

Abstract
During early telencephalic development, intricate processes of regional patterning and neural stem cell (NSC) fate specification take place. However, our understanding of these processes in primates, including both conserved and species-specific features, remains limited. Here, we profiled 761,529 single-cell transcriptomes from multiple regions of the prenatal macaque telencephalon. We deciphered the molecular programs of the early organizing centers and their cross-talk with NSCs, revealing primate-biased galanin-like peptide (GALP) signaling in the antero-ventral telencephalon. Regional transcriptomic variations were observed along the fronto-temporal axis during early stages of neocortical NSC progression and in neurons and astrocytes. Additionally, we found that genes associated with neuropsychiatric disorders and brain cancer risk might play critical roles in the early telencephalic organizers and during NSC progression.
One-Sentence Summary
Single-cell transcriptomics reveals molecular programs underlying regionalization of macaque telecephalon.
The development of the telencephalon entails a complex interplay of molecualr processes that govern the specification of distinct regions by early organizing centers and the commitment of radial glial cells (RG), which function as neural stem cells (NSCs) (1-6). In the pallium, RG generate glutamatergic excitatory neurons, eventually becoming gliogenic. Whereas these mechanisms have been extensively characterized in rodents (7), they remain elusive in primates (8, 9), limiting our understanding of the origins and dysfunctions of higher-order cognitive abilities.
Substantial progress has been made in elucidating the gene networks that guide human and non-human primate brain development by bulk tissue (10-14) and single-cell genomic profiling (15-17). Nonetheless, the early molecular events governing the spatiotemporal progression of NSCs and the diversification of telencephalic regions and cortical areas, as cells traverse neuronal and glial trajectories, are not fully understood.
We conducted single-cell RNA-sequencing on more than 761,000 cells of multiple regions of prenatal rhesus monkey telencephalon, ranging from the early phases when the organizers pattern different regional anlage before neurogenesis through to mid-gliogenesis. We performed comparison with mouse datasets revealing primate-biased expression of GALP neuropeptide in the antero-ventral organizer and evaluated its function in brain organoids. In addition, we defined the gene expression cascades underlying the early spatial divergence of the NSCs and the regional specification of cortical neurons and glia. Finally, we mapped the developmental expression of neuropsychiatric disorder- and brain cancer-associated genes showing that risk genes might have putative early roles in the telencephalic organizers and across NSC progression.
Spatiotemporal transcriptomic characterization of prenatal macaque telencephalic cells
Single-cell RNA-sequencing (scRNA-seq) was conducted on 82 samples collected from multiple prospective regions of 12 prenatal rhesus macaque telencephalons, from embryonic day (E)37, prior to neurogenesis, till mid-gliogenesis at E110 (14) (Fig. 1A; fig. S1A). The ganglionic eminence (GE), anterior (A)/frontal (FR), dorso-lateral (DL)/putative motor-somatosensory (MS), posterior (P, temporo-occipital)/occipital (OC) and putative temporal telencephalic walls were recognized at E37-E78. More refined areas were distinguished at E93 and E110 from GE, prospective prefrontal (PFC), primary motor (M1C), parietal (Par), primary visual (V1C), insula (Ins) and temporal (Tem) cortical walls (18). Stringent quality control resulted in 761,529 high-quality cells (fig. S1B and table S1).
Fig. 1. Cell atlas of macaque telencephalon.
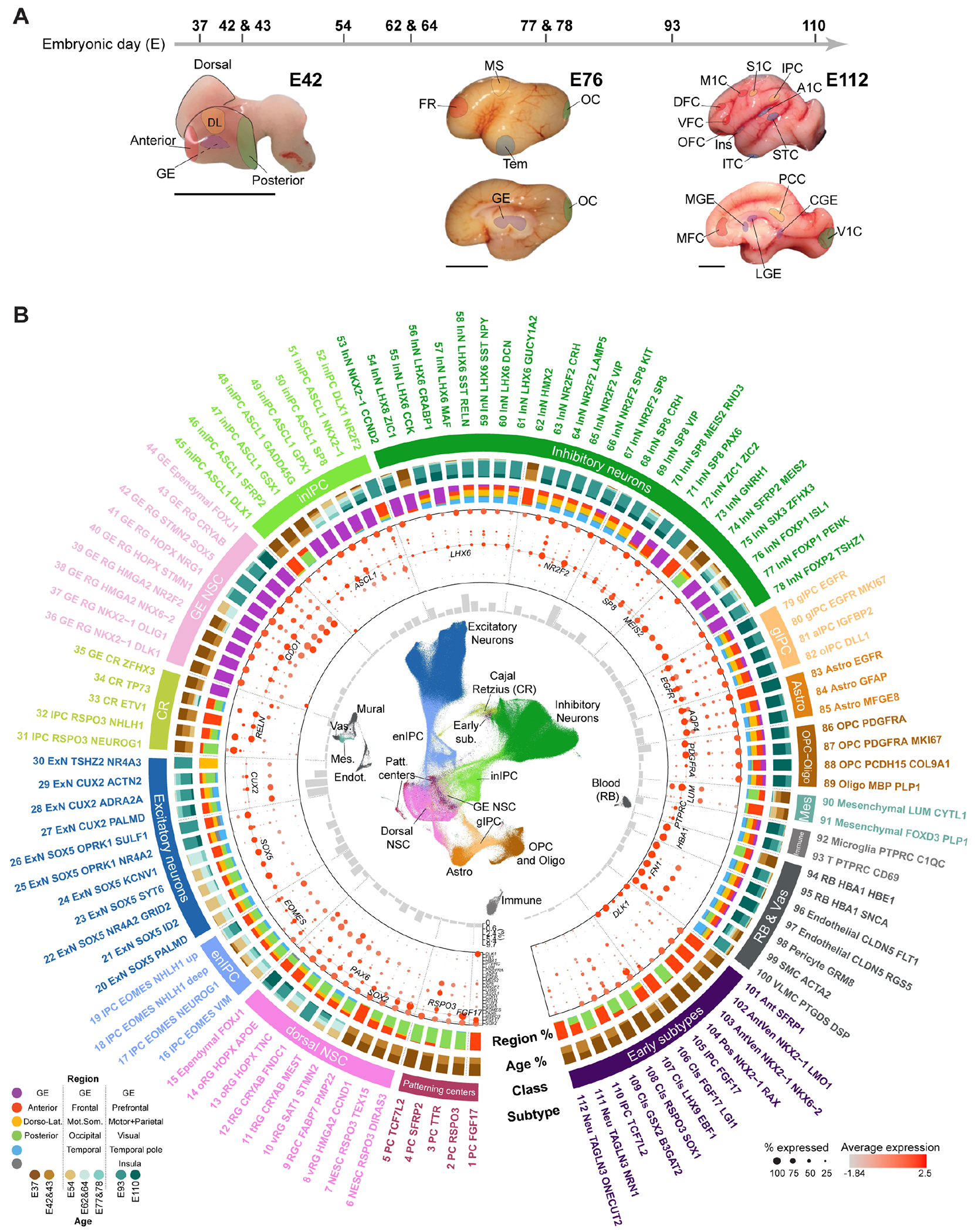
(A) Age scheme and representative macaque brains illustrating the regions/areas dissected. Scale bar: 1 cm. (B) From innermost to outmost: UMAP visualizing cell classes; subtype proportions; marker expression; region and age composition; cell classes; subtypes. The region nomenclature is based on the temporal development of the telencephalon (bottom left).
Unsupervised clustering and marker gene profiling identified 112 transcriptomically-defined cell subtypes, including progenitors of putative telencephalic patterning centers, dorsal and ventral neural stem cells (NSCs) traversing excitatory and inhibitory neurogenic lineages, respectively, gliogenic lineages and non-neural cells, which paralleled the sample spatiotemporal information on the uniform manifold approximation and projection (UMAP) layout of cells (Fig. 1B and fig. S2, A-E; table S2).
This resource depicting single-cell spatiotemporal transcriptomic dynamics of the developing macaque telencephalon is accessible at: http://resources.sestanlab.org/devmacaquebrain.
Transcriptomic signatures of macaque telencephalic patterning centers
We identified early domain-specific SOX2+/NES+ neuroepithelial progenitors representing putative telencephalic patterning centers (PC, also called organizers) (3, 4, 19) (Fig. 2, A-C, and fig. S3, A-C; tables S3 and S4). Three co-clustering subtypes, detected in the anterior region showed FGF8/17/18, SP8, FOXG1, NKX2-1 or SHH expression, representing putative anterior neural ridge/rostral patterning center (RPC PC FGF17) and AV progenitors (AV NKX2-1/NKX6-2 and AV NKX2-1/LMO1) (20, 21). Two NKX2-1+ subtypes found in the GE co-clustered with the AV progenitors, likely representing the organizer of the ventral forebrain (4, 19). ZIC genes (ZIC1/3/4) were detected in these anterior-ventral subtypes, forming a ventro-dorsal and antero-posterior gradient along with the AV patterning genes expressed in this domain. Two subtypes in the medio-posterior domain expressing LMX1A, WNT (RSPO3, WNT8B) and BMP (BAMBI) signaling members likely represent two states of the dorso-caudal organizer, namely hem (PC RSPO3) and hem/choroid plexus epithelium (CPe, PC TTR) (22, 23), whose domain was further characterized by ARX, FGFR3 and LHX9 expression (Fig. 2, A-C; fig. S3, B and C; S5E). Other posterior subtypes included one putative zona limitans intrathalamica (ZLI) (PC TCF7L2) and an antihem (PC SFRP2) (24). These cells were transient as barely detected after E43 (Fig. 2A) and their identities were further validated by transcriptomic comparisons with a mouse (25) and a macaque dataset (14) (fig. S3, D and E).
Fig. 2. Molecular signatures of putative telencephalic organizers.
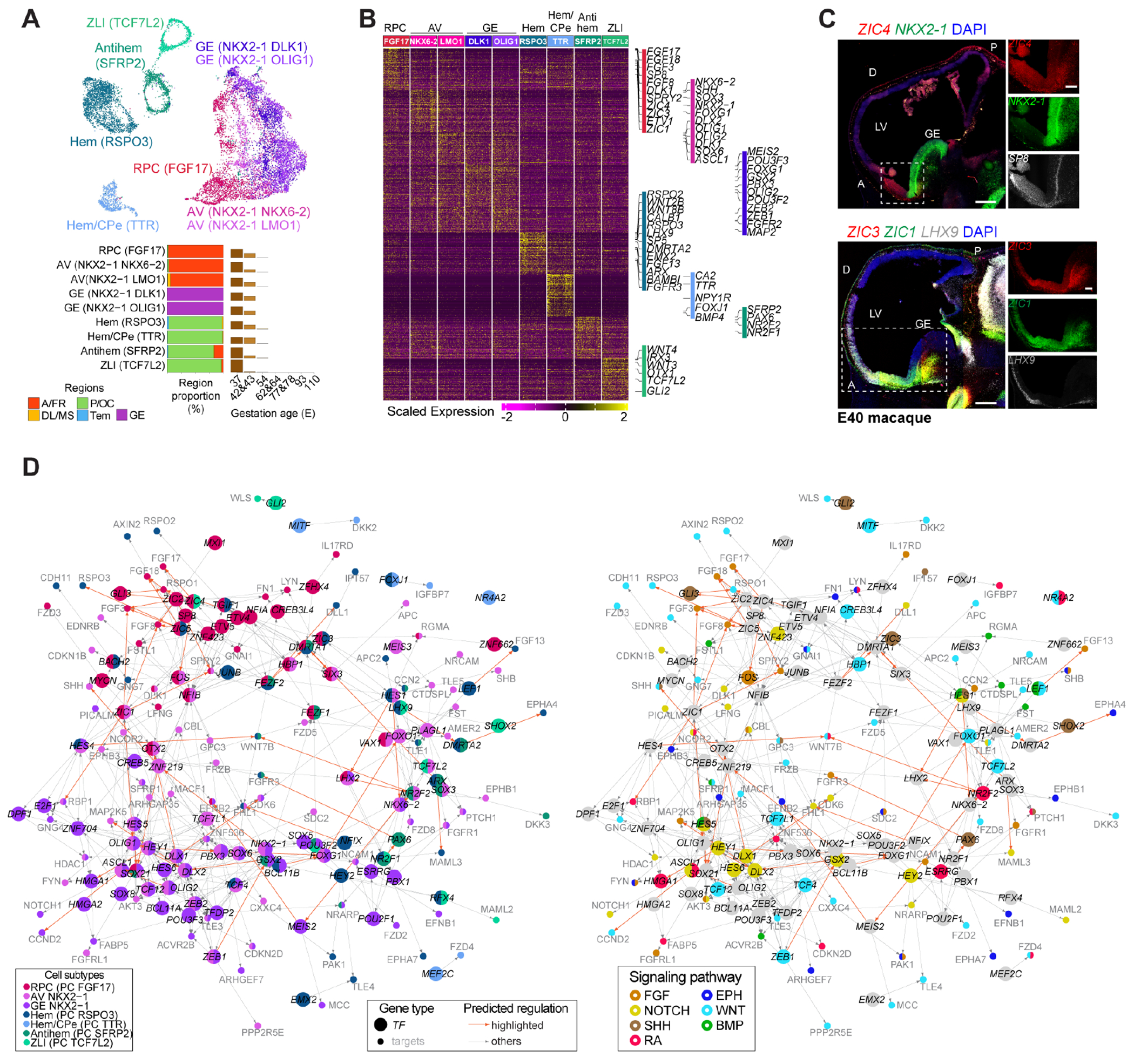
(A-B) Organizer cell subtypes (A) and their markers (B). (C) RNAscope of macaque sagittal brain sections. Scale bar: 500 μm (panoramic) and 200 μm (zoom-in). D: dorsal; LV: lateral ventricle. (D) Predicted TF regulatory network with nodes colored by subtype (left) or signaling (right).
Organizers secrete morphogens inducing gene expression gradients across the telencephalon (1, 4, 20, 23, 26). Integrating motif enrichment and gene coexpression, we predicted regulatory networks connecting transcription factors (TFs) with their putative target genes, including signaling components recruited across different domains (Fig. 2D; table S5). The RPC subtype putatively employed TF-target regulations involving ZIC genes upstream of FGF pathway-related genes, including FGF3/8/17/18 and SPRY2, whereas AV and GE progenitors exhibited both overlapping and subtype-specific regulations, including NKX2-1 linked to SHH through ZNF219. Likewise, posterior organizer subtypes showed specific regulations, including NFIX upstream of Notch and FGF signaling genes, as well as shared regulations, like ARX and LHX9. Different elements of the same signaling pathways exhibited divergent spatial activation (Fig. 2D and fig. S4A and B). For example, the BMP signaling members RGMA and FSTL1 were recruited anterior-ventrally, whereas LEF1 was in the hem. Together, these data denote the combinatorial interaction of TFs with signaling molecules orchestrating the patterning action of the organizer centers (1, 21).
Finally, RNA velocity inferred the potential neuronal lineage of the AV and cortical hem progenitors, generating LHX8+ and ONECUT1+/ONECUT2+ or TP73+ Cajal-Retzius neurons, respectively (fig. S4C). Taken together, these data highlight the molecular events underlying primate telencephalic organizer activities.
Signaling interaction between telencephalic organizers and neural stem cells
To define how the regional identity of the telencephalic RG is instructed by the organizers, we leveraged annotated signaling pathway-related ligand-receptor (L-R) pairs, inferring cell interactions between organizer subtypes, that is, RPC (FGF17+), AV (NKX2-1+), hem (RSPO3+) and hem/CPe (TTR+) expressing the ligands and region specific NSCs expressing the receptors, clustering them into modules (M1-M10) based on crosstalk patterns (Fig. 3, A and B, fig. S5, A–D). For example, M6 largely consisted of L-R pairs of FGF signaling and was characterized by FGF18-FGFR1 and FGF18-FGFR3 pairs, predicting signaling from the RPC expressing FGF18 ligand towards anterior, posterior and ventral NSCs expressing the receptor FGFR1, or selectively posterior and ventral NSCs expressing FGFR3. Other modules consisting of WNT (WNT5A-Frizzled receptor FZD5, M2; WNT5A-WNT signaling modulator PTPRK, M3) and BMP signaling-related L-R pairs predicted cross-talks between the cortical hem and region-specific NSCs. RNAscope supported the predicted interactions, outlining spatial expression patterns in the expected domains (Fig. 3B and fig. S5E). These data strongly suggest that brain organizers selectively signal to competent NSCs (27). Moreover, the results show regional expression of morphogens and paired receptors in macaques, supporting the hypothesis of a signaling code integrating the cross-talks between organizers and region-specific NSCs.
Fig. 3. Signaling cross-talks between organizers and NSCs.
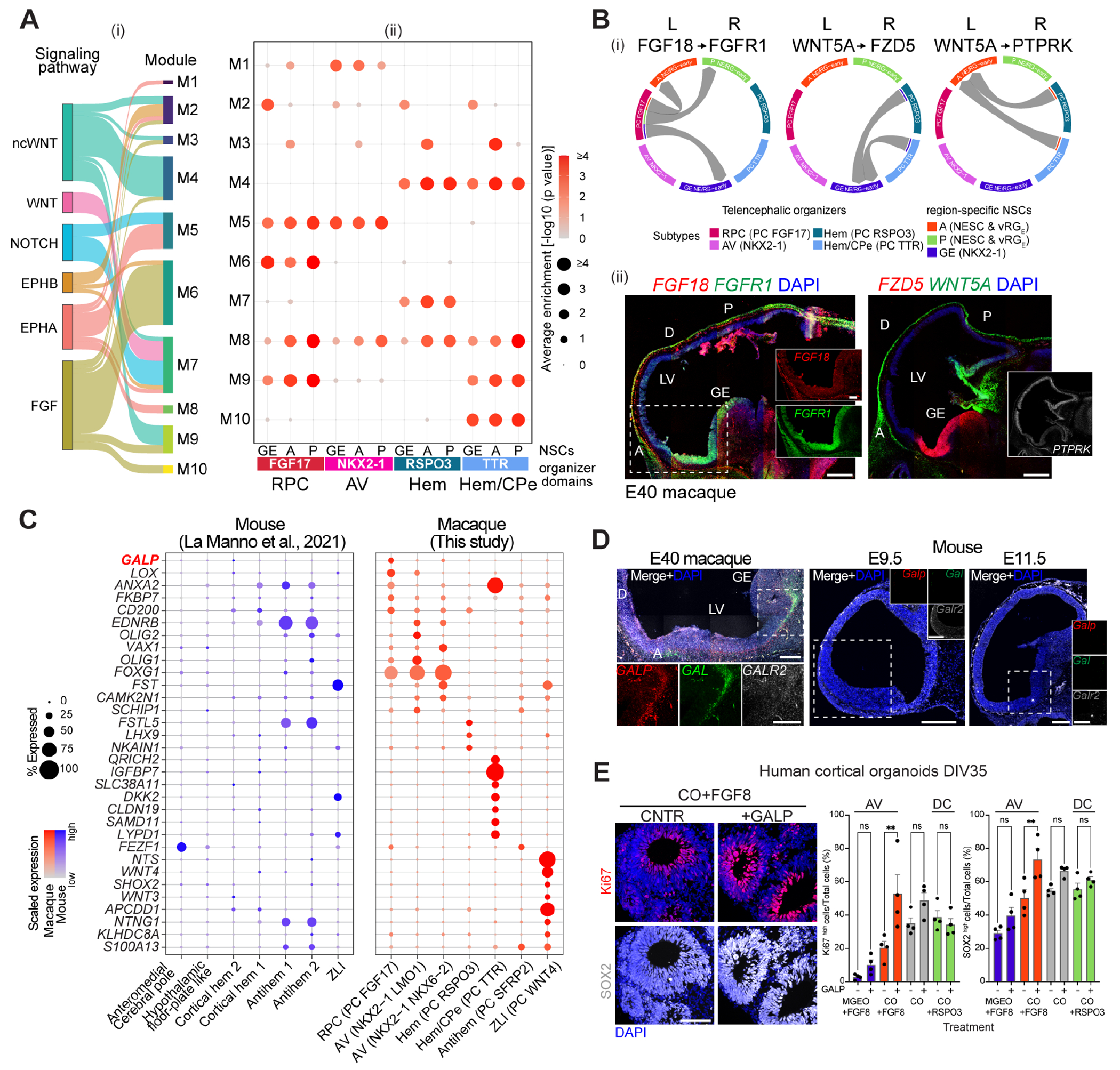
(A) L-R pair modules (M) for selected signaling pathways (i) mediating organizer-NSC cross-talks at E37-43 (ii). (B) Directed L-R mediated interactions (i). RNAscope of macaque sagittal brain sections (ii). Scale bar: 500 μm (panoramic) and 200 μm (zoom-in). (C) Organizer markers enriched in macaque versus mouse (25). (D) RNAscope of macaque and mouse sagittal brain sections. Scale bars as in B. (E) Immunohistochemistry of hCO exposed to FGF8 +/− GALP (left) and highthroughput quantification (± SD, right). Scale bar: 100 μm. One-way ANOVA, Dunnett’s multiple comparison (**: p < 0.01; ns: not significant). DIV: day in vitro.
Putative primate-specific proliferation signaling
Transcriptomic comparisons between macaque and mouse brain organizers (25) revealed similarities as well as notable differences between these species (Fig. 3C and fig. S6, A and B). Genes enriched in macaque organizers included the neuropeptide galanin-like peptide (GALP), known to be involved in hypothalamic functions in adult rodents (28). In macaque, we found GALP expressed by the RPC progenitors and their putative progeny lineages, decreasing after E43, which indicates a transient function at early phases (Fig. 3C and fig. S6C). RNAscope analysis validated the expression of GALP and its family-related Galanin (GAL) in the antero-ventral domain of E40 monkey telelencephalon, whereas they were not detected in mouse at the equivalent ages E9.5 and E11.5 (Fig. 3D). GALP/GAL receptor 2 (GALR2) was also evident in the monkey but weaker in the mouse telencephalon.
Human cortical (hCO) and medial GE (hMGEO) organoids were generated and further directed towards dorso-caudal or antero-ventral identity modulating, respectively, RSPO3 and FGF8 signaling during the patterning phase (29). Standard markers confirmed their regional bias; in addition, we observed higher expression of GAL, GALP, ZIC4 and SP8 in hMGEO than hCO, suggesting these are intrinsic features of antero-ventral neural cells (fig. S7, A and B). Furthermore, prolonged exposure of rhesus macaque cortical organoids (rmCO) to GALP, GAL or both ligands increased NSC proliferation and compromised neuronal differentiation (fig. S7, C and D).
Finally, exogenous GALP increased the number of Ki67+ and SOX2+ cells, which indicate proliferation and stem cell identity, respectively, in hCO with anterior identity (hCO+FGF8) rather than in anterior-ventral hMGEO (hMGEO+FGF8) or dorso-caudal hCO (hCO+RSPO3) (Fig. 3E and fig. S7E). Moreover, in utero intraventricular injection of GALP ligand in E11.5 mouse embryos, followed by EdU incorporation, resulted in higher proportion of EdU+, Sox2+ and Ki67+ cells in the rostral-medio dorsal telencephalon, but not in caudal nor ventral areas, relative to the PBS-injected controls, at E12.5 (fig. S8, A–C). Together, these data indicate that GALP preferentially induces proliferation of cortical RG with anterior identity.
Transcriptomic divergence in neural stem cell progression across cortical regions
Based on marker gene expression, cortical NSCs were distinguished into multiple subtypes showing different regional proportion and whose appearance correlated with developmental ages: two neuroepithelial stem cell (NESC) subtypes, two early (vRGE) and one late (vRGL) ventricular RG, two truncated (t)RG, one ependymal and two outer (o)RG subtypes (Fig. 4A and B; fig. S9, A and B). Pseudotime analysis further defined the progression of the ventricular NSCs up to ependymal cells distinguishing the oRG lineage (fig. S9C). Transcriptomic comparisons with human developing brain scRNA-seq datasets (15, 16) confirmed our annotation and identified earlier NSC states across the telencephalic regions (fig. S9D). Most of these subtypes were found in all the four main regions analyzed, however, an early RG subtype (vRGE PMP22+), highly expressing CYP26A1 and ZIC1/3/4, was found selectively enriched in the anterior region (Fig. 4B).
Fig. 4. Transcriptomic variation of NSC progression across cortical regions.
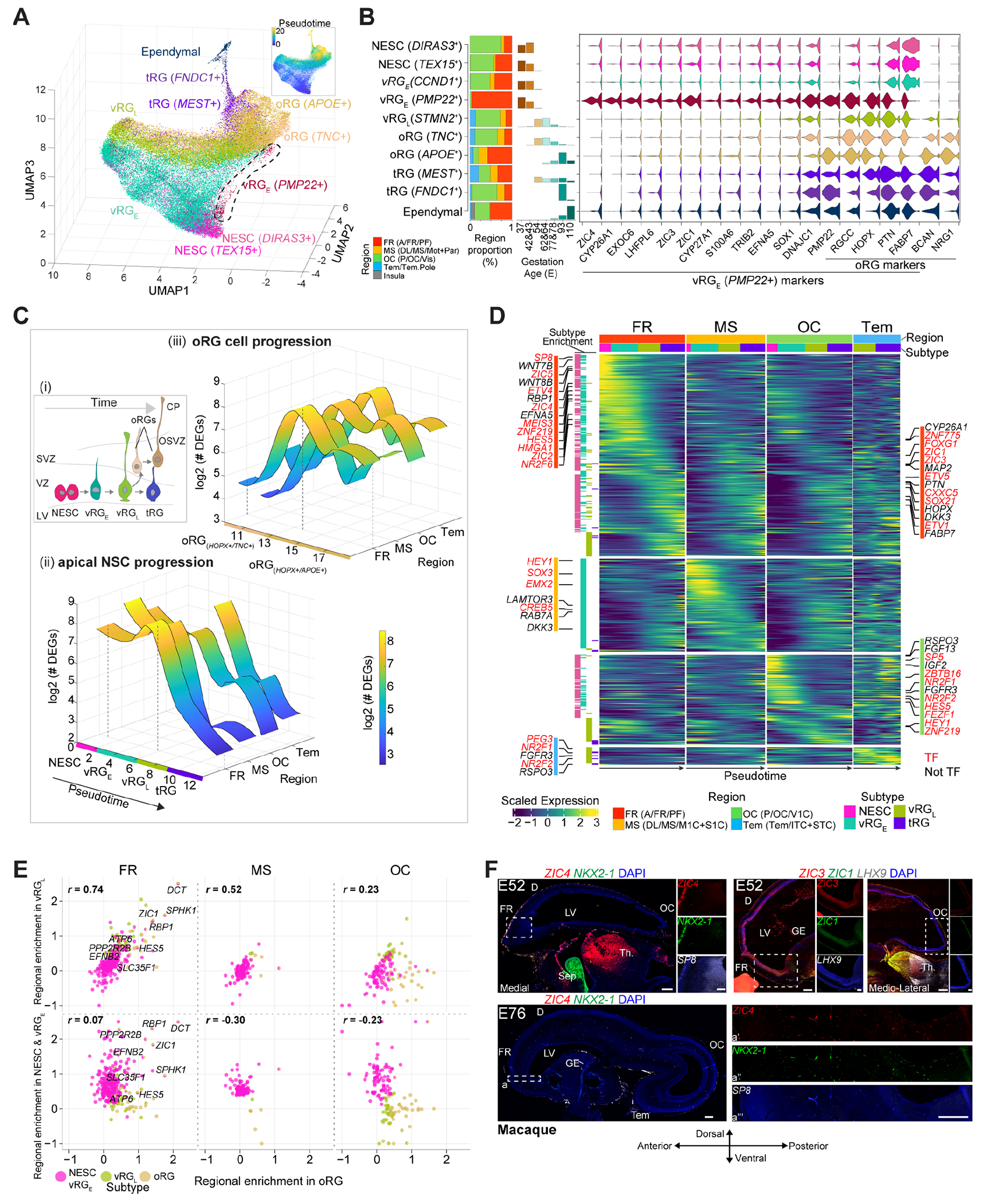
(A) Cortical NSC subtypes and pseudotime. (B) Region and age proportion of the NSCs (left). Markers for the anterior-enriched PMP22+ RG subtype (right). (C) Scheme of NSCs progression. OSVZ: outer SVZ; CP: cortical plate (i). Number of regional DEGs along ventricular NSCs (ii) and oRG (iii) progression. (D) Region-specific gene cascades along the ventricular NSC progression. (E) Region specificity correlation between early (NESCs and vRGE) or late (vRGL) NSCs versus oRG. Colors denote the subtypes showing region enrichment. (F) RNAscope of macaque brain sections. Scale bar: 500 μm (panoramic) and 200 μm (zoom-in). Sep: septum; Th: thalamus.
Within the region-shared subtypes, expression changes detected along the progression of ventricular NSCs and oRG were, for example, in chromatin remodeling factors (HMGA2 and JARID2), during the transition of the NESCs into vRG; in cilia-related genes (FOXJ1) in the observed transition from tRG to ependymal cells; in cell interaction (neurexins) and angiogenesis (VEGFA) genes, along the oRG development (fig. S9, E and F). Thus, this analysis determined the gene cascades underlying key cortical NSC state transitions.
Analysis of differentially expressed genes (DEGs) between the regions and along the progression of the NSCs of the ventricular zone (VZ; also called apical progenitors) and sub-ventricular zone (SVZ; also called basal progenitors), showed accentuated area diversification in the early NSCs (NESCs and vRGE) and in the late oRG (Fig. 4C). Within the ventricular NSC progression, regionally-enriched gene expression cascades resulted prominent at early phases of the anterior/frontal cells and included TFs like ZNF219, ZIC1/2/3/4/5 and SOX21, WNT members (WNT7B, WNT8B) and retinoic acid (RA) signaling proteins (CYP26A1 and RBP1) (Fig. 4D and fig. S10A; table S6). Some oRG marker genes (HOPX, PTN, FABP7 and PMP22) were enriched in these NSCs, representing unique features of this early anterior population, however, their expression became regionally comparable in more mature states (fig. S10, B and C). Both early posterior/occipital and temporal regions displayed higher expression of NR2F1/2, FGFR3, WNT (RSPO3) and Notch (HES5) signaling members (Fig. 4D; figs. S3C, S5E and S10A).
We also found expression enrichment of 130 genes shared by early anterior/frontal and posterior/occipital NSCs and 16 other genes, including the neuropeptide PENK, whose expression shifts from early anterior/frontal to late occipital NSCs (fig. S10, D-G; table S7).
Along the oRG lineage, DEGs across regions included RBP1 in the frontal; MEF2C and NPY neuropeptide in the occipital; the BDNF receptor NTRK2 in the temporal (fig. S10, H and I; table S8). Together, these results define temporally-regulated region-specific gene expression patterns along apical and basal NSC progression.
Finally, vRGL rather than early NSCs (NESC and vRGE) showed positive region-identity correlation with oRG, even more pronounced frontally, with few genes (for example, RBP1, ZIC1 and DCT) consistently expressed in all frontal cortical wall cell-types (Fig. 4E). This suggests that region-specific genetic programs of apical NSCs differ during early phases but are later relayed to oRG. However, all the frontal apical NSCs and oRG might share molecular mechanisms, including RA signaling response.
RNAscope of monkey tissue validated the expression pattern of several TFs at E40, E52 and E76, such as ZIC4, SP8, NKX2-1, LHX9, FEZF1 and NR2F1, which decreased over time in their respective regions, and others, like ZIC1/3, MEIS2 and PBX1 whose expression increased, spreading from the antero-ventral to the antero-posterior axis (Figs. 2C and 4F, fig. S3C). Thus, the early-generated gradients have been found to be transient, changing their spatial expression throughout development (4). In conclusion, primate telencephalic regions involve a code of sequentially-regulated genes, from NSCs throughout their progression along defined region-specific state transitions.
Transcriptomic diversification of the excitatory neurons across prospective cortical regions and areas
Unsupervised clustering and marker profiling identified distinct subtypes along the excitatory neuronal lineages, from EOMES+ intermediate precursor cells (IPCs) to deep (DL, SOX5+) and upper (UL, CUX2+) layer excitatory neurons (Fig. 5A, fig. S11, A and B). Integration with an adult macaque PFC dataset (30) further depicted these trajectories predicting mature identities of fetal neurons (Fig. 5B and fig. S11, A, C and D). DL neurons emerged at E37-43 and peaked at E54-64, promptly diversifying into subplate (L6B, from NR4A2+/GRID2+), corticothalamic (L6CT, from SYT6+) and intratelencephalic (L6IT-1 and L6IT-2, from OPRK1+) subtypes. The UL lineage was evident at E77-78 and enriched at E93, however their diversification into adult cell types was not clear, suggesting additional time required for their maturation. Neurogenesis dynamics also varied across cortical regions. For instance, occipital UL neurons emerged later than other regions, yet displaying faster maturation (fig. S12, A and B). These analyses define the cellular dynamics underlying the laminar organization of the different neocortical areas in primates.
Fig. 5. Spatiotemporal transcriptomic divergence of cortical neurogenesis.
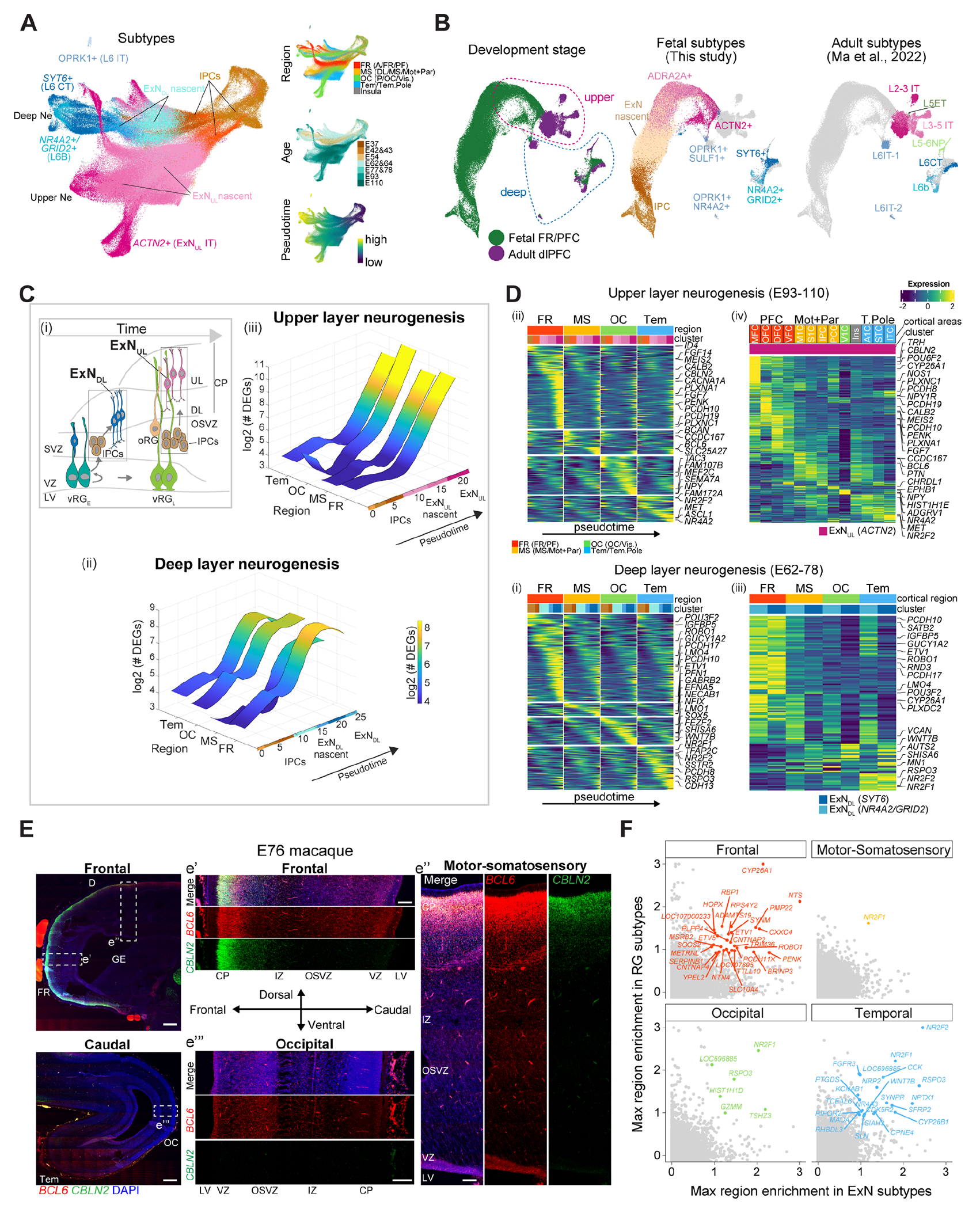
(A) UMAP showing IPCs generating excitatory neurons. (B) Transcriptomic integration of E54-110 and adult macaque PFC excitatory neurons (30). (C) Scheme of neurogenesis (i). Number of regional DEGs along the DL (ii) and UL (iii) neuron trajectories. (D) Region-specific gene cascades along neurogenesis (i and ii). Region/area-specific genes in late excitatory neurons (iii and iv). (E) RNAscope in macaque brain sections. Scale bar: 500 μm (panoramic) and 200 μm (zoom-in). (F) Shared region-specific genes between RG and excitatory neurons.
Differential expression and AUC score analyses along the excitatory neuron pseudotime and hierarchical clustering highlighted increasing regional/area divergence at late differentiation phase, identifying more region-specific genes in the frontal cortex (Fig. 5, C and D, fig. S12, C and D). Genes enriched in frontal DL neurons included protocadherins (PCDH10/17), whereas area divergence in UL neurons was defined by RA signaling members (CYP26A1, CBLN2 and MEIS2) in PFC and BCL6 in M1C, as previously reported (10, 31) (Fig. 5D). However, non-negligible regional differences were also detected in IPCs, likely representing earlier cell-autonomous events seeding neuron diversification. Furthermore, whereas region-specific signatures largely overlapped between DL neuron subtypes, they were scarcely shared between DL and UL neurons, suggesting distinct molecular programs governing the establishment of regional identities in inner versus outer cortical layers (fig. S12E). RNAscope on macaque brains further confirmed divergent expression patterns across regions outlining frontal-caudal gradient of BCL6 and CBLN2 (Fig. 5E). Finally, transcriptomic comparisons with age-matched prenatal human excitatory neurons (16) showed human-biased signatures, including RBP1 more prominent in the prefrontal cortex (fig. S12, F and G). These analyses delineate the developmental dynamics underlying regional diversification of cortical excitatory neurons, highlighting identity refinement during their maturation.
Next, we asked whether the protomap of the RG was related to the area-specificity of the excitatory neurons. Intersecting region-specific genes of early RG and neurons we found the genes expressed throughout whole the lineage progression. These included HOPX, CYP26A1 and RBP1 in frontal, NR2F1 and RSPO3 in occipital, NR2F2 and CYP26B1 in temporal regions, likely defining neural cells’ regional identity across development (Fig. 5F, and fig. S13A). In addition, bulk RNA-seq, across neurogenesis of monkey regional cortical NSCs differentiated in vitro (29), identified many region-specific genes expressed in neurons which were also present in the excitatory neurons in vivo, including RA signaling components (RBP1, CYP26A1, BRINP1 and CBLN2) and synaptic genes (LFRN2 and CAMKV) (fig. S13, B-E). As these in vitro neurons lack connectivity inputs from other brain regions, these genes might reflect the intrinsic events underlying neuronal regional divergence, and resulted more prominent in frontal than occipital cortex. Altogether these data denote the potential contribution of early and late cell-autonomous mechanisms in neuronal diversity across neocortical areas.
In contrast to the divergence of the excitatory neurons, the inhibitory neurons, which were distinguished in subtypes distributed across regions and ages, such as MGE (LHX6+)- and CGE (NR2F2+ and/or SP8+)-derived subtypes, showed limited cortical area differences (fig. S14, A-D). The area-specific genes expressed in LHX6+ interneurons were paralleled in NR2F2+/SP8+ subtypes and vice versa (fig. S14, E and F), indicating transcriptional overlapping of area identities between interneurons and suggesting that later cues might eventually contribute to their further diversification in the cortex (32).
Spatial transcriptomic divergence across gliogenesis
We next focused on gliogenesis trajectories. Through unsupervised clustering, cell trajectory reconstruction inference and pseudotime analysis, we distinguished late RG subtypes, which include vRGL, tRG and oRG, transitioning into excitatory neurons or EGFRhigh expressing glial intermediate precursors (gIPCs) (33), which diverge towards astrocytes or oligodendrocytes (Fig. 6, A and B, fig. S15A). Comparative analyses with multiple fetal and adult human, macaque and mouse datasets confirmed our annotation, and distinguished astrocytes in putative interlaminar (GFAP+) and protoplasmic (MFGE8+ and EGFR+) subtypes (fig. S15, B and C), suggesting that astrocytic adult identities emerge during mid-fetal stages in primates.
Fig. 6. Transcriptomic versatility of gliogenesis across cortical areas.
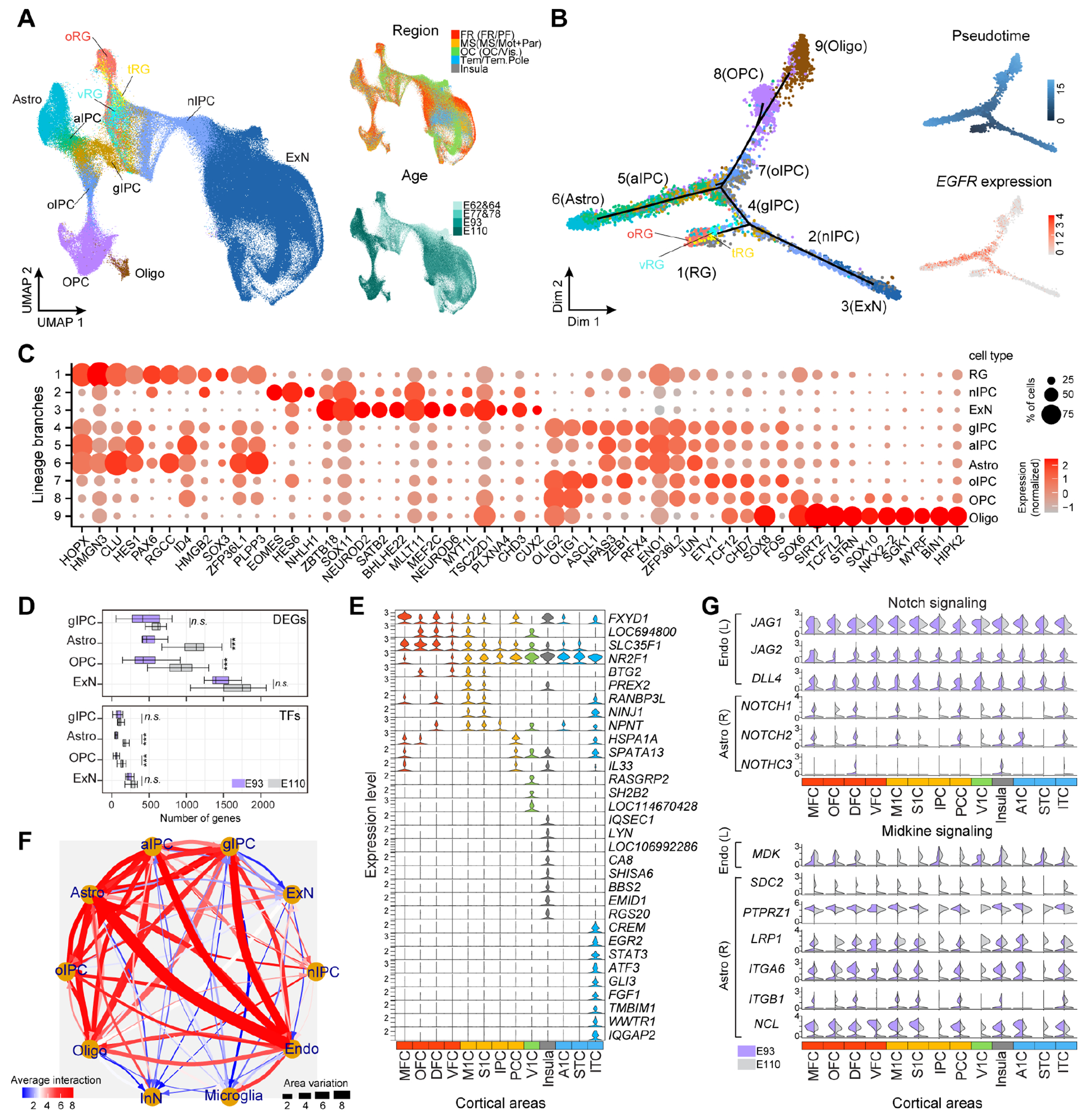
(A-B) UMAP (A) and Monocle2 (B) showing neurogenic and gliogenic trajectories from late RG. (C) Top 10 branch-specific genes. (D) Numbers of DEGs (top) and TFs (bottom) across areas, following one-sided t-test (P<0.001 ***, ns: not significant). (E) Curated area-specific genes in E110 astrocytes. (F) Predicted L-R mediated interactions. (G) Selected L and R expression. n/a/oIPC: neuron/astrocyte/oligodendrocyte IPC.
To define the transcriptomic programs underlying the switch of RG from neurogenic to gliogenic potential, we identified the top-ranked genes expressed at each lineage branch (Fig. 6C and fig. S15D; table S9). Known regulators of astrocytic fate such as the chromatin remodeling factors HMGN3 and HMGB2 and TFs such as PAX6, HES1 and SOX2/3/6 were expressed by late RG; OLIG1/2 and ASCL1 by gIPCs; SOX10 and NKX2-2 by oligodendrocytes, while astrocytes expressed many RG genes. These data reveal a temporally orchestrated combination of genes at the divergence from neurogenesis to gliogenesis during monkey corticogenesis (34, 35).
DEG analysis indicated that glial cells diverge across areas less than excitatory neurons. However, astrocytes displayed higher DEGs, including TFs, than gIPCs and oligodendrocytes at E110, expressing area-specific genes, including SLC35F1 in prefrontal and STAT3 in temporal cortex, denoting distinct spatial molecular features (Fig. 6, D and E; fig. S15E). Together, these results indicate that transcriptomic variation of the astrocytes across cortical areas is more accentuated during late differentiation.
L-R pair analysis among neural and non-neural cells predicted the highest number of putative interactions with most prominent area variation between endothelial cells and astrocyte IPCs or astrocytes (Fig. 6F). Endothelial cells showed low transcriptional variation among the areas (fig. S15F), suggesting that astrocytes might respond differently to their signals. We identified L-R pairs of diverse signaling pathways displaying variation across time and areas (fig. S15G). Notch and Midkine (MDK) signaling-related L-R pairs displayed similar expression of the ligands (JAG1/2, DLL4 and MDK, respectively) in endothelial cells, in contrast to the variation of the receptors found in the astrocytes (NOTCH1/2/3 and SDC2 and ITGB1, respectively) (Fig. 6G). These data suggest that astrocytes have area-specific competence to respond to the endothelial cells’ signals which might contribute to their transcriptomic variation across areas.
Spatiotemporal expression of disease-risk genes in early telencephalic development
Alterations of cortical development are implicated in neuropsychiatric diseases (36), however, little is known about the function of the risk-associated genes in the NSCs of the early telencephalon. We curated gene lists associated with major neuropsychiatric and neurodegenerative disorders, as well as brain cancers (table S10). Expression enrichment analysis showed that risk genes for multiple neuropsychiatric disorders were prominent in excitatory and inhibitory neurons, whereas glioblastoma-associated genes were highly expressed in oRG and glial precursors, as previously reported (18, 37-40) (Fig. 7A and fig. S16A).
Fig. 7. Expression of brain disease-risk genes in early telencephalon.
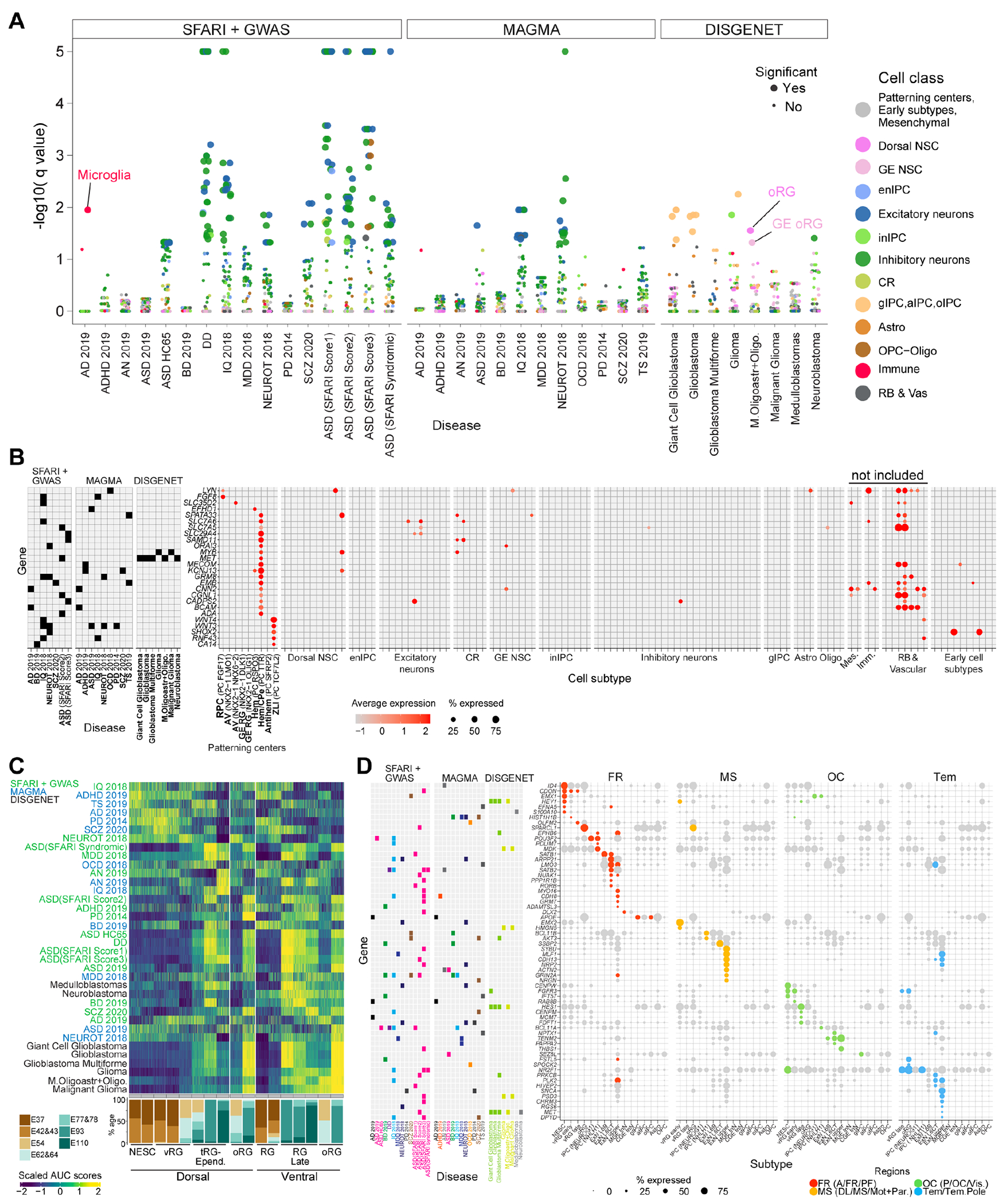
(A) Expression enrichment of disease genesets across cell subtypes (dots); Significance: q value < 0.05. (B) Expression of disease gene markers for a PC subtype expressed in no more than three other subtypes, excluding non-neural cells. Gene-disease association on the left. (C) Expression enrichment cascades of disease genesets along the temporal progression of dorsal and ventral NSCs. (D) Top regionally-enriched (dot colors) disease gene markers.
This analysis unveils the most salient signals in neurons, however, it might mask the gene expression patterns in brain organizer progenitors and RG, whose dysfunctions were suggested as early fetal risks for neuropsychiatric diseases (41). Although no enrichment of any disease gene set was observed in organizer domain subtypes (Fig. 7A), 26.1% to 36.2% of the genes from each list was expressed in these cells (in ≥10% cells) (fig. S16B). Among these risk genes, 132 overlapped with patterning center subtype markers, of which 26 were distinctive of these organizer domains and the other 106 also appear later in development in other cells (Fig. 7B, fig. S16C). The 26 genes included well-known patterning regulators, such as FGF8 expressed in the RPC, and other genes, for example, the autism spectrum disorder (ASD)- and glioma-associated gene MET, previously detected in late prenatal and postnatal human excitatory neurons (13, 30), that was now found expressed early selectively in the cortical hem (Fig. 7B). Many of these genes exhibited no or limited expression in late gestation and postnatal human cortex, further suggesting a function restricted to early developmental phases (fig. S17, A and B). These analyses indicate that certain risk genes are expressed earlier in the telencephalic organizers, with a putative role in regional patterning and NSC specification.
Similar analysis conducted for the NSCs showed enrichment of gene sets, including the multiple ASD- or glioblastoma-associated genes, in both dorsal and ventral late RG (tRG and/or oRG). However, other risk genes, associated with diseases incuding attention-deficit/hyperactivity disorder (ADHD), Tourette syndrome (TS), schizophrenia (SCZ), bipolar disorder (BD), exhibited expression in early dorsal and/or ventral NSCs (Fig. 7C, fig. S18, A and B). Thus, neurodevelopmental disorder-associated genes might function throughout the progression of dorsal and ventral telencephalic NSCs.
Intersecting disease-associated genes with top regionally-enriched subtype markers revealed spatial- and cell type-expression bias of the risk genes across primate telencephalic development (Fig. 7D and fig. S19A). For example, the ASD-associated gene CDON resulted preferentially expressed by antero/frontal NSCs, SATB1 by frontal DL neurons (37) while THSB1 by occipital UL neurons. Similarly, glioblastoma risk genes like HEY1 and HES1 were enriched in anterior/frontal or posterior/occipital NSCs, respectively. Thus, neuropsychiatric disorders and cancers might have region-specific patterns of risk.
In conclusion, these data point to telencephalic regional patterning and NSC progression as possible risk events for the origins of neurodevelopmental disorders. Moreover, the molecular programs underlying these NSC events might be dysfunctionally recapitulated in brain cancers (42).
DISCUSSION
This work reveals the dynamic transcriptomic programs and cellular events underlying the establishment of the telencephalic regional identity throughout macaque fetal brain development.
NSCs determine the layout of the cerebral cortex at the earliest stages of fetal development, by a coordinated expression of genes and enhancers’ activity in the protomap (1, 6). We characterized the transcriptome of the primate telencephalic organizers and the regulatory networks orchestrating their patterning function in discrete domains. Early regionalized morphogens likely induce region-specific gene expression cascades in competent NSCs which traverse intrinsic spatiotemporal state transitions.
Differences were observed between mouse and monkey organizers. GALP and GAL are expressed in the antero-ventral domain of the early monkey telencephalon but are not detected in the mouse at equivalent ages. As these neuropeptides are associated with more mature cortico-hypothalamic circuitries (43), our data suggest a role for both ligands as modulators of proliferation and likely differentiation of early fetal NSCs in primates. However, GALP enhances proliferation preferentially in frontal cortical RG, opening to potential mechanisms underlying patterning and evolutionary expansion of the primate cortex.
Our data highlight two waves of spatial diversification of the NSCs, in the early ventricular NSCs and in the late oRG. Neurons and astrocytes exhibit high regional divergence in their terminal differentiation phase, likely influenced by synaptic inputs or signaling from the vasculature (44, 45). However, we identified region-specific genes expressed from NSCs throughout their differentiation into neurons, as well as genes expressed in neurons despite the lack of brain area connections, as seen in vitro for the prefrontal-enriched RA signaling-associated CBLN2, suggesting that cell-intrinsic mechanisms might contribute to define regional neuronal diversity. Thus, these data support a model where cell-autonomous programs characterize the early specification of the NSCs and their spontaneous progression, and then an interplay of intrinsic and extrinsic cues might further shape the identity of neurons and astrocytes later during corticogenesis (46).
Several studies point to neuronal and glia dysfunction during mid-late corticogenesis and postnatal ages at the origins of many neuropsychiatric disorders (18, 37, 38). However, we found risk genes expressed in brain organizers and/or in dorsal and ventral NSCs, suggesting a potential earlier developmental origin for these disorders, which implicates dysfunctional patterning of the telencephalon and altered spatiotemporal identity of the RG along their neurogenic or gliogenic progression. We found that glioblastoma and neurodevelopmental disorders, like ASD and ADHD, share risk genes potentially implicated in brain organizer and NSC functions, suggesting that genetic lesions causing both diseases might converge to alter similar NSC gene networks (47, 48).
The absence of epigenomic data paralleling our transcriptomic analysis limits the understanding of gene expression regulation across regions and species. However, integrating these data with other datasets, as we showed, will help to better understand primate and human brain formation, evolution and diseases and even improve cellular systems for modeling neurogenesis and its disorders in vitro.
Materials and Methods
All procedures involving animals, including monkeys and mice, were carried out according to guidelines described in the Guide for the Care and Use of Laboratory Animals, and were approved by the Yale University Institutional Animal Care and Use Committee (IACUC).
Caesarean sections of the pregnant monkeys and collection of the fetal brains
Rhesus macaque monkeys were bred in Rakic and Sestan primate breeding colony at Yale. Timed-pregnant monkeys were subjected to caesarian section at the required gestational age, performed by Yale’s Veterinary Clinical Services (VCS). Monkeys were first sedated with ketamine (3 mg/kg) and atropine sulfate (Lily, 0.2 mg/kg). A butterfly catheter was introduced into the saphenous vein for continuous administration of fluids to prevent dehydration. Intravenous leads were secured subcutaneously for monitoring heart rate and respiration throughout the surgical procedure performed under isoflurane anesthesia and strict sterile procedures. A midline incision was made in the abdominal wall and the uterus gently exposed through the opening. The uterus was then incised between the primary and secondary lobes of the discoid placenta, the chorioallantoic membrane was punctured and the embryo delivered, decapitated still under the effects of anesthesia from maternal blood. The embryo head was transported to an adjacent room housing a BSL2 hood, brain tissue was dissected for single cell transcriptomics or fixed in paraformaldehyde (PFA). After delivery, the uterine and abdominal walls of the mother monkey was sutured in layers. Post-operatively, the animals were monitored several times a day until full recovery.
Fetal monkey brain single cell dissociation
Fetal macaque brains were isolated from E37 to E110, put on a dish containing PBS and sectioned. Telencephalic regions were identified and dissected using a blade. For E37-43 brains, the entire anterior and medio-posterior parts of the hemispheres were cut. Each tissue was incubated with HBSS-Papain (2 mg/ml, BrainBits, PAP) from 15 (for the early ages) to 30 (for more advanced ages) minutes at 37°C. The solution was removed and the tissue gently triturated in HBSS-DNase I 0.1mg/ml solution (STEMCELL Technologies, 07900) using a 2 ml pipette. Then, samples were filtered through 40 μm cell strainers (Falcon, 352340) and the cells counted with an automatic cell counter (ThermoFisher Scientific). Samples were diluted in HBSS to 1000 cells/microliter and processed for single cell RNA-seq analysis within 20 min at Yale Center for Genome Analysis (YCGA) core facility.
Dissection of the mice
Pregnant mice were delivered from Charles River to the animal facility at Yale. Animals were euthanized using a CO2 chamber. The embryos were harvested, decapitated and collected in PBS.
Fixation and sectioning of the brain tissue
Macaque and C57/BL6 mouse fetal brains were dissected and immerse in 4% PFA overnight (ON) at 4°C. Fixed brain blocks were immersed in step-gradients of sucrose/PBS up to 30% for 2-3 days at 4°C, then embedded in OCT and frozen at −80°C. Sagittal sections were cut 25 μm for the monkey brain tissue. Sagittal or coronal section were cut 15 μm for mouse brain tissue. Sections were prepared using a Leica CM3050S cryostat and stored at −80°C until use.
Construction of 10X Genomic Single Cell 3’ RNA-Seq libraries and sequencing
Sample Preparation. The first step for the construction of scRNA-Seq library involved the preparation of the single cell suspension described above.
GEM Generation and Barcoding. Single cell suspension in RT Master Mix is loaded on the Single Cell A Chip and partition with a pool of about 750,000 barcoded gel beads to form nanoliter-scale Gel Beads-In-Emulsions (GEMs). Each gel bead has primers containing (i) an Illumina® R1 sequence (read 1 sequencing primer), (ii) a 16 nt 10x Barcode, (iii) a 12 nt Unique Molecular Identifier (UMI), and (iv) a poly-dT primer sequence (30nt). Upon dissolution of the Gel Beads in a GEM, the primers are released and mixed with cell lysate and Master Mix. Incubation of the GEMs then produces barcoded, full-length cDNA from poly-adenylated mRNA.
Post GEM-RT Cleanup, cDNA Amplification and library construction. Silane magnetic beads are used to remove leftover biochemical reagents and primers from the post GEM reaction mixture. Full-length, barcoded cDNA is then amplified by PCR to generate sufficient mass for library construction. Enzymatic Fragmentation and Size Selection are used to optimize the cDNA amplicon size prior to library construction. R1 (read 1 primer sequence) are added to the molecules during GEM incubation. P5, P7, a sample index, and R2 (read 2 primer sequence) are added during library construction via End Repair, A-tailing, Adaptor Ligation, and PCR. The final libraries contain the P5 and P7 primers used in Illumina bridge amplification.
Sequencing libraries. The Single Cell 3’ Protocol produces Illumina-ready sequencing libraries. A Single Cell 3’ Library comprises standard Illumina paired-end constructs which begin and end with P5 and P7. The Single Cell 3’ 16 bp 10x Barcode and 12 bp UMI are encoded in Read 1, while Read 2 is used to sequence the cDNA fragment (91bp). Sequencing a Single Cell 3’ Library produces a standard Illumina BCL data output folder. The BCL data will include the paired-end Read 1 (containing the 16 bp 10x™ Barcode and 12 bp UMI) and Read 2 and the sample index in the i7 index read. Minimum sequencing depth is 20,000 read pairs per cell.
Single-cell RNA-seq data processing and filtering
Cellranger was applied to align the scRNA-seq reads to rhesus macaque genome assembly Mmul10 together with the gene annotation file from NCBI RefSeq (release 103), followed by barcode counting and unique molecular identifier (UMI) quantification. The resulted filtered gene by cell UMI count matrices were used for additional quality control and filtering.
An initial clustering was performed for each sample using Seurat (49) to spot potential low-quality cell clusters, which include cells with low number of UMIs and/or high percentage of mitochondria UMIs. The resulted count matrices were used in scrublet package (50) to predict the doublet score of each cell. Cell clusters with high doublet scores and exhibiting combinatory expression of two different cell type markers were considered as doublet clusters and removed. Because samples might behave differently, a sample-wise doublet score threshold were selected for each sample. To further remove such outlier cells, cells belonging to the same cell class across different batches were clustered together for additional rounds of quality control, which increased the power in detection of outliers. The filtered gene by cell UMI count matrices were utilized in the downstream analysis.
Normalization, clustering and dimension reduction of the scRNA-seq data
Filtered UMI counts in each cell was first log-normalized using the NormalizeData function in Seurat (49), with the scaling factor set as 10,000. To embed all cells across different development ages and brain regions in the same reduced dimension space, we applied fastMNN (51) and Harmony (52) to integrate the data. Here, both methods perform batch correction in the reduced dimensions and they overall exhibited very similar results. However, Harmony showed slightly better performance in preserving inter-cell heterogeneity and fastMNN were marginally better in recapitulation of cell differentiation lineages. And accordingly, Harmony was largely utilized for cell cluster identification and fastMNN for lineage visualization (for example, Fig. 1). Prior to batch correction, for each batch we identified the highly variable genes using the variance-stabilizing transformation implemented in the Seurat package. Because the default selction of the highly variable genes with the highest frequencies across all samples might lose some key signals in certain developmental ages with smaller sample sizes, we parcellated the developmental stages to three windows based on their transcriptomic similarity (E37-E43, E54-E78, E93-E110) and integrated the highly variable genes using the SelectIntegrationFeatures function in Seurat. The union of the genes across the three developmental periods were chosen for downstream analysis. Then, the normalized data were scaled separately in each batch (for Harmony), or together across all batches (for fastMNN), and used for principal components analysis (PCA), with elbow plots to select the significant principal components. The first 30 integrated reduced dimensions were used for UMAP visualization with the “umap-learn” method and the “correlation” metric.
Cell clustering was performed via “Louvain” algorithm based on the first 30 integrated reduced dimensions with the k-nearest neighbor set to 25. Dividing cells from different cell types (for example, early and late RG) might cluster together by cell cycle phases rather than their identities. Therefore, we categorized cell clusters into different cycling phases based on the expression of key cycling genes (for example, S phase – PCNA and MCM5; G2M phase – MKI67 and TOP2A) and gene set enrichment of cell cycling genes calculated via the Seurat CellCycleScoring function, and further sub-clustered cells within each phase to maximize the variance contribution from cell type heterogeneity rather than cell cycling differences. This method showed better performance than the traditional method that clusters cells using the data with cell cycle scores regressed.
Cell subtype annotation
Patterning center subtypes were identified based on several criteria: 1) temporal enrichment at E37 and E42-43; 2) high expression of canonical genes expressed in neuroepithelial stem cells such as SOX2 and NES, and low expression IPC makers (for example, ASCL1, NEUROG1 and EOMES); 3) combinatory expression of known patterning center markers: rostral patterning center (FGF8+, FGF17+) (20, 21), cortical hem and its following state hem/choroid plexus epithelium (RSPO3+, TTR+) (22, 23), antihem (SFRP2+, PAX6+) (53) and zona limitans (IRX1+, IRX2+, IRX3+, WNT3+, WNT4+) (24); 4) the spatial locations are consistent with the predicted identities. Furthermore, we spotted several early subtypes enriched in E37-43 and co-clustered with patterning center subtypes, which likely represent regional specialized domains. These included two NKX2-1+ subtypes (LMO1+ and NKX6-2+, respectively) detected in anterior samples representing anterior-ventral (AntVen) domain cells. Both subtypes were transcriptomically similar to the two NKX2-1+ early GE subtypes (GE RG NKX2–1 OLIG1 and GE RG NKX2–1 OLIG1) and all the four subtypes co-clustered with the rostral patterning center (FGF17+; Fig. 2). We also identified subtypes forming continuous manifold and connecting with the patterning center subtypes and those domain-specific subtypes, resembling cell differentiation lineages. These contained the anterior-ventral domain NKX2-1+/LMO1+ subtypes give rise to the DLX1+ IPC subtype (inIPC ASCL1 DLX1) which produces GNRH1+ interneurons and LHX8+/ZIC1+ interneurons. The rostral patterning subtypes seemed to form lineages with IPC FGF17 subtype and neuron subtype Neu TAGLN3 ONECUT2. The zona limitans subtype also led to a lineage with IPC TCF7L2 subtype. In addition, one NKX2-1+ subtype (RAX+) in posterior domain (Pos), and one SFRP1+ subtype with limited markers present in anterior domain (Ant) were also dectected. A few other early subtypes, including Cls FGF17 LGI1, Cls LHX9 EBF1, Cls RSPO3 SOX1, Cls GSX2 B3GAT2, were all small in sizes and their identities were left as unknown.
Neural stem cells in dorsal regions clustered together and were defined based on the expression profiles of PAX6+/SOX2+/NES+/EOMES−/EGFR−. These cells formed a circular shape on the UMAP layout representing cell cycling states and they were also organized along the temporal axis resembling the progression of their identities. This started with putative neuroepithelial stems cells, which were identified based on the high expression of RSPO3 and relatively lower expression of PAX6. Two early RG subtypes were defined at E37-43, one expressing FABP7 and PMP22, enriched at anterior regions, and the other one labeled by HMGA2 and CCND1 ubiquitously present in all regions analyzed. Late RG included two oRG (HOPX+/NRG1+) subtypes, two tRG (CRYAB+) subtypes and one vRG subtype directly connecting with early RG and showing low expression of oRG and tRG markers. There is also an ependymal subtype marked by FOXJ1 expression as well as well several CFAP genes (for example, CFAP45 and CFAP54). Neural stem cells in ventral regions also express SOX2 and NES, but only the late RG express PAX6. We also categorized them into different subtypes based on the similar genes we used for dorsal neural stem cells including HMGA2, HOPX and CRYAB as well as ventral-specific signatures such as NKX2-1, NKX6-2 and OLIG1.
Cajal Retzius cell lineages were extracted based on their evident RELN expression. Anterior-enriched Cajal Retzius cells showed higher expression of ETV1+ while the posterior-enriched ones had high TP73 expression (54). We also found two putative RSPO3+ IPC subtypes enriched in the posterior regions and they are marked by NEUROG1 and NHLH1 expression respectively.
Excitatory neuron lineages included IPCs marked by EOMES expression and post-mitotic neurons expressing NEUROD2. The IPCs consisted of three subtypes: VIM+ subtype transcriptomically more similar to radial glial cells, NEUROG1+ subtype dominating the cycling IPCs and also having non-cycling cells, and NHLH1+ postmitotic subtype coming from NEUROG1+ subtype. Subtypes of excitatory neurons were broadly categorized into deep and upper layer neurons based on their expression of SOX5 and CUX2, respectively. Deep layer neurons included two nascent subtypes (PALMD+ and ID2+), a corticothalamic subtype (SYT6+), two intratelencephalic subtypes (OPRK1+/SULF1+, OPRK1+/NR4A2+) and a L6B subtype (NR4A2+/GRID2+) (30). Upper layer neurons contained two nascent subtypes (PALMD+ and ADRA2A+) and one intratelencephalic type (ACTN2+) (30). We identified also a putative deep layer subtype (SOX5+/KCNV1+) which is transiently present at E62-64 and one excitatory neuron subtype enriched at posterior cingulate cortex (TSHZ2+/NR4A3+).
Interneurons were classified based on DLX1, DLX2, GAD1 and GAD2 expression. IPCs in the interneuron lineages were categorized based on their ASCL1 expression and their co-clustering on the UMAP connecting with post-mitotic interneurons. Putative identities of the postmitotic interneurons were classified by the expression of markers highly correlated with their developmental origins (MGE: LHX6; C/LGE: NR2F2, SP8, MEIS2) and transcriptomic integration with existing datasets with established identities including an independent developing macaque interneuron scRNA-seq data (55) and an adult macaque snRNA-seq data (30). The LHX6+ interneurons consisted of three major branches: CRABP1+ interneurons recently reported to be enriched in primates (55, 56), LHX8+ branch enriched at E37-43 likely contributing to cholinergic neurons (57), and LHX6+/CRABP1− interneurons dominating the MGE-derived interneurons and giving rise to majority of the SST and PVALB interneurons. Within the LHX6+/CRABP1− group, we spotted three potential sub-branches: SST+/NPY+ branch giving rise to long projecting inhibitory neurons (30, 32), GUCY1A2+/RELN+/DCN+ branch producing major PVALB and SST interneurons; CCK+ branch generating LAMP5 LHX6 interneurons that mapped to mouse hippocampus Ivy cells and were recently reported to show abundance enrichment in the primate neocortex (56, 58). The CGE- and LGE-derived interneurons encompassed two major branches marked by NR2F2/SP8 and MEIS2/SP8 expression, respectively. Within the MEIS2/SP8 lineage, cells were largely parcellated based on the expression of PAX6 (olfactory bulb neurons) and FOXP1/FOXP2 (striatum spiny projection neurons) (55). The NR2F2/SP8 lineage consists of cell subtypes giving rise to different adult interneuron subclasses (30): LAMP5+ interneurons becoming LAMP5 RELN subclass, KIT+ interneurons becoming ADARB2 KCNG1 subclass; VIP+ interneurons becoming VIP subclass.
The glia cells were classified into three major groups based on expression of OLIG2 (oligodendrocyte lineage-related cells), AQP4 (astrocyte lineage-related cells), and EGFR (glia precursor cells). The oligodendrocyte lineage-related cells were further divided by the expression of PDGFRA (oligodendrocyte precursor cells, OPCs), PDGFRA and MKI67 (oligodendrocyte precursor cells in proliferation stage, OPC PDGFRA MKI67), PCDH15 (late oligodendrocyte precursor cells), and MBP (oligodendrocytes). Notice the oligodendrocytes are less represented at the analyzed ages. The astrocyte lineage-related cells were divided into three subtypes by the expression of GFAP, EGFR, and MFGE8, respectively. Regarding glia precursor cells, we employed combinational expression of genes to sort them into astrocyte intermediate precursor cells (aIPCs, EGFR+/AQP4+/IGFBP2+), oligodendrocyte intermediate precursor cells (oIPCs, EGFR+/PDGFRA+/DLL1+), glia intermediate precursor cells (gIPCs, EGFR+/AQP4−/PDGFRA−), glia intermediate precursor cells in proliferation stage (EGFR+/MKI67+).
The rest of the immune cells and vascular-related cells were categorized using the following strategies. All immune cells were identified as PTPRC+, with microglia subtype further identified as C1QC+ and T cells identified as CD69+ (30). Two red blood lineage cell subtypes were classified based on the expression of HBA1 and their unique expression of HBE1 and SNCA, respectively. Vascular cells were characterized by their FN1 expression and specific expression of other subtype markers: endothelial cells (CLDN5+), pericytes (GRM8+), smooth muscle cells (ACTA2+) and vascular leptomeningeal cell (CEMIP+) (30). We also spotted two putative mesenchymal cell subtypes: one expressing LUM, consistent with the recently reported human mesenchymal cells in the human early developing telencephalon (59); the other is marked by FOXD3 and PLP1 expression, likely representing neural crest cells (60).
Identification of cell subtype markers and genes with region/area-divergent expression
To identify genes differentially expressed between cell subtypes or brain regions/areas, Wilcoxon Rank Sum test was utilized, and a minimum expression ratio of 0.1 and a Bonferroni-adjusted p value threshold of 0.01 were adopted. In certain analyses as detailed in the following sections, we incorporated additional requirements to filter the differentially expressed genes. For example, we inspected expression ratio fold changes, with a pseudo value of 0.01 adding to the gene expression ratios in the examined cell subtype (numerator) and background cells (denominator). Other criteria such as log fold changes of average expression and background expression ratios were also taken into consideration for gene filtering.
Inference of regulatory networks in telencephalic organizer cells and regional specific NSCs
The inference of transcription factor regulatory networks was based on the SCENIC workflow (61), by integrating motif enrichment in the promoter regions and gene-gene co-expression. The putative promoter region (upstream 2000 bases and downstream 500 bases of the transcription start site) of each gene, as well as the motifs of human transcription factors, were prepared (62). R package PWMEnrich as used to perform the motif enrichment analysis, with p value threshold set at 0.05 and raw score threshold set to 2.5 to only retain transcription factor genes with significant motif enrichment at the promoter of the given gene. This analysis generated raw regulons, which refers to a module of genes including a transcription factor gene and a list of putative targets. Then, within each relevant cell subtype, we calculated the top 200 subtype markers ranked by expression ratio fold changes using the FindMarkers function in Seurat (49) and intersected the markers with the raw regulons to filter transcription factors and targets. In this analysis, for simplicity, we merged the markers of the two anterior-ventral NKX2-1+ subtypes (AV NKX2-1 LMO1 and AV NK2-1 NKX6-2) and the markers of the two GE NKX2-1+ subtypes (GE RG NKX2-1 DLK1 and GE RG NKX2-1 OLIG1, respectively. To identify co-expressed transcription factor and targets, we correlated their expression across pseudobulk samples. Specifically, we ordered the cells along the UMAP-1 axis calculated via the Seurat RunUMAP function with a setting of dimension equal to one, parcellated cells into 30 equal-width bins along the axis and removed bins with less than 15 cells. Average expression was calculated in each bin, followed by assessing the Pearson correlation of the expression of transcription factors and the predicted targets. To further evaluate the robustness of the correlation, we also permutated the gene expression in each subtype, maintaining the average expression levels and variance of each gene but disrupting the gene-gene correlations. The permutation was repeated for 1000 times and the p value for the correlation of a given gene pair is defined as (n + 1) / (1000 + 1), where n represents the number of permuted correlation coefficients exceeding the actual correlation coefficient. Only transcription factor-target pairs with significant correlations (p value < 0.05 and coefficient > 0.3) were retained. The regulons in each cell subtype were then merged and visualized in a network with arrows indicating regulatory directions from transcription factor to targets and nodes colored by the cell subtypes showing the significant regulation. In addition, we curated several signaling pathways relevant to organizer functions and overlaid the information on the network (Fig. 2D).
To complement the analysis illustrating how signaling components are utilized across telencephalic domains, we performed the following two analyses: 1) measured the enrichment of signaling pathway genes in each organizer subtype by calculating the odds ratios of the overlapping between the signaling pathway genes and organizer subtype markers; 2) intersected the signaling pathway genes with organizer subtype markers.
Transcriptomic comparison between mouse and macaque organizer domain subtypes
To assess the transcriptomic similarity between macaque and mouse telencephalic organizer domains (25), we derived the subtype markers in each dataset using the FindMarkers function in Seurat and extracted the shared subtype markers between the two species. These included many key genes labeling homologous cell subtypes. Average expression of the shared subtype markers were calculated across subtypes followed by Pearson correlation coefficient measurement for each pair of subtypes between the two datasets. To avoid noise from background transcriptomic similarities, any correlation coefficients below the 80% quantile of all values were removed. The filtered subtype similarity was visualized in a Sankey plot, which illustrates subtype matching between the two species.
Alternatively, we calculated the enrichment of mouse subtype markers in this dataset through the AUCell algorithm (61). Because this method uses rank-based expression values to assess expression enrichment, it is robust to potential quality differences between the mouse and macaque datasets. For a given set of mouse subtype markers, we averaged its enrichment scores in each macaque subtype and visualized the results in a heat map, which recapitulated the subtype similarity patterns shown by the above correlation-based analysis.
In order to find species-specific expression patterns in homologous subtypes and avoid potential batch effects, we set a high threshold for the differential expression test. To identify macaque-enriched genes, we first extracted the top 100 markers for each macaque subtype and removed the genes that were also markers of the homologous mouse subtypes. Next, a minimum expression ratio of 0.2 was required in macaque subtypes whereas a maximum expression ratio of 0.05 was set in the mouse homologous subtypes. For each macaque subtype, the top 10 genes ranked by their expression fold changes between the given subtype and the background macaque cells were selected and visualized. The same approach was used to find mouse-enriched genes in homologous subtypes. Notice that the expression of three antihem canonical genes, TGFA, Neuregulin 1 (NRG1) and Neuregulin 3 (NRG3) (53), was not detected in the macaque putative antihem (PC SFRP2). Interestingly, we found these three genes be specific to mouse antihem subtypes (not shown). However, the expression of these genes was detected in other cell subtypes, ruling out genome annotation issue. In commensurate, realignment of the data to an independent genome annotation from ENSEMBL showed clear expression of SFRP2 in macaque PC SFRP2, but not for these three genes. Another possibility is that the sequencing depth for this subtype was too low to capture the signals. However, PC SFRP2 has an average of 2825 UMIs which is comparable to other subtypes expressing the three genes, suggesting that the sequencing depth might not be the issue. Thus, the undetectable level of these three genes in the monkey might be attributed to species differences in the timing of expression or in the developmental states of the antihem cells (27).
Ligand-receptor mediated cell-cell communication between patterning centers and early neural stem cells
We applied two complementary expression-based approaches, CellChat and CellphoneDB (63, 64), to infer putative cell-cell communications between organizer domain cell subtypes and early neural stem cells. In these analyses, we only included subtypes potentially secreting patterning ligands (RPC, anterior ventral domain subtypes, and cortical hem) and neural stem cell subtypes responding to these molecules (that is, NESC and early vRG from anterior, posterior regions and ganglionic eminence). Each subtype was down-sampled to have equal number of cells (1000 cells), otherwise subtype size differences could affect marker detection in CellChat and permutation in CellphoneDB. In both analyses, we set a minimum expression ratio of 0.05 and p value threshold at 0.05. For the resulted ligand-receptor interactions, we only considered the directions with ligands expressed in organizer domain subtypes and receptors in regional neural stem cells. Because the output results from the two analyses are largely shared, we mainly used the CellChat-based results and incorporated additional interactions reported only by CellphoneDB-based analysis.
To get a broad view of the interaction patterns between ligand-receptor pairs, we performed t-SNE analysis using the interaction matrix, with rows as ligand-receptor pair names and columns as cell subtype pairs. In addition, we clustered the ligand-receptor pairs based on their orchestrated cell-cell interaction patterns using robust sparse K-Means clustering algorithm. The resulted 10 clusters are well separated on the t-SNE layout and further confirmed the distinct cell-cell interaction patterns mediated by ligand receptor pairs.
Lineage construction from organizer domain progenitors to offspring cells
To define the lineage progression from organizer domain progenitors to their progeny cells and also delineate the gene cascades along the lineage, RNA velocity analysis using scVelo package (65) was applied. In each lineage, UMAP layout was first obtained via the RunUMAP function in Seurat and the resulted Seurat object was converted to anndata for scVelo anslysis. For simplicity and avoiding cell cycling genes driving the gene cascades, cycling cells were not included in the analysis. After data filtering, normalization, identification of highly variable genes and computing moments for velocity estimation, dynamic model was applied to compute the RNA velocity vectors and pseudotime. The top 300 genes showing transcriptional variation along the lineages were visualized on heat maps.
Transcriptomic comparisons with published data for neural stem cells and neurons
We applied the following two methods to evaluate cell subtype matching between datasets for neural stem cells and neurons. In the first method, cross-dataset cell subtype similarity was measured by Pearson correlation coefficients. Specifically, the intersection of the highly variable genes between this study and a given published dataset were selected, and the average expression of these genes across each cell subtypes were computed via the AverageExpression function in Seurat (49) followed by log-transformation. Pearson correlation coefficients were calculated on the log-transformed average expression for each pair of subtypes between this study and the given published dataset. In the other approach, cells from this study and a given published dataset were integrated and visualized on UMAP to evaluate the subtype alignment. Because there were prominent batch effects between this study and publish datasets, largely attributed to differences of species, developmental stages and technical approaches, we applied Seurat integration algorithm (49) as a stringent method to remove batch effects. Here, the intersection of the top 2,000 highly variable genes from each study were used for canonical correlation analysis, followed by anchor finding ad hierarchical integration of normalized data using the IntegrateData function. The integrated data were then scaled, used for principal components analysis and UMAP visualization.
Region-specific gene expression cascades
We used the following approaches to construct the region-specific expression cascades. For each subtype in a given region, we calculated the genes showing expression enrichment in this region compared to all other regions using Wilcoxon Rank Sum test. To avoid the influence of cell number differences, for each subtype we downsampled each region to have the same number of cells. The differential expression analysis results were further filtered based on expression ratios, fold changes of average expression, fold changes of expression ratio and Bonferroni-adjusted P values, to get genes with most salient regional enrichment. By leveraging the defined pseudotime that organize cells from different regions on the same scale, we parcellated cells from the four regions into bins with equal pseudotime width. We then calculated the average gene expression along the pseudotime bins for each region and fitted the expression into impulse models (linear, single sigmoid or double sigmoid) implemented in the URD package (66), which returned the pseudotime points representing where gene expression arises and diminishes. We thus ordered regionally enriched genes based on the earliest subtypes that they displayed regional enrichment, followed by the predicted pseudotime points denoting gene expression on and off. To gain a better visualization of the expression patterns, average expression along the pseudotime bins were smoothed using loess function in R.
Gene Ontology enrichment analysis
Gene ontology (GO) enrichment analysis was performed by the Bioconductor package ‘topGO’ (http://bioconductor.org/packages/release/bioc/html/topGO.html) using the Fisher’s exact test followed by FDR adjusting P values. Only GO terms under biological processes were included in the analyses and a threshold of FDR < 0.1 was selected to pick the significant terms.
Augur and differential gene expression analysis assessing transcriptomic divergence between the shared subtypes across different regions
In order to evaluate the magnitude of regional differences between different cell types, which might come from the same lineage (for example, IPCs, nascent and mature excitatory neurons) or belong to the same cell class (for example, LHX6+ versus NR2F2+/SP8+ interneurons), we applied the Augur algorithms to measure the transcriptomic separability of regions for each relevant subtype (67). To recapitulate regional variation as much as possible, for each batch we extracted the highly variable genes across the cells from all the analyzed brain regions and used the SelectIntegrationFeatures in Seurat to identify the top regionally variable features. In running Augur algorithms, these highly variable genes were directly used, with the mode set to “velocity” to avoid additional detection of highly variable genes. The Augur analysis was performed for each pair of regions, which provided a detailed view of transcriptomic divergence of cell types across brain regions.
In addition, we used the number of differentially expressed genes to evaluate regional difference changes along RG progression and excitatory neuron differentiation and maturation (Figs. 4C and 5C). We parcellated cells into different equal-width bins along the pseudotime and downsampled the cells from each region to have a balanced number of cells across bins and regions. Then we applied Wilcoxon Rank Sum test and calculated the number of differentially expressed genes between regions along the pseudotime bins followed by visualizing in log scale and smoothed via loess function.
Hierarchical clustering of excitatory neuron lineage subtypes across cortical regions/areas
To check whether the regional differences of the excitatory neurons are correlated with the anatomical proximity of the brain regions they populate, we leveraged the refined regions/areas sampled at E93 and E110. We first calculated the highly variables genes at each batch (here is individual) and used the Seurat SelectIntegrationFeatures function to capture the top 2000 genes with highest expression variability across analyzed brain regions. For a given excitatory neuron subtype, average expression was calculated in each region and cells from different regions were down-sampled to have the consistent number of cells (100 cells) prior to average expression calculation. The resulted average expression of the 2000 highly variable genes from the shared subtype in different regions were used for hierarchical clustering. Here, the distance matrices were defined as Pearson correlation coefficients subtracted from 1 and were subsequently used by the hclust function with “ward.D2” algorithm for clustering. Dendrogram visualization was achieved via the circlize R package.
To obtain robust estimate of region co-clustering, we generate 1000 bootstrap replicates from the above 2000 highly variable genes, randomly extracting 80% of genes for each replicate. For each replicate, the same subtype across regions were clustered using the above strategy followed by cluster separation via splitting the hierarchical tree to k=3 clusters. The frequencies of region co-clustering were measured and visualized by heat maps. In addition, we permutated the gene expression for 1000 replicates, keeping the gene-wise characteristics (for example, mean expression, variance) but destroying the gene-gene relationship. The average expression from the 1000 replicates of permuted data were used following above bootstrap clustering strategy to see how often subtypes from different regions cluster together. By comparing the co-clustering frequency in the actual data versus the permuted data, we confirmed the region co-clustering in excitatory neuron subtypes reflect true regional differences.
Correlation of regional gene expression specificity between cell types
To assess the effect of region-specific environment on cell type identities, we correlated the region-specificity of gene expression across multiple cell types. Specifically, we identified all the genes divergently expressed across brain regions and assessed their expression enrichment in each region. The gene expression fold changes were calculated by dividing the average expression in the given region by the average expression in other background regions, with a pseudo-value of 0.1 added to both the numerators and denominators. The resulted fold changes were log2 transformed, termed as the regional enrichment scores here. Within each region, the pairwise comparisons of such enrichment scores were visualized in dot plots and Pearson correlation coefficients were also calculated.
Analysis of bulk-tissue RNA-seq data
We applied STAR (68) to align the raw reads to the same genome we used for the scRNA-seq data, followed by FeatureCounts (69) to compute the read counts. DESeq2 (70) was utilized to perform differential expression tests between conditions, selecting only genes with false discovery rate smaller than 0.01 and log2 fold changes bigger than 1. In the differential expression analysis, we parcellated the in vitro time points to three stages (early: days in vitro [DIV] 1-5; middle: DIV 6-11; late: DIV 14-20).
We have conducted two types of analyses to correlate the in vitro results with the in vivo studies. First, we identified region-specific genes in the in vitro NSCs (DIV 1-5) and measured their regional enrichment in the age-matched NSCs in vivo via Wilcoxon Rank Sum test. We only considered genes to be significant if they have Bonferroni-correct P values smaller than 0.05, expression ratios larger than 0.05 and fold changes of expression larger than 0. The results were visualized in volcano plots and the genes displaying consistent regional enrichment between in vitro and in vivo were highlighted and labeled. This analysis showed many in vivo NSC regional identities were maintained in vitro in NSC stage. In the second analysis, we extracted genes showing enrichment in the late stage (DIV 14-20) but not in the early NSC stage (DIV 1-5). Similar regional enrichment tests were performed for these genes, with the exception that the tests were performed on age-matched excitatory neuron subtypes, that is E54-64 L6 CT (ExN SOX5 SYT6) and L6B (ExN SOX5 NR4A2 GRID2) for the regions-specific genes obtained in the differentiated neurons from E42 NSCs in vitro, and E93 upper layer excitatory neuron subtypes (ExN CUX2 ADRA2A and ExN CUX2 ACTN2) for those obtained in the differentiated neurons from E77 NSCs in vitro.
Evolutionary comparisons between human and macaque midfetal arealization signatures
We leveraged the transcriptomic-based age matching between human and macaque (13) to select the age-matched human and macaque scRNA-seq neocortical data for the evolutionary comparisons. Here, we used macaque E77-78 data in this study and the gestation week (GW) 18-19 data from a developing human scRNA-seq dataset (16), with only prefrontal and occipital regions included as there are more cells in these two regions for all the major excitatory neuron types. The human data were reprocessed and annotated in the same manner as we did for this study. To avoid potential sequencing depth bias, we subset the human and macaque data using ortholog genes and then downsampled each homologous cell subtype to have the same number of UMIs. Data normalization were performed again using only the the ortholog genes. Region specific genes were identified in each species using the FindMarkers function in Seurat. To identify the conserved region-specific signatures, we intersected the region-specific genes from the two species and visualized the top 20 genes ranked by their average expression ratio fold changes. We then used the same FindMarkers function to calculate the genes enriched in human or macaque homologous cell types, but with more stringent thresholds to avoild bias: minimum expression ratio of 0.1 in the enriched species, maximum expression ratio of 0.1 in the depleted species, expression ratio fold changes bigger than 1.5, and Bonferroni-adjusted p values smaller than 0.01. These genes were further intersected with region-specific genes in each species to obtain species- and region-specific signatures.
Hierarchical clustering of regional genes expression patterns for inhibitory neurons
Although Augur algorithm detected lower regional transcriptomic differences in inhibitory neurons, we could still find certain genes differentially expressed across cortical regions in the shared inhibitory neuron types. In order to obtain an overview of their global expression patterns across regions, we applied a hierarchical strategy clustering genes based on their expression similarity. Here, we only considered two major cortical inhibitory neuron groups: LHX6+ (excluding the CRABP1+ population not detected in neocortex) cells and NR2F2+/SP8+ cells. Regionally divergent genes were obtained through Wilcoxon Rank Sum test with the Bonferroni-corrected P value threshold set at 0.01. In case cell numbers affect the number of differentially expressed genes, we downsampled the cells in each region to the same level. For each region, we generated 20 pseudobulk samples each containing 200 random cells and calculated the average expression of the regionally-divergent genes across all pseudobulk replicates. The resulted expression matrix was used for hierarchical clustering, with the distance matrix defined as one minus Pearson correlation coefficients between gene pairs and clustering algorithm set as “ward.D2”.
Lineage inference of the switch between neurogenesis and gliogenesis
To explore the developmental transition from neurogenesis to gliogenesis, we employed two different approaches: unsupervised transcriptomic clustering by Seurat (49) analysis pipeline and cell lineage tracing by Monocle (version 2) analysis pipeline (71), to analyze the transcriptional association in-between radial glial cells, excitatory neurons and glia cells and define their lineage relationship.
In the Monocle analysis pipeline, we firstly recruited Seurat FindMarkers function to perform differential expression analysis for any pair of cell subtypes in each developmental stage. Subsequently, the differential expressed genes were accumulated and used to infer cell trajectory. Following up the recommended workflow, the suggested default parameters in Monocle were preferred, except that the ‘DDRTree’ method was use as reduction model and the batch correction was introduced. To compute the pseudotime for each cell along the cell trajectory tree, the tree root was manually selected through analyzing the tree structure and the distribution of cellular nature ages. Lastly, we used Seurat FindMarkers function to perform differential expression analysis for any pair of branches to identify genes specific to each tree branch, which led to a proxy for detecting the transcriptional program that dominates the segmentation and emergency of each tree branch.
Global cross-dataset comparison for glia cells
The glial cell types in the current study were compared to the external datasets, including datasets from prenatal humans, adult monkeys, and lifespan mice, to confirm the quality of cells and the precision of cell type annotation. For astrocytes, we defined three subtypes, which together with astrocyte precursor and glia precursor were compared among multiple brain regions to reveal their regional distribution. One external astrocyte dataset collected from different cortical layers in P14 mice (72) was compared with our astrocytes to show their laminar distribution, and another external astrocyte dataset collected from dorsolateral prefrontal cortex in adult macaque (30) were compared with our astrocytes to investigate the correspondence between immature and mature stages. We used specScore script, as previously detailed in (18), to compute specificity score for each gene in each subtype and correlated subtypes across datasets using the specificity scores, with the pairwise subtype similarity visualized by alluvial plots. On the other hand, the cross-dataset comparison was conducted by using abovementioned UMAP pipeline. Firstly, the different datasets were separately wrapped according to Seurat analysis workflow, which includes abovementioned a range of processes. Secondly, fastMNN (51) was used to correct unwanted batch variation by choosing the different datasets as the major source of systematic variation. Alternatively, other integration methods including Harmony (52) were considered to verify the results reported by fastMNN, whose analyses were not shown when an negligible difference of cell type correspondence was observed.
Predication of ligand-receptor mediated cell-cell communication across refined cortical areas in E93-110
We used the E93 and E110 macaque data for this analysis as more refined brain areas were sampled at this stage and most of neuron and glial cell types have emerged, in particular for astrocytes that gradually increase and interplay with other cells. The communication in-between cells can be inferred by correlating the expression of ligand and the corresponding receptor genes. We utilized CellChat (64) to compute the averaged communication in-between major cell types, including excitatory neurons, interneurons, astrocyte, enIPCs, OPC/oligodendrocyte, microglia and endothelial cells. The aggregation of cell subtypes into major cell types allow us to achieve high accuracy and reduce single cell noise. Cell-cell communication were computed in each brain region separately, and subsequently the interaction strength between any pair of cell types were averaged among brain regions to provide a proxy of the general tendency of cell-cell communication.
Compilation of brain disease risk gene lists
We compiled disease-risk genes from DISGENET (https://www.disgenet.org/) (73) and filtered-out those diseases having fewer than 30 risk genes associated. Genes from “Mixed oligoastrocytoma” and “oligodendroglioma” were combined in the “Mixed Oligoastrocytoma+Oligodendrogliomas” list (abbreviated as M. Oligoastr.+ Oligo.). Also, genes associated with any medulloblastoma were combined in the “Medulloblastomas” list.
Multiple genome wide associated studies (GWAS) were used to collect gene lists for the study: Alzheimer’s disease (AD) (74), anorexia nervosa (AN) (75), autistic spectrum disease (ASD) (76), bipolar disorder (BD) (77), intelligence quotient (IQ) (78), major depressive disorder (MDD) (79), neuroticism (NEUROT) (80), Parkinson’s disease (PD) (81) and schizophrenia (SCZ) (82).
Genes identified by running Multi-marker Analysis of GenoMic Annotation (MAGMA) (83) were also included for the following conditions: attention-deficit/hyperactivity disorder (ADHD), AD, AN, ASD, BD, IQ, MDD, NEUROT, obsessive-compulsive disorder (OCD) (84), PD, SCZ and Tourette syndrome (TS) (85). Only genes with a nominal p-value of less than 0.05 were used and we selected the top 200 genes according to their p-value.
Genes implicated in ASD susceptibility were also obtained from the SFARI database (https://gene.sfari.org). Only genes of SFARI categories S (syndromic), 1 (high confidence), 2 (strong candidate) and 3 (suggestive evidence) were employed.
Additionally, high-confidence ASD genes from (86), and a set of genes involved in developmental delay (DD) from the Deciphering Developmental Disorders Study consortium (87).
Here, we included all the regions for each cell subtype in majority of the disease risk gene analysis, except when intersecting disease risk genes with region-specific subtype markers.
Cell type enrichment of disease gene expression
scRNA-Seq data were preprocessed following the guidelines for EWCE analysis (88). In brief, normalization and variance stabilization of scRNA-seq data using regularized negative binomial regression was performed with the sctransform R library (89). SCT-corrected counts were computed for genes being marker genes of any subtype with an adjusted p-value < 0.05 and having a 1-to-1 matching human ortholog. Macaque-to-human orthologs were retrieved using the ortogene R library (https://github.com/neurogenomics/orthogene).
Then, we performed EWCE’s bootstrap enrichment test of the disease-associated gene lists previously defined (20000 repetitions, geneSizeControl=FALSE, controlledCT=NULL and mtc_method=‘BH’). Only the risk genes present in the dataset after SCT normalization were tested.
Identification of disease risk genes showing as organizer domain subtype markers
Marker genes were computed using the Wilcoxon Rank Sum test on log2-normalized data by FindAllMarkers from the Seurat package (49). We computed marker genes of all the cell subtypes and the organizer domain subtypes independently.
In those subsets, expression data were scaled. Averages of gene scaled expression were obtained for every gene in the disease-associated gene lists and we computed the median of averages per list. We also retained the number of genes from each list that were expressed in at least 10% of cells for each cell subtype.
Regarding patterning centers’ marker genes, we were interested in those that were expressed distinctively in one of the PC subtypes. For that, we selected the marker genes requiring them to be expressed in at least 10% of cells in the respective PC subtype and in less than 10% of cells of other organizer domain subtype cells. With those markers, we created 2x2 contingency tables for each disease-risk gene list and subtype with counts of genes being or not subtype markers, and genes being or not disease-associated. Using those tables, we estimated the log2 odd ratio of being disease-associated and subtype marker using the R function fisher.test.
Among those disease-risk genes showing as markers for a organizer domain subtype (132 genes), we also investigated whether their expression is restricted to the given organizer subtype versus other non-organizer cell subtypes. For this, we only considered the neural lineage cell subtypes (that is, all the subtypes excluding immune, red blood, vascular and mesenchymal cells). We required the genes to show as the marker of a given organizer subtype and also expressed in no more than three other neural lineage cell subtypes (genes expressing in more than 10% cells of a given subtype were considered to be expressed). This resulted 26 genes as shown in Fig. 7B.
Expression enrichment of disease risk genes across radial glial cells
To gain a detailed view of the expression enrichment patterns of disease risk genes, we applied AUCell (61) enrichment analysis on the disease gene list across neural stem cell subtypes. To have robust estimation of each subtype and further avoid the influence of certain low-quality cells, we generated 100 pseudobulk samples in each neural stem cells by randomly pooling 200 cells, followed by calculating average gene expression in each of the pseudobulk samples. AUCell analysis was directly performed on these pseudobulk samples together with the disease gene list, with the results visualized on a heat map. Diseases were ordered according to their peak enrichment along the x axis (the subtypes). Because we see many disease risk genes show high enrichment in outer radial glial cells, we wondered if it was caused by quality bias. Although AUCell package is robust to data quality differences, as it utilizes rank-based method to assess the separability of query genes versus background gene, we further subset the data to have 2000 UMIs per cell (cells with less than 2000 UMIs were not included) and generated pseudobulks and performed enrichment following the same strategy as described above. This overall lead to a similar enrichment pattern.
We next decided to identify the genes underlying the dynamics disease enrichment patterns. For each disease, we extracted its risk genes and correlated (Pearson correlation) their expression patterns with the given enrichment pattern of the disease. We set a threshold of 0.7 to capture the strongest signals and resulted genes were visualized in a heat map.
Intersection of disease risk genes and region-specific signatures
We set relatively high thresholds to identify disease genes showing prominent regional and cell-type enrichment. For each region, we calculated cell type markers and regionally-enriched genes using Wilcoxon Rank Sum test followed by setting the same thresholds for both analyses: Bonferrroni-corrected p value < 0.01, log fold changes of average expression > 0.25, expression ratio fold changes > 1.25, and expression ratio > 0.2. The intersection of the cell subtype markers and regionally-enriched genes were further overlapped with disease risk genes and the results were visualized in fig. S19. The intersection of the top 25 cell subtype markers and top 25 regionally-enriched genes ranked by fold changes of expression ratios were overlapped with disease risk genes and visualized in Fig. 7D.
Monkey fetal cortical NSC culture and in vitro differentiation
Fetal macaque brains were isolated from E42 and E77 feti. Telencephalic regions were identified and dissected and single cells isolated in HBSS, as described above. After centrifugation, HBSS was removed and cells were switched to DMEM/F12 medium with N2 supplement (described below). Monkey region-specific telencephalic cells were cultured using an adapted version of a protocol previously reported for mice (29). 3-5 x 106 cells were plated over 10 cm culture plates (Falcon, 35-3003), previously coated with poly-L-ornithine (PLO) (Sigma, P3655) and fibronectin (FN) (R&D Systems, 1030FN), and expanded at 37 °C, 5% O2 and 5% CO2 for 5-6 days in DMEM/F12 medium (Mediatech 16-405-CV) plus N2 supplement, containing 25 μg/ml bovine insulin (Sigma, I6634), 100 μg/mL apotransferrin (Sigma, T2036), 20 nM progesterone (Sigma, P8783), 100 μM putrescine (Sigma, P5780), 30 nM sodium selenite (Sigma, S5261), penicillin/streptomycin (Life Technology, 15140-122). We refer to DMEM/F12 medium with N2 supplement as N2 medium, hereafter. 20 ng/ml bFGF (R&D Systems, 4114-TC) was added daily and the medium was changed every other day. NSCs were lifted with Accutase (STEMCELL Technologies, 07920) and aliquots of 3-5 x 106 cells/500 μl of N2 were frozen at −80 °C. The in vitro neurogenesis experiment shown in fig.S13B was performed as following: thawed NSCs were expanded in presence of 20 ng/ml FGF2 for 5 days until confluence. Then region-specific NSCs were dissociated with Accutase and passaged into PLO/fibronectin-coated 6 well plates (Falcon, 35-3046), at a density of 300.000/well x 2 wells in N2 medium + 1 ng/ml FGF2, which favors neurogenesis (29). Cells were expanded from from DIV 1 to 5 and FGF2 was added daily. Differentiation of the NSCs was induced at DIV5 by FGF2 withdrawal, keeping the cells in N2. At DIV 7, N2 was replaced with NeuroBasal medium (NB) (Life Technology, 12348-017), containing 25 μg/ml insulin, 30 nM sodium selenite, Glutamax (Life Technology, 35050061), 1x B27 (Life Technologies, 17504-044), 10 ng/ml BDNF (R&D Systems, 248-BD) and 10 ng/ml NT-3 (R&D Systems, 267-N3) until the end of experiment at DIV17 or DIV20.
Bulk RNA-sequencing library preparation
At the time points across the in vitro differentiation indicated in fig.S13B, total RNA was extracted from 2 different wells/condition of the monkey primary region-specific cells, using RNeasy Mini Kit (Qiagen, 74104), according to manufacturer’s protocol.
RNA Seq Quality Control: Total RNA quality was determined by estimating the A260/A280 and A260/A230 ratios by nanodrop. RNA integrity was determined by running an Agilent Bioanalyzer gel, which measures the ratio of the ribosomal peaks. Samples with RIN values of 7 or greater were considered for library prep.
RNA Seq Library Prep: mRNA was purified from approximately 200ng of total RNA with oligo-dT beads and sheared by incubation at 94°C in the presence of Mg (Roche Kapa mRNA Hyper Prep Cat. KR1352). Following first-strand synthesis with random primers, second strand synthesis and A-tailing were performed with dUTP for generating strand-specific sequencing libraries. Adapter ligation with 3’ dTMP overhangs were ligated to library insert fragments. Library amplification amplifies fragments carrying the appropriate adapter sequences at both ends. Strands marked with dUTP were not amplified. Indexed libraries were quantified by qRT-PCR using a commercially available kit (Roch KAPA Biosystems Cat. KK4854) and insert size distribution determined by either the Agilent Bioanalyzer. Samples with a yield of ≥ 0.5 ng/ul and a size distribution of 150-300bp were used for sequencing.
Flow Cell Preparation and Sequencing: Sample concentrations were normalized to 1.2 nM and loaded onto an Illumina NovaSeq flow cell at a concentration that yields 25 million passing filter clusters per sample. Samples were sequenced using 100bp paired-end sequencing on an Illumina NovaSeq6000 according to Illumina protocols. The 10bp unique dual index was read during additional sequencing reads that automatically follow the completion of read 1. Data generated during sequencing runs were simultaneously transferred to the YCGA high-performance computing cluster. A positive control (prepared bacteriophage Phi X library) provided by Illumina was spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time.
Data Analysis and Storage: Signal intensities were converted to individual base calls during a run using the system’s Real Time Analysis (RTA) software. Base calls were transferred from the machine’s dedicated personal computer to the Yale High Performance Computing cluster via a 1 Gigabit network mount for downstream analysis. Primary analysis - sample de-multiplexing and alignment to the human genome - was performed using Illumina’s CASAVA 1.8.2 software suite.
Human and macaque telencephalic organoid culture
The human iPSC line Y6 was provided by Yale Stem Cell Center. This cell line was generated from neonatal skin fibroblasts using CytoTune™-iPS Reprogramming Kit (Invitrogen, A13780). Chromosome analysis was performed on cultured cells. Of the five metaphases examined, no structural and numerical abnormality was noted and the karyotype was consistent with that of a female (46, XX) complement. The pluripotency of the cells was confirmed by teratoma assay. Macaque iPSC line was generated by reprogramming E40 macaque lung fibroblasts using CytoTune-iPS 2.0 Sendai Reprogramming kit (ThermoFisher Scientific, A16517) and authenticated by morphology and karyotyping. All human and macaque iPSC lines were tested negative for mycoplasma contamination, checked monthly using the MycoAlert Mycoplasma Detection Kit (Lonza). For maintenance of pluripotency, cells were dissociated to single cells with Accutase (STEMCELL Technologies, 07920) and plated at a density of 1 × 105 cells per cm2 in Matrigel (BD, 354277)-coated 6-well plates (Corning, 3516) with mTeSR1 (STEMCELL Technologies, 85850) containing 5 μM Y27632, ROCK inhibitor (STEMCELL Technologies, 72302), as previously described (29). ROCK inhibitor was removed 24 h after plating, and cells were cultured for another four days before the next passage. Telencephalic organoids were generated by the directed differentiation protocol as previously described (31, 90). Human and macaque iPSCs were dissociated into single cells using Accutase. Neural induction was directed by dual SMAD and WNT inhibition (91) using a neural induction medium composited with 50% (v/v) DMEM/F12 (ThermoFisher Scientific, 11330032), 50% (v/v) Neurobasal medium (Thermo Fisher Scientific, 21103049), 1% (v/v) N2 (Thermo Fisher Scientific, 17502048), 2% (v/v) B27 minus vitamin A (Thermo Fisher Scientific, 12587010), 1% (v/v) MEM non-essential amino acid (Thermo Fisher Scientific, 11140050), 1% (v/v) GlutaMAX (Thermo Fisher Scientific, 35050061), 1% (v/v) Penicillin/Streptomycin (Thermo Fisher Scientific 15140122), 0.1 mM 2-Mercaptoethanol (Sigma-Aldrich, M3701), 1 μg/ml heparin (STEMCELL Technologies, 07980). The dissociated cells were reconstituted with the neural induction medium and plated at 10,000 cells per well in a 96-well v-bottom ultra-low-attachment plate (Sumitomo Bakelite, MS-9096V). To increase the cell survival and aggregate formation, 10 μM Y-27632 was added for the first day. For cortical organoids (COs), cells were cultured with the neural induction medium supplemented with 100 nM LDN193189 (STEMCELL Technologies, 72147), 10 μM SB431542 (Sigma-Aldrich, S4317), and 2 μM XAV939 (TOCRIS, 3748) for the first 4 days and then with the medium supplemented with 100 nM LDN193189 and 10 μM SB431542 for the next 4 days. For medial ganglionic eminence organoids (MGEOs), cells were cultured with the neural induction medium supplemented with 100 nM LDN193189, 10 μM SB431542, and 2 μM XAV939 for 8 days (90). After 8 days, both Cos and MGEOs were transferred to a 6-well ultra-low-attachment plate (Corning, CLS3471) and cultured with organoid growth medium on an orbital shaker (Thermo Fisher Scientific, 88881101) rotating at a speed of 90 rpm to enhance the nutrient and gas exchanges. COs were grown in organoid growth medium composited with 50% (v/v) DMEM/F12, 50% (v/v) Neurobasal medium, 1% (v/v) N2, 2% (v/v) B27 minus vitamin A, 1% (v/v) MEM non-essential amino acid, 1% (v/v) GlutaMAX, 1% (v/v) Penicillin/Streptomycin, 0.1 mM 2-Mercaptoethanol, 2 μg/ml heparin, 2.5 μg/ml human insulin (Sigma-Aldrich, I9278), and 200 ng/ml laminin (Thermo Fisher Scientific, 23017015). For MGEOs, 1X B27 plus vitamin A (Thermo Fisher Scientific, 17504044) was used for the organoid growth medium, and human SHH (R&D System, 1845-SH/CF) and 1 μM purmorphamine (TOCRIS, 4551) were additionally added for ventral specification. From day in vitro (DIV) 21, organoids were cultured with a neuronal maturation medium composited with Neurobasal medium, 1% (v/v) N2, 2% (v/v) B27 plus vitamin A, 1% (v/v) MEM non-essential amino acid, 1% (v/v) GlutaMAX, 1% (v/v) Penicillin/Streptomycin, 0.1 mM 2-Mercaptoethanol, 2 μg/ml heparin, 1% (v/v) Chemically defined lipid concentrate (Thermo Fisher Scientific, 11905031), 10 ng/ml BDNF (R&D System, 248-BD-025; or Peprotech, 450-02), 10 ng/ml GDNF (Peprotech, 450-10), 10 ng/ml NT3 (R&D System, 267-N3-025; or Peprotech, 450-03), 200 μM cAMP (Sigma-Aldrich, D0627) and 200 μM ascorbic acid (Sigma-Aldrich, A92902). For the regionalization of human COs and MGEOs, 100 ng/ml RSPO3 (R&D System, 3500-RS/CF) or 100 ng/ml FGF8b (R&D System, 423-F8/CF) were added to the organoid growth medium from day 8 to day 21. For the GAL and GALP treatment, macaque COs were exposed to 30 ng/ml of human GAL (Phoenix Pharmaceuticals, 026-01), 30 ng/ml human GALP (Phoenix Pharmaceuticals, 026-51), or both from DIV 46 to 60 and collected on DIV 60. Human COs and MGEOs were exposed to 30 ng/ml GALP from DIV 8 to 21 and collected on DIV 35. Brain organoids were collected and fixed in 4% PFA for 24 hours at 4 °C. Then, organoids were immersed in step-gradients of sucrose/PBS up to 30% for 2 days at 4 °C, embedded in OCT and frozen at −80 °C. Sections were prepared at 12 μm on a Leica CM3050S cryostat and stored at −80 °C until use.
Immuno-cytochemistry of the organoids
Immuno-cytochemistry of the human and monkey telencephalic organoids was started first rehydrating frozen slides in PBS. Blocking was done 1 hour RT in PBS- 10% normal donkey serum (Sigma-Aldrich, D9663) plus 0.5% tween-20. Incubation with primary antibodies was performed in PBS- 10% normal donkey serum plus 0.5% tween-20 at 4 °C, ON. The following primary antibodies were used at the concentration indicated: PAX6 (BioLegend, PRB-278P; 1:200); NKX2-1 (ABCAM, ab76013; 1:500); LMX1A (ABCAM, ab76013, 1:200); SP8 (ABCAM, ab73494; 1:200); ZIC4 (Lsbio, LS-B9905-50; 1:100); Galanin (Millipore Sigma, AB2233; 1:100); GALR2 (ABCAM, ab188753; 1:100); GALP (Novus biological, NBP2-84950; or Thermo Fisher Scientific, BS-11526R; 1:100); SOX2 (R&D Systems, AF2018; or MAB2018; 1:200); HuC/D (Thermo Fisher Scientific, A-21271; 1:500); KI67 (ABCAM, ab15580; 1:200); GABA (Sigma-Aldrich, A2052; 1:100); CTIP2 (ABCAM, ab18465; 1:1000). Secondary antibody incubation was performed in PBS 1 hour RT. Secondary antibodies were Alexa Fluor 488-, 594-, or 647-conjugated AffiniPure Donkey anti-IgG (1: 200; Jackson ImmunoResearch). Nuclei were counterstained with DAPI (Sigma, D8417). Finally, the slides were mounted using Vector mounting medium.
In utero intraventricular injection
Pregnant C57/BL6 mice were injected with pre-emptive analgesic Ethiqua (3.25 mg/Kg BW), 30 minutes before surgery, followed by injection of Ketamine-Xylazine mixture at 100 mg/kg and 10 mg/kg B/W, respectively. After 10-15 minutes animals were insentient, they were put dorsally on a sterile surgical cloth, kept over a heated pad (42 oC cycle of 30 min ON/OFF) throughout the procedure and limbs were fixed on the cloth using tape. Then, the ventral region of the animals were cleaned with an alcohol pad. A straight incision was made (~2-3mm) using a fine scissor. A surgical gauze with a circular hole (2-3 mm diameter) was soaked with sterile saline and kept over the site of incision. Using ring forceps, E11.5 embryos were taken out from the belly region and kept on gauze. In utero injection was performed with 100 ng of GALP peptide in 1μL (Phoenix Pharmaceuticals, 026-51; stock concentration: 1 μg/ml in PBS, diluted 1:10 in PBS + Fast Green 0.1%) injected into the lateral ventricle of the telencephalon through a glass capillary needle. The injection was seen going through the later ventricle with the Fast Green. After injection, the uterus was placed back in the peritoneal cavity and wound was sutured. A triple antibiotic paste was applied to the site of suture and the analgesic Meloxicam (5 mg/Kg B/W) was injected in the animal. The animals were placed on a heated pad (42 oC) under close monitoring until fully recovered. Embryos were harvested 24 hours after the injection. Two hours before harvesting the embryos, 100 mg/KG BW of EdU (Invitrogen, C10337) dissolved in saline solution was injected intra-peritoneally in the pregnant animals. Then the mice were sacrificed and the embryos collected and fixed as described above.
Immuno-histochemistry of the mouse brain tissue
Serial coronal sections (15 μm) were collected from the frontal to the caudal telencephalon. Each slide contained multiple serial sections of the mouse brain. Immuno-histochemistry (IHC) was performed initially rehydrating the frozen glass slides in PBS. Then the tissue was incubated with blocking solution, 10% normal donkey serum (NDS)/PBS including 0.1% tween-20 and 0.2% Triton X-100. Primary and secondary antibody incubations were diluited in blocking solution. Primary antibodies and dilutions: anti-goat Sox2 (R&D system, AF2018), dil: 1:200; anti-rabbit Ki67 (ABCAM, ab15580), dil:1:200. Secondary antibodies were Jackson DyLight donkey anti-(species) 488, 543, 647, and DAPI nuclear staining was Vector mounting medium H-1200. EdU staining (Invitrogen, C10337) was performed following the manufacturer’s protocol. Briefly, After drying the sections at room temperature for 10 minutes, the slides were in 0.5% Triton X100 for 20 minutes, then washed twice in 3% BSA for 10 minutes. Next, 500ul of reaction cocktail was added on each slide for 1hr at RT. After that, slides were washed in 3% BSA and 1 ml of DAPI (0.1μg/mL) was added for 5 minutes and mounted with Vector shield mounting media.
Single-molecule RNA in situ hybridization
RNA in situ hybridizations were performed by Advanced Cell Diagnostics, Newark, CA, using RNAscopeTM technology. Paired double-Z oligonucleotide probes were designed against target RNA using custom software. The probes used for rhesus macaque and mouse brain tissue samples are shown in table S11. RNAscope LS Fluorescent Multiplex Kit (Advanced Cell Diagnostics, Newark, CA, 322800) was used with custom pretreatment conditions following the instruction manual. Fixed frozen monkey and mouse fetal brain tissue slides were manually post-fixed in 10% neutral buffered formalin (NBF) at room temperature for 90 minutes. Then the slides were dehydrated in a series of ethanols and loaded onto the Leica Bond RX automated stainer, performing the reagent changes, starting with the pretreatments (protease), followed by the probe incubation, amplification steps, fluorophores, and DAPI counterstain. RNAscope 2.5 LS Protease III was used for 15 minutes at 40°C. Pretreatment conditions were optimized for each sample and quality control for RNA integrity was completed using probes specific to the housekeeping genes Polr2a, Ppib, and Ubc, which are low, moderate, and high expressing genes, respectively. Negative control background staining was evaluated using a probe specific to the bacterial dapB gene. Coverslipping was done manually using ProLong Gold mounting media at the end of each run.
Microscopy and imaging
Fluorescent monkey and mouse brain tissue specimens and organoids sections were imaged using a Zeiss LSM800 confocal microscope, or a Zeiss 510 Meta confocal microscope. Z-stack and tiled confocal images of brain slices and organoids were processed using Zeiss ZEN2009 and ImageJ (v.2.0.0-rc-69/1.52p). Slight artefactual defects of DAPI intensity were manually corrected with imageJ. When necessary, fluorescence intensity or contrast was slightly adjusted using the same parameters for all the spacimens.
Highthroughut quantification
Multiple images of the organoids and mouse sections were analyzed in batch using Volocity (v.6.3.1). The fluorescence intensities of all the channels were analyzed at single cell level obtaining the segmentation of the objects based on DAPI. Then Spotfire (v.12.3.0, TIBCO) software was used to identify the cells expressing high levels of a specific protein, setting the average or the third quartile (Q3) intensities of all objects as threshold. The final statistical analysis and visualization were performed using GraphPad Prism9.
Supplementary Material
Acknowledgements:
The authors thank the center for genome analysis (YCGA), animal resource center (YARC), veterinary clinical service and MacBrain Resource at Yale.
Funding:
This work was funded by: NIH grant R01MH113257 (to A.D.); grants MS20/00064 (ISCIII-MICINN/FEDER), PID2019-104700GA-I00 (/AEI/10.13039/501100011033), and Fundació LaMarató de TV3 (to G.S); NIH grant R01HG010898-01 (to G.S. and N.S.); NIH grants U01MH124619, MH122678, HG010898, MH116488 and DA053628 (to N.S.); NIH/NIDA DA023999 (to P.R.).
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability:
scRNA-seq data were deposited in NEMO Archive (RRID: SCR_002001) accessible at https://assets.nemoarchive.org/dat-fjx1jbr and NCBI GEO (GSE226451). The data can be visualized at http://resources.sestanlab.org/devmacaquebrain. Bulk RNA-seq data were deposited in NCBI GEO (GSE225482). Scripts are available at: https://github.com/sestanlab/Macaque_corticogenesis_scRNA-seq.
REFERENCES
- 1.Ypsilanti AR et al. , Transcriptional network orchestrating regional patterning of cortical progenitors. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N, The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 89, 248–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sur M, Rubenstein JL, Patterning and plasticity of the cerebral cortex. Science 310, 805–810 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Clowry GJ et al. , Charting the protomap of the human telencephalon. Semin Cell Dev Biol 76, 3–14 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR, Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Rakic P, Specification of cerebral cortical areas. Science 241, 170–176 (1988). [DOI] [PubMed] [Google Scholar]
- 7.O’Leary DD, Chou SJ, Sahara S, Area patterning of the mammalian cortex. Neuron 56, 252–269 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Rakic P, Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci 10, 724–735 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molnar Z et al. , New insights into the development of the human cerebral cortex. J Anat 235, 432–451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson MB et al. , Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang HJ et al. , Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JA et al. , Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y et al. , Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakken TE et al. , A comprehensive transcriptional map of primate brain development. Nature 535, 367–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowakowski TJ et al. , Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhaduri A et al. , An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trevino AE et al. , Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069 e5023 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Li M et al. , Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sousa VH, Fishell G, Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Curr Opin Genet Dev 20, 391–399 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cholfin JA, Rubenstein JL, Frontal cortex subdivision patterning is coordinately regulated by Fgf8, Fgf17, and Emx2. J Comp Neurol 509, 144–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storm EE et al. , Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831–1844 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Caronia-Brown G, Yoshida M, Gulden F, Assimacopoulos S, Grove EA, The cortical hem regulates the size and patterning of neocortex. Development 141,2855–2865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grove EA, Tole S, Limon J, Yip L, Ragsdale CW, The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125, 2315–2325 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Kiecker C, Lumsden A, The role of organizers in patterning the nervous system. Annu Rev Neurosci 35, 347–367 (2012). [DOI] [PubMed] [Google Scholar]
- 25.La Manno G et al. , Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Fukuchi-Shimogori T, Grove EA, Neocortex patterning by the secreted signaling molecule FGF8. Science 294, 1071–1074 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Martinez Arias A, Steventon B, On the nature and function of organizers. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shioda S, Kageyama H, Takenoya F, Shiba K, Galanin-like peptide: a key player in the homeostatic regulation of feeding and energy metabolism? Int J Obes (Lond) 35, 619–628 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Micali N et al. , Variation of Human Neural Stem Cells Generating Organizer States In Vitro before Committing to Cortical Excitatory or Inhibitory Neuronal Fates. Cell Rep 31, 107599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma S et al. , Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science, eabo7257 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibata M et al. , Regulation of prefrontal patterning and connectivity by retinoic acid. Nature 598, 483–488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim L, Mi D, Llorca A, Marin O, Development and Functional Diversification of Cortical Interneurons. Neuron 100, 294–313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W et al. , Origins and Proliferative States of Human Oligodendrocyte Precursor Cells. Cell 182, 594–608 e511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Bella DJ et al. , Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sloan SA, Barres BA, Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr Opin Neurobiol 27, 75–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poduri A, Evrony GD, Cai X, Walsh CA, Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willsey AJ et al. , Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schizophrenia C Working Group of the Psychiatric Genomics, Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weng Q et al. , Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell 24, 707–723 e708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhaduri A et al. , Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 26, 48–63 e46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariani J et al. , FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 162, 375–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couturier CP et al. , Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun 11, 3406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong W et al. , The neuropeptide landscape of human prefrontal cortex. Proc Natl Acad Sci U S A 119, e2123146119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadwell CR, Bhaduri A, Mostajo-Radji MA, Keefe MG, Nowakowski TJ, Development and Arealization of the Cerebral Cortex. Neuron 103, 980–1004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schober AL, Wicki-Stordeur LE, Murai KK, Swayne LA, Foundations and implications of astrocyte heterogeneity during brain development and disease. Trends Neurosci, (2022). [DOI] [PubMed] [Google Scholar]
- 46.Telley L et al. , Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, (2019). [DOI] [PubMed] [Google Scholar]
- 47.Sestan N, State MW, Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 100, 406–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willsey AJ et al. , The Psychiatric Cell Map Initiative: A Convergent Systems Biological Approach to Illuminating Key Molecular Pathways in Neuropsychiatric Disorders. Cell 174, 505–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T. et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolock SL, Lopez R, Klein AM, Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291 e289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haghverdi L, Lun ATL, Morgan MD, Marioni JC, Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korsunsky I et al. , Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Assimacopoulos S, Grove EA, Ragsdale CW, Identification of a Pax6-dependent epidermal growth factor family signaling source at the lateral edge of the embryonic cerebral cortex. J Neurosci 23, 6399–6403 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida M, Assimacopoulos S, Jones KR, Grove EA, Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development 133, 537–545 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Schmitz MT et al. , The development and evolution of inhibitory neurons in primate cerebrum. Nature 603, 871–877 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi Y et al. , Mouse and human share conserved transcriptional programs for interneuron development. Science 374, eabj6641 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer C et al. , Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krienen FM et al. , Innovations present in the primate interneuron repertoire. Nature 586, 262–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR, Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci 24, 584–594 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teng L, Mundell NA, Frist AY, Wang Q, Labosky PA, Requirement for Foxd3 in the maintenance of neural crest progenitors. Development 135, 1615–1624 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aibar S. et al. , SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lambert SA et al. , The Human Transcription Factors. Cell 175, 598–599 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Jin S et al. , Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ, Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Farrell JA et al. , Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skinnider MA et al. , Cell type prioritization in single-cell data. Nat Biotechnol 39, 30–34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trapnell C et al. , The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bayraktar OA et al. , Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci 23, 500–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pinero J, Sauch J, Sanz F, Furlong LI, The DisGeNET cytoscape app: Exploring and visualizing disease genomics data. Comput Struct Biotechnol J 19, 2960–2967 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jansen IE et al. , Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watson HJ et al. , Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat Genet 51, 1207–1214 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grove J et al. , Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stahl EA et al. , Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 51, 793–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Savage JE et al. , Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 50, 912–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wray NR et al. , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luciano M et al. , Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat Genet 50, 6–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nalls MA et al. , Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trubetskoy V et al. , Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Leeuw CA, Mooij JM, Heskes T, Posthuma D, MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.C. International Obsessive Compulsive Disorder Foundation Genetics, O. C. D. C. G. A. Studies, Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol Psychiatry 23, 1181–1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu D et al. , Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. Am J Psychiatry 176, 217–227 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanders SJ et al. , Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.S. Deciphering Developmental Disorders, Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skene NG, Grant SG, Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front Neurosci 10, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiang Y et al. , Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 21, 383–398 e387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chambers SM et al. , Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amiri A et al. , Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gordon A et al. , Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat Neurosci 24, 331–342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gelman DM, Marin O, Rubenstein JLR, in Jasper’s Basic Mechanisms of the Epilepsies, th et al. , Eds. (Bethesda (MD), 2012). [Google Scholar]
- 95.Fu Y et al. , Heterogeneity of glial progenitor cells during the neurogenesis-to-gliogenesis switch in the developing human cerebral cortex. Cell Rep 34, 108788 (2021). [DOI] [PubMed] [Google Scholar]
- 96.Li X et al. , Decoding Cortical Glial Cell Development. Neurosci Bull 37, 440–460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mi H, Haeberle H, Barres BA, Induction of astrocyte differentiation by endothelial cells. J Neurosci 21, 1538–1547 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herring CA et al. , Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution. Cell 185, 4428–4447 e4428 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq data were deposited in NEMO Archive (RRID: SCR_002001) accessible at https://assets.nemoarchive.org/dat-fjx1jbr and NCBI GEO (GSE226451). The data can be visualized at http://resources.sestanlab.org/devmacaquebrain. Bulk RNA-seq data were deposited in NCBI GEO (GSE225482). Scripts are available at: https://github.com/sestanlab/Macaque_corticogenesis_scRNA-seq.