

Abstract
BACKGROUND:
Left ventricular hypertrophy (LVH) is a bipolar response, starting as an adaptive response to the hemodynamic challenge, but over time develops maladaptive pathology partly due to microvascular rarefaction and impaired coronary angiogenesis. Despite the profound influence on cardiac function, the mechanotransduction mechanisms that regulate coronary angiogenesis, leading to heart failure, are not well known.
METHODS:
We subjected endothelial-specific knockout mice of mechanically activated ion channel, TRPV4 (TRPV4ECKO) to pressure-overload via transverse aortic constriction (TAC) and examined cardiac function, cardiomyocyte hypertrophy, cardiac fibrosis, and apoptosis. Further, we measured microvascular density and underlying TRPV4 mechanotransduction mechanisms using human microvascular endothelial cells (ECs), ECM gels of varying stiffness, unbiased RNA sequencing, siRNA, western blot, qPCR, and confocal immunofluorescence techniques.
RESULTS:
We demonstrate that endothelial-specific deletion of TRPV4 preserved cardiac function, cardiomyocyte structure and reduced cardiac fibrosis compared to TRPV4lox/lox mice, 28 days post-TAC. Interestingly, comprehensive RNA sequencing analysis revealed an upregulation of pro-angiogenic factors (VEGFα NOS3, and FGF2,) with concomitant increase in microvascular density in TRPV4ECKO hearts after TAC compared to TRPV4lox/lox. Further, an increased expression of VEGFR2 and activation of the YAP pathway were observed in TRPV4ECKO hearts. Mechanistically, we found that downregulation of TRPV4 in ECs induced matrix stiffness-dependent activation of YAP and VEGFR2 via the Rho/Rho kinase/LATS pathway.
CONCLUSIONS:
Our results suggest that endothelial TRPV4 acts as a mechanical break for coronary angiogenesis, and uncoupling endothelial TRPV4 mechanotransduction attenuates pathological cardiac hypertrophy by enhancing coronary angiogenesis.
Keywords: TRPV4, cardiac hypertrophy, coronary angiogenesis, endothelial, mechanotransduction, pressure overload
Graphical Abstract

Introduction
Left ventricular hypertrophy (LVH) is the heart’s response to a chronic hemodynamic challenge imposed by long-term hypertension or myocardial infarction. In a sense, LVH is a bipolar response, starting as an adaptive response to the hemodynamic challenge, but over time progressing towards a maladaptive pathology with a loss of ventricular function 1. A putative reason for this progression could be a maladaptive growth response of the coronary microcirculation (microvascular rarefaction and impaired coronary angiogenesis), leading to inadequate perfusion of the working myocardium. In fact, many studies of hypertrophied myocardium have revealed a decrease in coronary angiogenesis/capillary density, which is counterintuitive since the heart has increased metabolic needs in LVH 2. Importantly, the molecular mechanisms underlying this impaired coronary angiogenesis in LVH are unknown. To this end, we believe that the balance between mechanical and soluble (growth factor) stimuli that influence angiogenesis has been shifted in LVH to impede angiogenesis in response to the pathological stimuli 1–3. Although the heart is mechanically active as it stops beating only upon death, most of the research on coronary angiogenesis has focused on soluble factors and downstream signaling. However, mechanical forces are equally as important in controlling cardiac function, especially during cardiac injury and insult, and they are known to modulate soluble factor responses. Despite this profound influence on cardiac function, the mechanisms of mechanotransduction that regulate myocardial angiogenesis are not well known.
Transient Receptor Potential Vanilloid-4 (TRPV4) channel is a ubiquitously expressed, non-selective cation channel that’s been demonstrated as mechanically activated ion channel in endothelial cells and other cell types 4,5. We have shown that TRPV4 is activated in response to cyclic strain and matrix stiffness in vitro and in vivo, and negatively regulates angiogenesis and its absence enhances endothelial proliferation, migration, and tube formation 5–8. In the present study, we investigated the role of endothelial TRPV4 in coronary angiogenesis, cardiac function, and cardiac remodeling in response to TAC-induced pressure overload using endothelial specific knockout (TRPV4ECKO) mice.
Methods
The authors declare that all supporting data are available within the article (Online supplemental Material).
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Northeast Ohio Medical University (NEOMED) and The University of Toledo. TRPV4lox/lox, and TRPV4ECKO mice were fed a standard diet and water ad libitum and kept under a 12-h light/dark cycle.
Statistical analysis
Statistical analysis was performed using two-way ANOVA, followed by Tukey post hoc analysis or Student’s t-test, and the significance was set at *p≤0.05, *** p≤0.001 and **** p≤0.0001.
Results
Endothelial deletion of TRPV4 protects the myocardium following pressure-overload-induced hypertrophic stress
To confirm the role of endothelial TRPV4 in the regulation of cardiac remodeling, we first generated endothelial TRPV4 knockout mice (TRPV4ECKO) by crossing the TRPV4lox/lox (TRPV4 floxed) with Tie2-Cre. TRPV4 deletion was confirmed by genotyping the tail snips and measuring TRPV4 expression in isolated endothelial cells (EC) from TRPV4lox/lox and TRPV4ECKO mice using RT-PCR and functional Ca2+ imaging. Genotyping PCR data suggests that only homozygous TRPV4-Cre positive mice yielded a Cre specific band. Further, Pac-I digestion of lox-PCR product generated two bands in the homozygous TRPV4-Cre positive mice but not in heterozygous TRPV4-Cre mice (Figure 1A). To further confirm the deletion of TRPV4 in ECs, TRPV4 expression was analyzed in isolated ECs. RT-PCR data showed that TRPV4 expression was absent in EC isolated from TRPV4ECKO mice, when compared to TRPV4lox/lox mice (Figure 1B). To verify the functional disruption of TRPV4, we measured Ca2+ influx using a TRPV4-selective agonist, GSK1016790A (GSK101), in isolated ECs. As expected, isolated ECs from TRPV4ECKO failed to induce Ca2+ influx in response to GSK101, whereas ECs from TRPV4lox/lox robustly induced Ca2+ influx, confirming functional deletion of TRPV4 in ECs (Figure S2).
Figure 1. Endothelial specific deletion of TRPV4 (transient receptor potential cation channel subfamily V member 4) preserves cardiac structure and function, post transverse aortic constriction (TAC).

A, Genotyping of TRPV4ECKO mice. Genomic DNA was isolated from tail snips. Restriction endonuclease PacI digestion of lox-polymerase chain reaction (PCR) product (single band in undigested wild type mice (+/+)) generated two bands in the homozygous TRPV4-Cre positive mice (−/−) but not in heterozygous TRPV4-Cre mice (+/−). B, Reverse Transcription polymerase chain reaction (RT-PCR) analysis of TRPV4, CD31, GAPDH expression in isolated lung endothelial cells from TRPV4lox/lox and TRPV4ECKO mice. C, Representative images (20x) of WGA-stained heart sections from post-TAC and sham operated TRPV4lox/lox and TRPV4ECKO mice. D, Quantification of cardiomyocyte cross-sectional area from wheat Germ Agglutinin, Alexa Fluor™ 594 (WGA) stained TRPV4lox/lox and TRPV4ECKO heart sections (n=300 myocytes from 4–6 hearts per group). E and F Cardiac function was assessed via 2D-echocardiography at baseline and 28 days after TAC or sham surgeries. E, M-mode images showing cardiac function in TRPV4lox/lox and TRPV4ECKO mice subjected to sham or TAC surgeries. F, Quantitative analysis of cardiac function (% ejection fraction) in TRPV4lox/lox and TRPV4ECKO mice two-way ANOVA followed by Tukey post hoc analysis; Significance was set at *p<0.05; **p<0.01, ****p<0.0001).
Next, to explore if deletion of endothelial TRPV4 results in preserved/improved coronary function and structure following hypertrophic stress, we induced pressure overload in the hearts of TRPV4ECKO and TRPV4lox/lox mice via TAC. We found that TRPV4lox/lox mice showed an increased heart weight to body weight ratio after TAC compared to sham counter parts (Figure S3A). However, compared to TRPV4lox/lox, TRPV4ECKO showed a reduced heart weight to bodyweight ratio after TAC (Figure S3A). TRPV4lox/lox mice exhibited cardiomyocyte hypertrophy in response to TAC, as evidenced by increased cardiomyocyte cross-sectional area (Figure 1C). In contrast, we found no increase in the cardiomyocyte cross-sectional area in TRPV4ECKO-TAC mice (Figure 1C). Quantitative analysis revealed that there is no difference in cardiomyocyte cross-sectional area between sham groups, however there was a significant increase in cardiomyocyte cross-sectional area in TRPV4lox/lox-TAC mice with no increase in TRPV4ECKO-TAC (Figure 1D). Additionally, qPCR analysis of heart samples revealed increased expression of alpha-skeletal actin transcript in the TRPV4lox/lox compared to TRPV4ECKO mice after TAC (Figure S3B), suggesting that endothelial deletion of TRPV4 preserved the cardiac structure post TAC. Echocardiography revealed that TRPV4ECKO mice exhibited preserved cardiac function (ejection fraction and fractional shortening) compared to TRPV4lox/lox mice (Figure 1E, F and Table S1), 28 days post-TAC.
Loss of endothelial TRPV4 reduces cardiac fibrosis and cardiomyocyte apoptosis, post-TAC
To find out if improved cardiac function reflects cardiac remodeling, we measured cardiac fibrosis using Masson’s Trichrome staining (Figure 2A), and we found significantly less cardiac fibrosis in TRPV4ECKO hearts compared to TRPV4lox/lox hearts, post-TAC (Figure 2A and B). Since preserved cardiac structure, and reduced fibrosis was observed in TRPV4ECKO mice, we measured cardiomyocyte apoptosis. TUNEL staining of heart sections from TRPV4ECKO mice showed significantly lower number of apoptotic cells in TRPV4ECKO compared to TRPV4lox/lox, 28 days post-TAC (Figure 2C and D).
Figure 2. TRPV4ECKO mice exhibit reduced cardiac fibrosis and cardiomyocyte apoptosis following TAC.

A) Masson’s Trichrome stained images from the whole heart sections from TRPV4lox/lox and TRPV4ECKO mice, 28 days post sham or TAC surgeries. Inset shows magnified images of interstitial fibrosis. Scale bar=100 um. B) Quantification of percent of collagen deposition (fibrosis) in TRPV4lox/lox and TRPV4ECKO hearts, 28 days after sham or TAC surgeries (n=6). C) Representative images showing apoptosis of cardiomyocytes revealed by TUNEL staining from sham operated and post-TAC. TRPV4lox/lox and TRPV4ECKO hearts. D) Quantification of apoptotic cardiomyocytes from the hearts of TRPV4lox/lox and TRPV4ECKO mice, 28 days post-TAC. (n=5), Two-way ANOVA followed by Tukey post hoc analysis and Significance was set at *p<0.05, ** p<0.01; ***p<0.001 ****p<0.0001).
Deletion of endothelial TRPV4 enhances proangiogenic genes and coronary angiogenesis via YAP/VEGFR2 activation in response to pressure-overload
To identify potential molecular mechanisms by which TRPV4 regulates angiogenesis following hypertrophic stress, we performed unbiased RNA sequencing of the hearts from both TRPV4lox/lox and TRPV4ECKO mice after TAC. We found that 140 genes were differentially expressed in TRPV4lox/lox hearts after TAC compared to sham-operated animals. Importantly, we found that a large number of genes (797 genes) were differentially expressed in TRPV4ECKO-TAC hearts compared to other groups (Figure S4A). The heatmap of differentially expressed genes among sham and TAC hearts of TRPV4lox/lox and TRPV4ECKO mice showed 5 clusters of coregulated genes together with Gene Ontology (GO) terms (biological process: proangiogenic, angiogenesis, wound healing, extracellular matrix, and fibroblasts) related to the vascular development in heart (Figure S4B). A direct comparison of angiogenic genes revealed that the down regulation of pro-angiogenic and angiogenic regulators with concomitant upregulation of ECM genes in TRPV4lox/lox -TAC hearts compared to sham operated hearts (Figure S4C and Figure S4D). Notably, we found that endothelial deletion of TRPV4 significantly upregulated genes related to proangiogenic factors, angiogenic factors, and positive regulators of wound healing compared to TRPV4lox/lox after TAC (Figure S4C and Figure S4D). Furthermore, qPCR validation for gene expression for major proangiogenic factors revealed increased expression of VEGFα, FGF2, NOS3, and CYR61 in TRPV4ECKO hearts compared to TRPV4lox/lox hearts, after TAC (Figure 3 A–B; Figure S5A and Figure S5B). Next, we asked if the increase in these proangiogenic genes reflected in increased coronary angiogenesis. Indeed, we found that capillary density is significantly higher in TRPV4ECKO myocardium compared to TRPV4lox/lox mice after TAC, while it was unchanged in sham groups (Figure 3C; Figure S1). Quantitative analysis revealed that TAC-induced hypertrophic stress decreased capillary density in TRPV4lox/lox hearts compared to sham mice. In contrast, capillary density is significantly higher in TRPV4ECKO-TAC hearts compared to sham and TRPV4lox/lox-sham and TAC hearts (Figure 3D). Next, we measured the protein expression of VEGFR2 in the heart samples of TRPV4lox/lox-TAC and TRPV4ECKO-TAC, as TRPV4 siRNA downregulation was shown to activate YAP/VEGFR2 6,9. Indeed, we found that TRPV4ECKO-TAC hearts showed enhanced VEGFR2, and decreased phospho-YAP (S127) expression compared to TRPV4lox/lox-TAC and sham hearts, respectively (Figure 3E–F, and Figure 4A).
Figure 3. TAC induced proangiogenic factors and VEGFR2 receptors were enhanced in hearts of endothelial TRPV4 deleted mice after TAC.

A-B) Real time PCR of proangiogenic factors, VEGFα, and NOS3. C) Representative images of Isolectin IB4 stained heart sections from TRPV4lox/lox and TRPV4ECKO mice, 28 days post-TAC surgeries showing capillary density. n=6; Scale bar=75 um D) Quantification of capillary density from TRPV4lox/lox and TRPV4ECKO mice, 28 days post-TAC. E & F) Representative immunoblot and its quantification showing increased expression of VEGFR2 in TRPV4ECKO hearts after TAC compared to TRPV4lox/lox TAC (n=5). Note molecular weight markers are shown in the left side; Two-way ANOVA followed by Tukey post hoc analysis and Significance was set at *p<0.05, **p<0.01; ***p<0.001 ****p<0.0001).
Figure 4. TRPV4 regulates matrix stiffness dependent activation of VEGFR2 through YAP activation.
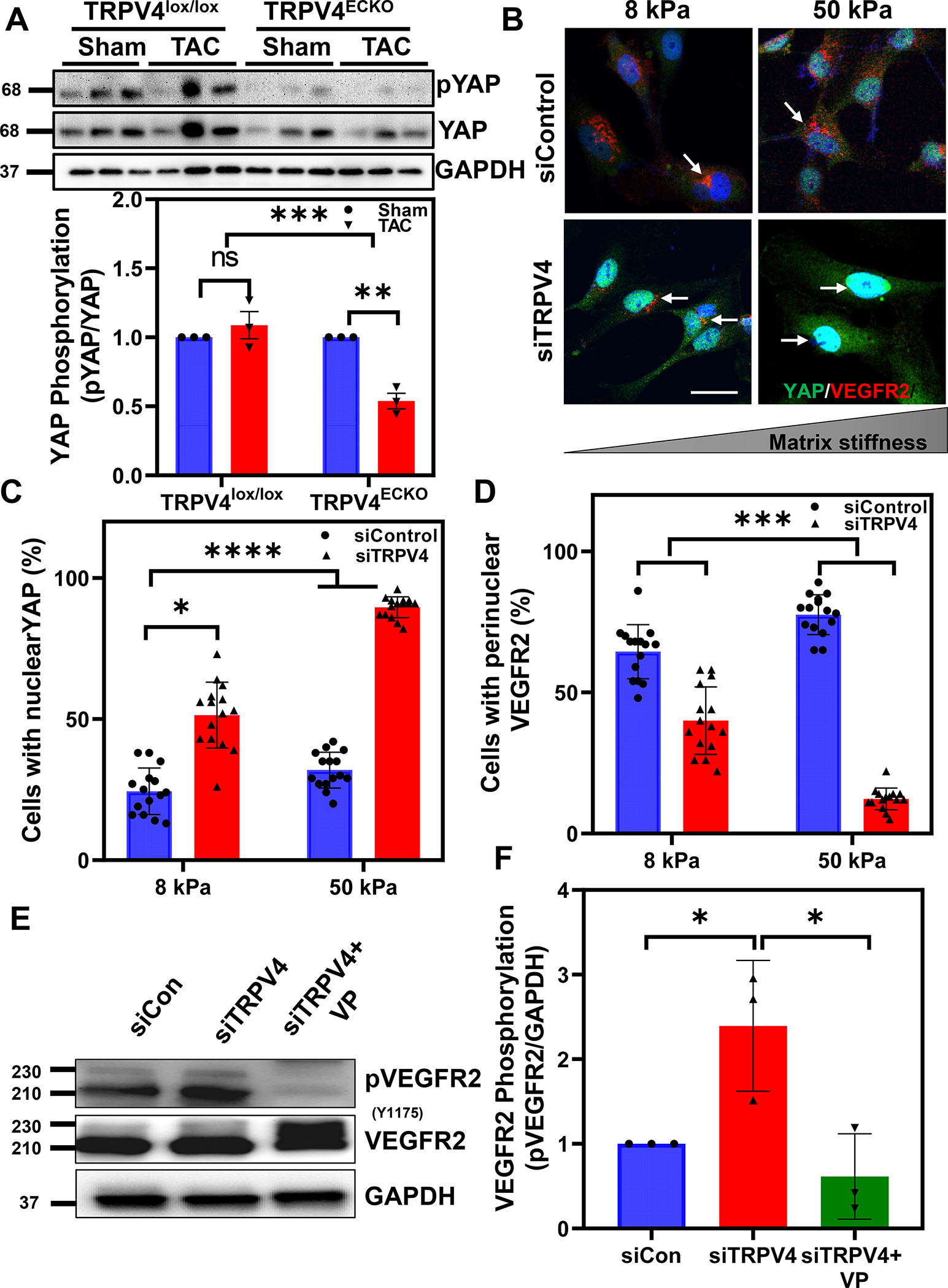
A) Representatives immunoblot and quantitative analysis showing decreased phosphorylation of YAP in TRPV4ECKO-TAC mice. Note molecular weight markers are shown in the left side B) Representative immunofluorescence images showing YAP (green) and VEGFR2 (red) in control and TRPV4 siRNA transfected human endothelial cells cultured on ECM gels mimicking normal heart stiffness (8kPa) and failing heart stiffness (50kPa). Note that perinuclear localization of VEGFR2 (red; arrows) in control siRNA-treated cells which is disappeared in TRPV4 siRNA-treated cells with concomitant nuclear translocation of YAP (green; arrows). C-D) Quantitative analysis showing the average number of cells/field (10–15 fields with around 200–300 total cells per condition from three independent experiments) with nuclear translocation of YAP and perinuclear VEGFR2. Representative western blots (E) and quantitative analysis (F) of phospho-VEGFR2 in TRPV4 siRNA downregulated EC in the presence or absence of YAP inhibitor, verteporfin (VP). Note molecular weight markers are shown in the left side; (n=3; Two-way ANOVA followed by Tukey post hoc analysis and Significance was set at *p<0.05; ***p<0.001 ****p<0.0001).
TRPV4 knockdown induces matrix stiffness dependent activation of VEGFR2 and YAP
To delineate the molecular mechanisms by which TRPV4 regulates VEGFR2 activation in response to pathological hypertrophy, we cultured control and TRPV4 siRNA-treated (Figure S6) endothelial cells on ECM stiffness gels that mimic the stiffness of normal healthy hearts (8 kPa) and failing hearts (50 kPa)4. We found that faint YAP nuclear staining in control siRNA-treated EC cultured on both 8 and 50 kPa gels. Notably, we found higher YAP staining in the nucleus of TRPV4 siRNA-treated EC on 8 kPa gels, which was further increased significantly in the EC cultured on 50 kPa gels (Figure 4B and C). In contrast to YAP, we found significantly high perinuclear VEGFR2 in control siRNA-treated EC on both 8 and 50 kPa gels, which disappeared in TRPV4 knockdown cells (Figure 4B and D). Further, we found that TRPV4 knockdown in EC increased VEGFR2 phosphorylation at Y1175 compared to control siRNA-treated cells, which was significantly attenuated by verteporfin (VP), small molecule inhibitor of YAP (Figure 4 E and F). Finally, we found TRPV4 siRNA knockdown in human cardiac microvascular endothelial cells (HMVEC-C) also enhanced YAP nuclear translocation and VEGFR2 membrane translocation compared to control siRNA cells (Figure S7).
TRPV4 regulates matrix-dependent activation of VEGFR2/YAP via modulation of Rho/Rho kinase/LATS1/2 pathway
To further explore the molecular mechanisms by which TRPV4 regulates activation of YAP/VEGFR2, we focused on Rho/Rho Kinase/LATS1/2, since LATS1/2 phosphorylation was shown to regulate YAP activation10. Previously, we have shown that TRPV4 negatively regulated Rho/Rho kinase activation 5,11, which are upstream of LATS/ YAP pathway. To investigate this, we measured Rho activation using Rho-GTP antibody and a Rho kinase inhibitor, Y27632. As shown in Figure 5A, we found that siRNA knockdown of TRPV4 significantly increased Rho-GTP (Figure 5 A and B), as well as Rho-dependent formation of actin stress fibers (Figure 5C). Next, immunostaining with phospho-LATS1/2 antibody revealed significantly decreased phosphorylation of LATS1/2 in TRPV4 knocked down EC compared to control siRNA-treated cells (Figure 5D and E). Further, western blotting revealed that TRPV4 knockdown significantly reduced phosphorylation of LATS at T1097, which was reversed upon treatment with Rho/Rho kinase inhibitor Y27632 (Figure 5F). Finally, to find out if Rho/Rho kinase mediates matrix-dependent activation of YAP/VEGFR2 downstream of TRPV4, we cultured control siRNA and TRPV4 siRNA- treated EC on 8 and 50 kPa gels in the presence or absence of Y27632. We found that while TRPV4 knockdown induced YAP nuclear translocation and disappearance of VEGFR2 on both 8 and 50 kPa stiffness gels in EC, Y27632 treatment attenuated this effect (Figure 6 A–C). Further, western blotting revealed that TRPV4 knockdown increased VEGFR2 phosphorylation at Y1175 compared to control siRNA treated EC, which was attenuated by Y27632 (Figure 6D).
Figure 5. Downregulation of TRPV4 inhibits LATS activation through modulation of Rho-actin pathway.

A) Representative images showing enhanced Rho activity (RhoGTP) in TRPV4 siRNA treated endothelial cells. B) Quantitative analysis showing the average fluorescence intensity of RhoGTP indicating that TRPV4 knockdown increased Rho activity. C) Representative images showing silencing of TRPV4 enhanced F-actin compared to control cells. D) Representative images showing TRPV4 knockdown reduced LATS1/2 phosphorylation compared to controls in endothelial cells. E) Quantitative analysis showing average fluorescence intensity was significantly reduced in TRPV4-si EC cells (Average number of cells/field (10–15 fields with around 200–300 total cells per condition from three independent experiments). F) Western blot and quantitative analysis showing decreased LATS phosphorylation in TRPV4 knockdown cells, which was reversed in response to Rho kinase inhibitor Y27362. Note molecular weight markers are shown in the left side; (n=3; Student’s T test, one-way ANOVA and Significance was set at *p<0.05, ** p<0.01; ****p<0.0001).
Figure 6. TRPV4 regulates matrix dependent activation of VEGFR2 via modulation of Rho kinase.

A) Representative images showing immunostaining of YAP and VEGFR2 on different stiffness gels after control and TRPV4 siRNA transfection in the presence or absence of Rho kinase inhibitor Y27632. Note that perinuclear localization of VEGFR2 (red) in control siRNA-treated cells which is disappeared in TRPV4 siRNA-treated cells with concomitant nuclear translocation of YAP in TRPV4 siRNA-treated cells (green). However, pre-treatment with Y27632 abolished the disappearance of VEGFR2 and nuclear translocation of YAP. B and C) Average number of cells/field (10–15 fields with around 200–300 total cells per condition from three independent experiments) indicating that TRPV4 knockdown enhanced YAP and VEGFR2 activation which was reduced upon treating with Rho kinase antagonist Y27632. D) Representative western blot and quantitative analysis showing the phosphorylation of VEGFR2 was enhanced in TRPV4 siRNA knockdowned EC which was reversed in response to Rho kinase antagonist Y27632. (n=3; Two-way ANOVA and Significance was set at *p<0.05, ** p<0.01; ****p<0.0001).
Discussion
In the present study, we demonstrated that the deletion of endothelial TRPV4 channels protects heart from pressure overload-induced pathological hypertrophy. We concluded this based on our findings that: 1) TAC-induced pressure-overload increased the expression of TRPV4 in the myocardium of WT mice (Figure S8), 2) Endothelial specific deletion of TRPV4 (with intact TRPV4 in cardiomyocytes and cardiac fibroblasts) preserved cardiac function and cardiomyocyte structure, and it reduced cardiomyocyte apoptosis and cardiac fibrosis, 3) RNA sequence and qPCR analysis revealed increased angiogenic genes, decreased negative regulators of angiogenesis with increased mRNA expression of VEGFα, Cyr61, FGF2 and NOX3; and protein expression of VEGFR2 and YAP in the hearts of endothelial specific TRPV4KO (TRPV4ECKO) mice subjected to TAC compared to TRPV4lox/lox, 4) Deletion of endothelial TRPV4 (TRPV4ECKO) increased coronary capillary density in response to TAC compared to TRPV4lox/lox, and 5) Mechanistically, TRPV4 knockdown in endothelial cells induced VEGFR2 activation through modulation of the Rho/LATS/YAP pathway in matrix stiffness-dependent manner.
TRPV4 channels have been shown to express in cardiomyocytes and cardiac fibroblasts of the heart4,12–14. Functionally, TRPV4 has been demonstrated to regulate cardiomyocyte calcium cycling and contractility, and increased TRPV4 expression contributes to damage in the aged heart following hypoosmotic stress 12,14. We have previously demonstrated that TRPV4 is mechanically activated in cardiac fibroblasts and mediates their differentiation into myofibroblasts, and that the deletion of TRPV4 protects the heart from myocardial infarction-induced adverse remodeling 4,13. All the previous work on TRPV4 in cardiac hypertrophy or fibrosis 4,13,15, is based on global TRPV4KO mice 4,15,16 and did not use cell-specific TRPV4KO mice.
Importantly, there are no studies on the role of endothelial TRPV4 in the regulation of either physiological or pathological cardiac remodeling. Although TRPV4 was demonstrated to regulate endothelial cell function, most of the work was focused on its role in hypertension and barrier function17,18. We have previously demonstrated that TRPV4 channels are mechanically activated by ECM stiffness and cyclic stretch in endothelial cells, and regulate tumor angiogenesis 5–9,19–23. In the present study, by using an endothelial specific TRPV4KO mice, we demonstrated that deletion of TRPV4, specifically in endothelial cells (but not in fibroblasts or cardiomyocytes), preserved cardiac function and reduced cardiac hypertrophy and cardiac fibrosis via increased coronary angiogenesis (Figure S9). Recent studies have shown that TAC induces a brief increase in proangiogenic factors, the expression of which continuously reduced after three weeks, leading to reduced microvascular density and increased cardiac fibrosis, which ultimately lead to a decline in myocardial function 24,25. In support of this idea, RNA sequencing and qPCR data revealed increased expression of extracellular matrix and negative regulators of angiogenesis genes in TRPV4lox/lox heart post-TAC, but a reduction in positive angiogenic regulators. In contrast, TRPV4ECKO hearts exhibited increased pro-angiogenic factors (VEGFα, FGF, NOS3 and CYR61) compared to TRPV4lox/lox post-TAC. Further, VEGFR2 expression was increased in TRPV4ECKO hearts after TAC compared to TRPV4lox/lox hearts, suggesting that increased VEGF/VEFR2 signaling may increase coronary angiogenesis in TRPV4ECKO mice. Previous studies have shown that a lack of VEGFR signaling promotes pressure overload and angiotensin II-induced cardiac hypertrophy 25,26, supporting a role for VEGF/VEGFR2 signaling in attenuating cardiac hypertrophy and pathological remodeling in TRPV4ECKO mice. However, the molecular mechanism by which TRPV4 regulates VEGF/VEGFR2 signaling in the heart is not known. Our findings showed that TRPV4 downregulation in EC increased the Rho/Rho Kinase pathway, enhancing F-actin formation. This, in turn, promoted VEGFR2 activation and increased endothelial proliferation and migration via YAP (Figure S9). Our data demonstrated that the absence of TRPV4 in endothelial cells completely abolished perinuclear VEGFR2 and enhanced YAP nuclear translocation at higher stiffness gels, which suggests that YAP nuclear translocation is required to activate VEGFR2. However, the molecular mechanisms by which TRPV4 regulates YAP/VEGFR2 activation are not well known.
YAP is an intracellular mechanosensitive transcription co-factor, which translocates into the nucleus in response to mechanical stress and plays a key role in survival, differentiation, proliferation, and other cellular functions 10. YAP activation can be regulated by both hippo dependent (LATS1/2 and MST) or independent pathways, however the upstream mechanical signaling that activates YAP is not known 27,28. Our findings demonstrated that TRPV4 regulated YAP nuclear translocation by dephosphorylating LATS1/2 via Rho/Rho Kinase. We found that TRPV4 knockdown increased YAP nuclear translocation by dephosphorylating LATS1/2. Next, we found that TRPV4 knockdown induced constitutive activation of Rho, which was responsible for the dephosphorylation of LATS1/2 by increasing the formation of F-actin stress fibers. In contrast, Rho kinase inhibitor Y27632 reverted the effects of TRPV4 knockdown induced LATS1/2 dephosphorylation, suggesting that Rho acts upstream of LATS1/2 in YAP nuclear translocation. Indeed, we found that inhibition of Rho kinase with Y27632 reduced YAP nuclear translocation and VEGFR2 activation in TRPV4 knocked-down EC cultured on ECM gels that mimic the stiffness of normal and failing hearts confirming that Rho/Rho kinase act upstream of YAP. YAP activation may then induce the localization of VEGFR2 from the perinuclear region to plasma membrane via Myosin 1c (Myo1c) 27.
Previous studies demonstrated that TRPV4 expression in cardiomyocytes and cardiac fibroblasts positively correlated with cardiac dysfunction and cardiac fibrosis, respectively 4,12–14. While TRPV4/Rho/MRTF-A signaling was implicated in MI-induced cardiac fibroblast differentiation and cardiac fibrosis4,13, cardiac hypertrophy appears to be mediated through TRPV4/CaMKII/NFkB/NLRP3 signaling in cardiomyocytes15. However, our study demonstrated that endothelial specific deletion of TRPV4 (with intact TRPV4 in cardiomyocyte and/or cardiac fibroblast) protected the heart from pressure-overload-induced cardiac hypertrophy via the activation of Rho/LATS/YAP/VEGFR2-mediated angiogenesis. We postulate that the increased angiogenesis in TRPV4ECKO TAC-hearts counteracts adverse remodeling by increasing the perfusion to the myocardium, thereby reducing cardiomyocyte apoptosis and cardiac fibrosis.
While our current study focused on the male mice, it is important to investigate if endothelial TRPV4 deletion protects myocardium from hypertrophic stress in female mice. Also, Tie-2 Cre may impact cells other than EC such as CD45 positive hemopoietic cells29. A recent study showed that endothelial TRPV4 channel knockout mice are mild hypertensive 30. In fact, impairment of endothelial TRPV4 activity in obesity via peroxynitrite could contribute to the obesity-induced hypertension30. The role of TRPV4 in vasodilation is further complicated because of recently observed contrasting roles of TRPV4 in smooth muscle (vasoconstrictor and vasodilator pools31. However, we have not measured blood pressure in our mice. We believe that the mild hypertensive phenotype of EC-specific KO mice will not have much effect on overall hypertrophic stress induced by TAC. In fact, we believe that this mild phenotype may be underestimating the overall protective effect of EC deletion of TRPV4 in the hearts subjected to pressure-overload-induced stress. Despite of these limitations, to our knowledge, our study represents the first evidence demonstrating that endothelial TRPV4 deletion preserves myocardial structure and function in response to pressure-overload-induced stress by reducing cardiac fibrosis and cardiomyocyte apoptosis via increased coronary angiogenesis.
Perspectives
One of the reasons for LV hypertrophy could be that reduced microvascular density due to microvascular rarefaction or impaired adaptive coronary angiogenesis can lead to inadequate myocardial perfusion and substrate delivery, contributing to the progression of heart failure in pressure-overload-induced hypertrophy or other ischemic heart diseases. Therefore, angiogenic therapy for ischemia/hypertrophic heart diseases such as gene therapy, delivery of growth factor proteins and stem cell implantation are appealing, there are limitations and concerns, including delivery modalities, uncontrolled angiogenesis, limited half-life of growth factors, and effects on other organs32. Notwithstanding these limitations, most therapeutic angiogenic strategies still focus on VEGF or other growth factor signaling. A potential alternative approach to therapeutic angiogenesis is physiological stimulation of vascular growth in the surviving myocardium. EC in the heart is continuously exposed to mechanical forces such as stretch, as well as changes in ECM stiffness due to cardiac remodeling. We speculate that mechanical forces exert tonic restraint on coronary angiogenesis more-so when myocardium is under stress, such as in pressure-overload. Indeed, our current study demonstrated that TRPV4-dependent mechanotransduction limited coronary angiogenesis by exerting such tonic restraint (Figure S9), and that endothelial deletion of TRPV4 released tonic restraint and increased coronary angiogenesis, which preserved cardiac function by reducing cardiac reducing cardiac hypertrophy and cardiac fibrosis. These findings also identify endothelial TRPV4 as a novel therapeutic target for pressure-overload-induced heart failure.
Supplementary Material
Novelty and Relevance.
What Is New?
We demonstrated that deletion of endothelial TRPV4 (endothelial transient receptor potential vanilloid family member 4) induces coronary angiogenesis in response to pressure-overload induced cardiac stress.
What Is Relevant?
Endothelial-specific deletion of TRPV4 exhibited preserved cardiac function, cardiomyocyte structure and reduced cardiomyocyte apoptosis and cardiac fibrosis following pressure-overload-induced stress compared to WT mice. Endothelial TRPV4/Rho/YAP/ VEGFR2 signaling axis plays a crucial role in cardio-protective roles against pressure-overload-induced stress.
Clinical/Pathophysiological Implications
TRPV4 is a mechanically activated ion channel in endothelial cells. Uncoupling endothelial TRPV4 mechanotransduction protects myocardium from the pressure-overload-induced stress and identifies endothelial TRPV4 as a novel target for heart failure.
Grant Support:
This work was supported by National Institutes of Health (NIH) (R01HL119705, R01HL148585 and R15CA202847; AHA-TPA-971237 CKT and R01AI144115; SP).
Nonstandard Abbreviations and Acronyms
- Ca2+
Calcium
- TRPV4
Transient receptor potential cation channel subfamily V member 4
- LVH
Left ventricular hypertrophy
- TAC
Transverse aortic constriction
- ECs
Endothelial cells
- VEGFR2
Vascular endothelial growth factor receptor 2
- YAP
Yes-associated protein
Footnotes
Conflict of Interest: The authors declare that they have no competing interests.
Data availability:
The datasets presented in the current study are available from the corresponding author on reasonable request.
References
- 1.Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y [DOI] [PubMed] [Google Scholar]
- 2.Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–571. doi: 10.1161/CIRCRESAHA.114.300507 [DOI] [PubMed] [Google Scholar]
- 3.Cuijpers I, Simmonds SJ, van Bilsen M, Czarnowska E, Gonzalez Miqueo A, Heymans S, Kuhn AR, Mulder P, Ratajska A, Jones EAV, et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol. 2020;115:39. doi: 10.1007/s00395-020-0798-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adapala R, Kanugula AK, Paruchuri S, Chilian WM, Thodeti CK. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res Cardiol. 2020;115:14. doi: 10.1007/s00395-020-0775-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adapala RK, Thoppil RJ, Ghosh K, Cappelli HC, Dudley AC, Paruchuri S, Keshamouni V, Klagsbrun M, Meszaros JG, Chilian WM, et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene. 2016;35:314–322. doi: 10.1038/onc.2015.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanugula AK, Adapala RK, Jamaiyar A, Lenkey N, Guarino BD, Liedtke W, Yin L, Paruchuri S, Thodeti CK. Endothelial TRPV4 channels prevent tumor growth and metastasis via modulation of tumor angiogenesis and vascular integrity. Angiogenesis. 2021;24:647–656. doi: 10.1007/s10456-021-09775-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb). 2010;2:435–442. doi: 10.1039/c0ib00034e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, Ingber DE. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104:1123–1130. doi: 10.1161/CIRCRESAHA.108.192930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanugula AK, Adapala RK, Midha P, Cappelli HC, Meszaros JG, Paruchuri S, Chilian WM, Thodeti CK. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019;33:195–203. doi: 10.1096/fj.201800509R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pocaterra A, Romani P, Dupont S. YAP/TAZ functions and their regulation at a glance. J Cell Sci. 2020;133. doi: 10.1242/jcs.230425 [DOI] [PubMed] [Google Scholar]
- 11.Thoppil RJ, Cappelli HC, Adapala RK, Kanugula AK, Paruchuri S, Thodeti CK. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget. 2016;7:25849–25861. doi: 10.18632/oncotarget.8405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones JL, Peana D, Veteto AB, Lambert MD, Nourian Z, Karasseva NG, Hill MA, Lindman BR, Baines CP, Krenz M, et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc Res. 2019;115:46–56. doi: 10.1093/cvr/cvy156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. doi: 10.1016/j.yjmcc.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veteto AB, Peana D, Lambert MD, McDonald KS, Domeier TL. Transient receptor potential vanilloid-4 contributes to stretch-induced hypercontractility and time-dependent dysfunction in the aged heart. Cardiovasc Res. 2020;116:1887–1896. doi: 10.1093/cvr/cvz287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou Y, Zhang M, Wu Q, Zhao N, Chen M, Yang C, Du Y, Han B. Activation of transient receptor potential vanilloid 4 is involved in pressure overload-induced cardiac hypertrophy. Elife. 2022;11. doi: 10.7554/eLife.74519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morine KJ, Paruchuri V, Qiao X, Aronovitz M, Huggins GS, DeNofrio D, Kiernan MS, Karas RH, Kapur NK. Endoglin selectively modulates transient receptor potential channel expression in left and right heart failure. Cardiovasc Pathol. 2016;25:478–482. doi: 10.1016/j.carpath.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YL, Sonkusare SK. Endothelial TRPV4 channels and vasodilator reactivity. Curr Top Membr. 2020;85:89–117. doi: 10.1016/bs.ctm.2020.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonkusare SK, Laubach VE. Endothelial TRPV4 channels in lung edema and injury. Curr Top Membr. 2022;89:43–62. doi: 10.1016/bs.ctm.2022.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, Thodeti CK. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H757–765. doi: 10.1152/ajpheart.00142.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cappelli HC, Guarino BD, Kanugula AK, Adapala RK, Perera V, Smith MA, Paruchuri S, Thodeti CK. Transient receptor potential vanilloid 4 channel deletion regulates pathological but not developmental retinal angiogenesis. J Cell Physiol. 2021;236:3770–3779. doi: 10.1002/jcp.30116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappelli HC, Kanugula AK, Adapala RK, Amin V, Sharma P, Midha P, Paruchuri S, Thodeti CK. Mechanosensitive TRPV4 channels stabilize VE-cadherin junctions to regulate tumor vascular integrity and metastasis. Cancer Lett. 2019;442:15–20. doi: 10.1016/j.canlet.2018.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guarino B, Katari V, Adapala R, Bhavnani N, Dougherty J, Khan M, Paruchuri S, Thodeti C. Tumor-Derived Extracellular Vesicles Induce Abnormal Angiogenesis via TRPV4 Downregulation and Subsequent Activation of YAP and VEGFR2. Front Bioeng Biotechnol. 2021;9:790489. doi: 10.3389/fbioe.2021.790489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarino BD, Adapala RK, Kanugula AK, Lenkey NM, Dougherty JA, Paruchuri S, Khan M, Thodeti CK. Extracellular Vesicles From Pathological Microenvironment Induce Endothelial Cell Transformation and Abnormal Angiogenesis via Modulation of TRPV4 Channels. Front Cell Dev Biol. 2019;7:344. doi: 10.3389/fcell.2019.00344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gogiraju R, Bochenek ML, Schafer K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front Cardiovasc Med. 2019;6:20. doi: 10.3389/fcvm.2019.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeriouh M, Sabashnikov A, Tenbrock A, Neef K, Merkle J, Eghbalzadeh K, Weber C, Liakopoulos OJ, Deppe AC, Stamm C, et al. Dysregulation of proangiogeneic factors in pressure-overload left-ventricular hypertrophy results in inadequate capillary growth. Ther Adv Cardiovasc Dis. 2019;13:1753944719841795. doi: 10.1177/1753944719841795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirronen A, Downes NL, Huusko J, Laakkonen JP, Tuomainen T, Tavi P, Hedman M, Yla-Herttuala S. The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death. Biomolecules. 2021;11. doi: 10.3390/biom11030452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L, et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev Cell. 2017;42:462–478 e467. doi: 10.1016/j.devcel.2017.08.002 [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Liang X, Zhou L, Liu S, Fang Z, Hu C, Hou Y, Guo Z. Yes-associated protein reacts differently in vascular smooth muscle cells under different intensities of mechanical stretch. Aging (Albany NY). 2022;14:286–296. doi: 10.18632/aging.203768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang EH, Vanhoutte PM. Endothelial dysfunction: a strategic target in the treatment of hypertension? Pflugers Arch. 2010;459:995–1004. doi: 10.1007/s00424-010-0786-4 [DOI] [PubMed] [Google Scholar]
- 30.Ottolini M, Hong K, Cope EL, Daneva Z, DeLalio LJ, Sokolowski JD, Marziano C, Nguyen NY, Altschmied J, Haendeler J, et al. Local Peroxynitrite Impairs Endothelial Transient Receptor Potential Vanilloid 4 Channels and Elevates Blood Pressure in Obesity. Circulation. 2020;141:1318–1333. doi: 10.1161/CIRCULATIONAHA.119.043385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen YL, Daneva Z, Kuppusamy M, Ottolini M, Baker TM, Klimentova E, Shah SA, Sokolowski JD, Park MS, Sonkusare SK. Novel Smooth Muscle Ca(2+)-Signaling Nanodomains in Blood Pressure Regulation. Circulation. 2022;146:548–564. doi: 10.1161/CIRCULATIONAHA.121.058607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, et al. Clinical trials in coronary angiogenesis: issues, problems, consensus: An expert panel summary. Circulation. 2000;102:E73–86. doi: 10.1161/01.cir.102.11.e73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in the current study are available from the corresponding author on reasonable request.