

Abstract
Background
The prognosis for Li–Fraumeni syndrome (LFS) patients with medulloblastoma (MB) is poor. Comprehensive clinical data for this patient group is lacking, challenging the development of novel therapeutic strategies. Here, we present clinical and molecular data on a retrospective cohort of pediatric LFS MB patients.
Methods
In this multinational, multicenter retrospective cohort study, LFS patients under 21 years with MB and class 5 or class 4 constitutional TP53 variants were included. TP53 mutation status, methylation subgroup, treatment, progression free- (PFS) and overall survival (OS), recurrence patterns, and incidence of subsequent neoplasms were evaluated.
Results
The study evaluated 47 LFS individuals diagnosed with MB, mainly classified as DNA methylation subgroup “SHH_3” (86%). The majority (74%) of constitutional TP53 variants represented missense variants. The 2- and 5-year (y-) PFS were 36% and 20%, and 2- and 5y-OS were 53% and 23%, respectively. Patients who received postoperative radiotherapy (RT) (2y-PFS: 44%, 2y-OS: 60%) or chemotherapy before RT (2y-PFS: 32%, 2y-OS: 48%) had significantly better clinical outcome then patients who were not treated with RT (2y-PFS: 0%, 2y-OS: 25%). Patients treated according to protocols including high-intensity chemotherapy and patients who received only maintenance-type chemotherapy showed similar outcomes (2y-PFS: 42% and 35%, 2y-OS: 68% and 53%, respectively).
Conclusions
LFS MB patients have a dismal prognosis. In the presented cohort use of RT significantly increased survival rates, whereas chemotherapy intensity did not influence their clinical outcome. Prospective collection of clinical data and development of novel treatments are required to improve the outcome of LFS MB patients.
Keywords: Li–Fraumeni syndrome, medulloblastoma, survival, TP53
Graphical Abstract
Graphical Abstract.
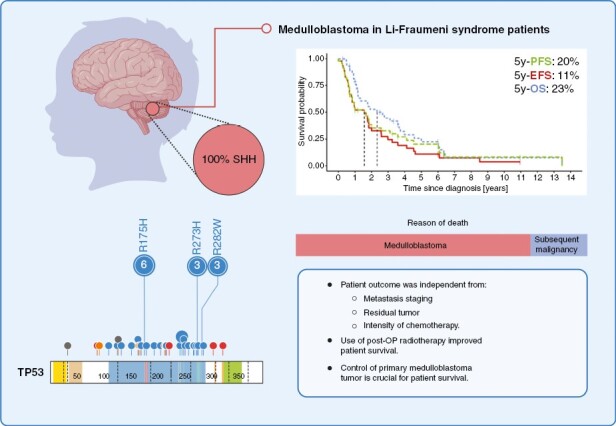
Key Points.
Li–Fraumeni syndrome patients with medulloblastoma have a dismal prognosis.
Use of radiotherapy in this patient group improves overall survival.
Control of the primary medulloblastoma tumor is critical for patient survival.
Importance of the Study.
Pediatric patients affected by the cancer predisposing Li–Fraumeni syndrome (LFS) are at risk of developing medulloblastoma (MB). Given LFS rarity, there is a paucity of published data on the biological features, clinical outcome and optimal therapy, leading to uncertainty regarding the most suitable therapeutic approach for these patients. In this retrospective study, we present molecular and clinical data about 47 pediatric LFS MB patients diagnosed in the years 1987–2022. LFS MB patients showed poor survival that was independent from clinical high-risk features such as residual tumor or metastases. Intensive treatment protocols including high-intensity chemotherapy did not result in a survival benefit for LFS MB patients in this cohort when compared with maintenance-type only chemotherapy, whereas application of RT seemed to improve survival. In order to verify these results and improve the outcome for LFS MB patients, molecular and clinical data should be collected in a prospective manner.
Medulloblastoma (MB) is one of the most common brain tumors in pediatric patients, with 2.0–5.8 cases per 1,000,000 annually.1–3 It consists of at least four different molecular groups (WNT, SHH, Group 3 and Group 4) with highly diverging clinical and molecular characteristics.4,5 Moreover, recent studies distinguished diverse molecular subgroups within these groups based on specific gene expression and DNA methylation patterns.6,7 For example, within the Sonic Hedgehog (SHH) group the 2021 WHO classification of CNS tumors recognizes subgroups SHH-1-4.3,5,8 In addition, SHH α-δ,5 SHH-Infant and -Child7,9 subgroups have been described in the literature.
TP53, a tumor suppressor gene, is somatically mutated in more than half of all cancers, yet somatic TP53 mutations are detected in only 10–16% MBs.10,11 Within specific MB groups, somatic TP53 mutations are most frequent in the SHH group (21%),11 in particular in the SHHα or SHH-3 subgroups.8,11 Moreover, somatically mutated TP53 (TP53mut) is highly enriched in the SHH-Child subgroup (27%), compared with the SHH-infant and adult subpopulations.11,12 Less frequently, somatic TP53 mutations are found in the WNT group (16%),6,11 and are very rarely observed in Group 3 and 4 tumors (0 and 0.8%, respectively).11
Li–Fraumeni syndrome (LFS), caused by inherited or de novo constitutional TP53 pathogenic variants, was attributed to 33–56% of TP53mut SHH-MB cases.7,11,13 Similar to other cancer types, both constitutional and somatic pathogenic TP53 mutations are found almost exclusively in the DNA binding domain.6,11,13,14 Based on the loss of p53 wildtype function, subsequent molecular hallmarks include higher mutational burden (number of nonsynonymous SNVs: range: 7–29, median: 19.5),12 and in MB the frequent occurrence of chromothripsis, a one-step catastrophic genetic event resulting in massive chromosomal rearrangements.13,15
Like in many other cancer types, both somatic, as well as constitutional TP53 alterations have a prognostic significance in MB patients. While in WNT-MB the impact of somatic TP53 mutations on survival is under debate,11,16,17 they indicate a significantly worse prognosis in SHH-MB patients. Zhukova et al. described 5-(y)ear OS+/−SE rates to be 81%+/−5% in TP53wt SHH-MB as opposed to 41%+/−9% in TP53mut SHH-MB.11 In addition, Schwalbe et al. described TP53 alterations (both somatic and constitutional) as an independent risk factor for progression-free survival (PFS) in the in MB SHH-Child cohort.7 It is therefore concluded that SHH-MB shows a differential prognosis according to the TP53 mutational status that renders poor survival in patients within this MB group.11 For these reasons, in the 2021 WHO classification of CNS tumors, TP53mut SHH-MB was described as a separate entity signifying a key diagnostic role for the TP53 gene in the “Medulloblastoma, SHH-activated” group18 and classified as a very high-risk disease.7,19
The highly increased incidence of relapses and secondary tumors contributes to the dismal prognosis in LFS MB patients.20–22 In the past decade, specific surveillance protocols dedicated for individuals affected by LFS have been implemented,23–25 and considerable survival benefit for LFS patients following surveillance was shown.26 In addition, radiotherapy and DNA damaging agents were recognized as potential causes of subsequent malignancies, and therefore their use in LFS patients needs to be carefully weighted in a risk-benefit assessment.25,27,28
Despite a high clinical need, comprehensive studies on the clinical course of LFS MB patients are lacking. Here, we present data on the LFS MB cohort, including analysis of molecular subgroups, TP53 alterations, treatment modalities, subsequent malignancies and patient survival.
Methods
Patient Cohort
This is a multinational, multicenter (31 centers), retrospective cohort study. The inclusion criteria were: I) diagnosis of MB at age below 21 years old, II) pathogenic (PV, class 5) or likely-pathogenic (LPV, class 4) TP53 constitutional variant.29 The mutational status of TP53, both constitutional and somatic in the tumor, was analyzed in all patients, as indicated in Table S1. Pathogenicity of constitutional TP53 variants was evaluated by two independent human genetics experts, S.H. and G.B., according to current recommendations.29 Patients were diagnosed in years 1987–2022 in the respective institutions; cases previously described in other publications were specified in Supplementary Table S1. Data collection was approved by institutional review boards in accordance with the local ethics committee’s requirements. Informed consent to collect data was obtained from the subjects after appropriate information was provided. Information on molecular characteristics was collected for all the included cases, complete survival and extended clinical data were available in 46/47 (98%) cases.
Nine sporadic TP53mut SHH-MB (Supplementary Table S2) used for comparative survival analysis were published in Waszak et al. 2018.13 The reference set of 389 SHH MB used for t-SNE analysis, provisionally grouped into four subgroups, was published.30
Molecular Subgroup Analysis
In 38/47 (80.9%) cases, molecular subgroup was determined by DNA methylation analysis as described previously in Waszak et al.13 (Supplementary Table S1). In 2/47 (4.3%) cases, determination of the MB group was based on immunohistochemistry (IHC) and in 1/47 (2.1%) based on Nanostring. The method of MB group determination was unknown in 6/47 (12.8%) cases.
Chemotherapy Intensity Stratification
Chemotherapy protocols used in MB treatment were divided into standard-intensity (SI) or high-intensity (HI). HI chemotherapy was defined as protocols that included thiotepa, melphalan or any high-dose chemotherapy except methotrexate (MTX). All treatment protocols that do not fulfil this criterion were classified as SI chemotherapy. The list of all chemotherapy protocols can be found in Supplementary Table S3.
Statistical Analysis
Survival time was calculated from the time of MB diagnosis. The endpoints were: I) MB progression or death without progression for PFS, II) MB progression, subsequent tumor or death for event-free survival (EFS), III) death for OS. In the recurrence analyses, patients who relapsed/progressed ≤ 6 months from diagnosis were classified as having refractory disease. In the survival analyses, relapses and progressions were included as events in PFS and EFS regardless of timepoint. Kaplan–Meyer analyses, including log-rank tests, were done using “survival” R package (v3.2-11). Cumulative incidence functions for competing reasons of death were estimated based on OS data (“cmprsk” R package, v2.2-11). Multivariable regression models were generated using a Cox proportional hazards regression model implemented the R package survival v3.2-7 (“coxph” function). All covariates were included based on clinical relevance and there was no exclusion of variables or specific model selection performed based on AIC or other metric. Hazard ratios (HRs) were calculated based on Cox regression models and included 95% confidence intervals (CI). To compare categorical variables, Fisher’s exact test was used.
DNA Methylation Profile Classification and t-SNE Visualization
DNA methylation profiles were analyzed using the DKFZ Molecular Neuropathology (MNP) classifier, version 12.5.30 Beta values for the 10 000 array probes used by the MNP classifier v12.5 were retrieved, dimensionality reduction was performed with t-SNE using the Rtsne package in R version 4.0.0. The parameters were set as theta = 0.5, eta = 200, and perplexity = 15.
Copy Number Variation Analysis
Copy number variation (CNV) analysis was carried out using the conumee bioconductor (http://www.bioconductor.org/packages/devel/bioc/html/conumee.html). The copy number segmentation was performed by applying the circular binary algorithm (CBS, R package: DNAcopy),31 as previously described and applied to methylation arrays.32 The averaged LRR (log2 copy number ratio) was calculated for each segment and sample and input into the GISTIC 2.033 to identify the most recurrent focal and broad events. For the selected genes, a threshold of LRR ≥ 0.4 and ≤ −0.4 was used to call amplifications and deletions, respectively. A chromosome was declared gained if the LRR ≥ 0.2, lost if the LRR ≤ −0.2, and balanced otherwise.
Results
Clinical Features of LFS MB Patients
Age at MB diagnosis ranged from 3 to 20.1 years (median: 9.1 years) (Figure 1A). The majority of patients were male (31/47, 66%) (Table 1). Gross total resection (GTR) was achieved in 32/45 (71.1%), and subtotal resection (STR) in 13/45 (28.9%) informative cases (Table 1). Tumors were staged M0 in 36/47 (76.6%) cases, whereas 11/47 (23.4%) were M + (M1-M3) (Table 1, Supplementary Table S1).
Figure 1:

Characteristics of the Li–Fraumeni syndrome (LFS) medulloblastoma (MB) patient cohort. (A) Age distribution at MB diagnosis; (B) TP53 constitutional variants in LFS MB patients; (C) TP53 variant pathogenicity; (D) Somatic TP53 mutational status in LFS-associated MB (LOH—loss of heterozygosity); (E) Origin of constitutional TP53 variants in LFS MB patients; (F) t-SNE clustering of DNA methylation profiles of LFS MBs with a reference SHH-MB cohort; (G) LFS-associated MB subgroups classified based on DNA methylation profile.
Table 1.
Li–Fraumeni syndrome medulloblastoma patients characteristics
| Variable | % | |
|---|---|---|
| Clinical characteristics | ||
| Total n of cases [n] | 47 | 100 |
| Sex [n] (n = 47): | ||
| Female | 16 | 34.0 |
| Male | 31 | 66.0 |
| Age at diagnosis [years] (n = 47): | ||
| Median | 9.1 | - |
| Minimum | 3 | - |
| Maximum | 20.1 | - |
| Timepoint of LFS diagnosis [n] (n = 42): | ||
| Before MB | 4 | 9.5 |
| After MB | 38 | 90.5 |
| Tumor characteristics | ||
| Method of subgroup determination [n] (n = 41): | ||
| IHC | 2 | 4.9 |
| Methylation array | 38 | 92.7 |
| 450k | 18 | 43.9 |
| EPIC/850k | 17 | 41.5 |
| Nanostring | 1 | 2.4 |
| Histology [n] (n = 42): | ||
| LCA | 22 | 52.4 |
| CMB | 8 | 19.0 |
| DMB | 4 | 9.5 |
| LCA/DMB | 2 | 4.8 |
| MBEN | 1 | 2.4 |
| Medulloepithelioma | 1 | 2.4 |
| NOS | 4 | 9.5 |
| M-Stage [n] (n = 47): | ||
| M0 | 36 | 76.6 |
| M+ | 11 | 23.4 |
| Medulloblastoma treatment information | ||
| Radicality of resection [n] (n = 45): | ||
| GTR | 32 | 71.1 |
| STR | 13 | 28.9 |
| Chemotherapy dose intensity [n] (n = 46): | ||
| No chemotherapy | 5 | 10.9 |
| Standard | 30 | 65.2 |
| High | 11 | 23.9 |
| RT [n] (n = 46): | ||
| Yes | 42 | 91.3 |
| No | 4 | 8.7 |
| Chemotherapy before RT [n] (n = 46): | ||
| Yes | 16 | 34.8 |
| No | 26 | 56.5 |
| Not applicable (No RT) | 4 | 8.7 |
| Maintenance chemotherapy [n] (n = 46): | ||
| Yes | 33 | 71.7 |
| No | 9 | 19.6 |
| Not applicable (No RT) | 4 | 8.7 |
| Survival information | ||
| Median survival [months] (n = 46) | ||
| PFS | 19 | - |
| EFS | 19 | - |
| OS | 28 | - |
| Reason of death [n] (n = 33): | ||
| Died of disease | 27 | 81.8 |
| Subsequent malignancy | 6 | 18.2 |
Abbreviations: LFS, Li–Fraumeni Syndrome; MB,medulloblastoma; IHC, Immunohistochemistry; LCA, large-cell anaplastic; CMB, classic medulloblastoma; DMB, desmoplastic/nodular MB; NOS, not otherwise specified; MEPL, medulloepithelioma; MBEN, MB with extensive nodularity; GTR, gross-total resection; STR, subtotal resection; PFS, progression-free survival; EFS, event-free survival; OS, overall survival.
TP53 Mutational Status and Molecular Features
Constitutional heterozygous PV or LPV in the TP53 gene were detected in all patients, confirming the LFS diagnosis. LFS was diagnosed prior to MB in 4/42 informative cases (9.5%) and after the MB diagnosis in 38/42 informative cases (90.5%) (Table 1). The majority of TP53 PV (74%) were characterized as missense variants, whereas nonsense (11%) and frameshift (11%) variants were less frequent. The majority of TP53 variants (41/47, 87%) were localized in the DNA-binding domain (DBD) (Figure 1B, Supplementary Table S4). The six (13%) variants found outside of the DBD coding region were either truncating (c.261_270del, c.273G>A, c.906del, c.960del) or intronic mutations (c.96 + 31_97-32del, c.376-2A>G). The most frequently identified amino acid substitutions were p.R175H (13%), p.R273H (6%) and p.R282W (6%) (Figure 1B). The constitutional TP53 variants were classified as pathogenic/class 5 (33/47, 70%) or likely pathogenic/class 4 (14/47, 30%) (Figure 1C, Supplementary Table S4). For 27/36 (75%) informative cases, TP53 loss of heterozygosity in MB was reported, and an additional LPV/PV was observed in MB tissue of 2/36 (6%) cases (Figure 1D). The origin of the constitutional TP53 variant allele was known in 21/47 (45%) cases: 8/21 (38%) variants were of maternal origin, 4/21 (19%) of paternal origin, 1/21 (5%) was assigned to one of the parents without further information, and in 8/21 cases (38%) the TP53 PV/LPVs were identified as de novo (Figure 1E).
Based on the methylome profile, IHC or Nanostring 41/41 (100%) MBs detected in the informative cases were classified as SHH group (Supplementary Table S1). Methylome profiles were available for 28/41 (68%). Random forest-based DNA methylation class prediction successfully classified 10/28 tumors (36%) with a score of ≥ 0.9 (all “SHH_3”), while 18/28 received highest scores < 0.9 (range: 0.42–0.85), in line with previous reports of tumors from patients with CPSs not always being readily classifiable (i.e. scoring ≥ 0.9) by random forest-based DNA methylation class prediction.34 Of the tumors with highest scores < 0.9, 14/18 (78%) were classified as “SHH_3” (range: 0.42–0.85), giving a total of 24/28 (86%) “SHH_3” MB, (range: 0.42–0.99). Three of 18 tumors (17%) with scores < 0.9 were classified as “SHH_4,” and 1/18 (6%) as “SHH_2” (Figures 1F, G, Supplementary Table S5). Based on local histological reports, more than half of the MB (22/42, 52.4%) were large cell anaplastic (LCA), less frequent histological variants were classic (CMB, 8/42, 19%), desmoplastic/nodular (DMB, 4/42, 9.5%), LCA/DMB (2/42, 4.8%), MB with extensive nodularity (MBEN, 1/42, 2.4%), medulloepithelioma (MEPL, 1/42, 2.4%) and MB not otherwise specified (MB NOS, 4/42, 9.5%) (Table 1, Supplementary Table S1).
Information on CNVs provided by methylome profiling (27 informative cases) was used to identify the most recurrent focal and broad copy number alterations. Deletions were observed in the following genes: PTEN (13/27, 48%), SUFU (13/27, 48%), TP53 (10/27, 37%), LHX1 (9/27, 33%), and PTCH1 (8/27, 30%) and amplifications in GLI2 (8/27, 30%), MYCN (7/27, 26%), and CCND2 (4/27, 15%) (Supplementary Figure S1A and B). Additionally, we found loss of chromosomes 3p (12/27, 44%), 9q (8/27, 30%), 10q (15/27, 56%), 14q (8/27, 30%), 17p (23/27, 85%), and 3q (1/27, 4%) (Supplementary Figure S1B). An amplification of chromosome 3q was detected in 6/27 cases (22%) (Supplementary Figure S1B). These results are consistent with genomic findings from previous large-scale SHH cohorts.6,9,12
Survival of LFS MB Patients
The median follow-up for 46/47 informative patients was 21 months (range: 3–161.8 months), with median PFS, EFS and OS of 19, 19, and 28 months, respectively (Figure 2A). The 2 and 5y-PFS were estimated as 35.5% ± 7.6% and 20.3% ± 6.8%, 2 and 5y-EFS 32.8% ± 7.5% and 10.9% ± 5.1%, and 2 and 5y-OS 52.6% ± 8.0% and 22.6% ± 7.2%, respectively (Figure 2A). A small number (6/46; 13%) of long-term survivors with a PFS ≥ 60 months was observed (Supplementary Table S6). When compared with SHH-MB patients with a somatic TP53mut,13 the overall survival in LFS SHH-MB patients (40/46 informative cases) was not statistically different (p = 0.28; Supplementary Figure S2). No difference in PFS, EFS, or OS were detected when comparing resection (n = 44; Figure 2B), metastatic status (n = 46; Figure 2C) or clinical risk stratification (standard vs. high risk; n = 46; Supplementary Figure S3).
Figure 2:
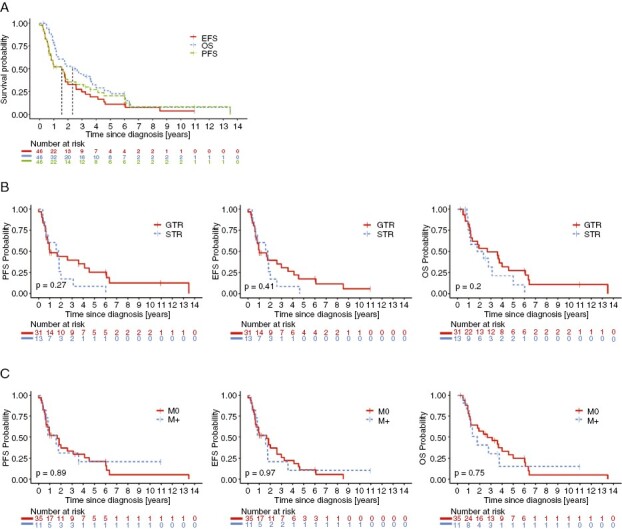
Survival of Li–Fraumeni syndrome (LFS) medulloblastoma (MB) patients: (A) Progression-free (PFS), event-free (EFS) and overall survival (OS) for LFS MB patients; (B) Impact of tumor resection radicality on survival of LFS MB patients (GTR—gross-total resection, STR—subtotal resection); (C) Impact of M-status on survival in LFS MB patients.
To assess the impact of the sequence of therapeutic elements, we analyzed the survival according to radiotherapy (RT) application (immediate post-operative RT: n = 26, chemotherapy before RT: n = 16, or no RT: n = 4; 46 informative cases). No significant survival difference was noted between application of immediate postoperative RT followed by chemotherapy (2y-PFS of 43.9% ± 10.5%, 2y-EFS of 43.9% ± 10.5% and 2y-OS of 59.7% ± 10.4%) and chemotherapy before RT (2y-PFS of 32.1% ± 12.6%, 2y-EFS of 24.1% ± 11.7% and 2y-OS of 47.6% ± 13.9%) (Figure 3A, Supplementary Table S7). Overall, 2y-PFS in irradiated patients was estimated for 39.3% ± 8.2%, 2y-EFS for 36.3% ± 8.1% and 2y-OS for 55.5% ± 8.4%. Patients who received postoperative RT showed significantly better outcome in terms of PFS, EFS or OS than patients who received no RT (2y-OS of 25% ± 21.1%, p < 0.05, 2y-PFS and EFS not estimated). Patients who received chemotherapy before RT compared to the group without RT showed significantly better OS without differences in PFS and EFS (Supplementary Table S7). It is noteworthy that of the four patients who did not receive RT, two did not receive any therapy with curative intent, based on parents’ decision. Craniospinal irradiation (CSI) dose did not influence patient survival (Supplementary Fig. S4).
Figure 3:

Impact of treatment modalities on survival of Li-Fraumeni syndrome (LFS) medulloblastoma (MB) patients: (A) Impact of relative chemotherapy (CT) and radiotherapy (RT) timepoints on survival of LFS MB patients; (B) Impact of overall chemotherapy intensity on survival of LFS MB patients (HI—high-intensity, SI—standard-intensity); (C) Impact of overall chemotherapy intensity on survival of LFS MB patients with distinguished PNET HR+ protocol.
High-intensity (HI) chemotherapy is commonly used in high-risk MB, but evidence of superior efficacy in LFS MB is lacking. Among all informative cases (46/47) 5/46 (10.9%) patients received no chemotherapy. Two of the five patients (LFS-MB13, LFS-MB19) who received no chemotherapy did not receive any therapy with curative intent. The remaining three patients (LFS-MB8, LFS-MB39, LFS-MB43) received photon RT only as a primary treatment, two of which were treated with chemotherapy once diagnosed with a subsequent malignancy (LFS-MB39) or a MB relapse (LFS-MB43). 30/46 (65.2%) patients received one of the chemotherapy protocols defined as standard intensity (SI), 11/46 (23.9%) patients received chemotherapy protocols defined as HI (Supplementary Tables S1 and S3). Overall, median PFS for no, SI- and HI-chemotherapy were 7.2, 10.7, and 18.8 months, respectively, and median OS 8, 15, and 33 months, respectively. No statistically significant difference in PFS, EFS or OS was noted between SI and HI, with a 2y-PFS of 35.2% ± 9.7% and 42.4% ± 15.6%, a 2y-EFS was 30.8% ± 9.4% and 42.4 ± 15.6%, and a 2y-OS 53.1% ± 10% and 67.5% ± 15.5%, respectively (Figure 3B, Supplementary Table S7). Nine out of 46 (19.6%) patients received a PNET HR + 5 chemotherapy,35 constituting one of the largest homogenously treated subgroups. Analysis of the patients treated with the PNET HR + 5 protocol as a separate group showed no statistically significant difference in comparison with other patient subgroups (Figure 3C, Supplementary Table S7). For patients who did not receive chemotherapy 2y-PFS, -EFS and -OS were estimated for 20% ± 17.9% because of one long-term survivor (LFS-MB39) in this group.
Impact of different treatment modalities, resection and metastatic staging was also evaluated in multivariate Cox regression analysis for PFS, EFS and OS (Supplementary Figure S5). The patient subgroup that did not receive RT had significantly inferior PFS (HR: 5.6, 95% CI: 1.69–18.6, p = 0.005), EFS (HR: 5.3, 95% CI: 1.62–17.6, p = 0.006) and OS (HR: 5.56, 95% CI: 1.67–18.5, p = 0.005) compared with the subgroup that received RT with or without maintenance chemotherapy. The remaining parameters did not show a statistically significant effect on patient survival. Supplementary Figure S6 presents LFS MB patient outcome with a subdivision into different therapeutic modalities.
In our cohort, 34/47 (72.3%) patients died during the observation period and the reason of death was known in 33/34 (97.1%) cases: 27/33 (81.8%) deaths were due to MB and in 6/33 (18.2%) subsequent malignancy (Table 1, Supplementary Table S1). The 2-year cumulative incidence of MB-related mortality was 63%, whereas for subsequent neoplasms the 2-year cumulative incidence was estimated for 6% (Supplementary Figure S7).
Recurrence Patterns
9/46 (20%) of tumors were refractory to treatments (best response: PD). In patients for whom the disease was not refractory (SD, PR or CR, 37/46, 80%) 21/37 (57%) experienced a recurrence (Figure 4A). By initial metastasis staging at MB diagnosis, a recurrence occurred in 15/28 (54%) M0 patients and in 6/9 (67%) M+ patients (Figure 4B). M0 patients had mostly local recurrences (57%), whereas M+ patients predominantly showed metastatic (33%) and combined (local plus metastatic; 33%) recurrences (Figure 4C, Fisher’s exact test: n.s.). Recurrences were detected during (3/21 informative cases; 14%) and after treatment (18/21 informative cases; 86%). Out of 36/37 (97%) patients who received RT, 20 (56%) experienced a recurrence. One patient who did not receive RT died from an early disease recurrence (Figure 4D). The majority of recurrences (18/19 informative cases; 95%) occurred inside the RT field (Figure 4E). With regard to extent of tumor resection, 13/25 (52%) patients with GTR had recurrence, whereas in the STR subgroup recurrence occurred in 8/11 (73%) cases (Figure 4F, Fisher’s exact test: n.s.).
Figure 4:

Medulloblastoma (MB) recurrence pattern in Li-Fraumeni syndrome (LFS) patients: (A) Recurrence frequency; (B) Impact of metastasis (M)-status on recurrence frequency; (C) Recurrence pattern according to M-status; (D) Impact of radiation treatment (RT) on recurrence frequency; (E) Recurrence location in relation to the RT field; F: Impact of resection staging on recurrence frequency (GTR—gross-total resection, STR—subtotal resection).
Other Malignancies Diagnosed in LFS MB Patients
Data on subsequent malignancies was available for 46/47 (98%) cases. Within the observation time, at least one subsequent malignancy occurred in 9/46 (19.6%) patients and was the reason of death in 6/46 (13%) (Supplementary Table S1). All 9 patients were treated with CSI, and 8/9 received heterogeneous chemotherapy treatment before a subsequent malignancy was detected (Table 2). The median time to subsequent malignancy was 41 months (range: 21–102.2 months).
Table 2.
Non-medulloblastoma malignancies diagnosed in LFS MB patients
| LFS MB patients with subsequent malignancies | |||||
|---|---|---|---|---|---|
| Patient ID | Subsequent malignancy | Time to subsequent malignancy [months after MB diagnonis] | Treatment of subsequent malignancy | Death of subsequent malignancy [months after MB diagnosis] | Chemotherapy protocol in primary MB treatment |
| LFS-MB2 | MDS-AML* | 31.1 | allogenic stem cell transplant | 37.4 | cisplatin/CCNU/VCR maintenance |
| LFS-MB3 | 1) rhabdoid tumour# 2) myelodysplastic syndrome* |
1) 35.8 2) 45 |
1) RHABDOID2007 2) allogenic stem cell transplant |
76.2 | cisplatin/CCNU/VCR maintenance |
| LFS-MB16 | Anaplastic astrocytoma III grade* | 73 | surgical resection | 73 | cisplatin/CCNU/VCR maintenance |
| LFS-MB30 | t-AML* | 37 | all-stem cell transplant | 44 | SJMB03 |
| LFS-MB35 | Supratentorial anaplastic astrocytoma* | 21 | stereotactic radiotherapy | No | PNET 5 SHH TP53 arm |
| LFS-MB39 | Angiosarcoma# | 102.2 | UNK | 161.8 | NA |
| LFS-MB41 | Osteosarcoma# | 55.6 | UNK | 72.2 | PNET HR + 5 |
| LFS-MB45 | Malignant peripheral nerve sheath tumor* | 49.5 | UNK | 55 (died of MB) | PNET HR + 5 |
| LFS-MB47 | IDH wildtype GBM* | 36 | resection (unknown extent) | No | ACNS0332 |
| LFS MB patients with malignancies diagnosed before MB | |||||
|---|---|---|---|---|---|
| Patient ID | Malignancy diagnosed prior to MB | [years before MB diagnosis] | Treatment of other malignancy | ||
| LFS-MB1 | Mediastinal T-cell non-Hodgkin lymphoma | 8 | ALL97 regimen B protocol | ||
| LFS-MB4 | Wilms tumour | 4 | no information | ||
| LFS-MB7 | Osteosarcoma | 1 | no information | ||
| LFS-MB20 | Pre-B-ALL | 3 | ALL-BFM-2000 regimen | ||
| LFS-MB27 | Osteosarcoma | 1 | COG AOST0331 protocol | ||
Abbreviations: LFS, Li–Fraumeni Syndrome; MB, medulloblastoma; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia; GBM, glioblastoma multiforme; CCNU, lomustine; VCR, vincristine; B-ALL, B-cell acute lymphoblastic leukemia; NA, not applicable; UNK, unknown.
* Inside craniospinal irradiation field, # outside craniospinal irradiation field.
Malignancies prior to MB diagnosis were reported in 5/46 (10.9%) patients (Table 2, Supplementary Table S1). Furthermore, our cohort also includes a patient who in addition to LFS-associated MB was also diagnosed with Fanconi Anemia (LFS-MB23, Supplementary Table S1, no information on the sequence of symptoms).
Family History of Cancer
Information about the family history of cancer were available in 33/47 (70.2%) cases, and cancer in one or more patients’ relatives were reported in 21/33 (63.6%) cases (Supplementary Table S1). Two of 42 patients in our cohort (LFS-MB1 and LFS-MB10, Supplementary Table S1) were diagnosed with MB during LFS-related surveillance.
Discussion
LFS is a relatively rare genetic disorder, thus, the number of LFS-associated MB cases in the general population is very low. We managed to collect information on 47 LFS-MB patients diagnosed in the years 1987–2022 from 17 countries. While this represents the largest cohort of pediatric LFS-MB patients that we are aware of, it still remains a relatively small cohort, potentially posing a challenge for statistical analyses. This series first confirms that SHH-MB with constitutional TP53 alterations have a much poorer prognosis compared to TP53wt SHH-MB.10,11 Several recent large prospective trials, ACNS0331,36 for average risk MB (R ≤ 1.5 cm2, M0, and no LCA), SJMB03,37 investigating for both average and high-risk MB, and ACNS033238 for high-risk MB (R > 1.5 cm2, M+ or LCA), have reported on survival data of MB with TP53 mutations. Of note, no separate detailed survival data on bona fide LFS-MB, i.e. SHH MB with constitutional TP53 mutations was reported in these trials. 5-year EFS for TP53 mutated MBs (mostly SHH) ranged between 14.3% (ACNS0331), 25% (SJMB03) and 35.7% (ACNS0332). While the cohorts (only standard or only high-risk vs. both) and the DNA analyzed (only somatic vs. constitutional) differed, the survival reported in those studies was comparable to our data with a 5-year EFS of 20%.
LFS MB are considered high-risk and consequently often treated using protocols including HI chemotherapy and stem cell rescue. However, our results showed that treatment including HI chemotherapy does not appear to improve survival of LFS-MB patients compared to SI chemotherapy. These results still need to be interpreted with caution, as this may not be independent of additional factors such as residual tumor and/or metastasis. But a major concern in long-term LFS-MB survivors remains subsequent malignancies,20–22,39 which can be related to the burden of mutagenic chemotherapies. This may weigh in the benefit/risk balance against HI.
Since RT may also lead to radiation-induced malignancies, avoidance or strict limitation of radiation is considered in many malignancies associated with LFS.20–22,39,40 In our cohort, patients who received RT during primary MB treatment had significantly better outcomes than the RT-untreated group. Therefore, our series does not support avoidance of RT in LFS-related MB. However, it should be noted that 1) two out of four patients without RT received no therapy with curative intent, and 2) our data on RT doses is insufficient to draw conclusions on RT-dosage-subsequent tumor dependencies.
Ten subsequent malignancies were detected in nine patients with a wide range of histologies. Similar to the general LFS population,22,39,41 we reported various types of sarcomas; the cancer spectrum also included AML and brain tumors (astrocytomas and glioblastoma), which are rarely described as subsequent neoplasms in LFS patients.22,39,41 Gajjar et al. reported on the LFS patient with SHH-MB in whom the high-grade glioma was observed as a second malignancy.37 Overall, the incidence rate (20%) of and mortality rate from subsequent malignancies (18% for entire cohort and 67% for cases with subsequent malignancies) in LFS MB patients were similar to the general LFS population (15-48% and 18%, respectively).22,41
Aside from subsequent neoplasms, LFS is characterized also by higher recurrence probability.21,22,39 In our study, MB recurrence was observed in the majority (57%) of cases, which is similar to the relapse frequency in the general TP53mut SHH-MB patient population (58.5-100%)9,10,36 and significantly higher than the relapse frequency for all SHH-MBs (28%).10,11,42 Altogether, since deaths due to subsequent malignancies occurred in general later and less frequently than MB-related deaths (Supplementary Figure S7), effectively treating the primary MB disease should be prioritized.
Considering all the aforementioned risks affecting LFS patients, the importance of genetic testing in children with SHH MB needs to be emphasized. Previous studies showed that up to 12.1% of childhood cancers can be associated with an underlying CPS.13,43–45 The updated Chompret criteria currently used for LFS diagnosis have limited sensitivity and specificity, especially in case of constitutional TP53 mutations arising de novo.24,46,47 Their frequency in the general population of LFS patients was estimated for 7–20%.48 However, in some cancer entities, the percentage of de novo TP53 mutations might be significantly higher, as exemplified by this study (38% of all informative cases) or a LFS osteosarcoma cohort (46%).49 A systematic screening for constitutional TP53 mutations in SHH MB is therefore justified.
In addition to the small cohort size, our study has several limitations to be considered when interpreting the results: first, the retrospective nature of the data presented carries an inherent risk of bias, potentially selective as well as confounding in nature. Second, the heterogenous nature of the treatments applied may limit the strength of the conclusions drawn. And third, due to the lack of systematic genetic testing for constitutional TP53 mutations in the past, as well as the nonpopulation-based cohort design, the number of cases in this cohort certainly does not reflect the true prevalence of LFS-associated MB. We propose to address these issues by systematic and prospective data collection within a study including homogenous treatments, such as the SHH-TP53 arm in the SIOP PNET5 trial (NCT02066220). Last, the conclusions can be also affected by the relatively short median follow-up time of 21 months that might be partially attributed to poor overall survival of LFS patients (28 months).
Finally, prognostic stratification of LFS-MB based on additional molecular features is highly desirable for future clinical trials. The lack of comprehensive molecular profiling in our cohort precludes the identification of such features e.g. in cases refractory to standard of care treatment, and will need to be identified in future studies.
In conclusion, our data confirms the dismal prognosis of LFS MB patients, and treating the primary MB disease remains the first priority. Further efforts should be focused on prospectively gathering clinical data on LFS MB cases to complement and verify previous conclusions. Our findings strongly support the need for exploration of alternative therapeutic approaches.
Supplementary Material
Acknowledgements
We thank Pierre Leblond, Ludovic Mansuy, Emilie de Carli, Olivier Ingster, Marion Gauthier-Villars, Isabelle Mortemousque, Gudrun Schleiermacher, Birgit Geoerger, Tiphaine Adam de Baumais, Laurence Brugieres, Anne-Isabelle Bertozzi, Sophie Julia, Céline de Bouyn-Icher, Marjolaine Willems, Gilles Paluenzela, Maria-Luisa Garrè, Valeria Capri, and the Associazione per la ricerca sui tumori cerebrali del bambino (ARTUCEBA) for discussion and provision of information.
Contributor Information
Anna S Kolodziejczak, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany.
Lea Guerrini-Rousseau, Department of Children and Adolescents Oncology, Gustave Roussy, Université Paris-Saclay, 94805 Villejuif, France; Molecular Predictors and New Targets in Oncology, Inserm U981 Team “Genomics and Oncogenesis of pediatric Brain Tumors,” Gustave Roussy, Université Paris-Saclay, Villejuif, France.
Julien Masliah Planchon, Department of Diagnostic and Theranostic Medicine, Somatic Genetics Unit, Institut Curie, Paris-Science Lettres University, Paris, France.
Jonas Ecker, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Florian Selt, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Martin Mynarek, Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Mildred Scheel Cancer Career Center HaTriCS4, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Denise Obrecht, Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Martin Sill, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Robert J Autry, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Eric Stutheit-Zhao, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Princess Margaret Cancer Centre, Toronto, Ontario, Canada.
Steffen Hirsch, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Elsa Amouyal, SIREDO Pediatric Oncology Center, Institut Curie, Paris-Science Lettres University, Paris, France.
Christelle Dufour, Department of Children and Adolescents Oncology, Gustave Roussy, Université Paris-Saclay, 94805 Villejuif, France; Molecular Predictors and New Targets in Oncology, Inserm U981 Team “Genomics and Oncogenesis of pediatric Brain Tumors,” Gustave Roussy, Université Paris-Saclay, Villejuif, France.
Olivier Ayrault, Institut Curie, PSL Research University, CNRS UMR, INSERM, Orsay, France Université Paris Sud, Université Paris-Saclay, CNRS UMR 3347, INSERM U1021, Orsay, France.
Jacob Torrejon, Institut Curie, PSL Research University, CNRS UMR, INSERM, Orsay, France Université Paris Sud, Université Paris-Saclay, CNRS UMR 3347, INSERM U1021, Orsay, France.
Sebastian M Waszak, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo and Oslo University Hospital, Oslo, Norway; Department of Neurology, University of California, San Francisco, CA, USA.
Vijay Ramaswamy, Division of Neurosurgery, Program in Developmental and Stem Cell Biology, Arthur and Sonia Labatt Brain Tumour Research Centre, Hospital for Sick Children, Toronto, Ontario, Canada; Division of Hematology and Oncology, Hospital for Sick Children, Toronto, Ontario, Canada.
Virve Pentikainen, Division of Hematology-Oncology and Stem Cell Transplantation, Children’s Hospital, Helsinki University Hospital, Helsinki, Finland.
Haci Ahmet Demir, Department of Pediatric Hematology-Oncology, Private Memorial Ankara Hospital, Ankara, Turkey.
Steven C Clifford, Wolfson Childhood Cancer Research Centre, Newcastle University Centre for Cancer, Newcastle upon Tyne, United Kingdom.
Ed C Schwalbe, Wolfson Childhood Cancer Research Centre, Newcastle University Centre for Cancer, Newcastle upon Tyne, United Kingdom; Applied Sciences, Northumbria University, Newcastle upon Tyne, United Kingdom.
Luca Massimi, Pediatric Neurosurgery, Fondazione Policlinico Universitario A. Gemelli IRCCS, Catholic University Medical School, Rome, Italy.
Matija Snuderl, Department of Pathology, New York University Langone Health, New York City, NY, USA.
Kristyn Galbraith, Department of Pathology, New York University Langone Health, New York City, NY, USA.
Matthias A Karajannis, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York City, NY, USA.
Katherine Hill, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York City, NY, USA.
Bryan K Li, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York City, NY, USA.
Mike Walsh, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York City, NY, USA.
Christine L White, Victorian Clinical Genetics Services, Parkville, Australia; Hudson Institute of Medical Research, Clayton, Australia; Department of Molecular and Translational Science, Monash University, Clayton, Australia.
Shelagh Redmond, Childhood Cancer Registry, Institute of Social and Preventive Medicine, University of Bern, Bern, Switzerland.
Loizou Loizos, Pediatric Oncology/Hematology/Immunology at the Medical School of the University of Nicosia, Nicosia, Cyprus.
Marcus Jakob, Department of Paediatric Haematology, Oncology and Stem-Cell Transplantation, University Hospital Regensburg, Regensburg, Germany.
Uwe R Kordes, Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Irene Schmid, Paediatric Haematology and Oncology, Dr. von Hauner Children’s Hospital, Ludwig Maximilians University Munich, Munich, Germany.
Julia Hauer, Pediatric Haematology and Oncology, University Hospital Carl Gustav Carus, Dresden, Germany.
Claudia Blattmann, Paediatric Haematology, Oncology and Immunology, Olgahospital, Klinikum Stuttgart, Stuttgart, Germany.
Maria Filippidou, Division of Pediatric Hematology-Oncology, First Department of Pediatrics, National and Kapodistrian University of Athens, “Aghia Sophia” Children’s Hospital, Athens, Greece.
Gianluca Piccolo, Neuro-Oncology Unit, IRCCS Istituto Giannina Gaslini, Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genoa, Genoa, Italy.
Wolfram Scheurlen, Paediatric Haematology and Oncology, Cnopfsche Paediatric Clinic, Nurnberg, Germany.
Ahmed Farrag, Department of Paediatric Haematology, Oncology and Stem-Cell Transplantation, Paediatric Clinic, University Hospital Aachen, Aachen, Germany; Department of Pediatric Oncology, South Egypt Cancer Institute, Assiut University, Egypt.
Kerstin Grund, Institute of Human Genetics, University Hospital Heidelberg, Heidelberg, Germany.
Christian Sutter, Institute of Human Genetics, University Hospital Heidelberg, Heidelberg, Germany.
Torsten Pietsch, Institute of Neuropathology, DGNN Brain Tumor Reference Center, University of Bonn Medical Center, Bonn, Germany.
Stephan Frank, Division of Neuropathology, Institute of Pathology, Basel University Hospital, Basel, Switzerland.
Denis M Schewe, Department of Pediatrics, Otto-von-Guericke University Magdeburg, Magdeburg, Germany.
David Malkin, Division of Hematology/Oncology, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada.
Myriam Ben-Arush, Pediatric Hematology Oncology, Rambam Medical Center, Haifa, Israel.
Astrid Sehested, Department of Paediatrics and Adolescent Medicine, Juliane Marie Centre, Copenhagen University Hospital, Copenhagen, Denmark.
Tai-Tong Wong, Division of Pediatric Neurosurgery, Department of Neurosurgery, Taipei Medical University Hospital, Taipei Medical University, Taipei 110, Taiwan.
Kuo-Sheng Wu, Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan.
Yen-Lin Liu, Department of Pediatrics, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan.
Fernando Carceller, Paediatric and Adolescent Oncology Drug Development Team, Children and Young People’s Unit, The Royal Marsden NHS Foundation Trust and The Institute of Cancer Research, Sutton, United Kingdom.
Sabine Mueller, Department of Neurology, Neurosurgery and Pediatrics, University of California, San Francisco, USA.
Schuyler Stoller, Department of Neurology, University of California, San Francisco, USA.
Michael D Taylor, Division of Neurosurgery, Program in Developmental and Stem Cell Biology, Arthur and Sonia Labatt Brain Tumour Research Centre, Hospital for Sick Children, Toronto, Ontario, Canada.
Uri Tabori, Division of Hematology and Oncology, Hospital for Sick Children, Toronto, Ontario, Canada.
Eric Bouffet, Division of Hematology and Oncology, Hospital for Sick Children, Toronto, Ontario, Canada; The Arthur and Sonia Labatt Brain Tumour Research Centre, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; Division of Haematology/ Oncology, The Hospital for Sick Children, Toronto, Ontario, Canada.
Marcel Kool, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands.
Felix Sahm, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, and CCU Neuropathology, German Cancer Institute (DKF), Heidelberg, Germany.
Andreas von Deimling, Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, and CCU Neuropathology, German Cancer Institute (DKF), Heidelberg, Germany.
Andrey Korshunov, Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, and CCU Neuropathology, German Cancer Institute (DKF), Heidelberg, Germany.
Katja von Hoff, Department of Pediatric Oncology and Hematology, Charité – Universitätsmedizin Berlin, Berlin, Germany; Department of Pediatrics and Adolescent Medicine, Aarhus University Hospital, Aarhus, Denmark.
Christian P Kratz, Pediatric Hematology and Oncology, Hannover Medical School, Hannover, Germany.
Dominik Sturm, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany; Pediatric Glioma Research, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
David T W Jones, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Pediatric Glioma Research, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Stefan Rutkowski, Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Cornelis M van Tilburg, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Olaf Witt, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Gaëlle Bougeard, Univ Rouen Normandie, Inserm U1245 and CHU Rouen, Department of Genetics, F-76000 Rouen, France.
Kristian W Pajtler, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Stefan M Pfister, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany; Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Franck Bourdeaut, SIREDO Pediatric Oncology Center, Institut Curie, Paris-Science Lettres University, Paris, France.
Till Milde, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; CCU Pediatric Oncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; National Center for Tumor Diseases (NCT), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
Funding
Bundesministerium für Bildung und Forschung ADDRess (01GM1909A, 01GM1909E to C.P.K., S.M.P., T.M.); Cancer Research UK to S.C.C. and E.C.S.; Physician Scientist Program of the Medical Faculty, University Heidelberg to JE; National Institutes of Health/ National Cancer Institute Cancer Center Support Grant (P30 CA008748 to M.A.K., K.H., B.K.L., M.W.); Research Council of Norway [187615], the University of Oslo, the South-Eastern Norway Regional Health Authority to S.M.W.; German Children Cancer Foundation grants (DKS 2006.03, 2009.19, 2011.01, and 2017.14. to T.P.); Deutsche Kinderkrebsstiftung (DKS 2021.25 to C.P.K., S.M.P.); Saint Baldrick’s Robert J.Arceci Innovation Award to O.A.; Deutsche Kinderkrebsstiftung to S.Ru.; Swiss Federal Office of Public Health to S. Re.; Cancer Australia, the Robert Connor Dawes Foundation, Carrie’s Beanies for Brain Cancer, the Victorian Government’s Operational Infrastructure Support Program to C.L.W.; Royal Marsden Cancer Charity (RMCC), Royal Marsden National Health Service Foundation Trust from the Hall-Hunter Foundation via the RMCC to F.C.; CIBC Children’s Foundation Chair in Child Health Research to D.M. Grants from the Friedberg Charitable Foundation, Making Headway Foundation and by NIH grants R56-NS122987, R01-NS122987 to M.S.
Conflict of Interest
O.W. and T.M. received research funding from Biomed Valley and Day One Therapeutics. C.M.v.T. participated in advisory boards for Novartis, Alexion and Bayer. U. K. participated in advisory board for Novartis and GBT. S.R. participated in advisory boards for Bayer, BMS, Novartis, and Roche, and in a DMSC for Cellgene. M.A.K. received research funding from Y-mAbs Therapeutics. M.S. is scientific advisor and shareholder of C2i Genomics and Halo Dx and received research funding from Lilly USA.
Authorship
Conceptualization: S.M.P., F.B., T.M.
Data generation: J.M.P., S.C.C., E.C.S., M.Sn., F.Sa., S.M.P.
Data analysis: A.S.K., L.G.-R., J.M.P., O.A., J.T.D., M. Si., R.J.A., E.S.-Z., M.M., M.W., F.B., T.M.
Writing of original draft: A.S.K., F.B., T.M.
Acquisition of patient samples and relevant clinical data: L.G-R, F.Se., M.M., D.O., S.H., S.M.W., V.R., V.P., H.A.D., S.C.C., E.C.S., L.M., M.Sn., K.Ga., M.A.K., K.H., B.L., C.L.W., S.Re., L.L., M.J., U.Kor., I.S., J.H., C.B., M.F., W.S., U.Kon., K.Gr., C.S., T.P., S.F., D.M.S., D.M., M.B.-A., A.M.S., Y.-L.L., T.-T.W., K.-S.W., F.C., S.M., S.S., M.D.T., U.T., E.B., M.K., F.Sa., A.K., K.V.H., D.S., D.T.W.J., G.B., F.B.
Writing, review, and editing: all authors;
Supervision: F.B., T.M.
This study was presented in part during the German GPOH meeting 2021 (10.12.2021) and the 20th International Symposium on Pediatric Neuro-Oncology (12-15.06.2022).
References
- 1. Johnson KJ, Cullen J, Barnholtz-Sloan JS, et al. Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev. 2014;23(12):2716–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18(7):958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia-Lopez J, Kumar R, Smith KS, Northcott PA.. Deconstructing sonic hedgehog medulloblastoma: molecular subtypes, drivers, and beyond. Trends Genet. 2021;37(3):235–250. [DOI] [PubMed] [Google Scholar]
- 9. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28(8):1345–1350. [DOI] [PubMed] [Google Scholar]
- 11. Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kool M, Jones DT, Jager N, et al. ; ICGC PedBrain Tumor Project. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Waszak SM, Northcott PA, Buchhalter I, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018;19(6):785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfaff E, Remke M, Sturm D, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010;28(35):5188–5196. [DOI] [PubMed] [Google Scholar]
- 15. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1-2):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lindsey JC, Hill RM, Megahed H, et al. TP53 mutations in favorable-risk Wnt/Wingless-subtype medulloblastomas. J Clin Oncol. 2011;29(12):e344–6; author reply e347. [DOI] [PubMed] [Google Scholar]
- 17. Goschzik T, Mynarek M, Doerner E, et al. Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol. 2022;144(6):1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wen PY, Packer RJT.. 2021 WHO Classification of Tumors of the Central Nervous System: clinical implications. Neuro Oncol. 2021;23(8):1215–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131(6):821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Limacher JM, Frebourg T, Natarajan-Ame S, Bergerat JP.. Two metachronous tumors in the radiotherapy fields of a patient with Li-Fraumeni syndrome. Int J Cancer. 2001;96(4):238–242. [DOI] [PubMed] [Google Scholar]
- 21. Heymann S, Delaloge S, Rahal A, et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol. 2010;5:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hendrickson PG, Luo Y, Kohlmann W, et al. Radiation therapy and secondary malignancy in Li-Fraumeni syndrome: a hereditary cancer registry study. Cancer Med. 2020;9(21):7954–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ballinger ML, Mitchell G, Thomas DM.. Surveillance recommendations for patients with germline TP53 mutations. Curr Opin Oncol. 2015;27(4):332–337. [DOI] [PubMed] [Google Scholar]
- 24. Kratz CP, Achatz MI, Brugières L, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. 2017;23(11):e38–e45. [DOI] [PubMed] [Google Scholar]
- 25. Frebourg T, Bajalica Lagercrantz S, Oliveira C, et al. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet. 2020;28(10):1379–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Villani A, Shore A, Wasserman JD, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016;17(9):1295–1305. [DOI] [PubMed] [Google Scholar]
- 27. Schuler N, Palm J, Schmitz S, Lorat Y, Rübe CE.. Increasing genomic instability during cancer therapy in a patient with Li-Fraumeni syndrome. Clin Transl Radiat Oncol. 2017;7:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thariat J, Chevalier F, Orbach D, et al. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol. 2021;22(12):e562–e574. [DOI] [PubMed] [Google Scholar]
- 29. Fortuno C, Lee K, Olivier M, et al. ; ClinGen TP53 Variant Curation Expert Panel. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat. 2021;42(3):223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olshen AB, Venkatraman ES, Lucito R, Wigler M.. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–572. [DOI] [PubMed] [Google Scholar]
- 32. Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125(6):913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sturm D, Capper D, Andreiuolo F, et al. Multiomic neuropathology improves diagnostic accuracy in pediatric neuro-oncology. Nat Med. 2023;29(4):917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dufour C, Foulon S, Geoffray A, et al. Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro Oncol. 2021;23(7):1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Michalski JM, Janss AJ, Vezina LG, et al. Children’s oncology group phase III trial of reduced-dose and reduced-volume radiotherapy with chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2021;39(24):2685–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gajjar A, Robinson GW, Smith KS, et al. Outcomes by clinical and molecular features in children with medulloblastoma treated with risk-adapted therapy: results of an International Phase III Trial (SJMB03). J Clin Oncol. 2021;39(7):822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leary SES, Packer RJ, Li Y, et al. Efficacy of carboplatin and isotretinoin in children with high-risk medulloblastoma: a randomized clinical trial from the Children’s Oncology Group. JAMA Oncol. 2021;7(9):1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le AN, Harton J, Desai H, et al. Frequency of radiation-induced malignancies post-adjuvant radiotherapy for breast cancer in patients with Li-Fraumeni syndrome. Breast Cancer Res Treat. 2020;181(1):181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bahar M, Kordes U, Tekautz T, Wolff J.. Radiation therapy for choroid plexus carcinoma patients with Li-Fraumeni syndrome: advantageous or detrimental? Anticancer Res. 2015;35(5):3013–3017. [PubMed] [Google Scholar]
- 41. Hisada M, Garber JE, Fung CY, Fraumeni JF, Li FP.. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–611. [DOI] [PubMed] [Google Scholar]
- 42. Hill RM, Richardson S, Schwalbe EC, et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: a multicentre cohort study. Lancet Child Adolesc Health. 2020;4(12):865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ripperger T, Bielack SS, Borkhardt A, et al. Childhood cancer predisposition syndromes-a concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A. 2017;173(4):1017–1037. [DOI] [PubMed] [Google Scholar]
- 44. Nguyen TMK, Behnert A, Pietsch T, Vokuhl C, Kratz CP.. Proportion of children with cancer that have an indication for genetic counseling and testing based on the cancer type irrespective of other features. Fam Cancer. 2021;20(4):273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ecker J, Selt F, Sturm D, et al. Molecular diagnostics enables detection of actionable targets: the Pediatric Targeted Therapy 2.0 registry. Eur J Cancer. 2022;180:71–84. [DOI] [PubMed] [Google Scholar]
- 46. Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol. 2015;33(21):2345–2352. [DOI] [PubMed] [Google Scholar]
- 47. Coffee B, Cox HC, Bernhisel R, et al. A substantial proportion of apparently heterozygous TP53 pathogenic variants detected with a next-generation sequencing hereditary pan-cancer panel are acquired somatically. Hum Mutat. 2020;41(1):203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet. 2009;46(10):689–693. [DOI] [PubMed] [Google Scholar]
- 49. Diessner BJ, Pankratz N, Hooten AJ, et al. Nearly half of TP53 germline variants predicted to be pathogenic in patients with osteosarcoma are de novo: A report from the children’s oncology group. JCO Precis Oncol. 2020;4:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.