

Abstract
Background
Diffuse intrinsic pontine glioma (DIPG) is a lethal childhood cancer with median survival of less than 1 year. Panobinostat is an oral multihistone deacetylase inhibitor with preclinical activity in DIPG models. Study objectives were to determine safety, tolerability, maximum tolerated dose (MTD), toxicity profile, and pharmacokinetics of panobinostat in children with DIPG.
Patients and Methods
In stratum 1, panobinostat was administered 3 days per week for 3 weeks on, 1 week off to children with progressive DIPG, with dose escalation following a two-stage continual reassessment method. After this MTD was determined, the study was amended to evaluate the MTD in children with nonprogressive DIPG/Diffuse midline glioma (DMG) (stratum 2) on an alternate schedule, 3 days a week every other week in an effort to escalate the dose.
Results
For stratum 1, 19 subjects enrolled with 17/19 evaluable for dose-finding. The MTD was 10 mg/m2/dose. Dose-limiting toxicities included thrombocytopenia and neutropenia. Posterior reversible encephalopathy syndrome was reported in 1 patient. For stratum 2, 34 eligible subjects enrolled with 29/34 evaluable for dose finding. The MTD on this schedule was 22 mg/m2/dose. DLTs included thrombocytopenia, neutropenia, neutropenia with grade 4 thrombocytopenia, prolonged intolerable nausea, and increased ALT.
Conclusions
The MTD of panobinostat is 10 mg/m2/dose administered 3 times per week for 3 weeks on/1 week off in children with progressive DIPG/DMG and 22 mg/m2/dose administered 3 times per week for 1 week on/1 week off when administered in a similar population preprogression. The most common toxicity for both schedules was myelosuppression.
Keywords: DIPG, brainstem glioma, midline glioma, HDAC inhibitors, epigenetics
Key Points.
Panobinostat tolerability in children with DIPG is limited; the major toxicity is myelosuppression.
Pharmacokinetics of panobinostat in this population varied from prior studies.
No significant clinical efficacy at tolerable doses was observed.
Importance of the Study.
Panobinostat is one of the most active agents against DIPG in preclinical studies. This clinical trial demonstrates that panobinostat was safe and tolerable at limited doses when administered on 2 different schedules to children with DIPG/DMG, with myelosuppression the primary toxicity. Pharmacokinetic analysis demonstrated varied exposure in this population compared to prior adult and pediatric studies, suggesting the need for population-based pharmacokinetic studies. Although some patients may have had modest clinical benefit, significant efficacy was not observed. Reasons for lack of observed efficacy may include altered pharmacokinetics in addition to limited exposure at the tumor site, the need for a multipronged approach, or undefined resistance mechanisms.
Diffuse intrinsic pontine glioma (DIPG) is an aggressive brainstem glioma, driven in the majority of cases by a recurrent point mutation in genes encoding Histone H3 (H3K27M) and consequent epigenetic dysregulation.1–3 Those with altered H3K27M are now classified by the World Health Organization (WHO) along with diffuse midline gliomas (DMG) in other neuroanatomical locations that harbor the H3K27M mutation or related alterations affecting epigenetic regulation at the H3K27 locus.4 Standard treatment for DIPG includes radiation therapy, but the antitumor effects are short-lived, and no chemotherapeutic agent has ever demonstrated significant efficacy.5 Despite many clinical trials and approaches, median progression-free survival (PFS) for children with DIPG is 6–8 months, and median postprogression overall survival is 2.3 months,6 resulting in a median overall survival from diagnosis of only 10–11 months.6,7
Panobinostat is a multi-histone deacetylases (HDAC) inhibitor of class I, II, and IV HDACs involved in the deacetylation of histone and non-histone proteins. In drug screens utilizing multiple patient-derived DIPG cell lines, and confirmed in patient-derived animal models, panobinostat was identified as one of the most active agents against DIPG, in a dose-dependent manner.8–12 Panobinostat inhibits total cellular HDAC activity and activities of most HDAC isoforms at nanomolar IC50.13,14 In H3K27M-mutant glioma, panobinostat restored H3K27-methylation and normalized gene expression, thereby decreasing tumor cell proliferation and increasing cell death.8 In addition, it induces expression of cell-cycle control genes, selectively inhibits the proliferation of various tumor cells compared to normal cells, and has shown antitumor activity in several other cancers.15–19 Clinical trials of panobinostat have been performed in adult and pediatric patients using different schedules, showing relative safety, tolerability, and some efficacy.20–22 Panobinostat was FDA-approved in combination with bortezomib and dexamethasone for adults with multiple myeloma at a dose of 20 mg 3 times per week for 2 weeks on/1 week off.23 Common adverse effects include myelosuppression and diarrhea; severe cardiac events have also been reported.23
Given the mechanisms of action of panobinostat and preclinical evidence of efficacy in patient-derived DIPG cell lines and animal models, we performed a phase I trial in children with DIPG/DMG. Initially, children with progressive DIPG (stratum 1) were enrolled. The study was later amended to include children with DIPG or thalamic DMG, H3K27M-mutated who had received radiation therapy and were eligible prior to first progression (stratum 2), using an alternate schedule in an effort to escalate the dose. The primary objectives for each stratum were to describe the toxicity profile and define the dose-limiting toxicities, to estimate the maximum-tolerated dose and recommended-phase 2 dose (RP2D), and to evaluate and characterize the plasma pharmacokinetics of panobinostat in this population. Secondary objectives included assessment of progression-free survival (PFS), overall survival (OS), and assessment of cell-free (cf)DNA.
Patients and Methods
The institutional review boards of each participating institution approved the protocol prior to enrollment, and continuing approval was maintained throughout the study.
Eligibility
For stratum 1, patients were required to have progressive DIPG, defined as one or more of the following: progressive neurologic abnormalities or worsening neurologic status not explained by causes unrelated to tumor progression, an increase in tumor bidimensional measurement, or the appearance of a new tumor lesion since diagnosis. Patients in stratum 2 were eligible if they had DIPG or thalamic H3K27M-mutated DMG, received standard radiation therapy and had not experienced disease progression (PD). Patients with a radiographically typical DIPG, defined as a tumor with a pontine epicenter and diffuse involvement of more than 2/3 of the pons, were eligible without histologic confirmation. Patients who did not meet these radiographic criteria were eligible upon histologic confirmation of malignant glioma involving the pons.
Additional inclusion criteria for both strata included age older than 2 years and less than 22 years, prior treatment with ≥54 Gy focal irradiation administered over approximately 42 days, ≥14 days since last fraction of radiation therapy, ability to swallow capsules, performance score (Lansky or Karnofsky) of ≥50, and adequate bone marrow function with absolute neutrophil count (ANC) ≥ 1000/μL and platelets ≥ 100 000/μL. Patients were required to have a left ventricular ejection fraction ≥50 or shortening fraction of ≥27%, a QTc interval <450 ms, and no significant ventricular arrythmia. Patients with overt renal, hepatic, cardiac, or pulmonary disease were excluded, as were patients who received >60 Gy total radiation to the pons, patients who received prior HDAC inhibitors or HSP90 inhibitors for treatment of their DIPG/DMG, patients who received valproic acid within 28 days prior to enrollment, and those who had prior bone marrow transplant. Patients or their legal guardians gave written informed consent, and assent when appropriate, at the time of enrollment.
Treatment Regimen and Dose Escalation
Panobinostat was initially supplied by Novartis and then by SecuraBio (IND#128437). All eligible and evaluable patients received panobinostat. Panobinostat, supplied as 5-, 10-, 15-, and 20 mg hard gelatin capsules and administered by mouth, was dosed based on assigned dose level with body surface area (BSA) calculated at the beginning of each course and rounded to the nearest deliverable dose. Dosing for stratum 1 was every other day, 3 times a week (eg, Monday, Wednesday, Friday, MWF) for 3 weeks on, 1 week off for each 28-day course. Dosing for stratum 2 was MWF every other week of each 28-day course. The starting dose for stratum 2 was based on the MTD determined in stratum 1. The panobinostat dose escalation schema is provided in Table 1. No intra-patient dose-escalation was allowed. Therapy was continued until development of unacceptable toxicity, PD, or completion of 26 courses of therapy.
Table 1.
Planned Panobinostat Dose Escalation Schema
| Dose level | Panobinostat dose | Frequency/schedule (9 doses/28-day course) |
|---|---|---|
| Stratum 1 | ||
| 0 | 5 mg/m2 | MWF, three weeks on, 1 week off |
| 1* | 10 mg/m2 | MWF, three weeks on, 1 week off |
| 2 | 16 mg/m2 | MWF, three weeks on, 1 week off |
| 3 | 22 mg/m2 | MWF, three weeks on, 1 week off |
| 4 | 28 mg/m2 | MWF, three weeks on, 1 week off |
| 5 | 36 mg/m2 | MWF, three weeks on, 1 week off |
| Dose level | Panobinostat dose | Frequency/schedule (6 doses/ 28-day course) |
| Stratum 2 | ||
| −1 | 5 mg/m2 | MWF every other week |
| 0 | 10 mg/m2 | MWF every other week |
| 1* | 16 mg/m2 | MWF every other week |
| 2 | 22 mg/m2 | MWF every other week |
| 3 | 28 mg/m2 | MWF every other week |
| 4 | 36 mg/m2 | MWF every other week |
*Starting dose.
Definition of MTD and DLT
The dose-finding period began with the initial dose of panobinostat and ended on the last day of course 1. Patients evaluable for toxicity included patients who received any panobinostat. Patients who received less than 8 of 9 prescribed doses in stratum 1 and less than 5 of 6 doses in stratum 2 for any reason other than toxicity were considered inevaluable for dose finding and were replaced. The MTD of panobinostat was estimated via a likelihood-based 2-stage continual reassessment method (CRM) with a target toxicity level set as 25%. A minimum of 6 patients were required to be treated at the MTD with a planned expansion to 12 subjects.
Toxicities were graded according to the NCI Common Terminology Criteria (CTCAE Version 4.0) scale. Hematologic DLT was defined as grade 4 thrombocytopenia, grade 3 thrombocytopenia with bleeding, grade 3 thrombocytopenia occurring twice within a treatment course, myelosuppression causing a >14-day delay between courses, grade 4 neutropenia, and grade 3 or 4 febrile neutropenia. Non-hematologic DLT was defined as any grade 2 toxicity persisting for >7 days and considered medically significant or intolerable that it required treatment interruption, any panobinostat-related toxicity that resulted in a delay of treatment >14 days between treatment courses, and any ≥ grade 3 toxicity with the specific exclusion of the following: grade 3 nausea or emesis controlled by antiemetics that resolved to ≤grade 2 within 5 days, grade 3 electrolyte abnormality that resolved to ≤grade 2 within 5 days, grade 3 rash not considered medically significant or intolerable by the patient, grade 3 diarrhea that resolved to ≤grade 1 with optimal use of antidiarrheal medication, grade 3 fever that resolved to≤ grade 2 within 5 days, and grade 3 infection that resolved to ≤grade 2 within 5 days.
Dose Modification for Toxicity
If a patient experienced dose-limiting hematological toxicity, panobinostat was held. If the toxicity resolved to meet on study parameters within 14 days of holding drug, the patient resumed therapy at one dose level lower. For dose-limiting nonhematologic toxicities, the following modifications were made: panobinostat was held for grade 3 diarrhea and restarted at one dose level lower when resolved to ≤grade 1 or baseline; panobinostat was discontinued if grade 4 diarrhea occurred. Panobinostat was also held until resolution and dose reduced for grade 3 or 4 emesis or grade 3 nausea that was not controlled or did not resolve to ≤grade 2 within 5 days despite the use of standard antiemetics, grade 3 fatigue that resolved in >7 days after holding drug, any grade 4 fatigue, grade 3 or 4 elevation in total bilirubin, increase in AST/SGOT, ALT/SGPT >5–10× institutional upper limit of normal (ULN) that resolved in >7 days or any elevation >10× ULN. Panobinostat was discontinued and not restarted if QTcF was >500 ms; if QTcF was ≥450 msec or above 60 msec from baseline for any predose ECG after the patient began treatment, the dose of panobinostat was omitted. If unresolved within 7 days, treatment was discontinued. If resolved within 7 days, panobinostat was resumed at the prior dose if it was the initial occurrence and dose reduced if it was recurrent.
Subjects dose-reduced for toxicity did not have their dose re-escalated. If toxicity did not resolve to meet on study parameters within 14 days of drug discontinuation, the patient was removed from protocol therapy. If any DLT occurred in a patient already dose reduced for toxicity, the patient was removed from protocol therapy.
Assessment of Antitumor Activity
PFS and OS were assessed as secondary objectives. Patients were evaluated by MRI after every 2 courses for the first 6 courses, then every 3 courses and when clinically indicated until PD or off-study criteria were met. Progressive disease was defined by one or more of the following: progressive neurologic abnormalities or worsening neurologic status not explained by causes unrelated to tumor progression, >25% increase in tumor bidimensional measurement taking as a reference the smallest disease measurement recorded, since the start of protocol therapy, or the appearance of a new tumor lesion. Although this was a phase I study, patients were assessed for objective response, with partial response (PR) defined as ≥50% reduction in tumor size by bidimensional measurement on axial FLAIR images, as compared with the baseline measurements, on a stable or decreasing dose of corticosteroids, accompanied by a stable or improving neurologic examination. Patients were considered to have stable disease (SD), if neurologic exam was at least stable and maintenance corticosteroid dose not increased, and MR/CT imaging met neither the criteria for PR nor the criteria for PD. Only those patients who had measurable disease present at baseline, received at least one course of therapy, and had their disease re-evaluated were evaluable for objective radiographic response. Patients who exhibited disease progression (clinical and/or radiographic) prior to the first scheduled MRI were still considered evaluable for response. PFS was defined as the interval of time between the time of treatment initiation until the time of progressive disease or death from any cause in patients with an event and until the time of last follow-up for patients who are progression-free at the time of analysis. OS was measured similarly with the endpoint of death.
Pharmacokinetics
Pharmacokinetic (PK) studies were performed on all enrolled patients. Samples were collected prior to the first dose of therapy (pretreatment) and then at 0.5, 1, 2, 4, 8 (±1), and 24 (±4) h after the first dose and on day 3 of course 1, prior to the second dose of panobinostat. Plasma concentrations of panobinostat were assayed using a validated LC-MS/MS method with a lower limit of quantitation (LLOQ) of 0.1 ng/mL, as previously reported.24 A noncompartmental analysis (NCA) was conducted to estimate pharmacokinetic (PK) parameters for each patient using WinNonlin 8.3 (Certara, Cary, NC), validated per FDA 21 CFR Part 11. The maximum plasma concentration (Cmax) and its corresponding time (Tmax) were reported as observed values. The area under the curve (AUC) was calculated using the linear up/log down trapezoidal rule from time of dosing to the last measurable concentration (AUC0-last), and to time infinity (AUCINF). AUCINF values with an extrapolated area exceeding 25% of the total were excluded. The elimination rate (lambda z) was estimated using at least 3 terminal points having a correlation (adjusted r2) > 0.8. Half-life (t1/2) was calculated as the natural log 2/lambda z. Clearance was calculated as dose/AUCINF.
Pharmacodynamic Studies: Cell-Free DNA
As an exploratory study in consenting patients, blood was collected to assess histone mutation status and changes with therapy in cell-free DNA via microfluidic analysis. Samples were collected on consenting patients prior to dosing of courses 1, 2, 4, 6, and 12.
Cell-free DNA extraction.
—Cell-free DNA (cfDNA) was manually extracted using the QIamp Circulating Nucleic Acid Extraction Kit (Qiagen) as per manufacturer’s protocol for most of the samples. The last samples were extracted using the automated Maxwell RSC ccfDNA Plasma Kit. Samples were then run on the 2100 BioAnalyzer (Agilent) using HS DNA chips.
Preamplification and ddPCR.
—For the H3F3A_K27M assay, samples were preamplified by Rnase H2-dependent PCR (rhPCR). The rhPCR reaction was carried out using 2.5 µl of cfDNA, 5 µl of SsoFast Evagreen mix (Biorad), 300 nM final of each primers, and 150 mU of P.a RnaseH2 (Integrated DNA Technologies). Conditions for the rhPCR were: 2 min at 98°C followed by 10 cycles of 5 sec at 98°C, 25 sec at 66°C. For Hist1H3B_K27M, samples were preamplified using SsoAdvanced PreAmp Supermix (Biorad). Reaction was carried out using 2.5 µl of cfDNA, 5 µl of SsoAdvanced mix (Biorad), and 50 nM final of each primers. Conditions for the reaction was: 3 min at 95°C followed by 10 cycles of 15 sec at 95°C, 4 min at 58°C. Preamplified products were diluted 1 in 5 in low-EDTA TE buffer, then 5 µl of diluted product was used for droplet digital PCR. The ddPCR reaction was carried out using 900 nmol primers, 250 nmol probes and 2× ddPCR Supermix for probes on the QX-100 (Biorad) following manufacturer’s instructions. PCR conditions were 10 min at 95°C, 50 cycles at 94°C for 30 sec, 1 min at 56°C, then 10 min for 98°C. Results were then analyxed using the Quantasoft software. For both H3F3A_K27M and Hist1H3B_K27M assays, the limit of detection (LOD) was calculated following the application note from Stilla Technologies (“How to characterize the LOB and LOD in Crystal Digital PCR”). The calculated LOD were 11 droplets for H3F3A_K27M and 9 droplets for Hist1H3B_K27M assays.
Statistical Considerations
Because panobinostat is an oral agent supplied in fixed capsule sizes, the deliverable doses based on BSA considerations were limited to the rounding constraints as set by the minimum capsule size. An algorithm was, therefore, developed that enabled us to choose dose levels that would avoid overlaps in BSA-adjusted doses in our patient cohorts. For the lower dose levels, minimum BSA restrictions were utilized for enrollment to avoid these overlaps.
Panobinostat dose finding was governed by a 2-stage CRM which was applied separately in the 2 strata.25–27 In the first stage, an approach similar to empirical methods, for example, 3 + 3 or Rolling-6, was used until the first DLT was observed; all patients had to complete the DLT assessment before escalation to the next dose level was considered. The second stage began once the first DLT was observed; a CRM design was then utilized using a likelihood-based algorithm via the empiric function (also called the power function, F(x,β)=xβ for 0 < x < 1) to guide dose finding.25 The maximum tolerated dose (MTD) was declared, as the dose level at which at least six patients were treated, and the algorithm did not recommend escalation or de-escalation based on a target toxicity level of 25%. Once the MTD was declared, 6 additional patients were enrolled at this dose level to better characterize the toxicity and pharmacokinetic profile. If the toxicities in this expanded cohort exceeded the target probabilities for the MTD, the dose level was reduced, and additional patients evaluated at the lower dose until 12 patients were treated at a dose level, and the model did not suggest de-escalation. Associations between PK parameters and clinical and demographic variables such as toxicity, response, and PFS were explored when feasible.
Secondary objectives of this trial included assessment of PFS and OS as well as identification of histone 3 K27M mutations in peripheral blood and evaluation of changes with treatment. Efficacy analyses were conducted separately for each stratum. For the cell-free DNA-based assays, we summarized the percentage of patients in whom this mutation was detected within each stratum and at each time point.
Results
Patient Demographics and Characteristics
Patient characteristics by stratum are listed in Table 2. Nineteen eligible patients were enrolled in stratum 1, with 17 of 19 evaluable for dose finding. The 2 inevaluable patients received less than the required dose of study drug. For stratum 2, 34 eligible patients enrolled, with 29 of 34 evaluable for dose finding. Inevaluable patients in stratum 2 included those who had PD prior to active treatment (n = 1), received less than required dose of study drug (n = 2), received insufficient labs to monitor for dose-limiting toxicities (n = 1), and patient withdrawal prior to beginning protocol therapy (n = 1). Patients who initiated treatment on the study were evaluable for all other safety and efficacy assessments.
Table 2.
Patient Demographics
| Stratum 1 | Stratum 2 | |
|---|---|---|
| Enrolled | 19 | 34 |
| Evaluable for dose-finding | 17 | 29 |
| Age, years (at consent) | ||
| Mean (±SD) | 9.0 (± 5.2) | 8.3 (± 3.6) |
| Median | 7.1 | 7.9 |
| Range | 3.2–21.4 | 3.5–17.3 |
| Gender | ||
| Male | 6 (31.6%) | 12 (35.3%) |
| Female | 13 (68.4%) | 22 (64.7%) |
| Diagnosis | ||
| DIPG | 19 (100.0%) | 34 (100.0%) |
| Thalamic DMG, H3K27M-altered | n/a ^ | 0 |
^ Thalamic DMG is eligible for Stratum 2 only.
Abbreviation: SD: standard deviation.
Toxicity
A total of 51 patients were evaluable for toxicity; 46 patients were evaluable for dose finding (Table 3). Of these, 12 DLTs were observed in 10 patients. The majority were myelosuppression, including neutropenia and thrombocytopenia. Additional DLTs in stratum 2 included grade 2 prolonged, intolerable nausea, and grade 3 increased ALT. No dose-limiting diarrhea or cardiac events were observed. The MTD was defined as 10 mg/m2/dose 3 times per week for 3 weeks on/1 week off in stratum 1 and 22 mg/m2/dose 3 times per week for 1 week on/1 week off in stratum 2.
Table 3.
Dose-Limiting Toxicities by Dose Level
| Dose level | Dose panobinostat (mg/m2) |
Eligible/evaluable for dose-finding (n) | # patients with DLT (n) |
DLT |
|---|---|---|---|---|
| Stratum 1 | ||||
| 0 | 5 | |||
| 1 | 10 | 12 | 0 | |
| 2 | 16 | 5 | 2 | gr 4 neutropenia with gr 4 thrombocytopenia (n = 1) gr 3 thrombocytopenia (n = 1) |
| 3 | 22 | |||
| 4 | 28 | |||
| 5 | 36 | |||
| Stratum 2. | ||||
| 1 | 16 | 3/3 | 0 | 0 |
| 2 | 22 | 2/10 | 1 | gr 3 thrombocytopenia |
| 3 | 28 | 12/11 | 4 | gr 4 neutropenia (n = 3) gr 4 neutropenia and gr 4 thrombocytopenia (n = 1) |
| 4 | 36 | 7/5 | 3 | gr 2 nausea (intolerable) (n = 1) gr 3 increased ALT (n = 1) gr 4 thrombocytopenia (n = 1) |
Toxicities at least possibly related to panobinostat are listed in Supplementary Table S1 as the highest grade reported per patient per toxicity. As anticipated, the most common toxicities were thrombocytopenia, neutropenia, and lymphopenia. Fatigue and GI toxicities, including diarrhea and nausea, were also common, but generally grade 1 or 2. Unexpected toxicities at least possibly attributed to panobinostat are listed in Supplement Table 2. These were all grade 1 or 2, with the exception of 1 patient in stratum 1, dose level 1, who had grade 3 posterior reversible encephalopathy syndrome.
Pharmacokinetics
Of the 53 enrolled patients, 50 patients (94%) had PK data available; all but 2 (48/50, 96%) of these patients had sufficient first-dose data for PK analysis. Panobinostat exhibited rapid absorption followed by a biphasic elimination (Figure 1), consistent with a prior report in pediatric patients.23 In general, there was good absorption from oral capsules resulting in dose-proportional increases in both Cmax and AUClast (Figures 2–3). Table 4 reports the statistical summaries of PK parameters by dose level. Of note, CMAX in this study was 30%–60% higher than in a pediatric study in acute myeloid leukemia (AML) patients (10 mg/m2: median 10.9 ng/mL vs. 8.4 ng/mL2; 16 mg/m2: 19.3 ng/mL vs. 11.8 ng/mL on 15 mg/m2; and 22 mg/m2: 31.8 ng/mL vs. 20.7 ng/mL on 20 mg/m2).28 In this study, the average half-life and clearance were 13 h (24% CV, n = 36) and 112 L/h (52% CV, n = 36).
Figure 1.
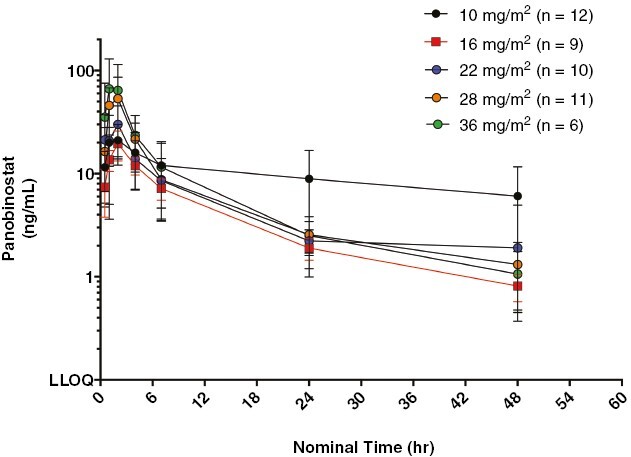
Mean log concentration of panobinostat versus time by dose level.
Figure 2.
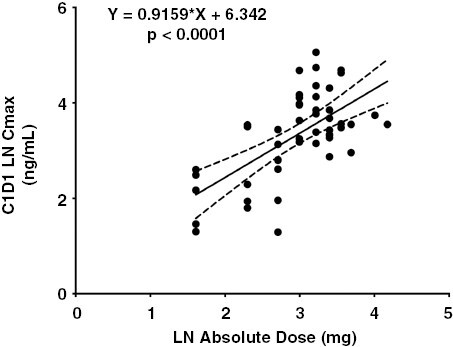
Dose proportional increases in panobinostat CMAX with absolute dose (mg).
Figure 3.
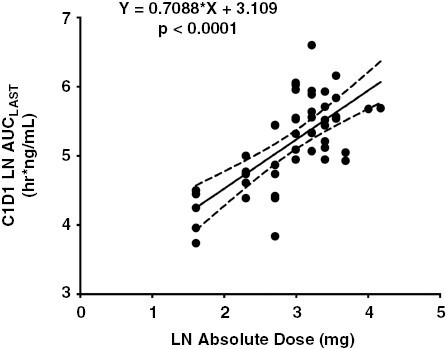
Dose Proportional increases in panobinostat AUCLAST with absolute dose (mg).
Table 4.
Noncompartmental Analysis of the Clinical Pharmacokinetics of Panobinostat
| First dose | CMAX (ng/mL) |
†CMAX/D (ng/mL/mg) |
TMAX (h) |
AUCLAST (h*ng/mL) |
†AUCLAST/D (h*ng/mL/mg) |
AUCINF (h*ng/mL) |
T1/2 (h) |
CL (L/h) |
Vd (L) |
|---|---|---|---|---|---|---|---|---|---|
| 10 mg/m2 (n = 12) a |
16.1 (78%) |
1.74 (59%) |
2.0 (0.6–6) |
102 (43%) |
11.8 (29%) |
110 (45%)a |
13.0 (31%)a |
87 (22%)a |
1704 (49%)a |
| 16 mg/m2 (n = 9) b |
22.5 (79%) |
1.0 (86%) |
2.0 (0.5–4.3) |
175 (70%) |
7.9 (71%) |
193 (74%)b |
12.4 (20%)b |
150 (55%)b |
2594 (50%)b |
| 22 mg/m2 (n = 10) c |
35.6 (41%) |
1.5 (54%) |
2.0 (0.7–7.0) |
239 (30%) |
10.4 (48%) |
224 (22%)c,d |
12.6 (22%)c,d |
115 (30%)c,d |
2141 (42%)c,d |
| 28 mg/m2 (n = 11) |
61.9 (48%) |
2.3 (45%) |
2.0 (1–2.2) |
285 (26%) |
10.7 (26%) |
297 (28%)e,f |
14.8 (13%)e,f |
93 (28%)e,f |
1967 (23%)e,f |
| 36 mg/m2 (n = 6) |
78 (67%) |
2.8 (88%) |
1.9 (1–2) |
372 (60%) |
12.9 (81%) |
376 (75%)g |
11.3 (33%)g |
129 (84%)g |
2487 (111%)g |
| All cohorts (n = 48) d |
n/a | 1.83 (72%) |
2.0 (0.5–7.0) |
n/a | 10.7 (50%) |
n/a | 13.0 (24%) |
112 (52%) |
2125 (57%) |
*Data presented as arithmetic means (%CV); TMAX reported as median (range).
†All dose-normalized metrics were scaled to the absolute dose values (in mg, not mg/m2).
aPatient 112049295 had unacceptable correlation (r2 < 0.8) for lambda z, hence cannot use values of T1/2, AUCINF, CL, and Vd.
bPatient 105049119 had insufficient data for lambda z estimation, hence no values of T1/2, AUCINF, CL, and Vd.
cPatient 125049981 had insufficient data for lambda z estimation, hence no values of T1/2, AUCINF, CL, and Vd.
dPatients 148049303 and 151049115 had unacceptable correlation (r2 < 0.8) for lambda z, hence cannot use values of T1/2, AUCINF, CL, and Vd.
ePatient 142049520 had insufficient data for lambda z estimation, hence no values of T1/2, AUCINF, CL, and Vd.
fPatients 130049176, 145049759, and 146049493 had unacceptable correlation (r2 < 0.8) for lambda z, hence cannot use values of T1/2, AUCINF, CL, and Vd.
gPatients 138049310 and 139049214 had unacceptable correlation (r2 < 0.8) for lambda z, hence cannot use values of T1/2, AUCINF, CL, and Vd.
Based on Wilcoxon rank sum test, there was not sufficient evidence to suggest an association between AUClast of panobinostat and DLT (P-value = .062). However, there was an association between panobinostat AUClast and incidence of myelosuppression (P-value = .018). On average, patients who had myelosuppression DLTs had higher panobinostat AUClast (median 281.4 vs. 164.6 h * ng/ml). In addition, patients with DLTs had higher Cmax values (median: 40.9 vs. 27.9 ng/ml, P = .0521), and patients with myelosuppression DLTs had higher Cmax values (median: 50.2 vs. 30.3 ng/ml, P = .0420). There was no association of AUC, Cmax, and clearance of panobinostat with use of dexamethasone. Based upon a Cox proportional hazards model for OS, there was no association between OS and AUClast for stratum 1 (P-value = .180) or stratum 2 (P-value = .585).
Assessment of Efficacy
For stratum 1, the median (range) number of courses administered was 2 (1–5); the median time to progression from initiation of protocol therapy was 2.0 months (range: 0.9–4.0 months), and median time to death from initiation of treatment was 5.2 months (range: 0.7–15.2 months). For stratum 2, the median (range) number of courses administered was 3 (1–12); 1 patient received 12 courses and another 1 received 11 courses of therapy. The median time to progression from initiation of protocol chemotherapy was 4.4 months (range: 1.0–11.0 months), and median time to death from initial diagnosis was 11.8 months (range: 4.8–25.0 months).
Radiographic Response
All patients had measurable disease at baseline. Forty-eight patients were evaluable for radiographic response. Two patients did not receive any protocol therapy. Three patients did not have disease re-evaluated by MRI due to patient withdrawal/refusal after beginning protocol therapy (n = 2) and death on study unrelated to study drug or disease progression (n = 1). One patient in stratum 1, dose level 1 (10 mg/m2), had ~35% decrease in tumor size but had to hold drug and then dose reduce due to an infection and subsequently had clinical progression and came off study treatment. One patient in stratum 2, dose level 2 (22 mg/m2) had a PR reportedatcourse 4 but PD at course 6. Two patients, both in stratum 2, had SD for >6 months, including 1 patient on dose level 16 mg/m2 with SD at 8.0 months and PD at 10.8 months and 1 patient on dose level 28 mg/m2 with SD at 8.1 months but PD at 10.7 months.
Exploratory Pharmacodynamic cfDNA Studies
A total of 74 plasma samples were collected from 34 unique patients. One sample was discarded due to a processing error. Six samples from 5 patients had levels above the level of detection (LOD). Within this limited number of patients, there was no apparent correlation with outcome. Given the limited number of samples above the LOD and the unknown mutation status of several patients, we cannot make conclusions regarding correlation of H3K27M status with clinical outcome.
Discussion
In this phase I study, panobinostat had limited tolerability in pediatric patients with DIPG/DMG. The major toxicity observed was myelosuppression, which limited drug administration and dose escalation. Please note that hematological dose-limiting toxicities were defined more stringently for this population of patients than the definitions used in previous pediatric AML studies.20,28 Panobinostat administered 3 times per week either 3 weeks on/1 week off or every other week is tolerated in pediatric patients with DIPG/DMG at doses below that estimated to be optimal for efficacy based on preclinical studies in animal models.
Although panobinostat demonstrated preclinical activity against DIPG, its efficacy in these studies was dose-related.8 Based on the preclinical studies of panobinostat in DIPG animal models, we aimed to achieve target dosing of at least 30 mg/m2/dose, which was achievable in clinical trials of panobinostat in pediatric patients with hematologic malignancies.20,28 In one study, heavily pretreated children with relapsed/refractory leukemia tolerated panobinostat doses up to 34 mg/m2/day, 3 days a week every week, without an MTD declared.20 In the second study, children with AML were treated with panobinostat prior to, and in combination with fludarabine and cytarabine, with panobinostat administered 3 times per week ×2 weeks for 1 cycle at doses up to 20 mg/m2; no DLTs were observed.28 However, in both studies, the definition of DLT was amended to account for the hematologic malignancy, that is, defined as failure to recover counts in the absence of leukemia by day 5628 and failure to recover a peripheral ANC >500/µl and platelet count >20 000/µl more than 42 days from Day 1 of cycle 1 in the absence of persistent leukemia or lymphoma20 thus was not comparable. In DIPG/DMG, which carries a significant risk of intratumoral hemorrhage29 thrombocytopenia in particular adds considerable potential risk to the patient, and therefore, was included in our definition of DLT.
A major question that remains for the use of panobinostat in DIPG/DMG is its optimal schedule. It is not known whether intermittent higher doses may be more efficacious than lower doses administered more frequently. Overall, the schedule employed in stratum 2 allowed for higher dosing to be administered and appeared better tolerated, although direct comparison of drug tolerability between schedules was constrained by the different eligibility criteria. However, while 16 mg/m2/dose was declared too toxic in stratum 1 due to dose limiting myelosuppression, no dose-limiting toxicities were observed in the 3 patients treated at this dose on the alternate schedule in stratum 2, suggesting a correlation between dose-density and toxicity. While we demonstrated no significant correlation between panobinostat exposure (AUClast) and DLT, patients with DLTs tended to have a higher Cmax with a significant association between Cmax and myelosuppression, suggesting intermittent higher dosing may be less tolerable.
Pharmacokinetic analysis on our study indicated a shorter half-life and different clearance of panobinostat in children compared to data on adults reported in the package insert. The prescribing information for Farydak® reports a half-life of 37 hr and a clearance of 160 L/hr, estimated from a population PK model based on data from adults with multiple myeloma.23 In our study of pediatric patients with a median age 7.8 yr (range 3.2 – 21.4 yr), the average half-life and clearance were 13 h (24%CV, n = 36) and 112 L/h (52%CV, n = 36), respectively. It is not yet clear why our pediatric population exhibits different pharmacokinetics. Panobinostat is a CYP3A substrate and CYP3A inducers, such as dexamethasone, may decrease panobinostat plasma concentrations.30 However, our PK parameters are in line with a study of single agent panobinostat in adult Japanese patients with advanced solid tumors (t1/2 17.8–18.4 h, CL 67–150 L/h).31 It is important to note that the adult Japanese study only conducted PK sampling out to 48 h, the same as this pediatric study, and both yielded comparable values for half-life and clearance. In addition, the half-life reported in a pediatric study of hematologic malignancies20 was very similar to ours at 12.8 ± 3.0 h. Clive et al.32 performed an extensive single dose study of radiolabeled panobinostat in adults to elucidate the metabolism and disposition, with dense serial sampling out to 168 h post dose and reported a half-life and clearance of 30.7 h and 209 L/h, respectively. Other reports, including the model estimates reported and used in the Farydak prescribing information23 were based on single dose studies with sampling out to 96 h or 168 h post dose. The variability in these pharmacokinetic studies suggest that the need for additional population pharmacokinetic studies of panobinostat.
No significant clinical benefit was observed in our study. It is assumed that the lack of efficacy observed in our study was due to insufficient dosing and exposure at the tumor site; it is unclear if the different PK profile in our population plays a role. Several preclinical studies have demonstrated limited penetration of panobinostat into the CNS (CSF and tissue),24 and CSF panobinostat levels were below the LOD in pediatric patients.20,33 Others have shown that effective concentrations may be achieved in a murine model.34 It remains unclear if species or tumor characteristics play a role in panobinostat CNS penetration, possibly accounting for these varied results. Alternative delivery of this agent may overcome this hurdle. Preclinical studies demonstrate efficacy of panobinostat delivery by convection enhanced delivery (CED),8 and a recent clinical trial of aqueous panobinostat delivery by CED in 7 subjects demonstrated tolerability and a median OS that compared favorably to historical controls in this small cohort.35 It is likely that a multipronged approach is needed to adequately suppress DIPG tumor growth. Given the promising preclinical activity of this agent in DIPG, panobinostat or similar agents may play a role as part of a multiagent strategy for these tumors,9 and alternative delivery methods such as CED should be studied further.
This is the first clinical trial assessing tolerability of systemically administered panobinostat in children with DIPG/DMG. The MTD differed between our 2 strata, which differed by schedule and disease status. Within the context of this phase 1 trial, no significant clinical benefit of panobinostat in the DIPG/DMG population was detected. Further development of this and other HDAC inhibitors for children with DIPG will likely necessitate combination therapy and alternative methods of delivery.
Supplementary Material
Acknowledgement
The authors thank Ashley Plant for helpful input. Presented in Part at: International Society of Pediatric Neuro-Oncology, Denver, 2018 and Hamburg, Germany, June 2022.
Contributor Information
Michelle Monje, Department of Neurology, Stanford University and Lucile Packard Children’s Hospital, Palo Alto, CA, USA.
Tabitha Cooney, Department of Pediatric Oncology, Dana Farber Cancer Institute/Boston Children’s Hospital, Boston, MA, USA.
John Glod, Pediatric Oncology, Pediatric Oncology Branch, National Cancer Institute, Bethesda, MD, US.
Jie Huang, Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Cody J Peer, Center for Cancer Research, Clinical Pharmacology Program, National Cancer Institute, Bethesda, Maryland, USA.
Damien Faury, Research Institute of the McGill University Health Center, Montreal, Quebec, CANADA.
Patricia Baxter, Pediatric Oncology, Texas Children’s Cancer Center, Houston, TX, USA.
Kim Kramer, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, USA.
Alicia Lenzen, Pediatric Hematology Oncology, Lurie Children’s Hospital, Chicago, IL, USA.
Nathan J Robison, Department of Pediatrics, Children’s Hospital, Los Angeles, CA, USA.
Lindsay Kilburn, Department of Oncology, Children’s National Hospital, Washington, DC, USA.
Anna Vinitsky, Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN, USA.
William D Figg, Center for Cancer Research, Clinical Pharmacology Program, National Cancer Institute, Bethesda, Maryland, USA.
Nada Jabado, Research Institute of the McGill University Health Center, Montreal, Quebec, CANADA.
Maryam Fouladi, Pediatric Hematology Oncology, Nationwide Children’s Hospital, Columbus, OH, USA.
Jason Fangusaro, Department: Pediatric Hematology/Oncology and Stem Cell Transplantation, Atlanta, GA, USA.
Arzu Onar-Thomas, Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Ira J Dunkel, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, USA.
Katherine E Warren, Department of Pediatric Oncology, Dana Farber Cancer Institute/Boston Children’s Hospital, Boston, MA, USA; Pediatric Oncology, Pediatric Oncology Branch, National Cancer Institute, Bethesda, MD, US.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number UM1CA081457. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the American Lebanese Syrian Associated Charities (ALSAC) which provides funding and infrastructure support for the PBTC Operations Core personnel but had no involvement in trial design, patient recruitment, data collection, analyses, or manuscript preparation. Lyla Nsouli Foundation also provided funding for data monitoring personnel and research studies. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Conflict of interest statement
Michelle Monje served on the scientific advisory board for Cygnal Therapeutics and her family holds equity in Maplight Therapeutics; Ira J. Dunkel serves on the advisory board for AstraZeneca and Bristol Meyers Squibb and is a consultant for DAY One Therapeutics, Fennel, Glaxo Smith Kline, Pyramid and QED; Jason Fangusaro serves on the advisory board for Alexion/AstraZeneca and Merck; Katherine E. Warren serves on the advisory board for DAY One Therapeutics and Glaxo Smith Kline. All other authors have nothing to disclose.
Authorship statement
MM,TC, MF, AOT, KEW participated in initial study design. JH, AOT participated in the statistical analysis of the data; MM, TC, JG, CJP, DF, PB, KK, AL, NJR, LK, AV, WDF, NJ, MF, JF, AOT, IJD, KEW participated in acquisition of the data; all authors provided critical review of the manuscript; all authors approved to submit the manuscript for publication.
Data Availability
Anonymized data are available to qualified research investigators. The policy and instructions can be viewed at https://www.pbtc.org/data-sharing.html.
References
- 1. Wu G, Broniscer A, McEachron T, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwartzentruber J, Korshunov A, Liu X, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. [DOI] [PubMed] [Google Scholar]
- 3. Khuong-Quang D, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropath. 2012;124(3):439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louis D, Perry A, Wesseling P, et al. The 2021 WHO Classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jansen M, van Vuurden D, Vandertop W, Kaspers G.. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev. 2012;38(1):27–35. [DOI] [PubMed] [Google Scholar]
- 6. Cooney T, Lane A, Bartels U, et al. Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro Oncol. 2017;19(9):1279–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffman L, Veldhuijzen van Zanten S, Colditz N, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol. 2018;36(19):1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grasso C, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21(6):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin G, Wilson K, Ceribelli M, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med. 2019;11(519):eaaw0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vitanza N, Biery M, Myers C, et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naïve diffuse midline glioma models. Neuro Oncol. 2021;23(3):376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hennika T, Hu G, Olaciregui N, et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS One. 2017;12(1):e0169485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ehteda A, Simon S, Franshaw L, et al. Dual targeting of the epigenome via FACT complex and histone deacetylase is a potent treatment strategy for DIPG. Cell Rep. 1089;35(2):108994. [DOI] [PubMed] [Google Scholar]
- 13. Crisanti M, Wallace A, Kapoor V, et al. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther. 2009;8(8):2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cha T-L, Chuang M-J, Wu S-T, et al. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009;15(3):840–850. [DOI] [PubMed] [Google Scholar]
- 15. José-Enériz E, Gimenez-Camino N, Agirre X, Prosper P.. HDAC inhibitors in acute myeloid leukemia. Cancers (Basel). 2019;11(11):1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maiso P, Carvajal-Vergara X, Ocio E, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66(11):5781–5789. [DOI] [PubMed] [Google Scholar]
- 17. Lemoine M, Derenzini E, Buglio D, et al. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood. 2012;119(17):4017–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grassadonia A, Cioffi P, Simiele F, et al. Role of hydroxamate-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment of solid malignancies. Cancers (Basel). 2013;5(3):919–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garrett L, Growdon W, Rueda B, Foster R.. Influence of a novel histone deacetylase inhibitor panobinostat (LBH589) on the growth of ovarian cancer. J Ovarian Res. 2016;9(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldberg J, Sulis M, Bender J, et al. A phase I study of panobinostat in children with relapsed and refractory hematologic malignancies. Pediatr Hematol Oncol. 2020;37(6):465–474. [DOI] [PubMed] [Google Scholar]
- 21. DeAngelo D, Spencer A, Bhalla K, et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013;27(8):1628–1636. [DOI] [PubMed] [Google Scholar]
- 22. Sharma S, Beck J, Mita M, et al. A phase I dose-escalation study of intravenous panobinostat in patients with lymphoma and solid tumors. Invest New Drugs. 2013;31(4):974–985. [DOI] [PubMed] [Google Scholar]
- 23. Novartis, FARYDAK (panobinostat) [package insert]. Novartis Pharmaceuticals Corporation. East Hanover, New Jersey: Novartis Pharmaceuticals Corporation; 2015. [Google Scholar]
- 24. Rodgers L, Lester McCully C, Odabas A, et al. Characterizing the pharmacokinetics of panobinostat in a non-human primate model for the treatment of diffuse intrinsic pontine glioma. Cancer Chemother Pharmacol. 2020;85(4):827–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheung Y. Dose Finding by the Continual Reassessment Method. CRC Press, 2011. [Google Scholar]
- 26. Lee S, Cheung Y.. Model calibration in the continual reassessment method. Clin Trial. 2009;6(3):227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wages N, Conaway M, O’Quigle J.. Performance of two-stage continual reassessment method relative to an optimal benchmark. Clin Trial 2013;10(6):862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karol S, Cooper T, Mead P, et al. Safety, pharmacokinetics, and pharmacodynamics of panobinostat in children, adolescents, and young adults with relapsed acute myeloid leukemia. Cancer. 2020;126(21):4800–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broniscer A, Laningham F, Kocak M, et al. Intratumoral hemorrhage among children with newly diagnosed, diffuse brainstem glioma. Cancer. 2006;106(6):1364–1371. [DOI] [PubMed] [Google Scholar]
- 30. Srinivas N. Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: review and perspectives. Xenobiotica. 2017;47(4):354–368. [DOI] [PubMed] [Google Scholar]
- 31. Fukutomi A, Hatake K, Matsui K, et al. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs. 2012;30(3):1096–1106. [DOI] [PubMed] [Google Scholar]
- 32. Clive S, Woo M, Nydam T, et al. Characterizing the disposition, metabolism, and excretion of an orally active pan-deacetylase inhibitor, panobinostat, via trace radiolabeled 14C material in advanced cancer patients. Cancer Chemother Pharmacol. 2012;70(4):513–522. [DOI] [PubMed] [Google Scholar]
- 33. Guntner A, Peyrl A, Mayr L, et al. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol Commun. 2020;8(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Homan M, Franson A, Ravi K, et al. Panobinostat penetrates the blood-brain barrier and achieves effective brain concentrations in a murine model. Cancer Chemother Pharmacol. 2021;88(3):555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mueller S, Kline C, Stoller S, et al. PNOC015: repeated convection enhanced delivery (CED) of MTX110 (aqueous panobinostat) in children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). Neuro Oncol. 2023;25(11):2074–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data are available to qualified research investigators. The policy and instructions can be viewed at https://www.pbtc.org/data-sharing.html.