl SARS-CoV-2 PLpro inhibitors.

(A) Design of the hybrid covalent inhibitor Jun11313 based on XR8–24 and Cp7. (B) Atomic model of the Jun11313 (in green sticks and spheres) binding site in PLpro (light grey sticks, residues within a 5 Å distance of the inhibitor), with hydrogen bonds displayed as black dashed lines. Jun11313 polder map (an unbiased difference map (36)) is displayed as a grey mesh with 4σ contour. (C) Superposition of the PLpro-Jun11313 structure to the structure of the PLpro-XR8–24 complex (PDB 7LBS), with XR8–24 in yellow sticks and spheres, with the relevant residues for binding of both compounds indicated. (D) Superimposed X-ray crystal structures of SARS-CoV-2 PLpro with Jun11313 (green) (PDB: 8UVM), XR8–24 (yellow) (PDB: 7LBS), and ubiquitin (orange) (PDB: 6XAA). (E) Generic chemical structure of the designed biarylphenyl PLpro inhibitor. Critical interactions are highlighted. (F) Flow chart for the lead optimziation of PLpro inhibitors. Jun12682 was selected as the in vivo lead candidate.
The lead covalent compound Jun11313 was co-crystallized with SARS-CoV-2 PLpro to visualize the key interactions for binding. We determined the co-crystal structure at 2.85 Å resolution (PDB: 8UVM). As expected, the fumarate ester electrophile forms a covalent bond with the catalytic Cys111 (Fig. 1B). Jun11313 forms hydrogen bonds with main-chain atoms of Leu162, Gly163, Tyr268, and Gly271, as well as with side-chain atoms of Trp106 and His272. Jun11313 also makes extensive van der Waals contacts with Leu162, Tyr264, Pro247, Pro248, and Thr301 (Fig. 1B).
Surprisingly, the thienyl group in Jun11313 is oriented towards the opposite side of the BL2 groove compared to the pyrrolidine-substituted thienyl group in XR8–24 (Fig. 1C). The phenylthiophene in XR8–24 makes extensive contacts with the BL2 groove, namely van der Waals interactions with residues surrounding the cavity (Pro248, Tyr264, and Tyr268; Fig. 1C). In comparison, the phenylthienyl group of Jun11313 is oriented towards Pro247, making van der Waals contacts with both Pro248 and Pro247, and CH–π and S–π interactions with Met208 (Fig. 1B). While the strength of these interactions is difficult to estimate and has been understudied in drug design (26, 27), here they may be the culprit for Jun11313’s unexpected conformation (Fig. 1B). If the methyl pyrrolidine group in XR8–24 were arranged as the phenylthienyl group of Jun11313, it should orient in a very solvent-exposed region to avoid a clash with Met208 (fig. S1A).
Superimposition of PLpro structures complexed with ubiquitin and Jun11313 revealed that the thienyl group occupies the same hydrophobic site as Val70 from ubiquitin (Fig. 1D). Therefore, we designate this pocket as the Val70Ub site. The comparison of the co-crystal structures of PLpro with Jun11313 and with ubiquitin suggests that this site is essential for binding both the ubiquitin substrate and this inhibitor. Similarly, the superimposition of PLpro structures complexed with ISG15 showed that the thienyl group interacts with the analogous Asn151-Leu152 site from ISG15 (fig. S1B).
Rational design of biarylphenyl PLpro inhibitors
The unexpected binding pose of Jun11313 in PLpro led us to hypothesize that potent PLpro inhibitors can be designed by simultaneously engaging the BL2 groove pocket and the Val70Ub hydrophobic site (Fig. 1E). It is noteworthy that the Val70Ub site has not been explored for PLpro inhibitor design (20). We designed and synthesized a library of 85 biarylphenyl benzamide compounds (Fig. S2). Aryl substitutions were installed on the 3 and 5 positions of the phenylethylamine ring to engage hydrophobic interactions with residues in the BL2 groove and the ubiquitin Val70Ub binding site (Fig. 1E). In addition, diverse amines were installed on the meta-position of the benzoic acid to engage electrostatic interactions with Glu167 (24). The central 1-phenylethyl benzamide core structure was kept intact to maintain the critical hydrogen bonds and π-π interactions with the BL2 loop. All synthesized compounds were initially tested in the FRET-based enzymatic assay and the cytotoxicity assay in Vero E6 cells (Fig. 1F). Promising lead compounds were then tested in a secondary FlipGFP PLpro assay and SARSCoV-2 antiviral assay (28). FlipGFP PLpro is a cell-based assay that validates intracellular PLpro target engagement (22). Next, leads were profiled for in vitro microsomal stability and in vivo oral pharmacokinetic (PK) properties in mice. Jun12682 was finalized as the in vivo lead for the SARS-CoV-2 infection mouse model study.
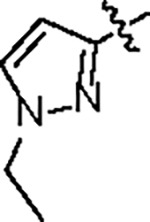
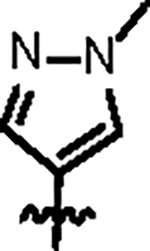
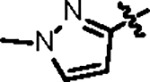
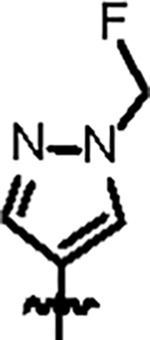
A complete list of the designed biarylphenyl PLpro inhibitors is shown in fig. S2, with representative examples in Table 1. Among the 85 compounds tested in the FRET assay, 26 had IC50 < 100 nM, 42 had IC50 between 100 – 200 nM, and 14 had IC50 between 200 – 400 nM. The control compound GRL0617 had IC50 values of 1.92 μM. The inhibitory constant Ki was determined for potent compounds (Table 1). The first designed compound, Jun11875, inhibited PLpro with a Ki of 13.2 nM, a 104-fold improvement over GRL0617 (Ki = 1,374 nM). However, thiophene-containing compounds were generally cytotoxic in Vero E6 cells (CC50 < 20 μM). Next, we examined several symmetric and asymmetric five and six-membered aromatic substitutions at the 3 and 5-positions to mitigate cellular cytotoxicity while maintaining enzymatic inhibition. Among the list of heterocycle substitutions examined, pyrazole was shown to confer potent PLpro inhibition and reduced cellular cytotoxicity. It is noted that despite the potent PLpro enzymatic inhibition for Jun1247 (IC50 = 165.3 nM), Jun121210 (IC50 = 81.9 nM), Jun121911 (IC50 = 73.2 nM), Jun12208 (IC50 = 91.6 nM), and Jun12242 (IC50 = 90.5 nM), their cellular activities were moderate to weak as shown by the FlipGFP assay (EC50 > 10 μM) (fig. S2), which might due to poor membrane permeability. The most potent compounds Jun12199 and Jun12197 had EC50 of 0.8 and 0.6 μM, respectively, more than 20-fold improved from GRL0617 (EC50 = 22.4 μM). The in vivo lead Jun12682 inhibited PLpro with a Ki of 37.7 nM and displayed an EC50 of 1.1 μM in the FlipGFP PLpro assay.
Table 1.
Representative biarylbenzamide series of SARS-CoV-2 PLpro inhibitors.
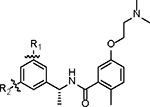
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | IC50 (nM) | Ki (nM) | CC50 (μM) | FlipGFP EC50 (μM) | Antiviral EC50 (μM)* | Microsomal Stability T1/2 (min) | PDB code |
| Jun11875 |
|
|
66.2 ± 2.0 | 13.2 ± 2.0 | 4.5 ± 1.6 | --- | --- | --- | |
| Jun12129 |
|
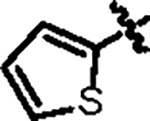
|
90.9 ± 4.6 | 75.5 ± 2.0 | 8.3 ± 1.2 | --- | --- | --- | 8UUY |
| Jun12303 |
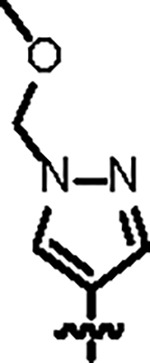
|
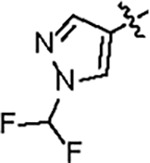
|
121.5 ± 4.0 | 88.2 ± 6.0 | 56.8 ± 7.4 | 2.6 ± 0.2 | 0.32 | 136 | 8UUG |
| Jun12763 |
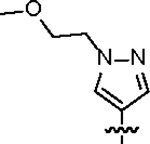
|
|
164.6 ± 23.0 | 37.2 ± 4.0 | > 125 | 2.0 ± 0.3 | 0.96 | --- | |
| Jun12395 |
|
|
121.9 ± 8.0 | 47.1 ± 4.0 | 109.4 ± 16.2 | 2.9 ± 0.3 | 1.15 | 88.3 | |
| Jun12713 |
|
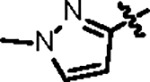
|
86.7 ± 6.0 | 34.0 ± 3.0 | 107.2 ± 6.2 | 2.7 ± 0.6 | 0.86 | --- | |
| Jun12602 |
|
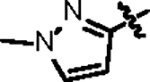
|
116.7 ± 9.2 | 46.9 ± 3.0 | > 125 | 14.7 ± 2.5 | 0.80 | > 145 | |
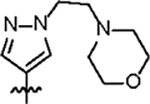
| Jun11941 |
|
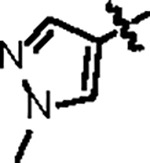
|
151.4 ± 4.0 | 34.3 ± 3.0 | 54.8 ± 8.3 | 1.8 ± 0.2 | 0.31 | > 145 | 8UUF |
| Jun12162 |
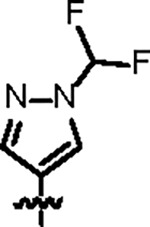
|
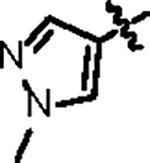
|
98.3 ± 7.0 | 33.6 ± 3.0 | 42.3 ± 2.3 | 2.1 ± 0.2 | 0.23 | 76.7 | 8UUU |
| Jun12199 |
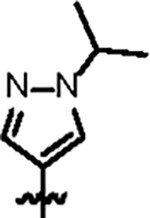
|
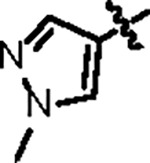
|
108.8 ± 10.2 | 47.6 ± 3.0 | 63.2 ± 11.3 | 0.8 ± 0.1 | 0.45 | 79.7 | 8UUH |
| Jun12197 |
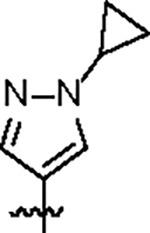
|
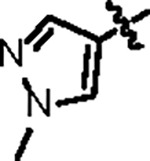
|
102.7 ± 10.2 | 33.2 ± 3.0 | 43.7 ± 5.1 | 0.6 ± 0.1 | 0.35 | 116.0 | 8UUV |
| Jun12351 |
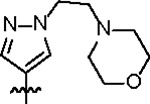
|
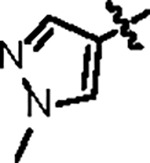
|
98.8 ± 5.2 | 29.3 ± 3.0 | > 125 | 2.0 ± 0.4 | 0.59 | > 145 | |
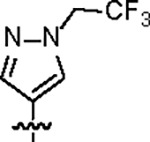
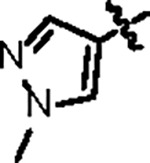
| Jun12603 |
|
|
112.2 ± 6.0 | 39.8 ± 4.0 | 61.0 ± 19.3 | 2.4 ± 0.4 | 0.50 | --- | |
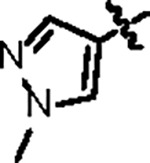
| Jun12145 |

|
|
108.5 ± 6.2 | 35.2 ± 2.0 | 31.3 ± 5.4 | 1.3 ± 0.3 | 0.58 | 26.3 | 8UUW |
| Jun12682 |
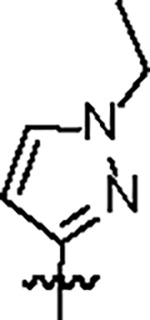
|
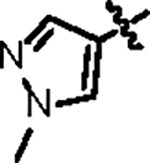
|
106.8 ± 5.0 | 37.7 ± 3.0 | 61.3 ± 4.5 | 1.1 ± 0.1 | 0.42 | 82.4 | 8UOB |
| GRL0617 |
|
1918.0 ± 150.0 | 1374 ± 58 | 107.3 ± 10.9 | 22.4 ± 2.3 | > 20 | --- | 7JRN | |
The positive control nirmatrelvir had an EC50 of 0.04 μM in the SARS-CoV-2 antiviral assay.
Prioritized lead compounds were tested in the SARS-CoV-2 antiviral assay and had EC50 from 0.23 to 1.15 μM. Jun12682 had EC50 values of 0.42 and 0.51 μM in the icSARS-CoV-2-nLuc reporter virus and plaque assays, respectively (fig. S3). To examine the potential of PLpro inhibitors in inhibiting SARS-CoV-2 variants and drug-resistant mutants, we selected two PLpro inhibitors Jun11941 and Jun12682 to test against SARS-CoV-2 delta and omicron variants and three recombinant SARS-CoV-2 viruses that are resistant to nirmatrelvir, rNsp5-S144M, rNsp5-L50F/E166V, and rNsp5-L50F/E166V/L167F (Fig. 2, A to C). S144M and E166V are nirmatrelvir-resistant mutations identified from enzymatic assays and viral passage experiments (9, 10). Jun11941 and Jun12682 showed consistent antiviral activity against these viruses with EC50 fold-increases less than 1.5 and 2.0, respectively, compared to wild-type (WT). In comparison, the rNsp5-S144M, rNsp5-L50F/E166V, and rNsp5-L50F/E166V/L167F showed significant resistance to nirmatrelvir with EC50 fold-increases of 12.5, 24.2, and 21.7, respectively, compared to WT.
Fig. 2. Antiviral activity of PLpro inhibitors against SARS-CoV-2 variants and mechanistic studies of Jun12682.

Antiviral activity of nirmatrelvir (A), Jun11941 (B), and Jun12682 (C) against SARS-CoV-2 WA1 strain (WT) (USA-WA1/2020), Omicron strain (BA.2 strain, lineage B.1.1.529, BA.2; HCoV-19/USA/CO-CDPHE-2102544747/2021), Delta strain (Lineage B.1.617.2; hCoV-19/USA/MD-HP05647/2021), and nirmatrelvir-resistant strains rNsp5-S144M, rNsp5-L50F/E166V, and rNsp5-L50F/E166V/L167F. (D) IC50 curve of Jun12682 in inhibiting SARS-CoV-2 PLpro hydrolysis of Ub-AMC. (E) Ki curve of Jun12682 in inhibiting SARS-CoV-2 PLpro hydrolysis of Ub-AMC. (F) IC50 curve of Jun12682 in inhibiting SARS-CoV-2 PLpro hydrolysis of ISG15-AMC. (G) Ki curve of Jun12682 in inhibiting SARS-CoV-2 PLpro hydrolysis of ISG15-AMC. (H) Counter screening of Jun12682 in inhibiting USP7 hydrolysis of Ub-AMC. (I) Counter screening of Jun12682 in inhibiting USP14 hydrolysis of Ub-AMC. (J) Differential scanning fluorimetry assay of Jun12682 and GRL0617 in stabilizing the SARS-CoV-2 PLpro.
Mechanism of action of Jun12682 in inhibiting SARS-CoV-2 PLpro
PLpro is known to antagonize the host innate immune response upon viral infection by hydrolyzing the isopeptide bond between ubiquitin and ISG-15 to lysine side chains of host proteins (17, 18). To characterize whether Jun12682 inhibits the deubiquitinating and deISGylating activities of SARS-CoV-2 PLpro, we performed the PLpro enzymatic assay using Ub-AMC and ISG15-AMC substrates (22). Jun12682 showed potent enzymatic inhibition with Ki values of 63.5 and 38.5 nM in the ubiquitin-AMC (7-amino-4-methylcoumarin) and ISG15-AMC FRET assays (Fig. 2, D to G). The results are consistent with GRL0617 and XR8–24 (22, 24). To profile the off-target effect, Jun12682 was tested against two closest human structural homologs of PLpro, USP7, and USP14 (24, 29). Jun12682 displayed no inhibition of USP7 and USP14-catalyzed Ub-AMC hydrolysis at up to 40 μM (Fig. 2, H and I). Jun12682 also showed dose-dependent stabilization of SARS-CoV-2 PLpro and was more potent than GRL0617 in the differential scanning fluorimetry assay (Fig. 2J).
X-ray crystal structures of SARS-CoV-2 PLpro with inhibitors
The X-ray co-crystal structures of SARS-CoV-2 PLpro were solved for eight biarylphenyl PLpro inhibitors, Jun11941, Jun12129, Jun12303, Jun12162, Jun12199, Jun12197, Jun12145, and Jun12682 (resolution range of 2.5–3.1 Å, Fig. 3, table S1). The phenylthienyl group of Jun11313 binds to the Val70Ub site of SARS-CoV-2 PLpro in the region where residues Val70Ub and Leu71Ub at the end of a β sheet in ubiquitin interact with PLpro (an analogous situation happens with residues Asn151 and Leu152 of ISG-15, fig. S1B) (Fig. 1B). This is a unique property of Jun11313 due to the unusual binding conformation of the phenylthienyl group, which we have subsequently exploited in a new compound series (Fig. 3), exemplified by Jun12682. The 2.52 Å resolution co-crystal structure of PLpro with Jun12682 reveals the “best of both worlds”, with the N-ethyl and N-methyl pyrazole substituents in the phenyl moiety binding toward the Met208/Pro247 direction (Val70Ub site) and the Pro248/Tyr264/Tyr268 direction (BL2 groove), respectively (Fig. 3A). While Met208 remains involved in CH–π and S–π interactions with the pyrazole ring, it also displays a van der Waals contact with the methyl substituent. Moreover, the phenyl group of Jun12682 makes a π–π interaction with the side chain of Tyr264 (Fig. 3A). Fig. 3B–I shows the unbiased electron density of PLpro inhibitors Jun12145, Jun12303, Jun12199, Jun12162, Jun12129, Jun12197, and Jun11941, in a similar conformation to Jun12682. The disubstituted phenyl group has two alternative conformations in the Jun12145 and Jun12129 complex structures, as indicated by the refined electron density (Fig. 3C, G; fig. S4). Despite the differences between compounds, the interactions depicted in Fig. 3A between PLpro and Jun12682 are conserved in all complexes (fig. S4). The crystallographic data and refinement statistics are listed in table S1.
Fig. 3. X-ray crystal structures of SARS-CoV-2 PLpro with biarylphenyl PLpro inhibitors.

(A) Atomic model of the Jun12682 (in orange sticks and spheres) binding site in SARS-CoV-2 PLpro (residues within 5 Å of the inhibitor are shown in light gray sticks), with hydrogen bonds displayed in black dashed lines, van der Waals contacts as red dashed lines, and π–π interactions as light green dashed lines. (B-I) Gallery of the polder maps of the indicated inhibitors, displayed as a grey mesh with contour levels between 2 and 4σ. The PLpro inhibitory constant Ki in the FRET enzymatic assay and the SARS-CoV-2 antiviral activity EC50 in Caco-2 cells were shown for each compound.
In vitro and in vivo pharmacokinetic (PK) profiling identified Jun12682 as an in vivo lead candidate
PLpro inhibitors with potent SARS-CoV-2 antiviral activity (EC50 ≤ 1 μM) and a high selectivity index (SI > 50) were selected for in vitro microsomal stability assay and in vivo oral PK studies in mice. Most pyrazole-containing PLpro inhibitors showed high stability in the mouse microsomal stability assay (T1/2 > 60 min) (Table 1). Next, ten compounds were advanced to the in vivo oral snap PK in C57BL/6J mice (Fig. 4A, B; table S2). Compounds were dosed to 3 male C57BL/6J mice per group at 50mg/kg through oral gavage, and blood samples were collected at 0.5, 1, 3, and 5 h, and the drug concentration was quantified by LC-MS/MS. Jun12199 and Jun12682 showed the highest in vivo plasma concentrations. Given the faster absorption of Jun12682 compared to Jun12199, Jun12682 was selected as an in vivo lead candidate. A 24-hour in vivo PK study was conducted to determine the oral bioavailability of Jun12682 (Fig. 4C; table S3). Jun12682 had a rapid absorption after p.o. administration (50 mg/kg) and reached the maximum plasma concentration at 1.67 h (Tmax), with a peak plasma concentration (Cmax) of 4537 ng/mL. The clearance of Jun12682 was moderate, with a half-life (t1/2) of 2.01 h (table S3). The plasma drug concentration was above the antiviral EC90 value (1.59 μM) for 7 h. The oral bioavailability of Jun12682 was 72.8%.
Fig. 4. In vitro and in vivo PK profiling of PLpro inhibitors, and in vivo antiviral efficacy of Jun12682.

(A) Plasma drug concentration of Jun12199, Jun12197, Jun12713, Jun12603, and Jun11941 in C57BL/6J mice (6 – 8 weeks old) following p.o. administration of 50 mg/kg of compound in 0.5% methylcellulose and 2% Tween 80 in water. (B) Plasma drug concentration of Jun12682, Jun12763, Jun12395, Jun12602, and Jun12351 in C57BL/6J mice (6 – 8 weeks old) following p.o. administration of 50 mg/kg of compound in 0.5% methylcellulose and 2% Tween 80 in water. (C) Plasma drug concentration of Jun12682 in C57BL/6J mice (6 – 8 weeks old) following p.o. administration of 50 mg/kg and i.v. injection of 10 mg/kg. (D) In vitro PK parameters of Jun12682. (E) Experimental design for the 5-day treatment experiment. Ten mice per group were intranasally (IN) inoculated with 5,600 PFU of SARS2-N501YMA30 and subsequently orally administered 500 mg/kg Jun12682 or vehicle twice a day (BID) for five days (BID_5). (F) Body weight loss and (G) survival rate of the BID_5 mice experiment. (H) Experimental design for the 3-day treatment experiment. Ten mice per group were intranasally (IN) inoculated with 5,600 PFU of SARS2-N501YMA30 and subsequently orally administered 125, 250 mg/kg Jun12682 or vehicle BID for three days (BID_3). (I) Body weight loss and (J) survival rate of the BID_3 mice experiment. Data in F, G, I, and J are pool of two independent experiments (n=10) and are shown as mean ± s.e.m. The p values in G and J were determined using a log-rank (Mantel–Cox) test. (K) Viral titers in lungs collected at 2 and 4 DPI from vehicle- or 250 mg/kg Jun12682-treated mice (n=5 each group). Data are mean ± s.e.m and analyzed with unpaired t test with Welch’s correction. *, p< 0.05; ***, p<0.001. (L-M) Quantitative PCR analysis of viral nucleocapsid gene (L) and cellular cytokines (M) in lungs collected at 2 DPI from vehicle- or 250mg/kg Jun12682-treated mice (n=5 each group). (N-Q) Lungs collected at 4 DPI from vehicle- or 250mg/kg Jun12682-treated mice (n=5 each group) were stained with haematoxylin and eosin (H&E) (N) or immunostained for SARS-CoV-2 nucleocapsid (P), and the pathological lesions and staining were quantified (O and Q, respectively). N, H&E stained lungs from vehicle-treated infected mice exhibited airway edema (asterisks), hyaline membranes (HM, arrowheads), and interstitial thickness (number sign). Scale bars, 100 μm (top) and 50 μm (bottom). O, Summary scores of lung lesions (n = 5 for each group). P, Lungs from vehicle- or Jun12682-treated mice (n = 5 for each treatment group) were immunostained to detect SARS-CoV-2 nucleocapsid protein. Scale bars, 100 μm (top) and 50 μm (bottom). Q, Summary scores of nucleocapsid immunostaining of lungs. Data in L, M, O and Q are mean ± sem and analyzed with unpaired t test with Welch’s correction. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Further profiling of the in vitro PK properties revealed that Jun12682 was highly stable in the human microsomes (T1/2 = 131.9 min, CLint(mic) = 10.5 μL/min/mg) and had high selectivity for CYP1A2, 2C9, 2C19, 2D6, and 3A-M (IC50 > 50.0 μM) (Fig. 4D). Jun12682 had a kinetic solubility of 180 μM and a thermodynamic solubility greater than 5mg/ml. The mouse plasma protein binding was 85.4%. Overall, Jun12682 showed favorable in vitro and in vivo PK properties amenable for the in vivo antiviral efficacy study.
In vivo antiviral efficacy of Jun12682 in a SARS-CoV-2 infection mouse model
To assess the in vivo antiviral efficacy of Jun12682, we utilized a lethal SARS-CoV-2 mouse model described in (30). This model involves infecting young Balb/c mice (9 to 12 weeks old) with a mouse-adapted SARS2-N501YMA30, resulting in severe lung disease resembling human patients’ lung injuries. This model has been widely used for evaluating SARS-CoV-2 therapeutics and vaccine candidates (31–34). To this end, young Balb/c mice were intranasally infected with 5,600 PFUs of SARS2-N501YMA30 and orally administered Jun12682 at 500 mg/kg twice daily for 5 days (Fig. 4E). The weight loss plot (Fig. 4F) illustrates that mice administered with the vehicle experienced rapid body weight loss exceeding 20%, leading to a 100% fatality rate by 5 DPI (day post inoculation) (Fig. 4G). In contrast, most mice treated with Jun12682 exhibited reduced weight loss, resulting in a significantly improved survival rate (0% vs. 70%, p < 0.0001) (n = 10, two independent studies). To further evaluate Jun12682’s in vivo efficacy, two lower dosages (125, 250 mg/kg) were tested with reduced dosing times from 5 days to 3 days twice daily (Fig. 4H). Mice treated with a dose of 250 mg/kg showed an average of 10% maximum weight loss, providing evident protection from infection compared to the vehicle- and 125 mg/kg-treated groups, which exhibited over 20% weight loss (Fig. 4I). Survival analyses demonstrated that Jun12682-treated mice had statistically higher survival rates compared to the vehicle group (0% survival): 125 mg/kg (20%, p = 0.0428), and 250 mg/kg (100%, p < 0.0001) (Fig. 4J). The three-day treatment of 250 mg/kg dose conferred even better protection than the five-day treatment of 500 mg/kg dose, possibly due to reduced drug toxicity. Lung viral load analyses revealed that at 2 DPI, the vehicle-treated mice had robust infections in the lungs (mean lung titer of log10 9.17 ± 0.124 PFUs/ml), while the 250 mg/kg Jun12682-treated mice had statistically lower lung viral titers (mean lung titers of log10 8.87 ± 0.194 PFUs/ml, p = 0.0199) (Fig. 4K). The antiviral effect of the 250 mg/kg treatment was more evident at 4 DPI with over a log viral titer reduction comparing to the vehicle treatment (mean lung titers of log10 7.05 ± 0.401 and log10 5.73 ± 0.528 PFUs/ml for the vehicle and 250 mg/kg groups, respectively, p = 0.00247) (Fig. 4K), corroborating the weight loss and survival data (Fig. 4, I and J).
Quantitative PCR analysis of the RNA samples extracted from 2 DPI mice lungs showed that the 250mg/kg treatment significantly reduced the viral N gene level (Fig. 4L) and the expression of multiple inflammatory cytokines, including IFN-β, IL-1β, IL-6, and CXCL10 (35) (Fig. 4M). Histopathological analysis revealed that lungs from the vehicle-treated, SARS2-N501YMA30–infected mice at 4 DPI exhibited multifocal pulmonary lesions, including lymphocytic perivascular cuffing, pulmonary edema, hyaline membrane formation, and interstitial thickening and inflammation compared with Jun12682-treated mice (Fig. 4N, O; fig. S5). Immunohistochemical analysis using a monoclonal antibody to detect SARS-CoV-2 nucleocapsid (N) in the lungs demonstrated strong and expansive antigen staining in lungs from vehicle-treated, infected mice, whereas Jun12682 treatment considerably decreased viral antigen staining levels with a few sporadic positive cells (Fig. 4P, Q; fig. S6), consistent with the lung viral titer results (Fig. 4K). Mock-infected lungs were negative for nucleocapsid staining. Overall, the reduced viral replication in the lung and the expression of inflammatory cytokines (Fig. 4K to M) corroborates with the reduced lung inflammation and N protein staining at 4 DPI (Fig. 4N to Q). In summary, these findings demonstrate that oral administration of our rationally designed PLpro inhibitor Jun12682 efficiently inhibited SARS-CoV-2 replication and mitigated SARS-CoV-2 induced lung lesions in vivo, and it represents a promising candidate for further development as orally bioavailable SARS-CoV-2 antivirals. PLpro inhibitors can be used alone or in combination with existing RdRp and Mpro inhibitors to combat SARS-CoV-2 variants and drug-resistant mutants.
Supplementary Material
Acknowledgments:
We thank Shannon Cowan and Caden Miller at the Immunopathology Core of the Oklahoma Center for Respiratory and Infectious Diseases for their technical assistance.
Funding:
National Institutes of Health grant U19AI171110 (J.W., E.A.)
National Institutes of Health grant R01AI158775 (J.W., X.D.)
National Institute of Food and Agriculture grants 2023-67015-39096 and 2023-70432-39482 (X.D.)
USDA-ARS Non-Assistance Cooperative Agreement 58-5030-3-047 (X.D.)
Center for Advancement of Science & Technology (OCAST) grant HR23-096 (X.D.)
Footnotes
Competing interests: Rutgers, the State University of New Jersey, has applied for PCT patents that cover the PLpro inhibitors reported in this manuscript and related compounds.
Data and materials availability:
All data are available in the main text or the supplementary materials. The PDB accession numbers for the coordinates of SARS-CoV-2 PLpro in complex with PLpro inhibitors are 8UUW (Jun12145), 8UUY (Jun12129), 8UUV (Jun12197), 8UUU (Jun12162), 8UUH (Jun12199), 8UUG (Jun12303), 8UUF (Jun11941), 8UOB (Jun12682), and 8UVM (Jun11313).
References and notes
- 1.Li G., Hilgenfeld R., Whitley R., De Clercq E., Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov 22, 449–475 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Consortium W. H. O. S. T., Remdesivir and three other drugs for hospitalised patients with COVID-19: final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 399, 1941–1953 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saravolatz L. D., Depcinski S., Sharma M., Molnupiravir and Nirmatrelvir-Ritonavir: Oral Coronavirus Disease 2019 Antiviral Drugs. Clin Infect Dis 76, 165–171 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owen D. R. et al. , An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Unoh Y. et al. , Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J Med Chem 65, 6499–6512 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukae H. et al. , Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin Infect Dis 76, 1403–1411 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu R. et al. , Evaluation of the Drug-Drug Interaction Potential of Ensitrelvir Fumaric Acid with Cytochrome P450 3A Substrates in Healthy Japanese Adults. Clin Drug Investig 43, 335–346 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimizu R. et al. , Safety, Tolerability, and Pharmacokinetics of the Novel Antiviral Agent Ensitrelvir Fumaric Acid, a SARS-CoV-2 3CL Protease Inhibitor, in Healthy Adults. Antimicrob Agents Chemother 66, e0063222 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iketani S. et al. , Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y. et al. , Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir. ACS Cent Sci 9, 1658–1669 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens L. J. et al. , Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci Transl Med 14, eabo0718 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gandhi S. et al. , De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Nat Commun 13, 1547 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuckerman N. S., Bucris E., Keidar-Friedman D., Amsalem M., Brosh-Nissimov T., Nirmatrelvir resistance - de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure - a case report. Clin Infect Dis doi: 10.1093/cid/ciad494, (2023). [DOI] [PubMed] [Google Scholar]
- 14.Sjaarda C. P. et al. , Prevalence of Low-Frequency, Antiviral Resistance Variants in SARS-CoV-2 Isolates in Ontario, Canada, 2020–2023. JAMA Netw Open 6, e2324963 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirotsu Y. et al. , Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med 4, 813–824.e4 (2023). [DOI] [PubMed] [Google Scholar]
- 16.Rut W. et al. , Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci Adv 6, eabd4596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin D. et al. , Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wydorski P. M. et al. , Dual domain recognition determines SARS-CoV-2 PLpro selectivity for human ISG15 and K48-linked di-ubiquitin. Nat Commun 14, 2366 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baez-Santos Y. M., St John S. E., Mesecar A. D., The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115, 21–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan H., Hu Y., Jadhav P., Tan B., Wang J., Progress and Challenges in Targeting the SARS-CoV-2 Papain-like Protease. J Med Chem 65, 7561–7580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei J., Kusov Y., Hilgenfeld R., Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res 149, 58–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma C. et al. , Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent Sci 7, 1245–1260 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh A. K., Mishevich J. L., Mesecar A., Mitsuya H., Recent Drug Development and Medicinal Chemistry Approaches for the Treatment of SARS-CoV-2 Infection and COVID-19. ChemMedChem 17, e202200440 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Z. et al. , Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J Med Chem 65, 2940–2955 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanders B. C. et al. , Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat Commun 14, 1733 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platzer G. et al. , PI by NMR: Probing CH-π Interactions in Protein-Ligand Complexes by NMR Spectroscopy. Angewandte Chemie 59, 14861–14868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beno B. R., Yeung K. S., Bartberger M. D., Pennington L. D., Meanwell N. A., A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J Med Chem 58, 4383–4438 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Tan H., Hu Y., Wang J., FlipGFP protease assay for evaluating in vitro inhibitory activity against SARS-CoV-2 M(pro) and PL(pro). STAR Protoc 4, 102323 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klemm T. et al. , Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 39, e106275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong L. R. et al. , Eicosanoid signalling blockade protects middle-aged mice from severe COVID-19. Nature 605, 146–151 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Kafrawy S. A. et al. , SARS-CoV-2-specific immunoglobulin Y antibodies are protective in infected mice. PLoS Pathog 18, e1010782 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L. et al. , Viral anti-inflammatory serpin reduces immuno-coagulopathic pathology in SARS-CoV-2 mouse models of infection. EMBO Mol Med 15, e17376 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J. et al. , RBD-mRNA vaccine induces broadly neutralizing antibodies against Omicron and multiple other variants and protects mice from SARS-CoV-2 challenge. Transl Res 248, 11–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fumagalli V. et al. , Nirmatrelvir treatment of SARS-CoV-2-infected mice blunts antiviral adaptive immune responses. EMBO Mol Med 15, e17580 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nobori H. et al. , Efficacy of ensitrelvir against SARS-CoV-2 in a delayed-treatment mouse model. J Antimicrob Chemother 77, 2984–2991 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liebschner D. et al. , Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol 73, 148–157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. The PDB accession numbers for the coordinates of SARS-CoV-2 PLpro in complex with PLpro inhibitors are 8UUW (Jun12145), 8UUY (Jun12129), 8UUV (Jun12197), 8UUU (Jun12162), 8UUH (Jun12199), 8UUG (Jun12303), 8UUF (Jun11941), 8UOB (Jun12682), and 8UVM (Jun11313).